

Abstract
The clinical phenotype of human dilated cardiomyopathy (DCM) encompasses a broad spectrum of etiologically distinct disorders. As targeting of etiology-related pathogenic pathways may be more efficient than current standard heart failure treatment, we obtained the genomic expression profile of a DCM subtype characterized by cardiac inflammation to identify possible new therapeutic targets in humans. In this inflammatory cardiomyopathy (DCMi), a distinctive cardiac expression pattern not described in any previous study of cardiac disorders was observed. Two significantly altered gene networks of particular interest and possible interdependence centered around the cysteine-rich angiogenic inducer 61 (CYR61) and adiponectin (APN) gene. CYR61 overexpression, as in human DCMi hearts in situ, was similarly induced by inflammatory cytokines in vascular endothelial cells in vitro. APN was strongly downregulated in DCMi hearts and completely abolished cytokine-dependent CYR61 induction in vitro. Dysbalance between the CYR61 and APN networks may play a pathogenic role in DCMi and contain novel therapeutic targets. Multiple immune cell-associated genes were also deregulated (e.g., chemokine ligand 14, interleukin-17D, nuclear factors of activated T cells). In contrast to previous investigations in patients with advanced or end-stage DCM where etiology-related pathomechanisms are overwhelmed by unspecific processes, the deregulations detected in this study occurred at a far less severe and most probably fully reversible disease stage.
Electronic supplementary material
Supplementary material is available in the online version of this article at http://dx.doi.org/10.1007/s00109-006-0122-9 and is accessible for authorized users.
Keywords: Human inflammatory cardiomyopathy, Genomic expression profiling, Molecular therapeutic targets, Molecular pathomechanisms, Dilated cardiomyopathy, Heart failure
Introduction
The term dilated cardiomyopathy (DCM) describes a cardiac phenotype characterized by dilation and dysfunction of the ventricles. From an etiologic perspective, DCM encompasses a spectrum of disease entities, some of which are due to monogenic defects in cardiac expressed genes [1], others triggered by exogenous factors including cardiotropic viruses [2]. Targeting of etiology-related pathogenic pathways in etiologically distinct DCM subtypes may be more efficient than current standard treatment protocols employed in heart failure of any origin. One important DCM subtype characterized by cardiac inflammation is designated as inflammatory cardiomyopathy (DCMi), often found in conjunction with cardiac viral infections. Spontaneous elimination of cardiac viruses was associated with significant hemodynamic improvement for various viruses [3], and interferon-β (IFN-β) therapy was useful to promote virus elimination and improve cardiac function and clinical status [4]. A search for new therapeutic targets in DCMi seems worthwhile due to the high prevalence of this condition and its still unsatisfactory long-term outcome. The current study combined in vivo and in vitro methods to identify possible DCMi-related gene regulatory networks that might be exploited to expand the therapeutic repertoire beyond the standard heart failure drugs currently used. The study does not address individual gene deregulations, but searches for disturbances of regulatory gene networks reflected by anomalous cardiac expression patterns. Genomic screening strategies require precise phenotyping of the study groups, as this will minimize interindividual differences and increase the likelihood of revealing distinctive pathomechanisms. Because, in advanced stages of DCM, such processes are most likely overwhelmed by unspecific reactions of the failing heart long after the initiating process has burnt out, we investigated patients with moderately severe disease in contrast to most previous microarray work in the field [5]. The standard clinical characterization was refined by endomyocardial biopsy (EMB)-based immunohistological and molecular virological analyses. In contrast to multiple fields of cancer research, restrictive disease phenotyping of this kind is rarely employed in cardiovascular research, mostly due to the difficult access to affected cardiovascular tissues in humans in vivo. The microarray part of our study was entirely based on RNA isolated from EMBs thus providing an approach suitable for follow-up investigations of clinical courses in relation to cardiac expression profiles. We report on a distinctive cardiac expression pattern in DCMi and two significantly altered gene networks centered around the cysteine-rich angiogenic inducer 61 (CYR61) and adiponectin (APN) gene.
Patients and methods
Characterization of study groups
The patients investigated by microarray analysis of cardiac gene expression displayed the distinct DCM subtype of DCMi as defined by the World Health Organization and characterized by its association with cardiac inflammation. The DCMi group of this study was derived from a large series of patients initially submitted to our clinic with symptoms and signs of DCM; in all of whom, EMBs from the right ventricular septum were obtained by standard procedure for histological, immunohistological, and molecular virological analyses. All patients had given written informed consent. After standard clinical, morphological, and functional patient characterization, their EMBs were assessed for myocardial inflammation by histological and immunohistological methods. Their cardiac virological status was assessed by polymerase chain reaction (PCR)/real-time polymerase chain reaction (RT-PCR) for the following viral genomes: enteroviruses, adenoviruses (ADV), Epstein–Barr virus, human herpesvirus 6 (HHV-6), and parvovirus B19 (PVB19). Eight DCMi patients with cardiac inflammation in association with cardiac PVB19 were selected for the microarray study (DCMi group, Table 1). This group was compared to a control group of four individuals submitted to our clinic and diagnosed to clarify a clinically suspected cardiomyopathy, in whom the full differential diagnostic workup finally revealed that their complaints were noncardiac in origin (Cont group, Table 1). They had normal cardiac morphology and function, were negative for cardiac inflammation or viral genomes, and were subjected to microarray analysis, too. The DCMi group patients were on standard heart failure medication and in New York Heart Association functional class II (Table 1).
Table 1.
Characterization of study groups
| Code | Group | Age (years) | EF (%) | LVEDD (mm) | INFLAM | PVB19 | EV | AdV | HHV6 | EBV |
|---|---|---|---|---|---|---|---|---|---|---|
| GA-1 | DCMi | 31 | 56 | 71 | + | + | − | − | − | − |
| HI-2 | DCMi | 39 | 31 | 61 | + | + | − | − | − | − |
| KA-3 | DCMi | 69 | 36 | 60 | + | + | − | − | − | − |
| PS-4 | DCMi | 60 | 34 | 76 | + | + | − | − | − | − |
| BA-5 | DCMi | 67 | 43 | 59 | + | + | − | − | − | − |
| DE-6 | DCMi | 31 | 52 | 57 | + | + | − | − | − | − |
| CH-7 | DCMi | 62 | 51 | 73 | + | + | − | − | − | − |
| GH-8 | DCMi | 45 | 34 | 62 | + | + | − | − | − | − |
| 52.5 ± 17 (31–69) | 42.0 ± 9.9 (31–56) | 62.8 ± 7.8 (57–73) | ||||||||
| RS-14 | Cont | 36 | 68 | 47 | − | − | − | − | − | − |
| SM-15 | Cont | 46 | 61 | 49 | − | − | − | − | − | − |
| KE-16 | Cont | 26 | 74 | 47 | − | − | − | − | − | − |
| TU-17 | Cont | 36 | 64 | 50 | − | − | − | − | − | − |
| 36.0 ± 8 (26–46) | 66.8 ± 5.6 (61–74) | 48.3 ± 1.2 (47–50) |
EF ejection fraction; LVEDD left ventricular end-diastolic diameter; INFLAM cardiac inflammation; PVB19 parvovirus B19; EV enterovirus; AdV adenovirus; HHV6 human herpesvirus 6; EBV Epstein–Barr virus
High density microarray studies of human cardiac biopsies
cDNA was synthesized from 200 ng of total RNA isolated from EMBs. The minimal amount of total RNA routinely available from human EMBs was 200 ng. cRNA targets were generated using the small sample protocol of Affymetrix. Briefly, in vitro transcribed unlabeled cRNA from the first round of amplification was subjected to a second round of amplification. cRNA was transcribed into cDNA using random primers. After RNAse H-mediated removal of surplus cRNA, a second strand was synthesized using a T7 primer. cRNA amount was determined by ultraviolet spectroscopy, and distribution of cRNA fragment sizes of both cRNA and fragmentation products was checked on a BioAnalyzer. Each cRNA sample was hybridized for 16 h at 45°C to an Affymetrix Human Genome U133 Plus 2.0 GeneChip Array. Chips were washed and stained with streptavidin–phycoerythrin using a fluidics station according to Affymetrix protocols. Probe arrays were scanned at 3-/1.56-μm resolution using the Affymetrix GeneChip System Confocal Scanner 3000. Raw data were analyzed with Affymetrix GeneChip Operating Software. The detection p value of a transcript determines the detection call indicating if the transcript is reliably detected (p < 0.05; present) or not detected (absent). To enable the comparison between chips, data were normalized to a global intensity of 500. The expression data can be obtained from the Gene Expression Omnibus (GEO) website http://www.ncbi.nlm.nih.gov/projects/geo/, GEO experimental series number: GSE4172. For TaqMan experiments to verify candidate genes, see next chapter.
Quantitative real-time PCR analysis of human cardiac biopsies
Total RNA was extracted using TRIzol reagent (Invitrogen). Quality and quantity was checked on a Agilent 2100 Bioanalyzer. A minimal amount of only 200 ng of total RNA was routinely available from human EMBs for cDNA synthesis to become reverse transcribed using the High Capacity cDNA Archive Kit (Applied) according to the manufacturers instructions. For determination of gene expression, quantitative PCR assays were performed on an ABI PRISM 7900 Sequence Detector (Applied) with TaqMan Universal PCR Master Mix and TaqMan Gene expression assays (Applied) consisting of a FAM™ dye-labeled TaqMan MGB probe and two PCR primers. The following oligonucleotide sets purchased from Applied Biosystems were used: human genes encoding APN (Hs00605917_m1), CYR61 (Hs00155479_m1), chemokine ligand 14 (CXCL14; Hs00171135_m1), Ras-responsive element protein 1 (RREB1; Hs00171486_m1), GTP binding protein overexpressed in skeletal muscle (GEM; Hs00170633_m1), membrane protein, palmitoylated 7 (MPP7; Hs00299584_m1), glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Hs99999905_m1), and hypoxanthine phosphoribosyltransferase 1 (HPRT1; Hs99999909_m1). Quantitative RT-PCR reactions were performed using standard cycle parameters determined by the company. Normalization and fold change were calculated with the ΔΔCt method with GAPDH and HPRT1 as reference mRNA species according to ABI Prism 7900 manufacturer’s instructions [6]. The values given in Supplementary Fig. 2 are the means ± SEM of two independent quantitative TaqMan PCRs performed on RNA isolated from the same EMB that was also used for the microarray analysis.
Cardiovascular cell cultures
Primary culture of neonatal rat cardiomyocytes (NRCMC): Cells were prepared from ventricular tissue of 1- to 3-day-old Wistar rat pups (Charles River) as described previously [7]. Briefly, the removed ventricles of 50–60 animals were placed into ice-cold calcium ion-free phosphate-buffered cell isolation medium (CIM) containing 120 mM NaCl, 4.56 mM KCl, 0.44 mM KH2PO4, 0.42 mM Na2HPO4, 25 mM NaHCO3, and 5.55 mM glucose, pH 7.5, as well as 0.5 mg/ml streptomycin and 5,000 IE/ml penicillin G (Biochrom). Ventricular tissue was transferred to a Petri dish and minced into pieces of approximately 1 mm3 in size using two sterile scalpels. Stepwise disaggregation for 15 min each of tissue pieces into single cells was performed at 37°C under continuous mixing with a magnetic stirrer at 150 rpm in a 50 ml Erlenmeyer flask containing 15-ml CIM supplemented with 0.12% porcine trypsin 1:250 (Belger). The first tissue digest was discarded. The following three to eight supernatants obtained after each 15-min digestion period were poured into single sterile glass centrifuge tubes each containing 4 ml of ice-cold, heat-inactivated fetal calf serum (FCS; Biochrom). Cells were gently sedimented at 400 g for 10 min. The sedimented cells were resuspended in cell growth medium CMRL 1415-ATM (CMRL-ATM, Connaught Medical Research Laboratories-atmospheric) that was supplemented with 10% FCS, 10% horse serum (HS), and 0.02 mg/ml gentamicin (all from Biochrom) and adjusted to pH 7.4 using 1 N NaOH. For enrichment of cardiomyocytes, 20 ml of this suspension containing approximately 1.5 × 108 cardiac cells were incubated in 175 cm2 plastic dishes, Corning culture flask in a water-saturated atmosphere for 90 min at 37°C. During this incubation, most of the nonmuscle cells comprising 30–40% of total cell number attached to the bottom surface of the culture flask. The cardiomyocyte-enriched supernatant was carefully removed and pooled in an Erlenmeyer flask. The number of cells was counted using a light microscope and a hemocytometer. The cell density was adjusted with additional growth medium to 1.2 × 106 cells/ml and the cells seeded to 6-well Nunc culture plates and incubated in a water-saturated atmosphere at 37°C. After 24 h of incubation, the FCS/HS-supplemented growth medium was replaced either by a 10% FCS-supplemented CMRL 1415-ATM or serum-free, hormone-supplemented CMRL 1415-ATM medium containing 2-μM fluorodeoxfyuridine (Sigma) for inhibition of proliferation of contaminating nonmuscle cells. Vascular endothelial cell line: The endothelial cell line EA.hy926 (a hybrid line derived from human umbilical vein endothelial cells and A549 cells) was cultured in Dulbecco’s modified Eagle’s medium supplemented with hypoxanthine–aminopterin–thymidine, 10% FCS, and 1% penicillin and streptomycin.
Quantitative real-time PCR analysis of activated cell cultures
To study regulatory factors of CYR61 and APN in cultured cardiovascular cells (Fig. 2a,b), NRCMCs were grown in 6-well and EA.hy926 cells in 12-well dishes for 48 h. Thereafter, they were incubated for 24 h with the following cytokines (concentrations): IFN-β1b (30 ng/ml), IFN-β1a (30 ng/ml), IFN-γ (30 ng/ml), IL-1β (30 ng/ml), IL-6 (100 ng/ml), tumor necrosis factor (TNF)-α (20 ng/ml), and TNF-β (50 ng/ml). In some experiments (Fig. 2a), EA.hy926 cells were preincubated for 1 h with APN (10 μg/ml) before the addition of the above cytokines. This APN concentration was chosen because the APN plasma range in humans is 3–30 μg/ml. Cells were harvested 24 h after cytokine addition. Of the total RNA, 1 to 3 μg were isolated from cell cultures to become reverse transcribed using the High Capacity cDNA Archive Kit, and TaqMan PCRs were then carried out employing the following primer systems (Applied) for the rat genes encoding APN (Acdc, Rn00595250_m1), CYR61 (Rn00580055_m1), and GAPD (Rn99999916_s1). mRNA amounts were standardized against the housekeeping gene GAPDH. The values given in Fig. 2a,b are the means±SEM of two independent experiments each performed in triplicate.
Fig. 2.
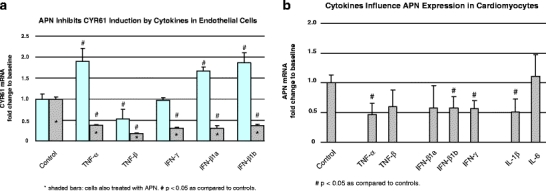
Cytokines regulating of APN and CYR61 in cardiovascular cells. We searched for possible triggers of cardiac APN and CYR61 deregulation in DCMi in both endothelial cells and cardiomyocytes. a, b Summarize real-time quantitative TaqMan PCR data on the regulatory potential of inflammatory cytokines in these cell cultures investigated as first approximation to the far more complex in vivo situation with multiple resident and infiltrating cell types (compare Fig. 3). a Endothelial cells expressed CYR61 and both APN receptors but no detectable amount of APN mRNA (data not shown). The inflammatory cytokines TNF-α and IFN-β induced CYR61 expression in these cells, whereas others showed no effect (IFN-γ) or even reduced CYR61 (TNF-β). To reveal possible interactions between the APN and CYR61 regulatory networks, CYR61 induction experiments were conducted in the presence (asterisksin shaded bars) vs the absence of APN. The effect of APN on CYR61 induction by cytokines was unexpectedly strong: induction of CYR61 by TNF-α or IFN-β was not only abolished but reverted resulting in a net reduction of CYR61 expression below that in untreated controls. The APN concentration at which effect was observed is in the range 3–30 μg/ml as observed in human plasma [14]. Error bars indicate the SEM from two independent experiments each performed in triplicate. b In contrast to endothelial cells, cardiomyocytes expressed APN and its receptors as well as CYR61. Several inflammatory cytokines (TNF-α, IFN-β,γ) significantly reduced APN expression in cardiomyocytes, whereas CYR61 expression was not significantly altered in this cell type (not shown). Error bars as in panel (a)
Network analysis
The networks shown in Fig. 1a,c,e were derived by the Ingenuity Pathways Analysis (IPA; http://www.ingenuity.com) software. The microarray dataset containing gene identifiers and corresponding expression values was uploaded as an Excel spreadsheet using the template provided in the application. Each gene identifier was mapped to its corresponding gene object in the Ingenuity Pathways Knowledge Base (IPKB). A cutoff of fold change ≥1.5 was set to identify genes whose expression was significantly differentially regulated; these were then used as the starting point for generating regulatory networks. Biological functions were assigned to each network or the overall analysis, respectively, by using the findings that have been extracted from the scientific literature and stored in the IPKB. The biological functions assigned to each network or the overall analysis, respectively, are ranked according to the significance of that biological function to the network or the overall analysis, respectively. A Fischer’s exact test is used to calculate a p value determining the probability that the biological function assigned to that network is explained by chance alone. The networks are displayed graphically as nodes (genes/gene products) and edges (the biological relationships between the nodes). Human, mouse, and rat orthologs of a gene are stored as separate objects in the knowledge base but are represented as a single node in the network. The intensity of the node color indicates the degree of up-(red) or down-(green) regulation. Nodes are displayed using various shapes that represent the functional class of the gene product. Edges are displayed with various labels that describe the nature of the relationship between the nodes (e.g., B for binding, T for transcription).
Fig. 1.
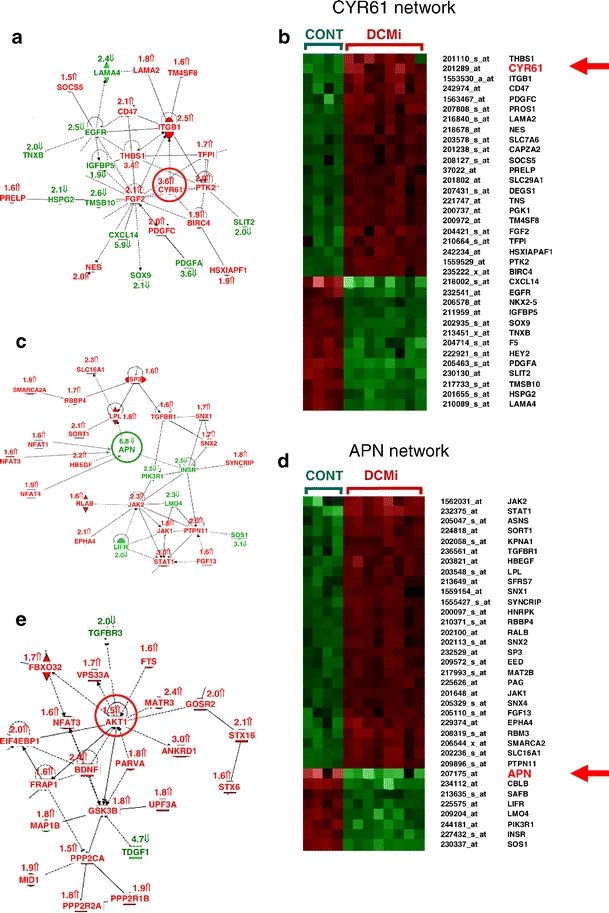
Altered gene expression profile and regulatory networks in DCMi. a, c, e The networks altered in DCMi are displayed graphically as nodes (genes/gene products) and edges (biological relationships between the nodes). Human, mouse, and rat orthologs of a gene are stored as separate objects in the knowledge base but are represented as a single node in the network. The intensity of the node color indicates the degree of up- (red) or down- (green) regulation. Nodes are displayed using various shapes that represent the functional class of the gene product. Continuous edge lines indicate protein–protein interactions, broken edge lines changes at the transcriptional level. The software further displays various labels that describe the nature of the relationship between the nodes (e.g., B for binding, T for transcription). b, d, f Show cluster analyses of the genes within the networks in (a), (c), (e), respectively. Here, analogous to the networks, red color denotes upregulation, green color denotes downregulation of a gene, whereas the brightness of the color indicates the extent of deregulation
Statistical analysis
For statistical analysis of microarray data, we used the significance analysis of microarrays (SAM). SAM statistics identifies significant changes in gene expression by performing a set of gene-specific t-tests. For each gene, a score is calculated on the basis of its change of expression relative to the standard deviation of repeated measurements for that gene. Genes with scores greater than a given threshold Δ are defined as significantly deregulated. Manual adjustment of this threshold Δ allows the identification of smaller or larger cohorts of genes. In addition, a false discovery rate can be estimated based on random permutations of all measurements. With respect to quantitative TaqMan PCRs, Student’s t-test was applied to demonstrate statistical significance. A value of p < 0.05 was considered statistically significant.
Results
Because, in advanced or end-stage DCM, any etiology-related pathomechanisms are most likely overwhelmed by unspecific reactions of the terminally failing myocardium long after the initiating process has burnt out, we studied patients with moderately severe disease (Table 1). The standard clinical characterization was refined by EMB-based immunohistological (Supplementary Fig. 1) and molecular virological analyses. The DCMi group was characterized by myocardial inflammation in association with PVB19. A distinctive expression profile was detected in the DCMi group by microarray analysis of the EMBs. The complete dataset is available from the website http://www.ncbi.nlm.nih.gov/projects/geo/. Genes significantly (q value < 5.0) and ≥1.5-fold deregulated are listed in Supplementary Table 1. Subsequently, a network analysis of the complete dataset with 380 genes ≥1.5-fold down- and 1,038 genes ≥1.5-fold up-regulated was performed as shown in Fig. 1a–f. Several of these genes were validated by quantitative TaqMan PCR (Table 2, Supplementary Fig. 2). Whereas confirmatory PCR analysis was desirable for a major number of DCMi-associated genes, the limited mRNA quantity available from human EMBs confined the number of such analyses. The minimal amount of total RNA available from EMBs for both microarrays and confirmatory PCR was only 200 ng. The genes selected for confirmatory PCRs are shown in Supplementary Fig. 2. They include CYR61, APN, CXCL14, and GEM as novel deregulated genes associated with DCMi, natriuretic peptide precursor A (NPPA), and natriuretic peptide precursor B (NPPB) as standard heart failure-associated genes and confirmed the microarray data in all cases. The microarray q values for each of the genes in Supplementary Fig. 2, the corresponding p values in the confirmatory TaqMan PCRs, and similar regulatory factors are proof of deregulation of these genes of particular interest.
Table 2.
Partial cardiac gene expression profile of human DCMi (further data at http://www.ncbi.nlm.nih.gov/projects/geo/ and in Supplementary Table 1)
| Network | Symbol | Regulation | Factor | q value | Gene Name |
|---|---|---|---|---|---|
| APN | APNa | Down | 6.8 | 0.605 | Adiponectin |
| 14.4 | |||||
| CYR | CXCL14 | Down | 5.9 | 1.943 | Chemokine (C–X–C motif) ligand 14 |
| 4.4 | |||||
| AKT | TDGF1 | Down | 4.7 | 0.605 | Teratocarcinoma-derived growth factor 1 |
| CYR | PDGFA | Down | 3.6 | 0.371 | Platelet-derived growth factor (PDGF)-α polypeptide |
| APN | SOS1 | Down | 3.1 | 0.371 | Son of sevenless homolog 1 (Drosophila) |
| CYR | TMSB10 | Down | 2.6 | 0.371 | Thymosin-β10 |
| APN | INSR | Down | 2.5 | 0.371 | Insulin receptor |
| CYR | EGFR | Down | 2.5 | 2.226 | Epidermal growth factor (EGF) receptor |
| APN | PIK3R1 | Down | 2.5 | 0.991 | Phosphoinositide-3-kinase regulatory subunit 1 (p85α) |
| CYR | LAMA4 | Down | 2.4 | 0.371 | Laminin α4 |
| APN | LMO4 | Down | 2.3 | 3.868 | Lin-11, Isl-1, Mec-3 (LIM) domain only 4 |
| CYR | HSPG2 | Down | 2.1 | 2.226 | Heparan sulfate proteoglycan 2 |
| CYR | SOX9 | Down | 2.1 | 0.371 | Sex determining region Y (SRY)-box 9 |
| APN | LIFR | Down | 2.0 | 1.632 | Leukemia inhibitory factor (LIF) receptor |
| CYR | TNXB | Down | 2.0 | 0.371 | Tenascin XB |
| AKT | TGFBR3 | Down | 2.0 | 0.895 | TGF-β receptor III (betaglycan) |
| AKT | EIF4EBP1 | Down | 2.0 | 2.345 | Eukaryotic translation initiation factor 4E binding protein 1 |
| CYR | SLIT2 | Down | 2.0 | 1.667 | Slit homolog 2 (Drosophila) |
| CYR | IGFBP5 | Down | 1.9 | 0.991 | Insulin-like growth factor binding protein 5 |
| CYR | CYR61a | Up | 3.6 | 1.943 | Cysteine-rich, angiogenic inducer, 61 |
| 3.3 | |||||
| CYR | THBS1 | Up | 3.4 | 3.868 | Thrombospondin 1 |
| APN | STAT1 | Up | 3.0 | 1.222 | Signal transducer and activator of transcription 1 |
| AKT | ANKRD1 | Up | 3.0 | 2.226 | Ankyrin repeat domain 1 (cardiac muscle) |
| CYR | ITGB1 | Up | 2.5 | 1.994 | Integrin β1 = β-subunit of fibronectin receptor (CD29) |
| AKT | BDNF | Up | 2.4 | 3.641 | Brain-derived neurotrophic factor |
| AKT | MATR3 | Up | 2.4 | 0.895 | Matrin 3 |
| APN | JAK2 | Up | 2.3 | 3.469 | Janus kinase 2 |
| APN | SLC16A1 | Up | 2.3 | 1.943 | Solute carrier family 16 member 1 |
| APN | HBEFG | Up | 2.2 | 3.179 | Heparin-binding EGF-like growth factor |
| CYR | CD47 | Up | 2.1 | 4.108 | Integrin-associated signal transducer |
| CYR | FGF2 | Up | 2.1 | 2.868 | Fibroblast growth factor (basic) |
| APN | SORT1 | Up | 2.1 | 1.632 | Sortilin |
| APN | EPHA4 | Up | 2.1 | 2.345 | EPH receptor A4 |
| AKT | STX16 | Up | 2.1 | 2.226 | Syntaxin 16 |
| CYR | NES | Up | 2.0 | 2.487 | Nestin |
| CYR | PDGFC | Up | 2.0 | 3.469 | Platelet derived growth factor C |
| CYR | PTK2 | Up | 2.0 | 1.571 | Protein tyrosine kinase 2 |
| APN | PTPN11 | Up | 2.0 | 1.632 | Protein tyrosine phosphatase non-receptor type 11 |
| AKT | GOSR2 | Up | 2.0 | 1.667 | Golgi SNAP receptor complex member 2 |
| APN | NFAT4a | Up | 1.9 | 2.345 | Nuclear factor of activated T cells 4 |
| CYR | BIRC4 | Up | 1.9 | 1.943 | Baculoviral IAP repeat-containing 4 |
| CYR | HSXIAPA1 | Up | 1.9 | 2.487 | XIAP associated factor-1 |
| AKT | PPP2R1B | Up | 1.9 | 3.641 | Protein phosphatase 2 regulatory subunit A β |
| APN | SMARCA2 | Up | 1.9 | 2.226 | SWI/SNF related matrix associated regulator of chromatin |
| AKT | MID1 | Up | 1.9 | 3.175 | Midline 1 |
| AKT | PARVA | Up | 1.8 | 2.621 | Parvin-α |
| APN | LPL | Up | 1.8 | 2.487 | Lipoprotein lipase |
| CYR | LAMA2 | Up | 1.8 | 1.994 | Laminin-α2 |
| AKT | GSK3B | Up | 1.8 | 0.991 | Glycogen synthase kinase 3β |
| AKT | PPP2R2A | Up | 1.8 | 1.222 | Protein phosphatase 2 regulatory subunit B α |
| APN | SYNCRIP | Up | 1.8 | 1.994 | Synaptotagmin binding RNA interacting protein |
| AKT | PPP2R2A | Up | 1.8 | 1.222 | Protein phosphatase 2 regulatory subunit B α |
| AKT | UPF3A | Up | 1.8 | 3.173 | UPF3 regulator of nonsense transcripts A |
| APN | ADIPOR1 | Up | 1.7 | 2.868 | Adiponectin receptor 1 |
| CYR | TFPI | Up | 1.7 | 3.868 | Tissue factor pathway inhibitor |
| APN | SNX1 | Up | 1.7 | 4.362 | Sorting nexin 1 |
| APN | SNX2 | Up | 1.7 | 4.467 | Sorting nexin 2 |
| APN | RBBP4 | Up | 1.7 | 2.345 | Retinoblastoma binding protein 4 |
| AKT | FBXO32 | Up | 1.7 | 2.487 | F-box protein 32 |
| AKT | VPS33A | Up | 1.7 | 2.226 | Vacuolar protein sorting 33A |
| APN | NFAT1a | Up | 1.6 | 5.091 | Nuclear factor of activated T cells 1 |
| AKT | NFAT3a | Up | 1.6 | 4.108 | Nuclear factor of activated T cells 3 |
| APN | JAK1 | Up | 1.6 | 1.994 | Janus kinase 1 |
| APN | SP3 | Up | 1.6 | 2.226 | Sp3 transcription factor |
| AKT | FRAP1 | Up | 1.6 | 4.362 | FK506 binding protein 12 |
| AKT | STX6 | Up | 1.6 | 1.994 | Syntaxin 6 |
| CYR | PRELP | Up | 1.6 | 4.757 | Proline arginine-rich end leucine-rich repeat protein |
| APN | RALB | Up | 1.6 | 3.173 | v-ral simian leukemia viral oncogene homolog B |
| APN | FGF13 | Up | 1.6 | 4.757 | Fibroblast growth factor 13 |
| APN | TGFBR1 | Up | 1.6 | 4.108 | Transforming growth factor-β receptor I |
| AKT | FTS | Up | 1.6 | 3.469 | Fused toes homolog |
| AKT | AKT1 | Up | 1.5 | 4.757 | v-akt murine thymoma viral oncogene homolog 1 |
| AKT | PPP2CA | Up | 1.5 | 4.362 | Protein phosphatase 2 catalytic subunit α |
All factors of deregulations are derived from the Affymetrix microarray analysis. For certain genes additional TaqMan analyses were conducted, which are then given in italics below the respective Affymetrix values (see also Supplementary Fig. 2).
aPlease note the synonyma APN = ADIPOQ, CYR61 = CCN1, NFAT1 = NFATC2IP, NFAT3 = NFATc4, NFAT4 = NFATc3
The network analysis of the microarray data [8] was conducted by the IPA software employing the genes significantly (q value < 5.0) and ≥1.5-fold deregulated. IPA is based on modeled biological networks derived from information of peer-reviewed scientific publications. Analysis of our data revealed several strongly altered regulatory networks. The gene networks shown in Fig. 1a,c,e have not been previously described in former microarray studies of DCM (details in discussion). One of these networks centered around the CYR61 or CCN1 gene (Fig. 1a,b), another one around the APN gene (Fig. 1c,d). These networks are related to cell-to-cell signaling and cellular compromise. Another network around the AKT1 gene (Fig. 1e,f) also deserves consideration because AKT1 is involved in the regulation of both CYR61 [9] and APN [10] and because APN strongly influences CYR61 expression in vitro (Fig. 2b). Table 2 summarizes all member genes of these three networks with full gene names, q values, and fold changes. Those two genes (CYR61, APN) that were among the most strongly deregulated and, at the same time, key components of complex deregulated networks were further investigated in cardiovascular cell cultures in vitro (Fig. 2a,b).
The APN regulatory network contains significantly deregulated (q value ≤ 5.0, ≥1.5-fold change) genes not previously reported in microarray studies of DCM (full gene names in Table 1): STAT1 3.0-fold up; JAK2 2.3-fold up; HBEGF 2.2-fold up; SORT1 2.1-fold up; PTPN11 2.0-fold up; LPL 1.8-fold up; JAK1 1.6-fold up; SP3 1.6-fold up; APN 6.8-fold down; INSR 2.5-fold down; PIK3R1 2.5-fold down; LIFR 2.0-fold down; TNXB 2.0-fold down; and TGFBR3 2.0-fold down. Three genes encoding the nuclear factors of activated T cells (NFAT), NFAT1 = NFATC2IP (1.6-fold up), NFAT3 = NFATC4 (1.6-fold up), NFAT4 = NFATC3 (1.9-fold up), are also linked to the APN network as negative regulators of APN expression [11]. Similarly, the ADIPOR1 gene (1.7-fold up) encoding the APN receptor 1 gene is part of the APN signal transduction system.
A second regulatory network around CYR61 contains further deregulated genes with q value ≤ 5.0 and ≥1.5-fold change (full gene names in Table 1): CYR61 = CCN1 3.6-fold up; THBS1 3.4-fold up; ITGB1 2.5-fold up; CD47 2.1-fold up; FGF2 2.1-fold up; NES 2.0-fold up; PDGFC 2.0-fold up; PTK2 2.0-fold up; BIRC4 1.9-fold up; HSXIAPA1 1.9-fold up; LAMA2 1.8-fold up; TFPI 1.7-fold up; CXCL14 5.9-fold down; PDGFA 3.6-fold down; TMSB10 2.6-fold down; EGFR 2.1-fold down; LAMA4 2.4-fold down; HSPG2 2.1-fold down; TNXB 2.0-fold; and IGFBP5 1.9-fold down. A recent publication also links CYR61 = CCN1 to Wnt signaling pathways [13] and several Wnt signal pathway genes were deregulated in DCMi: STX16 2.1-fold up; GCLM 2.0-fold up; CSNK1A1 1.9-fold up; PPP2R1B 1.9-fold up; GSK3B 1.8-fold up; INSR 2.5-fold down; FBXO40 2.1-fold down; and CLIC5 2.0-fold down.
In addition to the multifunctional CYR61 and APN genes, several other deregulated genes are known to be involved in immunological and inflammatory processes: The CXCL14 gene was 5.9-fold and the neurturin (NRTN) gene 5.8-fold downregulated in DCMi. The interleukin-17D (IL-17D) gene, a member of IL-17A to F gene family, was 2.6-fold induced. IL-17 serum levels were elevated in DCMi patients (not shown). Altered intracardiac immune regulation is further suggested by upregulation of the immediate early response 3 (IER3, 2.6-fold) and 5 (IER5, 2.3-fold) genes, ubiquitin-specific protease 39 (USP39, 3.4-fold), phospholipase A2-activating protein (PLAA, 2.6-fold), and IFN-related developmental regulator 1 (IFRD1, 2.4-fold). Induction of the neural cell adhesion molecule (NCAM1, 2.0-fold) gene [14] and the coxsackie virus–ADV-receptor (CXADR, 1.7-fold) gene [15] already known from immunohistochemical studies of DCMi hearts were recognized by the microarrays, too.
As a first step to elucidate the molecular mechanisms that deregulate CYR61 and APN expression in human DCMi, cell culture studies were conducted. Both genes were significantly deregulated in DCMi as components of complex gene networks. Figure 2a shows that certain inflammatory cytokines induced CYR61 expression in vascular endothelial cell cultures similarly as observed in the DCMi hearts. This CYR61 induction by TNF-α and IFN-β was completely abolished in the presence of APN (Fig. 2a). The APN plasma range in humans is 3–30 μg/ml [16], and the concentration therefore chosen for the cell culture investigations was 10 μg/ml. At this concentration, APN generally suppressed the CYR61 expression level in this cell type. In cardiomyocytes, TNF-α, IFN-β, IFN-γ, and IL-1β all significantly reduced the expression of APN (Fig. 2b), whereas CYR61 expression was not affected by these cytokines in this cell type. Several other deregulated genes of interest known to be involved in immunological and inflammatory processes were neither expressed in endothelial cells nor in cardiomyocytes, and therefore could not be studied in these cell biological models.
Discussion
Identification of DCM subtypes
DCM is the term used to describe any clinical phenotype characterized by dilation and dysfunction of the cardiac ventricles. From an etiologic perspective, however, DCM encompasses a diverse spectrum of disease entities, some of which are due to monogenic defects in cardiac expressed genes [1], others triggered by exogenous factors including cardiotropic viruses [2]. Therapies directed at etiology-related pathogenic pathways in these etiologically distinct DCM subtypes may be more efficient than current standard treatment protocols employed in heart failure of any origin. However, etiology-related early pathogenic pathways must be assumed to prevail only in the beginnings of the respective DCM subtype, whereas common unspecific molecular pathomechanisms will predominate in advanced heart failure irrespective of etiology. One important DCM subtype as defined by cardiac inflammation was investigated in the current study to further define it at the molecular level. Whereas previous studies focused on advanced or end-stage DCM [5] where specific pathomechanisms are overwhelmed by unspecific processes, we probed an earlier and restrictively defined phenotype (DCMi) to identify early and etiology-related disease processes. Importantly, our patients were not terminally ill nor on maximum supportive therapy as in previous studies of advanced DCM. Similarly, controls had no severe underlying condition as, e.g., heart transplantation donor hearts often used as controls for expression profiles but were free of cardiac disease after comprehensive exclusion of multiple cardiac differential diagnoses. Further, any search for etiology-related pathomechanisms is more likely successful if the study groups are highly homogenous. An excellent previous study has also defined its patient groups by clinical, morphological, and hemodynamic phenotyping [17] and derived interesting data on common expression profiles in disorders linked by common hemodynamic anomalies. We likewise defined a highly homogeneous study group by sequential stratification of a large DCM patient cohort, first, by standard clinical, morphological, and hemodynamic criteria, second, by biopsy-based detection of cardiac inflammation in conjunction with a cardiotropic virus. Cardiac inflammation in our patients was associated with the presence of PVB19 genomes in the myocardium. We think, however, that the observed anomalous expression pattern probably reflects a general inflammatory pathomechanism that may be triggered by diverse proinflammatory stimuli. After the current pilot study all the key genes of this anomalous pattern need to be further investigated in major patient groups with varying degrees of cardiac inflammation and hemodynamic impairment, to address issues of specificity and dynamic. In vivo biopsies on which the current study was entirely based further provide the perspective for follow-up studies of the clinical courses that would then add a third stratification criterion for patient selection to be used in the future.
Cardiac expression profile of DCMi
The anomalous cardiac expression pattern observed for human DCMi was not described in previous studies of human cardiac diseases or heart failure of any origin [5, 18–27]. Most network genes listed in Table 2 and, in particular, CYR61 and APN have, to our knowledge, not been found to be deregulated in human microarray studies and also not in animal models of DCM. A comprehensive recent review [5] summarizes that the major molecular pathway changes in human DCM, and other human cardiomyopathies encompass sarcomeric, cytoskeletal, extracellular matrix, Ca2+ homeostasis, apoptosis, and energy metabolism genes. Major deregulations of inflammation- and immune system-associated genes as in DCMi were not included. Because we investigated a DCM subtype associated with intracardiac inflammation, a recent microarray study of two giant cell myocarditis patients [28] is of specific interest for comparison. That study addressed an immunohistologically defined inflammatory cardiac phenotype clearly different from that in our study and found deregulated T cell activation genes, including IL-10 receptor-α, β2-integrin, chemokine receptor 4, and chemokine ligands 5, 9, 13, and 18. Remarkably, none of the latter genes was deregulated in DCMi but only CXCL14. Conversely, NFAT1 and NFAT4 gene upregulations and APN gene downregulation as observed in DCMi were not observed in the giant cell myocarditis study. This appears to reflect distinct immunological pathomechanisms.
The CYR61 gene and its associated network showed particularly strong deregulation in DCMi. CYR61 = CCN1 is a member of the CCN gene family [29] (also including connective tissue growth factor [CTGF] = CCN2) and has already attracted attention as a multifunctional, secreted protein able to modulate multiple cell functions. It induces apoptosis in fibroblasts [30] and mediates cell death in neurons [31] and in virus-infected cells [32]. From a therapeutic perspective, it is interesting that RNA interference-mediated silencing of the CYR61 gene protected cells against cell death, whereas CYR61 overexpression enhanced it [32]. Moreover, CYR61 coordinates genes that control inflammation (IL-1β), extracellular matrix remodeling (e.g., MMP1,3), cell–extracellular matrix interactions, and angiogenesis [33, 34]. CYR61 acts in a cell type- and context-dependent manner through direct binding to at least five distinct integrins, including α6β1 and heparan sulfate proteoglycans as co-receptors [35], integrins αvβ3 and αvβ5 [36], and integrin αMβ2 on blood monocytes [37]. CYR61 is involved in integrin-linked kinase-mediated AKT and β-catenin-T cell factor/Lef signaling [38]. It is also regulated by canonical Wnt signaling and involved in mesenchymal stem cell differentiation [13]. In contrast to this wealth of data on in vitro actions of CYR61, data on its physiological functions and pathogenic potential in humans are sparse. CYR61 upregulation has only recently been described in end-stage ischemic cardiomyopathy [39]. The current work shows, for the first time, CYR61 induction in an inflammatory cardiac disorder with far less severe clinical phenotype, where, most likely, no irreversible cardiac damage has yet occurred. Cardiac CYR61 expression is apparently induced by multiple stimuli [39], including inflammation in vivo and inflammatory cytokines in vitro (this study). Whereas transient CYR61 induction may be a temporarily beneficial response of the heart to various stress factors, chronic stimulation of the auto/paracrine signaling pathways activated by CYR61 may be maladaptive and pathogenic in itself by, e.g., sustaining exuberant tissue inflammatory and remodeling processes [33]. Our observation of CYR61 overexpression in DCMi suggests to investigate further, if modulation of CYR61-dependent auto/paracrine pathways might have therapeutic potential in cardiac diseases associated with local inflammation.
In the current study, we show, for the first time, that APN is synthesized by human hearts in situ, not only by adipose tissues as previously assumed, and that this local synthesis is strongly downregulated in a human cardiac disease. Cultured cardiomyocytes were already known to express APN and both of its receptors and secrete APN [42]. APN has encountered high interest in the cardiovascular field due to pioneering work in APN knockout mice, proving important functions of APN in the heart [40, 41]. These studies assumed, however, that cardioprotective effects were exerted by APN synthesized by adipose tissue and acting via the circulation. APN inhibits apoptosis and TNF-α production in cardiomyocytes, modulates hypertrophic signals in the heart [40], and protects it from ischemic injury in a mouse model [41]. APN regulation has not been previously investigated in human hearts in situ. Previous studies [40, 41] and the present work suggest that cardiac APN is influenced by multiple factors acting on components of the APN gene network. AKT1 is involved in the regulation of both CYR61 [9] and APN [10]. Interestingly, APN abolished CYR61 induction by inflammatory cytokines in endothelial cells (Fig. 2a). Because multiple other endothelial activation markers are induced in DCMi [54, 55], it is of interest to know the whole spectrum controlled by APN, but this is beyond the scope of the current study. In synopsis with a previous report that APN inhibits IL-8 synthesis by endothelial cells [10], these data suggest that local cardiac APN synthesis is required for adequate control of local inflammatory processes. If so, depression of cardiac APN synthesis by various mechanism may render the heart vulnerable to inflammatory challenges such as imposed by infiltrating immune cells in DCMi or under other conditions associated with enhanced cardiac inflammatory cytokine production [43, 44]. Modulation of the local cardiac APN system may therefore have therapeutic potential under certain conditions. Due to the complexities of the biological functions of both CYR61 and APN, it is impossible to predict the effects of their modulation in vivo. Synopsis of previous work and the current study suggests to investigate, however, if modulation of the balance between the CYR61 and APN networks may have therapeutic potential in cardiac diseases associated with local inflammation. One obvious approach would be cardiac APN gene therapy, another one RNA interference-mediated CYR61 suppression by cardiac shRNA expression.
In addition to the multifunctional CYR61 and APN proteins and interacting gene products such as the NFATs, several other deregulated genes are also directly involved in immunological and inflammatory processes. Two genes encoding proteins described as endothelial activation markers in DCMi [14, 15] were upregulated 2.0-fold (NCAM1) and 1.7-fold CXADR. CXCL14 (a component of the CYR61 network) belongs to a family of dendritic cell- and monocyte-attracting chemokine-like proteins [45, 46]. Loss of CXCL14 in tumors was associated with low infiltration by dendritic cells, whereas its restoration caused dendritic cell attraction in vitro and in vivo [47]. The downregulated NRTN gene belongs to the same family as glial cell line-derived neurotrophic factor (GDNF). GDNF and NRTN signaling is mediated by a two-component receptor: the signal-transducing component RET shared by both ligands and the ligand-specific binding components GFR-α1 (GDNF) or GFR-α2 (NRTN). Human T and B cells and monocytes produce RET and NRTN. GFR-α1 and GFR-α2 are transcribed in all immune cell subsets, with GFR-α2 being the dominant ligand-binding chain in T cells, B cells, and monocytes [48]. Therefore, NRTN downregulation may alter immune cell communication, whereas little is known about its functions in other cell types. IL-17D belongs to a family of secreted proteins that play an active role in inflammatory and autoimmune diseases [49]. Because IL-17D stimulates production of IL-6, IL-8, and granulocyte/macrophage colony-stimulating factor (GM-CSF) in human endothelial cells [50], its upregulation may enhance the activation of the endothelium, the primary attachment site for migrating immune cells. IL-17 functionally cooperates with TNF-α [51], enhances growth of vascular endothelial cells [52], and stimulates matrix metallo-proteinase-9 expression [53]. The upregulated PLAA activates mammalian PLA2s involved in inflammatory responses [54]. Thus, PLA2-IB is upregulated during inflammation, stimulates CXCL8 production via extracellular signal-regulated kinase (ERK) and nuclear factor-κB in neutrophils, and modulates inflammatory responses via the PLA2-IB receptor [55].
Working model of local cardiac inflammation control
A working model of a novel inflammation-associated cardiac pathomechanism is outlined in Fig. 3. The model proposes the existence and pathogenic relevance of a local inflammation control system in human hearts acting by local synthesis of anti-inflammatory proteins, a representative of which is APN. If local cardiac APN synthesis by cardiomyocytes is absent, as in knockout mice [40, 41], or suppressed, as in DCMi patients, inflammatory challenges, such as imposed by cardiac immune cell infiltrations (Supplementary Fig. 1), may result in grave damage to vascular endothelia and myocardium. The model assumes that cardiac inflammation control is predominantly mediated by auto/paracrine actions of locally synthesized APN, whereas, in the context of cardiac ischemic injury in APN knockout mice, a complementary systemic model of APN action has been proposed, in which APN secreted by adipose tissue acts hormone-like to protect the heart [41] and the endothelia via the circulation [10]. APN acts directly upon immune cells, such as natural killer (NK) cells [12], and on nonimmune cells, such as vascular endothelia [10]. One new target controlled by APN is CYR61 that is locally upregulated in DCMi hearts in situ and whose endothelial induction by cytokines is abolished by APN (Fig. 2a). Although CYR61 induces transient local inflammation and matrix remodeling during healing processes [33], its activity needs to be controlled so that it does not cause chronic tissue damage. In addition to such direct anti-inflammatory actions of APN upon non-immune targets (endothelial cells, cardiomyocytes), a recent study [12] shows that APN is also a potent negative regulator of IL-2-induced NK cell activation. IFN-γ production was suppressed by APN, accompanied by downregulation of the IFN-γ-inducible TNF-related apoptosis inducing ligand and Fas ligands. APN and cellular immunity are further linked by the fact that NFAT transcription factors are key regulators of T cell function [56] and also negative regulators of APN expression [11]. All local cardiac functions of CYR61, APN, and other factors in the model are thought to be mediated via auto/paracrine loops unrelated to the respective circulating plasma levels (3–30 μg/ml for APN in humans, unknown for CYR61).
Fig. 3.
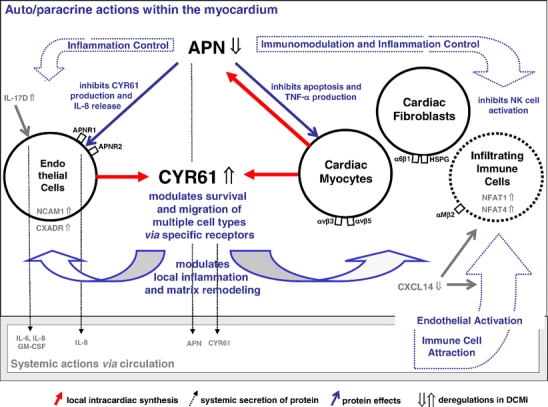
Working model of local cardiac inflammation control. The model proposes the existence of a inflammation control system in human hearts acting by local synthesis of anti-inflammatory proteins, a representative of which is APN. If local cardiac APN synthesis by cardiomyocytes is genetically absent [38, 39] or suppressed (this study), inflammatory challenges may result in aggravated damage to endothelia and myocardium. This local model assumes that inflammation control and other regulatory functions are mediated by auto/paracrine actions of APN synthesized locally by cardiomyocytes, whereas in the context of APN knockout mice [38, 39], a complementary systemic model of APN action was proposed in which APN secreted by adipose tissue acts hormone-like to protect heart and endothelia via the circulation. APN (downregulated in DCMi) acts directly upon immune cells [10] (right side of figure) and also on nonimmune cells such as vascular endothelia [8] (left side). One new target for control by APN is CYR61 that is locally upregulated in DCMi (center) and whose endothelial induction by cytokines is suppressed by APN (see Fig. 2a). In addition to such direct anti-inflammatory action of APN on vascular endothelia (left side), APN is a potent negative regulator of IL-2-induced NK cell activation (right side) [10]. APN and cellular immunity are further linked by NFATs (upregulated in DCMi), which are key regulators of T cell function [54], and negative regulators of APN expression [9]. The functions of CYR61, APN, and other factors deregulated in DCMi in this local model are thought to be mediated via auto/paracrine loops unrelated to the respective circulating plasma levels. CYR61-associated CXCL14 downregulation in DCMi (right side) may alter immune cell attraction to the heart. IL-17D induction in DCMi (left side) may stimulate IL-6, IL-8, and GM-CSF production by endothelial cells and thus activation of the endothelium, the primary attachment site for migrating immune cells
Limitations and perspective of the study
Gain/loss-of-function experiments are required to clarify which of the gene deregulations in DCMi are primary and causative or secondary and maladaptive. Conventional gain/loss-of-function experiments in cell cultures may contribute to solve these questions but are rather artificial as compared to the complex physiological situation in intact tissues composed of multiple interacting cell types. Cardiac overexpression of deregulated genes and silencing of these genes by shRNA-mediated RNA interference in vivo has recently become feasible, however, by intravenous injection of cardiotropic AAV8 and AAV9 vectors in mice [57, 58]. These systems have considerable potential for gene therapy [59]. Beyond the classical knockout technologies, this novel approach may also help to clarify pathogenic cause–effect relationships arising from genomic and proteomic screening studies in humans such as the current one based on EMBs of human hearts in situ.
Conclusions
Genomic expression profiling evolves as a useful tool to identify novel pathomechanisms in human cardiac disorders [5, 8] because most animal models, if available at all, display multiple crucial differences to the respective human disease. Restrictive disease phenotyping significantly improves the chance of genomic approaches to identify such mechanisms and therapeutic targets even in complex human disorders. The current work shows, for the first time, local APN expression in human hearts in situ and its downregulation in a human cardiac disease. It further demonstrates that APN efficiently inhibits the endothelial expression of CYR61 that is strongly induced in DCMi. Synopsis of previous work and these data suggests to investigate further, if modulation of the balance between the CYR61 and APN network may have therapeutic potential in cardiac diseases associated with inflammation.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
{kind=link}
The disease phenotyping of the DCMi patient group (Table 1 and Patients and methods) that were investigated by genomewide Affymetrix microarray expression profiling included (in addition to clinical and molecular virological characterization) an immunohistochemical characterization of cardiac immune cell infiltration and vascular endothelial activation. The immunohistochemical studies were done on EMBs obtained by the same standard procedure as those used for the Affymetrix microarray expression profiling and for the TaqMan quantitative RT-PCRs (264.6 KB) (226.1 KB) (159.6 KB)
Real-time quantitative TaqMan PCR data on shifts in gene expression in the DCMi patients versus the control group. TaqMan data on the above novel DCMi-associated genes were obtained by analysis of residual RNA extracted from the same EMB as already used for the genomic Affymetrix microarrays and confirmed the respective data from the microarray study reported in detail in Table 2, Fig. 2 (panels a–f), and http://www.ncbi.nlm.nih.gov/projects/geo/ in all cases. Sharp sign denotes p < 0.05 between the study groups. In addition to these new genes, the standard heart failure-associated genes NPPA and NPPB were significantly (p < 0.05) upregulated (18.7-fold and 39.0-fold, respectively) in the DCMi group as assessed by TaqMan PCR, confirming a trend observed in the microarrays (7.5-fold at q = 6.400 and 20.5-fold at q = 5.722; (PDF 22 kb).
Extension of Table 2 with all shifts in cardiac gene expression down to 1.5-fold change for DCMi patient group vs Cont control group, as assessed by Affymetrix microarray analysis of RNA extracted from EMB. For further details of the DCMi and Cont groups, see Table 1 and Patients and methods. The complete Affymetrix dataset is available from the GEO website http://www.ncbi.nlm.nih.gov/ projects/geo/. GEO experimental series number was assigned as GSE4172 (PDF 1 mb).
Acknowledgement
This work has been supported by Deutsche Forschungsgemeinschaft through SFB Transregio 19 (project grants C5 to WP and HF and Z1 to HPS) and through grant Po 378/6-1 to WP. It has also been supported by the Friede Springer Foundation, Berlin, Germany.
Abbreviations
- APN
adiponectin
- CYR61
cysteine-rich angiogenic inducer 61
- DCM
dilated cardiomyopathy
- DCMi
inflammatory cardiomyopathy
- EF
ejection fraction
- EMB
endomyocardial biopsy
- IFN-β,γ
interferon-β,γ
- IL
interleukin
- LVEDD
left ventricular end-diastolic diameter
- TNF-α/β
tumor necrosis factor-α/β
Biographies
Frank Wittchen works as physician and scientist at the Department of Cardiology and Pneumology of the Charité-Universitätsmedizin Berlin (Charité Centrum 11). His research interests are the cell and molecular biology of heart failure and the application of genomics for the analysis of human cardiomyopathies.

Wolfgang Poller is a Professor of Medicine at the Department of Cardiology and Pneumology of the Charité-Universitätsmedizin Berlin. His research interests include the molecular pathogenesis and treatment of cardiovascular and pulmonary disorders. A current field of interest is the application of genomics and proteomics to identify novel therapeutic targets in human cardiomyopathies and the development of new treatment strategies.

Footnotes
The mesenchymal stem cells protein DSC96 was 4.8-fold (q<0.371) downregulated in the DCMi group.
F. Wittchen and L. Suckau contributed equally to this paper.
Electronic supplementary material
Supplementary material is available in the online version of this article at http://dx.doi.org/10.1007/s00109-006-0122-9 and is accessible for authorized users.
References
- 1.Poller W, Kühl U, Tschoepe C, Pauschinger M, Fechner H, Schultheiss H-P (2005) Genome-environment interactions in the molecular pathogenesis of dilated cardiomyopathy. J Mol Med 83:579–586 [DOI] [PubMed]
- 2.Kühl U, Pauschinger M, Noutsias M, Seeberg B, Bock T, Lassner D, Poller W, Kandolf R, Schultheiss H-P (2005) High prevalence of viral genomes and multiple viral infections in the myocardium of adults with “Idiopathic” left ventricular dysfunction. Circulation 111:887–893 [DOI] [PubMed]
- 3.Kühl U, Pauschinger M, Seeberg B, Noutsias M, Poller W, Schultheiss H-P (2005) Virus persistence in the myocardium is associated with progressive cardiac dysfunction. Circulation 112:1965–1970 [DOI] [PubMed]
- 4.Kühl U, Pauschinger M, Schwimmbeck P, Seeberg B, Lober C, Noutsias M, Poller W, Schultheiss H-P (2003) Interferon-β treatment eliminates cardiotropic viruses and improves left ventricular function in patients with myocardial persistence of viral genomes and left ventricular dysfunction. Circulation 107:2793–2798 [DOI] [PubMed]
- 5.Sanoudou D, Vafiadaki E, Arvanitis DA, Kranias E, Kontrogianni-Konstantopoulos A (2005) Array lessons from the heart: focus on the genome and transcriptome of cardiomyopathies. Physiol Genomics 21:131–143 [DOI] [PubMed]
- 6.Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25:402–408 [DOI] [PubMed]
- 7.Vetter R, Kott M, Schulze W, Rupp H (1998) Influence of different culture conditions on sarcoplasmic reticular calcium transport in isolated neonatal rat cardiomyocytes. Mol Cell Biochem 188:177–185 [DOI] [PubMed]
- 8.Verducci JS, Melfi VF, Lin S, Wang Z, Roy S, Sen CK (2006) Microarray analysis of gene expression: considerations in data mining and statistical treatment. Physiol Genomics 25:355–363 [DOI] [PubMed]
- 9.Jin Y, Kim HP, Ifedigbo E, Lau LF, Choi AM (2005) Cyr61 protects against hyperoxia-induced cell death via Akt pathway in pulmonary epithelial cells. Am J Respir Cell Mol Biol 33:297–302 [DOI] [PubMed]
- 10.Kobashi C, Urakaze M, Kishida M, Kibayashi E, Kobayashi H, Kihara S, Funahashi T, Takata M, Temaru R, Sato A, Yamazaki K, Nakamura N, Kobayashi M (2005) Adiponectin inhibits endothelial synthesis of interleukin-8. Circ Res 97:1245–1252. See also related comment by Touyz RM, Circ Res 97:1216–1219 [DOI] [PubMed]
- 11.Kim HB, Kong M, Kim TM, Suh YH, Kim WH, Lim JH, Song JH, Jung MH (2006) NFATc4 and ATF3 negatively regulate adiponectin gene expression in 3T3-L1 adipocytes. Diabetes 55:1342–1352 [DOI] [PubMed]
- 12.Kim KY, Kim JK, Han SH, Lim JS, Kim KI, Cho DH, Lee MS, Lee JH, Yoon DY, Yoon SR, Chung JW, Choi I, Kim E, Yang Y (2006) Adiponectin is a negative regulator of NK cell cytotoxicity. J Immunol 176:5958–5964 [DOI] [PubMed]
- 13.Si W, Kang Q, Luu HH, Park JK, Luo Q, Song WX, Jiang W, Luo X, Li X, Yin H, Montag AG, Haydon RC, He TC (2006) CCN1/Cyr61 is regulated by the canonical Wnt signal and plays an important role in Wnt3A-induced osteoblast differentiation of mesenchymal stem cells. Mol Cell Biol 26:2955–2964 [DOI] [PMC free article] [PubMed]
- 14.Okamoto Y, Kihara S, Funahashi T, Matsuzawa Y, Libby P (2006) Adiponectin: a key adipocytokine in metabolic syndrome. Clin Sci (London) 110:267–278 [DOI] [PubMed]
- 15.Kaynak B, von Heydebreck A, Mebus S, Seelow D, Hennig S, Vogel J, Sperling HP, Pregla R, Alexi-Meskishvili V, Hetzer R, Lange PE, Vingron M, Lehrach H, Sperling S (2003) Genome-wide array analysis of normal and malformed human hearts. Circulation 107:2467–2474 [DOI] [PubMed]
- 16.Sharma UC, Pokharel S, Evelo CT, Maessen JG (2005) A systematic review of large scale and heterogeneous gene array data in heart failure. J Mol Cell Cardiol 38:425–432 [DOI] [PubMed]
- 17.Yang J, Moravec CS, Sussman MA, DiPaola NR, Fu D, Hawthorn L, Mitchell CA, Young JB, Francis GS, McCarthy PM, Bond M (2000) Decreased SLIM1 expression and increased gelsolin expression in failing human hearts measured by high-density oligonucleotide arrays. Circulation 102:3046–3052 [DOI] [PubMed]
- 18.Tan FL, Moravec CS, Li J, Apperson-Hansen C, McCarthy PM, Young JB, Bond M (2002) The gene expression fingerprint of human heart failure. Proc Natl Acad Sci USA 99:11387–11392 [DOI] [PMC free article] [PubMed]
- 19.Grzeskowiak R, Witt H, Drungowski M, Thermann R, Hennig S, Perrot A, Osterziel KJ, Klingbiel D, Scheid S, Spang R, Lehrach H, Ruiz P (2003) Expression profiling of human idiopathic dilated cardiomyopathy. Cardiovasc Res 59:400–411 [DOI] [PubMed]
- 20.Kittleson MM, Ye SQ, Irizarry RA, Minhas KM, Edness G, Conte JV, Parmigiani G, Miller LW, Chen Y, Hall JL, Garcia JG, Hare JM (2004) Identification of a gene expression profile that differentiates between ischemic and nonischemic cardiomyopathy. Circulation 110:3444–3451 [DOI] [PubMed]
- 21.Yung CK, Halperin VL, Tomaselli GF, Winslow RL (2004) Gene expression profiles in end-stage human idiopathic dilated cardiomyopathy: altered expression of apoptotic and cytoskeletal genes. Genomics 83:281–297 [DOI] [PubMed]
- 22.Kittleson MM, Minhas KM, Irizarry RA, Ye SQ, Edness G, Breton E, Conte JV, Tomaselli G, Garcia JG, Hare JM (2005) Gene expression analysis of ischemic and nonischemic cardiomyopathy: shared and distinct genes in the development of heart failure. Physiol Genomics 21:299–307 [DOI] [PubMed]
- 23.Margulies KB, Matiwala S, Cornejo C, Olsen H, Craven WA, Bednarik D (2005) Mixed messages: transcription patterns in failing and recovering human myocardium. Circ Res 96:592–599 [DOI] [PubMed]
- 24.Beisvag V, Lehre PK, Midelfart H, Aass H, Geiran O, Sandvik AK, Laegreid A, Komorowski J, Ellingsen O (2006) Aetiology-specific patterns in end-stage heart failure patients identified by functional annotation and classification of microarray data. Eur J Heart Fail 8:381–389 [DOI] [PubMed]
- 25.Lowes BD, Zolty R, Minobe WA, Robertson AD, Leach S, Hunter L, Bristow MR (2006) Serial gene expression profiling in the intact human heart. J Heart Lung Transplant 25:579–588 [DOI] [PMC free article] [PubMed]
- 26.Kittleson MM, Minhas KM, Irizarry RA, Ye SQ, Edness G, Breton E, Conte JV, Tomaselli G, Garcia JG, Hare JM (2005) Gene expression in giant cell myocarditis: altered expression of immune response genes. Int J Cardiol 102:333–340 [DOI] [PubMed]
- 27.Perbal B (2004) CCN proteins: multifunctional signalling regulators. Lancet 363:62–64 [DOI] [PubMed]
- 28.Todorovic V, Chen CC, Hay N, Lau LF (2005) The matrix protein CCN1 (CYR61) induces apoptosis in fibroblasts. J Cell Biol 171:559–568 [DOI] [PMC free article] [PubMed]
- 29.Kim KH, Min YK, Baik JH, Lau LF, Chaqour B, Chung KC (2003) Expression of angiogenic factor Cyr61 during neuronal cell death via the activation of c-Jun N-terminal kinase and serum response factor. J Biol Chem 278:13847–13854 [DOI] [PubMed]
- 30.Kim SM, Park JH, Chung SK, Kim JY, Hwang HY, Chung KC, Jo I, Park SI, Nam JH (2004) Coxsackie virus B3 infection induces cyr61 activation via JNK to mediate cell death. J Virol 78:13479–13488 [DOI] [PMC free article] [PubMed]
- 31.Chen CC, Mo FE, Lau LF (2001) The angiogenic factor Cyr61 activates a genetic program for wound healing in human skin fibroblasts. J Biol Chem 276:47329–47337 [DOI] [PubMed]
- 32.Nguyen N, Kuliopulos A, Graham RA, Covic L (2006) Tumor-derived Cyr61 (CCN1) promotes stromal matrix metalloproteinase-1 production and protease-activated receptor 1-dependent migration of breast cancer cells. Cancer Res 66:2658–2665 [DOI] [PubMed]
- 33.Grzeszkiewicz TM, Lindner V, Chen N, Lam SC, Lau LF (2002) The angiogenic factor cysteine-rich 61 (CYR61, CCN1) supports vascular smooth muscle cell adhesion and stimulates chemotaxis through integrin α6β1 and cell surface heparan sulfate proteoglycans. Endocrinology 143:1441–1450 [DOI] [PubMed]
- 34.Grzeszkiewicz TM, Kirschling DJ, Chen N, Lau LF (2001) CYR61 stimulates human skin fibroblast migration through Integrin αvβ5 and enhances mitogenesis through integrin αvβ3, independent of its carboxyl-terminal domain. J Biol Chem 276:21943–21950 [DOI] [PubMed]
- 35.Schober JM, Chen N, Grzeszkiewicz TM, Jovanovic I, Emeson EE, Ugarova TP, Ye RD, Lau LF, Lam SC (2002) Identification of integrin alpha(M)beta(2) as an adhesion receptor on peripheral blood monocytes for Cyr61 (CCN1) and connective tissue growth factor (CCN2): immediate-early gene products expressed in atherosclerotic lesions. Blood 99:4457–4465 [DOI] [PubMed]
- 36.Xie D, Yin D, Tong X, O’Kelly J, Mori A, Miller C, Black K, Gui D, Said JW, Koeffler HP (2004) Cyr61 is overexpressed in gliomas and involved in integrin-linked kinase-mediated Akt and beta-catenin-TCF/Lef signaling pathways. Cancer Res 64:1987–1996 [DOI] [PubMed]
- 37.Hilfiker-Kleiner D, Kaminski K, Kaminska A, Fuchs M, Klein G, Podewski E, Grote K, Kiian I, Wollert KC, Hilfiker A, Drexler H (2004) Regulation of proangiogenic factor CCN1 in cardiac muscle: impact of ischemia, pressure overload, and neurohumoral activation. Circulation 109:2227–2233 [DOI] [PubMed]
- 38.Shibata R, Ouchi N, Ito M, Kihara S, Shiojima I, Pimentel DR, Kumada M, Sato K, Schiekofer S, Ohashi K, Funahashi T, Colucci WS, Walsh K (2004) Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat Med 10:1384–1389 [DOI] [PMC free article] [PubMed]
- 39.Shibata R, Sato K, Pimentel DR, Takemura Y, Kihara S, Ohashi K, Funahashi T, Ouchi N, Walsh K (2005) Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat Med 11:1096–1103. See also related comment by Yuhki K et al, Nat Med 11:1048–1049 [DOI] [PMC free article] [PubMed]
- 40.Pineiro R, Iglesias MJ, Gallego R, Raghay K, Eiras S, Rubio J, Dieguez C, Gualillo O, Gonzalez-Juanatey JR, Lago F (2005) Adiponectin is synthesized and secreted by human and murine cardiomyocytes. FEBS Lett 579:5163–5169 [DOI] [PubMed]
- 41.Nian M, Lee P, Khaper N, Liu P (2004) Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res 94:1543–1553 [DOI] [PubMed]
- 42.Prabhu SD (2004) Cytokine-induced modulation of cardiac function. Circ Res 95:1140–1153 [DOI] [PubMed]
- 43.Pisabarro MT, Leung B, Kwong M, Corpuz R, Frantz GD, Chiang N, Vandlen R, Diehl LJ, Skelton N, Kim HS, Eaton D, Schmidt KN (2006) Cutting edge: novel human dendritic cell- and monocyte-attracting chemokine-like protein identified by fold recognition methods. J Immunol 176:2069–2073 [DOI] [PubMed]
- 44.Schaerli P, Willimann K, Ebert LM, Walz A, Moser B (2005) Cutaneous CXCL14 targets blood precursors to epidermal niches for Langerhans cell differentiation. Immunity 23:331–342 [DOI] [PubMed]
- 45.Shurin GV, Ferris R, Tourkova IL, Perez L, Lokshin A, Balkir L, Collins B, Chatta GS, Shurin MR (2005) Loss of new chemokine CXCL14 in tumor tissue is associated with low infiltration by dendritic cells (DC), while restoration of human CXCL14 expression in tumor cells causes attraction of DC both in vitro and in vivo. J Immunol 174:5490–5498 [DOI] [PubMed]
- 46.Vargas-Leal V, Bruno R, Derfuss T, Krumbholz M, Hohlfeld R, Meinl E (2005) Expression and function of glial cell line-derived neurotrophic factor family ligands and their receptors on human immune cells. J Immunol 175:2301–2308 [DOI] [PubMed]
- 47.Kolls JK, Linden A (2004) Interleukin-17 family members and inflammation. Immunity 21:467–476 [DOI] [PubMed]
- 48.Starnes T, Broxmeyer HE, Robertson MJ, Hromas R (2002) Cutting edge: IL-17D, a novel member of the IL-17 family, stimulates cytokine production and inhibits hemopoiesis. J Immunol 169:642–646 [DOI] [PubMed]
- 49.Ruddy MJ, Wong GC, Liu XK, Yamamoto H, Kasayama S, Kirkwood KL, Gaffen SL (2004) Functional cooperation between interleukin-17 and tumor necrosis factor-alpha is mediated by CCAAT/enhancer-binding protein family members. J Biol Chem 279:2559–2567 [DOI] [PubMed]
- 50.Takahashi H, Numasaki M, Lotze MT, Sasaki H (2005) Interleukin-17 enhances bFGF-, HGF- and VEGF-induced growth of vascular endothelial cells. Immunol Lett 98:189–193 [DOI] [PubMed]
- 51.Prause O, Bozinovski S, Anderson GP, Linden A (2004) Increased matrix metalloproteinase-9 concentration and activity after stimulation with interleukin-17 in mouse airways. Thorax 59:313–317 [DOI] [PMC free article] [PubMed]
- 52.Hite R, Seeds M, Safta A, Jacinto R, Gyves J, Bass D, Waite B (2004) Lysophospholipid generation and phosphatidylglycerol depletion in phospholipase A2-mediated surfactant dysfunction. Am J Physiol-Lung Cell Mol Physiol 288:L618–L624 [DOI] [PubMed]
- 53.Jo E, Lee H-Y, Lee Y-N, Kim J, Kang H-K, Park D-W, Baek S-H, Kwak J-Y, Bae Y-S (2004) Group IB secretory phospholipase A2 stimulates CXC chemokine ligand 8 production via ERK and NF-κB in human neutrophils. J Immunol 173:6433–6439 [DOI] [PubMed]
- 54.Noutsias M, Seeberg B, Schultheiss H, Kuhl U (1999) Expression of cell adhesion molecules in dilated cardiomyopathy: evidence for endothelial activation in inflammatory cardiomyopathy. Circulation 99:2124–2131 [DOI] [PubMed]
- 55.Noutsias M, Fechner H, Jonge Hd, Wang X, Dekkers D, Houtsmuller A, Pauschinger M, Bergelson J, Warraich R, Yacoub M, Hetzer R, Lamers J, Schultheiss H, Poller W (2001) Human coxsackie-adenovirus-receptor is co-localized with integrins αvβ3 and αvβ5 on the cardiomyocyte sarcolemma and upregulated in dilated cardiomyopathy-implications for cardiotropic viral infections. Circulation 104:275–280 [DOI] [PubMed]
- 56.Macian F (2005) NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol 5:472–484 [DOI] [PubMed]
- 57.Inagaki K, Fuess S, Storm TA, Gibson GA, McTiernan CF, Kay MA, Nakai H (2006) Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Molec Ther 14:45–53 [DOI] [PMC free article] [PubMed]
- 58.Wang Z, Zhu T, Qiao C, Zhou L, Wang B, Zhang J, Chen C, Li J, Xiao X (2005) Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat Biotechnol 23:321–328 [DOI] [PubMed]
- 59.Fechner H, Suckau L, Kurreck J, Sipo I, Wang X, Pinkert S, Loschen S, Rekittke J, Weger S, Dekkers D, Vetter R, Erdmann VA, Schultheiss H-P, Paul M, Lamers J, Poller W (2006) Highly efficient and specific modulation of cardiac calcium homeostasis by adenovector-derived short hairpin RNA targeting phospholamban. Gene Ther advance online publication, October 5 (in press) [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Below is the link to the electronic supplementary material.
The disease phenotyping of the DCMi patient group (Table 1 and Patients and methods) that were investigated by genomewide Affymetrix microarray expression profiling included (in addition to clinical and molecular virological characterization) an immunohistochemical characterization of cardiac immune cell infiltration and vascular endothelial activation. The immunohistochemical studies were done on EMBs obtained by the same standard procedure as those used for the Affymetrix microarray expression profiling and for the TaqMan quantitative RT-PCRs (264.6 KB) (226.1 KB) (159.6 KB)
Real-time quantitative TaqMan PCR data on shifts in gene expression in the DCMi patients versus the control group. TaqMan data on the above novel DCMi-associated genes were obtained by analysis of residual RNA extracted from the same EMB as already used for the genomic Affymetrix microarrays and confirmed the respective data from the microarray study reported in detail in Table 2, Fig. 2 (panels a–f), and http://www.ncbi.nlm.nih.gov/projects/geo/ in all cases. Sharp sign denotes p < 0.05 between the study groups. In addition to these new genes, the standard heart failure-associated genes NPPA and NPPB were significantly (p < 0.05) upregulated (18.7-fold and 39.0-fold, respectively) in the DCMi group as assessed by TaqMan PCR, confirming a trend observed in the microarrays (7.5-fold at q = 6.400 and 20.5-fold at q = 5.722; (PDF 22 kb).
Extension of Table 2 with all shifts in cardiac gene expression down to 1.5-fold change for DCMi patient group vs Cont control group, as assessed by Affymetrix microarray analysis of RNA extracted from EMB. For further details of the DCMi and Cont groups, see Table 1 and Patients and methods. The complete Affymetrix dataset is available from the GEO website http://www.ncbi.nlm.nih.gov/ projects/geo/. GEO experimental series number was assigned as GSE4172 (PDF 1 mb).