

Abstract
MicroRNAs (miRNAs) participate in diverse cellular and physiological processes through the post-transcriptional gene regulatory pathway. Hairpin is a crucial structural feature for the computational identification of precursor miRNAs (pre-miRs), as its formation is critically associated with the early stages of the mature miRNA biogenesis. Our incomplete knowledge about the number of miRNAs present in the genomes of vertebrates, worms, plants, and even viruses necessitates thorough understanding of their sequence motifs, hairpin structural characteristics, and topological descriptors. In this in-depth study, we investigate a comprehensive and heterogeneous collection of 2241 published (nonredundant) pre-miRs across 41 species (miRBase 8.2), 8494 pseudohairpins extracted from the human RefSeq genes, 12,387 (nonredundant) ncRNAs spanning 457 types (Rfam 7.0), 31 full-length mRNAs randomly selected from GenBank, and four sets of synthetically generated genomic background corresponding to each of the native RNA sequence. Our large-scale characterization analysis reveals that pre-miRs are significantly different from other types of ncRNAs, pseudohairpins, mRNAs, and genomic background according to the nonparametric Kruskal–Wallis ANOVA (p < 0.001). We examine the intrinsic and global features at the sequence, structural, and topological levels including %G+C content, normalized base-pairing propensity P(S), normalized minimum free energy of folding MFE(s), normalized Shannon entropy Q(s), normalized base-pair distance D(s), and degree of compactness F(S), as well as their corresponding Z scores of P(S), MFE(s), Q(s), D(s), and F(S). The findings will promote more accurate guidelines and distinctive criteria for the prediction of novel pre-miRs with improved performance.
Keywords: precursor microRNAs, minimum free energy of folding, Shannon entropy, Z-scores, second eigenvalue
INTRODUCTION
MicroRNAs (miRNAs) constitute an abundant class of endogenous and small (~21–23-nucleotides [nt]) regulatory ncRNA molecules that mediate post-transcriptionally the production of intracellular proteins in most eukaryotes (Ambros 2001; Bartel 2004; Mallory and Vaucheret 2004). The pioneers lin-4 and let-7 miRNAs were first discovered in 1993 and 2000 as key post-transcriptional modulators for the developmental transitions in early larval Caenorhabditis elegans (Banerjee and Slack 2002). Thereafter, an emerging body of experimental evidence substantiated that miRNAs are potential key regulators for diverse developmental and physiological processes such as C. elegans lsy-6 determining the left–right asymmetry of chemo-receptor expression (Johnston and Hobert 2003); Drosophila melanogaster miR-14 miRNA being involved in apoptosis, stress resistance, and fat metabolism (Xu et al. 2003); D. melanogaster bantam repressing the gene hid associated with apoptosis and proliferation (Brennecke et al. 2003); Mus musculus miR-181a modulating hematopoietic differentiation (Chen et al. 2004); M. musculus miR-196 inducing directed cleaving of Hox-B8 transcripts (Yekta et al. 2004); Arabidopsis thaliana miRNAs regulating the expression of transcription factor genes (Li and Zhang 2005); and viral-encoded miRNAs hijacking the host immune defense to sustain their viral replication and pathogenesis (Grey et al. 2005; Pfeffer et al. 2004, 2005; Samols et al. 2005). This dynamic range of biological findings underscores the functional importance of miRNAs and the need for expanding our limited knowledge concerning them.
The emerging model of miRNA maturation involves six (five) compartmentalized steps in vertebrates (plants) (Anthony and Peter 2005; Kim 2005). Briefly, (1) the majority of the primary miRNAs (pri-miRs) are transcribed by RNA polymerase II (Pol-II) into long primary transcripts from the polycistronic genes residing in the intergenic regions that overlap with the introns of protein-coding genes (Lee et al. 2002) or from the exons of the pseudo-ncRNA genes (Rodriguez et al. 2004). (2) These capped and polyadenylated pri-miRs are processed in the nucleus by an endonuclease RNase III Drosha/Pasha complex yielding ~60–120-nt intermediate precursor transcripts (pre-miRs) in vertebrates. Conversely, Dicer-like 1 enzyme (DCL1; a plant ortholog of Drosha) performs two cleavage steps in the nucleus: plant pri-miRs → ~80–200-nt pre-miRs → miR:miR* (Anthony and Peter 2005). (3) Vertebrate and plant pre-miRs possessing characteristic imperfect and extended hairpin structures are exported into the cytoplasm by the Exportin-5 in a Ran-GTP-dependent manner or by HASTY, the ortholog of Exportin-5 (Zhang et al. 2006a). (4) Cytoplasmic RNase III-type endonuclease Dicer excises the vertebrate pre-miR into ~22–23-nt asymmetric mature miRNA duplex miR:miR*. Plant miR:miR* contains more base-pairings and has tighter length distribution centering on 21 nt (Anthony and Peter 2005). (5) The strand miR with the less thermostable 5′ termini is favorably incorporated into a ribonucleoprotein to form an RNA-induced silencing complex (RISC) (Cullen 2004; Tijsterman and Plasterk 2004; Tang 2005). (6) The RISC represses post-transcriptionally the expression of the targeted gene by translational arrest of protein synthesis via imperfect complementarity at the 3′-untranslated regions (Moss et al. 1997; Reinhart et al. 2000; Doench and Sharp 2004) or mRNA cleavage degradation with near-perfect complementarity of ≤3 mismatches at the protein-coding regions of mRNAs (Yekta et al. 2004) primarily in vertebrates and plants, respectively (Anthony and Peter 2005).
Existing approaches for identifying pre-miRs or miRNA genes
Two broad strategies for identifying systematically novel miRNAs exist, in vivo and in silico screening (Ambros et al. 2003; Berezikov et al. 2006). The former, based on expression screening, commences with the isolation of distinct ~22-nt RNA transcripts. This is followed by intensive direct cloning and sequencing efforts of cDNA libraries derived from the size-fractionated small RNAs (Lagos-Quintana et al. 2001, 2002; Lau et al. 2001; Lee and Ambros 2001). Such experimental routes are neither exhaustive nor straightforward in discovering all the known miRNAs. Notably, not all miRNAs are well expressed in tissues, cell types, and developmental stages that have been sampled (Lagos-Quintana et al. 2001). Existing cloning methods are highly biased toward abundantly and/or ubiquitously expressed miRNAs that usually dominate the cloned products, rendering the isolation of novel miRNAs difficult (Lagos-Quintana et al. 2001, 2002; Lau et al. 2001; Lee and Ambros 2001). Novel miRNAs tend to be elusive, as they are expressed constitutively in low abundance or they have preferentially restrictive/specific temporal (cell-phase) and spatial (tissue-/cell-type) expression patterns. To express them sufficiently for cloning efforts under controlled cellular conditions and nonabundant cell types is technically involved. In principle, this issue can be overcome by running high-throughput deep-sequencing technology like massively parallel signature sequencing (MPSS) (Brenner et al. 2000) on appropriately pooled biological samples.
Computational strategies have been applied to C. elegans (Grad et al. 2003; Lim et al. 2003b), D. melanogaster (Lai et al. 2003), A. thaliana, Oryza sativa (Bonnet et al. 2004a; Jones-Rhoades and Bartel 2004; Wang et al. 2004; Adai et al. 2005), Homo sapiens (Berezikov et al. 2005, 2006; Lim et al. 2003a), and viruses (Pfeffer et al. 2004, 2005; Grey et al. 2005; Samols et al. 2005) for identifying candidate miRNAs. In part, they were extensively developed to overcome technical hurdles faced experimentally (Berezikov et al. 2006; Zhang et al. 2006a). Particularly, breakdown products of mRNA transcripts in the background and endogenous ncRNAs (e.g., tRNAs and rRNAs) as well as exogenous siRNAs are dominant players coexisting in the small RNA samples isolated from the cytoplasmic total RNA extracts. To thwart designating these fragments erroneously as novel miRNAs, cloned small RNAs are assessed computationally to identify their genomic location (Lai et al. 2003; Lim et al. 2003a,b; Adai et al. 2005; Fu et al. 2005; Cummins et al. 2006; Wheeler et al. 2006). A critical and necessary feature for mature miRNAs biogenesis is that they reside primarily on one arm of the pre-miRs that form characteristic imperfect hairpin structures. This criterion indicates that only those small RNA sequences occupying the ~20-nt matched regions on one arm of the hairpin precursors should be curated as novel miRNAs after experimentally validating them. The short sequence length of miRNAs, however, confers relatively low specificity whereby matching regions are readily encoded in an overwhelming number of unwanted genomic segments that can potentially fold into hairpin structures. Genome-wide screening for novel pre-miRs is technically complicated, considering that the hairpin structures are not unique to miRNAs exclusively. These dysfunctional inverted repeats (or pseudohairpins) are genomically prevalent in H. sapiens (1.1 × 107) (Bentwich et al. 2005) and C. elegans (4.4 × 104) (Pervouchine et al. 2003) genomes; only 462 and 114 bona fide pre-miRs, respectively, have been discovered according to miRBase 8.2 (Griffiths-Jones et al. 2006).
The majority of the pseudohairpins can be removed by comparative genomic techniques like MiRscan (Lim et al. 2003a,b), MIRcheck (Jones-Rhoades and Bartel 2004), miRFinder (Bonnet et al. 2004a), miRseeker (Lai et al. 2003), findMiRNA (Adai et al. 2005), PalGrade (Bentwich et al. 2005), and MiRAlign (Wang et al. 2005). Typically, conserved regions are first identified by aligning the entire genome of phylogenetically related species and masking out those regions most unlikely to be occupied by miRNAs (e.g., tRNAs and rRNAs). Sliding windows of the unmasked regions are folded at both strands by Mfold (Zuker 2003) or RNAfold (Hofacker 2003), two commonly used RNA secondary structure predictors. The folds are scored according to their minimum free energy of folding (MFE), length of the symmetric/asymmetric regions, and size of the terminal loop. The composite scores are thresholded, and those high-ranking ones deemed similar to pre-miRs published in miRBase (Griffiths-Jones et al. 2006) are then reserved for further experimental validation. Evidently, extensive genomics data sets for computationally intensive multiple genome alignments are involved, rendering identification of miRNAs impossible, especially for organisms whose closest relatives have partial or yet-to-start sequenced genomes. Furthermore, species-specific pre-miRs encoded in pathogenic viruses such as the Kaposi sarcoma-associated herpesvirus, Mouse gammaherpesvirus 68, and Human cytomegalovirus are likely to remain elusive to comparative-based detection, as they share little or no sequence homologies among themselves or with the host pre-miRs (Grey et al. 2005; Pfeffer et al. 2004, 2005; Samols et al. 2005).
Several (quasi) de novo state-of-the-art predictors have been extensively developed to aid the discovery of nonconserved pre-miRs and to surmount the technical drawbacks of comparative approaches. Typically, they first decompose the individual pre-miR into modularized RNA substructures comprising dangling termini, asymmetric or symmetric stem, and terminal loop. Derived from these specific regions is a complex array of sequences (e.g., nucleotide composition) and structural characteristics (e.g., thermodynamic stability). This is fashioned analogously to the protein-coding gene identification techniques that scan the genomic regions for signature signals of protein-coding genes without relying on external transcripts or genomic sequences. A supervised machine-learning classification algorithm, e.g., support vector machine (SVM), is trained on a binary-labeled positive set of genuine pre-miRs and a negative set of pseudohairpins. Through this inductive learning on their feature vectors, a classifier model and a set of decision rules are devised to discriminate between them.
An inaugural and definitive work (Pfeffer et al. 2005; Sewer et al. 2005) compiled 40 distinctive sequence and structural features from the hairpins without relying on comparative genomics information—stem length, length of the longest symmetrical region, number of complementary base pairs (bp) in the “relaxed symmetry” region, MFE, number of nucleotides in symmetrical and asymmetrical loops in the “relaxed symmetry” region, and the average size of the asymmetrical loops. The SVM classifier model trained with the experimental domain knowledge recovered 71.00% of the positive pre-miRs with a remarkably low false-positive rate of ~3.00%. The accuracy was improved to ~90.00% in human and up to 90.00% for other species by another de novo classifier, Triplet-SVM (Xue et al. 2005). It encoded the local contiguous structure-sequence features of known pre-miRs as a set of 32 triplet elements—a nucleotide type and three continuous substructures, e.g., “A(((” and “G(..”. Despite its methodological simplicity, promising performances, and independence of comparative genomics information, Triplet-SVM was largely limited to classifying RNA secondary structures not containing multiple loops. Another SVM-based approach, RNAmicro (Hertel and Stadler 2006), incorporating sequence and structural information as part of its feature vector, reported incredibly promising efficiencies of 91.16% and 99.47% for sensitivity and specificity, respectively. Still, its classification pipeline required computationally expensive multiple sequence alignments for inputs. ProMiR (Nam et al. 2005) took advantage of a probabilistic co-learning model, the hidden Markov model (HMM), to classify miRNA genes based on their pair-wise aligned sequences. It minimized the false-positive rate to as low as 4.00%, but compromised for a poorer performing sensitivity of only 73.00%. A relatively recent work, BayesMIRfinder (Yousef et al. 2006), adopted naive Bayesian induction (NBI) as its underlying classifier model. Notwithstanding its technical novelty, BayesMIRfinder relied on the comparative analysis of conserved genomics regions for post-processing to yield a considerably higher sensitivity of 97.00% and comparable specificity of 91.00% in mouse to existing algorithms.
Motivation and overview of study
Generally, the efficiency and reliability of classifiers for distinguishing species-specific and evolutionary well-conserved pre-miRs from genomic pseudohairpins and most types of ncRNAs depend largely on the size and selection of both the specific features and the relevant data samples. Existing (quasi) de novo attempts are still limited to and have far from satisfactory predictive performances, hampered largely by the difficulties of deriving and selecting appropriate features from pre-miRs. Proper feature selection should facilitate a more controllable generalization and scalability to new testing samples as well as provide more robust predictive ability to the underlying machine-learning algorithms.
To develop a true de novo predictor that can achieve highly accurate identification and classification of promising precursor transcripts as putative pre-miRs within a single genome, wholly independent of phylogenetic conservation, still entails numerous unforeseeable technical issues. Most notable of these is the previous lack of data and inconclusive findings from existing literature on the features that distinctively distinguish pre-miRs from pseudohairpins and other types of ncRNAs. Motivated by this incomplete knowledge and the many miRNAs present in the genomes of vertebrates, worms, plants, and even viruses not yet discovered, we conduct a large-scale characterization study. It comprehensively comprises a heterogeneous collection of 2241 published and nonredundant pre-miRs across 41 species (miRBase 8.2), 8494 pseudohairpins extracted from the human RefSeq genes, 12,387 nonredundant ncRNAs spanning 457 types (Rfam 7.0), 31 full-length mRNAs randomly selected from GenBank, and four sets of synthetically generated genomic background corresponding to each of the native RNA sequence. Hairpin is a crucial structural prerequisite for the computational identification of pre-miR, as its formation is critically associated with the early stages of the mature miRNA biogenesis. To elucidate the unique hairpin folding of an entire pre-miR, our in-depth statistical study focuses solely on their intrinsic and global features at the sequence, structural, and topological levels. The combinatoric features include %G+C content, normalized base-pairing propensity P(S), normalized minimum free energy of folding MFE(s), normalized Shannon entropy Q(s), normalized base-pair distance D(s), and degree of compactness F(S), as well as their corresponding Z scores of P(S), MFE(s), Q(s), D(s), and F(S). The findings will facilitate and promote more accurate guidelines and distinctive criteria for the prediction of authentic pre-miRs with improved performances.
RESULTS AND DICUSSION
Among the arthropoda, nematoda, vertebrata, viridiplantae, and viruses available from miRBase 8.2 (Griffiths-Jones et al. 2006), no orthologous miRNA gene shared by vertebrates and plants has ever been reported (Anthony and Peter 2005). Pathogenic viral-encoded pre-miRs present in K. sarcoma-associated virus, M. γ-herpesvirus 68, and H. cytomegalovirus should be treated as exceptions, although they have also been demonstrated to share significant sequence homology neither with known host pre-miRs nor among themselves (Grey et al. 2005; Pfeffer et al. 2005; Samols et al. 2005). Viral-encoded pre-miRs do not possess genes homologous to host miRNA processing proteins, e.g., Drosha, Dicer, and RISC, but are likely to hijack these proteins to facilitate their viral replication after infecting the host cells (Sarnow et al. 2006). Despite the apparent similarities of miRNA biogenesis between vertebrates and plants, their evolutionarily ancient processing pathways (≥400 million years ago) were not operating in a common ancestor and could have evolved independently from a more ancient system (Anthony and Peter 2005). We will focus on vertebrate and plant pre-miRs for discussion, as they are likely to exhibit distinct folding features that warrant careful structural analysis. Data are available to deduce conclusions about arthropoda, nematoda, and virus pre-miRs. (Supplemental Materials can be found at http://web.bii.a-star.edu.sg/~stanley/Publications.)
Vertebrate and plant pre-miRs have significantly distinct MFEI2, MFEI1, %G+C, P(S), MFE(s), Q(s), D(s), and F(S) from ncRNAs and mRNAs
Foremost, the sequence length (in nucleotides) differs considerably between and among pre-miRs (vertebrate, 90.4522 ± 0.4164 and plants, 137.9175 ± 2.0309), ncRNAs (frameshift, 53.2599 ± 0.2543 to IRES, 276.0841 ± 2.4342), and mRNA (332.3226 ± 16.3064) (Fig. 1A, Figure 3A, top heat map, see below; Supplemental Table S1). The sequence lengths of ncRNAs and mRNAs are strongly and positively correlated with their MFEs, as previously demonstrated (Seffens and Digby 1999; Bonnet et al. 2004b; Zhang et al. 2006b). Longer sequence length results in a greater degree of freedom such that the native RNA sequences can fold into complex secondary structures with corresponding higher thermostability or lower MFEs. By normalizing the MFE with the sequence length, the normalized MFE, MFE(s), ensures that it serves as a comparable measure without unduly penalizing the shorter pre-miRs or favoring the longer mRNAs (Seffens and Digby 1999; Freyhult et al. 2005; Zhang et al. 2006b). In agreement with earlier findings (Freyhult et al. 2005; Zhang et al. 2006b), vertebrate and plant pre-miRs possess statistically distinct MFE(s) of −0.4308 ± 0.0025 and −0.4456 ± 0.0038 (p < 0.001) and are the lowest except frameshift (−0.4814 ± 0.0023). Interestingly, a single criterion based on a variant of MFE(s) greater than a threshold value ε = 0.68 has been applied to genome-wide detection of C. elegans pre-miRs (Pervouchine et al. 2003). This yielded ~4.4 × 104 stable hairpins localized to ~4.00% of the genome, covering 64.29% (36/56) of the published ones (Lau et al. 2001).
FIGURE 1.

(Continued on next page)
FIGURE 1.

Distribution profiles for the 2241 nonredundant pre-miRs (Griffiths-Jones et al. 2006), 12,387 nonredundant ncRNAs (Griffiths-Jones et al. 2005), and 31 mRNAs (Freyhult et al. 2005). (A) Nine metrics are Length, MFEI2, MFEI1, %G+C, P(S), MFE(s), Q(s), D(s), and F(S). (B) zG, zQ, zD, zP, and zF, i.e., normalized forms of MFE(s), Q(s), D(s), P(S), and F(S) using the four sequence randomization algorithms. The horizontal dashed line indicates the Z score at zero. For A and B, box lines indicate the lower quartile, median, mean (statistical values are provided in Supplemental Tables S1 and S2), and upper quartile; whisker lines extend to the most extreme data value or at most 1.5 times the box height; outliers beyond the fifth and 95th percentiles are not shown.
FIGURE 3.

(A) Heat map of 1203 vertebrate and 606 plants pre-miRs versus 12,387 nonredundant ncRNAs (Griffiths-Jones et al. 2005) and 31 mRNAs (Freyhult et al. 2005). (B) Heat map of 2241 nonredundant pre-miRs (Griffiths-Jones et al. 2006) versus 8494 pseudohairpins (Xue et al. 2005). For A and B, e.g., zGM/D/Z/F denotes zG with respect to mono- and d-nucleotide shuffling, zero- and first-order Markov model; green represents statistically different median, red for no statistical difference, gray for ties, according to the nonparametric Kruskal–Wallis one-way ANOVA and Dunn's method of multiple comparisons tests at p < 0.001 and p < 0.01. (C) Correlation between Q(s), D(s), zQ, and zD for 2241 nonredundant pre-miRs; zQ, and zD correspond to dinucleotide shuffling; r indicates Pearson correlation coefficients Cp. The Pearson Cp, Spearman rank Cs (ranks based), and Kendall's Ck (relative ranks based) correlation coefficients for all the metrics and sequence randomization methods studied in this work are provided in Supplemental Table S3.
Vertebrate and plant pre-miRs possess the significantly highest normalized base-pairing propensity P(S) of 0.3518 ± 0.0009 and 0.3545 ± 0.0013 (p < 0.001), accounting for ~70.36–70.9% of their nucleotides forming complementary base pairings within their highly thermostable hairpin structures. A similar >72.00% for P(S) has also been reported, corroborating our findings, albeit a smaller data set of 513 plant pre-miRs across seven species was analyzed (Zhang et al. 2006b). The presence of more hydrogen bonds and base-pairings in the plant pre-miRs might benefit their recognition, processing, and nucleus-cytoplasm transport (Zhang et al. 2006b). Emerging experimental evidence also points to the hairpin motif of vertebrate pre-miRs as a critical feature for miRNA maturation (Zeng and Cullen 2004). Human pre-miR-30 binding by Exportin-5 involved recognition of almost the entire hairpin, except the terminal loop (Zeng and Cullen 2004). A hairpin structure >16 bp was required for detectable binding and >18 bp for high-affinity binding such that the stacking of contiguous paired nucleotides tended to reduce the MFE of the overall folded structure for greater thermostability. Contrary to the common belief that the unpaired regions tended to disrupt the RNA structure with greater MFE, deleting the 2-nt bulge of pre-miR-30 left the binding unaffected or reduced binding modestly, unless the stem length was suboptimal. There was negligible or no significant effect on the correct recognition for varying sizes of the terminal loop, until it was shortened from the normal 15–4 nt. Besides nuclear export of pre-miR, the binding of Exportin-5 served to stabilize the pre-miR in the nucleus and during export by inhibiting the in vitro exonucleolytic cleavage (Zeng and Cullen 2004).
Vertebrate and plant pre-miRs encode higher %A+U content than %G+C content of 48.3079 ± 0.2504 and 46.6719 ± 0.3513; similarly observed (Zhang et al. 2006b). The higher %A+U content in the plant pre-miRs (likewise for vertebrate pre-miRs) might possibly serve as a biochemical signal for miRNA biogenesis by the RISC (Zhang et al. 2006b). We also report that the %G+C contents for vertebrate and plant pre-miRs are not considerably different from mRNAs (50.4626 ± 1.4654) and common families of ncRNAs like cis-regulator (48.9672 ± 0.1188), frameshift (46.4785 ± 0.1477), riboswitch (50.5054 ± 0.3381), thermoregulator (42.6490 ± 3.2009), HACA-box snoRNA (46.3048 ± 0.3160), splicing RNA (47.6933 ± 0.3731), sRNA (46.3963 ± 0.3513), tRNA (48.2725 ± 0.3541), and intron (44.7871 ± 0.8350). Unlike the %G+C content, the MFEI1 [divides MFE(s) by %G+C content, a newly proposed folding energy score to analyze plant pre-miRs; Zhang et al. 2006b] for vertebrate and plant pre-miRs of −0.0091 ± 0.0001 and −0.0096 ± 0.0001 are statistically highest (p < 0.001) except anti-sense (−0.0083 ± 0.0001) and frameshift (−0.0104 ± 0.0000). Our finding and another (Zhang et al. 2006b) point to the MFEI1 as a potential discriminative criterion to distinguish pre-miRs from mRNAs and ncRNAs, which a recent comparative classifier RNAmicro has included into its feature set (Hertel and Stadler 2006).
Notably, vertebrate pre-miRs possess statistically higher normalized Shannon entropy Q(s) and normalized base-pair distance D(s) of 0.1161 ± 0.0025 and 0.0431 ± 0.0009 than plant pre-miRs of 0.1424 ± 0.0036 and 0.0502 ± 0.0011 (p < 0.001). Generally, RNA sequences having relatively high values of both advanced folding measures are either unstructured or long in length, which fold with the assistance of accessory proteins or have a repertoire of alternative (pseudoknot) structures (Freyhult et al. 2005). This suggests that vertebrate pre-miRs will likely fold into well-defined hairpins restricted to relatively fewer alternative conformations, possibly due to shorter sequence length (90.4522 ± 0.4164 nt) compared to plants (137.9175 ± 2.0309 nt). The different “structureness” of vertebrate and plant pre-miRs causes the former to display the significantly lowest and distinct Q(s) and D(s) (p < 0.001) except anti-sense (0.1336 ± 0.0061 and 0.0468 ± 0.0020). The latter is not significantly unique from cis-regulator (0.2124 ± 0.0021 and 0.0689 ± 0.0006), frameshift (0.1396 ± 0.0024 and 0.0552 ± 0.0009), anti-sense (0.1336 ± 0.0061 and 0.0468 ± .0020), snRNA (0.2305 ± 0.0260 and 0.0741 ± 0.0074), and intron (0.1802 ± 0.0089 and 0.0620 ± 0.0026). Maturation of the plant miR:miR* duplex is performed exclusively by Dicer-like 1 enzyme (DCL1) via two cleavage steps, pri-miR → pre-miR → miR:miR*, within the nucleus. In contrast to vertebrates (Anthony and Peter 2005; Zhang et al. 2006a), the two reactions are compartmentalized and directed separately by the nuclear Drosha (pri-miR → pre-miR) and cytoplasmic Dicer (pre-miR → miR:miR*). Moreover, plant pre-miRs are less conserved (conservation of plant mature miRNAs is well preserved) than those in vertebrates (Anthony and Peter 2005; Zhang et al. 2006a). Our structural analysis substantiates both experimental findings, pointing to the plant pre-miRs as very transient molecules (Zhang et al. 2006a) that possess less “structureness” indicative of lower Q(s) and D(s) compared to their vertebrate counterparts.
Finally, we analyzed two newly proposed topological measures, i.e., degree of compactness F(S) and MFEI2 [divides MFE(s) by number of stems S]. Vertebrate pre-miRs have a significantly higher F(S) of 0.2197 ± 0.0042 than plant pre-miRs of 0.1251 ± 0.0033 (p < 0.001). Generally, RNAs possessing lower F(S) have less structured folds (Barash 2003, 2004) like mRNAs (0.0391 ± 0.0059). Both vertebrate and plant pre-miRs fold into topologically distinct structures with F(S) being statistically different (p < 0.001) but not the extreme among mRNAs (0.0391 ± 0.0059) and common families of ncRNAs like frameshift (0.8865 ± 0.0079), IRES (0.0442 ± 0.0013), anti-sense (0.3734 ± 0.0133), rRNA (0.0933 ± 0.0020), snRNA (0.5372 ± 0.0415), and tRNA (0.5333 ± 0.0093). The other folding measure, MFEI2, was inspired by the formation of the critical hairpin structure in the early stages of miRNA maturation. Reasonably, MFE should be largely localized to the stem(s) within the hairpin such that the higher MFEI2 corresponds to greater thermostability per stem. The MFEI2 for vertebrate and plant pre-miRs of −0.0761 ± 0.0013 and −0.0539 ± 0.0010 are significantly different (p < 0.001) except anti-sense (−0.0811 ± 0.0030), snRNA (−0.0764 ± 0.0088), and tRNA (−0.0676 ± 0.0007), cis-regulator (−0.0793 ± 0.0017), snRNA (−0.0764 ± 0.0088), and intron (−0.0604 ± 0.0029).
In summary, the 1203 vertebrate and 606 plant pre-miRs are statistically distinct from 12,387 ncRNAs and 31 mRNAs according to the measures MFEI2, MFEI1, %G+C, P(S), MFE(s), Q(s), D(s), and F(S). Except two recent published works investigating 513 plant pre-miRs (Zhang et al. 2006b) and 135 pre-miRs from different species (Freyhult et al. 2005), we are unaware of any larger-scale and in-depth statistical analysis highlighting these results on the folding characteristics of published pre-miRs.
Vertebrate and plant pre-miRs have significantly distinct Z scores of MFE(s), Q(s), D(s), P(S), and F(S) compared to the ncRNAs and mRNAs
Evolutionarily conserved vertebrate and plant pre-miRs possess the considerably lowest zG (p < 0.001) except frameshift and anti-sense, regardless of the sequence randomization algorithms (Fig. 1B, Figure 3A, bottom heat map, see below; Supplemental Table S2). Our finding and another (Freyhult et al. 2005) affirm the hypothesis that pre-miRs fold into highly thermostable secondary structures with significantly lower MFEs relative to their synthetically generated sequence randomized controls (Workman and Krogh 1999; Bonnet et al. 2004b). Therefore this unique structural characteristic of vertebrate and plant pre-miRs is not expected to occur by chance; it is indispensable for correct recognition and processing by Dicer-like enzymes (Bonnet et al. 2004b). Earlier works (Workman and Krogh 1999; Bonnet et al. 2004b) were inconclusive, as their dinucleotide shuffling algorithms were heuristically based, and the resulting shuffled RNAs might not guarantee preserving the exact dinucleotide frequencies as the native RNAs (Clote et al. 2005). Instead, we used a considerably larger data set of pre-miRs and ncRNAs as well as the exact “Altschul–Erickson algorithm” (Altschul and Erickson 1985) for synthesizing 104 dinucleotide-shuffled RNAs. Two computational studies (Washietl and Hofacker 2004; Clote et al. 2005) also demonstrated that structural ncRNAs displayed lower MFEs than dinucleotide-shuffled RNAs, but pre-miRs were not analyzed.
Both zQ and zD of vertebrate and plant pre-miRs are statistically different (p < 0.001) and are the lowest except anti-sense, irrespective of the sequence randomization algorithms. A recent computational study reported that pre-miRs and ncRNAs (like hammerhead ribozyme type III and tRNAs) possessed significantly fewer k-locally optimal structures (potential kinetic traps) than their dinucleotide-shuffled RNAs (Clote 2005). Both findings suggest pre-miRs are likely to undergo evolutionary pressure in adopting relatively fewer alternative folds of significantly lower MFEs than the random background, in order to function properly in the post-transcriptional gene regulatory pathway.
Vertebrate and plant pre-miRs report the significantly highest zP (p < 0.001); i.e., more complementary base-pairings are present in their RNA secondary structures than the genomic background, irrespective of the sequence randomization methods. They also have statistically distinct zF (p < 0.001) except common families of ncRNAs like cis-regulator, IRES, thermoregulator, CD-box snoRNA, and HACA-box snoRNA, as well as mRNAs.
In summary, the 1203 vertebrate and 606 plant pre-miRs are significantly different from the 12,387 ncRNAs and 31 mRNAs, after examining their zG, zQ, zD, zP, and zF based on four sequence randomization algorithms and 104 random sequences corresponding to each native RNA sequence. This statistical finding confirms that to reliably identify pre-miRs from the genomic background requires more than their possessing characteristic and well-defined secondary structures of statistically significant MFEs (Rivas and Eddy 2000; Washietl and Hofacker 2004).
Comparison with previous studies on structural folding analysis of ncRNAs and mRNAs
For completeness of this large-scale study, we outline three notable points to revisit previous works investigating whether ncRNAs and mRNAs fold into statistically significant and thermodynamically stable secondary structures (Fig. 1B, Figure 3A, bottom heat map, see below; Supplemental Table S2). First, 51 mRNAs had significantly lower MFEs than their corresponding sets of 10 mononucleotide-shuffled RNAs (Seffens and Digby 1999) and a subset of 46 mRNAs did not display any statistically lower MFEs than their corresponding sets of 10 dinucleotide-shuffled RNAs (Workman and Krogh 1999). Our study (mononucleotide shuffling, −0.7223 ± 0.2089 and dinucleotide shuffling, 0.1021 ± 0.1625) and another using dinucleotide shuffling (Freyhult et al. 2005) support both previous conclusions (Seffens and Digby 1999; Workman and Krogh 1999). Unique to this work, we observe that the mRNAs have considerably lower MFEs than the genomic background for the zero-order Markov model (−0.4770 ± 0.1098), but not for the first-order Markov model (−0.0830 ± 0.0845).
Second, our investigated 1114 tRNAs possess significantly lower MFEs than the genomic background for the four sequence randomization methods. This finding agrees with earlier results (Washietl and Hofacker 2004; Clote et al. 2005; Freyhult et al. 2005) that relied on dinucleotide-shuffled RNAs, but differs from another work (Workman and Krogh 1999) in which the dinucleotide-shuffling algorithm was heuristically based, as previously explained (Clote et al. 2005). We report similar findings for hammerhead ribozyme type III (Washietl and Hofacker 2004; Clote et al. 2005), spliceosomal RNAs (Washietl and Hofacker 2004; Clote et al. 2005), riboswitches (Clote et al. 2005), and introns (Washietl and Hofacker 2004) that have considerably lower MFEs than corresponding sets of dinucleotide-shuffled RNA sequences.
Third, previously discussed (Workman and Krogh 1999; Bonnet et al. 2004b; Clote et al. 2005), the controls serving as the genomic background would give erroneous conclusions if they destroyed certain nonrandom compositions of the native sequence. Our results highlight that detectable systematic biases of zG distribution profiles exist among the four sequence randomization algorithms. Generally, the mean zG for pre-miRs, ncRNAs, and mRNAs are ordered from the lowest mononucleotide shuffling, marginally below those of dinucleotide shuffling, followed by the zero- and first-order Markov model. This result agrees with earlier works (Workman and Krogh 1999; Bonnet et al. 2004b; Clote et al. 2005) in which disrupting the naturally occurring biases in the inherent dinucleotide frequencies of the sequences base composition should be avoided for determining the significance of secondary structure. Preserving the dinucleotide frequencies of the native sequences is critical so as not to affect the critical energy contributions of stacked base pairs and the corresponding accuracy of the RNA structural predictions (Workman and Krogh 1999; Bonnet et al. 2004b; Clote et al. 2005).
Vertebrate and plant pre-miRs are significantly different from pseudohairpins
To elucidate the unique folding of pre-miRs present in vertebrates and plants, we repeat the preceding two statistical experiments by evaluating them against 8494 pseudohairpins instead of ncRNAs and mRNAs. Pseudohairpins are genomic inverted repeats extracted from the protein-coding regions of human RefSeq genes with no known alternative splicing (AS) events. They were first introduced as negative samples in Triplet-SVM (Xue et al. 2005), a de novo classifier based on triplet-encoding features, e.g., “A(((” and “G(..”. However, no structural analysis or comparison to published pre-miRs has been reported about them.
Generally, the vertebrate and plant pre-miRs have significantly higher P(S) and F(S) as well as lower MFEI2, MFEI1, %G+C, MFE(s), Q(s), and D(s) than pseudohairpins (p < 0.001) (Fig. 2A; Fig. 3B, top heat map; Supplemental Table S1). The distribution profiles of vertebrate and plant pre-miRs for zG, zQ, zD, and zP differ distinctively from pseudohairpins (p < 0.001), irrespective of the sequence randomization algorithms (Fig. 2B; Fig. 3B, bottom heat map; Supplemental Table S2). Unlike pseudohairpins, pre-miRs tend to fold into secondary structures with significantly higher thermodynamic structural stability (lower zG), fewer alternative folds (lower zQ and zD), and more base-pairings (higher zP). Except plants, vertebrate pre-miRs clearly have significantly higher zF (more compactness) than pseudohairpins (p < 0.001).
FIGURE 2.

Distribution profiles for the 2241 nonredundant pre-miRs (Griffiths-Jones et al. 2006) and 8494 pseudohairpins (Xue et al. 2005). (A) Nine metrics are Length, MFEI2, MFEI1, %G+C, P(S), MFE(s), Q(s), D(s), and F(S). (B) zG, zQ, zD, zP, and zF, i.e., normalized forms of MFE(s), Q(s), D(s), P(S), and F(S) using the four sequence randomization algorithms. The horizontal dashed line indicates Z-score at zero. For A and B, box lines indicate the lower quartile, median, mean (statistical values are provided in Supplemental Tables S1 and S2), and upper quartile; whisker lines extend to the most extreme data value or at most 1.5 times the box height; outliers beyond the fifth and 95th percentiles are not shown.
In summary, both findings invalidate conclusively the hypothesis that pseudohairpins share a comparable degree of structural folding characteristics with known vertebrate and plant pre-miRs. Our statistical results clearly point to the MFEI2, MFEI1, %G+C, P(S), MFE(s), Q(s), D(s), and F(S) as well as zG, zQ, zD, zP, and zF as potential discriminative descriptors. They effectively expand the triplet-encoding features in Triplet-SVM (Xue et al. 2005) to classify more accurately the genuine pre-miRs from pseudohairpins in genome-wide screens.
Correlation between folding measures
We conducted correlation tests on 2241 nonredundant known pre-miRs according to the following metrics: Length, MFEI2, MFEI1, %G+C, P(S), MFE(s), Q(s), D(s), and F(S) as well as the zG, zQ, zD, zP, and zF [normalized forms of MFE(s), Q(s), D(s), P(S), and F(S) using the four sequence randomization algorithms] (Fig. 3C; Supplemental Table S3). The Pearson correlation coefficients Cp are also validated against Spearman rank Cs (ranks based) and Kendall's Ck (relative ranks based) correlation coefficients, as Cs and Ck are extremely robust to non-normal distribution.
Generally, all of the metrics are weakly (|Cp| < 0.4) and moderately (0.4 < |Cp| < 0.9) correlated except Q(s), D(s), zQ, and zD, regardless of the sequence randomization algorithms. Both Q(s) and D(s) are computed from the McCaskill base-pair probability pij (Freyhult et al. 2005), explaining the strong quasilinear relationship (Cp ≥ 0.9) for the two pairs Q(s) and D(s) as well as their corresponding normalized form zQ and zD. There exist moderate Pearson correlations within the three pairs MFE(s) and zG, P(S), and zP, as well as F(S) and zF for the four sequence randomization algorithms. We initially expected Q(s) and zQ as well as D(s) and zD to behave similarly. Interestingly and currently unclear is why a strong association is observed within them. As a guide for future studies, especially where computational resources are limited, only Q(s) instead of D(s) should be included (Freyhult et al. 2005), while zQ and zD are extremely time consuming to compute beyond 103 random RNA sequences.
CONCLUSIONS
In this large-scale investigation characterizing the entire hairpin structure of known precursor miRNAs (pre-miRs), we have demonstrated that they (notably, vertebrate and plant pre-miRs) possess a set of 13 statistically significant global features. Our in silico findings have greatly advanced our understanding of miRNA functions and biogenesis in relation to their structural features and distinct folding patterns. A definitive criterion for identifying and classifying accurately promising precursor transcripts as bona fide pre-miRs while discriminating against abundant pseudohairpins within a single genome has not yet been discovered. Moreover, discriminative features used in existing (quasi) de novo classifiers have achieved far from satisfactory specificity and sensitivity, especially when cross-species conservation is unavailable. Our investigated features relating to the intrinsic folding and topological characteristics of pre-miRs can potentially serve as discriminative measures in improving the designs and performances of current de novo predictors. We have incorporated the 13 features into the development of a new and better performing de novo classifier for identifying species-specific and nonconserved pre-miRs, wholly independent of phylogenetic conservation.
MATERIALS AND METHODS
Biologically relevant data sets
Precursor miRNA sequences
We retrieved 4028 curated pre-miRs spanning 45 species from miRBase Registry Database version 8.2 (Griffiths-Jones et al. 2006) as of July 2006. As strong sequence homologies exist among pre-miRs both within a single and between different species, the original data set was filtered to 90% identity using a greedy incremental clustering algorithm (Li and Godzik 2006). Briefly, all the sequences were first sorted in order of decreasing length, where the longest one became the representative of the first cluster. Each remaining sequence was compared with the existing representatives and grouped into their cluster if the similarity with any representative was above a given threshold (i.e., 0.9); otherwise, that sequence became the representative of a new cluster. Consequently, we analyzed 2241 nonredundant pre-miRs spanning 41 species categorized into arthropoda, nematoda, vertebrata, viridiplantae, and viruses (Table 1); none belonging to Gorilla gorilla, Macaca nemestrina, Pan paniscus, and Pongo pygmaeus were retained.
TABLE 1.
Biologically relevant data sets and annotation information

TABLE 1.
Continued

Functional noncoding RNA sequences
We retrieved all available seed ncRNA sequences from Rfam Database version 7.0 (Griffiths-Jones et al. 2005) as of March 2005. After removing 46 types of pre-miRs, we analyzed 12,387 curated seed ncRNAs spanning 457 types categorized into 16 families (Table 1). These prokaryotic and eukaryotic ncRNAs have a length distribution similar to the known pre-miRs, and can fold with hairpins or stem–loops (Eddy 2001; Storz 2002; Svoboda and Cara 2006). Briefly, cis-regulatory elements are a well-conserved untranslated mRNA leader region capable of adopting alternate structural conformations that result in transcription termination or transcription elongation into the downstream region. For example, the T-box leader regulates transcription of many bacterial aminoacyl-tRNA synthetases, amino acid biosynthesis, and amino acid transport genes using uncharged tRNA as the effector (Winkler et al. 2001). The internal ribosome entry site (IRES) is a nucleotide sequence that allows for translation initiation in the middle of an mRNA. It mimics the 5′-cap structure, critical for the assembly of the initiation complex. Riboswitches are highly conserved RNA regulatory elements, embedded within the 5′-untranslated region (UTR) of biosynthesis genes or operons, and cis modulate their expressions upon binding to metabolite (e.g., guanine and thiamine pyrophosphate), without involving protein cofactors (Hesselberth and Ellington 2002; Lai 2003; Stormo 2003; Winkler and Breaker 2003; Mandal and Breaker 2004; Nudler and Mironov 2004; Soukup and Soukup 2004; Vitreschak et al. 2004). Thermoregulators are cis-regulatory elements commonly found in the 5′ UTR of mRNAs, whose secondary structure is regulated by temperature. For example, the structural motif of PrfA thermoregulator represses translation at 30°C by masking the Shine–Dalgarno sequence, but conformational change frees it for ribosome binding to allow maximal translation when the temperature rises to 37°C (Johansson et al. 2002). Anti-senses are characterized by a long hairpin structure interrupted by several unpaired residues or bulged loops, involved in negative regulation. For instance, the micF gene is a Escherichia coli stress response gene encoding an untranslated 93-nt anti-sense that binds to its target ompF mRNA (of the outer membrane porin gene) (Delihas and Forst 2001). It regulates ompF expression post-transcriptionally by causing translational repression. Ribozymes (e.g., the Hepatitis δ-virus ribozyme and Hammerhead ribozyme) possess endonuclease function and catalyze a range of reactions such as self-cleavage of hepatitis δ-virus transcript (Puerta-Fernandez et al. 2003). Small nucleolar RNAs (snoRNAs) can be functionally divided into C/D snoRNAs or H/ACA snoRNAs acting as guides for site-specific 2′-O-ribose methylation or as guides for pseudouridylation in the post-transcriptional processing of rRNAs (Weinstein and Steitz 1999). Spliceosomal RNAs (splicing RNAs), e.g., U1–2 and U4–6 (Storz et al. 2005), are small nuclear RNAs constituting the spliceosome that process pre-mRNA into mRNA by excising the intronic regions. Transfer RNAs (tRNAs) exist as ~54–93-nt hydrogen-bonded cloverleaf structures, involved in transporting amino acids to the site of protein synthesis during translation (Sprinzl and Vassilenko 2005). Group I/II intron RNAs are large self-splicing ribozymes catalyzing their own excision from mRNA, tRNA, and rRNA precursors (Cech 1990; Bonen and Vogel 2001).
mRNA sequences
We investigated 31 mRNA sequences that tend to fold into complex RNA structures with extremely negative MFEs (Table 1; Freyhult et al. 2005). They were randomly selected from the GenBank DNA database (Benson et al. 2005), as previously reported (Freyhult et al. 2005).
Pseudohairpin sequences
We analyzed 8494 pseudohairpins from the protein-coding regions (CDSs) according to the UCSC refGene annotation tables (Karolchik et al. 2003) and human RefSeq genes (Pruitt and Maglott 2001) without any known experimentally validated alternative splicing (AS) events, as described earlier (Xue et al. 2005). These genomic inverted repeats are analogous to but do not encode genuine human pre-miRs, by displaying similar distribution in terms of their length, hairpin structures, and MFEs. They possess ≥18 bp, including the GU wobble pairs, MFE ≤−15 kcal/mol, and fold without multiple loops in their RNA structures.
Random sequences
Four sets of R = 104 shuffled or randomized RNAs, r n = r1r2…rL, serving as the genomic background, are synthesized from each nth native RNA sequence s n = s1s2…sL, using four sequence randomization algorithms. L is the length of sequence in nucleotides and si∈∑ = (A, C, G, U) is the biochemical nucleotide at the ith position.
Mononucleotide shuffling
We implemented the “Fisher–Yates shuffle algorithm” that sequentially swaps the mononucleotides at all positions of s n with another at a randomly selected position. It consumes Θ(LlogL) bits and runs in linear time. The order of the shuffled nucleotides is truly random, preserving the mono- but not the dinucleotide frequencies.
Vars: s n ← r n.
For i ← L: 1, do
j ← uniform(1, i).
If i ≠ j, then swap(ri, rj).
Dinucleotide shuffling
Previous algorithms (Workman and Krogh 1999; Bonnet et al. 2004b) were heuristically based, and the shuffled RNA sequences might not guarantee to preserve correctly the exact mono- and dinucleotide frequencies as the native RNA. We implemented the exact “Altschul–Erickson algorithm” (Altschul and Erickson 1985) such that it shuffles s n while preserving exactly both the mono- and dinucleotide frequencies. The native and shuffled sequences always share the same first and last nucleotides (Coward 1999). The order of the shuffled nucleotides is “less random” due to fewer possible dinucleotide preserving permutations.
For each r ∈ r n, do
create an edge-list Lr of edge-pairs (r, x) with nucleotides r and x occurring as a dinucleotide rx in s n.
For each r ≠ rL ∈ r n, do
E(s n) ← select randomly an edge-pair from Lr. E(s n) contains at most three edge-pairs.
G ← (V, E) is the last-edge graph such that (r, x) ∈ V and (r, x) ∈ E(s n). If any vertex in G is not connected to rL, then go to (3). Else, go to (6) as all vertices are connected in G to rL.
For each r ∈ r n, do
permute the remaining edge-pairs in Lb − E(s n), Lr ← Lr ∪ E(s n).
Vars: r1 ← s1.
For i ← 1: L − 1, do
generate ri+1 such that (ri, ri+1) ∈ Lr.
Zero-order Markov model
A new random sequence r n is formed by iteratively adding nucleotide ri sampled with expected mononucleotide frequencies F(∑, s n). The sequence r n is “truly” random, and its mononucleotide frequencies fluctuate about the native ones.
Compute F(∑, s n) from s n.
For i ← 1: L, do
ri ← sampling with F(∑, s n).
First-order Markov model
A new random sequence r n is formed by first choosing a nucleotide r1 sampled with expected mononucleotide frequencies F(∑, s n). Iteratively add the next nucleotide ri+1 sampled with conditional probabilities P(ri+1|ri); i.e., the probability of occurrence of a nucleotide at a particular position depends only on the previous nucleotide. The sequence is “truly” random, and its dinucleotide frequencies fluctuate around the native ones.
Compute F(∑, s n) and G(∑ 1, ∑ 2, s n) from s n.
r1 ← sampling with F(∑, s n).
For i ← 2: L, do
ri ← sampling with P(∑ 2 |∑ 1) = G(∑ 1, ∑ 2, s n)/F(∑ 1, s n).
RNA folding measures
Normalized base-pairing propensity, P(S), measures the total number of base pairs present in the RNA secondary structure S normalized to the sequence length L (Schultes et al. 1999). P(S) removes the bias that a long sequence tends to have more base pairs. It ranges [0.0, 0.5], 0.0 for no base-pair interactions and 0.5 for a maximum of L/2 base pairs.
Normalized minimum free energy of folding
MFE(s), for sequence s is the lowest MFE for the most favorable conformation from a vast population of predicted RNA secondary structures, normalized to the sequence length L (Seffens and Digby 1999; Freyhult et al. 2005). MFE(s) removes the bias that a long sequence tends to have lower MFE. Alternatively, adjusted MFE (AMFE) refers to MFE(s)×100 nt (Zhang et al. 2006b).
MFE Index 1
MFEI1, is the ratio of MFE(s) and %G+C content (Zhang et al. 2006b).
Normalized Shannon entropy
Q(s) in Equation (1), characterizes the base-pairing probability distribution (BPPD) per base in a sequence s as a chaotic dynamical system (Huynen et al. 1997; Schultes et al. 1999; Freyhult et al. 2005). The local dominance of a single structure within the Boltzmann distribution of alternative secondary structures is strongly correlated with the reliability of the MFE structure. Low values of Q(s) correspond to the BPPD that are dominated by a single or a few base-pairing probabilities. These bases are better predicted than those having multiple alternative states.
 |
Here, the McCaskill base-pair probability pij is the probability of base-pairing between bases i and j. δijα = 1 if xi pairs with xj, 0 otherwise. RNAs exist in vivo as an ensemble of secondary structures Sα∈S(s) following the Boltzmann distribution probability P(Sα) (Mathews 2004).
Normalized base-pair distance
D(s) in Equation (2), is the base-pair distance for all pairs of structures Sα and Sβ on s (Moulton et al. 2000; Freyhult et al. 2005).
 |
Here, the number of base pairs not shared by them is given by 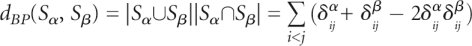 . The number of base pairs in Sα is
. The number of base pairs in Sα is 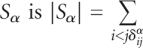 . Definitions of pij and δijα follow those of Q(s) in Equation (1).
. Definitions of pij and δijα follow those of Q(s) in Equation (1).
Second (or the Fiedler) eigenvalue
F(S) in Equation (3) measures the compactness of a tree-graph G = (V, E) (Fera et al. 2004; Gan et al. 2004). At the coarsest scale, each vertex v ∈ V represents a bulge loop, hairpin loop, internal loop, the 5′ and 3′ unpaired termini, or the multibranch loop; each edge e ∈ E denotes an RNA stem. F(S) is computed from the Laplacian matrix L(G), which is a mathematical representation of the tree-graph G. F(S) can be used as a similarity measure among a collection of RNA secondary structures.
MFE Index 2
MFEI2, is the ratio of MFE(s) and the number of stems S, which are structural motifs containing more than three contiguous base pairs.
Z score of RNA folding measure
The Z score of the RNA folding measure is described in Equation (4). The Z score Z(s n) for the structural biases observed in a native RNA is computed via a Monte Carlo randomization approach (Workman and Krogh 1999; Bonnet et al. 2004b; Clote et al. 2005). It normalizes the feature S(s n) of nth native RNA sequence s n in terms of the units of standard deviations by which S(s n) differs from the mean of inferred R = 104 randomized RNA sequences r n. The corresponding Z scores of MFE(s), Q(s), D(s), P(S), and F(S) are denoted as zG, zQ, zD, zP, and zF.
Here Si(r n) is the computed feature for the ith random RNA sequence of r n; μn and σn are the sample mean and the standard deviation of the feature S(s n) for R random RNA sequences r n.
Statistical analysis
Computing the RNA folding measures and Z scores
The most favorable RNA secondary structure for a given sequence and its normalized minimum free energy of folding MFE(s) are determined via RNAfold included in Vienna RNA Package 1.4 (Hofacker 2003), an implementation of Zuker's free energy minimization algorithm (Zuker and Stiegler 1981; Zuker 2003) with Turner energy parameters (Mathews et al. 1999). From the predicted structure, the intrinsic folding quantitative measures P(S), Q(s), and D(s) are computed by the perl script genRNAStats.pl interfaced to the module RNAlib of Vienna RNA Package 1.4 (Hofacker 2003). The topological descriptors S and F(S) are determined using an algorithm RNAspectral (see Supplemental materials for details). The normalized variants zP, zG, zQ, zD, and zF are computed in a similar manner using genRNARandomStats.pl, after generating the four sets of random RNA sequences with genRandomRNA.pl. All intensive computations are performed on three clusters of 192 dual-core computational nodes.
Statistical analysis measuring the differences inherent within pre-miRs’ global structural and intrinsic stability features
To compare the data sets and compute the probability that the samples are drawn from the same distribution, we conduct either nonparametric Kruskal–Wallis one-way analysis of variance (ANOVA) or nonparametric Mann–Whitney–Wilcoxon (Wilcoxon rank-sum). The former tests for statistically significant differences in the median values (p < 0.001) among the experimental groups against the control are greater than would be expected by chance. To isolate the groups that differ from the control, Dunn's method of multiple comparisons test is conducted at p < 0.01. It does not include an adjustment for ties but allows the sample sizes of the experimental groups to be different. The latter method tests for statistically significant differences in the median values between two experimental groups (p < 0.001). Unlike parametric statistical tests like Student's t-test, both ANOVA and Wilcoxon rank-sum compare the ranks of the data values instead of the actual data values. Thus, they are robust to samples drawn from populations with non-normal distribution or which have unequal variances (Systat SigmaPlot 9.0 and SigmaStat 3.11).
Correlation of quantitative metrics
To quantify the correlation between measures for native pre-miRs, the Pearson correlation coefficients Cp(f, g) in Equation (5) are computed, statistically significant at p < 0.001. We are aware that Cp is not robust to outliers and to non-Gaussian distributions, as it assumes a pseudo-Gaussian distribution of the data set. Thus, we also validate the results of Cp against those of nonparametric Spearman-rank Cs (ranks based) and Kendall's Ck (relative ranks based) correlation metrics. Both Cs and Ck are robust to samples containing outliers or drawn from populations with unequal variances, non-normality distribution, and nonlinearity (Mathworks Matlab 7.1).
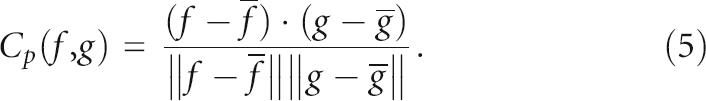 |
SUPPLEMENTAL DATA
The Supplemental details on RNAspectral and Tables S1–3, as well as the data sets (Fasta format files), raw statistical results (tab-delimited format and Excel files), and source codes are publicly available at http://web.bii.a-star.edu.sg/~stanley/Publications. Use of source codes is free purely for nonprofit or academic purpose adhering to the GNU General Public License (GPL) at http://www.gnu.org/copyleft/gpl.html.
ACKNOWLEDGMENTS
The authors are indebted to the Theoretical Biochemistry Group of the Institute for Theoretical Chemistry at the University of Vienna for their public release of the Vienna RNA software package, providing core components to our analysis. We thank the two anonymous reviewers for their thorough comments and constructive suggestions that greatly improved the quality and technical ideas developed in this article. Sincere appreciation to the technical team of BII's Clustering Group for their best effort in ensuring the three clusters of 192 dual-core computational nodes run smoothly. This work was supported by the Bioinformatics Institute. S.N.K.L. received Ph.D. scholarship funds from the Agency for Science, Technology and Research (A*STAR), Singapore. S.N.K.L. and S.K.M. conceived the initial ideas. S.N.K.L. designed and performed the experiments. S.N.K.L. and S.K.M. wrote this manuscript.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.223807.
REFERENCES
- Adai, A., Johnson, C., Mlotshwa, S., Archer-Evans, S., Manocha, V., Vance, V., Sundaresan, V. Computational prediction of miRNAs in Arabidopsis thaliana . Genome Res. 2005;15:78–91. doi: 10.1101/gr.2908205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S.F., Erickson, B.W. Significance of nucleotide sequence alignments: A method for random sequence permutation that preserves dinucleotide and codon usage. Mol. Biol. Evol. 1985;2:526–538. doi: 10.1093/oxfordjournals.molbev.a040370. [DOI] [PubMed] [Google Scholar]
- Ambros, V. microRNAs: Tiny regulators with great potential. Cell. 2001;107:823–826. doi: 10.1016/s0092-8674(01)00616-x. [DOI] [PubMed] [Google Scholar]
- Ambros, V., Bartel, B., Bartel, D.P., Burge, C.B., Carrington, J.C., Chen, X., Dreyfuss, G., Eddy, S.R., Griffiths-Jones, S., Marshall, M., et al. A uniform system for microRNA annotation. RNA. 2003;9:277–279. doi: 10.1261/rna.2183803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony, A.M., Peter, M.W. Plant and animal microRNAs: Similarities and differences. Funct. Integr. Genomics. 2005;5:129–135. doi: 10.1007/s10142-005-0145-2. [DOI] [PubMed] [Google Scholar]
- Banerjee, D., Slack, F. Control of developmental timing by small temporal RNAs: A paradigm for RNA-mediated regulation of gene expression. Bioessays. 2002;24:119–129. doi: 10.1002/bies.10046. [DOI] [PubMed] [Google Scholar]
- Barash, D. Deleterious mutation prediction in the secondary structure of RNAs. Nucleic Acids Res. 2003;31:6578–6584. doi: 10.1093/nar/gkg872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barash, D. Spectral decomposition for the search and analysis of RNA secondary structure. J. Comput. Biol. 2004;11:1169–1174. doi: 10.1089/cmb.2004.11.1169. [DOI] [PubMed] [Google Scholar]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Benson, D.A., Karsch-Mizrachi, I., Lipman, D.J., Ostell, J., Wheeler, D.L. GenBank. Nucleic Acids Res. 2005;33:D34–D38. doi: 10.1093/nar/gki063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentwich, I., Avniel, A., Karov, Y., Aharonov, R., Gilad, S., Barad, O., Barzilai, A., Einat, P., Einav, U., Meiri, E., et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat. Genet. 2005;37:766–770. doi: 10.1038/ng1590. [DOI] [PubMed] [Google Scholar]
- Berezikov, E., Guryev, V., van de, B.J., Wienholds, E., Plasterk, R.H., Cuppen, E. Phylogenetic shadowing and computational identification of human microRNA genes. Cell. 2005;120:21–24. doi: 10.1016/j.cell.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Berezikov, E., Cuppen, E., Plasterk, R.H. Approaches to microRNA discovery. Nat. Genet. 2006;38(Suppl):S2–S7. doi: 10.1038/ng1794. [DOI] [PubMed] [Google Scholar]
- Bonen, L., Vogel, J. The ins and outs of group II introns. Trends Genet. 2001;17:322–331. doi: 10.1016/s0168-9525(01)02324-1. [DOI] [PubMed] [Google Scholar]
- Bonnet, E., Wuyts, J., Rouze, P., Van de Peer, Y. Detection of 91 potential conserved plant microRNAs in Arabidopsis thaliana and Oryza sativa identifies important target genes. Proc. Natl. Acad. Sci. 2004a;101:11511–11516. doi: 10.1073/pnas.0404025101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet, E., Wuyts, J., Rouze, P., Van de Peer, Y. Evidence that microRNA precursors, unlike other non-coding RNAs, have lower folding free energies than random sequences. Bioinformatics. 2004b;20:2911–2917. doi: 10.1093/bioinformatics/bth374. [DOI] [PubMed] [Google Scholar]
- Brennecke, J., Hipfner, D.R., Stark, A., Russell, R.B., Cohen, S.M. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila . Cell. 2003;113:25–36. doi: 10.1016/s0092-8674(03)00231-9. [DOI] [PubMed] [Google Scholar]
- Brenner, S., Johnson, M., Bridgham, J., Golda, G., Lloyd, D.H., Johnson, D., Luo, S., McCurdy, S., Foy, M., Ewan, M., et al. Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nat. Biotechnol. 2000;18:630–634. doi: 10.1038/76469. [DOI] [PubMed] [Google Scholar]
- Cech, T.R. Self-splicing of group I introns. Annu. Rev. Biochem. 1990;59:543–568. doi: 10.1146/annurev.bi.59.070190.002551. [DOI] [PubMed] [Google Scholar]
- Chen, C.Z., Li, L., Lodish, H.F., Bartel, D.P. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–86. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- Clote, P. RNALOSS: A web server for RNA locally optimal secondary structures. Nucleic Acids Res. 2005;33:W600–W604. doi: 10.1093/nar/gki382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clote, P., Ferre, F., Kranakis, E., Krizanc, D. Structural RNA has lower folding energy than random RNA of the same dinucleotide frequency. RNA. 2005;11:578–591. doi: 10.1261/rna.7220505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coward, E. Shufflet: Shuffling sequences while conserving the k-let counts. Bioinformatics. 1999;15:1058–1059. doi: 10.1093/bioinformatics/15.12.1058. [DOI] [PubMed] [Google Scholar]
- Cullen, B.R. Transcription and processing of human microRNA precursors. Mol. Cell. 2004;16:861–865. doi: 10.1016/j.molcel.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Cummins, J.M., He, Y., Leary, R.J., Pagliarini, R., Diaz, L.A., Jr., Sjoblom, T., Barad, O., Bentwich, Z., Szafranska, A.E., Labourier, E., et al. The colorectal microRNAome. Proc. Natl. Acad. Sci. 2006;103:3687–3692. doi: 10.1073/pnas.0511155103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delihas, N., Forst, S. MicF: An anti-sense RNA gene involved in response of Escherichia coli to global stress factors. J. Mol. Biol. 2001;313:1–12. doi: 10.1006/jmbi.2001.5029. [DOI] [PubMed] [Google Scholar]
- Doench, J.G., Sharp, P.A. Specificity of microRNA target selection in translational repression. Genes & Dev. 2004;18:504–511. doi: 10.1101/gad.1184404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy, S.R. Non-coding RNA genes and the modern RNA world. Nat. Rev. Genet. 2001;2:919–929. doi: 10.1038/35103511. [DOI] [PubMed] [Google Scholar]
- Fera, D., Kim, N., Shiffeldrim, N., Zorn, J., Laserson, U., Gan, H., Schlick, T. RAG: RNA-As-Graphs web resource. BMC Bioinformatics. 2004;5:88. doi: 10.1186/1471-2105-5-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyhult, E., Gardner, P., Moulton, V. A comparison of RNA folding measures. BMC Bioinformatics. 2005;6:241. doi: 10.1186/1471-2105-6-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, H., Tie, Y., Xu, C., Zhang, Z., Zhu, J., Shi, Y., Jiang, H., Sun, Z., Zheng, X. Identification of human fetal liver miRNAs by a novel method. FEBS Lett. 2005;579:3849–3854. doi: 10.1016/j.febslet.2005.05.064. [DOI] [PubMed] [Google Scholar]
- Gan, H.H., Fera, D., Zorn, J., Shiffeldrim, N., Tang, M., Laserson, U., Kim, N., Schlick, T. RAG: RNA-As-Graphs database—Concepts, analysis, and features. Bioinformatics. 2004;20:1285–1291. doi: 10.1093/bioinformatics/bth084. [DOI] [PubMed] [Google Scholar]
- Grad, Y., Aach, J., Hayes, G.D., Reinhart, B.J., Church, G.M., Ruvkun, G., Kim, J. Computational and experimental identification of C. elegans microRNAs. Mol . Cell. 2003;11:1253–1263. doi: 10.1016/s1097-2765(03)00153-9. [DOI] [PubMed] [Google Scholar]
- Grey, F., Antoniewicz, A., Allen, E., Saugstad, J., McShea, A., Carrington, J.C., Nelson, J. Identification and characterization of human cytomegalovirus-encoded microRNAs. J. Virol. 2005;79:12095–12099. doi: 10.1128/JVI.79.18.12095-12099.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones, S., Moxon, S., Marshall, M., Khanna, A., Eddy, S.R., Bateman, A. Rfam: Annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005;33:D121–D124. doi: 10.1093/nar/gki081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones, S., Grocock, R.J., van Dongen, S., Bateman, A., Enright, A.J. miRBase: MicroRNA sequences, targets, and gene nomenclature. Nucleic Acids Res. 2006;34:D140–D144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertel, J., Stadler, P.F. Hairpins in a haystack: Recognizing microRNA precursors in comparative genomics data. Bioinformatics. 2006;22:e197–e202. doi: 10.1093/bioinformatics/btl257. [DOI] [PubMed] [Google Scholar]
- Hesselberth, J.R., Ellington, A.D. A (ribo) switch in the paradigms of genetic regulation. Nat. Struct. Biol. 2002;9:891–893. doi: 10.1038/nsb1202-891. [DOI] [PubMed] [Google Scholar]
- Hofacker, I.L. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynen, M., Gutell, R., Konings, D. Assessing the reliability of RNA folding using statistical mechanics. J. Mol. Biol. 1997;267:1104–1112. doi: 10.1006/jmbi.1997.0889. [DOI] [PubMed] [Google Scholar]
- Johansson, J., Mandin, P., Renzoni, A., Chiaruttini, C., Springer, M., Cossart, P. An RNA thermosensor controls expression of virulence genes in Listeria monocytogenes. Cell. 2002;110:551–561. doi: 10.1016/s0092-8674(02)00905-4. [DOI] [PubMed] [Google Scholar]
- Johnston, R.J., Hobert, O. A microRNA controlling left/right neuronal asymmetry in Caenorhabditis elegans . Nature. 2003;426:845–849. doi: 10.1038/nature02255. [DOI] [PubMed] [Google Scholar]
- Jones-Rhoades, M.W., Bartel, D.P. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol. Cell. 2004;14:787–799. doi: 10.1016/j.molcel.2004.05.027. [DOI] [PubMed] [Google Scholar]
- Karolchik, D., Baertsch, R., Diekhans, M., Furey, T.S., Hinrichs, A., Lu, Y.T., Roskin, K.M., Schwartz, M., Sugnet, C.W., Thomas, D.J., et al. The UCSC Genome Browser Database. Nucleic Acids Res. 2003;31:51–54. doi: 10.1093/nar/gkg129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, V.N. MicroRNA biogenesis: Coordinated cropping and dicing. Nat. Rev. Mol. Cell Biol. 2005;6:376–385. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana, M., Rauhut, R., Lendeckel, W., Tuschl, T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana, M., Rauhut, R., Yalcin, A., Meyer, J., Lendeckel, W., Tuschl, T. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 2002;12:735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- Lai, E.C. RNA sensors and riboswitches: Self-regulating messages. Curr. Biol. 2003;13:R285–R291. doi: 10.1016/s0960-9822(03)00203-3. [DOI] [PubMed] [Google Scholar]
- Lai, E., Tomancak, P., Williams, R., Rubin, G. Computational identification of Drosophila microRNA genes. Genome Biol. 2003;4:R42. doi: 10.1186/gb-2003-4-7-r42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau, N.C., Lim, L.P., Weinstein, E.G., Bartel, D.P. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans . Science. 2001;294:858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- Lee, R.C., Ambros, V. An extensive class of small RNAs in Caenorhabditis elegans . Science. 2001;294:862–864. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- Lee, Y., Jeon, K., Lee, J.T., Kim, S., Kim, V.N. MicroRNA maturation: Stepwise processing and subcellular localization. EMBO J. 2002;21:4663–4670. doi: 10.1093/emboj/cdf476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W., Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- Li, X., Zhang, Y.Z. Computational detection of microRNAs targeting transcription factor genes in Arabidopsis thaliana . Comput. Biol. Chem. 2005;29:360–367. doi: 10.1016/j.compbiolchem.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Lim, L.P., Glasner, M.E., Yekta, S., Burge, C.B., Bartel, D.P. Vertebrate microRNA genes. Science. 2003a;299:1540. doi: 10.1126/science.1080372. [DOI] [PubMed] [Google Scholar]
- Lim, L.P., Lau, N.C., Weinstein, E.G., Abdelhakim, A., Yekta, S., Rhoades, M.W., Burge, C.B., Bartel, D.P. The microRNAs of Caenorhabditis elegans . Genes & Dev. 2003b;17:991–1008. doi: 10.1101/gad.1074403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallory, A.C., Vaucheret, H. MicroRNAs: Something important between the genes. Curr. Opin. Plant Biol. 2004;7:120–125. doi: 10.1016/j.pbi.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Mandal, M., Breaker, R.R. Gene regulation by riboswitches. Nat. Rev. Mol. Cell Biol. 2004;5:451–463. doi: 10.1038/nrm1403. [DOI] [PubMed] [Google Scholar]
- Mathews, D.H. Using an RNA secondary structure partition function to determine confidence in base pairs predicted by free energy minimization. RNA. 2004;10:1178–1190. doi: 10.1261/rna.7650904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews, D.H., Sabina, J., Zuker, M., Turner, D.H. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- Moss, E.G., Lee, R.C., Ambros, V. The cold shock domain protein LIN-28 controls developmental timing in C. elegans and is regulated by the lin-4 RNA. Cell. 1997;88:637–646. doi: 10.1016/s0092-8674(00)81906-6. [DOI] [PubMed] [Google Scholar]
- Moulton, V., Zuker, M., Steel, M., Pointon, R., Penny, D. Metrics on RNA secondary structures. J. Comput. Biol. 2000;7:277–292. doi: 10.1089/10665270050081522. [DOI] [PubMed] [Google Scholar]
- Nam, J.W., Shin, K.R., Han, J., Lee, Y., Kim, V.N., Zhang, B.T. Human microRNA prediction through a probabilistic co-learning model of sequence and structure. Nucleic Acids Res. 2005;33:3570–3581. doi: 10.1093/nar/gki668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nudler, E., Mironov, A.S. The riboswitch control of bacterial metabolism. Trends Biochem. Sci. 2004;29:11–17. doi: 10.1016/j.tibs.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Pervouchine, D.D., Graber, J.H., Kasif, S. On the normalization of RNA equilibrium free energy to the length of the sequence. Nucleic Acids Res. 2003;31:e49. doi: 10.1093/nar/gng049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer, S., Zavolan, M., Grasser, F.A., Chien, M., Russo, J.J., Ju, J., John, B., Enright, A.J., Marks, D., Sander, C., et al. Identification of virus-encoded microRNAs. Science. 2004;304:734–736. doi: 10.1126/science.1096781. [DOI] [PubMed] [Google Scholar]
- Pfeffer, S., Sewer, A., Lagos-Quintana, M., Sheridan, R., Sander, C., Grasser, F.A., van Dyk, L.F., Ho, C.K., Shuman, S., Chien, M., et al. Identification of microRNAs of the herpesvirus family. Nat. Methods. 2005;2:269–276. doi: 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- Pruitt, K.D., Maglott, D.R. RefSeq and LocusLink: NCBI gene-centered resources. Nucleic Acids Res. 2001;29:137–140. doi: 10.1093/nar/29.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puerta-Fernandez, E., Romero-Lopez, C., Barroso-delJesus, A., Berzal-Herranz, A. Ribozymes: Recent advances in the development of RNA tools. FEMS Microbiol. Rev. 2003;27:75–97. doi: 10.1016/S0168-6445(03)00020-2. [DOI] [PubMed] [Google Scholar]
- Reinhart, B.J., Slack, F.J., Basson, M., Pasquinelli, A.E., Bettinger, J.C., Rougvie, A.E., Horvitz, H.R., Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans . Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- Rivas, E., Eddy, S.R. Secondary structure alone is generally not statistically significant for the detection of noncoding RNAs. Bioinformatics. 2000;16:583–605. doi: 10.1093/bioinformatics/16.7.583. [DOI] [PubMed] [Google Scholar]
- Rodriguez, A., Griffiths-Jones, S., Ashurst, J.L., Bradley, A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14:1902–1910. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samols, M.A., Hu, J., Skalsky, R.L., Renne, R. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi's sarcoma-associated herpesvirus. J. Virol. 2005;79:9301–9305. doi: 10.1128/JVI.79.14.9301-9305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarnow, P., Jopling, C.L., Norman, K.L., Schutz, S., Wehner, K.A. MicroRNAs: Expression, avoidance and subversion by vertebrate viruses. Nat. Rev. Microbiol. 2006;4:651–659. doi: 10.1038/nrmicro1473. [DOI] [PubMed] [Google Scholar]
- Schultes, E.A., Hraber, P.T., LaBean, T.H. Estimating the contributions of selection and self-organization in RNA secondary structure. J. Mol. Evol. 1999;49:76–83. doi: 10.1007/pl00006536. [DOI] [PubMed] [Google Scholar]
- Seffens, W., Digby, D. mRNAs have greater negative folding free energies than shuffled or codon choice randomized sequences. Nucleic Acids Res. 1999;27:1578–1584. doi: 10.1093/nar/27.7.1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewer, A., Paul, N., Landgraf, P., Aravin, A., Pfeffer, S., Brownstein, M., Tuschl, T., van Nimwegen, E., Zavolan, M. Identification of clustered microRNAs using an ab initio prediction method. BMC Bioinformatics. 2005;6:267. doi: 10.1186/1471-2105-6-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soukup, J.K., Soukup, G.A. Riboswitches exert genetic control through metabolite-induced conformational change. Curr. Opin. Struct. Biol. 2004;14:344–349. doi: 10.1016/j.sbi.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Sprinzl, M., Vassilenko, K.S. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 2005;33:D139–D140. doi: 10.1093/nar/gki012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stormo, G.D. New tricks for an old dogma: Riboswitches as cis-only regulatory systems. Mol. Cell. 2003;11:1419–1420. doi: 10.1016/s1097-2765(03)00240-5. [DOI] [PubMed] [Google Scholar]
- Storz, G. An expanding universe of noncoding RNAs. Science. 2002;296:1260–1263. doi: 10.1126/science.1072249. [DOI] [PubMed] [Google Scholar]
- Storz, G., Altuvia, S., Wassarman, K.M. An abundance of RNA regulators. Annu. Rev. Biochem. 2005;74:199–217. doi: 10.1146/annurev.biochem.74.082803.133136. [DOI] [PubMed] [Google Scholar]
- Svoboda, P., Cara, A.D. Hairpin RNA: A secondary structure of primary importance. Cell. Mol. Life Sci. 2006;63:901–908. doi: 10.1007/s00018-005-5558-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, G. siRNA and miRNA: An insight into RISCs. Trends Biochem. Sci. 2005;30:106–114. doi: 10.1016/j.tibs.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Tijsterman, M., Plasterk, R.H. Dicers at RISC; the mechanism of RNAi. Cell. 2004;117:1–3. doi: 10.1016/s0092-8674(04)00293-4. [DOI] [PubMed] [Google Scholar]
- Vitreschak, A.G., Rodionov, D.A., Mironov, A.A., Gelfand, M.S. Riboswitches: The oldest mechanism for the regulation of gene expression? Trends Genet. 2004;20:44–50. doi: 10.1016/j.tig.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Wang, X.J., Reyes, J., Chua, N.H., Gaasterland, T. Prediction and identification of Arabidopsis thaliana microRNAs and their mRNA targets. Genome Biol. 2004;5:R65. doi: 10.1186/gb-2004-5-9-r65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X., Zhang, J., Li, F., Gu, J., He, T., Zhang, X., Li, Y. MicroRNA identification based on sequence and structure alignment. Bioinformatics. 2005;21:3610–3614. doi: 10.1093/bioinformatics/bti562. [DOI] [PubMed] [Google Scholar]
- Washietl, S., Hofacker, I.L. Consensus folding of aligned sequences as a new measure for the detection of functional RNAs by comparative genomics. J. Mol. Biol. 2004;342:19–30. doi: 10.1016/j.jmb.2004.07.018. [DOI] [PubMed] [Google Scholar]
- Weinstein, L.B., Steitz, J.A. Guided tours: From precursor snoRNA to functional snoRNP. Curr. Opin. Cell Biol. 1999;11:378–384. doi: 10.1016/S0955-0674(99)80053-2. [DOI] [PubMed] [Google Scholar]
- Wheeler, G., Ntounia-Fousara, S., Granda, B., Rathjen, T., Dalmay, T. Identification of new central nervous system specific mouse microRNAs. FEBS Lett. 2006;580:2195–2200. doi: 10.1016/j.febslet.2006.03.019. [DOI] [PubMed] [Google Scholar]
- Winkler, W.C., Breaker, R.R. Genetic control by metabolite-binding riboswitches. ChemBioChem. 2003;4:1024–1032. doi: 10.1002/cbic.200300685. [DOI] [PubMed] [Google Scholar]
- Winkler, W.C., Grundy, F.J., Murphy, B.A., Henkin, T.M. The GA motif: An RNA element common to bacterial antitermination systems, rRNA, and eukaryotic RNAs. RNA. 2001;7:1165–1172. doi: 10.1017/s1355838201002370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman, C., Krogh, A. No evidence that mRNAs have lower folding free energies than random sequences with the same dinucleotide distribution. Nucleic Acids Res. 1999;27:4816–4822. doi: 10.1093/nar/27.24.4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, P., Vernooy, S.Y., Guo, M., Hay, B.A. The Drosophila MicroRNA Mir-14 suppresses cell death and is required for normal fat metabolism. Curr. Biol. 2003;13:790–795. doi: 10.1016/s0960-9822(03)00250-1. [DOI] [PubMed] [Google Scholar]
- Xue, C., Li, F., He, T., Liu, G.P., Li, Y., Zhang, X. Classification of real and pseudo microRNA precursors using local structure-sequence features and support vector machine. BMC Bioinformatics. 2005;6:310. doi: 10.1186/1471-2105-6-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekta, S., Shih, I.H., Bartel, D.P. MicroRNA-directed cleavage of HOXB8 mRNA. Science. 2004;304:594–596. doi: 10.1126/science.1097434. [DOI] [PubMed] [Google Scholar]
- Yousef, M., Nebozhyn, M., Shatkay, H., Kanterakis, S., Showe, L.C., Showe, M.K. Combining multi-species genomic data for microRNA identification using a Naive Bayes classifier. Bioinformatics. 2006;22:1325–1334. doi: 10.1093/bioinformatics/btl094. [DOI] [PubMed] [Google Scholar]
- Zeng, Y., Cullen, B.R. Structural requirements for pre-microRNA binding and nuclear export by Exportin 5. Nucleic Acids Res. 2004;32:4776–4785. doi: 10.1093/nar/gkh824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, B., Pan, X., Cobb, G.P., Anderson, T.A. Plant microRNA: A small regulatory molecule with big impact. Dev. Biol. 2006a;289:3–16. doi: 10.1016/j.ydbio.2005.10.036. [DOI] [PubMed] [Google Scholar]
- Zhang, B., Pan, X., Cox, S., Cobb, G., Anderson, T. Evidence that miRNAs are different from other RNAs. Cell. Mol. Life Sci. 2006b;63:246–254. doi: 10.1007/s00018-005-5467-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker, M., Stiegler, P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 1981;9:133–148. doi: 10.1093/nar/9.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]