

Abstract
Central to the replication of poliovirus and other positive-strand RNA viruses is the virally encoded RNA-dependent RNA polymerase. Previous biochemical studies have suggested that direct polymerase– polymerase interactions might be important for polymerase function, and the structure of poliovirus polymerase has revealed two regions of extensive polymerase–polymerase interaction. To explore potential functional roles for the structurally observed polymerase–polymerase interactions, we have performed RNA binding and extension studies of mutant polymerase proteins in solution, disulfide cross-linking studies, mutational analyses in cells, in vitro activity analyses and RNA substrate modeling studies. The results of these studies indicate that both regions of polymerase–polymerase interaction observed in the crystals are indeed functionally important and, furthermore, reveal specific functional roles for each. One of the two regions of interaction provides for efficient substrate RNA binding and the second is crucial for forming catalytic sites. These studies strongly support the hypothesis that the polymerase– polymerase interactions discovered in the crystal structure provide an exquisitely detailed structural context for poliovirus polymerase function and for poliovirus RNA replication in cells.
Keywords: macromolecular assembly/protein–protein interactions/RNA binding/RNA polymerase/RNA virus
Introduction
Poliovirus, a small positive-strand RNA virus, is the prototype picornavirus, and is closely related to several medically important viruses including rhinoviruses, hepatitis A virus, coxsackieviruses and echoviruses. Upon infection of a cell with poliovirus, the viral RNA genome is released from the particle and translated into a polyprotein, which is then cleaved by virally encoded proteases to yield new viral capsid proteins and proteins required for RNA replication (Kitamura et al., 1981; Racaniello and Baltimore, 1981; Wimmer et al., 1993). Replication of the RNA occurs within large replication complexes that are associated with the membranes of virus-induced cytoplasmic vesicles (Baltimore et al., 1963; Caliguiri and Tamm, 1969; Takeda et al., 1986; Bienz et al., 1992). These replication complexes are known to include a variety of viral and, possibly, host proteins (reviewed in Richards and Ehrenfeld, 1990; Wimmer et al., 1993), including the virally encoded RNA-dependent RNA polymerase, the enzyme most central to the replication process.
Although viral replication occurs in large complexes in cells, purified poliovirus polymerase alone, in the absence of any other viral or host proteins, is capable of elongating primed template RNAs in solution (Van Dyke and Flanegan, 1980). Highly purified poliovirus polymerase has been observed to exhibit a high degree of cooperativity with regard to both RNA binding and efficient template utilization, suggesting that polymerase–polymerase interactions may be important for function (Pata et al., 1995; Beckman and Kirkegaard, 1998). Polymerase–polymerase interactions have also been detected using the yeast two-hybrid system (Hope et al., 1997; Xiang et al., 1998), glutaraldehyde cross-linking studies in solution (Pata et al., 1995) and in the crystal structure of poliovirus polymerase (Hansen et al., 1997).
The X-ray crystal structure of Mahoney type 1 poliovirus polymerase (Figure 1A) (Hansen et al., 1997) shows that the polymerase molecules interact within the crystal lattice via two extensive polymerase–polymerase interfaces, referred to as Interface I and Interface II (Figure 1B). These interactions are much more extensive than those typically observed for crystal packing interactions, with Interface I (Figure 1C) alone burying a total of 2180 Å2 of solvent-accessible surface area. These two sets of interactions give rise to an intriguing higher order structure that might provide uniquely detailed structural insights into the context of viral RNA replication in cells.

Fig. 1. Poliovirus polymerase–polymerase interactions as observed in the structure. (A) Ribbon representation of the structure of poliovirus polymerase. The thumb, fingers and palm subdomains are labeled. The conserved A and C sequence and structural motifs are colored red and yellow, respectively. The polypeptide regions involved in interactions at Interface I are blue and the N-terminal regions involved in interactions at Interface II are light gray. (B) Four polymerase molecules as packed within the crystals. Interfaces I and II are labeled. (C) Stereo view of the polymerase–polymerase interactions at Interface I. Side chain carbon atoms of residues in the thumb are shown in blue and those from the back of the palm are shown in yellow, oxygen atoms are colored red and nitrogen atoms blue. (D) Polymerase–polymerase interactions at Interface II. The N-terminal polypeptide regions are in light gray and the thumb subdomain is in dark gray. The three hydrophobic residues of the N-terminal strand that wedge into the hydrophobic core of the thumb subdomain are labeled. The Ca2+ site is shown in yellow and labeled.
To explore potential functional roles for the polymerase–polymerase interactions observed in the crystals, we have carried out the following experiments. (i) We have constructed mutant polymerases with specific amino acid substitutions at each of the two interfaces observed in the crystals and evaluated these mutant polymerases for their ability to bind substrate RNAs and elongate primed template RNAs in solution. (ii) We have carried out an in vitro disulfide cross-linking analysis at Interface II to determine whether the catalytically essential N-terminal amino acids are contributed intramolecularly or intermolecularly, by a second polymerase molecule associated via Interface II. (iii) We have evaluated the enzymatic activity of poliovirus polymerase as a function of polymerase concentration to determine whether the specific activity of the polymerase displays the concentration dependence that would be expected if polymerase–polymerase interactions are important for activity in solution. (iv) Finally, we have evaluated the ability of Interface I mutants in vivo to give rise to viral progeny. Each of these studies indicates that, indeed, the polymerase–polymerase interactions observed in the crystals are important for function in solution and in cells. More specifically, they indicate that interactions via Interface I are important for efficient RNA binding but are not essential for catalytic activity and that interactions via Interface II are crucial for catalytic activity.
Results
Interface I mutant polymerases
Seven different mutant polymerases containing specific amino acid substitutions designed to attenuate and disrupt the protein–protein interactions at Interface I were generated for our studies. These included singly substituted L342A, D349R, L446A, R455D and R456D mutant polymerases and doubly substituted L446A:R455D and R455A:R456A (AL28, previously reported by Diamond and Kirkegaard, 1994) mutant polymerases (Figure 1). These substitutions are widely dispersed along both surfaces of Interface I to reduce difficulties in interpretation potentially arising from specific local effects. Single point mutations have previously been observed to attenuate or disrupt protein–protein interactions in complexes with interfaces of comparable size (reviewed in Bogan and Thorn, 1998).
All seven of these mutant polymerases are stable, soluble proteins that can be expressed at levels comparable to that of wild-type protein in Escherichia coli. Precise characterization of the polymerase–polymerase interactions in solution by gel filtration, light scattering, etc. have been consistently frustrated by the apparently complex, heterogeneous and highly cooperative nature of the poliovirus polymerase associations, which cause the enzyme to precipitate over a very narrow range of concentrations. Each of the Interface I mutant polymerases exhibits significantly greater solubility than wild-type polymerase, consistent with the hypothesis that Interface I interactions contribute to the insolubility and oligomerization in solution. Furthermore, extensive two-dimensional lattices observed by electron microscopy for wild-type poliovirus polymerase are disrupted by the R455A:R456A (AL28) mutant (J.Lyle, E.Bullitt and K.Kirkegaard, manuscript in preparation). These results argue that mutations at Interface I, as predicted, disrupt extensive oligomerization and precipitation in solution.
RNA binding
To identify suitable RNA substrates for binding analyses, we first evaluated several different RNAs for their ability to bind wild-type poliovirus polymerase. Hairpin RNAs and short duplex RNAs (<10 bp) bound weakly and gave Km values >100 µM in enzymatic assays. In contrast, RNAs containing 20 bp duplex regions exhibited much higher affinities for wild-type poliovirus polymerase and gave Km values of ∼0.2 µM (data not shown). When the self-complementary duplex RNAs (shown in Figure 2A) were denatured by heating in low salt solutions, they bound poliovirus polymerase >10-fold more weakly than when they were annealed (Figure 2B), indicating that poliovirus polymerase binds duplex RNAs more tightly than single-stranded or alternatively folded RNAs. Interestingly, RNAs containing blunt ends (0.20) or 5′ single-strand extensions (5.20) bound more tightly than RNAs that contained 3′ single-strand extensions (0.20.5), indicating that binding is specific for a base-paired 3′-end (Figure 2B). Although the blunt end 0.20 RNA and the 5′-extended 5.20 RNA have nearly identical affinities for the polymerase in the conditions used in Figure 2B, differences in binding exist between these substrates. For example, removing the 5′-terminal phosphate from the 0.20 RNA results in an increase in RNA affinity, whereas phosphatasing the 5.20 RNA has no apparent effect on RNA affinity (data not shown).

Fig. 2. Binding and extension analysis of wild-type and Interface I mutant polymerases. (A) Sequences of the self-complementary 0.20, 5.20 and 0.20.5 RNAs shown as duplexes. (B) Binding curves for wild-type polymerase with the annealed and single-stranded 0.20, 5.20 and 0.20.5 RNAs. (C) Binding curves for wild-type and Interface I mutant polymerases with annealed duplex 0.20 RNA. (D) Binding curves for wild-type and Interface I mutant polymerases with annealed duplex 5.20 RNA. (E) Substrate utilization by wild-type and Interface I mutant polymerases as measured by [α-32P]UMP incorporation into oligo(dT16)-primed poly(A) template. The values are normalized versus wild-type incorporation. (F) Native polyacrylamide gel analysis of extension reactions for wild-type and Interface I mutant polymerases using the 5.20 RNA substrate. The lower band is unelongated RNA, the middle band results from RNA that has been elongated on only one end and the upper band contains RNA that has been elongated on both ends, as indicated by the schemes to the right of the gel.
Mutations at Interface I had a striking effect on RNA binding. Each Interface I mutant polymerase bound the 0.20 RNA much more weakly than wild-type polymerase (Figure 2C). The apparent Kds were 8- to >20-fold higher for each of the singly mutant polymerases. The D349R mutation was least disruptive, the L342A and L446A mutations were more disruptive, and the R455D and R456D mutations were most disruptive.
Likewise, each of the Interface I mutant polymerases bound the 5.20 RNA more weakly than wild-type polymerase (Figure 2D). Although these differences were not as dramatic as for the 0.20 RNA, perhaps because of interactions with the single-stranded region of this template, the apparent Kds of the mutant polymerases were consistently and reproducibly higher than that of wild-type polymerase. For binding the 5.20 RNA, the L342A mutant was least disruptive, the D349R mutation was slightly more disruptive and the R456D mutation was most disruptive. The doubly mutant AL28 and L446A:R455D polymerases bound the 5.20 RNA with lower affinity than each of the singly mutant polymerases. In contrast, mutant polymerases in which the first six (D6) or even 65 (D65) residues have been deleted from the N-terminus bound the 5.20 RNA indistinguishably from wild-type polymerase (Figures 2D and 6B).

Fig. 6. (A) Interactions between the N-terminal strand (light gray) and the thumb subdomain of poliovirus polymerase. The positions of mutations introduced at Interface II are labeled and shown in yellow, modeled as the mutated amino acid. (B) Binding curves for wild-type, D6 and D65 mutant polymerases with the 5.20 RNA substrate. (C) Incorporation of [α-32P]UMP into oligo(dT16)-primed poly(A) template by wild-type and Interface II mutant polymerases. The values are normalized versus wild-type incorporation. (D) Intra- and intermolecular distances between residues 35 and 69 in crystals of poliovirus polymerase. (E) Non-reducing SDS–PAGE of the disulfide cross-linking reactions with wild-type and cysteine mutant polymerases. Individual lanes are as labeled. The reactions in lanes 13 and 14 were carried out for only 6 h because of extensive formation of larger cross-linked species for the A29C:I441C double mutant.
RNA substrate utilization
Extension of primed template RNA substrates by wild-type and mutant polymerases was evaluated using two different substrates: the 5.20 RNA used in the binding studies and oligo(dT16)-primed poly(A) to ensure that the observed effects were not template specific.
Although the Interface I mutant polymerases extend oligo(dT16)-primed poly(A) substrate less well than wild-type polymerase (Figure 2E), each of the singly mutant polymerases was able to achieve a significant level of incorporation at high concentrations of polymerase, with D349R, R456D and L342A showing the highest levels of incorporation. Incorporation by the doubly mutant polymerases was further reduced from those of the singly mutant polymerases.
Extension of the 5.20 RNA template was evaluated using labeled RNA substrates and native polyacrylamide gels, so that the actual fraction of substrate utilized in the extension reactions could be observed directly. Extension was evaluated for wild-type and mutant polymerases at concentrations of 75, 375 and 1500 nM (Figure 2F). Native polyacrylamide gels resolve three RNA species corresponding to unelongated duplex 5.20 RNA, a duplex RNA with one of the two 3′-sites elongated and a duplex RNA with both 3′-sites elongated. The identity of these products was verified by isolating the band from a native gel and evaluating the RNAs by denaturing PAGE (data not shown). Once again the Interface I mutant polymerases utilized the 5.20 RNA substrate less efficiently than wild-type polymerase, but at high concentrations of enzyme the mutant polymerases show substantial enzymatic activity.
Enzymatic assays of wild-type and mutant polymerases
The studies described above were carried out with limiting substrate rather than limiting enzyme in order to correlate binding with substrate utilization. We also evaluated the specific activities of wild-type and mutant polymerases using standard enzymatic assays in which substrate was in excess and enzyme was limiting. These analyses show that the specific activities of the Interface I mutant polymerases under conditions of excess substrate are within 3-fold of that of the wild-type enzyme (Figure 3A), indicating that mutations at Interface I do not greatly affect catalytic activity.
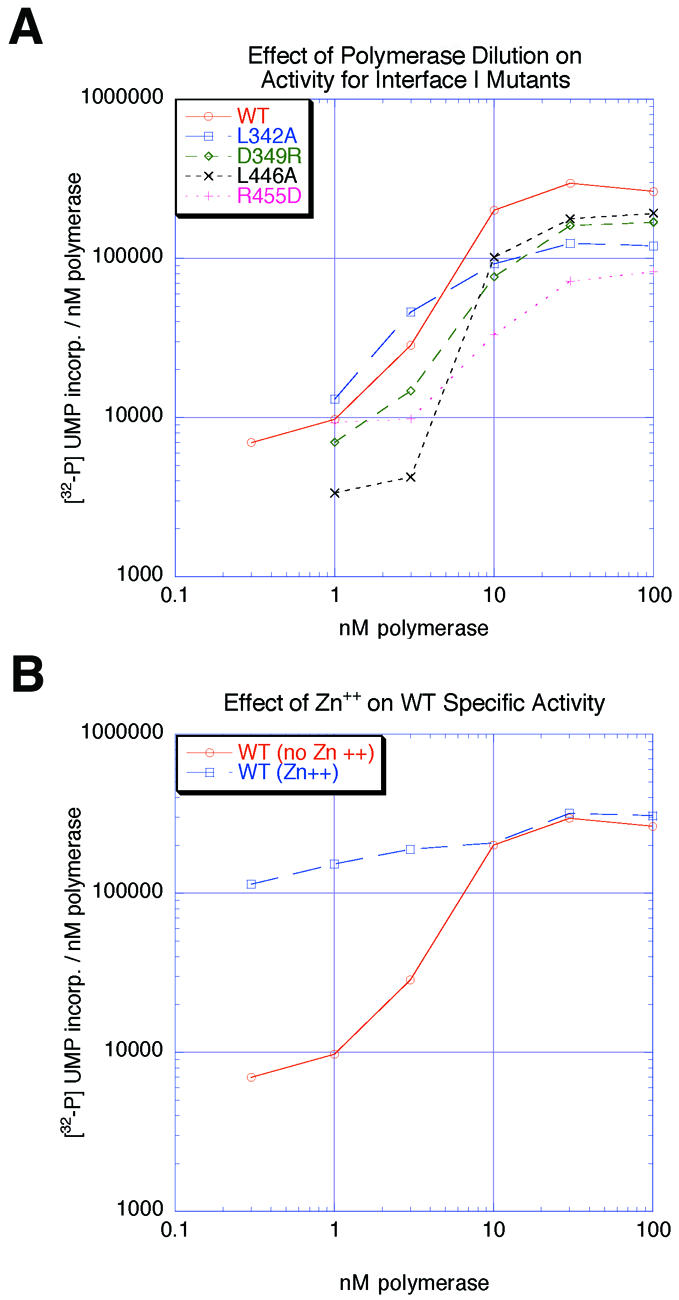
Fig. 3. (A) Plot of specific activity versus concentration of wild-type and Interface I mutant polymerases. (B) Plot of specific activity versus concentration of wild-type polymerase in the presence and absence of 60 µM Zn2+.
It is apparent from Figure 3A that the specific activity ([α-32P]UMP incorporation/nM polymerase) of wild-type poliovirus polymerase decreased dramatically when enzyme concentration was reduced from 10 to 1 nM. This result indicates that the active form of poliovirus polymerase is sensitive to protein concentrations as, for example, would occur if polymerase–polymerase interactions were important for poliovirus polymerase activity. This change in specific activity versus enzyme concentration is similar for wild-type and Interface I mutant polymerases, indicating that polymerase associations via Interface I are not responsible for the concentration dependence of specific activity between 1 and 10 nM, but that another interaction, perhaps Interface II, is limiting in these enzymatic assays.
At polymerase concentrations >10 nM, or at all concentrations tested in the presence of Zn2+, the specific activity varied <3-fold over the entire concentration range tested (Figure 3B), indicating that at concentrations >10 nM or in the presence of Zn2+, polymerase– polymerase interactions are not limiting for polymerase activity under these conditions. The ability of Zn2+ to suppress the concentration dependence of the specific activity (Figure 3B), possibly by stabilizing Interface II, will be discussed later.
Interface I mutational analysis in tissue culture
Each of the Interface I mutations (L342A, D349R, L446A, R455D, R456D and L446A:R455D) was also introduced into the coding region of 3D in the poliovirus genome. To determine the phenotypes of the resulting viruses, full-length cDNAs under the control of a T7 promoter were transfected into cells that constitutively express T7 RNA polymerase (Nugent et al., 1999). Under conditions in which wild-type cDNA gave rise to thousands of plaques, L446A, R455D, R456D and L446A:R455D yielded no plaques, indicating that viruses containing these mutations are non-viable. However, transfection of D349R cDNA yielded similar numbers of plaques at all temperatures tested, and L342A yielded similar numbers of plaques at 32.5°C but not at 39°C. Plaque-purified D349R and L342A viruses were found to display wild-type and temperature-sensitive growth phenotypes, respectively (Figure 4). Thus, the order of decreasing phenotype severity is D349R > L342A > L446A, R455D, R456D, L446A:R455D. Note that D349R and L342A were also the least disruptive mutants in the RNA binding studies. These results indicate that the polymerase–polymerase interactions at Interface I discovered structurally and biochemically are indeed relevant to viral growth.

Fig. 4. Plaque assays from transfected cDNAs of wild type, L342A, D349R and mock transfection into KJT7 cells at 32.5, 37 and 39.5°C. Transfection of cDNAs that contained the R455D, R456D, L446A, L446A:R455D and V33A:F34A mutations did not give rise to viable virus.
Modeling of RNA contacts at Interface I
Several structures of polymerases bound to nucleic acid substrates are now known, including that of HIV-1 reverse transcriptase (RT) bound to dsDNA (Huang et al., 1998). Since the structure of the poliovirus polymerase active site closely resembles that of these other polymerases (Hansen et al., 1997) and especially that of HIV-1 RT, we can model primed template RNA into the structure of poliovirus polymerase based on the structure of the HIV-1 RT–dsDNA complex.
This model (Figure 5) predicts that a monomer of poliovirus polymerase would interact with dsRNA over only one-half of a turn of the double helix. Interestingly, association of a second poliovirus polymerase molecule via Interface I extends the potential dsRNA binding surface in a manner similar to that of the connection and RNase H subdomains and p51 subunit in the HIV-1 RT heterodimer. This model further supports the observations that mutations at Interface I reduce the affinity of the polymerase for primed template RNAs.

Fig. 5. Model of a poliovirus polymerase–dsRNA complex based on the structure of HIV-1 RT complexed to dsDNA (Huang et al., 1998).
Mutational analysis of Interface II and the N-terminal strand interactions
Since the N-terminal residues of poliovirus polymerase are critical for elongation activity (Plotch et al., 1989), and because the N-terminal polypeptide segment is intimately associated with the top of the thumb subdomain at Interface II (Figure 1D), we wished to explore the importance of this unusual interaction. We therefore introduced point mutations into the N-terminal strand (A29C and F30A) and into the top of the thumb subdomain (I441C and G442L) (Figure 6A), and also introduced deletions of the first six (D6) and 65 (D65) N-terminal amino acids of poliovirus polymerase.
Interestingly, binding of the 5.20 RNA by the N-terminal deletion mutants (Figure 6B) was indistinguishable from that of wild-type enzyme. Even deleting the first 65 residues, which eliminates the N-terminal strand and, therefore, a significant portion of Interface II, does not affect RNA affinity.
In contrast, the N-terminal deletion mutations and the point mutations in the N-terminal interaction region all resulted in polymerases that were unable to incorporate a significant amount of [α-32P]UTP into oligo(dT16)- primed poly(A) substrate (Figure 6C). That mutations at Interface II dramatically reduce catalytic activity but do not affect RNA affinity contrasts with the results of mutations at Interface I, which affect RNA binding but do not significantly affect catalytic activity when RNA concentration is not limiting.
Disulfide cross-linking analysis of the N-terminal strand interaction
Interface II involves the N-terminal polypeptide regions of poliovirus polymerase. Residues 13–35 form a strand that begins in the active site cleft and loops over the top of the thumb subdomain (Figure 1A). Three hydrophobic residues, F30, V33 and F34, appear to anchor this N-terminal strand by wedging into the hydrophobic core near the top of the thumb subdomain (Figure 1D). Residues 69–97 form an α-helix positioned at the bottom of the fingers subdomain (Figure 1A). Unfortunately, the residues connecting these two ordered portions of the polypeptide chain (residues 36–68) are disordered in the crystals. Curiously, residues 35 and 69 are on opposite sides of the molecule, such that residues 36–68 would need to span the entire molecule, a distance of >44 Å, to connect in an intramolecular manner (Figure 6D). Although this is possible, these residues might also connect by an intermolecular interaction via Interface II, spanning a distance of <32 Å (Figure 6D). In this case, the catalytically essential N-terminal residues of poliovirus polymerase would be contributed in trans by an adjacent polymerase molecule associated via Interface II.
We designed a disulfide cross-linking experiment to determine whether residues 13–35 are provided by an intra- or an intermolecular interaction (Figure 6D). Three separate mutant polymerases were constructed: A29C, in which a Cys residue was introduced into the N-terminal strand; I441C, in which a Cys residue was introduced at the top of the thumb subdomain; and the double mutant A29C:I441C, which contained both of these Cys mutations. Residues A29 and I441 are positioned directly across from each other at Interface II, such that the predicted positions of the sulfur atoms would be <4.0 Å apart (Figure 6A).
The results of this cross-linking experiment are shown in Figure 6E. Lanes 3–6 contain samples in which 50 mM dithiothreitol (DTT) was added just before the cross-linking reaction. These lanes display only monomer-length products, indicating that under reducing conditions no higher molecular weight products formed. Lanes 7–12 contained samples in which wild-type and mutant polymerases were incubated individually and together in the absence of reducing agent. Note that these are quite mild oxidizing conditions in that no oxidizing agents were introduced. Several diffuse bands with greater than dimer-length mobilities were observed for the individual mutants and for the mixtures with wild-type enzyme, which is not surprising since there are five naturally occurring cysteine residues in poliovirus polymerase, providing ample opportunity for formation of a variety of disulfide cross-linked products. In the mixture of A29C and I441C mutant proteins, however (Figure 6E, lane 12), a discrete dimer-length product was observed, consistent with the formation of a disulfide cross-link between the A29C and I441C mutant polymerases. The amount of the dimer-sized product, ∼15% of the initial material, was significant considering that the yield was likely to be limited by formation of other disulfide products and incomplete reaction. This result indicates that in solution a substantial portion of the polymerase associates intermolecularly at Interface II. Unfortunately, we could not adequately evaluate enzymatic activity during the course of the cross-linking experiment because the activity decreased steadily, probably due to competing processes such as formation of other disulfide bonds in the bulk reaction.
Cross-linking data for the A29C:I441C doubly mutant polymerase (Figure 6E, lanes 13 and 14) showed a dimer-length product of the same molecular weight as the mixture of the A29C and I441C individual mutant polymerases. However, the yield of the dimeric product was lower and additional higher molecular weight products were also observed. These larger products likely arose from formation of the trimeric, tetrameric and larger disulfide-cross-linked products that become possible with the A29C:I441C double mutant. Importantly, no other products of intermediate or faster mobility, as would be expected from intramolecularly disulfide cross-linked products, were observed for the doubly mutant polymerase, indicating little if any intramolecular association of the N-terminal strand.
To test the effect of disrupting Interface II on poliovirus viability, V33 and F34 were changed to alanine by site-directed mutagenesis of the full-length poliovirus cDNA clone. As was the case with most of the Interface I mutations, no viable virus was recovered after transfection of the mutated cDNA under conditions in which thousands of plaques resulted from wild-type cDNA transfection (Materials and methods). Therefore, the V33A:F34A mutations, predicted to disrupt the Interface II interaction, destroyed viral viability.
Discussion
Mutational analysis of Interface I
Polymerase–polymerase associations at Interface I involve two distinct surfaces of the polymerase which interact via 21 direct amino acid side chain interactions, at least five water-mediated interactions and one direct backbone– backbone hydrogen bond (Figure 1). One of these surfaces is positioned at the bottom of the thumb subdomain and the other at the back of the palm subdomain (highlighted in blue in Figure 1A). Importantly, these interactions do not define a single discrete oligomeric state (e.g. a dimer, trimer, etc.); rather, Interface I interactions give rise to a directional fiber in which the polymerase molecules associate in a head-to-tail fashion that might vary greatly in length. Interestingly, nearly identical Interface I interactions are observed in two other, very different crystal forms of poliovirus polymerase as well, arguing that interactions at Interface I are robust and probably form in solution as well (S.D.Hobson, J.L.Hansen and S.C.Schultz, manuscript in preparation).
Mutations designed to disrupt Interface I had little or no effect on catalytic activity at high polymerase concentrations, indicating that these mutations do not greatly affect stable folding or catalytic activity (Figure 3). However, mutations at Interface I clearly affect the RNA binding properties of the polymerase. Each of the mutant polymerases bound the 0.20 RNA and 5.20 RNA less tightly than wild-type polymerase (Figure 2). For the blunt-ended 0.20 RNA, the effect of the mutations was large, with the apparent Kd for D349R, the least attenuated of the mutants, 8-fold higher than that of the wild-type polymerase; the apparent Kd for the rest of the mutants was >20-fold higher. For the 5.20 RNA, the effects were less dramatic but significant. These smaller differences are likely to result from interactions with the single-stranded regions or the duplex–single strand junction in the 5.20 substrate RNA.
RNA extension analyses show that when RNA substrate is limiting, the Interface I mutant polymerases incorporate NTPs into primed template RNAs less efficiently than wild-type polymerase. Each Interface I mutant was, however, able to achieve a significant level of incorporation at high polymerase concentrations. This result shows that mutations at Interface I do not seriously cripple the catalytic processes of the polymerase. Interestingly, the relative efficiency of utilization of 5.20 RNA by each Interface I mutant (Figure 2F) was approximately the same as the relative binding affinities for the 5.20 RNA (Figure 2D), arguing that the different efficiencies of RNA utilization are a direct consequence of differences in RNA binding.
Analysis of the in vivo phenotype of the Interface I mutants yielded results consistent with those observed for both RNA binding and extension in vitro. Specifically, the mutations that destroyed viral viability (R455D, R456D, L446A and L446A:R455D) were those that were most disruptive to RNA binding and catalytic activity under conditions of limiting RNA (Figure 2). Mutation D349R, the least disruptive mutation in vitro, gave rise to virus indistinguishable in phenotype from wild-type virus, whereas the L342A mutation, which was intermediate in its effects on RNA binding and polymerization (Figure 2), gave rise to temperature-sensitive virus (Figure 4). The distributed nature of the mutations across both surfaces of Interface I argues for a general effect due to disruption of Interface I, rather than specific local effects. Whether Interface I interactions are required for RNA pre-initiation complex formation, initiation of RNA synthesis, negative-strand synthesis, positive-strand synthesis, elongation, packaging or any other function of the RNA replication complex remains to be determined.
Analysis of the N-terminal interactions and Interface II
To evaluate the importance of the association between the N-terminal strand of poliovirus polymerase and the thumb subdomain at Interface II (Figure 1), we constructed two different deletion mutants, which remove the first six (D6) or 65 (D65) N-terminal amino acids, and also a number of point mutations (A29C, F30A, I441C and G442L), which should disrupt the interaction between the N-terminal strand and the top of the thumb subdomain. Each of these mutant polymerases exhibited dramatically reduced polymerase activity (Figure 6C), indicating that direct association of the N-terminal strand with the top of the thumb subdomain is critical for activity. The distributed nature of these mutations on both sides of Interface II argues against a specific local effect. Mutation of two residues in the N-terminal strand intricately involved in interactions at Interface II (V33A:F34A) destroys viral infectivity, suggesting that Interface II interactions are important for virus viability. In contrast to Interface I mutant polymerases, the N-terminal mutant polymerases exhibited solubility properties similar to that of wild-type polymerase, and bound the 5.20 RNA with apparent Kds essentially identical to that of wild-type polymerase (Figure 6B).
Since residues 36–68 are disordered in the structure, it was unclear whether the catalytically essential N-terminal strand derives from intra- or intermolecular interactions (Figure 6D). Therefore, we carried out a disulfide cross-linking experiment in which Cys residues were introduced into the top of the thumb subdomain (I441C) and into the N-terminal strand (A29C). Mixing these two mutant polymerases together should, upon oxidation, yield a discrete dimeric product if the N-terminal strand association is in trans. This was, indeed, the result, indicating that in solution a significant amount of poliovirus polymerase associates via intermolecular interactions at Interface II. Since the double mutant (A29C:I441C) polymerase yielded only larger molecular weight products and no additional bands, as would be expected for an intramolecular cross-link, this suggests that the N-terminal strand associates exclusively in trans.
This intermolecular interaction between the N-terminal strand of one polymerase molecule and the thumb subdomain of another buries a total of 2270 Å2 of solvent-accessible surface area, comparable to that of Interface I. The nature of the interactions at Interfaces I and II, however, are quite different. Interface I involves two, at least partially structured surfaces coming together, whereas Interface II involves an extended strand with little apparent structure of its own folding together with the thumb subdomain of the adjacent molecule (Figure 1D). The very different nature of these interactions will likely have important implications with regard to the relative kinetics and stabilities of these two different types of interaction in solution and in cells.
The cooperative transition observed in Figure 3A, in which the specific activity decreased greatly when poliovirus polymerase concentration was decreased from 10 to 1 nM, was insensitive to mutations at Interface I (Figure 3A). We suggest that the polymerase–polymerase interactions responsible for this cooperative transition result from donation of the N-terminal strand intermolecularly across Interface II. The elimination of this cooperative transition in the presence of 60 µM Zn2+ suggests that protein–protein interactions at Interface II are facilitated by Zn2+. Interestingly, a Ca2+ ion is observed in the crystal structure at Interface II (Figure 1D). Although Ca2+ was included in the crystallization of poliovirus polymerase, it does not support catalysis in the absence of Mg2+. Perhaps other metals such as Zn2+ could coordinate in this site and stabilize interactions at Interface II.
Are higher order polymerase structures used by other viruses?
Sequence alignments of picornaviral polymerases reveal a significant degree of conservation among amino acid residues within and around both Interfaces I and II, suggesting that these interactions might also be important in other picornaviruses such as other enteroviruses, rhinoviruses, coxsackieviruses and hepatitis A virus. However, non-picornaviral positive-strand RNA polymerases do not share these particular regions of sequence homology, suggesting that if higher order polymerase structures are important in these other viruses, they might be different to those in poliovirus.
The structure of the RNA-dependent RNA polymerase of hepatitus C virus (HCV) has recently been reported (Ago et al., 1999; Bressanelli et al., 1999; Lesburg et al., 1999). Unlike poliovirus polymerase crystals, which display extensive polymerase–polymerase interactions, only limited protein–protein interactions are observed within the HCV polymerase crystals.
The structure of the HCV polymerase is similar to that of poliovirus polymerase, but it also exhibits several interesting differences. The N-terminal polypeptide segment of HCV polymerase does not interact in the active site cleft as in poliovirus polymerase; rather, a peptide loop from the HCV polymerase thumb subdomain (residues 441–456), which is not present in poliovirus polymerase, extends down into its active site cleft, apparently replacing the N-terminal strand interaction of poliovirus polymerase.
The first three α-helices of the HCV polymerase thumb subdomain form a structure similar to that of poliovirus polymerase, but the HCV thumb subdomain contains five additional helices, none of which corresponds to the C-terminal α-helix of poliovirus polymerase involved in Interface I interactions. Therefore, since the regions of poliovirus polymerase that form Interfaces I and II are both different in HCV polymerase, it would seem that HCV polymerase can not form the same type of higher order structure as poliovirus and probably other picornaviral polymerases.
Structural model of the poliovirus polymerase oligomer
The research presented here supports and extends our previous hypothesis (Pata et al., 1995; Hansen et al., 1997; Beckman and Kirkegaard, 1998) that poliovirus polymerase forms a specific higher order structure that is important for function. In the three-dimensional structure, both Interface I and II form in a directional manner, leading to the possibility of extended oligomers along either or both interfaces.
Representations of two possible higher order polymerase structures are shown in Figure 7. In the first (Figure 7A), two fibers, each containing four polymerase molecules interacting via Interface I, are shown interacting via Interface II. RNAs are modeled into the two active sites formed by the Interface II interactions to illustrate how this higher order structure can yield multiple active sites when two Interface I fibers interact. One might imagine frequent associations via Interface II to give a tightly packed core of catalytic centers, or less frequent associations via Interface II to give widely spaced catalytic centers. In the second representation (Figure 7B), two fibers, each containing five polymerase molecules interacting via Interface II, are shown contacting each other via Interface I. Additional sites for Interface I interactions are shown in blue patches along the Interface II fibers. In this representation, potential active sites are present in all molecules except the ones at the top left and bottom right. RNA is modeled only into the lower set of molecules. Recent studies with poliovirus polymerase indicate that potentially up to 80% of polymerase molecules have RNA extension ability in vitro (Arnold and Cameron, 2000). This result supports a model with frequent interactions via Interface II. These representations are of the two extremes: Interface I fibers interacting via Interface II and, conversely, Interface II fibers interacting via Interface I. Functional complexes are likely to be somewhere between these extremes.

Fig. 7. Structural representations of poliovirus polymerase oligomers. (A) Stereo image of two Interface I fibers interacting via Interface II. Each Interface I fiber contains four polymerase molecules colored orange and light gray in one strand and brown and dark gray in the second strand. The Interface I interaction surfaces are shown in blue. The N-terminal strands interacting via Interface II are shown in white. Duplex RNA, shown in red, is modeled into each fiber. (B) Stereo image of two Interface II fibers interacting via Interface I. Each Interface II fiber contains five polymerase molecules colored as in (A). The Interface I interaction surfaces are shown in blue and, therefore, illustrate additional sites of potential interactions via Interface I. The thumb subdomain of each polymerase molecule is indicated in stereo with a T. Duplex RNA, shown in red, has been modeled into the bottom two molecules of each Interface II fiber.
The association of polymerase molecules in this manner could serve a number of functions. For example, a higher order polymerase structure might sequester and protect the RNA within the replication complex. It has been shown that treating poliovirus replication complexes with RNase A does not result in RNA degradation (Bienz et al., 1992), indicating that the RNA is protected from degradation within the RNA replication complex. A higher order polymerase structure might also serve to mediate the coupling observed between viral RNA replication and packaging (Nugent et al., 1999) in that capsid precursors might assemble onto an oligomeric polymerase structure. In addition, RNA recombination, which has been shown to occur by means of a copy-choice mechanism during poliovirus replication (Kirkegaard and Baltimore, 1986; Arnold and Cameron, 1999), might be facilitated by higher order structures in which multiple RNAs are replicated in close proximity.
Since poliovirus is known to replicate its RNA in large replication complexes in cells, perhaps the higher order poliovirus polymerase structures described here form the heart of an efficient ordered machinery that carries out poliovirus RNA replication in cells.
Materials and methods
Mutagenesis and polymerase purification
Mutations were introduced into the sequence of the Mahoney type 1 strain of poliovirus by PCR mutagenesis and were verified by DNA sequencing. Wild-type and mutant polymerases were expressed using a T7-based expression system (pKKT7-derived plasmids) in E.coli BL21(DE3) pLysS. Cells containing the expression plasmid were grown in 2× YT medium at 37°C to an OD600 of ∼0.2. The cells were then cooled to room temperature, grown to an OD600 of ∼0.4, and production of the polymerase was induced by the addition of 0.5 mM isopropyl-β-d-thiogalactopyranoside followed by incubation for an additional 16–18 h at room temperature. The polymerase was purified as described previously (Hansen et al., 1997), except that an FPLC S-Sepharose column was used instead of using a gravity S-Sepharose column. The protein was eluted from the S-Sepharose column using a linear gradient of NaCl from 0.1 to 0.38 M. Typical yields of the wild-type and mutant polymerases are 12 mg/10 g of cells (wet weight). Polymerase concentrations were determined by absorbance at 280 nm (A280) using an extinction coefficient of 72 430 M–1 cm–1 (66 740 and 62 900 M–1 cm–1 for the D6 and D65 polymerase mutants, respectively), which was calculated according to Gill and von Hippel (1989).
Mass spectroscopic analysis indicated that the N-terminal Met was efficiently removed. The N-terminal endoproteinase Lys-C fragment missing the N-terminal Met was clearly present, whereas no peptides were detected that contained the N-terminal Met. Extension activities, RNA binding, etc. of 3D polymerase purified as described above were indistinguishable from 3D polymerase generated by cleavage of 3CD, which also indicates that the N-terminal Met is efficiently removed and/or does not interfere with the activities of the polymerase evaluated here.
RNA preparation
Poly(A) and oligo(dT16) were purchased from Pharmacia Biotech. The 0.20 RNA and 5.20 RNA were generated by T7 transcription. The primer and template DNAs were purchased from Operon Technologies Inc. The T7 transcription reaction contained: 0.2 µM single-strand template DNA, 0.22 µM complementary promoter DNA, 40 mM Tris pH 8.0, 25 mM Mg(OAc)2, 0.01% Triton X-100, 10 mM DTT, 1 mM spermidine, 2 mM each of ATP, GTP and UTP, 1 mM CTP and 0.12 µCi/µl [α-32P]CTP (3000 Ci/mmol; NEN). The transcription reactions were incubated at 37°C for 3 h. The RNAs were purified by electrophoresis in polyacrylamide gels containing 8.3 M urea. Concentrations were determined by absorbance at 260 nm (A260) using the average extinction coefficient of 0.11 mM/nucleotide/OD.
RNA binding assays
RNA binding was evaluated using a nitrocellulose filter binding assay. The binding reactions contained 25 mM MES pH 6.5, 2.5 mM Mg(OAc)2, 2 mM DTT, 10% glycerol, 40 µM ZnSO4, 1 mM each of GTP, CTP and UTP, 10 nM α-32P-labeled 5.20 RNA and polymerase at various concentrations from 7.5 nM to 3.0 µM. The 30 µl binding reactions were incubated for 15 min on ice and then loaded onto a 96-well filter apparatus containing one layer each of nitrocellulose membrane (Schleicher and Schuell), a 25 µm filter paper (Whatman) for separation, a positively charged nylon membrane (Hybond-N+, Amersham) and Gel Blot paper (GB002, Schleicher & Schuell). All filters and paper were equilibrated in 50 mM MES pH 6.5 and 10 mM EDTA for at least 30 min before use. The amount of RNA bound to each membrane was quantified using a PhosphorImager (Molecular Dynamics). Apparent Kds were determined according to the equation fraction RNA bound = ([3D]/(Kd+[3D]).
Elongation assays
Elongation of oligo(dT16) primer in the presence of poly(A) template was evaluated by measuring incorporation of [α-32P]UTP into RNA products. Various concentrations of polymerase from 7.5 nM to 3.0 µM were incubated with 2.5 µg/ml poly(A), 46 nM dT16 and elongation buffer {25 mM MES pH 6.5, 2.5 mM Mg(OAc)2, 50 mM NaCl, 2 mM DTT, 50 µM UTP, 10% glycerol and 0.01 µCi/µl [α-32P]UTP} for 30 min at 30°C. Incorporation of the radiolabel was evaluated by filtering 27 µl of the reaction through Hybond-N+ nylon membrane in a 96-well filter apparatus. Each well was washed with a total of 3 ml of 5% w/v sodium phosphate, 2% w/v sodium pyrophosphate to remove unincorporated label. The amount of 32P that remained bound to the Hybond paper was quantified by PhosphorImager analysis.
Elongation assays of the 5.20 RNA were performed as follows. The polymerase was incubated with 25 mM MES pH 6.5, 2.5 mM Mg(OAc)2, 25 mM NaCl, 2.0 mM DTT, 10% glycerol, 40 µM ZnSO4, 0.5 mM of each NTP and 10 nM α-32P-labeled 5.20 RNA at 30°C for 15 min. After the incubation, the samples were placed on ice and 1/10 volume (2 µl) of 50 mM Tris pH 7.5, 20 mM EDTA and 2% SDS was added to each sample. Elongated products were then separated by electrophoresis on a 20% native polyacrylamide (19:1)/1.0× TBE gel. The relative amounts of elongated and unelongated products were evaluated by PhosphorImager analysis.
Dilution studies
Enzymatic elongation assays for wild-type and mutant polymerases were carried out in 50 mM HEPES pH 7.2, 0.5 mM each of ATP, GTP and CTP, 50 µM UTP (for assays containing 0.3–10 nM 3D) or 500 µM UTP (for additional assays containing 10–100 nM 3D), 4 µCi of [α-32P]UTP/incubation (30 µl; NEN; 3000 Ci/mmol), 4 mM DTT; 3 mM magnesium acetate, 0.1% NP-40, 1 µM poly(A) and 20 µM oligo(U) (for assays containing 0.3–10 nM 3D) or 3 µM poly(A) and 60 µM oligo(U) (for assays containing 10–100 nM 3D), 60 µM ZnCl2 (where indicated) and 3D (0.3–100 nM, dilutions in 50 mM HEPES pH 7.2, 0.1% NP-40, 50 mM KCl and 5 mM β-mercaptoethanol). Reaction mixes were prepared at 4°C and the mixes were pre-incubated at 4°C for 60 min. The samples were incubated at 30°C for 10 min in order to obtain initial linear rates and were performed in duplicate for any given set of assays. Assays were repeated two or three times for each 3D polymerase preparation. Aliquots were removed at 0 and 10 min and assayed for acid-precipitable material. Zero time counts/min were subtracted from 10 min values to obtain net counts/min for each assay. Finally, specific activities were plotted versus polymerase concentration to illustrate any deviation from a constant specific activity.
Disulfide cross-linking experiments
For the disulfide cross-linking reactions, 2 mM DTT (285 mM for the reduced samples) was added to the protein and incubated on ice for 15 min. This mix was then diluted to 50 µl so that final concentrations were: 7.5 µM total protein, 50 mM PIPES pH 6.6, 1 mM ATP, 80 mM NaCl and 0.35 mM DTT (50 mM DTT for the reduced samples), and incubated at 30°C for 8 h. The reaction was quenched by adding 21 µl of gel loading buffer (50 mM Tris pH 6.8, 4.4% SDS and 8% glycerol) and the samples were boiled for 2 min. These samples were loaded onto SDS gels and the proteins were visualized by Coomassie staining.
Cells and viruses
The Interface I mutations were introduced into T7pGempolio (Sarnow, 1989), which contains the full-length Mahoney type 1 poliovirus cDNA, using PCR mutagenesis. BglII–EcoRI fragments were ligated into the BglII- and EcoRI-cut T7pGempolio plasmid. Codon usage for each of the mutations was as follows: L342A, GCG; D349R, CGT; L446A, GCT; R455D, GAC; R456D, GAC; L446A:R455D, GCT (Ala), GAC (Asp); V33A:F34A, GCC (Ala), GCG (Ala). The sequences for each mutant were confirmed by DNA sequencing. Two independent isolates of each mutant were generated.
KJT7 cells (Nugent et al., 1999), which express T7 RNA polymerase, were transfected with mutant or wild-type plasmid DNA using LipofectaminePLUS (Gibco) following the manufacturer’s instructions. Transfected cell monolayers were overlaid with Dulbecco’s modified Eagle’s medium containing 10% calf serum and 1% agar. Plaques were allowed to develop for 72 h at 32.5°C or 48 h at 37 or 39.5°C. Transfections were performed for two clones each of wild-type and all mutant cDNAs. For wild-type, L342A and D349R, 0.4 µg of DNA per plate yielded 10–50 plaques, and 4 µg of DNA per plate yielded thousands of plaques that destroyed the monolayer. For L446A, R455D, R456D, L446A:R455D or V33A:F34A, neither DNA concentration yielded detectable virus in duplicate transfections.
Single plaques of wild-type, L342A and D349R viruses were isolated at 32.5°C and used to infect HeLa cells to prepare high-titer virus stocks. Phenotypes of high-titer stocks were identical to those of the original plaque isolates, as determined by plaque assay performed as described (Kirkegaard, 1990).
Acknowledgments
Acknowledgements
We thank Jeff Hansen, Janice Pata, John Lyle, Esther Bullitt, Martin Horvath, Olve Peersen, Andy Berglund, Scott Classen and Doug Theobald for assistance and helpful discussions during this work. This work was funded by the Colorado Advanced Technology Institute through a grant from the Colorado RNA Center (to S.C.S.) and by DARPA (to K.K.).
References
- Ago H., Adachi,T., Yoshida,A., Yamamoto,M., Habuka,N., Yatsunami,K. and Miyano,M. (1999) Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Struct. Fold. Des., 7, 1417–1426. [DOI] [PubMed] [Google Scholar]
- Arnold J.J. and Cameron,C.E. (1999) Poliovirus RNA-dependent RNA polymerase (3Dpol) is sufficient for template switching in vitro. J. Biol. Chem., 274, 2706–2716. [DOI] [PubMed] [Google Scholar]
- Arnold J.J. and Cameron,C.E. (2000) Poliovirus RNA-dependent RNA polymerase (3D(pol)). Assembly of stable, elongation-competent complexes by using a symmetrical primer-template substrate (sym/sub). J. Biol. Chem., 275, 5329–5336. [DOI] [PubMed] [Google Scholar]
- Baltimore D., Eggers,H.J., Franklin,R.M., and Tamm,I. (1963) Poliovirus induced RNA polymerase and the effects of virus-specific inhibitors on its production. Proc. Natl Acad. Sci. USA, 49, 843–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman M.T.L. and Kirkegaard,K. (1998) Site size of cooperative RNA binding by poliovirus RNA-dependent RNA polymerase. J. Biol. Chem., 273, 6724–6730. [DOI] [PubMed] [Google Scholar]
- Bienz K., Egger,D., Pfister,T. and Troxler,M. (1992) Structural and functional characterization of the poliovirus replication complex. J. Virol., 66, 2740–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogan A.A. and Thorn,K.S. (1998) Anatomy of hot spots in protein interfaces. J. Mol. Biol., 280, 1–9. [DOI] [PubMed] [Google Scholar]
- Bressanelli S., Tomei,L., Roussel,A., Incitti,I., Vitale,R.L., Mathieu,M., De Francesco,R. and Rey,F.A. (1999) Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc. Natl Acad. Sci. USA, 96, 13034–13039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caliguiri L.A. and Tamm,I. (1969) Membranous structures associated with translation and transcription of poliovirus RNA. Science, 166, 885–886. [DOI] [PubMed] [Google Scholar]
- Diamond S.E. and Kirkegaard,K. (1994) Clustered charged-to-alanine mutagenesis of poliovirus RNA-dependent RNA polymerase yields multiple temperature-sensitive mutants defective in RNA synthesis. J. Virol., 68, 863–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill S.C. and von Hippel,P.H. (1989) Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem., 182, 319–326. [DOI] [PubMed] [Google Scholar]
- Hansen J.L., Long,A.M. and Schultz,S.C. (1997) Structure of the RNA-dependent RNA polymerase of poliovirus. Structure, 5, 1109–1122. [DOI] [PubMed] [Google Scholar]
- Hope D.A., Diamond,S.E. and Kirkegaard,K. (1997) Genetic dissection of interaction between poliovirus 3D polymerase and viral protein 3AB. J. Virol., 71, 9490–9498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H., Chopra,R., Verdine,G.L. and Harrison,S.C. (1998) Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science, 282, 1669–1675. [DOI] [PubMed] [Google Scholar]
- Kirkegaard K. (1990) Mutations in VP1 of poliovirus specifically affect both encapsidation and release of viral RNA. J. Virol., 64, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkegaard K. and Baltimore,D. (1986) The mechanism of RNA recombination in poliovirus. Cell, 47, 433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura N. et al. (1981) Primary structure, gene organization and polypeptide expression of poliovirus RNA. Nature, 291, 547–553. [DOI] [PubMed] [Google Scholar]
- Lesburg C.A., Cable,M.B., Ferrari,E., Hong,Z., Mannarino,A.F. and Weber,P.C. (1999) Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nature Struct. Biol., 6, 937–943. [DOI] [PubMed] [Google Scholar]
- Nugent C.I., Johnson,K.L., Sarnow,P. and Kirkegaard,K. (1999) Functional coupling between replication and packaging of poliovirus replicon RNA. J. Virol., 73, 427–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pata J.D., Schultz,S.C. and Kirkegaard,K. (1995) Functional oligomerization of poliovirus RNA-dependent RNA polymerase. RNA, 1, 466–477. [PMC free article] [PubMed] [Google Scholar]
- Plotch S.J., Palant,O. and Gluzman,Y. (1989) Purification and properties of poliovirus RNA polymerase expressed in Escherichia coli. J. Virol., 63, 216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racaniello V.R. and Baltimore,D. (1981) Molecular cloning of poliovirus cDNA and determination of the complete nucleotide sequence of the viral genome. Proc. Natl Acad. Sci. USA, 78, 4887–4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards O.C. and Ehrenfeld,E. (1990) Poliovirus RNA replication. Curr. Top. Microbiol. Immunol., 161, 89–119. [DOI] [PubMed] [Google Scholar]
- Sarnow P. (1989) Role of 3′-end sequences in infectivity of poliovirus transcripts made in vitro. J. Virol., 63, 467–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda N., Kuhn,R.J., Yang,C.F., Takegami,T. and Wimmer,E. (1986) Initiation of poliovirus plus-strand RNA synthesis in a membrane complex of infected HeLa cells. J. Virol., 60, 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyke T.A. and Flanegan,J.B. (1980) Identification of poliovirus polypeptide P63 as a soluble RNA-dependent RNA polymerase. J. Virol., 35, 732–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer E., Hellen,C.U.T. and Cao,X. (1993) Genetics of poliovirus. Annu. Rev. Genet., 27, 353–436. [DOI] [PubMed] [Google Scholar]
- Xiang W., Cuconati,A., Hope,D., Kirkegaard,K. and Wimmer,E. (1998) Complete protein linkage map of poliovirus P3 proteins: interaction of polymerase 3Dpol with VPg and with genetic variants of 3AB. J. Virol., 72, 6732–6741. [DOI] [PMC free article] [PubMed] [Google Scholar]