

Abstract
The tumor suppressor p53 is stabilized and activated in response to cellular stress through post-translational modifications including acetylation. p300/CBP-mediated acetylation of p53 is negatively regulated by MDM2. Here we show that MDM2 can promote p53 deacetylation by recruiting a complex containing HDAC1. The HDAC1 complex binds MDM2 in a p53-independent manner and deacetylates p53 at all known acetylated lysines in vivo. Ectopic expression of a dominant-negative HDAC1 mutant restores p53 acetylation in the presence of MDM2, whereas wild-type HDAC1 and MDM2 deacetylate p53 synergistically. Fibroblasts overexpressing a dominant negative HDAC1 mutant display enhanced DNA damage-induced p53 acetylation, increased levels of p53 and a more pronounced induction of p21 and MDM2. These results indicate that acetylation promotes p53 stability and function. As the acetylated p53 lysine residues overlap with those that are ubiquitylated, our results suggest that one major function of p53 acetylation is to promote p53 stability by preventing MDM2-dependent ubiquitylation, while recruitment of HDAC1 by MDM2 promotes p53 degradation by removing these acetyl groups.
Keywords: acetylation/HDAC1/MDM2/p53/ubiquitylation
Introduction
The activation of the tumor suppressor p53, which triggers growth arrest and apoptosis in cells that are in danger of becoming cancerous, involves the regulation of p53 stability. In unstressed cells, p53 is maintained at low levels by its key negative regulator MDM2 (Freedman et al., 1999). Although p53 is required for active tumor suppression, the negative regulation of p53 by MDM2 is fundamentally important. This point is best illustrated by a genetic study in mice, where the loss of MDM2 led to early embryonic lethality due to uncontrolled p53 levels and activity (Montes de Oca Luna et al., 1995). The importance of this negative regulation is further supported by the observation that expression of MDM2 is positively regulated by p53 (Wu et al., 1993), and thus establishes a tight negative feedback loop. MDM2 is an E3 ligase that ubiquitylates a defined set of lysine residues at the C-terminus of p53 (Honda et al., 1997; Nakamura et al., 2000; Rodriguez et al., 2000). The MDM2-mediated ubiquitylation of p53 is believed to trigger rapid degradation of p53 by proteasomes (Freedman et al., 1999), or to promote its nuclear export (Boyd et al., 2000; Geyer et al., 2000). Thus, MDM2 functions as a key negative regulator for p53, at least in part, by controlling the p53 ubiquitylation status (Zhang and Xiong, 2001).
p53 is transiently stabilized and activated in response to various cellular insults. The stabilization and activation of p53 are thought to be mediated by post-translational modification events, such as phosphorylation (Giaccia and Kastan, 1998; Appella and Anderson, 2001), which was proposed to interfere with the ability of MDM2 to negatively regulate p53 (Shieh et al., 1997; Unger et al., 1999). Although multiple serine residues are phosphorylated in response to genotoxic stress (Shieh et al., 1997; Canman et al., 1998; Hirao et al., 2000), p53 activation is not always accompanied by the specific phosphorylation events proposed to be important for its function (Ashcroft et al., 2000). In contrast to phosphorylation, we have reported that p53 invariably becomes acetylated in response to a wide variety of cellular insults that are known to be potent p53-activating agents (Ito et al., 2001). Importantly, stress-induced p53 acetylation is transient and reversible, suggesting the existence of negative regulators that keep p53 acetylation in check (Ito et al., 2001). Supporting this hypothesis, we and others have found that MDM2 negatively regulates p53 acetylation (Kobet et al., 2000; Ito et al., 2001). This result suggests a functional link between MDM2, an E3 ubiquitin ligase, and acetylation. The importance of this observation is further supported by the finding that the inhibitory effect of MDM2 on p53 acetylation can be reversed by the tumor suppressor p14ARF (Ito et al., 2001), which binds to MDM2 and inhibits its activity (Pomerantz et al., 1998; Zhang et al., 1998b; Honda and Yasuda, 1999). Together, these observations support the notion that the regulation of acetylation is a central target of the p53–MDM2–p14ARF feedback loop. However, the mechanism by which MDM2 controls p53 acetylation is not completely understood. It was previously shown that MDM2 can suppress CREB binding protein (CBP) acetyltransferase activity in vitro. This result supports the idea that MDM2 inhibits p53 acetylation, at least in part, by binding and reducing p300/CBP acetyltransferase activity (Kobet et al., 2000; Ito et al., 2001). However, the observation that the deacetylase inhibitor TSA can restore p53 acetylation in the presence of MDM2 suggests a more complex mechanism, wherein MDM2 might also utilize a deacetylase pathway to control p53 acetylation level (Ito et al., 2001). Uncovering this pathway will be essential to understanding how p53 acetylation is regulated and is the subject of this study.
The acetylation of p53 is mainly catalyzed by the acetyltransferases p300 and CBP in vivo (Ito et al., 2001). Interestingly, acetylation occurs at multiple lysine residues (six in total, see below) clustered at the C-terminus of p53 (Gu and Roeder, 1997; Sakaguchi et al., 1998; Liu et al., 1999). It is not known why there are so many lysine residues targeted for acetylation. The precise function of acetylation and the mechanism by which acetylation controls p53 also remain to be established. Acetylation was previously correlated with p53-dependent senescence and its ability to induce apoptosis (Luo et al., 2000; Pearson et al., 2000). As acetylation of p53 enhances its DNA-binding activity in vitro (Gu and Roeder, 1997; Sakaguchi et al., 1998), it has been widely assumed that acetylation functions by enhancing p53 transcriptional activity. However, there may be additional mechanisms by which acetylation could control p53 function. For example, since the activation of p53 involves its stabilization, we have previously suggested that acetylation might control the stability of p53. Indeed, our observation that inhibition of p53 deacetylation by the deacetylase inhibitor trichostatin A (TSA) is accompanied by an increase in p53 stability is consistent with this hypothesis (Ito et al., 2001). However, how acetylation controls p53 stability is not known.
In this report, we present evidence that acetylation controls p53 stability by potentially interfering with MDM2-mediated ubiquitylation. We found that MDM2 and the deacetylase HDAC1 form a complex that controls p53 acetylation in a cooperative fashion, thus providing a molecular link between the ubiquitylation and the deacetylation machinery. We provide evidence that stable overexpression of a dominant-negative HDAC1 mutant in fibroblasts leads to markedly enhanced p53 acetylation and p53 stability in response to DNA damage, suggesting that one function of acetylation is to promote p53 stability. Consistent with this conclusion, we found that acetylation and ubiquitylation occur at an overlapping set of lysine residues in p53. Our results suggest a simple model wherein acetylation promotes p53 stability by competing with MDM2-mediated ubiquitylation. The MDM2– HDAC1 interaction thus provides an efficient coupling of deacetylation and ubiquitylation machinery to negatively regulate p53 function.
Results
HDAC1 specifically interacts with MDM2
Our previous studies have shown that MDM2 can inhibit p300-induced p53 acetylation, in part, by repressing p300 acetyltransferase activity. However, the inhibitory effect of MDM2 on p53 acetylation can also be reversed by a pan-HDAC deacetylase inhibitor TSA (Ito et al., 2001), suggesting the involvement of active deacetylation. To further address the potential mechanism by which MDM2 negatively regulates p53 acetylation, we first considered the possibility that MDM2 may induce p53 deacetylation by recruiting a putative p53 deacetylase, since MDM2 itself does not possess TSA-sensitive deacetylase activity (data not shown). To test this hypothesis, we investigated whether MDM2 interacts with any of the known HDAC family members. Through a co-immunoprecipitation assay, we found that among HDAC1–7, only HDAC1 strongly co-immunoprecipitated with MDM2, while the others did not (Figure 1A; data not shown). This result indicates that MDM2 selectively interacts with HDAC1. To determine whether MDM2 binds HDAC1 directly, the ability of bacterially-expressed recombinant GST HDAC1 and MDM2 proteins to interact was investigated by pull-down assay (Figure 1B). As expected, recombinant MDM2 bound GST–p53 (Figure 1B, lane 3); however, MDM2 failed to bind GST–HDAC1 under the same conditions (Figure 1B, lane 2). Thus, these results indicate that MDM2 specifically interacts with HDAC1 via an indirect mechanism or that a specific modification of MDM2 or/and HDAC1 is required for this interaction.

Fig. 1. Interaction between MDM2 and HDAC1. (A) 293T cells were cotransfected with 10 µg of MDM2 and 5–10 µg of either Flag-tagged HDAC1 (lane 1), HDAC2 (lane 2), HDAC3 (lane 3), HDAC4 (lane 4) or HDAC5 (lane 5). Cellular extracts were immunoprecipitated with anti-Flag antibody followed by immunoblotting with anti-MDM2 antibody (top panel) or anti-Flag antibody (middle panel). Total MDM2 protein was detected with anti-MDM2 antibody (bottom panel). (B) Either GST (lane 1), GST–HDAC1 (lane 2) or GST–p53 (lane 3) were incubated with recombinant MDM2 protein followed by immunoblotting with anti-MDM2 antibody. (C) H1299 cells were transfected with Flag-tagged HDAC1 and the cellular extracts were incubated with either GST (lane 1) or GST–MDM2 (lane 2) followed by immunoblotting with anti-Flag antibody. (D) H1299 cells were transfected with MDM2 alone (lane 1) or cotransfected with Flag-tagged HDAC1 (lane 2) and cellular extracts were immunoprecipitated with anti-MDM2 antibody followed by immunoblotting with anti-Flag antibody. (E) A549 cells were exposed to UV-B irradiation (75 J/m2) for 6 h and cellular extracts were immunoprecipitated with either anti-MDM2 antibody (lanes 1 and 2) or mouse IgG as a control (lane 3) followed by immunoblotting with anti-HDAC1 antibody (top panel) or anti-MDM2 antibody (bottom panel). Total HDAC1 and MDM2 protein were detected with either anti-HDAC1 antibody (top panel, lanes 4 and 5) or anti-MDM2 antibody (bottom panel, lanes 4 and 5).
As MDM2 binds p53 directly, we investigated whether the binding of MDM2 and HDAC1 was mediated by p53. To test this possibility, an HDAC1 expression plasmid was transfected into H1299 cells (p53–/–) and the interaction of MDM2 and HDAC1 was determined by pull-down assay using GST–MDM2, followed by immunoblotting to visualize the associated HDAC1. As shown in Figure 1C, GST–MDM2, but not GST, interacts with HDAC1 expressed in H1299 cells. A similar conclusion regarding HDAC1–MDM2 interaction in H1299 cells was obtained by co-transfecting HDAC1 and MDM2 expression plasmids, which were assayed by co-immunoprecipitation (Figure 1D). These results demonstrate a specific and p53-independent interaction between MDM2 and HDAC1.
We investigate further the interaction between the endogenous HDAC1 and MDM2. As shown in Figure 1E, at the basal state in which the MDM2 level is low, a co-immunoprecipitation assay revealed a weak but reproducible interaction of MDM2 and HDAC1. However, upon UV irradiation, which induces MDM2 expression (∼4-fold), a marked increase of MDM2 and HDAC1 association (∼3-fold) was observed. These results demonstrate an endogenous MDM2–HDAC1 interaction that can be stimulated by UV irradiation, likely due to an increase of the MDM2 protein level in response to DNA damage.
MDM2–HDAC1 deacetylates p53 cooperatively
The physical interaction of MDM2 and HDAC1 supports the hypothesis that MDM2 recruits HDAC1 to deacetylate p53. Consistent with this hypothesis, it was previously proposed that HDAC1 could function as a p53 deacetylase (Luo et al., 2000). However, although the ectopic expression of HDAC1-associated MTA2/PID can induce p53 deacetylation, whether or not HDAC1 is a p53 deacetylase in vivo was not directly tested, nor did the study address whether other HDAC family members could deacetylate p53. To investigate these issues and determine whether the specific association with MDM2 is functionally correlated with the ability of an HDAC to function as a p53 deacetylase, expression plasmids for p300, HDAC1–5 were cotransfected into 293T cells and the p53 acetylation status on lysine 382 was assessed using an acetyl-Lys382-specific antibody. As shown in Figure 2A, consistent with its unique association with MDM2, only HDAC1 but not other HDACs efficiently induced p53 deacetylation on lysine 382 (Figure 2A, lanes 3–7). Importantly, ectopic expression of HDAC1 also led to deacetylation at lysine 320 and 373 (Figure 2B), both of which are also known to become acetylated upon p53 activation (Sakaguchi et al., 1998; Liu et al., 1999). Thus, in vivo, HDAC1 can deacetylate all three known acetylated lysine residues in p53. An identical conclusion that HDAC1, but not other HDAC members, possesses strong p53 deacetylase activity can be demonstrated directly by an in vitro assay using HDACs immunoprecipitated from cells transfected with the HDAC expression plasmids (Figure 2C), while all HDAC family members possess a deacetylase activity toward histones (Figure 2 D). Thus, among the HDAC members tested, HDAC1 uniquely associates with MDM2 and can specifically function as a p53 deacetylase, supporting its role for mediating MDM2-dependent p53 deacetylation.

Fig. 2. Deacetylation of p53 by HDAC1. (A) 293T cells were transfected with an empty vector (lane 1) or cotransfected with either 2 µg of p300 alone (lane 2) or cotransfected with 2 µg of Flag-tagged HDAC1 (lane 3), 2 µg of HDAC2 (lane 4), 2 µg of HDAC3 (lane 5), 4 µg of HDAC4 (lane 6) or 4 µg of HDAC5 (lane 7). The levels of endogenous acetylated p53, total p53, and each HDAC were detected by immunoblotting with anti-acetyl ated p53 (Lys382) (top panel), anti-p53 antibody (middle panel) and anti-Flag antibody (bottom panel). (B) H1299 cells (p53–/–) were transfected with 0.2 µg of p53 alone (lane 1) or cotransfected with either 2 µg of p300 alone (lane 2), with 2 µg of HDAC1 wild-type (lane 3) or 2 µg of enzyme-dead H141A mutant (lane 4). The level of acetylated p53 was assessed using either antibody specific for acetylated Lys382 (Ac382), acetylated Lys373 (Ac373) or acetylated Lys320 (Ac320). The levels of total p53 and Flag-tagged HDAC1 were detected as described in (A). (C and D) 293T cells were transfected with empty vector or Flag-tagged HDACs and cellular extracts were prepared as described in Materials and methods. Deacetylase activity was measured against acetylated GST–p53 (C) or histone H4 peptide (D) in the presence or absence of TSA. Results are representative of three independent experiments. Note that all HDAC family members possess deacetylase activity towards histones, but only HDAC1 can efficiently deacetylate p53.
To determine whether MDM2 is required for HDAC1 to function as a p53 deacetylase, we investigated the ability of HDAC1 to deacetylate p53 in MDM2-deficient cells. As shown in Figure 3A, overexpression of HDAC1 still results in p53 deacetylation in MDM2–/–;p53–/– mouse embryonic fibroblasts (MEFs). Although MDM2 is not required for high levels of HDAC1 to deacetylate p53, it is possible that the interaction with MDM2 could facilitate HDAC1’s activity as a p53 deacetylase. To test this possibility, low concentrations of MDM2 and HDAC1 were co-expressed with p53 and p300 in MDM2–/–; p53–/– MEFs, and the p53 acetylation status was assessed. Under these conditions, neither MDM2 nor HDAC1 alone had an appreciable effect on the level of p53 acetylation (Figure 3B, lanes 2 and 4). However, co-expression of both MDM2 and HDAC1 dramatically reduced the level of p53 acetylation (Figure 3B, lane 5). These results demonstrate that MDM2 and HDAC1 function synergistically to induce p53 deacetylation. To elucidate the potential mechanism underlying this synergistic activity, we asked whether MDM2 affects the interaction between p53 and HDAC1. As shown in Figure 3C, in MDM2-deficient MEFs, HDAC1 only interacts weakly with p53 (Figure 3C, lane 2). However, this interaction is dramatically stimulated upon the re-introduction of wild-type MDM2 (Figure 3C, lane 3). The p53-binding-deficient MDM2 mutant is less efficient in facilitating the p53–HDAC1 interaction. (Figure 3C, lane 4). Thus, the reduced p53 binding is correlated with reduced recruitment of HDAC1 by MDM2. Together, these results indicate that MDM2 promotes the interaction between HDAC1 and p53 and forms a ternary complex, allowing efficient p53 deacetylation.
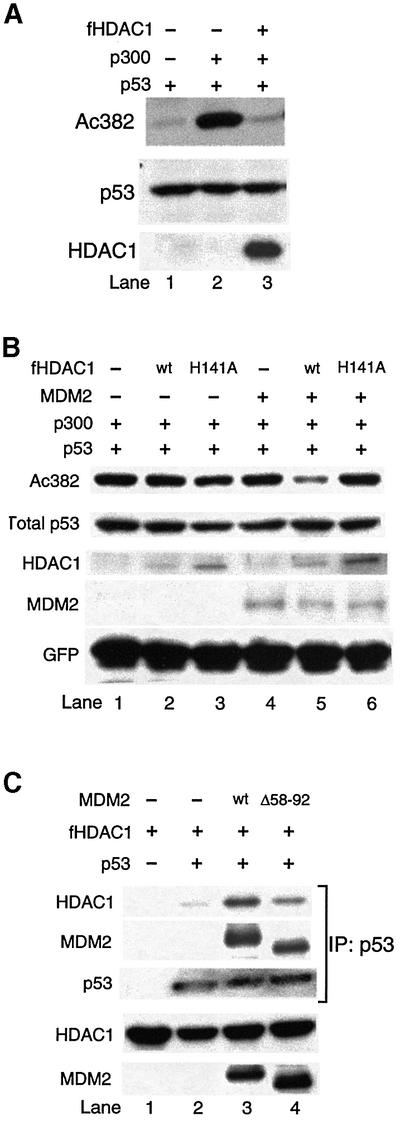
Fig. 3. HDAC1 and MDM2 work cooperatively to deacetylate p53. (A) MEF (p53–/–;MDM2–/–) cells were transfected with 0.1 µg of p53 (lane 1), or cotransfected either with 2 µg of p300 alone (lane 2) or with p300 and 2 µg of Flag-tagged HDAC1 (lane 3). The level of total p53 (middle panel) and acetylated p53 (top panel) were detected as described in Figure 2. (B) MEF (p53–/–;MDM2–/–) cells were transfected with either 0.1 µg of p53, 2 µg of p300 and 0.5 µg of internal control GFP (lane 1), or cotransfected either with 12.5 ng of Flag-tagged HDAC1 wild-type (lane 2), with 12.5 ng of Flag-tagged HDAC1 H141A mutant (lane 3), with 0.5 µg of MDM2 (lane 4), with MDM2 and 12.5 ng of Flag-tagged HDAC1 wild-type (lane 5) or 0.5 µg of MDM2 and 12.5 ng of Flag-tagged HDAC1 H141A mutant (lane 6). The levels of indicated proteins were determined by immunoblotting. Of note, we used 160× the amount of Flag-tagged HDAC1 expression vectors in (A) when compared with (B). (C) MEF (p53–/–;MDM2–/–) cells were transfected with either 1 µg of Flag-tagged HDAC1 wild-type alone, or cotransfected either with 0.3 µg of p53, with 0.3 µg of p53 and 4µg of MDM2 wild-type, or 0.3 µg of p53 and 4µg of p53-binding-deficient MDM2 mutant (Δ58–92). Cells were also treated 24 h post-transfection with the protease inhibitor LLnV (10 µM) for 4 h to inhibit MDM2-mediated p53 degradation. Cellular extracts were immunoprecipitated with anti-goat p53 antibody followed by immunoblotting with Flag antibody (top panel), anti-MDM2 antibody (second panel) or anti-p53 antibody (third panel). Total HDAC1 and MDM2 protein were detected with either anti-Flag antibody (fourth panel) or anti-MDM2 antibody (bottom panel). Of note, the interaction between p53 and p53-binding-deficient MDM2 mutant (Δ58–92) is likely mediated by HDAC1 through the ternary complex formation (second panel, lane 4).
HDAC1 mediates MDM2-dependent p53 deacetylation
We next examined whether or not HDAC1 was responsible for MDM2-mediated p53 deacetylation. The observation that HDAC1 binds MDM2 indirectly (Figure 1) and that bacterially-expressed recombinant HDAC1 does not possess p53 deacetylase activity (data not shown) suggests that the p53 deacetylase activity of HDAC1 requires additional factors. We reasoned that as HDAC1 normally functions in a multi-protein complex (Zhang et al., 1998a), a catalytically inactive (enzyme-dead) HDAC1 could potentially titrate away the cofactors needed for the active complex and thus function as a dominant-negative mutant. To this end, we asked whether a catalytically inactive HDAC1 mutant (H141A) could reverse the effect of MDM2 on p53 acetylation. As shown in Figure 4, overexpression of MDM2 causes a dramatic reduction of p53 acetylation induced by p300 (Figure 4, lane 3). However, upon co-expression of the HDAC1 H141A mutant, the effect of MDM2 is partially lost and p53 acetylation restored (Figure 4, lane 7). This observation supports the idea that HDAC1 H141A acts as a dominant-negative mutant and that MDM2-induced p53 deacetyl ation is mediated by HDAC1. Together, these observations provide evidence that MDM2 recruits HDAC1 into a multi-protein complex, which promotes p53 deacetylation.

Fig. 4. MDM2 induces p53 deacetylation through HDAC1. H1299 cells were transfected with 0.2 µg of p53 wild-type and 0.5 µg of GFP (all lanes), and cotransfected with one or more of the following; 2 µg of p300 (lanes 2–7), 2 µg of MDM2 (lanes 3, 6 and 7), 2 µg of Flag-tagged HDAC1 wild-type (lanes 4 and 6), 2 µg of Flag-tagged HDAC1 H141A mutant (lanes 5 and 7). The levels of indicated proteins were determined by immunoblotting.
A common set of lysines are modified by acetylation and ubiquitylation
To begin to address the potential function of p53 acetylation, we first generated p53 mutants that can not be acetylated (Figure 5A). By analyzing the acetylation pattern of recombinant p53 acetylated by CBP, we found that mutation of all three known acetylated lysines (320, 373 and 382) does not eliminate p53 acetylation completely (Figure 5B, lane 2). However, additional mutations of lysines 370, 372 and 381 essentially abolished the acetylation by CBP (Figure 5B, lane 3). Thus, there are at least six lysine residues in p53 that can be acetylated by CBP in vitro. To generate the non-acetylatable p53 mutant, all six lysine residues were mutated to arginine (6KR), in order to minimize the structural impact by maintaining the positive charges at these residues.

Fig. 5. Mutations of lysine residues of acetylation sites prevent MDM2-mediated p53 degradation. (A) Schematic structure of p53 mutations of C-terminal lysines to alanines or arginines. (B) GST–p53 wild-type (lane 1), GST–p53 3KA mutant with mutated lysine residues 320, 373 and 382 to alanine residues (lane 2), or GST–p53 6KR mutant with mutated lysine residues 320, 370, 372, 373, 381 and 382 to arginine residues (lane 3) were acetylated by recombinant CBP in the presence of the [ 14C]acetyl-CoA and analyzed by SDS–PAGE followed by autoradiograph. Acetylated p53 and CBP are indicated by arrows. As a negative control, wild-type GST–p53 was incubated without recombinant CBP in the presence of the [14C]acetyl-CoA (lane 4). (C) Acetylation sites of p53 overlap with ubiquitylation sites. The lysine residues susceptible to acetylation and ubiquitylation in the C-terminus of p53 are indicated by arrows. (D) H1299 cells were transfected with 0.5 µg of an expression plasmid encoding GFP as an internal control, 0.2 µg of wild-type p53 or p53 6KR mutant, together with empty pcDNA3 vector or the indicated amount of MDM2 vector. Thirty-six hours after transfection, cell extracts were prepared and analyzed by immunoblotting with anti-p53, anti-MDM2 and anti-GFP antibodies. (E) The band intensity of p53 and GFP protein levels was measured with NIH imaging software. p53 (empty circles) and 6KR mutant (filled circles) levels were normalized to GFP levels and were set to 1 in the absence of MDM2.
Our previous study established a positive correlation between p53 acetylation and its total protein level (Ito et al., 2001); we therefore examined the potential function of acetylation in regulating p53 stability. As both acetylation and ubiquitylation modify the ε amino group of lysine residues, we first evaluated whether there is a relationship between p53 acetylation and ubiquitylation. Recent studies on p53 ubiquitylation have identified several lysine residues important for MDM2-mediated ubiquitylation (Nakamura et al., 2000; Rodriguez et al., 2000). When compared with the acetylated lysine residues identified in this report, we found that lysine residues targeted for ubiquitylation overlap those that can be acetylated in vitro (Figure 5C). Consistent with this idea, the acetylation-deficient 6KR mutant is completely resistant to MDM2-mediated degradation (Figure 5D and E). Thus, a common set of lysine residues is targeted by both p300/CBP-mediated acetylation and MDM2-mediated ubiquitylation. This result suggests the possibility that the acetylation of C-terminal lysine residues might prevent their ubiquitylation and consequently lead to p53 stabilization.
A dominant-negative HDAC1 mutant promotes both p53 acetylation and stability in response to DNA damage
Despite its ability to affect p53 acetylation levels, HDAC1 and its H141A mutant showed little effect on p53 protein levels when acetylation was induced by overexpression of p300 (Figure 4). It is possible that there may be only a very small portion of total p53 becoming acetylated under this artificial condition, and consequently, the balance of acetylation and ubiquitylation is not faithfully reflected in this system. To circumvent this problem and to investigate the role of p53 acetylation in a physiological setting, we generated NIH 3T3 lines that stably expressed HDAC1 or its dominant-negative mutant by retrovirus-mediated gene transfer. A physiological level of p53 acetylation can be induced by DNA-damaging agents in these cell lines, and p53 stability and activity can then be evaluated. To this end, control NIH 3T3 cells and their derivatives overexpressing wild-type HDAC1 and dominant-negative HDAC1 were irradiated with UV, and the levels of p53 and its acetylation were determined. Importantly, overexpression of HDAC1 or its dominant-negative mutant had no effect on the basal level of p53 or its acetylation (Figure 6A). This indicates that there is little non-specific effect on p53 activation caused by HDAC1 or its mutant. After UV treatment, however, the p53 acetylation level was significantly enhanced in NIH 3T3 cells overexpressing dominant negative HDAC1 and reduced in the wild-type HDAC1-expressing cells (Figure 6A and C). This result demonstrates that HDAC1 can modulate p53 acetylation status in response to DNA damage. Consistent with the hypothesis that acetylation promotes p53 stability, the level of stabilized p53 protein is dramatically increased in the H141A mutant cell line in response to UV irradiation (Figure 6A and B). This conclusion is further supported by the observation of an increase in p53 half-life in mutant HDAC1 H141A-expressing cells and a decrease in wild-type HDAC1-expressing cell when compared with control cell lines (Figure 6D and E). Importantly, the UV-induced, p53-dependent induction of p21 and MDM2 also reaches a much higher level in HDAC1 H141A mutant cell lines than in control and wild-type HDAC1 lines, demonstrating that an increase in p53 acetylation is accompanied by both p53 stabilization and enhanced function (Figure 6A and B). Together, these results support the idea that HDAC1 regulates p53 stability and function by modulating its acetylation levels.

Fig. 6. Effect of HDAC1 on p53 acetylation, stability and activity in response to DNA damage. (A) NIH 3T3 cells infected with mock vector (control), pBabe-HDAC1 wild-type (HDAC1-wt), or pBabe-HDAC1 H141A mutant (HDAC1 H141A) were exposed to UV-B (75 J/m2). Cells were harvested at the indicated times. The levels of total p53 (panel 1), acetylated p53 (panel 2), p21 (panel 3), MDM2 (panel 4) and the internal control α-tubulin (panel 5) were assessed by immunoblotting. (B and C) The band intensity of p53, acetylated p53 and α-tubulin protein levels in all three cell lines were measured with NIH imaging software. The levels of p53 (B) and acetylated p53 (C) were normalized to α-tubulin and the highest intensity levels of p53 or acetylated p53 were set to 1. (B) and (C) are representative results of three (B) and two (C) independent experiments. (D) All three stable cell lines were exposed to UV-B (75 J/m2) and 2 h post-irradiation, cyclohexamide (10 µg/ml) was added to inhibit new p53 protein synthesis (designated 0 h). Cells were harvested at the time points indicated after cyclohexamide treatment. The level of total p53 (upper panel) and α-tubulin (lower panel) was determined. (E) The band intensity of p53 and α-tubulin protein levels were measured by NIH imaging software. p53 levels were normalized to α-tubulin levels and calculated against the amount of p53 present at time point 0, which was set at 100%. Results are representative of three independent experiments.
Discussion
We have demonstrated previously that acetylation of p53 is invariably and transiently induced upon its activation (Ito et al., 2001). The transient nature of p53 acetylation suggests the presence of negative regulators for p53 acetylation. In this report, we identify MDM2 and HDAC1 as the key components that function cooperatively to control p53 deacetylation. We found that acetylation functions, at least in part, by promoting p53 stability. The realization that p300/CBP-mediated acetylation and MDM2-mediated ubiquitylation occur at a common set of lysine residues provides a potential molecular mechanism by which acetylation controls p53 stability by competing with ubiquitylation. The identification of an MDM2–HDAC1 interaction thus provides a novel mechanism to couple the regulation of acetylation and ubiquitylation for the efficient control of p53 levels.
We have previously demonstrated that p53 acetylation is negatively regulated by MDM2 and that this activity can be reversed by p14ARF (Ito et al., 2001). We and others have shown that MDM2 suppresses p53 acetylation, at least in part, by inhibiting the acetyltransferase activity of p300 and CBP (Kobet et al., 2000; Ito et al., 2001). However, we also observed that the inhibitory activity of MDM2 toward p53 acetylation can be reversed by the deacetylase inhibitor TSA (Ito et al., 2001). This observation led us to propose that MDM2 must utilize additional mechanisms to regulate p53 acetylation. The identification of a specific interaction between MDM2 and HDAC1 now provides evidence for a second mechanism employed by MDM2 to control p53 acetylation. Consistent with the idea that MDM2 recruits HDAC1 to downregulate p53 acetylation, HDAC1, but not other members of the HDAC family (2, 3, 4, 5 and 7), has the capacity to function as a p53 deacetylase towards all three known acetylated lysine residues of p53 in vivo (Figure 2; data not shown). Interestingly, HDAC2 has been found to co-exist with HDAC1 in several complexes (Zhang et al., 1998a), but does not interact with MDM2 appreciably and has no significant p53 deacetylase activity. This surprising observation raises an interesting possibility that HDAC1 might reside in a different complex to function as a p53 deacetylase. An earlier report demonstrated that HDAC1 acts as a p53 deacetylase based on the observation that overexpression of HDAC1-associated PID/MTA-2 protein can induce p53 deacetylation (Luo et al., 2000). Our study, however, provides the first direct evidence that HDAC1 indeed functions as a p53 deacetylase in vivo (Figure 2). It is worth noting that our data suggest that MDM2 and HDAC1 may interact via an intermediate factor; however, given the important role of PID/MTA-2 in p53 deacetyl ation (Luo et al., 2000), it remains to be tested whether PID plays a role in mediating the HDAC1–MDM2 interaction. The important role of HDAC1 in MDM2-mediated p53 deacetylation is further substantiated by the finding that a dominant-negative HDAC1 mutant (H141A) can restore p53 acetylation levels in the presence of MDM2 (Figure 4). The identification of an MDM2–HDAC1 complex not only provides a novel mechanism by which p53 acetylation is regulated, but it also revealed an unexpected link between the acetylation and ubiquitylation machinery (see below for more discussion).
Although high levels of HDAC1 can induce p53 deacetylation in the absence of MDM2, low levels of HDAC1, which likely reflect physiological conditions, fail to do so (Figure 3A and B). However, the ability of HDAC1 to function as a p53 deacetylase, at low levels, is dramatically induced in the presence of MDM2 (Figure 3B). This result suggests that MDM2 facilitates the functional interaction between HDAC1 and p53. In support of this hypothesis, we showed that the physical association between HDAC1 and p53 was enhanced in the presence of MDM2 (Figure 3C). As the level of HDAC1 itself appears to be constant and not subject to regulation (Figure 1E; A.Ito, unpublished observation), the interaction with MDM2, whose level is regulated by p53 in response to various stresses, provides a means to control the activity of HDAC1 toward p53. Under this scenario, the p53 deacetylase activity of HDAC1 becomes activated when the MDM2 levels are increased by active p53 in response to stress. Consistent with this idea, the endogenous interaction between HDAC1 and MDM2 is enhanced after DNA damage when the level of MDM2 is induced (Figure 1E). Thus, together with MDM2, HDAC1 becomes a key component of a p53 negative feedback loop. Supporting this idea, inhibition of HDAC1 activity by an HDAC1 dominant-negative mutant leads to a dramatic enhancement of DNA damage-induced p53 acetylation, p53 stability and activity (Figure 6). The cooperative activity of MDM2 and HDAC1 toward p53 could be achieved through the stimulation of p53–HDAC1 complex formation as shown in Figure 3C. Another interesting possibility is that, as MDM2 can ubiquitylate substrates other than p53 (Shenoy et al., 2001), MDM2 might regulate the activity of HDAC1 by ubiquitylating HDAC1 or a component of the HDAC1 complex. Such a possibility is consistent with our previous observation that p14ARF, which is known to inhibit the MDM2 E3 ligase activity toward p53 (Honda and Yasuda, 1999), can reverse MDM2-mediated p53 deacetylation (Ito et al., 2001).
Regardless of the mechanism, our results support the idea that the recruitment of HDAC1 by MDM2 plays an important role in regulating p53 deacetylation and function. However, it is likely that HDAC1 is not the only deacetylase that regulates p53 acetylation. We have evidence that a portion of p53 deacetylation could be carried out in the cytoplasm whereas HDAC1 resides in the nucleus (A.Ito, Y.Kawaguchi and T.P.Yao, unpublished observation). Furthermore, recent studies have shown that the NAD-dependent and TSA-insensitive histone deacetylase Sir-2, can also deacetylate p53 (Luo et al., 2001; Vaziri et al., 2001). However, unlike HDAC1, which is able to deacetylate p53 at all three known acetylated Lys residues, Sir-2 was shown to mainly deacetylate Lys 382 (Vaziri et al., 2001). Further studies will be required to elucidate the individual role of these deacetylases in regulating p53 function. However, the participation of multiple deacetylases further supports the idea that acetylation is a critical mechanism for regulation of p53.
We have previously shown that acetylation appears to be a critical modification, as it invariably accompanies p53 activation and is the target of key p53 regulators, like MDM2, p14ARF and p300/CBP (Ito et al., 2001). However, the exact function and mechanism by which acetylation controls p53 activation remains unclear. As acetylation stimulates p53 DNA-binding activity in vitro (Gu and Roeder, 1997; Sakaguchi et al., 1998), it was hypothesized that acetylation of p53 promoted its transcriptional activity. Supporting this idea, a recent study shows that, although it does not play a major role on p53 association with target promoters in vivo, p53 acetylation is involved in the recruitment of transcriptional co-activators (Barlev et al., 2001). Our current study provides evidence that, in addition to a role in p53 transcriptional activity, there exists another novel function for p53 acetylation. We have reported previously the parallel kinetics of p53 acetylation and stabilization and the enhancement of p53 stability by inhibition of its deacetylation (Ito et al., 2001). Both observations are consistent with the idea that one function of acetylation is to promote p53 stability. By assessing the p53 status in fibroblasts stably overexpressing a dominant negative mutant of HDAC1, we have obtained further evidence that enhanced acetylation is associated with an increase in p53 protein stability in response to DNA damage (Figure 6). Although the complete in vivo acetylation sites in p53 still remain to be established, the in vitro mapping effort reveals that both p300/CBP-mediated acetylation (Figure 5B) and MDM2-mediated ubiquitylation (Nakamura et al., 2000; Rodriguez et al., 2000) occurred at an overlapping set of lysine residues (Figure 5C). This observation offers a further link between p53 acetylation and stability. The potential competition for acetylation and ubiquitylation of these lysine residues provides a plausible molecular mechanism by which acetylation promotes p53 stability. In this model (Figure 7), at the p53 basal state, a set of C-terminal lysine residues of p53 are subject to MDM2-mediated ubiquitylation, which leads to p53 degradation. Upon its activation by stress signals, these lysine residues become acetylated by p300/CBP and are no longer available for MDM2-dependent ubiquitylation, leading to p53 stabilization. Stabilized p53 functions as a tumor suppressor and induces high levels of MDM2, which in turn, promotes p53 deacetylation by recruiting a p53 deacetylase, HDAC1. The unmodified lysine residues can then serve as the substrates for MDM2-mediated ubiquitylation resulting in p53 degradation. The interaction of MDM2 and HDAC1 provides a novel molecular mechanism for an efficient coupling of deacetylation and ubiquitylation of p53 that allows MDM2 to effectively inactivate and degrade p53 and complete the negative feedback loop. The potential functional interaction between the acetylation and ubiquitylation machinery described in this model would also suggest a broader and more general role for reversible acetylation in the regulation of protein stability and other ubiquitylation-dependent biological processes.

Fig. 7. A model for the regulation and interplay of p53 acetylation and ubiquitylation. Under unstressed conditions, p53 is ubiquitylated on lysine residues by MDM2 and targeted for degradation (A). Upon its activation by various cellular insults, p53 becomes acetylated by p300/CBP at the same set of lysine residues also targeted by MDM2 (B). Acetylation on lysine residues thus prevents MDM2-mediated ubiquityl ation and leads to p53 stabilization (C). The stabilized p53 functions as a tumor suppressor and also induces MDM2 (D). The high level of MDM2, in turn, recruits the p53 deacetylase HDAC1 and triggers p53 deacetylation (E). The deacetylated p53 with unmodified lysine residues is now ready to be ubiquitylated by MDM2 and ultimately degraded.
Materials and methods
Cell lines and transfection
293T, H1299, A549 and MDM2–/–;p53–/– double null MEF cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, while NIH 3T3 cells were maintained in DMEM supplemented with 10% calf serum. All cells were grown at 37°C in a humidified atmosphere of 5% CO2. All transfections were performed by either the calcium phosphate method as described previously (Yao et al., 1992) or the lipofectamine method (Invitrogen). Retroviruses were produced by transient transfection of a pBabe Puro vector construct into phoenix cells. NIH 3T3 cells were infected with retrovirus containing media in the presence of 8 µg/ml of polybrene overnight. Thirty-six hours later, cells were selected in the presence of 1.5 µg/ml puromycin and kept under selection in medium containing 1.5 µg/ml puromycin during experiments.
Plasmids
p53, MDM2, and p300 vectors were described previously (Ito et al., 2001). HDAC3 cDNA was provided by Dr F.Dangond (Harvard Medical School) and cloned into the EcoRI/ NotI sites of pcDNA3 vector with a Flag tag. HDAC4 and 5 cDNAs were provided by Dr S.L.Schreiber (Harvard University) and described in Grozinger et al. (1999). HDAC1 H141A mutant cDNA (provided by Dr S.L.Schreiber) was subcloned into the BamHI/EcoRI sites of pcDNA3 vector. For retroviral constructs, HDAC1 wild-type and H141A mutant cDNAs were cloned into the Bam HI/EcoRI sites of the pBabe Puro vector. p53–3KA mutant cDNA, mutated lysines 320, 373 and 382 to alanine, 6KR mutant, and mutated lysines 320, 370, 372, 373, 381 and 382 to arginine cDNAs were generated by site-directed mutagenesis and cloned into the pcDNA3 or the pGEX vector (Amersham).
DNA-damage treatment
DNA damage was performed by exposing cells to a 310 nm wavelength UV source and cells were harvested at indicated time points.
Immunoprecipitation and immunoblotting
Cells were lysed in buffer (20 mM Tris–HCl pH 7.6, 170 mM NaCl, 1 mM EDTA, 0.5% NP-40, 1 mM DTT) supplemented with 5 µM TSA and protease inhibitors. For immunoprecipitation with anti-goat p53 antibody, equal amounts of lysate (containing 100–300 µg of total cellular protein) were incubated with 1 µg of goat anti-p53 antibody (FL-393; Santa Cruz) and protein G–Sepharose (Pharmacia) for 3 h at 4°C. To detect acetylated mouse p53, equal amounts of lysate (containing 300–500 µg of total cellular protein) were incubated with agarose-conjugated anti-p53 antibody (Pab421) overnight at 4°C. To detect endogenous MDM2 and HDAC1 interaction, 2 mg of cellular lysate was incubated with 1 µg of anti-MDM2 antibody (SMP14; Santa Cruz) and protein A and G–Sepharose mixtures (Pharmacia) overnight at 4°C. When immunoprecipitation was not performed, 20–50 µg of total extracts were analyzed. Proteins were detected by chemiluminescent ECL kit (Amersham) with one of the following antibodies: anti-human p53 antibody (Ab-6; Calbiochem), anti-p53 antibody for detecting mouse p53, anti-human acetylated (Lys320) p53 antibody (Sakaguchi et al., 1998), anti-human acetylated (Lys373) p53 antibody, anti-human acetylated (Lys382) p53 antibody (Calbiochem), anti-mouse acetylated (Lys382) p53 antibody, anti-human MDM2 antibody (SMP14; Santa Cruz), anti-Flag antibody (M2; Sigma), anti-HDAC1 antibody (H-11; Santa Cruz), anti-α-tubulin antibody (DM1A; Sigma), anti-p21 antibody (H164; Santa Cruz) or anti-GFP antibody (Boehringer Mannheim).
In vitro deacetylation assay
The expression vectors for Flag-tagged HDACs (10–15 µg) were transfected into 293T cells and the cells were lysed in low stringency buffer (50 mM Tris–HCl pH 7.5, 120 mM NaCl, 0.5 mM EDTA, 0.5% NP-40) in the presence of protease inhibitors. After pre-clearing with protein A beads, the extracts were immunoprecipitated with anti-Flag antibody in the presence of rabbit anti-mouse antibody and protein A beads for 5 h at 4°C and then the beads were washed three times with low stringency buffer, twice with low stringency buffer containing 0.5 M NaCl, and twice with deacetylase buffer (10 mM Tris–HCl pH 8.0, 10 mM NaCl, 10% glycerol). For inhibition studies, the immune complexes were pre-incubated with 400 nM of TSA in deacetylase buffer for 30 min at 4°C. The immune complexes were incubated with 10 000 c.p.m. of 14C-labeled acetylated GST–p53 or 3H-labeled acetylated histone 4 peptide in 200 µl of deacetylase buffer for 2 h at 37°C and the release of [14C] or [3H]acetate was quantified by scintillation counting.
In vitro acetylation assay
The in vitro acetylation assay was performed as described previously (Ito et al., 2001). Briefly, recombinant CBP protein (1 µg), purified from sf9 insect cells infected with baculovirus expressing CBP, was incubated with 1 µg of wild-type GST–p53 or GST–p53 mutants in the presence of 50 nCi [14C]acetyl-coenzyme A in 30 µl of reaction buffer (50 mM Tris–HCl pH 8.0, 10% glycerol, 1 mM dithiothreitol, 100 µM EDTA, 1 mM PMSF) for 1 h at 37°C. Acetylation was analyzed by SDS–PAGE followed by autoradiography.
Acknowledgments
Acknowledgements
We thank Dr S.L.Schreiber for his generous gift of the HDAC1 H141A, HDAC4 and HDAC5 cDNAs, Dr F.Dangond for the HDAC3 cDNA, Dr C.W.Wu for the GST–HDAC1 construct and Dr G.Lozano for the MDM2–/–;p53–/– double null MEF cells. We are grateful to Drs C.Anderson, B.Harvat, A.R.Means, X.F.Wang, Ms A.Guardiola and Mr T.A.Bolger for critically reading the manuscript. This work is supported by funding from the Damon Runyon-Walter Winchell Cancer Foundation (DRS 20), National Institutes of Health (CA85676-01A1) and Department of Defense (DAMD 17-01-1-0054) to T.-P.Y.
References
- Appella E. and Anderson,C.W. (2001) Post-translational modifications and activation of p53 by genotoxic stresses. Eur. J. Biochem., 268, 2764–2772. [DOI] [PubMed] [Google Scholar]
- Ashcroft M., Taya,Y. and Vousden,K.H. (2000) Stress signals utilize multiple pathways to stabilize p53. Mol. Cell. Biol., 20, 3224–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlev N.A., Liu,L., Chehab,N.H., Mansfield,K., Harris,K.G., Halazonetis,T.D. and Berger,S.L. (2001) Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol. Cell, 8, 1243–1254. [DOI] [PubMed] [Google Scholar]
- Boyd S.D., Tsai,K.Y. and Jacks,T. (2000) An intact HDM2 RING-finger domain is required for nuclear exclusion of p53. Nat. Cell Biol., 2, 563–568. [DOI] [PubMed] [Google Scholar]
- Canman C.E., Lim,D.S., Cimprich,K.A., Taya,Y., Tamai,K., Sakaguchi,K., Appella,E., Kastan,M.B. and Siliciano,J.D. (1998) Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science, 281, 1677–1679. [DOI] [PubMed] [Google Scholar]
- Freedman D.A., Wu,L. and Levine,A.J. (1999) Functions of the MDM2 oncoprotein. Cell. Mol. Life Sci., 55, 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer R.K., Yu,Z.K. and Maki,C.G. (2000) The MDM2 RING-finger domain is required to promote p53 nuclear export. Nat. Cell Biol., 2, 569–573. [DOI] [PubMed] [Google Scholar]
- Giaccia A.J. and Kastan,M.B. (1998) The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev., 12, 2973–2983. [DOI] [PubMed] [Google Scholar]
- Grozinger C.M., Hassig,C.A. and Schreiber,S.L. (1999) Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl Acad. Sci. USA, 96, 4868–4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W. and Roeder,R.G. (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell, 90, 595–606. [DOI] [PubMed] [Google Scholar]
- Hirao A., Kong,Y.Y., Matsuoka,S., Wakeham,A., Ruland,J., Yoshida,H., Liu,D., Elledge,S.J. and Mak,T.W. (2000) DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science, 287, 1824–1827. [DOI] [PubMed] [Google Scholar]
- Honda R. and Yasuda,H. (1999) Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J., 18, 22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda R., Tanaka,H. and Yasuda,H. (1997) Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett., 420, 25–27. [DOI] [PubMed] [Google Scholar]
- Ito A., Lai,C.H., Zhao,X., Saito,S., Hamilton,M.H., Appella,E. and Yao,T.P. (2001) p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J., 20, 1331–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobet E., Zeng,X., Zhu,Y., Keller,D. and Lu,H. (2000) MDM2 inhibits p300-mediated p53 acetylation and activation by forming a ternary complex with the two proteins. Proc. Natl Acad. Sci. USA, 97, 12547–12552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Scolnick,D.M., Trievel,R.C., Zhang,H.B., Marmorstein,R., Halazonetis,T.D. and Berger,S.L. (1999) p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell. Biol., 19, 1202–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J., Su,F., Chen,D., Shiloh,A. and Gu,W. (2000) Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature, 408, 377–381. [DOI] [PubMed] [Google Scholar]
- Luo J., Nikolaev,A.Y., Imai,S., Chen,D., Su,F., Shiloh,A., Guarente,L. and Gu,W. (2001) Negative control of p53 by Sir2α promotes cell survival under stress. Cell, 107, 137–148. [DOI] [PubMed] [Google Scholar]
- Montes de Oca Luna R., Wagner,D.S. and Lozano,G. (1995) Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature, 378, 203–206. [DOI] [PubMed] [Google Scholar]
- Nakamura S., Roth,J.A. and Mukhopadhyay,T. (2000) Multiple lysine mutations in the C-terminal domain of p53 interfere with MDM2-dependent protein degradation and ubiquitination. Mol. Cell. Biol., 20, 9391–9398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson M. et al. (2000) PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature, 406, 207–210. [DOI] [PubMed] [Google Scholar]
- Pomerantz J. et al. (1998) The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell, 92, 713–723. [DOI] [PubMed] [Google Scholar]
- Rodriguez M.S., Desterro,J.M., Lain,S., Lane,D.P. and Hay,R.T. (2000) Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Mol. Cell. Biol., 20, 8458–8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi K., Herrera,J.E., Saito,S., Miki,T., Bustin,M., Vassilev,A., Anderson,C.W. and Appella,E. (1998) DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev., 12, 2831–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy S.K., McDonald,P.H., Kohout,T.A. and Lefkowitz,R.J. (2001) Regulation of receptor fate by ubiquitination of activated β2-adrenergic receptor and β-arrestin. Science, 294, 1307–1313. [DOI] [PubMed] [Google Scholar]
- Shieh S.Y., Ikeda,M., Taya,Y. and Prives,C. (1997) DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell, 91, 325–334. [DOI] [PubMed] [Google Scholar]
- Unger T., Juven-Gershon,T., Moallem,E., Berger,M., Vogt Sionov,R., Lozano,G., Oren,M. and Haupt,Y. (1999) Critical role for Ser20 of human p53 in the negative regulation of p53 by Mdm2. EMBO J., 18, 1805–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaziri H., Dessain,S.K., Eaton,E.N., Imai,S.I., Frye,R.A., Pandita,T.K., Guarente,L. and Weinberg,R.A. (2001) hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell, 107, 149–159. [DOI] [PubMed] [Google Scholar]
- Wu X., Bayle,J.H., Olson,D. and Levine,A.J. (1993) The p53-mdm-2 autoregulatory feedback loop. Genes Dev., 7, 1126–1132. [DOI] [PubMed] [Google Scholar]
- Yao T.P., Segraves,W.A., Oro,A.E., McKeown,M. and Evans,R.M. (1992) Drosophila ultraspiracle modulates ecdysone receptor function via heterodimer formation. Cell, 71, 63–72. [DOI] [PubMed] [Google Scholar]
- Zhang Y. and Xiong,Y. (2001) Control of p53 ubiquitination and nuclear export by MDM2 and ARF. Cell Growth Differ., 12, 175–186. [PubMed] [Google Scholar]
- Zhang Y., LeRoy,G., Seelig,H.P., Lane,W.S. and Reinberg,D. (1998a) The dermatomyositis-specific autoantigen Mi2 is a component of a complex containing histone deacetylase and nucleosome remodeling activities. Cell, 95, 279–289. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Xiong,Y. and Yarbrough,W.G. (1998b) ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell, 92, 725–734. [DOI] [PubMed] [Google Scholar]