

Abstract
DNA interstrand crosslinks (ICLs) present formidable blocks to DNA metabolic processes and must be repaired for cell survival. ICLs are induced in DNA by intercalating compounds such as the widely used therapeutic agent psoralen. In bacteria, both nucleotide excision repair (NER) and homologous recombination are required for the repair of ICLs. The processing of ICLs in mammalian cells is not clearly understood. However, it is known that processing can occur by NER, which for psoralen ICLs can be an error-generating process conducive to mutagenesis. We show here that another repair pathway, mismatch repair (MMR), is also involved in eliminating psoralen ICLs in human cells. MMR deficiency renders cells hypersensitive to psoralen ICLs without diminishing their mutagenic potential, suggesting that MMR does not contribute to error-generating repair, and that MMR may represent a relatively error-free mechanism for processing these lesions in human cells. Thus, enhancement of MMR relative to NER may reduce the mutagenesis caused by DNA ICLs in humans.
Keywords: DNA interstrand crosslinks, DNA repair, mismatch repair, psoralen
Introduction
A single DNA crosslink, if not repairable, can be lethal because it prevents essential DNA metabolic functions (Huang et al, 1996). It has been estimated that ∼40 interstrand crosslinks (ICLs) can kill a repair-deficient mammalian cell, whereas ∼2,500 ICLs can be lethal to a repair-proficient mammalian cell (Akkari et al, 2000). Unfaithful repair of ICLs can lead to mutations, resulting in cell death or tumorigenesis. Conversely, deliberate induction of ICLs is a proven strategy for the treatment of hyperproliferative skin disorders, including psoriasis and skin cancer (Momtaz & Fitzpatrick, 1998). Psoralen, a photoactivatable DNA-crosslinking agent, is commonly used for this purpose. Psoralen intercalates into the DNA and, on exposure to ultraviolet (UVA) radiation, can form covalent ICLs with thymines preferentially at 5′-TpA sites in the genome, inducing apoptosis. Psoralen plus UVA (PUVA) therapy has shown considerable clinical efficacy. Unfortunately, a side effect of PUVA treatment is a higher risk of skin cancer (Momtaz & Fitzpatrick, 1998). Thus, a better understanding of psoralen ICL repair should contribute to improved therapies for these patients. Although the repair of psoralen ICLs is well characterized in bacteria and yeast (Cole, 1973; Magana-Schwencke et al, 1982; Van Houten et al, 1986; Dronkert & Kanaar, 2001), their repair is poorly understood in mammalian cells. In bacteria, the repair of psoralen ICLs is carried out by both nucleotide excision repair (NER) and homologous recombination (HR). Our recent report has shown a new pathway for the processing of ICLs in mammalian cells that is dependent on the mismatch repair (MMR) protein complex MutSβ (Zhang et al, 2002). Although the differential susceptibility of MMR-deficient cells to crosslinking agents has been controversial and may reflect clonal differences in tumour cell lines (Bignami et al, 2003; Papouli et al, 2004), our results with the isogenic MMR-proficient and MMR-deficient human cells used in this study are consistent with many previous demonstrations that MMR-deficient mammalian cell lines are sensitive to crosslinking agents (Aquilina et al, 1998; Fiumicino et al, 2000; Lin et al, 2001; Lan et al, 2004).
Whereas TA sequences are the preferred crosslinking sites of psoralen, flanking sequences have little effect on psoralen reactivity, and thus we used triplex-forming oligonucleotides (TFOs) to direct psoralen ICLs to specific sites to study their processing. TFOs are single-stranded DNA molecules that bind sequence specifically in the major groove of duplex DNA by Hoogsteen hydrogen bonding (Vasquez & Glazer, 2002). Using triplex technology, we and others have targeted chromosomal genes in mammalian cells and in animals, for gene modification (Majumdar et al, 1998; Vasquez et al, 1999, 2000). Psoralen-conjugated TFOs can bind their target duplex sequence and induce sitespecific photoadducts on exposure to UVA radiation at 365 nm (Vasquez et al, 1996; Perkins et al, 1999).
In this study, we used a triplex-directed site-specific psoralen ICL as a model substrate to explore the molecular mechanisms of psoralen ICL repair in human cells. It is known that NER is involved in a mutagenic repair of psoralen ICLs in mammalian cells (Wang et al, 1996; Faruqi et al, 2000; Datta et al, 2001; Vasquez et al, 2002; Christensen et al, 2004). Here, we show that the human MMR protein MSH2 is also crucial for efficient processing and error-free repair of psoralen ICLs. Cells deficient in the MSH2 protein show increased sensitivity to psoralen ICLs and a reduced level of psoralen ICL processing. Using a psoralen ICL-induced mutagenesis assay, we show that MSH2 does not contribute to the mutagenic repair of triplex-directed ICLs, suggesting that MSH2 participates in a relatively error-free repair pathway for this type of damage in human cells.
Results and Discussion
Human repair-deficient cells are sensitive to ICLs
To determine whether functional MMR is required for psoralen ICL repair, we measured the sensitivity of MMR-proficient and MMR-deficient human cell lines to psoralen ICLs. As shown in Fig 1A, the MMR-deficient human colon cancer cell line HEC59 (MSH2-deficient) showed an ∼2.5-fold greater sensitivity to the UVA-activated psoralen derivative 4′-hydroxymethyl-4,5′,8′-trimethylpsoralen (HMT) than its isogenic MMR-proficient control cell line HEC59+Chr2. The cells were exposed to a range of HMT concentrations from 10−9 to 10−5 M and irradiated at a dose of 1.8 J/cm2 UVA to generate psoralen ICLs in the DNA. Cell sensitivity to the psoralen ICLs was assessed by cell survival using a 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay. A similar experiment was performed with matched human cell lines deficient (XPA−/−) or proficient (XPA+/+) in NER. The NER-deficient line showed an approximately fourfold greater sensitivity to the HMT+UVA treatment (Fig 1B). The sensitivity of cells deficient in MMR or NER to psoralen ICLs is a measure of the involvement of this gene product in ICL repair. Thus, these results show that both functional MMR and NER are involved in the processing and removal of psoralen ICLs in intact human cells.
Figure 1.
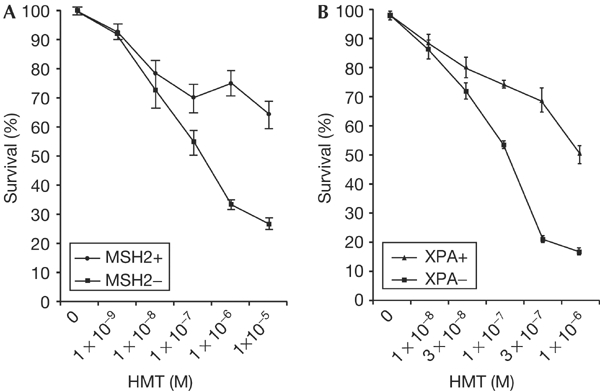
Sensitivity of MMR-deficient and NER-deficient cells to psoralen ICLs. Survival curves are shown for cells treated with HMT and UVA at 365 nm (to allow ICL formation). (A) MSH2+ (HEC59+Chr2), MSH2− (HEC59). (B) XPA+ (GM05566), XPA− (GM05509C). Cell survival was determined by an MTT assay performed in triplicate. The bars represent the standard error of the means.
MSH2 is required for efficient processing of psoralen ICLs
To study the role of MMR proteins in the repair process, we used a psoralen-modified TFO to target a site-specific psoralen ICL to a 5′-TpA site located in the supF mutation reporter gene of the pSupFG1 shuttle vector (Vasquez et al, 2001). The efficiency of sitespecific psoralen ICL formation in the supF gene on the plasmid was ∼78%. When the plasmid was incubated with a scrambled control TFO, pSCR30 (that does not bind the supF triplex target site), nonspecific psoralen ICLs were formed in only ∼5% of the total plasmid population (supplementary Fig 1 online).
To determine the role of the human MMR proteins in the processing of psoralen ICLs, DNA repair assays were performed on the psoralen-crosslinked pSupFG1 plasmid in MMR-proficient and MMR-deficient human cell-free extracts. The ICL-damaged plasmid was subjected to cell-free extracts deficient in MSH2 (HEC59) and in the isogenic control repair-proficient HEC59+Chr2 extract together with [α-32P]dCTP, unlabelled dNTPs and an ATP-regenerating system. Incorporation of radioactivity from [α-32P]dCTP into the damaged plasmid indicates the occurrence of DNA synthesis in association with repair. As shown in Fig 2, radiolabelled nucleotides were incorporated into the damaged plasmid in the repair-proficient HEC59+Chr2 cell extract. However, incorporation was significantly reduced (∼50%) in the MSH2-deficient (HEC59) cell extract, indicating that 50% of the DNA repair synthesis of psoralen ICLs requires the MMR protein MSH2. A UVC-damaged plasmid served as a control for extract activity (as UVC damage is known to be repaired by NER) and, as expected, this plasmid was repaired similarly in the MMR-deficient cell extracts, suggesting that extracts from both cell lines were proficient in NER. This result shows that TFO-directed psoralen ICLs require the MMR protein MSH2 for their efficient processing and repair in human cell extracts. This finding shows that human cells handle DNA ICLs in a way that is strikingly different from that used by bacteria and yeast, which use NER and HR to remove these lesions.
Figure 2.
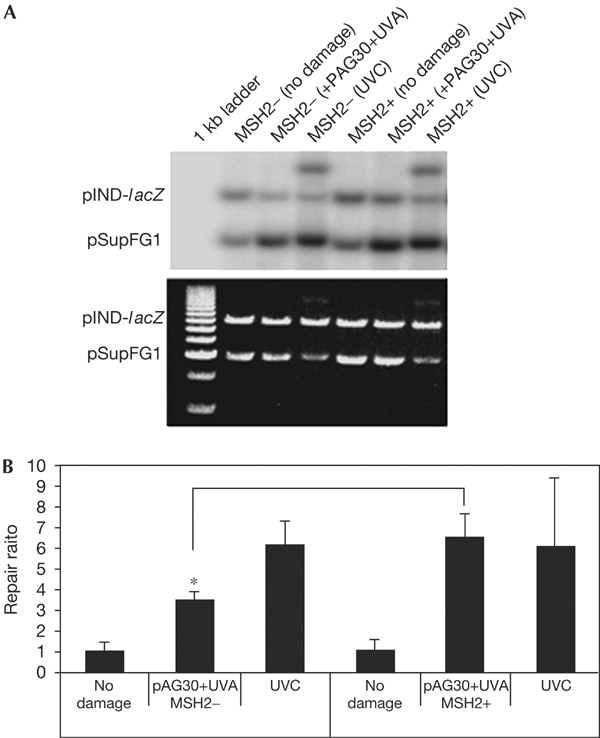
Repair efficiency of TFO-targeted psoralen ICLs in MMR-deficient and MMR-proficient human cell extracts. (A) Autoradiogram (upper panel) and ethidium-bromidestained gel (lower panel) showing DNA repair efficiencies measured by incorporation of radiolabelled nucleotides into the damaged plasmid. pSupFG1 is the damaged plasmid and pIND-lacZ is an undamaged control plasmid. (B) Quantification of repair efficiency.
MMR is not required for ICL-induced mutagenesis
It is known that bacteria and yeast use both NER and HR in the repair of psoralen ICLs and that the NER-dependent pathway can lead to mutations in the DNA, with the predominant psoralen mutation being T:A to A:T transversions. Our group and others have shown that NER is involved in the error-generating repair of TFO-directed lesions in mammalian cells (Wang et al, 1996; Datta et al, 2001; Vasquez et al, 2002). The data in Table 1 show the requirement for NER in the mutagenic processing of TFO-directed psoralen ICLs. For example, in normal fibroblasts, triplex-directed damage induces mutations ten- to 100-fold above background levels, but this induction is reduced to less than twofold in NER-deficient cells (Wang et al, 1996). Because we know that NER is involved in the error-generating repair of psoralen ICLs and we have now found MMR to be involved in their repair, we speculated that perhaps MMR is involved in error-free processing of ICLs. To determine whether the MMR proteins are involved in the mutagenic repair of ICLs, we subjected psoralen-damaged plasmids to a mutagenesis assay in MMR-proficient and MMR-deficient human cells. The psoralen-crosslinked plasmid was transfected into MMR-deficient cell lines HEC59 (MSH2 deficient) and LoVo (MSH2 deficient) and the control MMR-proficient cell line HEC59+Chr2. Plasmids were collected from the human cells after 48 h to allow time for replication and repair, and were transfected into Escherichia coli to screen for supF gene mutations. If MMR was involved in an error-generating repair pathway for psoralen ICLs, we would expect the induced mutation frequency to be significantly reduced in the MMR-deficient cell lines compared with that in the MMR-proficient cell lines, as seen in NER-deficient cells (Table 1). The mutation frequency observed in pSupFG1 plasmids rescued from the MMR-proficient cell line (HEC59+Chr2) is 4.6%. When compared with undamaged plasmid, TFO-directed psoralen ICLs induced the mutation frequency ∼110-fold above the background mutation frequency of 0.04%. Surprisingly, in the MMR-deficient cell lines, TFO-directed psoralen ICLs still induce mutation frequencies ∼100-fold above the background levels (Fig 3). These data clearly show that functional MMR is not required for the error-generating repair of psoralen ICLs in human cells, as the MMR deficiency does not reduce the mutagenic potential of the ICLs. Our results show that human cell lines deficient in MSH2 are sensitive to psoralen-induced DNA ICLs, but do not have lower frequencies of crosslink-induced mutations. Restoration of the missing MMR protein restores survival rates without increasing mutation frequencies. Together with our previously published results indicating that the NER proteins are necessary for error-generating repair of these lesions, these results indicate that the MMR machinery can carry out essentially error-free repair of these lesions in human cells. However, the relative contributions of MMR and NER in the removal of psoralen ICLs are not clear. The similar sensitivities of the human MMR- and NER-deficient cell lines to psoralen ICLs (Fig 1) suggest that both pathways may contribute to the repair of these lesions at similar levels.
Table 1.
Summary of triplex-forming oligonucleotide-induced mutation frequencies in repair-proficient and repair-deficient mammalian cells
Figure 3.
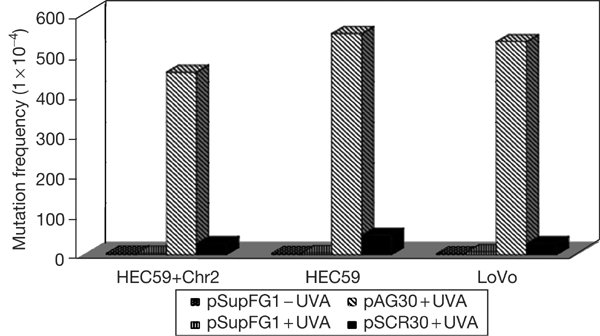
Psoralen-induced mutagenesis in MMR-deficient and MMR-proficient human cell lines. MMR-proficient (HEC59+Chr2) and MMR-deficient cell lines (HEC59, LoVo) were transfected with the pSupFG1 mutation reporter plasmid and mutations in the supF reporter gene were measured 48 h after transfection. pSupFG1−UVA represents undamaged plasmid in the absence of irradiation; pSupFG1+UVA represents undamaged plasmid in the presence of UVA irradiation at 1.8 J/cm2; pAG30+UVA represents pSupFG1 plasmid treated with the psoralen-modified TFO (pAG30) and then UVA irradiation to produce psoralen ICLs in the supF mutation reporter gene; and pSCR30 represents plasmid that was incubated with the psoralen-modified control (pSCR30) oligonucleotide and UVA irradiated. The mutation frequency of the supF gene was determined as the number of mutant colonies (white colonies) to the total colonies (blue+white colonies).
Psoralen ICL-induced mutations in human cell lines
DNA sequence analysis of the mutations in the supF gene shows that, in the MMR-proficient HEC59+Chr2 cell line, 95% (17 out of 18) of the mutations were in the targeted region and 33% (6 out of 18) were T:A → A:T transversions. This result was expected because T:A → A:T transversions are the typical mutation induced by psoralen ICLs. Other mutations include 11% (2 out of 18) T:A → G:C transversions, 40% (7 out of 18) single- or double-base mutations at adjacent sites and 11% (2 out of 18) deletions encompassing the targeted sites as shown in Fig 4A. The DNA-damage-induced mutation spectrum in the MMR-deficient HEC59 (Fig 4B) is similar to that found in the MMR-proficient HEC59+Chr2 cell line. This result indicates that MMR does not contribute to the mutations produced in the targeted supF gene. Collectively, our findings implicate MMR in error-free processing of psoralen ICLs in human cells and, more generally, provide evidence that proteins from several repair pathways (e.g. NER, HR and MMR) work together to remove complex lesions from the mammalian genome. Our working model for ICL processing and removal in human cells is depicted in Fig 5. Here, we suggest that error-generating repair is the result of an NER-dependent incision on one strand followed by translesion synthesis. In the error-free repair pathway, we suggest that the substrate is processed by doublestrand breaks followed by high-fidelity repair by HR. The formation of double-strand breaks after ICL damage has been shown in yeast and human cells (Dardalhon et al, 1998; De Silva et al, 2000) and may be the result of a replication fork collapse at the site of damage. The low error rate associated with MMR implies that treatments to enhance MMR relative to NER may prove beneficial for reducing mutagenesis in response to psoralen ICLs in experimental or therapeutic contexts.
Figure 4.
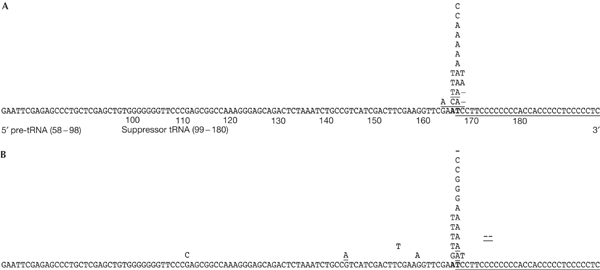
Characterization of psoralen-induced mutations in human cells. Mutation spectrum of the psoralen ICL-induced mutations in the supF gene in the (A) MSH2-proficient HEC59+Chr2 cell line and (B) MSH2-deficient HEC59 cell line. Base substitutions are listed above the supFG1 sequence. Single-base deletions are indicated by a ‘–'. Multiple mutations in the same plasmid are underlined. The TFO-binding site is underlined. The targeted TA-crosslinking site is indicated by boldface type.
Figure 5.
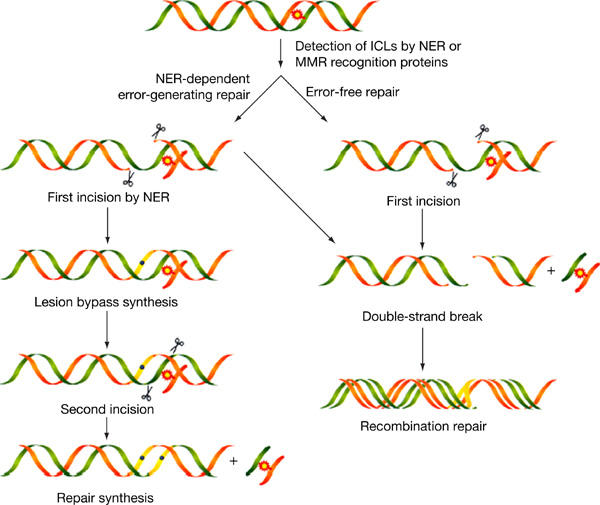
Model for psoralen ICL repair in human cells involving nucleotide excision repair, mismatch repair and homologous recombination.
Concluding remarks
Many anticancer agents, including mitomycin C, cisplatin, nitrogen mustards and psoralen, induce ICLs in DNA. Unfortunately, after chemotherapy, residual unrepaired ICLs in normal cells can be mutagenic and may lead to development of secondary malignancies. The mechanisms by which ICLs are repaired in human cells are poorly understood despite the importance of these lesions to human health. In this study, we made the surprising discovery that MMR proteins are involved in a relatively error-free pathway of psoralen ICLs in human cells, thus identifying a new paradigm for their error-free repair. This finding should help to develop a better understanding of cancer aetiology in relation to crosslinked DNA and may lead to improved therapies for the treatment and/or prevention of human cancer.
Methods
Cell lines. The HEC59 (MSH2-deficient) and HEC59+Chr2 (MSH2-complemented) human colon cancer cell lines were cultured in Dulbecco's modified Eagle's medium/F12 medium plus 10% fetal bovine serum (FBS). The medium for the HEC59+Chr2 cell line also contained 100 μg/ml G418. The LoVo cell line was cultured in F12K medium plus 10% FBS. The GM05509C (XPA−/−) and GM05566 (XPA+/+) human fibroblast cell lines were cultured in MEM Eagle–Earle's balanced salt solution medium supplemented with 2 × essential amino acids and non-essential amino acids, vitamins, 2 mM L-glutamine and 15% FBS.
In vitro cytotoxicity assay. The sensitivity of the human cell lines to PUVA treatment was evaluated using an MTT assay (CellTiter 96 cell proliferation assay kit, Promega, Madison, WI, USA). Briefly, 2 × 104 cells were seeded in 96-well microplates and incubated at 37°C. After 18 h, the medium was replaced with serum-free medium containing various concentrations of HMT (Sigma, St Louis, MO, USA). After incubation for 1 h, the cells were irradiated (UVA, 1.8 J/cm2). Triplicate cultures were established for each treatment. Cytotoxicity was evaluated 72 h after irradiation, using an MTT assay.
Oligonucleotides and mutation reporter plasmid, pSupFG1. 5′-Psoralen-modified, 3′-amine-modified oligonucleotides were synthesized by the Midland certified reagent company (Midland, TX, USA). The pSupFG1 plasmid used in this study contains an supF mutation reporter gene, an ampicillin resistance gene, a pBR327 replication origin and a simian virus 40 viral replication origin to permit facile blue/white screening of mutants generated in mammalian cells.
In vitro repair assay. Human cell extracts were prepared from frozen cell pellets using the NucBusterTM protein extraction kit (Novagen, Madison, WI, USA). The final salt concentration was 200 mM. Supercoiled plasmids, pSupFG1 (∼5 kb) and a larger control plasmid, pIND-lacZ (∼8.2 kb, not containing the TFO-binding site), were incubated with psoralen-modified TFOs and exposed to UVA to induce psoralen ICLs. The DNA (300 ng) was then added to whole-cell extracts (80 μg protein) in a buffer containing near-physiological salt concentration, as described previously (Wood et al, 1988; Holmes et al, 1990; Iaccarino et al, 1998). Visualization of plasmid DNA and the incorporated [α-32P]dCTP was achieved by ethidium bromide staining and autoradiography; quantification was performed using a PhosphorImager. The ‘repair ratio' was calculated by dividing the amount of radioactivity in the damaged pSupGF1 plasmid (normalized to the amount of damaged pSupFG1 DNA loaded) by the amount of radioactivity incorporated into the control plasmid pIND-lacZ (normalized to the amount of pIND-lacZ loaded).
Mutagenesis assay. Psoralen-crosslinked pSupFG1 plasmid was transfected into human cells using Gene-PORTER (Gene Therapy System Inc., San Diego, CA, USA). About 5 μg of plasmid DNA was used per 5 × 105 human colorectal cells. The cells were incubated 48 h before their transformation into E. coli MBMB7070 indicator strain, as described previously (Vasquez et al, 2001). The mutation frequency was determined as the number of mutant colonies to total colonies.
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Methods
Acknowledgments
We acknowledge Dr R.A. Finch and Dr T.G. Wensel for advice, Dr K.E. Gray and Dr B.D. Hammann for technical assistance and Dr S.P. Henninger for help in the preparation of the manuscript. This work was supported by National Institutes of Health/NCI grants to K.M.V. (CA93729 and CA97175) and an NIEHS Center grant (ES07784).
References
- Akkari YM, Bateman RL, Reifsteck CA, Olson SB, Grompe M (2000) DNA replication is required to elicit cellular responses to psoralen-induced DNA interstrand cross-links. Mol Cell Biol 20: 8283–8289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aquilina G, Ceccotti S, Martinelli S, Hampson R, Bignami M (1998) N-(2-chloroethyl)-N′-cyclohexyl-N-nitrosourea sensitivity in mismatch repair-defective human cells. Cancer Res 58: 135–141 [PubMed] [Google Scholar]
- Bignami M, Casorelli I, Karran P (2003) Mismatch repair and response to DNA-damaging antitumour therapies. Eur J Cancer 39: 2142–2149 [DOI] [PubMed] [Google Scholar]
- Chen et al. (2003) Carcinogenesis 24: 1111–1121 [DOI] [PubMed] [Google Scholar]
- Christensen LA, Conti CJ, Fischer SM, Vasquez KM (2004) Mutation frequencies in murine keratinocytes as a function of carcinogenic status. Mol Carcinog 40: 122–133 [DOI] [PubMed] [Google Scholar]
- Cole RS (1973) Repair of DNA containing interstrand crosslinks in Escherichia coli: sequential excision and recombination. Proc Natl Acad Sci USA 70: 1064–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dardalhon M, de Massy B, Nicolas A, Averbeck D (1998) Mitotic recombination and localized DNA doublestrand breaks are induced after 8-methoxypsoralen and UVA irradiation in Saccharomyces cerevisiae. Curr Genet 34: 30–42 [DOI] [PubMed] [Google Scholar]
- Datta HJ, Chan PP, Vasquez KM, Gupta RC, Glazer PM (2001) Triplex-induced recombination in human cell-free extracts. Dependence on XPA and HsRad51. J Biol Chem 276: 18018–18023 [DOI] [PubMed] [Google Scholar]
- De Silva IU, McHugh PJ, Clingen PH, Hartley JA (2000) Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol Cell Biol 20: 7980–7990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dronkert ML, Kanaar R (2001) Repair of DNA interstrand cross-links. Mutat Res 486: 217–247 [DOI] [PubMed] [Google Scholar]
- Faruqi AF, Datta HJ, Carroll D, Seidman MM, Glazer PM (2000) Triple-helix formation induces recombination in mammalian cells via a nucleotide excision repair-dependent pathway. Mol Cell Biol 20: 990–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiumicino S, Martinelli S, Colussi C, Aquilina G, Leonetti C, Crescenzi M, Bignami M (2000) Sensitivity to DNA cross-linking chemotherapeutic agents in mismatch repair-defective cells in vitro and in xenografts. Int J Cancer 85: 590–596 [DOI] [PubMed] [Google Scholar]
- Holmes J Jr, Clark S, Modrich P (1990) Strandspecific mismatch correction in nuclear extracts of human and Drosophila melanogaster cell lines. Proc Natl Acad Sci USA 87: 5837–5841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LC, Clarkin KC, Wahl GM (1996) Sensitivity and selectivity of the DNA damage sensor responsible for activating p53-dependent G1 arrest. Proc Natl Acad Sci USA 93: 4827–4832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaccarino I, Marra G, Palombo F, Jiricny J (1998) hMSH2 and hMSH6 play distinct roles in mismatch binding and contribute differently to the ATPase activity of hMutSα. EMBO J 17: 2677–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan L et al. (2004) Functional and physical interactions between ERCC1 and MSH2 complexes for resistance to cis-diamminedichloroplatinum(II) in mammalian cells. DNA Repair (Amst) 3: 135–143 [DOI] [PubMed] [Google Scholar]
- Lin X, Ramamurthi K, Mishima M, Kondo A, Christen RD, Howell SB (2001) P53 modulates the effect of loss of DNA mismatch repair on the sensitivity of human colon cancer cells to the cytotoxic and mutagenic effects of cisplatin. Cancer Res 61: 1508–1516 [PubMed] [Google Scholar]
- Magana-Schwencke N, Henriques JA, Chanet R, Moustacchi E (1982) The fate of 8-methoxypsoralen photoinduced crosslinks in nuclear and mitochondrial yeast DNA: comparison of wild-type and repair-deficient strains. Proc Natl Acad Sci USA 79: 1722–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar A et al. (1998) Targeted gene knockout mediated by triple helix forming oligonucleotides. Nat Genet 20: 212–214 [DOI] [PubMed] [Google Scholar]
- Momtaz K, Fitzpatrick TB (1998) The benefits and risks of long-term PUVA photochemotherapy. Dermatol Clin 16: 227–234 [DOI] [PubMed] [Google Scholar]
- Papouli E, Cejka P, Jiricny J (2004) Dependence of the cytotoxicity of DNA-damaging agents on the mismatch repair status of human cells. Cancer Res 64: 3391–3394 [DOI] [PubMed] [Google Scholar]
- Perkins BD, Wensel TG, Vasquez KM, Wilson JH (1999) Psoralen photo-cross-linking by triplex-forming oligonucleotides at multiple sites in the human rhodopsin gene. Biochemistry 38: 12850–12859 [DOI] [PubMed] [Google Scholar]
- Van Houten B, Gamper H, Holbrook SR, Hearst JE, Sancar A (1986) Action mechanism of ABC excision nuclease on a DNA substrate containing a psoralen crosslink at a defined position. Proc Natl Acad Sci USA 83: 8077–8081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez KM, Glazer PM (2002) Triplex-forming oligonucleotides: principles and applications. Q Rev Biophys 35: 89–107 [DOI] [PubMed] [Google Scholar]
- Vasquez KM, Wensel TG, Hogan ME, Wilson JH (1996) High-efficiency triple-helix-mediated photo-cross-linking at a targeted site within a selectable mammalian gene. Biochemistry 35: 10712–10719 [DOI] [PubMed] [Google Scholar]
- Vasquez KM, Wang G, Havre PA, Glazer PM (1999) Chromosomal mutations induced by triplex-forming oligonucleotides in mammalian cells. Nucleic Acids Res 27: 1176–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez KM, Narayanan L, Glazer PM (2000) Specific mutations induced by triplex-forming oligonucleotides in mice. Science 290: 530–533 [DOI] [PubMed] [Google Scholar]
- Vasquez KM, Dagle JM, Weeks DL, Glazer PM (2001) Chromosome targeting at short polypurine sites by cationic triplex-forming oligonucleotides. J Biol Chem 276: 38536–38541 [DOI] [PubMed] [Google Scholar]
- Vasquez KM, Christensen J, Li L, Finch RA, Glazer PM (2002) Human XPA and RPA DNA repair proteins participate in specific recognition of triplex-induced helical distortions. Proc Natl Acad Sci USA 99: 5848–5853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Seidman MM, Glazer PM (1996) Mutagenesis in mammalian cells induced by triple helix formation and transcription-coupled repair. Science 271: 802–805 [DOI] [PubMed] [Google Scholar]
- Wood RD, Robins P, Lindahl T (1988) Complementation of the xeroderma pigmentosum DNA repair defect in cell-free extracts. Cell 53: 97–106 [DOI] [PubMed] [Google Scholar]
- Zhang N, Lu X, Zhang X, Peterson CA, Legerski RJ (2002) hMutSβ is required for the recognition and uncoupling of psoralen interstrand cross-links in vitro. Mol Cell Biol 22: 2388–2397 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Methods