

Abstract
XIAP is a potent suppressor of apoptosis that directly inhibits specific members of the caspase family of cysteine proteases. Here we demonstrate a novel role for XIAP in the control of intracellular copper levels. XIAP was found to interact with MURR1, a factor recently implicated in copper homeostasis. XIAP binds to MURR1 in a manner that is distinct from that utilized by XIAP to bind caspases, and consistent with this, MURR1 did not affect the antiapoptotic properties of XIAP. However, cells and tissues derived from Xiap-deficient mice were found to contain reduced copper levels, while suppression of MURR1 resulted in increased intracellular copper in cultured cells. Consistent with these opposing effects, XIAP was observed to negatively regulate MURR1 protein levels by the formation of K48 polyubiquitin chains on MURR1 that promote its degradation. These findings represent the first described phenotypic alteration in Xiap-deficient mice and demonstrate that XIAP can function through MURR1 to regulate copper homeostasis.
Keywords: copper, MURR1, ubiquitin, XIAP
Introduction
The inhibitor of apoptosis (iap) genes were originally identified in baculoviruses (Crook et al, 1993), and homologs have subsequently been identified in a wide range of genomes. The antiapoptotic activity of IAPs was the first property of these proteins to be noted, and this has been largely attributed to their ability to directly inhibit members of the caspase family (Devereaux et al, 1997). However, IAPs have been implicated in a number of diverse cellular functions seemingly unrelated to caspase inhibition. In particular, several IAPs are known to participate in cell division and cytokinesis (Uren et al, 2000), while other family members have been shown to function in distinct signal transduction pathways (Rothe et al, 1995; Yamaguchi et al, 1999; Birkey Reffey et al, 2001). IAPs contain between one and three imperfect repeats of an approximately 65-residue motif termed the baculoviral IAP repeat (BIR). Many IAPs also contain a carboxy-terminal RING finger domain that possesses E3 ubiquitin ligase activity and has been implicated in proteasome-mediated degradation of various protein targets (Yang et al, 2000; Li et al, 2002).
One mammalian member of this family is X-linked IAP (XIAP), a 56-kDa protein comprised of three amino-terminal BIR domains and a RING domain at the carboxyl terminus (Holcik and Korneluk, 2001). XIAP has potent antiapoptotic properties, and has been shown to directly suppress caspases through elements within and immediately adjacent to its second and third BIRs (Chai et al, 2001; Huang et al, 2001; Riedl et al, 2001). The antiapoptotic activity of XIAP can itself be suppressed by two mitochondrial proteins, Smac/DIABLO and Omi/HtrA2 (Du et al, 2000; Verhagen et al, 2000; Hegde et al, 2001; Martins et al, 2001; Suzuki et al, 2001a), which are released from mitochondria into the cytoplasm during apoptosis. These molecules bind to the same domains in XIAP that mediate its interactions with caspases and act as negative regulators of the protective properties of XIAP by releasing XIAP from caspases (Liu et al, 2000; Wu et al, 2000; Srinivasula et al, 2001), or in the case of Omi/HtrA2, also by promoting the proteolytic cleavage of XIAP (Srinivasula et al, 2003; Yang et al, 2003). In addition to the involvement of XIAP in caspase inhibition, other roles for this molecule have been demonstrated. XIAP has been implicated in several intracellular signaling cascades, including the c-Jun N-terminal kinase (JNK) pathway (Sanna et al, 1998, 2002), the nuclear factor-κB (NF-κB) pathway (Hofer-Warbinek et al, 2000; Levkau et al, 2001), and the transforming growth factor-β (TGF-β) pathway (Yamaguchi et al, 1999; Birkey Reffey et al, 2001). Many of these signaling cascades are also involved in the cellular response to stress, including oxidative stress (Mercurio and Manning, 1999; Martindale and Holbrook, 2002). In addition, upregulation of XIAP expression during cellular stress has been observed, thought to result from Cap-independent translation mediated by an internal ribosomal entry site present in its mRNA (Holcik et al, 1999). Therefore, besides the ability of XIAP to inhibit apoptotic cell death, a more general role for this molecule can be proposed as a factor that participates in the response to cellular stress.
Copper is essential for the enzymatic activity of a number of housekeeping genes, such as superoxide dismutase, and it plays a role in oxidative stress damage through free radical formation (Puig and Thiele, 2002). Important insights into the regulation of copper homeostasis in mammalian cells were gleaned after the isolation of copper transporters, some of which are responsible for human diseases (Mercer, 2001). Recently, a novel gene, MURR1, has been implicated in the regulation of copper homeostasis in mammals. This gene was found by positional cloning to be responsible for an autosomal recessive form of copper toxicosis that affects a specific canine strain (Bedlington terriers) (van De Sluis et al, 2002). Dogs affected were found to harbor a mutation in this gene that leads to an mRNA lacking exon 2 (van De Sluis et al, 2002), and MURR1 protein was undetectable in affected animals (Klomp et al, 2003). The mechanism by which MURR1 is involved in copper regulation remains unknown, although the ability of this protein to interact with the copper transporter ATP7B has been recently reported (Tao et al, 2003).
Here we describe a novel function of XIAP in the regulation of cellular copper levels that is mediated by its ability to function as an E3 ubiquitin ligase for MURR1. An interaction between XIAP and MURR1 was identified and reduced copper levels were found in liver tissue and fibroblasts derived from Xiap-deficient mice. XIAP was observed to function as a negative regulator of MURR1 protein levels and to direct the ubiquitination of MURR1 by K48 polyubiquitin chains. Taken together, these data reveal a previously unrecognized role for XIAP in copper homeostasis.
Results
Identification of MURR1 as an XIAP-associated factor
To identify novel XIAP binding proteins, full-length XIAP was used to screen a yeast two-hybrid library prepared from human liver cDNA. This screen led to the identification of MURR1 as an XIAP binding protein. Coprecipitation studies were performed with lysates from 293 cells to determine whether endogenous XIAP and MURR1 proteins interact in mammalian cells. XIAP was readily detected in MURR1 immunoprecipitates (Figure 1A), but not in immunoprecipitates prepared using a control serum, indicating a specific interaction between MURR1 and XIAP. The MURR1 antibody efficiently immunodepleted endogenous MURR1 but not β-actin from the cell lysates used (Figure 1A), confirming the specificity of this antibody (Klomp et al, 2003).
Figure 1.
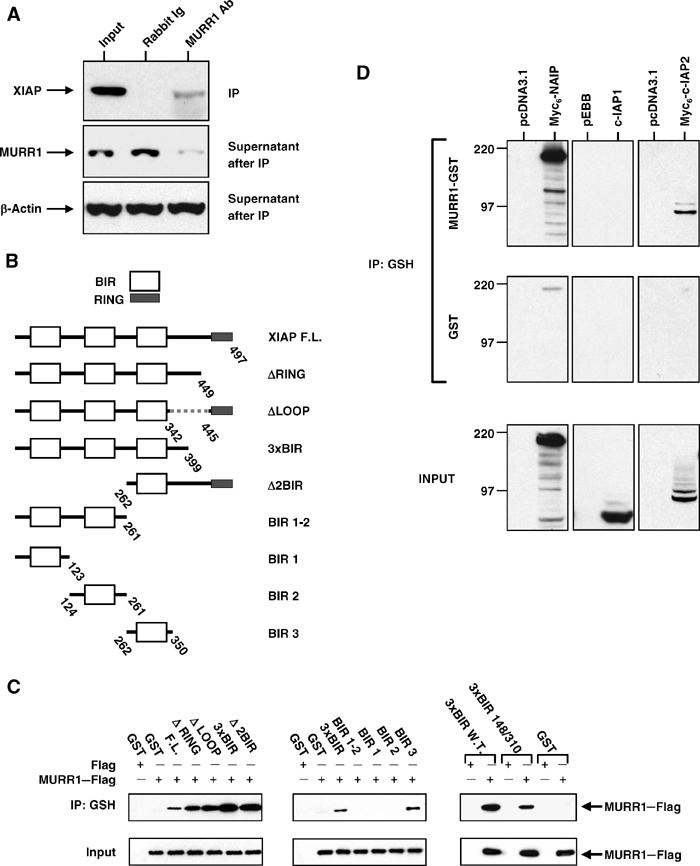
XIAP and MURR1 interactions in mammalian cells. (A) Coprecipitation of endogenous MURR1 and XIAP: 293 cells were lysed in a buffer containing 0.5% Triton X-100. Immunoprecipitation of endogenous proteins was performed using a rabbit polyclonal antibody against MURR1 or normal rabbit Ig. The precipitated material was used for a Western blot for XIAP (top panel) and the supernatants after immunoprecipitation were immunoblotted for MURR1 (middle panel) and β-actin (bottom panel). (B) Schematic of deletion constructs: truncations in XIAP as shown in this schematic were expressed in mammalian cells in fusion with GST and used as shown in (C). (C) Deletion mapping: 293 cells were cotransfected with C-terminal Flag-tagged MURR1 together with vectors expressing GST fusion proteins with XIAP full-length and various truncated forms as indicated. In addition, GST-3 × BIR wild-type, or GST-3 × BIR D148A/W310A were also used in these experiments (right panels). Cell lysates prepared with a buffer containing 0.5% Triton X-100 were used for precipitations with glutathione beads. The precipitated material was immunoblotted with Flag antibodies. (D) Coprecipitation of other 3-BIR containing IAPs: 293 cells were transfected with either MURR1–GST or GST together with Myc6-c-IAP2 or Myc6-NAIP. Cell lysates prepared with a buffer containing 0.5% Triton X-100 were used for precipitations using glutathione sepharose beads. The precipitated material was immunoblotted with Myc antibody or c-IAP1 antibody.
In order to delineate the domains involved in the interaction between MURR1 and XIAP, further coprecipitation experiments were performed. Full-length XIAP or various truncated forms that lack known domains (Figure 1B) were expressed in 293 cells as GST fusion proteins and tested for their ability to bind MURR1. The three BIR domains of XIAP were found to be sufficient for interaction with MURR1, while the spacer region between the BIRs and the RING domain, as well as the RING itself, were not required (Figure 1C, left panels). To more finely map the interacting domain in XIAP responsible for MURR1 binding, precipitation experiments were subsequently performed utilizing individual BIRs expressed in fusion with GST. These experiments demonstrated that BIR 3 was sufficient for binding to MURR1; neither BIR 1 nor 2 bound to MURR1 (Figure 1C, middle panels). To determine whether XIAP utilizes the same mechanism to bind caspases and MURR1, a double point mutant of XIAP that we have previously shown to be incapable of binding caspases (Bratton et al, 2002; Silke et al, 2002) was tested for its ability to bind MURR1. This mutant readily coprecipitated MURR1 (Figure 1C, right panels), indicating that the mechanism utilized by XIAP to bind MURR1 is distinct from that used to bind caspases.
The ability of MURR1 to bind to other 3-BIR-containing IAPs was also examined. MURR1 in fusion with GST (MURR1–GST) or GST alone were transfected into 293 cells and tested for their ability to coprecipitate c-IAP1, c-IAP2, or NAIP. MURR1–GST was able to bind to c-IAP2 and NAIP, but not to c-IAP1 (Figure 1D).
Cellular localization of MURR1
In order to determine the cellular localization of MURR1 and XIAP, fusions of both proteins with either YFP or CFP were generated and expressed in cells and subsequently examined by fluorescence confocal microscopy. A MURR1–YFP fusion protein exhibited a cytoplasmic localization with accumulation of the protein in discrete perinuclear foci; coexpression of MURR1–YFP with CFP–XIAP revealed the colocalization of these two proteins within these foci, in addition to a more diffuse, cytoplasmic pattern (Figure 2A, upper panels). CFP was distributed diffusely throughout the cell and did not colocalize in perinuclear foci with MURR1–YFP (Figure 2A, middle panels). Similarly, CFP–XIAP when coexpressed with YFP demonstrated a diffuse predominantly cytoplasmic pattern and no perinuclear foci were noticeable (Figure 2A, lower panels). These data are consistent with specific colocalization of MURR1–YFP and CFP–XIAP within these foci.
Figure 2.
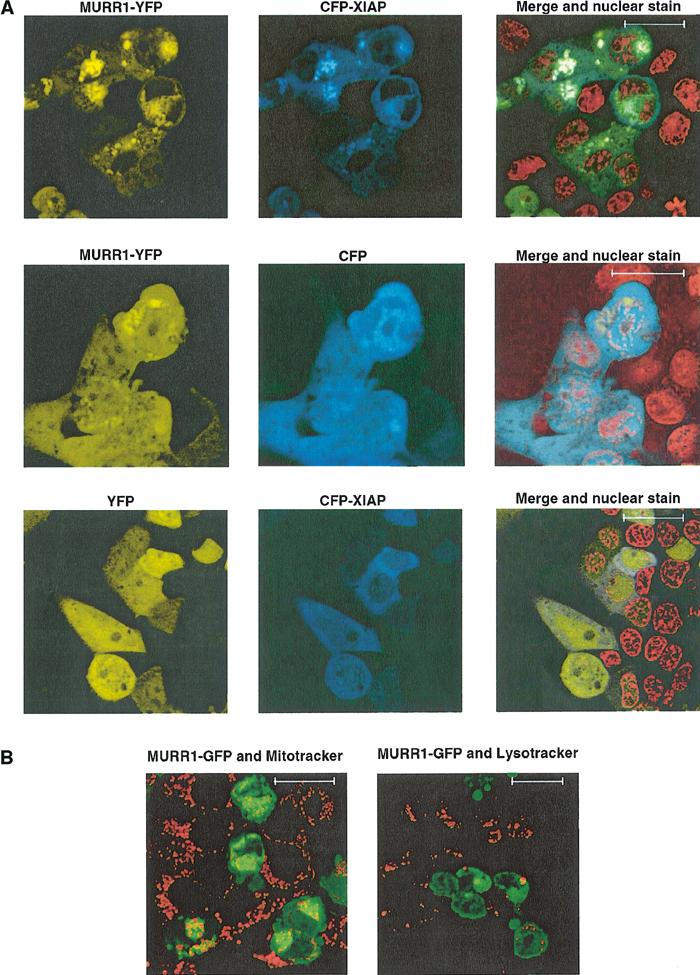
Localization of fluorescently tagged MURR1. (A) Colocalization of fluorescently tagged MURR1 and XIAP: 293 cells plated onto coverglass chamber slides were transfected with MURR1 fused with YFP (MURR1–YFP) and XIAP fused with CFP (CFP–XIAP) (top panels), or MURR1–YFP and CFP (middle panels), or YFP and CFP–XIAP (lower panels). Counterstaining of the nuclei was also performed using Hoechst 33342 (displayed in red for clarity). The scale bar shown corresponds to 18 μm. (B) MURR1 does not colocalize with mitochodrial or lysosomal markers: MURR1–GFP was transfected into 293 cells seeded on coverglass chambered slides. After 24 h the cells were stained with the vital dyes Mitotracker (left panel) or Lysotracker (right panel) (Molecular Probes). Scale bars correspond to 18 μm.
To further characterize the subcellular distribution of MURR1, staining experiments were performed with cell-permeable dyes specific for mitochondria or lysosomes in cells transfected with MURR1–GFP. As shown in Figure 2B, MURR1 was found to localize in a perinuclear compartment that does not represent mitochondria or lysosomes.
MURR1 is not involved in the regulation of caspase-mediated cell death
Since XIAP is a strong suppressor of apoptosis due to its caspase-inhibiting properties, the possibility that MURR1 might function to modulate the protective properties of XIAP was tested. Using experimental systems in which XIAP has been previously shown to inhibit cell death effectively (Richter et al, 2001), no modulatory activity of MURR1 was detected, as evaluated by morphological criteria for cell viability or enzymatic evidence of caspase activation. This was the case for the induction of apoptosis by ectopic expression of Bax (Figures 3A and B) or Fas (Figures 3C and D). These data suggest that MURR1 does not play a significant role in regulating the ability of XIAP to inhibit caspase-dependent cell death following these stimuli.
Figure 3.
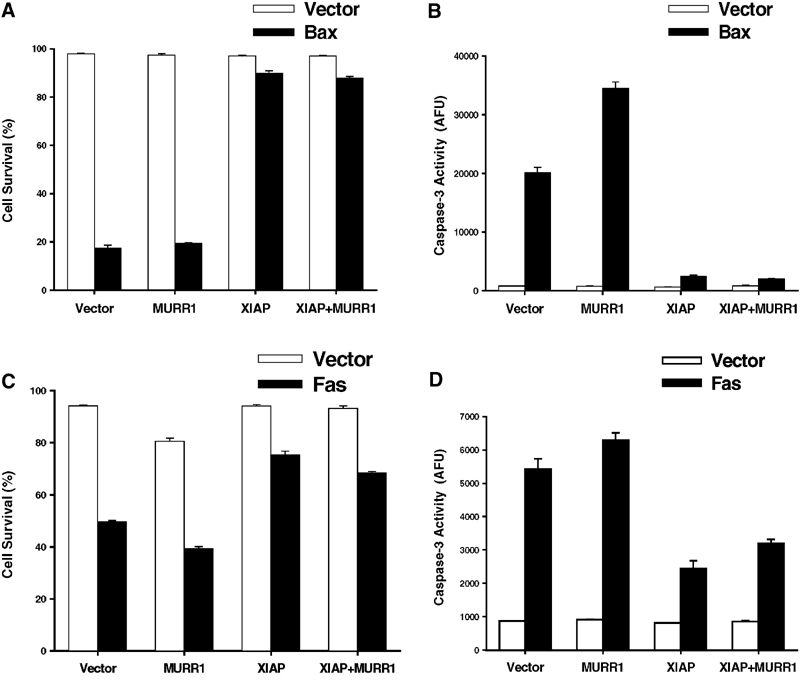
MURR1 does not affect the antiapoptotic activities of XIAP. Expression vectors encoding Bax (A and B) or Fas (C and D) were transfected into 293 cells either with empty vector controls or XIAP, MURR1 or both, together with a plasmid expressing EGFP as a marker of transfection. Cell viability was assessed by morphology of EGFP-positive cells 16 h (A) or 30 h (C) post-transfection, and caspase-3 activity was determined in the same cells (B and D). Data presented represent the average and s.e.m. of three measurements per group.
XIAP affects intracellular copper levels
The identification of the interaction between XIAP and MURR1, a factor whose mutation results in copper toxicosis in dogs (van De Sluis et al, 2002; Klomp et al, 2003), led us to investigate whether XIAP itself could modulate copper homeostasis. First, the effect of decreased MURR1 protein expression in cultured cells was examined. Transfection of short interfering RNA (siRNA) oligonucleotides targeting MURR1 resulted in increased intracellular copper levels in 293 cells (Figure 4A), an effect consistent with the observed phenotype in animals carrying mutations in this gene. The effects of changes in XIAP protein levels on copper homeostasis were subsequently examined. Ectopic expression of XIAP resulted in increased copper levels (Figure 4B). In addition, transformed fibroblasts derived from Xiap-deficient mice contained reduced copper levels when compared to transformed fibroblasts derived from wild-type controls (Figure 4C). Similarly, the copper content in liver tissue from Xiap-deficient mice was lower when compared to sex- and age-matched wild-type control animals (Figure 4D). Therefore, decreased XIAP expression led to decreased copper levels in cultured cells and animal tissues.
Figure 4.
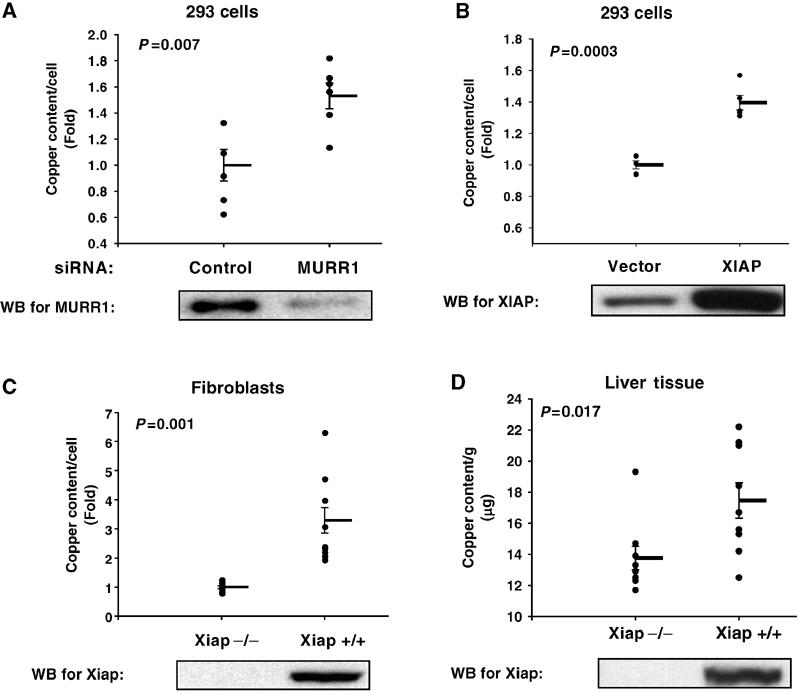
Copper levels are regulated by XIAP. 293 cells were transiently transfected with siRNA oligonucleotides or plasmids as indicated. Copper levels were measured by atomic absorption and corrected for cell number in each sample. The effect of RNAi of MURR1 is shown in (A) and the effect of transient transfection of XIAP is shown in (B). Similar experiments were performed with transformed fibroblasts derived from wild-type and Xiap-deficient mice (C). Copper content in liver tissue obtained from Xiap-deficient mice and age- and sex-matched wild-type controls is presented in (D). Each graph depicts all data points from multiple independent experiments. The mean and s.e.m. bars are also shown. A Student's t-test was performed, and the P-value is indicated in the graph panel. Western blots for MURR1 or XIAP are included below each panel as indicated.
XIAP promotes the ubiquitination and degradation of MURR1
The data shown in Figure 4 demonstrate that XIAP expression levels were directly proportional to intracellular copper levels. In contrast, MURR1 protein levels inversely correlated with intracellular copper content. These findings suggested the possibility that XIAP could be a negative regulator of MURR1. In the course of our experiments, it was noticed that alterations in XIAP protein levels in cells resulted in changes in MURR1 expression. Transfection of XIAP led to decreased MURR1 protein levels and, conversely, a reduction in endogenous XIAP protein levels by RNAi led to increased levels of MURR1 protein (Figure 5A). Similarly, transfection of an XIAP mutant (H467A) that lacks E3 ubiquitin ligase activity (Yang et al, 2000) also resulted in increased MURR1 protein levels, mimicking the effects induced by RNAi of XIAP. These observations suggested that, under basal conditions, MURR1 protein levels are regulated by ubiquitination and proteasomal degradation in a manner that is stimulated by the E3 ubiquitin ligase activity of XIAP.
Figure 5.
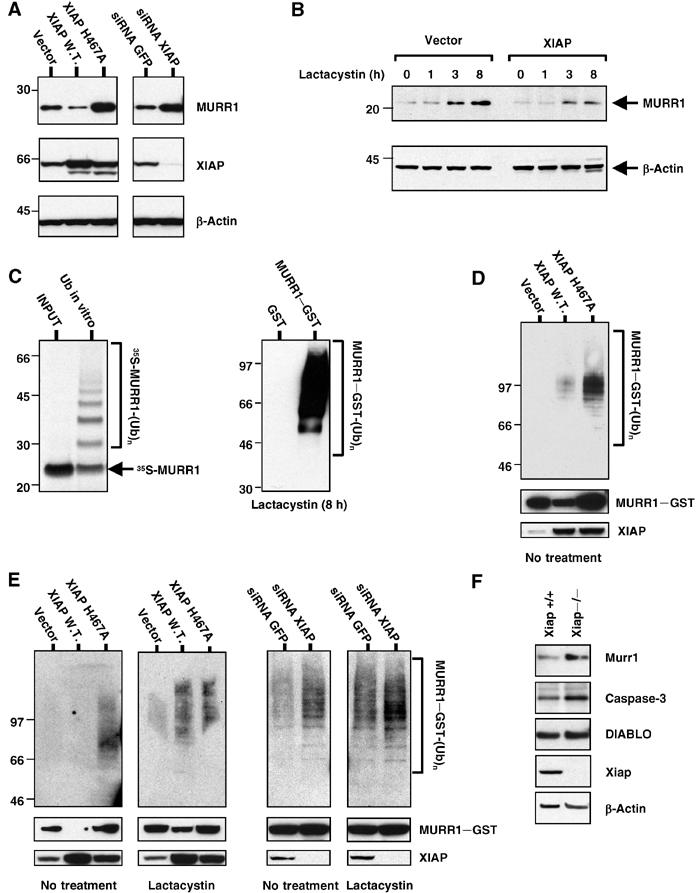
XIAP targets MURR1 for ubiquitination and degradation. (A) MURR1 protein levels are regulated by XIAP: 293 cells were transfected with pEBB–XIAP, pEBB–XIAP H467A or vector control or with siRNA oligonucleotides targeting GFP or XIAP. MURR1–Flag was cotransfected and expression was determined by Western blot using Flag antibodies. The same membrane was probed for β-actin and for XIAP. (B) MURR1 protein levels are regulated by proteasomal degradation: 293 cells were transfected with MURR1–Flag and protein levels were determined by Western blot using Flag antibodies. The cells were cotransfected with XIAP or a vector control and treated with lactacystin (10 μM) for the time indicated. (C) MURR1 is ubiquitinated in vitro and in intact cells: in vitro translated and 35S-labeled MURR1 was used as substrate for in vitro ubiquitination (left). Ubiquitination was also studied in intact cells. 293 cells were transfected as indicated. One day after transfection, cells were treated with lactacystin (10 μM) for 8 h, and lysed in RIPA buffer. Lysates were precipitated with glutathione sepharose beads and analyzed by Western blot using ubiquitin antibodies (right). (D) Effect of XIAP on MURR1 ubiquitination: 293 cells were transfected as indicated. One day after transfection, cells were lysed in RIPA buffer and precipitation was performed using glutathione sepharose beads. The precipitated material was used for Western blot with ubiquitin antibodies. (E) Effect of XIAP in the accumulation of ubiquitinated MURR1 after lactacystin treatment: 293 cells were transfected as indicated; 1 day later, half of the cultures were treated with lactacystin (10 μM) for an additional 3 h. Cell lysates were prepared with RIPA buffer and precipitated with glutathione sepharose beads. The precipitated material was used for Western blot with ubiquitin antibodies. (F) Murr1 protein levels in Xiap-deficient fibroblasts: cell lysates from wild-type and Xiap-deficient fibroblasts were used for Western blot and immunoblotted as indicated.
The effect of proteasomal blockade on MURR1 protein levels was examined next. Treatment with lactacystin, a highly specific proteasome inhibitor, led to progressive accumulation of MURR1; in cells transfected with XIAP the accumulation of MURR1 protein also occurred but to a lesser extent (Figure 5B, left). These data indicate that basal levels of MURR1 are regulated by proteasomal degradation and that XIAP accelerates this process.
To determine whether MURR1 is indeed ubiquitinated, in vitro translated and radiolabeled MURR1 was incubated with a mammalian ubiquitin conjugating enzyme fraction extracted from HeLa cell lysates, and subsequently resolved by polyacrylamide gel electrophoresis and fluorography. A ladder comprised of MURR1 species with progressively higher molecular weights was detected (Figure 5C, left), indicating that MURR1 can be readily ubiquitinated in vitro. The ability of MURR1 to be ubiquitinated in intact cells was also examined. A mammalian expression vector encoding MURR1–GST was expressed in 293 cells and these cells were treated with lactacystin for 8 h. Lysates were precipitated with glutathione sepharose beads under stringent buffer conditions (RIPA buffer), and the precipitated material was immunoblotted for ubiquitin. High levels of ubiquitinated MURR1–GST were detected, while no ubiquitination of the control GST protein was observed (Figure 5C, right). These data indicate that MURR1 is ubiquitinated.
The ability of XIAP to promote the ubiquitination of MURR1 was examined next. A mammalian expression vector encoding MURR1–GST was transfected into 293 cells along with XIAP or a vector control. Under these conditions, which did not include the addition of proteasome inhibitors, ubiquitinated material of progressively higher molecular weight was observed when MURR1 was cotransfected with XIAP but not with the empty control vector (Figure 5D, top panel). In addition, a significant reduction of MURR1–GST protein levels after XIAP transfection was also observed (Figure 5D, middle panel), consistent with the findings shown in Figure 5A.
Interestingly, transfection of XIAP H467A led to greater recovery of ubiquitinated MURR1 when compared to transfection of vector or wild-type XIAP (Figure 5D, top panel). However, MURR1–GST protein levels were observed to increase following coexpression of XIAP H467A (Figure 5D, middle panel), consistent with the observed effect shown in Figure 5A. Similarly, in the absence of proteasomal inhibition, RNAi of XIAP also led to accumulation of ubiquitinated MURR1 (see the third panel in Figure 5E), while as shown before, MURR1 protein levels actually increased.
These findings raised the possibility that the ubiquitinated species accumulating in cells transfected with XIAP H467A or after RNAi of XIAP are not being targeted for proteasomal degradation. To test this hypothesis, the accumulation of ubiquitinated species after proteasomal inhibition was investigated. Cells transfected with wild-type XIAP and treated with lactacystin for 3 h accumulated ubiquitinated MURR1 to a greater extent than vector transfected cells (Figure 5E, first and second panels, top). In addition, MURR1–GST protein levels, which were greatly reduced in untreated cells and only noticeable upon long exposures, were stabilized after proteasomal blockade (Figure 5E, first and second panels, middle).
While transfection of XIAP H467A led to accumulation of ubiquitinated material in untreated cells, 3 h of lactacystin treatment caused no further increase in ubiquitinated MURR1 or MURR1–GST input levels in cells transfected with XIAP H467A (Figure 5E, first and second panels, top and middle). Similarly, RNAi of XIAP also led to the accumulation of ubiquitinated MURR1 in untreated cells, but ubiquitinated material increased very little after proteasomal inhibition, in contrast to control transfected cells (Figure 5E, third and fourth panels). Taken together, these data indicate that wild-type XIAP accelerates the formation of ubiquitinated material that is targeted to the proteasome for degradation. Blockade of XIAP-mediated ubiquitination by transfection of XIAP H467A or RNAi of XIAP therefore revealed the existence of an alternative pathway of MURR1 ubiquitination that is insensitive to proteasomal blockade and that does not lead to degradation of MURR1.
The effect of complete absence of Xiap on Murr1 protein levels was investigated using fibroblasts derived from the Xiap-deficient mice. When compared to wild-type fibroblasts, Xiap −/− fibroblasts demonstrate a modest but reproducible increase in Murr1 protein levels (Figure 5F). Other previously described targets of XIAP-mediated ubiquitination (Suzuki et al, 2001b; MacFarlane et al, 2002) also showed modest increases (caspase-3) or no increase (DIABLO).
K48-independent polyubiquitination of MURR1
As noted above, blockade of XIAP-mediated ubiquitination of MURR1 by transfection of the catalytically inactive mutant XIAP H467A or after RNAi of XIAP leads to the accumulation of ubiquitinated MURR1 that is not targeted for proteasomal-mediated degradation. This situation has been observed previously for unrelated proteins, and would occur if the polyubiquitin chains on MURR1 are not linked at lysine 48 (K48) of ubiquitin (Aguilar and Wendland, 2003).
To test this hypothesis, MURR1 ubiquitination was evaluated by a method that allowed us to compare the incorporation into polyubiquitinated MURR1 of wild-type ubiquitin and a K48R point mutant of ubiquitin that cannot be conjugated into K48 polyubiquitin chains. In this approach, MURR1–Flag was cotransfected with His6-tagged ubiquitin expression vectors, and nickel-coated agarose beads were used to precipitate ubiquitinated proteins. Immunoblotting of the precipitated material with Flag antibody was performed to detect ubiquitin-conjugated MURR1. Using wild-type ubiquitin in the experiment, transfection of wild-type XIAP resulted in increased recovery of ubiquitinated MURR1 (Figure 6A, left). Similar to the results shown in Figure 5D, transfection of XIAP H467A along with wild-type ubiquitin resulted in even greater levels of ubiquitinated MURR1. Following transfection of ubiquitin K48R, ubiquitinated MURR1 was recovered, demonstrating that non-K48 polyubiquitin chains could also be added to MURR1. In contrast to the pattern of ubiquitination observed with wild-type ubiquitin, the amount of polyubiquitinated MURR1 containing the K48R ubiquitin mutant was not affected by XIAP (Figure 6A, right). These results demonstrate that in addition to K48 polyubiquitin chains targeting MURR1 to proteasomal degradation, non-K48 polyubiquitination of MURR1 can also be detected and this pathway is independent of XIAP.
Figure 6.
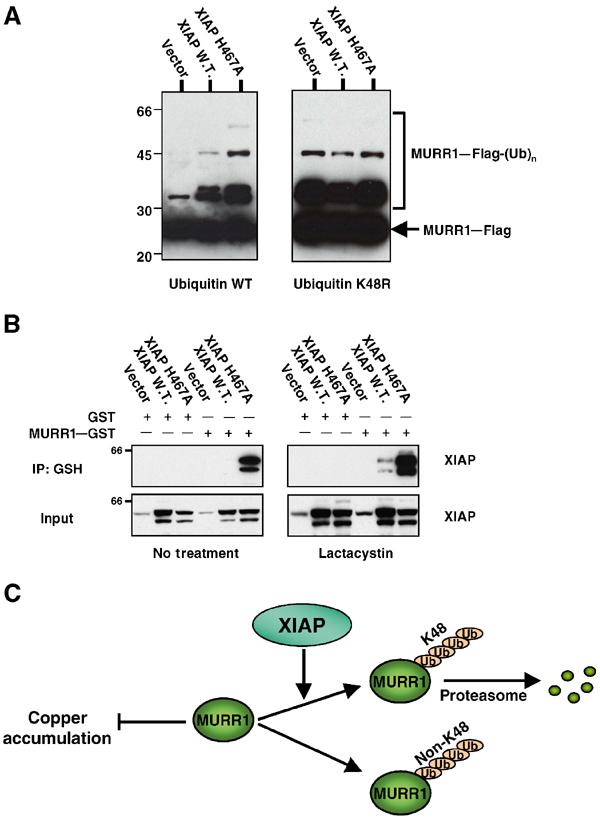
Non-K48 ubiquitination of MURR1. (A) Non-K48-linked ubiquitination of MURR1: 293 cells were transfected with either wild-type His6-tagged ubiquitin or a K48R mutant of ubiquitin along with MURR1–Flag. In addition, XIAP wild-type, XIAP H467 or a vector control were also transfected, as indicated. Two days after transfection, cell lysates were prepared and used for precipitation with Ni–NTA agarose beads. The precipitated material was immunoblotted with Flag antibodies. Molecular weight markers are in kilodaltons. (B) Binding of XIAP H467A to MURR1 and the effects of proteasome inhibition: 293 cells were transfected as indicated and 1 day later half of the cultures were treated with lactacystin (10 μM) for an additional 6 h. Cell lysates were then prepared with RIPA buffer and precipitated with glutathione sepharose beads. The precipitated material was immunoblotted for XIAP. (C) Schematic representation of MURR1 ubiquitination pathways and the role of XIAP.
This alternative pathway of ubiquitination was uncovered by RNAi of XIAP and similarly by transfection of XIAP H467A. The ability of this catalytically inactive mutant to act as a dominant-negative molecule could result from binding to MURR1 in a manner that prevents its ubiquitination by endogenous wild-type XIAP. Indeed, under stringent immunoprecipitation conditions (RIPA buffer), MURR1 was found to bind much more strongly to XIAP H467A than to wild-type XIAP (Figure 6B, left). Interestingly, proteasomal blockade also led to increased binding to MURR1of both wild-type XIAP and XIAP H467A, although the binding to XIAP H467A remained stronger (Figure 6B, right).
Discussion
In this study, we have characterized a novel property of XIAP in copper homeostasis through its ability to ubiquitinate the copper regulating factor, MURR1. An association between these two proteins was first identified in a yeast two-hybrid screen, and was subsequently confirmed by coprecipitation experiments with both endogenous and ectopically expressed proteins. Deletion mapping of XIAP revealed that BIR 3 is necessary and sufficient for binding to MURR1. MURR1 was found to bind to XIAP even after critical residues required for interaction with caspase-3 (D148) and caspase-9 (W310) had been mutated, thus abrogating caspase binding (Bratton et al, 2002; Silke et al, 2002) (Figure 1D). This suggests that the mechanism involved in MURR1 binding to XIAP is distinct from that utilized by caspases, Smac/DIABLO and Omi/HtrA2 (Shi, 2002). Processed caspase-9, Smac/DIABLO and Omi/HtrA2 bind to a defined region in BIR 3 through an amino-terminal tetrapeptide element present in the mature forms of these proteins. Consistent with our observation that a critical mutation in the caspase-9 binding pocket (W310A) does not affect MURR1 binding, we have not identified a canonical tetrapeptide IAP binding motif in MURR1. In addition, MURR1 can bind to other IAPs, such as c-IAP2 and NAIP, although the functional significance of such associations remains to be elucidated.
MURR1 has been implicated in the regulation of copper homeostasis in mammals. This gene was found by positional cloning to be responsible for an autosomal recessive form of copper toxicosis that affects a specific canine strain (van De Sluis et al, 2002). The MURR1 mutation results in loss of protein expression in affected animals (Klomp et al, 2003). Copper is essential for the enzymatic activity of a number of housekeeping genes, including Cu,Zn superoxide dismutase, and it can play a pathophysiologic role in oxidative stress damage through its ability to induce free radical formation (Mercer, 2001). The regulation of copper homeostasis in mammalian cells is a complex process involving high-affinity copper transporters that can mediate copper uptake or efflux from the cell, as well as copper chaperones involved in the transfer of copper ions to certain copper-binding proteins (Puig and Thiele, 2002). The precise mechanism by which MURR1 is involved in copper regulation remains unknown, although an association between MURR1 and the copper transporter ATP7B has been recently described (Tao et al, 2003). Utilizing RNAi to decrease the endogenous levels of MURR1, we show here the first biochemical evidence that deficiency of this protein can indeed lead to copper accumulation in cultured cells. Given that ATP7B expression is largely restricted to the liver and brain (Puig and Thiele, 2002), the effects on cellular copper levels observed in our studies using 293 cells and fibroblasts suggest that mechanisms involving other molecular targets are possibly implicated as well.
Although we did not find a role for MURR1 in the control of apoptosis by XIAP, we did find an effect of XIAP expression on copper levels. Increased levels of XIAP expression led to copper accumulation in several cell models, and Xiap deficiency in mice led to decreases in copper content in hepatic tissue. These findings support a physiological role for the interaction between MURR1 and XIAP. In addition, this represents the first phenotypic alteration reported in Xiap-deficient mice, which do not have detectable defects in the regulation of apoptotic cell death (Harlin et al, 2001).
The effect of XIAP expression on copper regulation was the opposite of that observed for MURR1. Consistent with this, our studies demonstrate that XIAP negatively regulates MURR1 protein levels. Expression of wild-type XIAP led to reductions in MURR1 levels. Conversely, reduction of endogenous levels of XIAP by RNAi or expression of XIAP H467A, harboring a point mutation that abolishes its E3 ubiquitin ligase activity, led to increased MURR1 protein levels. The studies presented here demonstrate that the ability of XIAP to reduce MURR1 protein levels is mediated by ubiquitination and proteasomal degradation of MURR1. Proteasomal blockade with lactacystin led to increased MURR1 levels demonstrating that under basal conditions, MURR1 protein levels are subject to a constant rate of degradation. In addition, this effect also occurred in XIAP-transfected cells, although to a lesser extent, suggesting that XIAP accelerates the process of proteasomal degradation of MURR1. Indeed, increased amounts of ubiquitinated MURR1 could be recovered after transfection of wild-type XIAP, and after proteasomal blockade this material accumulated to a greater extent in XIAP-transfected cells, indicating that the ultimate fate of this ubiquitinated material is its degradation by the proteasome. Finally, consistent with a role for XIAP in the regulation of basal levels of MURR1, Xiap-deficient fibroblasts had modest increases in Murr1 protein levels. The modest changes observed in protein levels may represent the upregulation of yet undetermined compensatory changes in these cells, and other presumed targets of XIAP-mediated ubiquitination (Suzuki et al, 2001b; MacFarlane et al, 2002) such as Smac/DIABLO or caspase-3 are either unchanged or only minimally elevated in these cells.
Interestingly, an increase in ubiquitinated MURR1 was also observed following transfection of XIAP H467A or RNAi of endogenous XIAP, although total MURR1 protein levels also increased, strongly suggesting that this polyubiquitinated material was not being targeted for proteasomal degradation. Consistent with this interpretation, proteasomal blockade did not lead to further accumulation of ubiquitinated MURR1 in cells transfected with XIAP H467A or after RNAi of XIAP. Proteasome-mediated degradation of proteins occurs when polyubiquitin chains of four or more ubiquitin molecules linked at lysine 48 of ubiquitin are added to the target protein (Thrower et al, 2000). However, other forms of polyubiquitin branches that are generally not degraded by the proteasome have been described; these may instead play distinct roles in signal transduction (Deng et al, 2000). As shown here, polyubiquitination of MURR1 in a manner that does not require K48 of ubiquitin was found, and our data indicate that this process is XIAP-independent, likely resulting from (an) as yet unidentified E3(s) that target(s) MURR1 (Figure 6C). The nature of the lysine involved in the branching of these polyubiquitin chains and their functional significance remain to be determined. In addition, the data suggest that, under basal conditions, XIAP regulates the rate of MURR1 ubiquitination through this alternative pathway, perhaps through substrate competition. When basal XIAP activity is suppressed, as a result of RNAi of endogenous XIAP or transfection of the catalytically inactive H467A mutant, nondegradable ubiquitinated MURR1 readily accumulates, likely as a result of the unimpeded activity of this alternative ubiquitination pathway. Depending on the functional significance of non-K48 polyubiquitinated MURR1, the regulation of the cellular pool of this material might be another level at which XIAP can regulate MURR1 activity.
The ability of XIAP H467A to have a dominant-negative effect on XIAP-mediated regulation of MURR1 could result from the ability of this molecule to bind to MURR1 with even greater affinity than wild-type XIAP. The reason for this increased affinity is unclear, but could implicate a contribution of the RING finger domain in the stability of the interaction. Alternatively, a factor(s) that stabilize(s) MURR1–XIAP interactions might also accumulate after transfection of XIAP H467A. This possibility is partially supported by the fact that proteasomal inhibition also increases the affinity of the interaction to a degree that cannot be simply explained by accumulation of the proteins.
Overall, these results are consistent with a model in which XIAP regulates copper levels through its ability to negatively regulate MURR1 by directing the ubiquitination and proteasomal degradation of this molecule. The data also demonstrate the presence of an alternative ubiquitination pathway for MURR1 that is K48-independent and that, under basal conditions, is likely suppressed by XIAP. While a role for XIAP can be envisaged as a general sensor of cellular stress, possible relationships between its seemingly unrelated functions, including caspase inhibition, TGF-β signaling and copper regulation, remain to be elucidated.
Materials and methods
Yeast two-hybrid screening
A GAL4-based system was used (Fields and Song, 1989) (Matchmaker GAL4 two-hybrid system 3, Clontech). Full-length XIAP was cloned into pGBKT7; this plasmid was transformed into yeast and no spontaneous transactivation was noted. The transformed yeast were expanded and mated with yeast pretransformed with a liver cDNA library (Clontech). A total of 4 × 106 transformants were screened.
Plasmids and siRNA oligonucleotides
The MURR1 clone obtained from the cDNA library containing amino acids 4–190 of the protein (pAct2-MURR1) was subcloned into pCMV-HA (Clontech). The coding sequence for the first three amino acids was cloned in frame using synthetic oligonucleotides. pEBB–MURR1–Flag and pEBB–MURR1–GST were constructed by PCR of MURR1 using pCMV–HA full-length MURR1 as template. pcDNA3.1(+) MURR1 and pMURR1–DsRed2 were constructed by inserting full-length MURR1 into pcDNA3.1(+) and pDsRed2-N1, respectively. pEBB–CFP and pEBB–YFP were constructed by inserting PCR fragments for CFP and YFP into pEBB. pEBB–CFP–XIAP and pEBB–MURR1–YFP were similarly constructed by inserting CFP and YFP into the 5′ end of pEBB–XIAP and the 3′ end of pEBB–MURR1–Flag, respectively. pEBG–BIR 1, pEBG–BIR 2, pEBG–BIR 3 and pEBG–BIR 1–2 were generated by PCR using pEBB–XIAP as template, with the boundaries for each construct as indicated in Figure 1B. Other pEBB-derived plasmids used here have been described previously (Duckett et al, 1998; Richter et al, 2001). Plasmid pCW7-His6-Ub has also been described (Yu and Kopito, 1999); the K48R mutation in ubiquitin was introduced by site-directed mutagenesis. For RNA interference (RNAi), double-stranded RNA oligonucleotides (Xeragon/Qiagen) targeting the following gene-specific sequences were used: AAGTGGTAGTCCTGTTTCAGC (XIAP coding sequence nucleotides 111–131), AAGTCTATTGCGTCTGCAGAC (MURR1 coding sequence nucleotides 178–198) and AAGACCCGCGCCGAGGTGAAG (GFP coding sequence nucleotides 322–342).
Cell culture and transfection
Human embryonic kidney 293 cells and transformed fibroblasts were cultured in DMEM supplemented with 10% FBS and 2 mM L-glutamine. A standard calcium phosphate transfection protocol (Duckett et al, 1997) was used to transfect 293 cells; this was the case for both plasmids and siRNA oligonucleotides.
Caspase assays
Floating and attached cells were harvested and caspase-3 assays were performed using the ApoTarget protease assay kit (BioSource) according to the manufacturer's instructions. Caspase activity was measured for 2 h at 90-s intervals on a Cytofluor 4000 fluorescence plate reader (Perseptive Biosystems) as described previously (Richter et al, 2001).
In vitro ubiquitination assay
MURR1 was translated in vitro using pcDNA3.1 (+) MURR1 as template in the presence of 35S-labeled methionine and cysteine (Amersham) following the manufacturer's instructions (Promega). The in vitro ubiquitination reaction utilized 35S-labeled MURR1 as substrate for a mammalian ubiquitin conjugating enzyme fraction extracted from HeLa cell lysates containing a mixture of E1, E2 and E3 enzymes with the addition of ubiquitin aldehyde (Boston Biochem) according to the manufacturer's instructions.
Western blot, antibodies and immunoprecipitation
Cell lysates were prepared as previously described (Richter et al, 2001). The lysis buffers used were RIPA (PBS, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) and a buffer containing 0.5% Triton X-100 (25 mM HEPES, 100 mM NaCl, 1 mM EDTA, 10% glycerol), as indicated in each experiment. In both cases, the lysis buffer was supplemented with 1 mM Na3VO4, 1 mM PMSF, one protease inhibitor tablet (Roche) per 10 ml of buffer, 10 mM DTT and 1 mM NaF.
Protein samples were resolved using 4–12% gradient Novex Bis-Tris gels (Invitrogen), transferred to nitrocellulose membranes (Invitrogen) and blocked with 5% milk solution in TBS containing 0.05–0.2% Tween-20 (depending on the antibody). The membranes were incubated with primary antibodies as indicated, followed by incubation with HRP-conjugated sheep anti-mouse or donkey anti-rabbit secondary antibodies (Amersham). Antibody detection was performed by an enhanced chemiluminescence (ECL) Western blot detection system (Amersham) according to the manufacturer's instructions.
A polyclonal MURR1 antiserum was used; further characterization of this antibody has been described elsewhere (Klomp et al, 2003). A previously described mouse monoclonal antibody raised against a fragment of c-IAP1 lacking the first BIR was used (Harlin et al, 2001). Mouse monoclonal antibodies against XIAP (Transduction Labs, 59520), Flag (Sigma, A8592), β-actin (Sigma, A5441), Myc (Covance, 9E10) and ubiquitin (Affinity, FK2) were also used.
For MURR1 immunoprecipitation, polyclonal antibody was added to the cell lysate and incubated at 4°C. After 1 h, protein G agarose beads (Invitrogen) were added and the samples were incubated for an additional 2 h. For precipitations of GST-tagged proteins and for precipitation of His6-ubiquitin conjugated proteins, glutathione sepharose beads (Amersham) or nickel agarose beads (Invitrogen) were added and the samples were incubated at 4°C for 3–6 h, respectively. In all precipitation experiments, the beads were washed four times with lysis buffer and pelleted material was resuspended in LDS loading buffer (Invitrogen) and used for immunoblotting as described above.
Confocal and fluorescence microscopy
Morphological assays for cell viability were performed by observing EGFP fluorescent cells with a Leica DM IRB inverted fluorescence microscope. Images for MURR1 localization experiments were obtained from live cells plated onto coverglass chambers utilizing a Zeiss Axiovert 100M confocal microscope equipped with a Zeiss LSM 510 Meta spectrometer. Nuclear counterstaining was performed by adding Hoechst 33342 to phenol-red-free culture media to a final concentration of 5 μg/ml.
Copper measurements
Cells were harvested and washed three times in PBS. After the last wash, the cells were counted twice and pelleted. The cell pellet was digested by refluxing in Ultrex nitric acid for 2 h. The excess nitric acid was removed by evaporation and the residue was dissolved in 0.5% nitric acid. Copper levels were determined by atomic absorption in duplicate or triplicate for each sample using an IL451 single-beam flame spectrophotometer. The copper content obtained was expressed as micrograms of copper per million cells, and these values were used to calculate fold relationships between samples. For measurements of copper content in liver tissue, samples were first dried in a 65°C vacuum oven and then processed in a similar way as cell pellets. The copper content obtained from these samples was expressed as micrograms of copper per gram of dry tissue.
Acknowledgments
We are grateful to AS Wilkinson, J Lewis and B Balliu for valuable technical assistance. We would also like to thank P Liston for providing the expression plasmid for NAIP used in our studies. This work was supported by NIH/NCI Cancer Biology Training Grant (T32 CA09676) to JCW, the Michigan Gastrointestinal Hormone Research Core Center (5 P30 DK34933-18) and an American Gastroenterological Association Research Scholar Award granted to EB.
References
- Aguilar RC, Wendland B (2003) Ubiquitin: not just for proteasomes anymore. Curr Opin Cell Biol 15: 184–190 [DOI] [PubMed] [Google Scholar]
- Birkey Reffey S, Wurthner JU, Parks WT, Roberts AB, Duckett CS (2001) X-linked inhibitor of apoptosis protein functions as a cofactor in transforming growth factor-β signaling. J Biol Chem 276: 26542–26549 [DOI] [PubMed] [Google Scholar]
- Bratton SB, Lewis J, Butterworth M, Duckett CS, Cohen GM (2002) XIAP inhibition of caspase-3 preserves its association with the Apaf-1 apoptosome and prevents CD95- and Bax-induced apoptosis. Cell Death Differ 9: 881–892 [DOI] [PubMed] [Google Scholar]
- Chai J, Shiozaki E, Srinivasula SM, Wu Q, Dataa P, Alnemri ES, Shi Y (2001) Structural basis of caspase-7 inhibition by XIAP. Cell 104: 769–780 [DOI] [PubMed] [Google Scholar]
- Crook NE, Clem RJ, Miller LK (1993) An apoptosis-inhibiting baculovirus gene with a zinc finger-like motif. J Virol 67: 2168–2174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ (2000) Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103: 351–361 [DOI] [PubMed] [Google Scholar]
- Devereaux QL, Takahashi R, Salvesen GS, Reed JC (1997) X-linked IAP is a direct inhibitor of cell-death proteases. Nature 388: 300–304 [DOI] [PubMed] [Google Scholar]
- Du C, Fang M, Li Y, Li L, Wang X (2000) Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102: 33–42 [DOI] [PubMed] [Google Scholar]
- Duckett CS, Gedrich RW, Gilfillan MC, Thompson CB (1997) Induction of nuclear factor κB by the CD30 receptor is mediated by TRAF1 and TRAF2. Mol Cell Biol 17: 1535–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckett CS, Li F, Wang Y, Tomaselli KJ, Thompson CB, Armstrong RC (1998) Human IAP-like protein regulates programmed cell death downstream of Bcl-xL and cytochrome c. Mol Cell Biol 18: 608–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields S, Song O (1989) A novel genetic system to detect protein–protein interactions. Nature 340: 245–246 [DOI] [PubMed] [Google Scholar]
- Harlin H, Reffey SB, Duckett CS, Lindsten T, Thompson CB (2001) Characterization of XIAP-deficient mice. Mol Cell Biol 21: 3604–3608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde R, Srinivasula SM, Zhang Z, Wassell R, Mukattash R, Cilenti L, DuBois G, Lazebnik Y, Zervos AS, Fernandes-Alnemri T, Alnemri ES (2001) Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts IAP–caspase interaction. J Biol Chem 277: 432–438 [DOI] [PubMed] [Google Scholar]
- Hofer-Warbinek R, Schmid JA, Stehlik C, Binder BR, Lipp J, de Martin R (2000) Activation of NF-κB by XIAP, the X chromosome-linked inhibitor of apoptosis, in endothelial cells involves TAK1. J Biol Chem 275: 22064–22068 [DOI] [PubMed] [Google Scholar]
- Holcik M, Korneluk RG (2001) XIAP, the guardian angel. Nat Rev Mol Cell Biol 2: 550–556 [DOI] [PubMed] [Google Scholar]
- Holcik M, Lefebvre C, Yeh C, Chow T, Korneluk RG (1999) A new internal-ribosome-entry-site motif potentiates XIAP-mediated cytoprotection. Nat Cell Biol 1: 190–192 [DOI] [PubMed] [Google Scholar]
- Huang Y, Park YC, Rich RL, Segal D, Myszka DG, Wu H (2001) Structural basis of caspase inhibition by XIAP. Differential roles of the linker versus the BIR domain. Cell 104: 781–790 [PubMed] [Google Scholar]
- Klomp AEM, van de Sluis B, Klomp LWJ, Wijmenga C (2003) The ubiquitously expressed MURR1 protein is absent in canine copper toxicosis. J Hepatol 39: 703–709 [DOI] [PubMed] [Google Scholar]
- Levkau B, Garton KJ, Ferri N, Kloke K, Nofer JR, Baba HA, Raines EW, Breithardt G (2001) XIAP induces cell-cycle arrest and activates nuclear factor-κB: new survival pathways disabled by caspase-mediated cleavage during apoptosis of human endothelial cells. Circ Res 88: 282–290 [DOI] [PubMed] [Google Scholar]
- Li X, Yang Y, Ashwell JD (2002) TNF-RII and c-IAP1 mediate ubiquitination and degradation of TRAF2. Nature 416: 345–347 [DOI] [PubMed] [Google Scholar]
- Liu Z, Sun C, Olejniczak ET, Meadows RP, Betz SF, Oost T, Herrmann J, Wu JC, Fesik SW (2000) Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature 408: 1004–1008 [DOI] [PubMed] [Google Scholar]
- MacFarlane M, Merrison W, Bratton SB, Cohen GM (2002) Proteasome-mediated degradation of Smac during apoptosis: XIAP promotes Smac ubiquitination in vitro. J Biol Chem 277: 36611–36616 [DOI] [PubMed] [Google Scholar]
- Martindale JL, Holbrook NJ (2002) Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol 192: 1–15 [DOI] [PubMed] [Google Scholar]
- Martins LM, Iaccarino I, Tenev T, Gschmeissner S, Totty NF, Lemoine NR, Savopoulos J, Gray CW, Creasy CL, Dingwall C, Downward J (2001) The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a Reaper-like motif. J Biol Chem 277: 439–444 [DOI] [PubMed] [Google Scholar]
- Mercer JF (2001) The molecular basis of copper-transport diseases. Trends Mol Med 7: 64–69 [DOI] [PubMed] [Google Scholar]
- Mercurio F, Manning AM (1999) NF-kappaB as a primary regulator of the stress response. Oncogene 18: 6163–6171 [DOI] [PubMed] [Google Scholar]
- Puig S, Thiele DJ (2002) Molecular mechanisms of copper uptake and distribution. Curr Opin Chem Biol 6: 171–180 [DOI] [PubMed] [Google Scholar]
- Richter BWM, Mir SS, Eiben LJ, Lewis J, Reffey SB, Frattini A, Tian L, Frank S, Youle RJ, Nelson DL, Notarangelo LD, Vezzoni P, Fearnhead HO, Duckett CS (2001) Molecular cloning of ILP-2, a novel member of the inhibitor of apoptosis protein (IAP) family. Mol Cell Biol 21: 4292–4301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl SJ, Renatus M, Schwarzenbacher R, Zhou Q, Sun C, Fesik SW, Liddington RC, Salvesen GS (2001) Structural basis for the inhibition of caspase-3 by XIAP. Cell 104: 791–800 [DOI] [PubMed] [Google Scholar]
- Rothe M, Pan M-G, Henzel WJ, Ayres TM, Goeddel DV (1995) The TNFR2–TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell 83: 1243–1252 [DOI] [PubMed] [Google Scholar]
- Sanna MG, da Silva Correia J, Ducrey O, Lee J, Nomoto K, Schrantz N, Deveraux QL, Ulevitch RJ (2002) IAP suppression of apoptosis involves distinct mechanisms: the TAK1/JNK1 signaling cascade and caspase inhibition. Mol Cell Biol 22: 1754–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna MG, Duckett CS, Richter BWM, Thompson CB, Ulevitch RJ (1998) Selective activation of JNK1 is necessary for the anti-apoptotic activity of hILP. Proc Natl Acad Sci USA 95: 6015–6020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y (2002) A conserved tetrapeptide motif: potentiating apoptosis through IAP-binding. Cell Death Differ 9: 93–95 [DOI] [PubMed] [Google Scholar]
- Silke J, Hawkins CJ, Ekert PG, Chew J, Day CL, Pakusch M, Verhagen AM, Vaux DL (2002) The anti-apoptotic activity of XIAP is retained upon mutation of both the caspase 3- and caspase 9-interacting sites. J Cell Biol 157: 115–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasula SM, Gupta S, Datta P, Zhang Z, Hegde R, Cheong N, Fernandes-Alnemri T, Alnemri ES (2003) Inhibitor of apoptosis proteins are substrates for the mitochondrial serine protease Omi/HtrA2. J Biol Chem 278: 31469–31472 [DOI] [PubMed] [Google Scholar]
- Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J, Lee RA, Robbins PD, Fernandes-Alnemri T, Shi Y, Alnemri ES (2001) A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature 410: 112–116 [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K, Takahashi R (2001a) A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell 8: 613–621 [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Nakabayashi Y, Takahashi R (2001b) Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death. Proc Natl Acad Sci USA 98: 8662–8667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao TY, Liu F, Klomp L, Wijmenga C, Gitlin JD (2003) The copper toxicosis gene product Murr1 directly interacts with the Wilson disease protein. J Biol Chem 278: 41593–41596 [DOI] [PubMed] [Google Scholar]
- Thrower JS, Hoffman L, Rechsteiner M, Pickart CM (2000) Recognition of the polyubiquitin proteolytic signal. EMBO J 19: 94–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uren AG, Wong L, Pakusch M, Fowler KJ, Burrows FJ, Vaux DL, Choo KH (2000) Survivin and the inner centromere protein INCENP show similar cell-cycle localization and gene knockout phenotype. Curr Biol 10: 1319–1328 [DOI] [PubMed] [Google Scholar]
- van De Sluis B, Rothuizen J, Pearson PL, van Oost BA, Wijmenga C (2002) Identification of a new copper metabolism gene by positional cloning in a purebred dog population. Hum Mol Genet 11: 165–173 [DOI] [PubMed] [Google Scholar]
- Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL (2000) Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 102: 43–53 [DOI] [PubMed] [Google Scholar]
- Wu G, Chai J, Suber TL, Wu JW, Du C, Wang X, Shi Y (2000) Structural basis of IAP recognition by Smac/DIABLO. Nature 408: 1008–1012 [DOI] [PubMed] [Google Scholar]
- Yamaguchi K, Nagai S, Ninomiya-Tsuji J, Nishita M, Tamai K, Irie K, Ueno N, Nishida E, Shibuya H, Matsumoto K (1999) XIAP, a cellular member of the inhibitor of apoptosis protein family, links the receptors to TAB1–TAK1 in the BMP signaling pathway. EMBO J 18: 179–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang QH, Church-Hajduk R, Ren J, Newton ML, Du C (2003) Omi/HtrA2 catalytic cleavage of inhibitor of apoptosis (IAP) irreversibly inactivates IAPs and facilitates caspase activity in apoptosis. Genes Dev 17: 1487–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Fang S, Jensen JP, Weissman AM, Ashwell JD (2000) Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science 288: 874–877 [DOI] [PubMed] [Google Scholar]
- Yu H, Kopito RR (1999) The role of multiubiquitination in dislocation and degradation of the α subunit of the T cell antigen receptor. J Biol Chem 274: 36852–36858 [DOI] [PubMed] [Google Scholar]