

Abstract
The PrP gene of the host exerts a major influence over the outcome of transmissible spongiform encephalopathy (TSE) disease, but the mechanism by which this is achieved is not understood. We have introduced a specific mutation into the endogenous murine PrP gene using gene targeting to produce transgenic mice with a single amino acid alteration (proline to leucine) at amino acid position 101 in their PrP protein (P101L). The effect of this alteration on incubation time, targeting and PrPSc formation has been studied in TSE-infected animals. Transgenic mice carrying the P101L mutation in PrP have remarkable differences in incubation time and targeting of central nervous system pathology compared with wild-type littermates, following inoculation with infectivity from human, hamster, sheep and murine sources. This single mutation can alter incubation time across three species barriers in a strain-dependent manner. These findings suggest a critical role for the structurally ‘flexible’ region of PrP in agent replication and targeting of TSE pathology.
Keywords: P101L PrP/prion diseases/species barrier/transmissible spongiform encephalopathies/vCJD
Introduction
Transmissible spongiform encephalopathies (TSEs) are a group of infectious neurodegenerative diseases which include Creutzfeldt–Jakob disease (CJD) and Gerstmann– Straussler–Scheinker syndrome (GSS) in humans, scrapie in sheep, and bovine spongiform encephalopathy (BSE) in cattle. During disease, cellular PrP converts from its normal, protease-sensitive α-helical form (PrPC) to a predominantly β-sheet conformation (PrPSc), which is detergent insoluble and partially resistant to protease digestion (Prusiner, 1996). The prion hypothesis proposes that this disease-associated isoform of PrP (PrPSc) constitutes infectivity (Prusiner, 1982) and acts as a seed or a template in the conversion of host PrPC to PrPSc (Jarrett and Lansbury, 1993; Prusiner, 1996; Weissmann, 1996). Alternatively, it has been argued that while PrPC may act as a receptor for the infectious agent, the conversion of PrPC to PrPSc is secondary to the infectious process and the TSE agent remains to be identified (reviewed in Chesebro, 1999).
Whatever the molecular nature of the infectious agent, it is clear that the PrP gene of the host has a major influence over the outcome of TSE disease. Polymorphisms in the PrP gene have been shown to alter incubation time and TSE susceptibility in mice (Moore et al., 1998), sheep (Goldmann et al., 1994) and man (Palmer et al., 1991; Goldfarb et al., 1992). These polymorphisms may control the rate of replication of the infectious agent (Dickinson and Outram, 1979; Prusiner, 1999), but the mechanism by which this is achieved is not known. In addition, mutations in the human PrP gene have been linked with ‘familial’ forms of TSE. It has been proposed that these mutations lead to a destabilization of the PrPC isoform and favour the formation of a disease-associated β-sheet conformation (Prusiner and Scott, 1997). If these mutations do indeed cause spontaneous TSE disease, as has been suggested by mice overexpressing a murine Prnp gene carrying the 101L mutation (Hsiao et al., 1990, 1994; Telling et al., 1996a), it would provide powerful evidence that the PrP protein in some conformation constitutes infectivity in these diseases.
Transgenic mice that over- and under-express different PrP genes have demonstrated that the amount of PrP expressed in the brain influences the incubation time of disease (Scott et al., 1989; Westaway et al., 1991; Bueler et al., 1993; Manson et al., 1994). In addition, changing the species of PrP expressed can alter the susceptibility of the mice to different strains of agent (Scott et al., 1989, 1999). While it is clear that host PrP influences the outcome of TSE disease, it is not yet clear how this is achieved. The key to understanding the infectious process of these diseases may, therefore, lie in understanding the mechanisms by which host PrP can modulate the TSE disease phenotype.
It has been proposed that efficient transmission of TSE disease requires similar primary amino acid sequence in the PrP of both the donor and the recipient of infectivity, allowing the ‘incoming’ PrPSc to interact specifically with and convert the host’s PrPC into the disease-associated form. In this scenario, any sequence differences between PrPC and PrPSc at the interacting sites would be expected to reduce the efficiency of this conversion reaction (Prusiner et al., 1990; Scott et al., 1993; Cohen and Prusiner, 1999). Such sequence differences have been proposed to be the cause of the barrier to TSE transmission between species (Prusiner et al., 1990; Scott et al., 1992), resulting in prolonged incubation times when infectivity passes from one species to another.
This prerequisite for sequence homology between exogenous and endogenous PrP molecules has been demonstrated in transgenic mice expressing the hamster PrP gene, which were found to be more susceptible to hamster and less susceptible to murine strains of scrapie than wild-type mice (Scott et al., 1989). This requirement for PrP amino acid identity has also been supported by in vitro experiments in which PrPC is converted into a protease-resistant form (PrP-res) by PrPSc. The highest efficiency of conversion occurs between homologous PrPC and PrPSc molecules (Kocisko et al., 1994). In these in vitro assays, non-homologous PrPC molecules will bind to PrPSc from other species, but they are not converted to a protease-resistant form, and appear to block the conversion of any homologous PrPC present (Horiuchi et al., 2000). The efficiency of conversion in these assays has been shown to correlate with PrP amino acid sequence specificity and thus it has been suggested that this accounts for species specificity in TSE transmissions.
Despite the results obtained from in vitro conversion experiments, PrP sequence identity does not always lead to increased susceptibility to TSE agents in vivo. Transgenic mice overexpressing human PrP are less susceptible to human variant CJD (vCJD) than wild-type mice (Hill et al., 1997). Moreover, TSE transmissibility has previously been demonstrated in the presence of heterologous PrP molecules with experimental transmissions of BSE to both wild-type mice (Bruce et al., 1994) and sheep (Goldmann et al., 1994), and transmission of vCJD, BSE and sheep scrapie to transgenic mice expressing only bovine PrP (Scott et al., 1999). These findings suggest that factors other than PrP sequence identity alone must underlie transmission of TSE disease.
The definition of strains of TSE agents has been suggested to be based on the structural conformation of PrP (Bessen and Marsh, 1992; Telling et al., 1996b; Caughey et al., 1998; Safar et al., 1998). It is possible, therefore, that the secondary and tertiary structure, rather than the primary structure, of host PrP may be important in determining the transmissibility of an agent in a specific host. One region of PrP that appears to be involved in modulating TSE disease is the N-terminal region, which has been described by NMR analysis as a ‘flexible tail’ with no definitive structure (Donne et al., 1997; Hornemann et al., 1997; Zahn et al., 2000). Mutations in this region of PrP do appear to have a strong influence on the disease process. Mutations at amino acids 102 (Hsiao et al., 1989), 105 (Kitamoto et al., 1993) and 117 (Doh-ura et al., 1989) are associated with familial TSE disease in man, and transgenic mouse studies have shown that mutations in this region can alter the incubation time of disease (Moore et al., 1998; Manson et al., 1999). In one transgenic model, a proline to leucine mutation at amino acid 101 in the endogenous murine PrP protein alters the incubation time of two strains of TSE, decreasing the incubation time of P102L GSS and increasing that of ME7 (Manson et al., 1999). Recent reports have also shown that overexpression of a mutant murine PrP molecule containing methionine at amino acids 108 and 111 causes prolonged incubation times with four murine strains of TSE (Supattapone et al., 2001). The effect of these mutations on incubation time may reflect the lack of either sequence or structural identity between host and agent PrP. Such incompatibility may affect the conversion efficiency of PrPC to PrPSc and result in prolonged incubation times.
To examine further the role of this ‘flexible’ N-terminal region of PrP in determining the outcome of TSE disease, we have investigated the effect of the proline to leucine mutation at amino acid 101 in murine PrP on incubation time of disease, targeting of pathology, the production of PrPSc and the susceptibility of the host to infectivity from four different species. A comparison of the incubation times and pathological lesions in 101PP and 101LL mice demonstrated that the 101L mutation in PrP has a profound effect on incubation time, producing surprising and dramatic alterations across three species barriers in a strain-specific manner. In addition, this single point mutation in PrP can dramatically alter the targeting of individual strains, thus demonstrating a major involvement of host PrP in the neuronal targeting of TSE pathology in the central nervous system (CNS).
Results
The incubation time of the hamster 263K strain in mice is reduced by the 101L mutation
Following intracerebral inoculation with 263K, all four 101LL mice were culled with TSE disease between 356 and 383 days, with a mean incubation time of 374 ± 6 days (Table I). None of the 101PL mice showed clinical or pathological signs of TSE disease. One of four 101PP mice was culled with clinical TSE disease at 707 days, showing vacuolar pathology in the brain but no detectable PrPSc by western analysis. The transmission of 263K to inbred strains of non-transgenic mice has been observed previously within our laboratory, with clinical disease usually occurring in only a small number of mice after an incubation period of between 700 and 800 days. This observed reduction in incubation time in 101LL mice infected with 263K has been confirmed in a subsequent experiment using a different isolate of 263K. In this experiment, 14 out of 16 101LL mice were culled with clinical and pathologically confirmed TSE disease between 373 and 559 days post-infection. Two animals culled at 324 and 554 days on welfare grounds were found to be pathologically negative. No clinical or pathological signs of TSE disease have yet been detected in 101PL or 101PP mice after 600 days (Table I). Thus, despite the PrP sequence differences between species (hamster PrP differs from mouse PrP at 12 amino acid residues between 23 and 231; Figure 1) and at amino acid 101 between donor (hamster 101P) and recipient (mouse 101L), the transmission of 263K in 101LL mice occurs with dramatically shorter incubation times than in wild-type 129/Ola mice.
Table I. Susceptibility of P101L mice to TSE infectivity from different species.
| Inoculum (10–2 dilution) | PrP genotype of source animal | Mouse straina | TSE diseaseb | Incubation time (days ± SEM) |
|---|---|---|---|---|
| Human vCJD | 102PP | 101PP | 24/24 | 373 ± 8 |
| 101PL | 14/20 | 450 ± 10 | ||
| 101LL | 8/18 | 556 ± 12 | ||
| Hamster 263K (#1) | 101PP | 101PP | 1/4 | 707 |
| 101PL | 0/3 | >570c | ||
| 101LL | 4/4 | 374 ± 6 | ||
| Hamster 263K (#2) | 101PP | 101PP | 0/20 | >600d |
| 101PL | 0/20 | >600d | ||
| 101LL | 14/16 | 464 ± 18 | ||
| Sheep SSBP-1 | 105PP | 101PP | 0/18 | >400d |
| 101LL | 14/16 | 346 ± 5 | ||
| Mouse 22A | 101PP | 101PP | 18/18 | 493 ± 6 |
| 101PL | 0/18 | >716c | ||
| 101LL | 9/16 | 527 ± 28 | ||
| VM | 6/6 | 193 ± 2 |
The PrP genotype of the source animal at amino acid 102 or 101 is shown.
aAll 101 mice are Prnpa, VM mice are Prnpb.
bNumber with TSE disease/total inoculated.
cIncubation time in excess of last death in group.
dExperiments still under way.

Fig. 1. Sequence differences between mouse PrP 23–231 and human, hamster and sheep PrP. Differences in the signal peptide and glycosylphosphatidylinositol anchor sequence are not shown, as these are known to be species specific. Numbering is with respect to murine PrP.
‘Adaptation’ of 101LL/263K on subsequent passage in 101LL mice
Brains of 101LL mice infected with 263K were subpassaged into 101PP, 101LL and 101PL mice (Table II). Each of the four 101LL mice that had succumbed to TSE disease in the first pass of 263K was subpassaged separately (labelled a–d in Table II). Samples from all four brains transmitted to 101LL mice with a further reduction in incubation time compared with primary passage. Three of the four subpassages resulted in transmission to all three genotypes of mice [Table II, 101LL/263K(a–c)], with the shortest incubation periods being observed in the 101LL mice (isolate 1). However, passage of the fourth 101LL brain infected with 263K resulted in transmission to all 101LL and 101PL mice, but not to 101PP mice (Table II, isolate 2). Vacuolar targeting in the 101LL mice infected with isolate 2 was different to that in those infected with isolate 1 and therefore different lesion profiles were obtained for the two isolates (Figure 2A). The two distinct patterns of vacuolar targeting indicate that two different TSE isolates were derived during the primary passage of 263K in 101LL mice. Despite the difference in lesion profiles, the two isolates displayed no difference in size or ratio of PrP glycoforms by western analysis (data not shown). We are currently investigating whether two strains were present in the primary inoculum or whether different strains have emerged on passage through the 101LL mice. Analysis of incubation times and lesion profiles has determined that neither of the isolates resembles mouse scrapie strains 139A and 79A, which were originally identified early in the passage history of 263K (Kimberlin et al., 1989). The possibility that the two strains resulted from contamination of 263K with a second strain of agent in the laboratory has been considered, but thought to be most unlikely, since the only source of infectivity that is known to produce a similar disease phenotype is brain homogenate from a 101LL mouse infected with GSS, and experiments with this source were conducted several years before those with 263K. The 101L mutation has therefore permitted the transmission of 263K to a mouse, a species that does not normally succumb to disease with this strain of agent. Once in the mouse, the 263K strain can undergo a further divergent ‘adaptation’ upon mouse-to-mouse passage, which is also affected by the 101L mutation.
Table II. Incubation times on subpassage of 101LL/263K from four mice with clinical and pathological signs of TSE disease.
| Inoculum (10–2 dilution) | Isolate typea | Mouse strain | TSE diseaseb | Incubation period (days ± SEM) |
|---|---|---|---|---|
| 101LL/263K (a) | 1 | 101PP | 10/10 | 262 ± 3 |
| 101PL | 8/8 | 251 ± 15 | ||
| 101LL | 7/7 | 131 ± 3 | ||
| 101LL/263K (b) | 1 | 101PP | 10/10 | 269 ± 8 |
| 101PL | 16/16 | 208 ± 2 | ||
| 101LL | 11/12 | 113 ± 1 | ||
| 101LL/263K (c) | 1 | 101PP | 10/10 | 263 ± 4 |
| 101LL | 14/14 | 161 ± 3 | ||
| 101LL/263K (d) | 2 | 101PP | 0/10 | >520c |
| 101PL | 11/11 | 302 ± 4 | ||
| 101LL | 9/9 | 129 ± 2 |
101LL, 101PL and 101PP mice were inoculated intracerebrally with 1% brain homogenate from 263K-infected 101LL mice which had been killed at the terminal stage of TSE disease.
aIsolate type based on lesion profile and incubation times in 101LL, 101PL and 101PP mice.
bNumber with TSE disease/total inoculated.
cIncubation time in excess of last death in group.

Fig. 2. The extent of the vacuolar change in the brain was assessed semi-quantitatively in nine areas of grey matter and three areas of white matter by lesion profiling as described (Fraser and Dickinson, 1967). Animals were scored on a scale of 0–5 in each specific area, and mean scores (calculated from a minimum of six animals) are shown graphically (error bars ± SEM). Grey matter scoring areas: 1, dorsal medulla; 2, cerebellar cortex; 3, superior colliculus; 4, hypothalamus; 5, medial thalamus; 6, hippocampus; 7, septum; 8, cerebral cortex; 9, forebrain cerebral cortex. White matter scoring areas: 1*, cerebellar white matter; 2*, midbrain white matter; 3*, cerebral peduncle. (A) Mice inoculated with brain homogenate from 101LL/263K mice. Blue diamonds, 101LL/263K(d); red squares, 101LL/263K(a) (see Table II). (B) Mice inoculated with brain homogenate from a patient with confirmed vCJD. Blue diamonds, 101PP mice; pink squares, 101PL mice; green circles, 101LL mice.
Reduction in incubation time across a second species barrier due to the 101L mutation
The surprising finding that the P101L mutation in murine PrP could reduce the incubation time of a TSE agent from another species was demonstrated more dramatically using a sheep-passaged strain of scrapie. A panel of 101LL and 101PP mice was inoculated by both intracerebral and intraperitoneal routes with SSBP/1 (#24), which is a pool of natural sheep scrapie that has been passaged through sheep 24 times. This source of infectivity was derived from and passaged in sheep encoding amino acid 105P in PrP, which is equivalent to 101P in murine PrP. In the 101LL group, 14/16 mice succumbed to TSE disease in 346 ± 5 days. At the time of writing (400 days), none of the 101PP mice inoculated with SSBP/1 has succumbed to clinical and pathological TSE disease (Table I). Previous experiments, in which C57BL and RO mice (both of which are 101PP) were inoculated with SSBP/1 using combined intraperitoneal and intracerebral routes of challenge, have produced incubation times of 586 ± 12 and 500 ± 15 days, respectively, but with only 40% of each group ever succumbing to disease (J.Foster, unpublished data). Incubation times and susceptibilities similar to those described above for the C57BL and RO mice would, therefore, be expected in the wild-type 101PP mice used in the current experiments. Thus, as with the 263K challenge, the 101L mutation dramatically reduced the incubation time of SSBP/1 in mice, despite both species-specific differences (sheep PrP differs from mouse at 22 amino acid residues between 23 and 231; Figure 1) and amino acid 101 incompatibility between donor and recipient PrP molecules.
The 101L mutation prolongs incubation time for the murine 22A strain of scrapie
A panel of 101LL, 101PL, 101PP and wild-type (VM) mice was inoculated with the murine 22A strain of scrapie, which had previously been passaged in mice carrying the Prnpb allele. 101PP (129Ola) mice carrying the Prnpa allele all succumbed to TSE disease in 493 ± 6 days, whereas wild-type mice (VM) carrying the Prnpb allele were culled with TSE disease at 193 ± 2 days (Table I). These incubation times were as expected for Prnpa and Prnpb alleles with 22A scrapie. Nine out of sixteen 101LL mice (Prnpa background) were culled with clinical and pathological TSE disease between 400 and 650 days, with an average incubation time of 527 ± 28 days, which is slightly prolonged when compared with the 101PP mice. Seven clinically negative 101LL mice were culled due to intercurrent illness between 455 and 655 days, but only 2 (killed at 503 and 665 days) showed signs of TSE pathology in the brain. The 101PL mice were all culled due to intercurrent illness or senility between 500 and 716 days post-inoculation. Only one animal in this group showed a small amount of TSE vacuolation at 716 days.
The 101L mutation is, therefore, able to extend the incubation time of disease with the murine 22A strain of TSE. The identification of TSE pathology in one of the 101PL mice killed at 716 days due to senility suggests that the incubation time of disease is extended beyond the lifespan of the mouse in the presence of both 101P and 101L alleles. This observation is similar to the overdominance described in Prna × Prnpb heterozygotes infected with the same strain of agent (Dickinson and Meikle, 1971).
The 101L mutation can also prolong incubation time of disease across a species barrier
A panel of 101PP, 101PL and 101LL mice was inoculated intracerebrally with brain homogenate from a patient with confirmed vCJD (CJD Surveillance Unit, Edinburgh, UK). All 24 101PP mice were culled with TSE disease between 322 and 497 days post-inoculation, with a mean incubation time of 373 ± 8 days (Table I). Fourteen out of twenty 101PL mice were culled between 392 and 511 days with clinical and pathological signs of TSE disease, and a mean incubation time of 450 ± 10 days. The six remaining 101PL animals, which died or were culled between 412 and 467 days, showed no evidence of TSE pathology on analysis of brain tissue. Clinical and pathological signs of TSE disease were not observed in 101LL mice until after 500 days, with an average incubation time of 556 ± 12 days. Eight 101LL mice, which were culled between 504 and 597 days, displayed TSE disease pathology and two 101LL mice remained pathologically negative at 546 and 574 days post-inoculation (Table I).
The 101L mutation in the murine PrP gene therefore extends the incubation time of vCJD in mice by at least 130 days. This prolonged incubation time demonstrates that incubation time is defined by the strain of agent rather than by any inherent instability of the host 101L PrP protein, which would be expected to lead to a consistent shortening of incubation time irrespective of the strain of agent.
The 101L mutation alters targeting of pathology with vCJD
Despite the difference of >70 days between the mean incubation times of 101PP and 101PL mice infected with vCJD, the same pattern of PrP deposition and vacuolation was observed in the brains of both genotypes. However, the targeting of pathological lesions in the 101LL mice was altered from that observed in the 101PP and 101PL mice, in both the lesion profile (Figure 2B) and the distribution of PrP deposition (Figure 3). Striking differences were observed in the hippocampal regions, where PrP deposition was evident throughout the entire hippocampus of the 101LL mice, in contrast to the CA2 targeting seen in 101PP and 101PL mice (Figure 3). Although the distribution of vacuolar and PrP pathology was similar in all the 101LL mice with clinical TSE disease, the degree of vacuolation and PrP deposition was variable. This variability in PrP deposition did not show any correlation with incubation time of disease since two animals culled with clinical TSE disease at 504 and 525 days showed a higher degree of vacuolation and much heavier PrP deposition than animals culled after 525 days (Figure 3). Western blot analysis confirmed that the 101LL mouse showing high levels of deposition contained far more protease-resistant PrP than 101PP, 101PL and low deposition 101LL mice (Figure 5). Although glycoform ratios are difficult to assess from this blot, the unglycosylated band in the 101LL mice does appear to be slightly larger than that seen in both 101PP and 101PL mice. We have been unable to resolve this further due to the limited availability of tissue; however, this property will be analysed further on subsequent passage of the material, which is now under way. Thus, TSE pathology in the CNS is not determined solely by the strain of agent, since a single amino acid alteration in host PrP can have a pronounced effect on both the incubation time and the targeting of the pathological lesions in the brain.

Fig. 3. PrP detected by immunocytochemistry in P101L mice inoculated with vCJD. Sections from the brains of P101L transgenic mice showing PrP deposition in the hippocampus. Sections probed with monoclonal antibody 6H4. (A) 101PP mouse; (B) 101PL mouse; (C) 101LL mouse with high levels of deposition killed at 525 days post-infection; (D) 101LL mouse with low levels of deposition killed at 560 days post-infection. Bars, 100 µm.
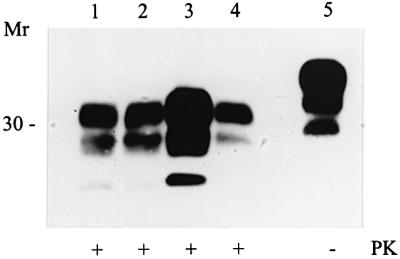
Fig. 5. Western analysis of vCJD brain homogenates for PrPSc using monoclonal antibody 8H4. Lane 1, 101PP; lane 2, 101PL; lane 3, 101LL with high levels of PrP deposition; lane 4, 101LL with low levels of deposition. All samples were digested with proteinase K at 20 µg/ml. Approximately 200 µg of brain homogenate were loaded per well on both gels.
PrP containing the 101L mutation can produce PrPSc during scrapie infection
It was difficult to detect PrPSc in the 101LL mice infected with 263K by either immunocytochemistry (data not shown) or western analysis using both monoclonal (8H4, Figure 4) and polyclonal antibodies. This result is similar to that observed on inoculation of 101LL mice with P102L GSS (Manson et al., 1999). These data might suggest that PrP containing the 101L mutation is less able to produce PrPSc than the wild-type 101P PrP molecule. However, on primary passage with vCJD (Figure 5) and secondary passage of ME7 and P102L GSS in 101LL mice (Manson et al., 1999), PrPSc was readily detected. Thus, although the formation of PrPSc does not always appear to be a dominant feature of 101LL mice infected with TSE at primary passage, there is no inherent failure of the 101L PrP protein to form PrPSc. The ability of 101L PrP to form PrPSc during infection therefore appears to be determined by the strain of agent rather than by any properties of the mutant PrP molecule.
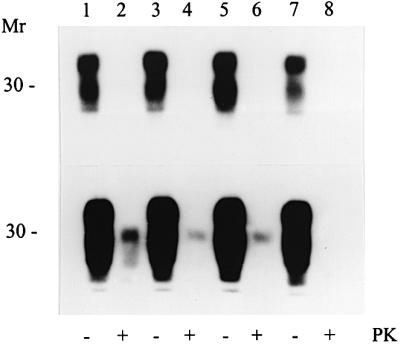
Fig. 4. Western blot analysis of 263K brain homogenates for PrPSc using monoclonal antibody 8H4. Lanes 1 and 2, 101LL/263K(d); lanes 3 and 4, 101LL/263K(a); lanes 5 and 6, 101LL/263K(b); lanes 7 and 8, 101LL/263K(c). Lanes 2, 4, 6 and 8 were digested with proteinase K at 20 µg/ml. Exposure times of 10 s (top) and 10 min (bottom) are shown to demonstrate the extremely low levels of PrPSc.
Discussion
The data presented have shown that the specific alteration of amino acid 101 from proline to leucine in murine PrP has a major influence on the incubation time of disease across three species barriers. The remarkable finding that this single point mutation can allow TSE disease that originated in another species to transmit readily to mice indicates the importance of this region of PrP in TSE transmissibility, and also suggests a major role in agent replication. Although alterations at amino acids 108 (leucine to methionine) and 111 (valine to methionine) have been shown to be associated with a non-specific prolongation of TSE disease in mice (Supattapone et al., 2001), we have shown here that the 101L mutation can both increase or decrease incubation time, depending on the strain of agent inoculated. The mechanism by which these alterations in incubation time occur does not rely on sequence identity between donor and recipient PrP.
If primary sequence compatibility between donor and recipient PrP does not underlie transmissibility, it may be, as suggested by others, that secondary or tertiary structural identity of the two PrP molecules is the key component to transmissibility of a TSE agent (Telling et al., 1996b; Caughey et al., 1998; Safar et al., 1998). Spectroscopic data have predicted that amino acids 23–124 of PrPC form an unstructured ‘flexible tail’ (Zahn et al., 2000), but it has been shown that this N-terminal region may possess some degree of secondary structure when present in a membrane-rich environment (Morillas et al., 1999). Specific deletions in this area can prevent the acquisition of proteinase K resistance (Muramoto et al., 1996), and reduce the efficiency of disease transmission (Flechsig et al., 2000; Supattapone et al., 2001). Antibody epitopes in the ‘flexible tail’ become obscured after conversion to PrPSc (Kascsak et al., 1987; Peretz et al., 1997; Kanyo et al., 1999), suggesting that a conformational change occurs in this region during disease. Thus, while it seems likely that structural conformations and alterations in this region are important components of the infectious process, it is not clear whether structural identity in donor and recipient PrP is the key to transmissibility in the 101LL mice infected with 263K and SSBP/1. Whether the introduction of leucine at amino acid 101 in murine PrP can induce a conformation with more identity to sheep and hamster PrP than wild-type murine PrP will be addressed in structural studies. It may be, however, that this region of host PrP does not enhance transmissibility through interactions with structurally homologous PrP molecules, but through interaction with other regions of host PrP, or with molecules other than PrP. The introduction of other specific mutations into this region of PrP has been undertaken to investigate this further.
The barrier to transmission of TSE disease from one species to another is thought to reside in the differences in PrP sequence between species (Prusiner et al., 1990; Scott et al., 1992; Kocisko et al., 1995; Raymond et al., 2000). NMR structural analysis of recombinant human (Zahn et al., 2000), hamster (Donne et al., 1997) and mouse (Riek et al., 1997) PrP has identified specific sequence differences involving α-helix 3 and the nearby loop of residues 167–171, which have been proposed to affect species specificity by altering the binding efficiency of protein X. This protein is hypothesized to bind to the C-terminus of PrP (Telling et al., 1995; Kaneko et al., 1997) and catalyse the conversion of PrPC to PrPSc. However, here we have demonstrated that a single mutation in the N-terminal flexible region of PrP can permit a strain of agent to transmit efficiently across a species barrier to animals that would not normally readily succumb to disease. This transmission occurs despite the fact that no alterations have been made to the region of PrP thought to be involved in species specificity and binding protein X. One of the keys to understanding the susceptibility of a host to TSE infection may, therefore, reside in understanding interactions at the N-terminal region of host PrP. The effect of the 101L mutation described here may be due to a direct interaction between the agent and this region of PrP. Alternatively, the mutation may alter interactions between the N-terminus and other regions of the PrP molecule, and thus alter agent binding or replication at these sites. We have now targeted mutations into different structural regions of murine PrP both separately, to assess the effect of each region, and together, to assess the interaction between different regions of host PrP in the infectious process.
The P102L mutation is associated with familial TSE disease in humans. The prion hypothesis predicts that this and other mutations in PrP destabilize the molecule and facilitate conversion to PrPSc (Cohen et al., 1994; Huang et al., 1994; Harrison et al., 1997). The 101L mutation in mice might therefore be expected to be unstable and convert to PrPSc more readily than wild-type murine PrP, leading to a general reduction in incubation time of disease with all strains. However, this is not the case. Indeed, with the vCJD and 22A strains described here and ME7 described previously (Manson et al., 1999), we have demonstrated much longer incubation times in mice carrying this mutation than in wild-type mice. The alterations in incubation time caused by the 101L mutation therefore occur in a strain-specific manner, and are not due to any inherent instability of the PrP molecule carrying the 101L mutation. Furthermore, results presented here and previously (Manson et al., 1999) demonstrate that the 101L PrP protein does not show an inherent ability to form PrPSc in the infected mice, since the detection of PrPSc in the terminal stages of disease in 101LL mice infected with some TSE agents has proved difficult.
The major alteration in the disease phenotype of vCJD in mice carrying the 101L mutation is an important finding of this work. Changes in the targeting of both vacuolation and PrP deposition, and possibly also in glycoform profile, were identified in the brains of the 101LL mice when compared with wild-type (101PP) mice. These differences cannot be due to differential expression of the 101L transgene compared with wild-type PrP since the 101L mutation has been introduced directly into the endogenous murine Prnp gene. These results demonstrate that single amino acid substitutions in host PrP can have a profound effect on the targeting of TSE strains in the CNS, and can alter the disease pathology obtained with specific TSE agents. No cases of vCJD have yet been identified in individuals carrying PrP with 129V rather than 129M. This has been a cause for concern in studying the vCJD epidemic. It has not been possible to assess whether such individuals may present with a different disease phenotype to those carrying 129M. The data presented here are the first indication that differences in host PrP can indeed alter the disease profile of vCJD. Although these data are associated with polymorphisms in PrP that may not be directly relevant to vCJD presentation in man, they do demonstrate the potential for different PrP alleles to determine different phenotypes and incubation times of vCJD.
The original aim of introducing the 101L mutation into murine PrP by gene targeting was to study the ability of this mutation to produce an unstable PrP molecule, which might lead to a spontaneous TSE disease. To date we have found no evidence for such a spontaneous disease in these mice, although we continue to investigate for CNS alterations that may be consistent with neurodegenerative disease. In addition we have shown no inherent ability of the 101L PrP protein to form PrPSc; indeed, PrPSc has proved difficult to detect in 101LL mice infected with some strains of TSE. We have, however, demonstrated a remarkable ability for the 101L mutation to alter unpredictably the incubation time of disease with several different TSE agents from other species. We have therefore been unable to substantiate the existence of infectious PrP protein, but have provided evidence of altered incubation times of TSE disease in mice carrying the 101L mutation in their PrP protein.
Materials and methods
Preparation of inoculum
Inocula were prepared from human brain tissue from a patient with pathologically confirmed vCJD, a hamster with 263K scrapie, 263K-inoculated 101LL mice showing clinical signs of disease, sheep brain infected with SSBP1, and a VM mouse inoculated with 22A murine scrapie. A 1% homogenate of each sample was prepared in normal saline prior to use as an inoculum. P101L transgenic mice (Manson et al., 1999) were genotyped and grouped prior to intracerebral inoculation with 20 µl of inoculum under halothane anaesthesia.
Scoring of clinical TSE disease
The presence of clinical TSE disease was assessed as described (Dickinson et al., 1968). Incubation times were calculated as the interval between inoculation and cull due to terminal TSE disease. Mice were killed by cervical dislocation at the terminal stage of disease, at termination of the experiment (between 600 and 700 days), or due to intercurrent illness. Half-brains were fixed in 10% formol saline for 48 h, followed by decontamination in 98% formic acid for 1 h. The remaining half-brain was frozen at –70°C for western analysis. Fixed brain tissue was dehydrated in alcohol and impregnated in wax during a 7 h automated processing cycle. Sections were cut coronally at four levels and mounted on Superfrost slides.
Lesion profiles
Sections were haematoxylin & eosin stained and scored for vacuolar degeneration on a scale of 0–5 in nine standard grey matter areas and three standard white matter areas as described previously (Fraser and Dickinson, 1967).
Immunocytochemical analysis
Sections were immunostained using standard procedures. Briefly, sections were blocked with normal rabbit serum and probed overnight with monoclonal antibody 6H4 (Prionics) at a dilution of 1:1000. A parallel panel of sections was also probed with normal mouse serum as a control. Antibody binding was detected with biotinylated rabbit anti-mouse secondary antibody (Jacksons) and the Vectastain Elite ABC Kit (Vector Laboratories). Reaction products were visualized with diaminobenzidine (DAB), and sections were lightly counterstained with haematoxylin.
Western analysis
For the detection of PrPSc in transgenic and wild-type mice, 10% (w/v) homogenates of frozen brain tissue were prepared in NP-40 buffer [0.5% (v/v) NP-40, 0.5% (w/v) sodium deoxycholate, 150 mM NaCl, 50 mM Tris–HCl pH 7.5]. Homogenates were centrifuged at 11 000 g for 15 min at 10°C to remove cellular debris. Supernatant samples (100 µl) were incubated with or without proteinase K (20 µg/ml) for 1 h at 37°C, and the reaction terminated by the addition of phenylmethylsulfonyl fluoride (PMSF). Samples were incubated at 90°C for 30 min in SDS–PAGE sample buffer, and ∼200 µg of homogenate were separated on either 10–15% gradient acrylamide gels using the Neville buffer system (Neville, 1971) or 12% Novex Tris–glycine acrylamide gels (Invitrogen). Proteins were transferred onto PVDF membrane by electroblotting, and incubated for 2 h at room temperature with monoclonal antibody 8H4 (Zanusso et al., 1998) diluted 1:20 000. Proteins were visualized with horseradish peroxidase (HRP)-conjugated rabbit anti-mouse secondary antibody (Jacksons) and a chemiluminescence detection kit (Roche Diagnostics). Membranes were exposed to X-ray film for periods ranging from 10 s to 10 min.
Genotyping
All P101L transgenic mice were genotyped after termination of experiments to confirm genotype. A 765 bp fragment containing the entire Prnp open reading frame was generated using a 5′ primer (5′-ATGGCGAACCTTGGCTACTGGCTG-3′; position 107–130; DDBJ/EMBL/GenBank accession No. M18070) and a 3′ primer (5′-TCATCCCACGATCAGGAAGATGAG-3′; position 871–848; DDBJ/EMBL/GenBank accession No. M18070). Cycle conditions were 94°C for 3 min, followed by 30 cycles of 30 s at 94°C, 30 s at 62°C and 1 min at 72°C. This was followed by a final 10 min at 72°C (Biometra Triblock). The presence or absence of a DdeI site within the PCR product provided a marker for the codon 101P-L alteration.
Acknowledgments
Acknowledgements
The authors would like to thank M.Bruce and J.Almond for discussions concerning the 101L mice, F.Robertson and E.Murdoch for care and scoring of the animals, A.Boyle and W.-G.Liu for lesion profile data, E.Gall for technical assistance, and Christopher Bostock and Jan Fraser for review and comment on the manuscript. This work was supported by a grant from BBSRC.
References
- Bessen R.A. and Marsh,R.F. (1992) Biochemical and physical properties of the prion protein from 2 strains of the transmissible mink encephalopathy agent. J. Virol., 66, 2096–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce M., Chree,A., McConnell,I., Foster,J., Pearson,G. and Fraser,H. (1994) Transmission of bovine spongiform encephalopathy and scrapie to mice—strain variation and the species barrier. Philos. Trans. R. Soc. Lond. B Biol. Sci., 343, 405–411. [DOI] [PubMed] [Google Scholar]
- Bueler H., Aguzzi,A., Sailer,A., Greiner,R.A., Autenried,P., Aguet,M. and Weissmann,C. (1993) Mice devoid of PrP are resistant to scrapie. Cell, 73, 1339–1347. [DOI] [PubMed] [Google Scholar]
- Caughey B., Raymond,G.J. and Bessen,R.A. (1998) Strain-dependent differences in β-sheet conformations of abnormal prion protein. J. Biol. Chem., 273, 32230–32235. [DOI] [PubMed] [Google Scholar]
- Chesebro B. (1999) Prion protein and the transmissible spongiform encephalopathy diseases. Neuron, 24, 503–506. [DOI] [PubMed] [Google Scholar]
- Cohen F.E. and Prusiner,S.B. (1999) Structural studies of prion proteins. In Prusiner,S.B. (ed.), Prion Biology and Diseases. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Cohen F.E., Pan,K.M., Huang,Z., Baldwin,M., Fletterick,R.J. and Prusiner,S.B. (1994) Structural clues to prion replication. Science, 264, 530–531. [DOI] [PubMed] [Google Scholar]
- Dickinson A.G. and Meikle,V.M. (1971) Host-genotype and agent effects in scrapie incubation: change in allelic interaction with different strains of agent. Mol. Gen. Genet., 112, 73–79. [DOI] [PubMed] [Google Scholar]
- Dickinson A.G. and Outram,G.W. (1979) The scrapie replication-site hypothesis and its implications for pathogenesis. In Prusiner,S.B. and Hadlow,W.J. (eds), Slow Transmissible Diseases of the Central Nervous System. Vol. 2. Academic Press, New York, NY, pp. 13–32.
- Dickinson A.G., Meikle,V.M. and Fraser,H. (1968) Identification of a gene which controls the incubation period of some strains of scrapie agent in mice. J. Comp. Pathol., 78, 293–299. [DOI] [PubMed] [Google Scholar]
- Doh-ura K., Tateishi,J., Sasaki,H., Kitamoto,T. and Sakaki,Y. (1989) Pro-leu change at position-102 of prion protein is the most common but not the sole mutation related to Gerstmann–Straussler syndrome. Biochem. Biophys. Res. Commun., 163, 974–979. [DOI] [PubMed] [Google Scholar]
- Donne D.G., Viles,J.H., Groth,D., Mehlhorn,I., James,T.L., Cohen,F.E., Prusiner,S.B., Wright,P.E. and Dyson,H.J. (1997) Structure of the recombinant full-length hamster prion protein PrP(29–231): the N-terminus is highly flexible. Proc. Natl Acad. Sci. USA, 94, 13452–13457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flechsig E., Shmerling,D., Hegyi,I., Raeber,A.J., Fischer,M., Cozzio,A., von Mering,C., Aguzzi,A. and Weissmann,C. (2000) Prion protein devoid of the octapeptide repeat region restores susceptibility to scrapie in PrP knockout mice. Neuron, 27, 399–408. [DOI] [PubMed] [Google Scholar]
- Fraser H. and Dickinson,A.G. (1967) Distribution of experimentally induced scrapie lesions in the brain. Nature, 216, 1310–1311. [DOI] [PubMed] [Google Scholar]
- Goldfarb L.G. et al. (1992) Fatal familial insomnia and familial Creutzfeldt–Jakob disease: disease phenotype determined by a DNA polymorphism. Science, 258, 806–808. [DOI] [PubMed] [Google Scholar]
- Goldmann W., Hunter,N., Smith,G., Foster,J. and Hope,J. (1994) Prp genotype and agent effects in scrapie—change in allelic interaction with different isolates of agent in sheep, a natural host of scrapie. J. Gen. Virol., 75, 989–995. [DOI] [PubMed] [Google Scholar]
- Harrison P.M., Bamborough,P., Daggett,V., Prusiner,S.B. and Cohen,F.E. (1997) The prion folding problem. Curr. Opin. Struct. Biol., 7, 53–59. [DOI] [PubMed] [Google Scholar]
- Hill A.F., Desbruslais,M., Joiner,S., Sidle,K.C.L., Gowland,I., Collinge,J., Doey,L.J. and Lantos,P. (1997) The same prion strain causes vCJD and BSE. Nature, 389, 448–450. [DOI] [PubMed] [Google Scholar]
- Horiuchi M., Priola,S.A., Chabry,J. and Caughey,B. (2000) Interactions between heterologous forms of prion protein: binding, inhibition of conversion, and species barriers. Proc. Natl Acad. Sci. USA, 97, 5836–5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornemann S., Korth,C., Oesch,B., Riek,R., Wider,G., Wuthrich,K. and Glockshuber,R. (1997) Recombinant full-length murine prion protein, mPrP(23–231): purification and spectroscopic characterization. FEBS Lett., 413, 277–281. [DOI] [PubMed] [Google Scholar]
- Hsiao K., Baker,H.F., Crow,T.J., Poulter,M., Owen,F., Terwilliger,J.D., Westaway,D., Ott,J. and Prusiner,S.B. (1989) Linkage of a prion protein missense variant to Gerstmann–Straussler syndrome. Nature, 338, 342–345. [DOI] [PubMed] [Google Scholar]
- Hsiao K.K., Scott,M., Foster,D., Groth,D.F., DeArmond,S.J. and Prusiner,S.B. (1990) Spontaneous neurodegeneration in transgenic mice with mutant prion protein. Science, 250, 1587–1590. [DOI] [PubMed] [Google Scholar]
- Hsiao K.K. et al. (1994) Serial transmission in rodents of neuro degeneration from transgenic mice expressing mutant prion protein. Proc. Natl Acad. Sci. USA, 91, 9126–9130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z.W., Gabriel,J.M., Baldwin,M.A., Fletterick,R.J., Prusiner,S.B. and Cohen,F.E. (1994) Proposed 3-dimensional structure for the cellular prior protein. Proc. Natl Acad. Sci. USA, 91, 7139–7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett J.T. and Lansbury,P.T.,Jr (1993) Seeding ‘one-dimensional crystallization’ of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell, 73, 1055–1058. [DOI] [PubMed] [Google Scholar]
- Kaneko K., Zulianello,L., Scott,M., Cooper,C.M., Wallace,A.C., James,T.L., Cohen,F.E. and Prusiner,S.B. (1997) Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc. Natl Acad. Sci. USA, 94, 10069–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanyo Z.F., Pan,K., Williamson,R.A., Burton,D.R., Prusiner,S.B., Fletterick,R.J. and Cohen,F.E. (1999) Antibody binding defines a structure for an epitope that participates in the PrPc to PrPSc conformational change. J. Mol. Biol., 293, 855–863. [DOI] [PubMed] [Google Scholar]
- Kascsak R.J., Rubenstein,R., Merz,P.A., Tonnademasi,M., Fersko,R., Carp,R.I., Wisniewski,H.M. and Diringer,H. (1987) Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J. Virol., 61, 3688–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimberlin R.H., Walker,C.A. and Fraser,H. (1989) The genomic identity of different strains of mouse scrapie is expressed in hamsters and preserved on reisolation in mice. J. Gen. Virol., 70, 2017–2025. [DOI] [PubMed] [Google Scholar]
- Kitamoto T., Amano,N., Terao,Y., Nakazato,Y., Isshiki,T., Mizutani,T. and Tateishi,J. (1993) A new inherited prion disease (prp-p105L mutation) showing spastic paraparesis. Ann. Neurol., 34, 808–813. [DOI] [PubMed] [Google Scholar]
- Kocisko D.A., Come,J.H., Priola,S.A., Chesebro,B., Raymond,G.J., Lansbury,P.T. and Caughey,B. (1994) Cell-free formation of protease-resistant prion protein. Nature, 370, 471–474. [DOI] [PubMed] [Google Scholar]
- Kocisko D., Priola,S.A., Raymond,G., Chesebro,B., Landsbury,P.T. and Caughey,B. (1995) Species specificity in the cell-free conversion of prion protein to protease-resistant forms: a model for the scrapie species barrier. Proc. Natl Acad. Sci. USA., 92, 3923–3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manson J.C., Clarke,A.R., McBride,P.A., McConnell,I. and Hope,J. (1994) Prp gene dosage determines the timing but not the final intensity or distribution of lesions in scrapie pathology. Neurodegeneration, 3, 331–340. [PubMed] [Google Scholar]
- Manson J.C. et al. (1999) A single amino acid alteration (101L) introduced into murine PrP dramatically alters incubation time of transmissible spongiform encephalopathy. EMBO J., 18, 6855–6864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore R.C., Hope,J., McBride,P.A., McConnell,I., Selfridge,J., Melton,D.W. and Manson,J.C. (1998) Mice with gene targetted prion protein alterations show that Prnp, Sinc and Prni are congruent. Nature Genet., 18, 118–125. [DOI] [PubMed] [Google Scholar]
- Morillas M., Swietnicki,W., Gambetti,P. and Surewicz,W.K. (1999) Membrane environment alters the conformational structure of the recombinant prion protein. J. Biol. Chem., 274, 36859–36865. [DOI] [PubMed] [Google Scholar]
- Muramoto T., Scott,M., Cohen,F.E. and Prusiner,S.B. (1996) Recombinant scrapie-like prion protein of 106 amino acids is soluble. Proc. Natl Acad. Sci. USA, 93, 15457–15462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neville D.M. (1971) Molecular weight determination of protein-dodecyl sulfate complexes by gel electrophoresis in a discontinuous buffer system. J. Biol. Chem., 246, 6328–6334. [PubMed] [Google Scholar]
- Palmer M.S., Dryden,A.J., Hughes,J.T. and Collinge,J. (1991) Homozygous prion protein genotype predisposes to sporadic Creutzfeldt–Jakob disease [published erratum appears in Nature, 352, 547]. Nature, 352, 340–342. [DOI] [PubMed] [Google Scholar]
- Peretz D. et al. (1997) A conformational transition at the N-terminus of the prion protein features in formation of the scrapie isoform. J. Mol. Biol., 273, 614–622. [DOI] [PubMed] [Google Scholar]
- Prusiner S.B. (1982) Novel proteinaceous infectious particles cause scrapie. Science, 216, 136–144. [DOI] [PubMed] [Google Scholar]
- Prusiner S.B. (1996) Molecular biology and pathogenesis of prion diseases. Trends Biochem. Sci., 21, 482–487. [DOI] [PubMed] [Google Scholar]
- Prusiner S.B. (1999) Transmission and replication of prions. In Prusiner,S.B. (ed.), Prion Biology and Diseases. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Prusiner S.B. and Scott,M.R. (1997) Genetics of prions. Annu. Rev. Genet., 31, 139–175. [DOI] [PubMed] [Google Scholar]
- Prusiner S.B. et al. (1990) Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell, 63, 673–686. [DOI] [PubMed] [Google Scholar]
- Raymond G.J. et al. (2000) Evidence of a molecular barrier limiting susceptibility of humans, cattle and sheep to chronic wasting disease. EMBO J., 19, 4425–4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riek R., Hornemann,S., Wider,G., Glockschuber,R. and Wuthrich,K. (1997) NMR characterization of the full-length recombinant murine prion protein, mPrP(23–231). FEBS Lett., 413, 282–288. [DOI] [PubMed] [Google Scholar]
- Safar J., Wille,H., Itrri,V., Groth,D., Serban,H., Torchia,M., Cohen,F.E. and Prusiner,S.B. (1998) Eight prion strains have PrPSc molecules with different conformations. Nature Med., 4, 1157–1165. [DOI] [PubMed] [Google Scholar]
- Scott M. et al. (1989) Transgenic mice expressing hamster prion protein produce species-specific scrapie infectivity and amyloid plaques. Cell, 59, 847–857. [DOI] [PubMed] [Google Scholar]
- Scott M.R., Kohler,R., Foster,D. and Prusiner,S.B. (1992) Chimeric prion protein expression in cultured cells and transgenic mice. Protein Sci., 1, 986–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott M., Groth,D., Foster,D., Torchia,M., Yang,S.L., DeArmond,S.J. and Prusiner,S.B. (1993) Propagation of prions with artificial properties in transgenic mice expressing chimeric PrP genes. Cell, 73, 979–988. [DOI] [PubMed] [Google Scholar]
- Scott M.R., Will,R., Ironside,J., Nguyen,H.O.B., Tremblay,P., DeArmond,S.J. and Prusiner,S.B. (1999) Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proc. Natl Acad. Sci. USA, 96, 15137–15142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supattapone S., Muramoto,T., Legname,G., Mehlhorn,I., Cohen,F.E., DeArmond,S.J., Prusiner,S.B. and Scott,M.R. (2001) Identification of two prion protein regions that modify scrapie incubation time. J. Virol., 75, 1408–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telling G.C., Scott,M., Mastrianni,J., Gabizon,R., Torchia,M., Cohen,F.E., Dearmond,S.J. and Prusiner,S.B. (1995) Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell, 83, 79–90. [DOI] [PubMed] [Google Scholar]
- Telling G.C., Haga,T., Torchia,M., Tremblay,P., DeArmond,S. and Prusiner,S. (1996a) Interactions between wild-type and mutant prion proteins modulate neurodegeneration in transgenic mice. Genes Dev., 10, 1736–1750. [DOI] [PubMed] [Google Scholar]
- Telling G.C. et al. (1996b) Evidence for the conformation of the pathological isoform of the prion protein enciphering and propagating prion diversity. Science, 274, 2079–2082. [DOI] [PubMed] [Google Scholar]
- Weissmann C. (1996) Molecular biology of transmissible spongiform encephalopathies. FEBS Lett., 389, 3–11. [DOI] [PubMed] [Google Scholar]
- Westaway D. et al. (1991) Paradoxical shortening of scrapie incubation times by expression of prion protein transgenes derived from long incubation period mice. Neuron, 7, 59–68. [DOI] [PubMed] [Google Scholar]
- Zahn R. et al. (2000) NMR solution structure of the human prion protein. Proc. Natl Acad. Sci. USA, 97, 145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanusso G. et al. (1998) Prion protein expression in different species: analysis with a panel of new mAbs. Proc. Natl Acad. Sci. USA, 95, 8812–8816. [DOI] [PMC free article] [PubMed] [Google Scholar]