

Abstract
The murine cytomegalovirus (MCMV) fcr-1 gene codes for a glycoprotein located at the surface of infected cells which strongly binds the Fc fragment of murine immunoglobulin G. To determine the biological significance of the fcr-1 gene during viral infection, we constructed MCMV fcr-1 deletion mutants and revertants. The fcr-1 gene was disrupted by insertion of the Escherichia coli lacZ gene. In another mutant, the marker gene was also deleted, by recombinase cre. As expected for its hypothetical role in immunoevasion, the infection of mice with fcr-1 deletion mutants resulted in significantly restricted replication in comparison with wild-type MCMV and revertant virus. In mutant mice lacking antibodies, however, the fcr-1 deletion mutants also replicated poorly. This demonstrated that the cell surface-expressed viral glycoprotein with FcR activity strongly modulates the virus-host interaction but that this biological function is not caused by the immunoglobulin binding property.
DNA viruses have developed numerous strategies to modulate or evade the immune system control of the host. Immune system modulators encoded within viral genomes include proteins that interrupt the complement cascade, act as cytokines or cytokine antagonists (34, 37, 40), inhibit the effector mechanisms mediated through antibodies, or interfere with antigen processing and presentation pathways (reviewed in references 15 and 28). Cytomegaloviruses (CMV) genomes encode several genes whose products modify the efficiency of host immune system control. Both human and mouse CMVs (HCMV and MCMV) contain genes which code for proteins that interfere with antigen processing and presentation in the major histocompatibility complex class I pathway and abolish the efficient recognition of CMV-infected cells by cytotoxic T lymphocytes and natural killer (NK) cells (2, 12, 17, 31, 39).
Viral proteins that directly interact with host immunoglobulins (Ig) have a potential to interfere with the humoral effector arm of the immune system and to modify the antibody response of the host. Viral Fc receptors (FcR) have been described for herpes simplex virus type 1 (HSV-1) and type 2 (HSV-2) (1, 10) and varicella-zoster virus (24). The binding of human IgG to HCMV-infected cells has been described previously (19), but the gene responsible for this phenomenon has not been identified. We have recently shown that a 88-kDa early glycoprotein of MCMV has FcR properties and have identified the corresponding gene, designated fcr-1 (38).
The biological role of herpesvirus FcRs is not clear. They are functional but not structural homologs of cellular FcRs. The HSV-1 glycoproteins E (gE) and I (gI) form an FcR complex, which protects infected cells from complement-mediated lysis and antibody-dependent cellular cytotoxicity in vitro (11). The mechanism is explained by bipolar bridging of specific IgG (13). However, protective effects mediated by the binding of the Ig Fc fragment have not yet been demonstrated in vivo. There is evidence for other in vivo functions of the HSV-1 gE-gI heterodimer, as well as its pseudorabies virus homolog. These functions are associated with direct cell-to-cell spread of the virus (3, 8, 9).
To investigate the biological role of the MCMV-encoded FcR, we constructed fcr-1 deletion mutants. The in vitro growth kinetics of the recombinant viruses was comparable to that of wild-type virus, indicating that the FcR is dispensable for viral growth in cell culture. After infection of experimental animals, FcR deletion mutants exhibited reduced growth in various organs. We analyzed whether this loss of virulence in vivo is caused by an increased virus sensitivity to specific antibody. Virus replication in antibody-deficient mice demonstrated the attenuating effect of the fcr-1 deletion is not linked to its IgG-specific function.
MATERIALS AND METHODS
Cells and virus.
Mouse NIH 3T3 cells (ATCC CRL1658) were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% (vol/vol) newborn calf serum. A immortalized cell line of mouse fibroblasts (B12 cells) (7) was grown in minimum essential medium with 10% fetal calf serum. Third-passage mouse embryo fibroblasts, prepared from BALB/cJ mice and grown in minimum essential medium supplemented with 10% fetal calf serum, were used for virus infection. The Smith strain (ATCC VR-194) of MCMV was used as a tissue culture-grown virus.
Recombinant plasmids.
Plasmid cloning was done by standard methods (26). To generate the recombination plasmid pJAR4, a lacZ cassette driven by the Rous sarcoma virus promoter from plasmid pATLacZ (a gift of G. Darai, University of Heidelberg, Heidelberg, Germany) was inserted between the BssHII and BglII sites of the phagemid pREG16. This plasmid contains a 2.4-kb BamHI-BamHI subfragment of the MCMV HindIII J fragment encompassing the fcr-1 gene (38).
To flank the lacZ marker with loxP sites (35), a polylinker (5′-CTAGGAAGCTTGATATCGAATTCCTGCAGATCTG-3′) was inserted into a XbaI site between tandem loxP sites of plasmid pP4 (a gift from T. Boehm, DKFZ, Heidelberg, Germany) and then the lacZ cassette was cloned between the HindIII and BglII sites. The loxP-flanked lacZ cassette was released by digestion with NotI and BamHI and inserted into the BssHII and BglII sites of plasmid pREG16 by using a BssHII-NotI adapter (5′-GGCCACATGCCGATGG-3′), resulting in the recombination plasmid pIC4.
The Kunkel method of site-directed mutagenesis (22) was used to introduce a silent mutation into the fcr-1 gene. To delete the XbaI site of the pREG16 phagemid, single-stranded DNA was purified from phages produced in the ung Escherichia coli strain CJ236. The oligonucleotide 5′-GTCTCTCTGGACCTCTC-3′ was annealed to the single-stranded DNA, and the complementary strand was synthesized by T7 DNA polymerase. After electroporation of the double-stranded phagemids into ung+ E. coli, mutagenized phagemids were identified by the absence of the XbaI site. The correct alteration was confirmed by sequencing of the mutated region.
Generation of a recombinase cre+ cell line (N2).
A plasmid designated pneocre2 was generated by introducing a XhoI fragment containing the recombinase cre gene from pMC-Cre (16; a gift from K. Rajewsky, Cologne, Germany) into the XhoI site of the plasmid pUC21neo, which contains a neomycin resistance gene driven by the simian virus 40 enhancer-promoter. The cre recombinase-encoding plasmid pneocre2 (10 μg) was transfected into NIH 3T3 cells, and G418 resistant clones were isolated and tested for cre recombinase activity.
Construction of recombinant MCMV.
The linearized recombination plasmid (30 fmol) and intact viral DNA (1.5 μg) were cotransfected into NIH 3T3 fibroblasts by the calcium phosphate precipitation technique (27). Recombinant viruses were isolated from the resulting virus progeny by at least five rounds of limiting dilution followed by 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) staining. The lacZ mutants were generated by a single passage through the recombinase cre+ cell line (N2) and purified by limiting dilution after X-Gal color screening.
DNA analysis.
MCMV DNA was prepared from infected NIH 3T3 cells. Five days after infection, the cells were rinsed with phosphate-buffered saline, overlaid with lysis buffer (100 mM NaCl, 10 mM Tris [pH 8.0], 10 mM EDTA, 0.1% sodium dodecyl sulfate, 0.25 mg of proteinase K per ml), and incubated for 3 h at 37°C. Lysates were extracted twice with an equal volume of phenol and then once with phenol-chloroform-isoamyl alcohol (25:24:1) and chloroform-isoamyl alcohol (24:1). After addition of 2 volumes of isopropanol, the DNA was spooled on a glass rod, dried, and resuspended in 10 mM Tris–1 mM EDTA (pH 7.5).
For Southern blot analysis, approximately 1 μg of viral DNA was digested with restriction enzymes, separated by gel electrophoresis, blotted to positive-charged nylon membrane (Qiagen, Hilden, Germany), and hybridized with a digoxigenin-labeled probe. The bound probe was detected with an alkaline phosphatase-coupled anti-digoxigenin antibody and visualized by a chemiluminiscence method (Boehringer, Mannheim, Germany).
Flow cytometry.
For detection of FcR on the cell surface, MCMV-infected cells (16 h postinfection [p.i.]) were detached from the plastic with PBS–2 mM EDTA and incubated for 30 min with murine IgG (Dianova, Hamburg, Germany) at 100 μg/ml. Fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG F(ab′)2 (Dianova) served as a secondary reagent.
Immunoprecipitation.
B12 cells were infected either with wild-type MCMV or with FcR recombinants at a multiplicity of infection of 20. At 16 h p.i., the cells were pulse-labelled for 45 min with 150 μCi of [35S]methionine (Amersham, Braunschweig, Germany) per ml in methionine-free minimal essential medium supplemented with 5% dialyzed fetal calf serum. Labelled cells were lysed in a buffer containing 10 mM Tris-HCl (pH 7.4), 100 mM NaCl, 1% Nonidet P-40, 1 mM EDTA, and 1 mM phenylmethylsulfonyl fluoride. Cytoplasmic extracts were precleared twice with protein A-coupled Sepharose (Pharmacia, Uppsala, Sweden) and precipitated with murine IgG-coated protein A-Sepharose. Bound complexes were eluted by heating at 96°C for 5 min in reducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis sample buffer and analyzed by polyacrylamide gel electrophoresis (9% polyacrylamide).
Animals and infection conditions.
Newborn BALB/c mice (H-2d), B-cell-deficient mice (μMT/μMT), and their heterozygous littermates (μMT/+) (H-2b) (20) from the breeding colony at the Medical Faculty, University of Rijeka, were injected intraperitoneally with 103 PFU of either wild-type or recombinant MCMV. Homozygous and heterozygous B-cell-deficient animals were selected by an enzyme-linked immunosorbent assay for the presence of the IgM in sera, as described previously (18).
Adult, immunocompetent BALB/c mice were subcutaneously injected with 105 PFU of wild-type or recombinant viruses.
C57BL/6 mice (6 to 8 weeks old) were depleted of immune cells and injected by either the intravenous or subcutaneous (hind footpad) route with 105 PFU of wild-type or mutant virus. The depletion was performed by weekly injection of 1 mg of purified monoclonal antibodies to CD4 (YTS 191.1.2), CD8 (YTS 169.4.2) (6), and NK1.1 (PK136) (21).
Adult B-cell-deficient mice and their heterozygous littermates were depleted of CD4+, CD8+, and NK1.1+ cells and additionally treated with cyclophosphamide (150 mg/kg).
The animals were sacrificed at the indicated time p.i. Virus titers in organ homogenates were determined on mouse embryo fibroblasts by a standard plaque assay (29).
RESULTS
Plasma membrane expression of the MCMV FcR.
In previous experiments, the intracellular location of the FcR function was used to identify the gene (38). To fulfill an immunomodulatory function during infection, the FcR activity has to be present at the cell membrane. Therefore, we tested the surface expression of the viral FcR under various conditions. We found that trypsin addition to cells for removal of adherent cells destroyed the plasma membrane expression of the FcR activity. In the absence of trypsin, however, the binding of monomeric murine IgG can be demonstrated on the surface of infected fibroblasts (Fig. 1). Thus, the FcR activity is located at the site of potential immunological function.
FIG. 1.

Plasma membrane expression of the MCMV FcR function after infection. Mock-infected and MCMV-infected cells were incubated with murine IgG. Bound IgG was visualized with FITC-conjugated anti-mouse IgG F(ab′)2. The dashed line indicates the FITC control in the absence of mouse IgG. The plasma membrane expression was assessed by fluorescence-activated cell sorter analysis.
Generation of recombinant MCMV.
To analyze the role of the MCMV FcR in virus-host interactions, we constructed several FcR deletion mutants (illustrated in Fig. 2a). The recombinant virus ΔMS94.4 was generated by homologous recombination with the recombination plasmid pJAR4. In this plasmid, the 1.35-kb BssHII-BglII fragment of the fcr-1 gene, including the promoter and the coding region for first 416 amino acids of the open reading frame, was deleted and replaced by the E. coli lacZ gene. The recombinant virus was isolated by five rounds of limiting dilution prior to generation of virus stocks.
FIG. 2.

Characterization of MCMV recombinants. (a) Schematic structure of recombinant MCMV. Shown is the HindIII cleavage map of the MCMV genome (top) and the expanded HindIII-J region of wild-type and recombinant viruses with HindIII (H) and XbaI (X) cleavage sites indicated (below). The position and orientation of the fcr-1 gene is indicated by an arrow. The open box depicts a 1.3-kb fragment of the fcr-1 gene deleted in the ΔMS94.4, ΔMC95.15, and ΔMC95.16 recombinants, and the hatched boxes represent homologous regions used for recombination. The positions of the loxP sites are indicated by asterisks. The probe used for Southern blot analysis is represented by a bar. The expected sizes of the HindIII and XbaI restriction fragments are indicated. (b and c) Southern blot analysis of the recombinant viruses. DNA was isolated from infected NIH 3T3 cells and digested with HindIII (b) and XbaI (c). The sizes of the DNA fragments are indicated in kilobases.
To generate a revertant virus, we reinserted the fcr-1 gene in the ΔMS94.4 deletion mutant. To be able to differentiate between wild-type and revertant rMS95.9, we introduced a silent mutation into the coding sequence of the fcr-1 gene. The introduced mutation (A → G) eliminated a single restriction site (XbaI) and changed the restriction pattern of the rescued virus. The resulting mutant is easily distinguishable from the wild-type virus (Fig. 2c).
Since the fcr-1 deletion mutant carries the foreign gene lacZ, a revertant to wild-type properties would not formally distinguish biological effects caused by the deletion of the fcr-1 gene from effects caused by the insertion of lacZ into that gene position. We therefore created a second recombination plasmid, pIC4. In this plasmid, the lacZ gene is flanked by loxP sites (32). The deletion mutant ΔMC95.16 was isolated as above and served two purposes. It was used as a second fcr-1 deletion mutant prepared independently and was also used to create a deletion mutant without a marker gene. To this end, the ΔMC95.16 recombinant was passaged in the recombinase cre+ cell line N2. As expected, the marker gene was efficiently excised from the viral genome and the recombinant virus ΔMC95.15, without the lacZ gene, was generated.
Southern blot analyses of the isolated and plaque-cloned viruses confirmed that the recombination occurred at the expected position (Fig. 2b). In all deletion mutants, the original HindIII J fragment (8.2 kb) was replaced by new HindIII fragments. The sizes of the fragments corresponded to the sizes predicted from the distribution of HindIII sites in the MCMV genome and in the recombination plasmids. In the ΔMS94.4 recombinant, new HindIII fragments of 1.7 and 9.7 kb were present, while in ΔMC95.15 a 6.9-kb fragment was found and in ΔMC95.16 5.1- and 6.2-kb fragments were found. The difference between the fragment pattern of wild-type and rescued virus rMS95.9 after XbaI digestion is shown in Fig. 2c. As expected, the XbaI N (4.1 kb) and G (13.0 kb) fragments, visible in the wild-type virus, were replaced by a single 17.1-kb fragment in rMS95.9. The comparison of the HindIII, EcoRI, and XbaI restriction fragment patterns of the recombinants with those of wild-type MCMV confirmed that the recombinant viruses were free of detectable deletions or insertions in any other region of the viral genome (data not shown).
Expression of MCMV FcR in wild-type, deletion mutant, and rescued virus.
To verify that the recombinant viruses with the deletion of the fcr-1 gene do not express a protein with FcR properties, proteins from MCMV-infected and [35S]methionine-labelled cells were precipitated with mouse IgG. In cells infected with wild-type MCMV and with fcr-1 rescued virus, proteins with molecular masses of 86 to 88 kDa and 105 kDa, corresponding to the MCMV FcR (38), were detected (Fig. 3). These proteins were absent in cells infected with ΔMS94.4, ΔMC95.15, and ΔMC95.16, confirming the correct deletion of the FcR-encoding gene. In addition, it indicated the absence of any other proteins with FcR properties encoded by MCMV whose function could be disguised in the wild-type virus by the fcr-1 gene product.
FIG. 3.

Presence of glycoproteins with the FcR property in MCMV and mutants. B12 cells infected with wild-type MCMV and the different FcR recombinants were pulse-labelled for 45 min with 150 μCi of [35S]methionine per ml. Precipitation from cytoplasmic extracts was done with purified mouse IgG and protein A-Sepharose.
In vitro growth of MCMV mutants.
To assess whether the fcr-1 gene affects the growth of virus in cell culture, single-step growth curves of recombinant and wild-type viruses were determined. After infection of NIH 3T3 fibroblasts at a multiplicity of infection of 0.1 PFU per cell, the replication kinetics of recombinants were indistinguishable from that of the wild-type MCMV (Fig. 4). Also, the size and morphology of viral plaques were identical in recombinant and parental viruses, indicating that the fcr-1 gene is dispensable for growth in cultured fibroblasts.
FIG. 4.
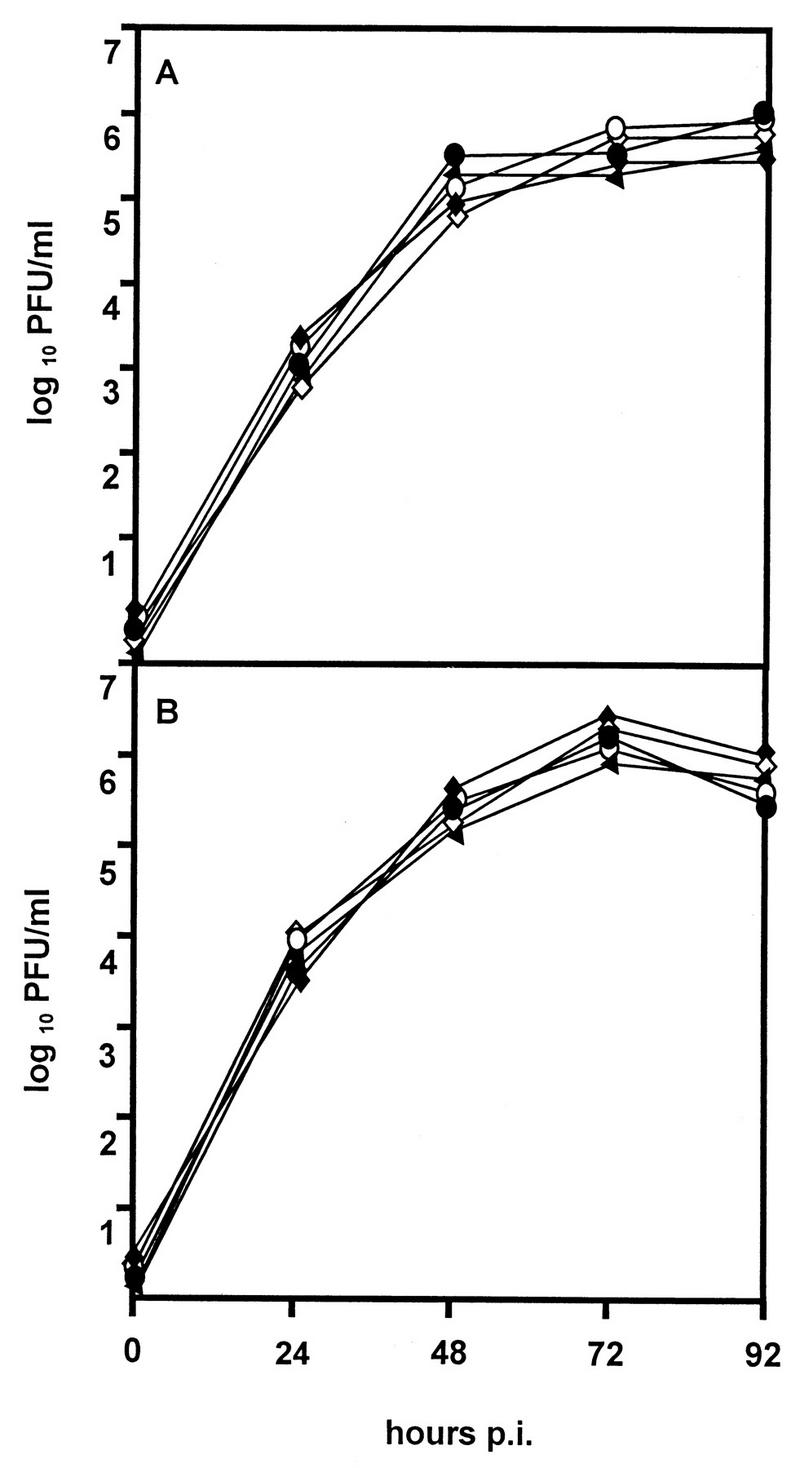
In vitro growth of MCMV recombinants. NIH 3T3 cells were infected with wild-type MCMV (open circles), ΔMS94.4 (solid triangles), rMS95.9 (open diamonds), ΔMC95.15 (solid circles), and ΔMC95.16 recombinants (solid diamonds) at a multiplicity of infection of 0.1. Supernatants (A) and cells (B) were harvested at different time points p.i., and virus titers were determined. Standard deviations (not shown) were less than 15% of the mean values.
Reduced growth of fcr-1 deletion mutants in newborn mice.
To investigate the influence of the fcr-1 gene deletion on in vivo replication, we infected 6-week-old BALB/c mice with wild-type and fcr-1 deletion mutant viruses. The tissues of mice infected with the fcr-1 deletion mutants contained very low virus titers that made quantitative comparisons difficult (data not shown). Therefore, we studied virus replication in the immunologically immature newborn mice, which are highly susceptible to MCMV infection (30). At 8 days p.i. the wild-type virus reached high titers in the organs of new-born mice (Fig. 5). At the same time, we observed a more than 100-fold-reduced replication of the fcr-1 deletion mutants in the lungs, liver, and spleen. Propagation of the rMS95.9 revertant virus was indistinguishable from that of the wild-type virus, confirming that the attenuation of the ΔMS94.4 recombinant virus is entirely due to the site-specific mutation.
FIG. 5.
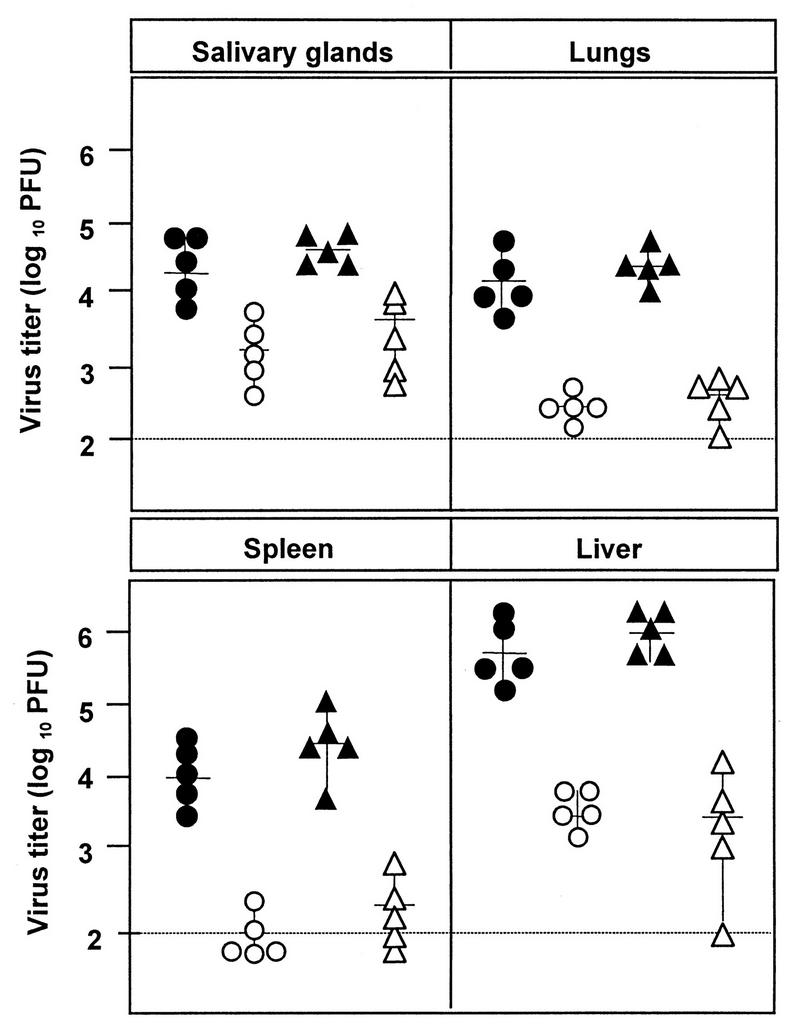
Reduced growth of fcr-1 deletion mutants in mice. Newborn BALB/c mice from timed pregnancies were inoculated intraperitoneally on day 0 with 103 PFU of wild-type (solid circles), ΔMS94.4 (open circles), rMS95.9 (solid triangles), and ΔMC95.15 (open triangles) recombinant MCMV. Virus titers were determined 8 days p.i. for individual mice (symbols), and median values (horizontal bars) were calculated. The solid line represents the detection limit.
Altered biological properties of a virus mutant can be caused by the deletion or the insertion of a marker gene or both. Reduced growth in salivary glands of the lacZ+ MCMV recombinant without any other mutation has been described by Stoddard et al. (36). The authors excluded the immune response to β-galactosidase as a basis of reduced replication, but the mechanism of the observed phenomenon remained unclear. Therefore, the deletion mutant lacking the marker gene was tested. There was no difference between the titers of fcr-1 lacZ+ (ΔMS94.4) and fcr-1 lacZ (ΔMC95.15) recombinants (Fig. 5). The ΔMC95.16 recombinant virus, which was used to create the marker-free mutant, reached titers comparable to those of the other two fcr-1 deletion mutants (data not shown). Thus, at least in this case, the lacZ marker gene did not contribute to the attenuation of the mutant.
Growth of the fcr-1 deletion mutant in B-cell-deficient mice.
According to the hypothesis of bipolar bridging (13), the FcR should protect the virus and virus-infected cells from effects mediated through the Fc portion of Igs after binding to specific antigen. We reasoned that if the attenuated phenotype of the fcr-1 deletion mutants was due to the loss of an immunoevasive function mediated by antibody binding, the mutant virus should grow comparably to wild-type MCMV in mutant mice that lack B cells (20). Immunocompetent littermates heterozygous for the μ-chain mutation (μMT/+) severely restricted the growth of the MCMV mutant. The MCMV-specific antibody response of newborn heterozygous animals on day 21 p.i. was comparable to the response of adult mice (data not shown). Notably, the growth of the fcr-1 deletion mutant was inhibited to the same degree in μMT/μMT mice, which completely lack B cells and Igs (Fig. 6). Thus, the capacity to bind the Fc portion of Ig cannot represent the principle which dictates the attenuated phenotype of the fcr-1 deletion mutants.
FIG. 6.
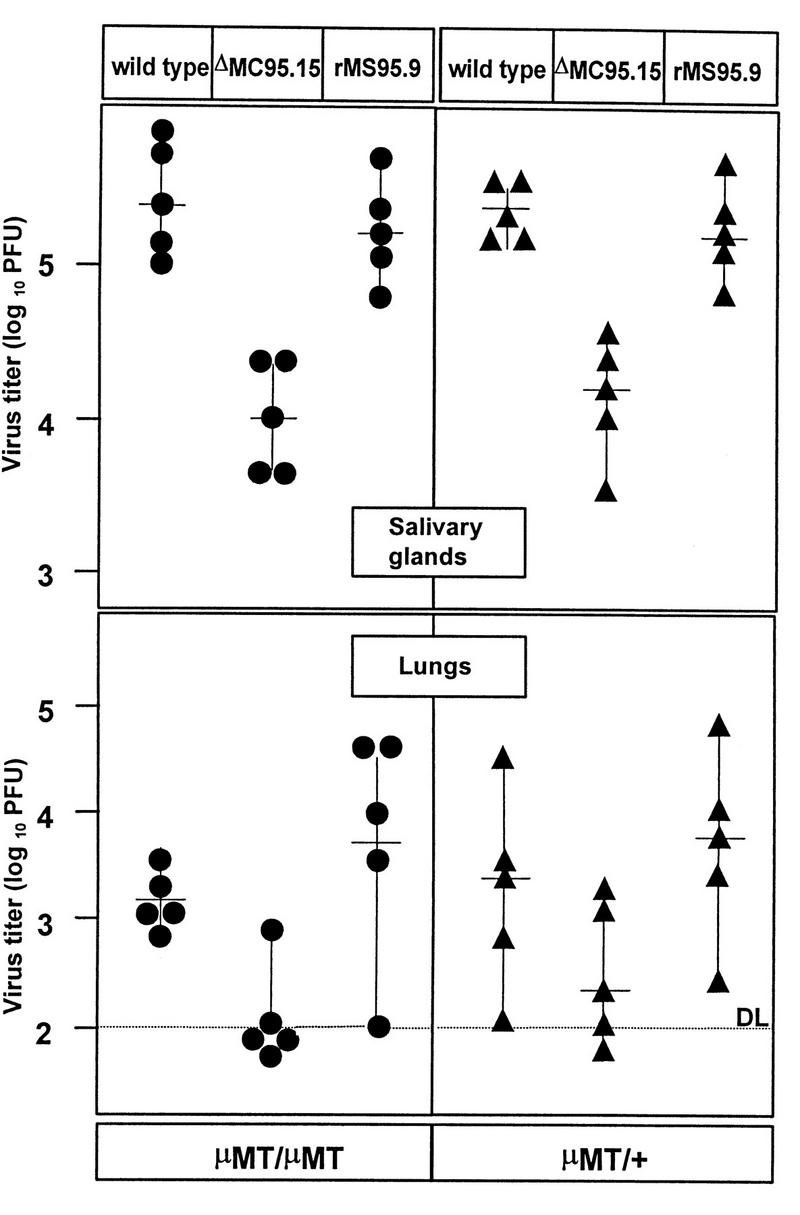
Growth of fcr-1 deletion mutants in B-cell-deficient mice. Newborn mice heterozygous (μMT/+) (circles) or homozygous (μMT/μMT) (triangles) for the μ exon mutation (20) were infected with 103 PFU of wild-type, ΔMC95.15, and rMS95.9 MCMV. Virus titers were determined 3 weeks p.i. (symbols), and median values were calculated (horizontal bars).
Reduced growth of fcr-1 deletion mutants in T- and NK-cell-depleted mice.
After depletion of T cells and NK cells, the C57BL/6 mice became increasingly susceptible to MCMV infection. Although the mutant virus showed marginal growth in immunodepleted juvenile mice, the significant difference between the growth of mutant and wild-type viruses and the growth of revertant virus remained evident. This suggested that the viral FcR does not interfere with the function of T cells and NK cells. Another explanation for reduced virulence could be the inability of the mutant virus to spread from the site of primary infection to the target organs. Therefore, we compared the virus titer that viruses reached in different organs after footpad and intravenous infection. The infection of the visceral organs and the salivary gland by the fcr-1 gene deletion mutant occurred at a low rate irrespective of the route of infection (Fig. 7). We therefore concluded that an inability of the mutant virus to spread hematogenously is not likely to explain the data.
FIG. 7.
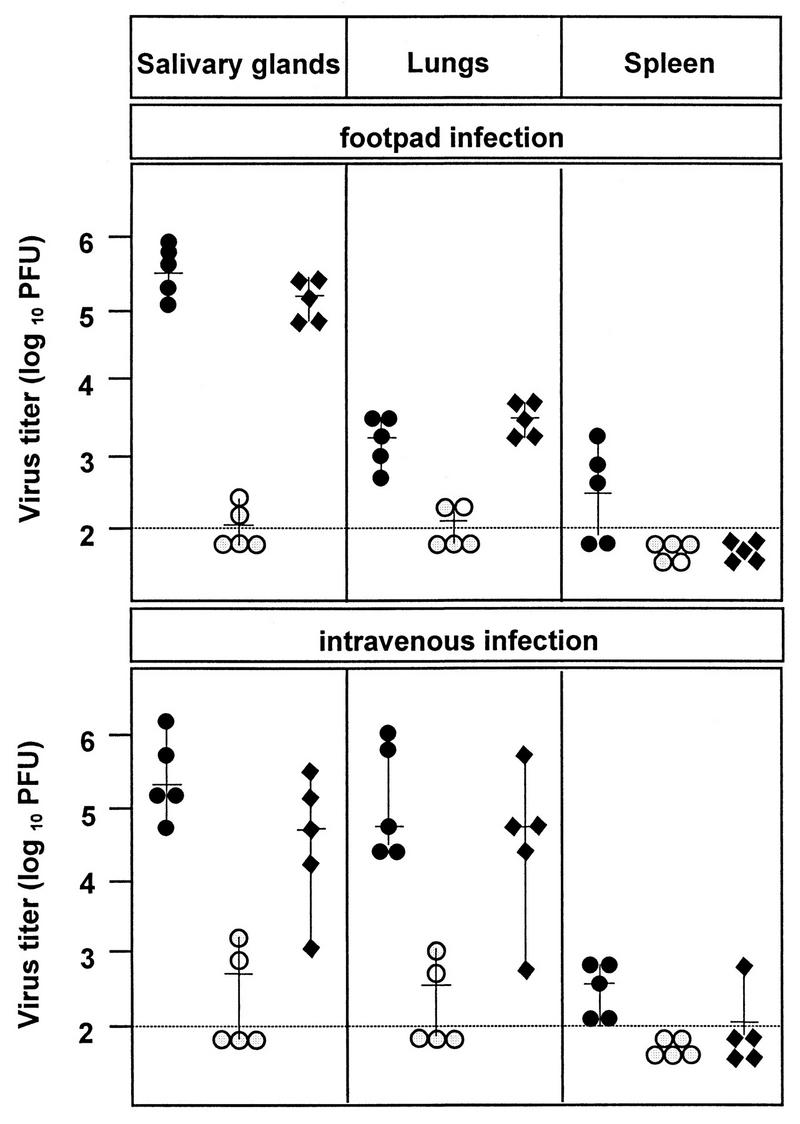
Lack of effect of T cells, NK cells, and route of infection on the in vivo phenotype. C57BL/6 mice (6 weeks old) were depleted of CD4+, CD8+, and NK1.1+ cells and infected subcutaneously (footpad) or intravenously with 105 PFU of wild-type (solid circles), ΔMS94.4 (open circles), and rMS95.9 (solid diamonds) MCMV. At 2 weeks p.i., virus titers were determined. Titers for individual mice (symbols) and median values (horizontal bars) are shown.
DISCUSSION
We previously identified an MCMV protein with an FcR property that is highly selective for mouse Igs and subsequently identified and localized the corresponding gene (fcr-1) (38). Here, we analyzed its function during the in vivo infection. Due to the IgG binding property, this protein has a potential to modulate immune responses of the host that are triggered through the Fc fragment of the Ig. This potential could enable efficient replication and spread of the virus in the presence of an active immune system producing specific antibodies.
To investigate the biological role of the MCMV FcR, we generated MCMV mutants by deleting major parts of the open reading frame of the fcr-1 gene. By creating a revertant virus and a deletion mutant lacking the marker gene, we unambiguously connected the in vivo phenotype with the lack of the fcr-1 gene product expression. The fcr-1 gene is nonessential for replication in cell culture and has no phenotype in vitro. Herpesvirus functions have been shown to modulate virus-host interactions such as antigen presentation and immune system control (2, 4, 12, 15, 17, 31, 34, 37, 39–41), tissue tropism (3–5, 23, 27), virus spread (9, 25, 42), and the establishment of latency (33). Most of these functions are encoded by nonessential genes. In accordance with such an in vivo function, the growth of viruses that do not express the fcr-1 gene was significantly restricted in all organs tested. The differences were in the range between 1.5 and 3.5 log10 PFU. This indicated that the fcr-1 gene product has an important function in vivo.
Considering the in vitro interaction of the viral FcR with the Fc fragment of mouse IgG (38), it was reasonable to presume that the reduced virulence of the fcr-1 deletion resulted from the more effective clearance through antibody-mediated mechanisms. Such protective effects have been suggested for FcRs in HSV-1 and varicella-zoster virus. Under in vitro conditions, they protect virus from neutralization and virus-infected cells from lysis mediated by antibody and complement and by antibody-dependent cellular cytotoxicity (10, 11, 13, 14). Our experiments showed, however, that the reduced replication capacity of fcr-1 deletion mutants is not resolved in the Ig-deficient host. Therefore, the interaction of MCMV FcR with the Ig Fc fragment either is irrelevant during in vivo infection or is minor in comparison with the effect of a strong second antibody-independent function. This mechanism is also T-cell and NK-cell independent, since depletion of these cell populations did not selectively improve the in vivo replication of the deletion mutant.
MCMV recombinants with deletions in the HindIII I and J fragments including the fcr-1 gene have been described recently (4), although a mutant with a singular deletion of the fcr-1 gene was not generated. In view of our results, the reduced in vivo virulence of these recombinants can now be explained at least in part by the lack of the fcr-1 gene. It has been shown that the deletion mutant missing both the fcr-1 gene and the m137 gene grew normally in the IC-21 macrophage cell line (4). This indicates that the FcR acts differently from the neighboring genes M140 and/or M141, to which macrophages growth deficiency was mapped.
An in vivo role of alphaherpesvirus proteins with FcR properties appears to be facilitation of infection through the direct cell-to-cell route (3, 9, 42). The HSV-1 gE− and gI− mutants fail in axonal spread due to the inefficient neuron-to-neuron transmission (9). They also form significantly smaller plaques than the wild type does (8, 42). Impaired cell-to-cell spread in polarized retinal epithelium cells is a characteristic of an HCMV US9 deletion mutant (25). Although the MCMV FcR shows no significant sequence homology to alphaherpesvirus FcRs or HCMV US9, it cannot yet be excluded that the MCMV FcR acts as a mediator of viral spread in certain tissues. The spread of the virus to several organs including the salivary gland is not inhibited after local injection, and also the clearance kinetics reflects, although at a lower level, that of the wild-type infection (data not shown). Thus, the phenotype of the infection in vivo does not yet provide a clue to the function of this glycoprotein, which clearly contributes to the virulence of the virus.
Although the deletion of the fcr-1 gene results in severely reduced viral growth in vivo, we could falsify the hypothesis concerning the underlying mechanism. Thus, potential immune system evasion principles, which appear obvious by studying isolated gene functions in vitro after high expression of the respective function, need to be confirmed by the analysis of appropriate mutants in vivo. The molecular basis for the reduced in vivo replication of fcr-1 deletion mutants is again unresolved. Attempts to identify the putative cellular ligand, perhaps a member of the Ig superfamily, are in progress.
ACKNOWLEDGMENTS
We thank T. Boehm, G. Darai, and K. Rajewsky for providing plasmids and T. Flohr for performing site-directed mutagenesis. The skilful technical assistance of Jelena Dirlić and Dijana Lukić is greatly appreciated.
This work was supported by grants from DFG and the BMBF to U.K. and by the Croatian Ministry of Science and Technology (project 006204).
REFERENCES
- 1.Baucke R B, Spear P G. Membrane proteins specified by herpes simplex viruses. V. Identification of an Fc-binding glycoprotein. J Virol. 1979;32:779–789. doi: 10.1128/jvi.32.3.779-789.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beersma M F C, Bijlmakers M J E, Ploegh H L. Human cytomegalovirus down-regulates HLA class I expression by reducing the stability of class I heavy chains. J Immunol. 1993;51:4455–4464. [PubMed] [Google Scholar]
- 3.Card J P, Whealy M E, Robbins A K, Enquist L W. Pseudorabies virus envelope glycoprotein gI influences both neurotropism and virulence during infection of the rat visual system. J Virol. 1992;66:3032–3041. doi: 10.1128/jvi.66.5.3032-3041.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cavanaugh V J, Stenberg R M, Staley T L, Virgin IV H W, McDonald M R, Paetzold S, Farrell H E, Rawlinson W D, Campbell A E. Murine cytomegalovirus with a deletion of genes spanning HindIII-J and -I displays altered cell and tissue tropism. J Virol. 1996;70:1365–1374. doi: 10.1128/jvi.70.3.1365-1374.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chou J, Kern E R, Whitley R J, Roizman B. Mapping of herpes simplex virus-1 neurovirulence to γ134.5, a gene nonessential for growth in culture. Science. 1990;250:1262–1266. doi: 10.1126/science.2173860. [DOI] [PubMed] [Google Scholar]
- 6.Cobbold S P, Jayasuriya A, Nash A, Prospero T D, Waldman H. Therapy with monoclonal antibodies by elimination of T cell subsets in vivo. Nature (London) 1984;312:548–551. doi: 10.1038/312548a0. [DOI] [PubMed] [Google Scholar]
- 7.Del Val M, Schlicht H J, Ruppert T, Reddehase M J, Koszinowski U K. Efficient processing of an antigenic sequence for presentation by MHC class I molecules depends on its neighboring residues in the protein. Cell. 1991;66:1145–1153. doi: 10.1016/0092-8674(91)90037-y. [DOI] [PubMed] [Google Scholar]
- 8.Dingwell K S, Brunetti C R, Hendricks R L, Tang Q, Tang M, Rainbow A J, Johnson D C. Herpes simplex virus glycoproteins E and I facilitate cell-to-cell spread in vivo and across junctions of cultured cells. J Virol. 1994;68:834–845. doi: 10.1128/jvi.68.2.834-845.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dingwell K S, Doering L C, Johnson D C. Glycoproteins E and I facilitate neuron-to-neuron spread of herpes simplex virus. J Virol. 1995;69:7087–7098. doi: 10.1128/jvi.69.11.7087-7098.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dowler K W, Veltri R W. In vitro neutralisation of HSV-2: inhibition by binding of normal IgG and purified Fc to virion Fc receptor (FcR) J Med Virol. 1984;13:251–259. doi: 10.1002/jmv.1890130307. [DOI] [PubMed] [Google Scholar]
- 11.Dubin G, Socolof E, Frank I, Friedman H M. Herpes simplex virus type 1 Fc receptor protects infected cells from antibody-dependent cellular cytotoxicity. J Virol. 1991;65:7046–7050. doi: 10.1128/jvi.65.12.7046-7050.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farell H E, Vally H, Lynch D M, Fleming P, Shellam G R, Scalzo A A, Davis-Poynter N J. Inhibition of natural killer cells by a cytomegalovirus MHC class I homologue in vivo. Nature (London) 1997;386:510–514. doi: 10.1038/386510a0. [DOI] [PubMed] [Google Scholar]
- 13.Frank I, Friedman H M. A novel function of the herpes simplex virus type 1 Fc receptor: participation in bipolar bridging of antiviral immunoglobulin G. J Virol. 1989;63:4479–4488. doi: 10.1128/jvi.63.11.4479-4488.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friedman H M, Cohen G H, Eisenberg R J, Siedel C A, Clines D B. Glycoprotein C of herpes simplex virus 1 acts as a receptor for the complement component C3b on infected cells. Nature (London) 1984;309:633–635. doi: 10.1038/309633a0. [DOI] [PubMed] [Google Scholar]
- 15.Gooding L R. Virus proteins that counteract host immune defences. Cell. 1992;71:5–7. doi: 10.1016/0092-8674(92)90259-f. [DOI] [PubMed] [Google Scholar]
- 16.Gu H, Zou Y R, Rajewsky K. Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell. 1993;73:1155–1164. doi: 10.1016/0092-8674(93)90644-6. [DOI] [PubMed] [Google Scholar]
- 17.Jones T R, Hanson K, Sun L, Slater S, Stenberg R M, Campbell A E. Multiple independent loci within the human cytomegalovirus unique short region down-regulate expression of major histocompatibility complex class I heavy chains. J Virol. 1995;69:4830–4841. doi: 10.1128/jvi.69.8.4830-4841.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jonjic S, Pavic I, Polic B, Crnkovic I, Lucin P, Koszinowski U H. Antibodies are not essential for the resolution of primary cytomegalovirus infection but limit dissemination of reccurent virus. J Exp Med. 1994;179:1713–1717. doi: 10.1084/jem.179.5.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keller R, Peitchel R, Goldman J N, Goldman M. An IgG-Fc receptor induced in cytomegalovirus infected fibroblasts. J Immunol. 1976;116:772–777. [PubMed] [Google Scholar]
- 20.Kitamura D, Roes J, Kühn R, Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin μ chain gene. Nature (London) 1991;350:423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- 21.Koo G C, Dumond F J, Tutt M, Hackett J, Kumar V. The NK 1.1-mouse: a model to study differentiation of murine NK cells. J Immunol. 1986;137:3742–3747. [PubMed] [Google Scholar]
- 22.Kunkel T A. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci USA. 1985;82:488–497. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lagenaur L A, Manning W C, Viera J, Martens C L, Mocarski E S. Structure and function of the murine cytomegalovirus sgg1 gene: a determinant of viral growth in salivary gland acinar cells. J Virol. 1994;68:7717–7727. doi: 10.1128/jvi.68.12.7717-7727.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Litwin V, Jackson W, Grose C. Receptor properties of two varicella-zoster virus glycoproteins, gpI and gpIV, homologous to herpes simplex virus gE and gI. J Virol. 1992;66:3643–3651. doi: 10.1128/jvi.66.6.3643-3651.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maidji E, Tugizov S, Jones T, Zheng Z, Pereira L. Accessory human cytomegalovirus glycoprotein US9 in the unique short component of the viral genome promotes cell-to-cell transmission of virus in polarized epithelial cells. J Virol. 1996;70:8402–8410. doi: 10.1128/jvi.70.12.8402-8410.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maniatis T, Fritsch E, Sambrook J. Molecular cloning: a laboratory manual. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1982. [Google Scholar]
- 27.Manning W C, Stoddart C A, Langenaur L A, Abenes G B, Mocarski E S. Cytomegalovirus determinant of replication in salivary glands. J Virol. 1992;66:3794–3802. doi: 10.1128/jvi.66.6.3794-3802.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McFadden G, Kane K. How DNA viruses perturb functional MHC expression to alter immune recognition. Adv Cancer Res. 1994;63:117–209. doi: 10.1016/s0065-230x(08)60400-5. [DOI] [PubMed] [Google Scholar]
- 29.Reddehase M J, Weiland F, Münch K, Jonjic S, Lüske A, Koszinowski U H. Interstitial murine cytomegalovirus pneumonia after irradiation: characterization of cells that limit viral replication during established infection of the lungs. J Virol. 1985;55:264–273. doi: 10.1128/jvi.55.2.264-273.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reddehase M J, Balthesen M, Rapp M, Jonjic S, Pavic I, Koszinowski U H. The conditions of primary infection define the load of latent viral genome in organs and the risk of recurrent cytomegalovirus disease. J Exp Med. 1994;179:185–193. doi: 10.1084/jem.179.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reyburn H T, Mandelboim O, Vales-Gomez M, Davis D M, Pazmany L, Strominger J. The class I homologue of human cytomegalovirus inhibits attack by natural killer cells. Nature (London) 1997;386:514–518. doi: 10.1038/386514a0. [DOI] [PubMed] [Google Scholar]
- 32.Sauer B, Henderson N. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc Natl Acad Sci USA. 1988;85:5166–5170. doi: 10.1073/pnas.85.14.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sears A E, Halliburton I W, Maigneir B, Silver S, Roizman B. Herpes simplex virus 1 mutant deleted in the α22 gene: growth and gene expression in permissive and restrictive cells and establishment of latency in mice. J Virol. 1985;55:338–346. doi: 10.1128/jvi.55.2.338-346.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spriggs M K, Hruby D E, Maliszewski C R, Pickup D J, Sims J E, Buller R M, VanSlyke J. Vaccinia and cowpox viruses encode a novel secreted interleukin-1-binding protein. Cell. 1992;71:145–152. doi: 10.1016/0092-8674(92)90273-f. [DOI] [PubMed] [Google Scholar]
- 35.Stenberg N, Hamilton D. Bacteriophage P1 site-specific recombination. I. Recombination between loxP sites. J Mol Biol. 1981;150:467–486. doi: 10.1016/0022-2836(81)90375-2. [DOI] [PubMed] [Google Scholar]
- 36.Stoddart C A, Cardin R D, Boname J M, Manning W C, Abenes G B, Mocarski E S. Peripheral blood mononuclear phagocytes mediate dissemination of murine cytomegalovirus. J Virol. 1994;68:6243–6253. doi: 10.1128/jvi.68.10.6243-6253.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Swaminham S, Hesselton R, Sullivan J, Kieff E. Epstein-Barr virus recombinants with specifically mutated BCRF1 genes. J Virol. 1993;67:7406–7413. doi: 10.1128/jvi.67.12.7406-7413.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thäle R, Lucin P, Schneider K, Eggers M, Koszinowski U K. Identification and expression of a murine cytomegalovirus early gene coding for an Fc receptor. J Virol. 1994;68:7757–7765. doi: 10.1128/jvi.68.12.7757-7765.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thäle R, Szepan U, Hengel H, Geginat G, Lucin P, Koszinowski U K. Identification of the mouse cytomegalovirus genomic region affecting major histocompatibility complex class I molecule transport. J Virol. 1995;69:6098–6105. doi: 10.1128/jvi.69.10.6098-6105.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Upton C, Mossman K, McFadden G. Encoding of a homolog of the IFN-γ receptor by myxoma virus. Science. 1992;258:1369–1372. doi: 10.1126/science.1455233. [DOI] [PubMed] [Google Scholar]
- 41.Ziegler H, Thäle R, Lucin P, Muranyi W, Flohr T, Hengel H, Farell H, Rawlinson W, Koszinowski U. A mouse cytomegalovirus glycoprotein retains MHC class I complexes in the ERGIC/cis-Golgi compartments. Immunity. 1997;6:57–63. doi: 10.1016/s1074-7613(00)80242-3. [DOI] [PubMed] [Google Scholar]
- 42.Zsak L, Zuckermann F, Sugg N, Ben-Porat T. Glycoprotein gI of pseudorabies virus promotes cell fusion and virus spread via direct cell-to-cell transmission. J Virol. 1992;66:2316–2325. doi: 10.1128/jvi.66.4.2316-2325.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]