

Abstract
CD40, a cell surface receptor in the tumor necrosis factor receptor family, first identified and functionally characterized on B lymphocytes, is also expressed on epithelial and other cells and is now thought to play a more general role in immune regulation. Overexpression of the NFκB activator 1 (Act1) leads to the activation of both NFκB and Jun kinase in epithelial cell lines. Endogenous Act1 is recruited to the CD40 receptor in human intestinal (HT29) and cervical (HeLa) epithelial cells upon stimulation with CD40 ligand, indicating that Act1 is involved in this signaling pathway. Act1 also interacts with tumor necrosis factor receptor-associated factor 3, a component involved in CD40-activated pathway. Furthermore, transfection of Act1 into C33A cervical epithelial cells, which do not express it, renders these cells sensitive to CD40 ligand-induced NFκB activation and protects them from CD40 ligand-induced apoptosis. We conclude that Act1 plays an important role in CD40-mediated signaling in epithelial cells.
CD40, a member of the tumor necrosis factor receptor superfamily, was first identified on B lymphocytes; CD40 ligand (CD40L) was originally thought to be restricted to activated CD4+ T lymphocytes (1). Until recently, the primary actions of CD40 and CD40L were considered restricted to B and T lymphocytes and their interactions required for thymus-dependent humoral responses (1). However, current work from many groups expands this view of the CD40/CD40L dyad as a mere mediator of lymphocyte communication (2, 3). Although many cell types were shown to express CD40L, a variety of nonlymphocytic cell types also express the CD40 receptor, including monocytes, basophils, dendritic cells, fibroblasts, smooth muscle cells, endothelial cells, and epithelial cells (3–6). The ligation of CD40 mediates a variety of inflammatory responses, including the expression of cellular adhesion molecules, cytokines, matrix-degrading enzymes, and apoptotic mediators (7). Consequently, CD40-dependent signaling has been associated with pathogenic processes of autoimmune and chronic inflammatory diseases, such as graft vs. host disease, neurodegenerative disorders, asthma, and atherosclerosis (7).
Despite the impressive amount of data generated within recent years, our knowledge regarding CD40-mediated signal transduction pathways and the associated transducers remains incomplete and controversial. Most studies have used B lymphocytes. CD40 ligation leads to activation of the transcription factors NFκB and AP1 (1). NFκB is kept inactive in the cytoplasm through its interaction with the IκB inhibitory proteins. After stimulation with CD40L or other inflammatory mediators, the IκB proteins are phosphorylated by IκB kinase (IKK) (8–12). IKK is composed of the catalytic subunits IKKα and IKKβ and the regulatory subunit IKKγ. Phosphorylated IκB is then rapidly ubiquitinated and degraded (13, 14). The liberated NFκB translocates to the nucleus and activates transcription. Recent studies have shown that IKK also leads to the phosphorylation of NFκB itself, which is required for the full transcriptional activity of NFκB (15). Transforming growth factor-β-activated kinase 1 (TAK1; a mitogen-activated protein kinase kinase kinase) and its two binding proteins, TAB1 and TAB2 (transforming growth factor-β-activated kinase 1 binding proteins 1 and 2), have been implicated in linking tumor necrosis factor (TNF) receptor-associated factor (TRAF)-6 to IKK in IL-1-mediated signaling (16–23). TAK1 is also activated after stimulation with TNF, lipopolysaccharide, and receptor activator of NFκB ligand (RANKL) (24, 25), indicating that TAK1 is probably used by different NFκB-dependent signaling pathways. The role of TAK1 in CD40-mediated NFκB activation has not yet been identified. NFκB-inducing kinase (NIK) was also shown to play a role in activating IKK (13). Although NIK-null cells display normal NFκB DNA-binding activity in response to many stimuli, NIK is required for CD40L-induced NFκB activation in B cells and for the expression of NFκB-dependent genes in response to lymphotoxin-β (26, 27).
Because a direct association of specific kinases or phosphatases with CD40 has not been demonstrated, the association of intermediary CD40-binding proteins has been considered crucial in CD40-mediated signal transduction (1, 28). Several members of the TRAF family, including TRAFs 2, 3, 5, and 6, interact with CD40 and play important roles in CD40-mediated signaling pathways (1). Deletion mutants lacking intracellular domains of CD40 localized the association of TRAF2, TRAF3, and TRAF5 to one region (PVQET, residues 250–254), whereas TRAF6 is associated with a separate domain (QEPQEINF, residues 231–238). TRAF3 can initiate signaling pathways that lead to the activation of p38 and Jun kinase (JNK) but not NFκB (29, 30). Induced overexpression of wild-type TRAF3 in B cells dramatically inhibits CD40-stimulated secretion of Ab (31). Furthermore, TRAF3 has a proapoptotic role in epithelial cells (32, 33). TRAF3-null mice die after birth and are defective in T-cell dependent immune responses (34). However, the precise function of TRAF3 in CD40-dependent signaling is still unclear. Different TRAF3 splice variants have been identified and the overexpression of some of the variants induces NFκB activation, revealing a complex role for TRAF3 in CD40-mediated pathways (35, 36). Overexpression of TRAF2, -5, or -6 can activate both NFκB and JNK (28, 37). In TRAF2-null mice, Ig isotype switching in response to virus infection is defective and CD40-mediated proliferation and NFκB activation are impaired (38). Studies with TRAF6-null mice have shown that it is required for CD40-induced NFκB and JNK activation (39). However, CD40-mediated NFκB and JNK activation is not defective in TRAF5-null mice. Instead, TRAF5-null B cells have defects in proliferation and in up-regulating various surface molecules, including CD23, CD54, and Fas, in response to CD40L (40).
Several studies have suggested that one (or more) unknown CD40-specific cofactor is required for CD40L-induced IKK activation. Homozygosity for the aly point mutation in NIK results in alymphoplasis in mice (41). The activation of NFκB by Epstein–Barr virus latent membrane protein 1 or LTβR was selectively inhibited by a wild-type dominant negative NIK (DN-NIK) and not by aly DN-NIK. In contrast, CD40-mediated NFκB activation is inhibited by both wild-type and aly DN-NIK, suggesting that CD40 signaling through the IKK complex might be mediated by an unknown protein that can be blocked by either wild-type or aly DN-NIK (42). The requirement of a CD40-specific cofactor for activating the IKK complex is supported further by studies of patients with X-linked hyper-IgM syndrome and hypohydrotic ectodermal dysplasia carrying mutations in a putative zinc-finger domain of IKKγ that result in hyper-IgM syndrome with hypohydrotic ectodermal dysplasia (43). These mutations prevent CD40 ligand-mediated NFκB activation, whereas lipopolysaccharide (LPS)-mediated NFκB activation is normal in these patients. Because both LPS and CD40L use TRAF6 to activate IKK, the above results strongly suggest that TRAF6 utilizes different stimulus-specific cofactors and that the mutations in the putative zinc-finger domain of IKKγ prevent the recruitment of CD40L-restricted cofactors needed to activate the IKK complex.
We have previously developed a strategy for cloning new components of NFκB-dependent signaling pathways. In the human embryonic kidney cell line 293, an NFκB-dependent promoter (E-selectin) was used to drive the expression of a zeocin-resistance gene (Zeo) and the Herpes simplex thymidine kinase gene (TK) (44). We introduced a retroviral cDNA library made from human keratinocyte HaCat cells into 293-TK/Zeo cells and then selected the infected cells in Zeo. A 2.6-kb cDNA insert was recovered by reverse transcription–PCR from one of the obtained clones (45). This 60-kDa (574-aa) polypeptide is named NFκB activator 1 (Act1). Simultaneously, Act1 was also cloned by Leonardi et al. (46) and named CIKS, through a yeast-two hybrid screening, based on its interaction with IKKγ.
We found that major sites of Act1 expression are epithelial cells of the respiratory and gastrointestinal tract, and its expression is augmented by proinflammatory mediators (Z.Z. and X.L., unpublished data). Act1 does not have any enzymatic domain; instead, it contains a helix–loop–helix at the N terminus and a coiled-coil at the C terminus. In addition, Act1 contains two TRAF binding sites, EEESE (residues 38–42) and EERPA (residues 333–337). The data reported in this article show that the activation of NFκB by Act1 is through the TAK1 and IKK signaling cascade. Importantly, we found that endogenous Act1 is recruited to the CD40 receptor in epithelial cells after stimulation with CD40L, mediating CD40L-induced NFκB activation and protection against CD40L-induced apoptosis in epithelial cells. We conclude that Act1 functions as an adapter in CD40-mediated pathways in epithelial cells during inflammation.
Materials and Methods
Biological Reagents and Cell Culture.
Rabbit anti-TAK1 and anti-TAB1 polyclonal Abs were described (17, 19). An Ab to cleaved poly(ADP-ribose) polymerase (PARP) was from Cell Signaling (Beverly, MA). Anti-α-tubulin, anti-IKKα/β, and anti-CD40 were from Santa Cruz Biotechnology. Anti-Act1 was produced by using a purified peptide expressing residues 1–300 of human Act1 as the antigen. HeLa, HT29, and C33A cells were maintained in DMEM, supplemented with 10% FCS, penicillin G (100 μg/ml), and streptomycin (100 μg/ml). A549 cells were maintained in minimum essential media, supplemented with 10% FCS, penicillin G (100 μg/ml), and streptomycin (100 μg/ml). Wild-type primary mouse embryonic fibroblasts (MEFs), IKKα-null, and IKKβ-null MEFs (from I. Verma, Salk Institute, La Jolla, CA) were maintained in DMEM, supplemented with 10% FCS, G418 (400 μg/ml) penicillin G (100 μg/ml), and streptomycin (100 μg/ml). The membrane-bound CD40L prepared from Hi 5 cells infected with baculoviral CD40L was kindly provided by H. Wesche (Tularik, South San Francisco).
Recombinant Plasmids.
The pCMV-Act1 (CMV, cytomegalovirus) expression constructs [flag-tagged and hemagglutinin (HA)-tagged] and pE-selectin-luc have been described (45, 47). Dominant negative TAK1 (DNTAK1-K66W) was a kind gift from K. Matsumoto (Nagoya University, Nagoya, Japan). Mammalian expression constructs for TRAFs 1–6 (flag-tagged) and CD40 (flag-tagged) were kindly provided by H. Wesche.
Coimmunoprecipitation and Immunoblotting.
For coimmunoprecipitations, cells untreated or treated with 100 μg/ml of CD40L were lysed in a Triton-containing buffer (0.5% Triton X-100/20 mM Hepes, pH 7.4/150 mM NaCl/12.5 mM β-glycerophosphate/1.5 mM MgCl2/10 mM NaF/2 mM DTT/1 mM sodium orthovanadate/2 mM EGTA/20 μM aprotinin/1 mM phenylmethylsulfonyl fluoride). Cell extracts were incubated with 1 μg of Ab or preimmune serum (negative control) for 2 h, followed by incubation for 2 h with 20 μl of protein A-Sepharose beads (prewashed and resuspended in PBS at a 1:1 ratio). After incubations, the beads were washed 4 times with lysis buffer, separated by SDS/PAGE, and analyzed by immunoblotting.
Transfection and Reporter Assays.
For stable transfections, 2 × 105 cells were seeded onto a 10-cm plate and cotransfected the following day by the calcium phosphate method with 10 μg of each expression vector and 1 μg of pBabePuro vector. After 48 h, the cells were selected with 1 μg/ml of puromycin until clones appeared. For reporter assays, 2 × 105 cells were transfected by the same procedure with 1 μg of pE-selectin-luc, 1 μg of pSV2-β-gal, and 100 ng of each expression construct. After 48 h, the cells were split onto two 35-mm plates and, the next day, were stimulated with CD40L for 4 h before harvest. Luciferase and β-galactosidase activities were determined by using the luciferase assay system and chemiluminescent reagents from Promega.
Gel-Shift Assays.
An NFκB-binding site (5′-GAGCAGAGGGAAATTCCGTAACTT-3′) from the IP-10 gene was used as a probe (48). Complementary oligonucleotides, end-labeled with polynucleotide kinase (Roche Molecular Biochemicals) and [γ-32P]ATP, were annealed by slow cooling. Approximately 20,000 cpm of probe were used per assay (49). Whole-cell extracts were used for the assay. The binding reaction was carried out at room temperature for 20 min in a total volume of 20 μl containing 20 mM Hepes buffer (pH 7.0), 10 mM KCl, 0.1% Nonidet P-40, 0.5 mM DTT, 0.25 mM phenylmethanesulfonyl fluoride, and 10% glycerol.
Results
Act1 Activates NFκB Through the TAK1 and IKK Signaling Cascade.
Many signaling pathways involving NFκB activation converge on IKK. We have shown that IKK is activated constitutively in cells transfected with pCMV-Act1 (45), indicating that Act1 probably activates NFκB through IKK. Furthermore, Act1 is bound to IKKγ in coimmunoprecipitation experiments, strongly suggesting that it forms a complex with IKK (45). To confirm those interactions, we tested Act1-mediated NFκB activation in IKKα- and IKKβ-null primary MEFs. Act1 activated NFκB in wild-type MEFs but failed to activate it in IKKα- and IKKβ-null MEFs, indicating that both IKKα and IKKβ are required for Act1-mediated NFκB activation (Fig. 1).
Figure 1.
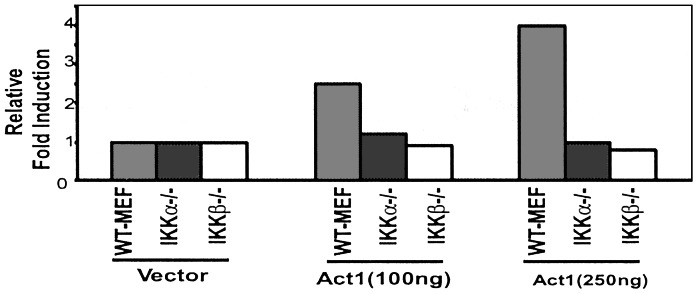
Act1 function in IKK-null cells. pCMV-Act1 (Act1, 100 or 250 ng) or control vector (250 ng) was transiently cotransfected with 5 × IP-10-luc (5 × NFκB sites from the IP10 promoter, 100 ng) into wild-type, IKKα-, and IKKβ-null MEFs. After 48 h, the cells were harvested, followed by a luciferase reporter assay. The data are presented as fold induction of luciferase activity of the cells transfected with Act1 compared with wild-type MEFs transfected with control vector.
TAK1, another mitogen-activated protein kinase kinase kinase, has been implicated in activating IKK (16). TAB1, a TAK1 substrate, is phosphorylated in cells transfected with pCMV-Act1, compared with control cells (Fig. 2A). To determine whether Act1 forms a complex with TAK1, extracts of 293-TK/Zeo cells transfected with flag-tagged Act1 (CMV-Act1-flag) were immunoprecipitated with anti-TAK1 and probed with anti-flag (M2). Act1 was immunoprecipitated with TAK1, strongly suggesting that these two proteins form a complex (Fig. 2B). Moreover, kinase-dead dominant negative TAK1 inhibited Act1-dependent induction of the E-selectin promoter, indicating that Act1 functions upstream of TAK1 (Fig. 2C). Taken together, these results suggest that Act1 activates NFκB through the TAK1 and IKK signaling cascade.
Figure 2.

Interaction of Act1 with TAB1 and TAK1. (A) TAB1: Extract from 293-TK/Zeo cells with or without expression of flag-Act1 was either untreated (−) or treated (IL-1, 10 ng/ml, 30 min), followed by Western analysis with anti-TAB1. (B) TAK1: Extract from 293-TK/Zeo cells was immunoprecipitated with anti-TAK1 and probed with anti-flag (M2). Preimmune serum was used as a negative control. (C) Effect of DN-TAK1 on Act1-mediated NFκB activation: Increasing amounts of the kinase-dead mutant pCMV-TAK (K66-W) were transiently cotransfected with pCMV-Act1 and E-selectin-Luc into 293-TK/Zeo cells followed by a luciferase reporter assay 72 h later. The data presented are averages and SDs from three independent experiments.
Act1 Interacts with TRAF3.
To find a home for this orphan activator, we examined the interaction of Act1 with known receptor-proximal signaling components in NFκB-dependent signaling pathways. Because Act1 contains several putative TRAF-binding sites, we examined its interaction with TRAFs 1–6. We transfected flag-tagged TRAFs 1–6 into 293 cells already expressing HA-tagged Act1. Cell extracts were coimmunoprecipitated with anti-HA, followed by Western analysis with Ab against the flag-tag (M2). Although HA-tagged Act1 was immunoprecipitated with TRAF3 (Fig. 3), weak interactions of TRAFs 1 and 5 with Act1 were also observed. Because TRAF3 participates in signaling pathways mediated by CD40, LTβR, and several other members of the tumor necrosis factor receptor superfamily, the above result suggests that Act1 may play a role in pathways mediated by different receptors.
Figure 3.

Act1-interacting proteins. Flag-tagged TRAFs 1–6 in the pCMV vector were transfected transiently into 293 cells containing HA-Act1. Extracts of these transfected cells were immunoprecipitated with anti-HA and probed with anti-flag (M2) and anti-HA. Whole-cell extracts were examined directly by Western analysis with the above Abs as well, to show the expression of these signaling components after transfection.
Endogenous Act1 Is Recruited to CD40 After Stimulation with CD40L.
We transfected flag-tagged CD40 into 293 cells containing HA-tagged Act1. Extracts of the transfected cells, with or without stimulation, were coimmunoprecipitated with anti-HA and probed with anti-flag. Interestingly, whereas Act1 interacted weakly with CD40 in untreated cells, stimulation with CD40L greatly induced the interaction with CD40 (Fig. 4A). These results suggest that Act1 is recruited to the CD40 receptor after stimulation.
Figure 4.

Recruitment of Act1 to CD40 after CD40L stimulation. (A) Overexpression of Act1 and CD40: Flag-tagged CD40 was transfected into 293 cells transfected with HA-tagged Act1. Extracts from the transfected cells, untreated (0) or treated with CD40L (30 min), were immunoprecipitated with anti-HA and probed with anti-flag (M2) and anti-HA. (B) Overexpression of CD40: Extracts from HeLa cells transfected with flag-tagged CD40, untreated (0) or treated with CD40L for different times, were immunoprecipitated with anti-Act1 and probed with anti-flag (M2) and anti-Act1. (C) Western analysis of CD40: Extracts from various epithelial cells were analyzed by the Western procedure with anti-CD40. (D) Endogenous Act1 and CD40: Extracts from HeLa and HT29 cells, untreated (0) or treated with CD40L for different times, were immunoprecipitated with anti-Act1 and probed with anti-CD40 and anti-Act1.
Because the previous experiments were done with overexpressed Act1 and CD40, it was critical to examine whether endogenous Act1 can also interact with the stimulated CD40 receptor. Our recent studies indicate that Act1 is expressed well in epithelial cells of the respiratory and gastrointestinal tracts, and that its expression is augmented by proinflammatory mediators (Z.Z. and X.L., unpublished data). Therefore, we decided to use epithelial cell lines as models to study the functions of Act1. We transfected flag-tagged CD40 stably into HeLa cells, in which endogenous Act1 is highly expressed. Extracts of these transfected cells were immunoprecipitated with anti-flag (M2) and probed with anti-Act1 or vice versa. Endogenous Act1 is clearly recruited to the transfected CD40 after stimulation with CD40L. Furthermore, this recruitment is transient (Fig. 4B).
To test whether endogenous Act1 can be recruited to endogenous CD40, we first identified cell lines in which both endogenous Act1 and CD40 are expressed. By Western analysis, we found that both are expressed at high levels in HeLa and HT29 cells (Figs. 4C and 5B). Extracts of HeLa or HT29 cells, untreated or treated with CD40L, were immunoprecipitated with anti-Act1 and probed with anti-CD40. Importantly, we found that endogenous Act1 does indeed interact with CD40, both in HeLa and HT29 cells stimulated with CD40L (Fig. 4D). The interaction is transient, peaking at 5 min and disappearing after 30 min. Taken together, the above data clearly show that Act1 is recruited to CD40 after stimulation with CD40L, strongly suggesting that Act1 plays a critical role in CD40-mediated signaling.
Figure 5.

Role of Act1 in CD40-mediated signaling. (A) NFκB gel-shift assay: Extracts of C33A, HT29, A549, and HeLa cells untreated (−) or treated with CD40L (+, 30 min) were used for gel-shift assays. An NFκB-binding site from the IP-10 gene was used as a probe (Materials and Methods). (B) Western analysis: Extracts of various cell lines were used for Western analysis with anti-Act1. (C) NFκB gel shift: Extracts of C33A and C33A cells stably transfected with pCMV-Act1 (C33/Act1) and untreated (−) or treated (+, 30 min) with CD40L were used for gel-shift assay. (D) Western analysis: Extracts of C33A and C33A/Act1 cells were used for Western analysis with anti-Act1. (E) DNA fragmentation: Genomic DNA from C33A cells untreated (−) or stimulated (+) with CD40L for 24 h were analyzed on 2% agarose gel and visualized by ethidium bromide staining. (F) Western analysis: Extracts from various cell lines, untreated (−) or treated (+, 30 min) with CD40L for 24 h, were used for Western analysis with an Ab that recognizes the cleaved poly(ADP-ribose) polymerase (PARP).
Expression of Act1 Confers CD40L-Induced NFκB Activation in Act1-Null C33A Cells.
The CD40-mediated signaling pathway has been studied extensively in B cells, but the signaling events in this pathway are poorly understood in epithelial cells. We examined CD40-induced NFκB activation in these epithelial cells in a gel-shift assay. Whereas NFκB is strongly activated by CD40L in HT29 and HeLa cells, where Act1 is highly expressed and slightly activated in A549 cells that express a low level of Act1, CD40L failed to activate NFκB in C33A cells that do not express Act1 (Fig. 5 A and B). It is clear that the level of CD40L-induced NFκB activation correlates well with the levels of Act1 in these cells, suggesting that Act1 might be required for CD40-mediated NFκB activation.
To test this hypothesis, we examined whether the expression of Act1 in the natural Act1-null C33A cells can confer these cells sensitive to CD40L-induced NFκB activation by stably transfecting pCMV-Act1 into them. To avoid overexpression, we selected for clones that express very little Act1 (Fig. 5D). Stable transfection of pCMV-Act1 into C33A cells did render them susceptible to CD40L-induced NFκB activation (Fig. 5C), showing that Act1 can mediate this process.
Expression of Act1 Protects Act1-Null C33A Cells from CD40-Mediated Apoptosis.
Previous studies showed that the ligation of CD40 can lead to apoptosis in some epithelial cells (3). HT29, HeLa, A549, and C33A cells were treated with CD40L for 24 h and analyzed for the activation of caspase 3 by the Western method, with an Ab that recognizes caspase 3-cleaved poly(ADP-ribose) polymerase (PARP). CD40L activates apoptosis in C33A or A549 cells but not in HT29 or HeLa cells (Fig. 5F). The CD40L-induced apoptosis in C33A cells was confirmed by signal-induced DNA fragmentation (Fig. 5E). Taken together, these results show a correlation between the expression of Act1 and protection against apoptosis, which is likely to have resulted from Act1-mediated NFκB activation after stimulation with CD40L. To test this possibility further, we examined C33A cells stably transfected with pCMV-Act1 and found that the expression of Act1 suppressed CD40L-induced apoptosis (Fig. 5 D and F).
TRAF3 Inhibits Act1-Mediated NFκB Activation.
TRAF3 has been implicated in CD40-mediated JNK activation and may have proapoptotic activity but it is not involved in the activation of NFκB (29–33). Furthermore, previous studies showed that overexpression of TRAF3 inhibited CD40-mediated NFκB activation (31, 50, 51). Because we found that Act1 interacts with TRAF3 after stimulation with CD40L, we tested the effect of overexpressing it on Act1-mediated NFκB activation. Increasing amounts of TRAF3 were cotransfected with pCMV-Act1 and E-selectin luciferase into 293 cells, followed by a luciferase reporter assay. Interestingly, the overexpression of TRAF3 effectively inhibited NFκB activation mediated by Act1 (Fig. 6A) but had no effect on IRAK-induced NFκB activation (Fig. 6B).
Figure 6.

TRAF3 inhibits Act1-mediated NFκB activation. pCMV-Act1 (250 ng, A) or pTK-IRAK (250 ng, B) was transiently cotransfected with E-selectin-luc and increasing amounts of pCMV-TRAF3 into 293 cells, followed by a luciferase reporter assay. The fold induction is relative to that in cells transfected with vector DNA.
Discussion
Our previous studies showed that the overexpression of Act1 led to constitutive activation of both NFκB and JNK. We now find that NFκB is activated by overexpression of Act1 through the TAK1 and IKK signaling cascade. In addition, Act1 binds to TRAF3. Endogenous Act1 is recruited to the CD40 receptor in epithelial cells after stimulation with CD40L, strongly suggesting that it is involved in CD40-mediated signaling. Importantly, expression of Act1 in Act1-null C33A cells confers these cells CD40-mediated NFκB activation and protects them from CD40L-induced apoptosis. Taken together, our results indicate that Act1 functions as an important adapter in the CD40-mediated pathway.
Act1-mediated NFκB activation is regulated by one of the Act1-interacting proteins, TRAF3 (Fig. 6). Both TRAF3 and Act1 are recruited to the CD40 receptor after stimulation with CD40L and they also interact with each other. Previous studies showed that overexpression of TRAF3 inhibits CD40-mediated NFκB activation (31, 50, 51) and we now find that TRAF3 also inhibits NFκB activation, probably through its interaction with Act1. Importantly, TRAF3 has been shown to play an important role in LTβ-mediated apoptosis in epithelial cells (33), suggesting that TRAF3 may also be required for CD40-mediated apoptosis. The above information suggests that the levels of Act1 and TRAF3 may play key roles in determining whether CD40L activates NFκB or leads to apoptosis.
Although we have clearly shown that endogenous Act1 is recruited to CD40 after stimulation with CD40L, it is not clear how Act1 is recruited. As discussed in the introduction, several TRAF proteins, including TRAFs 2, 3, 5, and 6, have been shown to interact with CD40 and to play critical roles in CD40-mediated pathways in B cells. Act1 also contains two TRAF binding sites (EEESE, residues 38–42; EERPA, residues 333–337) and is shown in the current work to interact strongly with TRAF3 and weakly with TRAFs 1 and 5 after overexpression. The TRAFs may function as adapters, linking Act1 to the CD40 receptor after stimulation. Although TRAF3 seems to play a negative role in Act1-dependent CD40-mediated NFκB activation, the other TRAFs may help Act1 to mediate CD40-induced NFκB and JNK activation. Furthermore, the facts that different TRAF3 splice variants have been identified and that the overexpression of some of these variants induce NFκB activation reveal a complicated role for TRAF3 in CD40-mediated pathways (35, 36). Nevertheless, the work presented here leads us to propose a working model for role of Act1 in the CD40 pathway (Fig. 7). Act1 functions as a receptor proximal-adapter involved in CD40-mediated NFκB and JNK activation and inhibits CD40L-induced apoptosis in epithelial cells. Act1 is activated by its recruitment to the CD40 receptor through its interaction with TRAF proteins. Activated Act1 then links TRAFs to the TAK1/IKK signalsome, mediating the activation of NFκB, and to an unknown mitogen-activated protein kinase complex to activate JNK. The Act1-mediated activation of NFκB protects cells from apoptosis. These Act1-mediated signaling events in the CD40 pathway are regulated by the various interactions of Act1 with different TRAFs. The relative levels of Act1 and TRAFs may play critical roles in the life-or-death decisions of epithelial cells when stimulated with CD40L.
Figure 7.

A model of CD40-mediated signaling. TRAFs 2, 3, 5, and 6 are intermediary CD40-binding proteins. Act1 functions as an adapter, linking TRAF proteins to TAK1/IKK to activate NFκB and to mitogen-activated protein kinase complex to activate JNK. TRAF3 inhibits Act1-dependent CD40-mediated NFκB activation and initiates CD40L-induced apoptosis. Act1-dependent CD40-mediated NFκB activation in turn protects the cells from CD40L-induced apoptosis.
Acknowledgments
We thank Dr. George Stark for critical comments and careful editing of the manuscript. We thank Dr. Hoger Wesche for providing us with the mammalian expression constructs for TRAFs 1–6 and CD40 and CD40L. We thank Dr. Inder Verma for providing us with the IKKα-null and IKKβ-null MEFs. We thank Dr. Jun Ninomiya-Tsuji (Nagoya University, Nagoya, Japan) and Dr. Kunihiro Matsumoto (Nagoya University) for sending us the dominant negative mutant TAK1. This work was supported by National Institutes of Health Grant GM 600020 (to X.L.).
Abbreviations
- IKK
IκB kinase
- JNK
Jun kinase
- NIK
NFκB-inducing kinase
- Zeo
zeocin
- TK
thymidine kinase
- TRAF
tumor necrosis factor receptor-associated factor
- CMV
cytomegalovirus
- Act1
NFκB activator 1
- CD40L
CD40 ligand
- TAK1
transforming growth factor-β-activated kinase 1
- TAB1 and 2
transforming growth factor-β-activated kinase 1 binding protein 1 and 2
- MEF
mouse embryonic fibroblast
- HA
hemagglutinin
- DN
dominant negative
References
- 1.Banchereau J, Bazan F, Blanchard D, Briere F, Galizzi J P, van Kooten C, Liu Y J, Rousset F, Saeland S. Annu Rev Immunol. 1994;12:881–922. doi: 10.1146/annurev.iy.12.040194.004313. [DOI] [PubMed] [Google Scholar]
- 2.van Kooten C, Banchereau J. Int Arch Allergy Immunol. 1997;113:393–399. doi: 10.1159/000237614. [DOI] [PubMed] [Google Scholar]
- 3.Young L S, Eliopoulos A G, Gallagher N J, Dawson C W. Immunol Today. 1998;19:502–506. doi: 10.1016/s0167-5699(98)01340-1. [DOI] [PubMed] [Google Scholar]
- 4.Mach F, Schonbeck U, Sukhova G K, Bourcier T, Bonnefoy J Y, Pober J S, Libby P. Proc Natl Acad Sci USA. 1997;94:1931–1936. doi: 10.1073/pnas.94.5.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Young L S, Dawson C W, Brown K W, Rickinson A B. Int J Cancer. 1989;43:786–794. doi: 10.1002/ijc.2910430508. [DOI] [PubMed] [Google Scholar]
- 6.Stamenkovic I, Clark E A, Seed B. EMBO J. 1989;8:1403–1410. doi: 10.1002/j.1460-2075.1989.tb03521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biancone L, Cantaluppi V, Camussi G. Int J Mol Med. 1999;3:343–353. doi: 10.3892/ijmm.3.4.343. [DOI] [PubMed] [Google Scholar]
- 8.DiDonato J A, Hayakawa M, Rothwarf D M, Zandi E, Karin M. Nature (London) 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 9.Mercurio F, Zhu H, Murray B W, Shevchenko A, Bennett B L, Li J, Young D B, Barbosa M, Mann M, Manning A, et al. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 10.Regnier C H, Song H Y, Gao X, Goeddel D V, Cao Z, Rothe M. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- 11.Woronicz J D, Gao X, Cao Z, Rothe M, Goeddel D V. Science. 1997;278:866–869. doi: 10.1126/science.278.5339.866. [DOI] [PubMed] [Google Scholar]
- 12.Zandi E, Rothwarf D M, Delhase M, Hayakawa M, Karin M. Cell. 1997;91:243–252. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 13.Karin M, Ben Neriah Y. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 14.Ghosh S, May M J, Kopp E B. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 15.Sizemore N, Lerner N, Dombrowski N, Sakurai H, Stark G R. J Biol Chem. 2002;277:3863–3869. doi: 10.1074/jbc.M110572200. [DOI] [PubMed] [Google Scholar]
- 16.Kishimoto K, Matsumoto K, Ninomiya-Tsuji J. J Biol Chem. 2000;275:7359–7364. doi: 10.1074/jbc.275.10.7359. [DOI] [PubMed] [Google Scholar]
- 17.Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K. Nature (London) 1999;398:252–256. doi: 10.1038/18465. [DOI] [PubMed] [Google Scholar]
- 18.Irie T, Muta T, Takeshige K. FEBS Lett. 2000;467:160–164. doi: 10.1016/s0014-5793(00)01146-7. [DOI] [PubMed] [Google Scholar]
- 19.Takaesu G, Kishida S, Hiyama A, Yamaguchi K, Shibuya H, Irie K, Ninomiya-Tsuji J, Matsumoto K. Mol Cell. 2000;5:649–658. doi: 10.1016/s1097-2765(00)80244-0. [DOI] [PubMed] [Google Scholar]
- 20.Takaesu G, Ninomiya-Tsuji J, Kishida S, Li X, Stark G R, Matsumoto K. Mol Cell Biol. 2001;21:2475–2484. doi: 10.1128/MCB.21.7.2475-2484.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qian Y, Commane M, Ninomiya-Tsuji J, Matsumoto K, Li X. J Biol Chem. 2001;276:41661–41667. doi: 10.1074/jbc.M102262200. [DOI] [PubMed] [Google Scholar]
- 22.Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen Z J. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 23.Wang C, Deng L, Hong M, Akkaraju G R, Inoue J, Chen Z J. Nature (London) 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 24.Lee J, Mira-Arbibe L, Ulevitch R J. J Leukocyte Biol. 2000;68:909–915. [PubMed] [Google Scholar]
- 25.Mizukami J, Takaesu G, Akatsuka H, Sakurai H, Ninomiya-Tsuji J, Matsumoto K, Sakurai N. Mol Cell Biol. 2002;22:992–1000. doi: 10.1128/MCB.22.4.992-1000.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yin L, Wu L, Wesche H, Arthur C D, White J M, Goeddel D V, Schreiber R D. Science. 2001;291:2162–2165. doi: 10.1126/science.1058453. [DOI] [PubMed] [Google Scholar]
- 27.Garceau N, Kosaka Y, Masters S, Hambor J, Shinkura R, Honjo T, Noelle R J. J Exp Med. 2000;191:381–386. doi: 10.1084/jem.191.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bishop G A, Hostager B S. Immunol Res. 2001;24:97–109. doi: 10.1385/IR:24:2:097. [DOI] [PubMed] [Google Scholar]
- 29.Grammer A C, Swantek J L, McFarland R D, Miura Y, Geppert T, Lipsky P E. J Immunol. 1998;161:1183–1193. [PubMed] [Google Scholar]
- 30.Dadgostar H, Cheng G. J Biol Chem. 2000;275:2539–2544. doi: 10.1074/jbc.275.4.2539. [DOI] [PubMed] [Google Scholar]
- 31.Hostager B S, Bishop G A. J Immunol. 1999;162:6307–6311. [PubMed] [Google Scholar]
- 32.Eliopoulos A G, Dawson C W, Mosialos G, Floettmann J E, Rowe M, Armitage R J, Dawson J, Zapata J M, Kerr D J, Wakelam M J, et al. Oncogene. 1996;13:2243–2254. [PubMed] [Google Scholar]
- 33.VanArsdale T L, VanArsdale S L, Force W R, Walter B N, Mosialos G, Kieff E, Reed J C, Ware C F. Proc Natl Acad Sci USA. 1997;94:2460–2465. doi: 10.1073/pnas.94.6.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu Y, Cheng G, Baltimore D. Immunity. 1996;5:407–415. doi: 10.1016/s1074-7613(00)80497-5. [DOI] [PubMed] [Google Scholar]
- 35.van Eyndhoven W G, Frank D, Kalachikov S, Cleary A M, Hong D I, Cho E, Nasr S, Perez A J, Mackus W J, Cayanis E, et al. Mol Immunol. 1998;35:1189–1206. doi: 10.1016/s0161-5890(98)00099-6. [DOI] [PubMed] [Google Scholar]
- 36.Gamper C, Omene C O, van Eyndhoven W G, Glassman G D, Lederman S. Hum Immunol. 2001;62:1167–1177. doi: 10.1016/s0198-8859(01)00284-1. [DOI] [PubMed] [Google Scholar]
- 37.Schonbeck U, Libby P. Cell Mol Life Sci. 2002;58:4–43. doi: 10.1007/PL00000776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nguyen L T, Duncan G S, Mirtsos C, Ng M, Speiser D E, Shahinian A, Marino M W, Mak T W, Ohashi P S, Yeh W C. Immunity. 1999;11:379–389. doi: 10.1016/s1074-7613(00)80113-2. [DOI] [PubMed] [Google Scholar]
- 39.Lomaga M A, Yeh W C, Sarosi I, Duncan G S, Furlonger C, Ho A, Morony S, Capparelli C, Van G, Kaufman S, et al. Genes Dev. 1999;13:1015–1024. doi: 10.1101/gad.13.8.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakano H, Sakon S, Koseki H, Takemori T, Tada K, Matsumoto M, Munechika E, Sakai T, Shirasawa T, Akiba H, et al. Proc Natl Acad Sci USA. 1999;96:9803–9808. doi: 10.1073/pnas.96.17.9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shinkura R, Kitada K, Matsuda F, Tashiro K, Ikuta K, Suzuki M, Kogishi K, Serikawa T, Honjo T. Nat Genet. 1999;22:74–77. doi: 10.1038/8780. [DOI] [PubMed] [Google Scholar]
- 42.Luftig M A, Cahir-McFarland E, Mosialos G, Kieff E. J Biol Chem. 2001;276:14602–14606. doi: 10.1074/jbc.C100103200. [DOI] [PubMed] [Google Scholar]
- 43.Jain A, Ma C A, Liu S, Brown M, Cohen J, Strober W. Nat Immunol. 2001;2:223–228. doi: 10.1038/85277. [DOI] [PubMed] [Google Scholar]
- 44.Li X, Commane M, Burns C, Vithalani K, Cao Z, Stark G R. Mol Cell Biol. 1999;19:4643–4652. doi: 10.1128/mcb.19.7.4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li X, Commane M, Nie H, Hua X, Chatterjee-Kishore M, Wald D, Haag M, Stark G R. Proc Natl Acad Sci USA. 2000;97:10489–10493. doi: 10.1073/pnas.160265197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leonardi A, Chariot A, Claudio E, Cunningham K, Siebenlist U. Proc Natl Acad Sci USA. 2000;97:10494–10499. doi: 10.1073/pnas.190245697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schindler U, Baichwal V R. Mol Cell Biol. 1994;14:5820–5831. doi: 10.1128/mcb.14.9.5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Majumder S, Zhou L Z, Chaturvedi P, Babcock G, Aras S, Ransohoff R M. J Neurosci Res. 1998;54:169–180. doi: 10.1002/(SICI)1097-4547(19981015)54:2<169::AID-JNR5>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 49.Kessler D S, Veals S A, Fu X Y, Levy D E. Genes Dev. 1990;4:1753–1765. doi: 10.1101/gad.4.10.1753. [DOI] [PubMed] [Google Scholar]
- 50.Takaori-Kondo A, Hori T, Fukunaga K, Morita R, Kawamata S, Uchiyama T. Biochem Biophys Res Commun. 2000;272:856–863. doi: 10.1006/bbrc.2000.2860. [DOI] [PubMed] [Google Scholar]
- 51.Urbich C, Mallat Z, Tedgui A, Clauss M, Zeiher A M, Dimmeler S. J Clin Invest. 2001;108:1451–1458. doi: 10.1172/JCI13620. [DOI] [PMC free article] [PubMed] [Google Scholar]