

Abstract
Interferon regulatory factor (IRF)7 is a key transcription factor required for establishment of antiviral resistance. In response to infection, IRF7 is activated by phosphorylation through the action of the non-canonical IκB kinases, IκB kinase-ε and TANK-binding kinase 1. Activation leads to nuclear retention, DNA binding, and de-repression of transactivation ability. Clusters of serine residues located in the carboxyl-terminal regulatory domain of IRF7 are putative targets of virus-activated kinases. However, the exact sites of phosphorylation have not yet been established. Here, we report a comprehensive structure-activity examination of potential IRF7 phosphorylation sites through analysis of mutant proteins in which specific serine residues were altered to alanine or aspartate. Phosphorylation patterns of these mutants were analyzed by two-dimensional gel electrophoresis, and their transcriptional activity was monitored by reporter assays. Essential phosphorylation events were mapped to amino acids 437–438 and a redundant set of sites at either amino acids 429–431 or 441. IRF7 recovered from infected cells was heterogeneously phosphorylated at these sites, and greater phosphorylation correlated with increased transactivation. Interestingly, a distinct serine cluster conserved in the related protein IRF3 was also essential for IRF7 activation and distal phosphorylation. However, the essential role of this motif did not appear to be fulfilled by phosphorylation. Rather, these serine residues and an adjacent leucine were required for phosphorylation at distal sites and may determine a conformational element required for function.
Production of type I interferon in response to a wide variety of bacterial and viral infections is a major component of innate immunity and is essential to host defense against microbial invasion. Type I interferon (IFN)1 family consists of a single IFNβ gene and multiple IFNα genes that are clustered on mouse chromosome 4 (1) and transcriptionally activated in virus-infected cells. Transcriptional induction of the IFNα/β genes is a rapid biphasic process in most cell types; initial induction of IFNα4 and -β is dependent on the constitutively expressed transcription factor IRF3 and subsequent induction of the other members of the IFNα family (IRF7-dependent) is achieved after positive feedback resulting in IRF7 protein production and activation (2, 3).
Similar to many signaling pathways, the activating switch for transcription is achieved by phosphorylation of specific transcription factors. Transcription of the IFNβ gene is controlled by an enhanceosome composed of at least the AP1 transcription factor (c-jun-ATF2), phosphorylated by the c-Jun kinase, NFκB, released from its inhibitor IκB after phosphorylation-induced degradation through the action of the IκB kinase (composed of IKKα, IKKβ, and IKKγ/NEMO), and IRF3 and IRF7, activated by phosphorylation. The kinases thought responsible for phosphorylating IRF3 and IRF7 have been recently identified as distant members of the IKK family, TBK1 and IKK-ε(or IKKi) (4, 5). Similarly to IRF3 and IRF7, TBK1 is constitutively expressed, whereas IKK-ε is mainly induced by stress stimuli, such as tumor necrosis factor α and lipopoly-saccharide (6–8). Phosphorylation takes place on a stretch of serine/threonine residues in the carboxyl-terminal regulatory domain of the proteins. The primary structure of these two proteins is closely related, and, notably, an SSL motif, which has been shown to be essential for transcriptional activity, is conserved between IRF3 and IRF7 of every studied species. However, the IRF7 regulatory domain contains many more serine/threonine residues, and the exact phosphorylation sites have yet to be determined precisely. Although considerable work has been reported concerning the target sites of IRF3, much less has been done regarding the identification of IRF7 phosphorylation sites. As it has been described for IRF3, our previous observation showed that mutation of the proximal set of serine residues 425 and 426 of murine IRF7 into alanine abrogated virus-induced phosphorylation and transcriptional activity (2). However, we could not exclude the possibility that these residues were indispensable for proper conformation of the regulatory domain and that other residues were the bona fide targets of the virally induced kinases. Most recent work by Lin et al. (9) and Yang et al. (10) on human IRF7 suggested that distal serine residues downstream of serines 425 and 426 could also play a role in virus-induced activation. However, Ser479, one residue believed to be a major target, is not conserved in murine IRF7 or IRF3.
In this study, we investigated the relative importance of the proximal and distal sets of serine residues in the carboxyl-terminal regulatory domain of IRF7. We show that multiple serine residues are target for phosphorylation. In particular, two distinct groups of serine, Ser425 and Ser426 and Ser437 and Ser438, are indispensable for virally induced activation of IRF7 in a partially redundant manner. Furthermore, we demonstrate that secondary phosphorylation sites synergize with the primary targets, Ser437 and Ser438, to achieve maximal activation in response to viral infection. Moreover, we provide several lines of evidence that the essential nature of the proximal group of serine residues may not be mediated by phosphorylation. Instead, these residues are crucial for phosphorylation at distal sites possibly by maintaining the proper conformation of the carboxyl-terminal transactivation domain or by serving as a recognition motif for the virally activated kinases.
MATERIALS AND METHODS
Cell Culture
Human embryonic kidney 293T cells, monkey kidney COS cells, and murine TBK1-deficient immortalized embryo fibroblasts (a kind gift from W.-C. Yeh) were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% calf serum.
Transfections, Luciferase Assays, and Viral Infections
DNA transfections of 293T and COS cells were carried out by using standard calcium phosphate coprecipitation and DEAE-dextran methods, respectively. Transfection of TBK1-deficient fibroblasts was performed using Lipofectamine 2000 (Invitrogen). Luciferase activities were measured in cell lysates using commercial reagents as recommended by the manufacturer (Promega) and were normalized to β-galactosidase activity of a cotransfected CMV-lacZ plasmid measured on a luminescent substrate (Tropix). Each construct was tested in duplicate in at least three independent experiments. Results shown are from a single experiment representative of results obtained. Newcastle disease virus (NDV), Manhattan strain, was grown in 10-day embryonated chicken eggs (a kind gift from A. Garcia-Sastre), and viral infections were performed as previously described (2).
Plasmid Constructs and Mutagenesis
FLAG-tagged full-length murine IRF7 cDNA cloned into pcDNA3 and IFNα6-luciferase constructs have been described previously (2). FLAG-tagged IKKε and FLAG-tagged IKKεK38A expression vectors were kind gifts from T. Maniatis (7), and the TBK1 expression vector was a kind gift from D. Baltimore (6). The expression constructs encoding different IRF7 point mutants (M1–M18) were generated by site-directed mutagenesis (Stratagene) by using Pfu Turbo polymerase and were confirmed by DNA sequencing.
In Vitro Kinase Assay
In vitro kinase assays for IKKε were performed after overexpression of the FLAG-tagged protein in 293T cells. After immunoprecipitation using anti-FLAG (M2) beads (Sigma), the recovered protein was incubated in 20 μl of kinase buffer (50 mm Hepes, pH 7.6, 50 mm NaCl, 10 mm -glycerophosphate, 1 mm MgCl2, 20 mm β- glycerophosphate NaF, 1 mm dithiothreitol) in the presence of 10 μCi of [γ-32P]ATP and 1 μl of WT or mutant peptide at a final concentration of 0, 50, 100, or 200 μm at 30 °C for 30 min and analyzed by 10% SDS-PAGE and quantification using a PhosphorImager. Peptides were custom synthesized at the Keck Foundation, Yale University, using standard Fmoc (N-(9-fluorenyl)methoxycarbonyl) chemistry.
Two-dimensional Gel Analysis
Nuclear extracts for isoelectric focusing were prepared as described previously (11). Extracts were loaded onto acrylamide gel strips containing 15 m urea, 3.5% Nonidet P-40, and 8% Pharmalyte™ 5–8 ampholins (Amersham Biosciences) and run at 200 V for 15 min, 300 V for 30 min, and 400 V overnight, followed by 800 V for 1 h. Gel strips were then equilibrated in 125 mm Tris-HCl, pH 6,8, 1% SDS, and 20% glycerol for 1 h at room temperature prior to sealing on 7.5% SDS-PAGE for the second dimension electrophoresis. Gels were transferred to polyvinylidene difluoride membranes and processed by Western blotting using anti-FLAG (M2) antibodies.
Electrophoretic Mobility Shift Assay
Nuclear extracts of 293T transfected cells were prepared as previously described (11). The electrophoretic mobility shift assay was performed by incubating nuclear extracts of each sample (2 μg) with a 32P-labeled double stranded oligonucleotide corresponding to the IFN-stimulated response element sequence derived from the ISG15 gene (12), as described previously (13).
RESULTS
IRF7 Is Phosphorylated on Multiple Serine Residues within Its Carboxyl-terminal Regulatory Domain
Virally induced phosphorylation is a prerequisite to initiate the presumed conformational changes responsible for unmasking the potent transactivation ability of IRF7. Previous work has mapped the phosphorylation events to a serine/threonine-rich domain in the carboxyl terminus of IRF7 and shown that a truncated mutant lacking the last 34 amino acids was unresponsive to viral infection (2, 3). Targeted phosphorylation was further confirmed by phospho-amino acid analysis and phospho-peptide mapping of tryptic digests of IRF7. Virus infection led to an increase in IRF7 phosphoserine and the appearance of a single predominant novel V8 phosphopeptide.2 The 34-amino acid regulatory domain of IRF7 contains 8 serine residues that are potential phosphorylation sites (Fig. 1A). However, which of these residues becomes phosphorylated has not been determined.
Fig. 1. Two-dimensional gel analysis of IRF7.
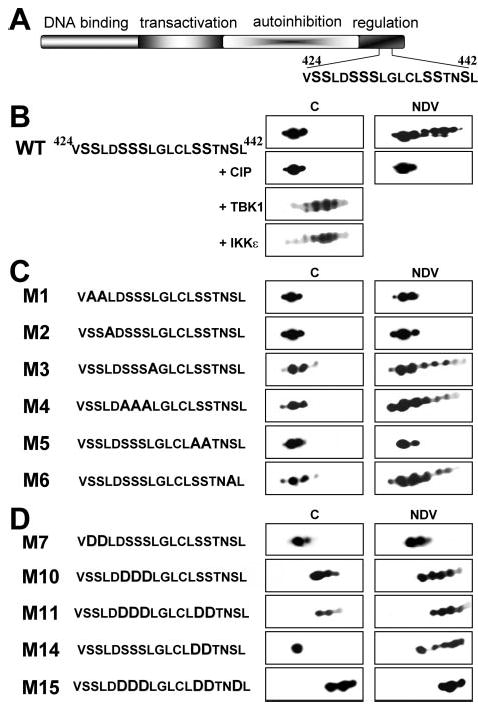
A, diagram of IRF7 functional domains, indicated by different shading and labeled above. The sequence of amino acids 424–442 containing the serine-rich domain is shown. B, two-dimensional gel analysis of WT IRF7. Nuclear extracts from IRF7-Flag-transfected, control (C) or NDV-infected (NDV) 293T cells were subjected to two-dimensional gel analysis and revealed after Western blotting using anti-FLAG antibodies. The panel indicated +CIP shows the same extracts after treatment with alkaline phosphatase. The two lower panels show a two-dimensional gel analysis of extracts from 293T cells cotransfected with WT IRF7 and TBK1 or IKK-ε (as indicated). C and D, same as B except that 293T cells were transfected with different IRF7 mutants (as indicated). The sequence of the serine-rich domain of WT and each mutant of IRF7 is indicated on the left of the figure.
One of the assays commonly used to monitor IRF7 phosphorylation status is the appearance of a slower migrating band on SDS-PAGE. Although sensitive to qualitative changes in phosphorylation, this method cannot distinguish among multiple potential sites of phosphorylation. An alternative technique to detect different post-translational variants of the same protein is two-dimensional gel analysis. As shown in Fig. 1B, WT IRF7 from uninfected cells can be resolved into three distinct electrophoretic subspecies. Although these different spots are very likely due to post-translational modifications, treatment with alkaline phosphatase did not affect their distribution, suggesting that phosphorylation is not the cause of the observed pattern. However, when a similar analysis was performed on extracts from NDV-infected cells, 4–5 additional spots were resolved. These additional isoforms of IRF7 were indeed due to phosphorylated species, because they disappeared after phosphatase treatment, collapsing into the basal pattern. This result shows that IRF7 can be phosphorylated at multiple distinct sites. It also indicates that virus infection does not achieve complete phosphorylation of the protein, leading to coexistence of several species bearing different degrees of phosphorylation.
Interestingly, overexpression of either TBK1 or IKKε, kinases responsible for IRF7 phosphorylation, led to complete phosphorylation, shown by almost total disappearance of the pre-existing spots (Fig. 1B). This result showed that each of the three pre-existing species could be phosphorylated and that action of either kinase was sufficient to produce the same bulk phosphorylation pattern. Notably, overexpression of both kinases together did not lead to further alteration of the two-dimensional pattern (data not shown).
Two Groups of Serine Are Necessary for Virally Induced Phosphorylation of IRF7
To determine the major targets of virally induced phosphorylation, we mutated each contiguous group of serine residues in the regulatory domain into alanine, a residue that cannot be phosphorylated. The status of phosphorylation of the different mutants was monitored by two-dimensional gel analysis before and after viral infection. This analysis revealed that loss of two distinct sets of serine residues, Ser425 and Ser426 and Ser437 and Ser438, rendered IRF7 unresponsive to viral infection. In contrast, mutation of the other two sets of serines, Ser429–Ser431 and Ser441, did not show a significant alteration in the overall phosphorylation pattern, although the efficiency of phosphorylation appeared to be reduced (Fig. 1C). We noted that leucine residues are associated with these serine residues, suggesting a potential phosphorylation motif. Specifically, each group of serine residues is either preceded or followed by a leucine residue. To address the importance of these leucine residues, we mutated individually Leu427 and Leu432 into alanines. As shown in Fig. 1C, mutation of Leu427 abrogated virally induced phosphorylation, whereas mutant L432A showed the same phosphorylation pattern as WT IRF7. Notably, this result is consistent with what was observed when the adjacent serine sets were altered, in other words, it seems that the motif SSL (amino acids 425–427) has a preponderant role in virally induced phosphorylation, whereas the next motif SSSL (amino acids 429–432) has a minor role, if any.
Ser437 and Ser438 Are the Primary Targets of Phosphorylation
To better determine the respective roles of each group of serines, we undertook another series of mutations. The different sets of serine residues were altered to aspartate, a residue that mimics the charge of a phosphorylated serine and can often substitute for its function. Analysis by two-dimensional gel confirmed that these mutants had an altered charge. In addition, we observed that most mutants were still further phosphorylated in response to viral infection, as attested by the appearance of new spots toward the acidic end of the pH gradient (Fig. 1D). Interestingly, the pattern of mutant M7 (S425D/S426D) was not modified after viral infection suggesting that either this mutation rendered the protein unresponsive to viral infection or that these two serines were the only targets for phosphorylation. It is unlikely that Ser425 and Ser426 are the only sites of phosphorylation given the multiple isoelectric species detected after phosphorylation of the wild type protein (Fig. 1B), indicative of more than two phosphorylated residues. Moreover, mutant M15, where all the serines except Ser425 and Ser426 were mutated into aspartate, showed a dramatic shift toward the acidic pH, resembling the fully phosphorylated protein and an unchanged pattern when analyzed from NDV-infected extracts. One hypothesis that reconciles these observations is that the distal serine residues are major targets for virus-induced phosphorylation, whereas Ser425 and Ser426 are necessary for kinase targeting but are either not themselves phosphorylated or their function is not correctly mimicked by aspartate substitution.
Transcriptional Activity of IRF7 Mutants
Although informative, two-dimensional gel migration patterns were not sufficient to give an answer concerning the functionality of the different residues, especially considering the limitations of phosphomimetic mutants. To complement this study, it was essential to determine the level of activity of each mutant in control and infected cells. For this purpose, we cotransfected WT IRF7 and the different mutants in COS cells along with a luciferase reporter construct driven by the IFNα6 promoter, a typical target gene of IRF7 (2). As shown previously, WT IRF7 transactivation ability was greatly enhanced (over 8-fold) following viral infection (2). However, mutation of Ser425 and Ser426 or Ser437 and Ser438 totally abrogated virally induced activity (Fig. 2A). Moreover, mutant M2 (L427A) also showed a complete loss of viral responsiveness. The other mutants, where Ser429–Ser431, Ser441, or Leu432 were mutated, were not significantly impaired in their viral sensitivity compared with WT. These results are fully consistent with what was observed with the phosphorylation status, reaffirming that virally induced phosphorylation is the key regulatory event to derepress transactivation ability.
Fig. 2. Transcriptional activity of IRF7 mutants.
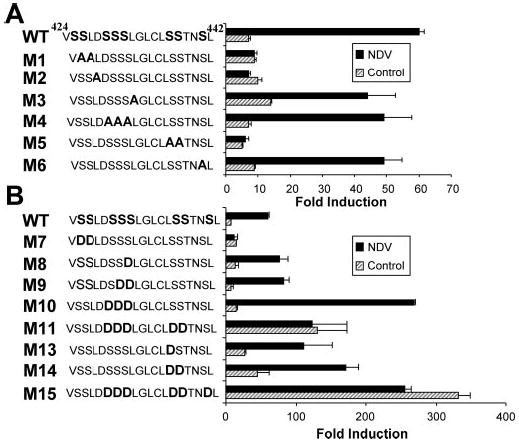
A and B, transactivation of the IFNα6 promoter by different mutants of IRF7. COS cells were transfected with WT IRF7 or the indicated IRF7 mutants along with a luciferase reporter driven by the IFNα6 promoter. At 24 h after transfection, cells were mock-infected (hatched bars) or infected with NDV for 12 h (solid bars) before being assayed for luciferase activity. The values are expressed as -fold induction relative to cells transfected with empty vector after normalization to cotransfected β-galactosidase. Mean values from a single representative experiment performed in duplicate are shown. Each mutant was tested in at least three separate experiments. The sequence of the serine-rich domain of WT and each mutant of IRF7 is indicated on the left of the figure.
A more complicated situation was observed when the activity of the phosphomimetic mutants was measured. Consistent with the finding that mutation of Ser429–Ser431 to Ala did not have a noticeable effect on the virus-induced pattern or transactivation potential, mutation of the same residues into Asp, either individually or taken together (mutants M8, M9, and M10), did not render the mutants constitutively active. However, M10 reached a level of activity 4-fold higher than WT in the presence of virus (Fig. 2B). This acute sensitivity to viral infection suggests that these residues (Ser429–Ser431) are not, or at most very inefficiently, phosphorylated in response to viral infection. However, if they were to become phosphorylated, as mimicked by the aspartate mutation, they could cooperate with the residues that are primary targets for the virus-activated kinases, yielding higher transactivation potential. Again, as predicted by the phenotype observed after mutation into alanine, M14 (S437D/S438D) showed a significant constitutive activity in absence of virus, 7-fold higher than the WT level, consistent with these residues being the major targets for virally induced phosphorylation. Interestingly, additional mutation of serine residues 429–431 (M11) rendered the mutant highly constitutively active, reaching levels of activity in the range of the wild type protein following viral infection. Furthermore, when we generated a mutant (M15) where Ser441 was mutated into aspartate in addition to the five mutations present in M11, we obtained a protein that displayed even a greater activity, reaching levels 5-fold higher than WT IRF7 activated by viral infection. It is important to note that activity of both M11 and M15 was not further increased by viral infection, suggesting that no additional phosphorylation could take place. We conclude that phosphorylation of Ser437 and Ser438 is required for virus-induced transactivation, whereas phosphorylation of Ser429–Ser431 and/or Ser441 can augment transactivation and/or the efficiency of distal phosphorylation.
This notion raised the question of the physiological role of the first set of serines, Ser425 and Ser426, that were previously found to be essential to virally induced phosphorylation and activity (Ref. 2 and Figs. 1C and 2A). Surprisingly, when Ser425 and Ser426 were mutated into aspartate, the resulting mutant (M7) did not exhibit constitutive activity but rather became insensitive to viral infection. This phenotype is consistent with our previous finding that this mutant could not be phosphorylated in response to viral infection (Fig. 1D) and suggests that aspartate cannot mimic the essential role of these serine residues.
Transactivation ability is the ultimate indication of IRF7 activity. Nevertheless, lack of or increased activity of the mutants could be because of alteration of other parameters that are a prerequisite to IRF7 transactivation ability, namely dimerization, retention in the nucleus, or DNA binding. We therefore tested the DNA binding ability of several mutants. As previously described (14), DNA binding by WT IRF7 was dramatically increased in NDV-infected extracts. Interestingly, constitutively active mutants M14 and M15 showed highly increased DNA binding capabilities in control extracts compared with WT. Whereas the DNA binding ability of M14 was further enhanced after viral infection, M15 exhibited the same degree of DNA binding in control and NDV-infected extracts (Fig. 3). These findings are reminiscent of what was observed when we monitored IRF7 activity by luciferase assay, showing a strong correlation between stimulation of DNA binding and transcriptional activity.
Fig. 3. DNA binding ability of IRF7 mutants.
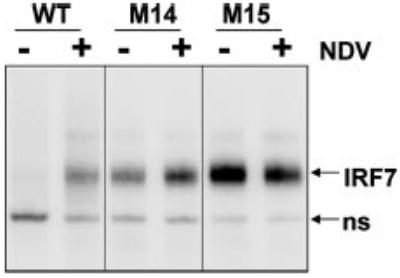
Nuclear extracts derived from 293T cells transfected with WT IRF7 or mutants M14 and M15 (as indicated) that had been mock- or NDV-infected for 8 h were subjected to electrophoretic mobility shift assay using an interferon-stimulated response element probe derived from the ISG15 gene.
Essential Functions of Ser425 and Ser426
The fact that the mutation of Ser425 and Ser426 to aspartate did not confer increased activity suggested that these residues might not be targets of phosphorylation, although they played an essential role in IRF7 function. We considered whether their critical role impacted proper conformation of the regulatory domain or served as a recognition site for the virally activated kinases, IKK-ε and TBK1.
To test this hypothesis, we mutated the proximal group of serines, Ser425 and Ser426 either to alanine or aspartate, in the context of the constitutively active phosphomimetic mutants M11 and M15. We reasoned that, if the only function of the proximal set of serines was to provide a docking site for the kinases that will subsequently phosphorylate the distal group of serine (437 and 438), their mutation in the context of constitutively active mutants would have a limited impact. Unexpectedly, as shown in Fig. 4A, these mutations partially impaired the activity of the constitutively active mutants (compare mutant M12 with M11 and mutants M16 and M17 with M15). These results indicated that Ser425 and Ser426 were critical, even when all other serine residues in the regulatory domain available for phosphorylation were already mutated into aspartate, suggesting either that these serines are obligatory phosphorylation targets or that they are mandatory for adequate folding of the carboxyl-terminal transactivation domain. To determine whether these residues were phosphorylated in the context of M15, we compared the two-dimensional pattern of M15 with M17 where Ser425 and Ser426 are mutated into alanine. As shown in Fig. 4B, M17 lost the more acidic minor spot present in M15, suggesting that M15 may indeed be partially phosphorylated on residues Ser425 and Ser426. However, mutation of Ser425 and Ser426 into either Asp or Ala reduced but did not abrogate the constitutive activity of mutant M11 and M15. Mutants M12 and M17 still exhibited a transactivation ability 5–8-fold higher than WT IRF7 in control cells, reaching levels of activation close to that of viral activated WT IRF7 (Fig. 4A). This result indicates that although Ser425 and Ser426 are important to achieve maximal activity, these residues are not compulsory to obtain a constitutively active mutant.
Fig. 4. Ser425 and Ser426 are critical but cannot be substituted by phosphomimetic residues.
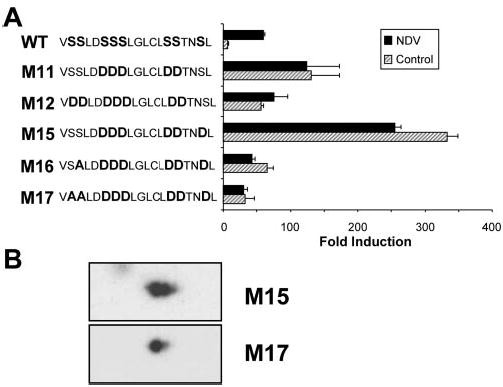
A, COS cells were transfected with WT IRF7 or the indicated IRF7 mutants along with a luciferase reporter driven by the IFNα6 promoter. At 24 h after transfection, cells were mock-infected (hatched bars) or infected with NDV for 12 h (solid bars) before being assayed for luciferase activity. The values are expressed as fold induction relative to cells transfected with empty vector after normalization to cotransfected β-galactosidase. Mean values from a single representative experiment performed in duplicate are shown. Each mutant was tested in at least three separate experiments. The sequence of the serine-rich domain of WT and each mutant of IRF7 is indicated on the left of the figure. B, two-dimensional gel analysis of IRF7 mutants M15 and M17. Nuclear extracts from IRF7M15-Flag and IRF7M17-Flag-transfected 293T cells were subjected to two-dimensional gel analysis and revealed after Western blotting using anti-FLAG antibodies.
Nevertheless, this result did not definitively exclude the possibility that these residues also serve as a recognition motif in the context of the WT protein. To address this point, we designed a synthetic peptide encompassing the sequence surrounding the proximal SSL motif, i.e. amino acids 422–435, fused to the protein transduction domain (amino acid 47–57) of the human immunodeficiency virus type 1 transactivator protein Tat (15). As a control, we also synthesized a mutant peptide bearing the same sequence except that the SSL motif was mutated into triple alanine (Fig. 5A). The potential interaction between the peptides and the IRF7 kinase, IKK-ε, was tested by quantifying the level of IKK-ε autophosphorylation in an in vitro kinase assay in presence or absence of the WT or mutant peptide (Fig. 5B). WT peptide strongly inhibited IKK-ε autophosphorylation in a dose-dependent manner, suggesting that the kinase efficiently interacted with the peptide. The corresponding mutant peptide also showed some degree of inhibition, although this inhibition did not show a clear dose-response effect, suggesting that it was likely to be low affinity or nonspecific (Fig. 5B).
Fig. 5. Phosphorylation-independent role of Ser425 and Ser426.
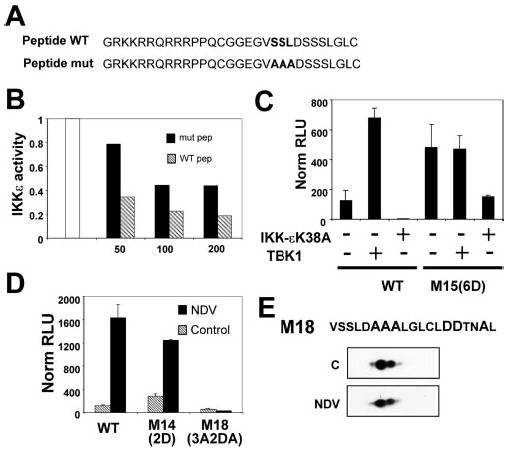
A, sequence of the synthetic peptides used in B. The peptides are composed of the sequence surrounding the proximal SSL motif i.e. amino acids 422–435 (either WT or mutant (mut) as indicated) fused to the transduction domain (amino acids 47–57) of the human immunodeficiency virus type 1 transactivator protein TAT. B, activity of the kinase IKK-ε was monitored by in vitro kinase assay in presence or absence of increasing amounts (50, 100, 200 μm as indicated) of the WT (hatched bars) or mutant (solid bars) synthetic peptides (pep) described in A, as indicated. The results are expressed as fractions of the activity in absence of peptide normalized to 1 (white bar). C, TBK1 deficient fibroblasts were transfected with WT IRF7 or the mutant M15 along with a luciferase reporter driven by the IFNα6 promoter and expression plasmids encoding either WT TBK1 or a dominant negative form of IKK-ε, IKK-εK38A, as indicated. Luciferase activity was assayed 48 h after transfection. The values are expressed as relative luciferase units after normalization to cotransfected β-galactosidase. Mean values of duplicates from a single representative of three independent experiments are shown. D, COS cells were transfected with WT IRF7 or the IRF7 mutants, M14 and M18, as indicated, along with a luciferase reporter driven by the IFNα6 promoter. At 24 h after transfection, cells were mock-infected (hatched bars) or infected with NDV for 12 h (solid bars) before being assayed for luciferase activity. The values are expressed as fold induction relative to cells transfected with empty vector after normalization to cotransfected β-galactosidase. Mean values from a single representative experiment performed in duplicate are shown. Each mutant was tested in at least three separate experiments. E, two-dimensional gel analysis of IRF7 mutant M18. Nuclear extracts from IRF7M18-Flag-transfected, control or NDV-infected 293T cells (C and NDV, respectively) were subjected to two-dimensional gel analysis and revealed after Western blotting using anti-FLAG antibodies. The sequence of the serine-rich domain of M18 (amino acids 424–442) is shown on top.
Mori et al. (16) recently proposed that constitutive activity of IRF3-5D (analogous to mutant M15 in this study) was because of the constitutive phosphorylation of the equivalent proximal set of serine residues (Ser385 and Ser386 in IRF3). To examine the possibility of a similar phenomenon in IRF7, we assayed IRF7 WT and M15 transactivation ability in the IFNα6-luciferase reporter assay using cells that could be depleted of the kinase activity responsible for phosphorylating IRF7, namely TBK1 and IKK-ε. For this purpose, we used TBK1-deficient fibroblasts from gene-targeted mice and transfected them with a dominant negative form of IKK-ε, IKK-εK38A (6), to impair its activity. In these cells virtually devoid of the relevant kinase activity capable of targeting IRF7 carboxyl-terminal regulatory domain, WT IRF7 lost its low basal activity, whereas M15 retained robust constitutive activity (Fig. 5C). As a control, TBK1-deficient cells reconstituted with TBK1 showed a high constitutive activity of WT IRF7, reaching levels ~1000-fold higher than in the absence of kinases. In contrast, M15 activity was only 4-fold greater in cells transfected with TBK1. This result strongly suggested that M15 activity was not because of constitutive phosphorylation of Ser425 and Ser426 by TBK1 or IKK-ε, whereas the basal activity of WT IRF7 is probably because of low levels of constitutive IKK catalytic activity.
In another tactic to determine whether Ser425 and Ser426 were targets of phosphorylation, we designed a mutant, M18, derived from the phosphomimetic mutant M14 (S437D/S438D). As discussed above, M14 is weakly constitutively active but still strongly activated by virus. Moreover, our results indicated that Ser429–Ser431 and Ser441 were not essential residues to achieve viral-induced phosphorylation (Figs. 1C and 2A, M4 and M6). A mutant combining the mutations of M14, M4, and M6 only leaves Ser425 and Ser426 as potential phosphorylation targets in response to viral infection. We tested the ability of such a mutant to be activated in response to viral infection. As shown in Fig. 5D and E, mutant M18 was not responsive to NDV infection, either by increased transcriptional activity or by altered electrophoretic mobility.
Interestingly, however, M18 resolved as a major isoelectric species plus a minor, more acidic species (Fig. 5E) that was lost when Ser425 and Ser426 were mutated to Ala in M17, suggesting a degree of phosphorylation (Fig. 4B). This result is consistent with the conclusion that Ser425 and Ser426 are not primary targets for virally induced phosphorylation, although they may become secondarily phosphorylated and then contribute to the overall phosphorylation-dependent activity of IRF7. Furthermore, although the groups of Ser429–Ser431 and Ser441 seem dispensable when mutated individually, the combined mutation of both sets rendered IRF7 totally inactive. This result indicates that these serine residues are redundant with each other but that the presence of at least one set, Ser429–Ser431 or Ser441, is crucial for viral responsiveness of IRF7.
DISCUSSION
Virally induced phosphorylation is the crucial event that stimulates the strong transactivation ability of IRF7. One of the characteristics of IRF7 is that it contains numerous potential phosphoacceptor sites clustered in its carboxyl-terminal regulatory domain. To date, reports have been conflicting concerning which residues are essential for virally dependent IRF7 activity and whether this requirement necessarily implies that these residues are phosphorylated in vivo (9, 10). In this study, we provide evidence that Ser437 and Ser438 are the primary targets for phosphorylation but that additional phosphorylation on either some residues of the group of Ser429–Ser431 or on Ser441 is required to render IRF7 fully active in virus-infected cells. Furthermore, our data strongly suggest that the proximal group of Ser425 and Ser426 is essential for function, albeit not solely as phosphorylation targets but also for a different critical function during activation, which facilitates phosphorylation at the major distal sites. This function may be as a recognition motif for the virally activated kinases or as a critical determinant for proper conformation of IRF7 regulatory domain, presumably for dimer formation. Indeed, a non-phosphorylation-dependent critical role for Ser425 and Ser426 is supported by several lines of evidence. 1) The phosphomimetic mutant S425D/S426D is not constitutively active. 2) The mutant M15 that carries all of the remaining serines mutated in aspartate is strongly constitutively active, and its activity is not further enhanced by viral infection. 3) The mutant M15 still exhibits a strong constitutive activity in the absence of active IRF7 kinases, suggesting that its constitutive activity is not because of constitutive phosphorylation of Ser425 and Ser426, whereas the basal activity of the WT protein is dependent on these kinases. 4) The mutant M18, where the only targets left for phosphorylation are Ser425 and Ser426, cannot be activated in response to viral infection. Taken together, our data support a model in which phosphorylation of Ser425 and Ser426 is neither necessary nor sufficient for transcriptional activity, unlike the major phosphorylation targets, Ser437 and Ser438. However, it does not rule out the possibility that Ser425 and Ser426 become phosphorylated secondarily to the phosphorylation of Ser437 and Ser438 and contribute to the overall phosphorylation and therefore the overall transcriptional magnitude of activated IRF7. Nonetheless, these serine residues along with Leu427 are required for the TBK/IKK-mediated phosphorylation of the distal serine residues, possibly serving as a recognition motif for TBK1 and IKK-ε. Kinase interaction might lead to a subsequent conformational change, exposing the distal group of serines and allowing for phosphorylation of the primary targets Ser437 and Ser438 followed by phosphorylation of secondary targets among Ser429–Ser431 and Ser441, and possibly also Ser425 and Ser426. Interestingly, the serine residues analogous to Ser425 and Ser426 in IRF3 lie at an exposed apex at the dimer interface, as revealed in the recently solved IRF3 crystal structure, suggesting that these residues would be available to interact with other proteins (17, 18).
Our findings are reminiscent of what has been reported for IRF3, with some striking differences. The major target of phosphorylation identified in this study is the distal group of Ser437 and Ser438. One of these residues, Ser438, is conserved between IRF3 and IRF7 in both the mouse and human. Nevertheless, this residue would correspond to Ser398 in human IRF3, which has been shown to play only a minor role, because mutant S398A still interacts with CREB-binding protein after viral stimulation and the phosphomimetic counterpart S398D interacts very inefficiently with CREB-binding protein and is consequently a poor constitutive transactivator of IRF3 target genes (19). Conversely, Ser396, which has been identified as the major target for in vivo phosphorylation of IRF3, is not present in IRF7 (19). However, these findings have been debated by Mori et al. (16), who support a model in which the proximal group Ser386 is the target of the IRF3 kinases. In either case, IRF3 and IRF7 would appear to be phosphorylated on distinct, non-homologous residues.
One of the distinctive features of IRF7 compared with IRF3 is its basal activity when transfected, even in absence of viral infection. We show here that this activity is likely because of nonspecific activation of the IRF3/IRF7 kinases during the transfection process, possibly through a double stranded RNA-dependent pathway, because WT IRF7 does not display any activity when cotransfected along with dominant negative IKKε in TBK1-deficient cells (see Fig. 5C). Interestingly, under the same conditions, the activity of the phosphomimetic mutant M15 is only marginally decreased (4-fold difference over a 200-fold activation of the IFNα6 promoter). This observation suggests that M15 constitutive activity is not due to phosphorylation of the proximal group of serine, Ser425 and Ser426, unless phosphorylation of this set of serines is achieved by an unknown kinase distinct from TBK1 and IKKε. However, if phosphorylation of Ser425 and Ser426 by a yet to be identified kinase is the critical step for IRF7 activation, as argued by Mori et al. (16) for IRF3, the profound defect in IFNα induction displayed in TBK1-deficient cells would be difficult to explain (20–23). Interestingly, an alternative mechanism for IRF7 action was recently proposed, in which its transcriptional activity is modulated by direct interaction with MyD88 and TRAF6 in response to Toll-like receptor stimulation (24, 25). In this case, however, TBK1/IKK-ε kinases appeared not to be required, and a role for carboxyl-terminal phosphorylation was not established. Moreover, it is unlikely that Toll-like receptor proteins are involved in the response of IRF7 to viral infection (26).
In this study, analysis of phospho-IRF7 by two-dimensional gel was performed for the first time. We have been able to determine that several species bearing different degrees of phosphorylation coexisted after viral infection in the cell. The data provided by the phosphomimetic mutants imply that IRF7 transcriptional activity is directly proportional to the degree of phosphorylation of the protein, with the final transcriptional activity of IRF7 resulting from the summation of the individual phosphorylation events. A striking prediction from our results is that the magnitude of responses mediated by IRF7 may be malleable, dependent on the extent of phosphorylation. We propose that the activity of IRF7 can be modulated as a function of its degree of phosphorylation as a way of adapting the host response, contingent on the severity of an ongoing viral infection as interpreted by the level and extent of kinase activity.
Acknowledgments
We thank W.-C. Yeh, A. Garcia-Sastre, T. Maniatis, and D. Baltimore for the gift of cell line, virus, and plasmids. We thank Eric Smith for unpublished data, Gregory David for helpful discussions and critical comments, and Hélène Collandre for valuable assistance.
Footnotes
This work was supported by grants from the Association sur la Recherche contre le Cancer (to I. J. M.), by Grants R01 A146503, P01 AI48204, and U54 AI05715801 from the National Institutes of Health, and Grant-in-aid 0256165T from the American Heart Association (to D. E. L.).
The abbreviations used are: IFN, interferon; IRF, interferon regulatory factor; ISG, interferon-stimulated gene; WT, wild type; NDV, Newcastle disease virus; TBK, TANK-binding kinase; IKK, IκB kinase.
References
- 1.Kelley, K. A., and Pitha, P. M. (1985) Nucleic Acids Res.13, 805–823 [DOI] [PMC free article] [PubMed]
- 2.Marié, I., Durbin, J. E., and Levy, D. E. (1998) EMBO J.17, 6660–6669 [DOI] [PMC free article] [PubMed]
- 3.Sato, M., Hata, N., Asagiri, M., Nakaya, T., Taniguchi, T., and Tanaka, N. (1998) FEBS Lett.441, 106–110 [DOI] [PubMed]
- 4.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 5.Sharma, S., tenOever, B. R., Grandvaux, N., Zhou, G. P., Lin, R., and Hiscott, J. (2003) Science300, 1148–1151 [DOI] [PubMed]
- 6.Pomerantz, J. L., and Baltimore, D. (1999) EMBO J.18, 6694–6704 [DOI] [PMC free article] [PubMed]
- 7.Peters RT, Liao SM, Maniatis T. Mol Cell. 2000;5:513–522. doi: 10.1016/s1097-2765(00)80445-1. [DOI] [PubMed] [Google Scholar]
- 8.Shimada T, Kawai T, Takeda K, Matsumoto M, noue J, Tatsumi Y, Kanamaru A, Akira S. Int Immunol. 1999;11:1357–1362. doi: 10.1093/intimm/11.8.1357. [DOI] [PubMed] [Google Scholar]
- 9.Lin R, Mamane Y, Hiscott J. J Biol Chem. 2000;275:34320–34327. doi: 10.1074/jbc.M002814200. [DOI] [PubMed] [Google Scholar]
- 10.Yang H, Lin CH, Ma G, Baffi MO, Wathelet MG. J Biol Chem. 2003;278:15495–15504. doi: 10.1074/jbc.M212940200. [DOI] [PubMed] [Google Scholar]
- 11.Silvennoinen, O., Ihle, J. N., Schlessinger, J., and Levy, D. E. (1993) Nature366, 583–585 [DOI] [PubMed]
- 12.Levy, D. E. (1998) Methods15, 167–174 [DOI] [PubMed]
- 13.Silvennoinen, O., Schindler, C., Schlessinger, J., and Levy, D. E. (1993) Science261, 1736–1739 [DOI] [PubMed]
- 14.Marié I, Smith E, Prakash A, Levy DE. Mol Cell Biol. 2000;20:8803–8814. doi: 10.1128/mcb.20.23.8803-8814.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holzberg D, Knight CG, Dittrich-Breiholz O, Schneider H, Dorrie A, Hoffmann E, Resch K, Kracht M. J Biol Chem. 2003;278:40213–40223. doi: 10.1074/jbc.M304058200. [DOI] [PubMed] [Google Scholar]
- 16.Mori M, Yoneyama M, to T, Takahashi K, nagaki F, Fujita T. J Biol Chem. 2004;279:9698–9702. doi: 10.1074/jbc.M310616200. [DOI] [PubMed] [Google Scholar]
- 17.Takahasi K, Suzuki NN, Horiuchi M, Mori M, Suhara W, Okabe Y, Fukuhara Y, Terasawa H, Akira S, Fujita T, Inagaki F. Nat Struct Biol. 2003;10:922–927. doi: 10.1038/nsb1001. [DOI] [PubMed] [Google Scholar]
- 18.Qin BY, Liu C, Lam SS, inath H, Delston R, Correia JJ, Derynck R, Lin K. Nat Struct Biol. 2003;10:913–921. doi: 10.1038/nsb1002. [DOI] [PubMed] [Google Scholar]
- 19.Servant MJ, Grandvaux N, tenOever BR, Duguay D, Lin R, Hiscott J. J Biol Chem. 2003;278:9441–9447. doi: 10.1074/jbc.M209851200. [DOI] [PubMed] [Google Scholar]
- 20.Hemmi H, Takeuchi O, Sato S, Yamamoto M, Kaisho T, Sanjo H, Kawai T, Hoshino K, Takeda K, Akira S. J Exp Med. 2004;199:1641–1650. doi: 10.1084/jem.20040520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perry AK, Chow EK, Goodnough JB, Yeh WC, Cheng G. J Exp Med. 2004;199:1651–1658. doi: 10.1084/jem.20040528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McWhirter SM, Fitzgerald KA, Rosains J, Rowe DC, Golenbock DT, Maniatis T. Proc Natl Acad Sci U S A. 2004;101:233–238. doi: 10.1073/pnas.2237236100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.tenOever, B. R., Sharma, S., Zou, W., Sun, Q., Grandvaux, N., Julkunen, I., Hemmi, H., Yamamoto, M., Akira, S., Yeh, W. C., Lin, R., and Hiscott, J. (2004) J. Virol.78, 10636–10649 [DOI] [PMC free article] [PubMed]
- 24.Kawai T, Sato S, shii KJ, Coban C, Hemmi H, Yamamoto M, Terai K, Matsuda M, noue J, Uematsu S, Takeuchi O, Akira S. Nat Immunol. 2004;5:1061–1068. doi: 10.1038/ni1118. [DOI] [PubMed] [Google Scholar]
- 25.Honda K, Yanai H, Mizutani T, Negishi H, Shimada N, Suzuki N, Ohba Y, Takaoka A, Yeh WC, Taniguchi T. Proc Natl Acad Sci U S A. 2004;101:15416–15421. doi: 10.1073/pnas.0406933101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levy DE, Marie IJ. Nat Immunol. 2004;5:699–701. doi: 10.1038/ni0704-699. [DOI] [PubMed] [Google Scholar]