

Abstract
Nanohydrogels combine advantages of hydrogels and nanoparticles. In particular, they represent promising drug delivery systems. Nanogel synthesis by oxidative condensation of polyglycidol prepolymers, that are modified with thiol groups, results in crosslinking by disulfide bonds. Hereby, biomolecules like the antidiabetic peptide RS1‐reg, derived from the regulatory protein RS1 of the Na+‐D‐glucose cotransporter SGLT1, can be covalently bound by cysteine residues to the nanogel in a hydrophilic, stabilizing environment. After oral uptake, the acid‐stable nanogels protect their loading during gastric passage from proteolytic degradation. Under alkaline conditions in small intestine the nanohydrogels become mucoadhesive, pass the intestinal mucosa and are taken up into small intestinal enterocytes by endocytosis. Using Caco‐2 cells as a model for small intestinal enterocytes, by confocal laser scanning microscopy and structured illumination microscopy, the colocalization of fluorescent‐labeled RS1‐reg with markers of endosomes, lysosomes, and trans‐Golgi‐network after uptake with polyglycidol‐based nanogels formed by precipitation polymerization is demonstrated. This indicates that RS1‐reg follows the endosomal pathway. In the following, the design of bespoken nanohydrogels for specific targeting of RS1‐reg to its site of action at the trans‐Golgi network is described that might also represent a way of targeted transport for other drugs to their targets at the Golgi apparatus.
Keywords: drug delivery, nanohydrogels, regulation of the Na+‐D‐glucose cotransporter SGLT1 in intestine, regulatory protein RS1, targeted transport
The design of polyglycidol‐based nanohydrogels for selective organelle targeting of therapeutic agents is reported with the example of the targeted retrograde transport of the antidiabetic peptide RS1‐reg to its site of action at the trans‐Golgi network, demonstrating that endosomal escape is not always required for specific intracellular drug transport.
1. Introduction
Hydrogels are 3D polymer networks, that are able to swell and expand in an aqueous medium. Hereby they absorb large amounts of water or biological liquids in the range of 20% up to more than 10 times of their volume without dissolving.[ 1 ] The mechanical and physiochemical similarity with components of the native extracellular matrix and the high water storage capacity conveys their biocompatibility.[ 2 ] Depending on their size, polymer gels can be divided into micro‐ and nanogels, which can be produced in a targeted manner by controlling the crosslinking conditions during their synthesis.[ 3 ] Limitations for the application of hydrogels result from their macroscopic dimensions and the rapid elution of active ingredients from the swollen hydrogel matrix. The use of nanohydrogels is a very promising solution here.[ 4 ] Nanohydrogels are 3D polymer networks, composed of hydrophilic, cross‐linked macromolecule chains with a particle diameter on the nanoscale.[ 5 ] They combine advantages of hydrogels (hydrophilicity, biocompatibility, high water content, flexibility, adjustable physicochemical properties) and nanoparticles (high surface area, size in subcellular range).[ 6 ] One area of application for nanohydrogels is their use as “drug delivery systems.” Here they show many advantages over micro and macro systems such as good injectability, longer circulation times in the blood without being surrounded by macrophages, the ease of penetration into the tissue through receptors, capillaries, and biological membranes and the ability to be easily recognized by cells. Nanohydrogels are also characterized by a high and sustained therapeutic effect up to a period of days to weeks in the target area.[ 7 ] They can transport a large number of active substances (hydrophilic and hydrophobic, high or low molecular weight) and are suitable for different forms of administration.[ 8 ] A new trend is the development of “smart” hydrogels that respond to external stimuli, e.g., by swelling or shrinking in response to changes in their environment (e.g., changes in temperature, pH, redox state, ionic strength, electric or magnetic fields).[ 9 ] Nanohydrogels that respond to external stimuli are particularly promising for use as controlled, site‐directed drug delivery systems. In case of changes in environmental conditions, they react much faster than macroscopic hydrogels due to, e.g., faster diffusion processes, based on their smaller dimensions.[ 10 , 11 ]
In our group, the synthesis of “intelligent,” redox‐sensitive nanohydrogels based on thiol‐functionalized, hydrophilic polyglycidol prepolymers was optimized by crosslinking via oxidative coupling in inverse miniemulsion and by inverse nanoprecipitation polymerization. The chemical structure of the polyglycidol‐based‐nanogels, including disulfide bonds between polyglycidol prepolymers and between nanogel and the cargo TAT peptide were already characterized before.[ 12 , 13 , 14 ] The example TAT peptide can be seen generic for other peptides or proteins. Thus, biomolecules like proteins and peptides can be covalently bound by cysteine residues to the nanogel matrix in a hydrophilic, stabilizing environment. The formation of nanohydrogels with a defined particle size was demonstrated by dynamic light scattering (DLS), cryo‐scanning electron microscopy (cryo‐SEM), and atomic force microscopy (AFM). The formed nanohydrogels were stable in PBS buffer (pH 7.4) but decomposed under reducing conditions in the presence of glutathione.[ 12 ] Cell viability tests according to ISO 10.993‐5 showed that the nanohydrogels are not toxic to L929 fibroblasts. Biocompatibility, intracellular decomposability, polymer building blocks with a molecular weight suitable for renal clearance and the possibility of adapting the polyglycidol prepolymers in terms of structure and functionalization make this system a versatile and promising “drug delivery platform.” According to our model, after their oxidative coupling, protein or peptide drugs are protected from enzymatic degradation during gastric passage by the enveloping nanohydrogel, which is stable under acidic conditions. In the alkaline environment of the small intestine, on the other hand, decomposition of the nanohydrogel particles begins, which thereby develop mucoadhesive properties. After adhesion and passage of the mucosa, the nanohydrogel particles including the drug are absorbed into the enterocytes via endocytosis. Under the reducing conditions in the presence of glutathione, the nanohydrogel particles dissolve inside the cell, releasing the active ingredient.
Within the scope of this work drug uptake via nanogels and intracellular trafficking to its site of action at the trans‐Golgi network (TGN) was analyzed with the antidiabetic RS1‐reg peptide as example cargo, which is derived from the regulator protein RS1. RS1 is a 67 kDa protein, that occurs for the first time in mammals and is encoded by the intron‐free gene RSC1A1 in humans. It is involved in both slow transcriptional and rapid post‐translational regulation of the Na+‐D glucose cotransporters SGLT1, which is located in the brush border membrane of small intestinal epithelial cells, is significantly involved in the absorption of glucose in the small intestine and plays an important role in the glucose‐dependent secretion of enterohormones.[ 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 ] RS1 inhibits the release of SGLT1‐containing vesicles from the TGN at low intracellular glucose concentrations.[ 16 , 17 , 18 , 24 ] The rapid Golgi‐dependent downregulation of the transport activity of SGLT1 by hRS1 is thus based on a decrease in the amount of SGLT1 in the plasma membrane and can be offset by higher intracellular glucose concentrations.[ 16 ] An N‐terminal domain in hRS1 (amino acid 16‐98, called hRS1‐reg) with 15 potential phosphorylation sites has been identified, which is responsible for the rapid post‐translational downregulation of hSGLT1 at the TGN.[ 25 ] Depending on the phosphorylation state, hRS1‐reg inhibits the release of SGLT1‐containing vesicles from TGN glucose‐dependent via the enzyme ornithindecaboxylase 1 (ODC1).[ 26 ] As with RS1 total protein, the inhibitory effect of hRS1‐reg present at low intracellular glucose concentrations is canceled when the intracellular glucose concentration is increased. In the case of a RS1‐reg mutant, in which serine in the QSP motif was exchanged for glutamate (hRS1‐reg(S20E) in humans and mRS1‐reg(S19E) in mice), the inhibitory effect on SGLT1 is maintained even at elevated intracellular glucose concentrations (see scheme in Figure S1, Supporting Information).[ 25 , 26 , 27 ]
We have thus demonstrated in previous work that nanogels do facilitate the transport of hRS1‐reg into enterocytes after oral administration, and that the peptide remains active after release. The mechanism of this process and the mode of action of the nanogels has, however, so far not been understood. The goal of this study has thus been to generate a better understanding of the delivery process and evaluate whether the specificity of the delivery to the TGN can be improved. Using Caco‐2 cells as model for small intestinal enterocytes, we show by confocal laserscanning microscopy (CLSM) and structured illumination microscopy (SIM) a colocalization of fluorescent‐labeled RS1‐reg with markers of endosomes, lysosomes and the trans‐Golgi‐network after uptake via polyglycidol‐based nanogels formed by nanoprecipitation polymerization, indicating that RS1‐reg follows the endosomal pathway. We hereby provide evidence for a targeted transport of RS1‐reg to its site of action at the TGN without endosomal escape. Thus, our approach might also represent a way of targeted delivery for other drugs to their targets at the trans‐Golgi network or, more generally, the Golgi apparatus.
2. Results
From previous studies, it is known that the antidiabetic peptide RS1‐reg, derived from the N‐terminal part of the regulatory protein RS1 of the Na+‐D glucose cotransporters SGLT1, can be taken up via polyglycidol‐based nanohydrogels, prepared in inverse miniemulsions, into mouse small intestinal enterocytes and inhibits glucose uptake by blocking the release of SGLT1‐containing vesicles from the trans‐Golgi network.[ 25 ] Here we investigated the uptake of RS1‐reg through the mediation of polyglycidol‐based nanohydrogels into Caco‐2 cells as model of small intestinal enterocytes and the intracellular targeting to its place of action at the TGN in more detail. Thereby we studied the influence of different nanogel preparation methods (by inverse miniemulsion versus inverse nanoprecipitation) to evaluate whether different ways of preparing nanohydrogels out of the same chemical compounds result in different intracellular localization of the cargo RS1‐reg and the presence or absence of the cell‐penetrating TAT‐peptide as transfection reagent.[ 12 , 28 , 29 ]
2.1. Expression of hRS1‐Reg(S20E) and mRS1‐Reg(S19E) in E. coli
E. coli BL21 STAR cells were transformed by inserting the pET21a plasmid containing the genes for hRS1‐reg(S20E) or mRS1‐reg(S19E). After recombinant expression, cell harvest and disruption, the supernatant following a high‐speed centrifugation was used for protein affinity purification via His‐Tag. Figure 1A shows a SDS‐PAGE analysis of fractions, collected in the course of purification with Ni2+‐NTA‐Agarose. In the eluates, the purified peptides hRS1‐reg(S20E) and mRS1‐reg(S19E) showed a molecular weight of approximately 9 kDa, which corresponds well to the predicted MW of 9.3 kDa. For some follow‐up experiments RS1‐reg peptides were labeled via lysine residues with NHS‐ATTO488, NHS‐Alexa488 or NHS‐Alexa647 conjugates according to manufacturer´s protocol.
Figure 1.
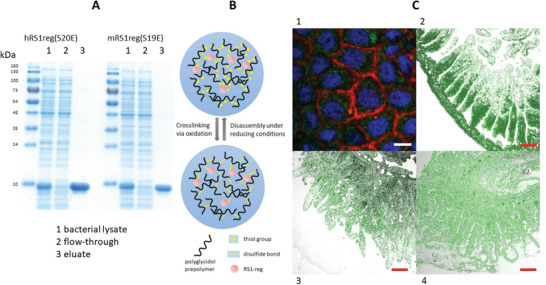
A) SDS‐PAGE showing purification of recombinant hRS1‐reg(S20E) and mRS1‐reg(S19E) expressed in E. coli. After protein expression for 3 h at 30°C, bacteria were lysed and centrifuged for 1 h at 100 000 × g. The supernatant (bacterial lysate) was incubated for 1 h with Ni2+‐NTA agarose, the suspension was poured into an empty gravity flow column and the flow‐through was removed. The resin was washed and purified RS1‐reg peptides were eluted with 500 × 10−3 m imidazole (eluate). The samples were subjected to SDS‐PAGE analysis. The gel was stained with Coomassie Brilliant Blue. Per lane, 10 µg (lysates and flow‐through) or 2 µg of protein (purified RS1‐reg) was applied. B) Scheme of nanohydrogel preparation by oxidative coupling of thiol‐functionalized polyglycidol prepolymers via crosslinking by formation of disulfide bridges. A cargo (for example the RS1‐reg peptide) can be coupled to the matrix via cysteine residues. Under the reducing conditions in the cytosol after cellular uptake, the nanogels are degraded under drug release (for further chemical details of the components see Figure S5, Supporting Information). C) Confocal laser scanning microscopy analysis, demonstrating uptake of ATTO488‐labeled RS1‐reg peptide (green fluorescence) after preincubation with polyglycidol‐based nanohydrogel, formed in inverse miniemulsion, into Caco‐2 cells (1) (staining of nuclei with Hoechst 33 342, staining of plasma membranes with Claret Far Red, Bar = 15 µm) and into small intestine of mouse (2), pig (3) and human (4) (Red Bars = 200 µm).
2.2. RS1‐Reg Peptides Are Taken Up via Polyglycidol‐Based Nanohydrogels into Caco‐2 Cells and Small Intestinal Mucosa of Mouse, Pig, and Human
Redox‐sensitive nanohydrogels based on thiol‐functionalized, hydrophilic polyglycidol prepolymers were formed by crosslinking via oxidative coupling in inverse miniemulsion or by inverse nanoprecipitation polymerization.[ 12 , 28 ] In this context the hRS1‐reg(S20E) peptide or the mRS1‐reg(S19E) peptide and for some experiments also the cell‐penetrating TAT‐peptide were covalently bound by cysteine residues to the nanogel matrix in a hydrophilic, stabilizing environment (see scheme in Figure 1B; for further chemical details of the components see Figure S5, Supporting Information). Nanogel concentrations and sizes were measured using nanoparticle tracking analysis (NTA). Furthermore nanogel size distribution was measured by DLS and ζ potential by electrophoretic light scattering using a Zetasizer Nano ZSP (Malvern Instruments) instrument (see Figure S2, Supporting Information). Depending on the method (NTA or DLS) and nanogel charge a monodisperse size distribution (PdI < 0,2) with mean diameter in the range of 200 –280 nm was obtained for nanogels from inverse miniemulsions, and 250– 300 nm for nanohydrogels formed by nanoprecipitation. Interestingly, the ζ potential values showed no significant difference and were in the range of ‐20 to ‐25 mV. Cryo‐SEM analysis confirmed that nanogels from both manufacturing methods are present as individual colloidal particles in solution. To study the encapsulation efficiency regarding RS1reg or rather the TAT‐peptide, nanohydrogels were prepared using defined amounts of ATTO488‐RS1‐reg in presence or absence of Alexa647‐labeled TAT peptide. After washing and reductive degradation of the nanogels in presence of DTT the amount of released peptides was determined by fluorescence measurements, employing ATTO488‐RS1reg or rather Alexa647‐TAT calibration grades. The encapsulation efficiency (mass percentage of the drug loaded relative to the drug fed) of RS1reg without TAT or in presence of TAT was 83% or rather 78%. For TAT an encapsulation efficiency of 93 percent was calculated. The loading efficiency (mass percentage of the drug loaded relative to the dried nanogel) of TAT was 9%. To investigate whether the payload TAT was bound merely in the peripherie of nanogel particles or coupled to the matrix throughout the entire particle volume, the TAT peptide was fluorescently labeled with Alexa 647 and incorporated into nanoprecipitation nanogels. The sample, and especially particles in it with a much higher diameter than the mean to get a clear result, were analyzed using a ZEISS LSM 900 confocal microscope with Airyscan 2 in Airyscan mode with a laser at a wavelength of 640 nm. Z‐stacks of large particles revealed presence of the coupled TAT throughout the entire particle volume (Figure S6, Supporting Information).
Stability analysis of nanogels out of precipitation polymerization and inverse miniemulsion was performed by DLS and fluorescence measurements (Figure S3, Supporting Information ). DLS measurements of nanohydrogel stability were done after 1:100 dilution in 100 × 10−3 m HCl (pH 1), PBS (pH 7.4) and 20 × 10−3 m Tris/HCl (pH 8.5) for 0–60 min, mimicking pH conditions in stomach (pH 1), in small intestinal lumen and mucosa after fasting (neutral pH range, exact value varies from patient to patient, animal to animal, study to study and position in small intestine) and in presence of digestive juices within small intestine (pH 7.5–8.5) after a meal.[ 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 ] While the nanogels were quite stable for 1 h under acidic conditions (pH 1) in the stomach and neutral pH conditions (pH 7.4) of small intestinal lumen and mucosa after fasting, nanogel size decreased significantly under slightly more alkaline conditions (pH 8.5) as within small intestine in presence of digestive juices. DLS analysis of nanohydrogel stability was conducted for 0, 10, 20, and 30 min after 1:100 dilution in PBS in absence or presence of 10 × 10−3 m DTT, mimicking the reducing conditions in the cytoplasm. Under reducing conditions the nanogels rapidly degraded. Furthermore, fluorescence measurements of the stability of precipitation polymerization based nanohydrogels loaded with ATTO488‐RS1reg were carried out for 10, 20, and 30 min after 1:100 dilution in PBS with or without DTT under liberation of the labeled peptide. In presence of DTT a pronounced peptide release was detectable.
To get proof for differences regarding stability and cross‐linking density of miniemulsion nanogels versus nanoprecipitation nanogels, we performed time‐dependent turbidity measurements of samples of both nanogel types with equal particle density (1 × 1011 particles mL−1) following supplementation with 10 × 10−3 m DTT (Figure S7, Supporting Information) for 1 h. The nanogels without DTT used as controls showed stable transmission over the 60 min, whereby the transmission value of the nanogels from precipitation polymerization was lower than that of the nanogels from inverse miniemulsion. The control approach with 10 × 10−3 m DTT alone showed a stable transmission of about 1 over the whole measurement period. The sample of nanogels from inverse emulsion with DTT showed an earlier, faster increase in transmission until the plateau value of slightly less than 1 was reached than the analog sample with precipitation nanogels. This is consistent with the assumption that the degree of disulfide cross‐linking is lower in the nanogels from inverse emulsion, the particles are therefore less stable and decompose more easily or more quickly in the presence of DTT than in case of nanoprecipitation nanogels.
To demonstrate that Caco‐2 cells can serve as a model for enterocytes of the intestinal mucosa in respect to peptide uptake via nanohydrogels in subsequent immunofluorescence experiments, Caco‐2 cells and segments of small intestinal mucosa of mouse, pig and human were preincubated with nanohydrogel, harboring ATTO488‐labeled RS1‐reg peptide. Confocal laser scanning microscopy analysis confirmed uptake of the green fluorescent peptide in all cases (Figure 1C).
Confocal LSM studies (Figure S4, Supporting Information) to localize the uptake of ATTO488‐RS1‐reg via nanohydrogels out of inverse miniemulsion administered by gavage to a group of mice on sections of small intestinal segments one hour after administration showed strong accumulation in the initial, proximal region of the small intestine, 3 to 10 cm after gastric outlet. A weak signal was seen in the segment 15 cm after the gastric outlet, no more uptake in the later, distal segments (20 and 25 cm after the gastric outlet). No signal of the ATTO488‐RS1‐reg peptide was detected in stomach. These findings support the model concept that the uptake of the cargo occurs after gastric passage in the small intestine, where the nanogels develop mucoadhesive properties under the alkaline conditions, adhere to the mucosa of the small intestine and are absorbed there.
2.3. Glucose Uptake Is Inhibited in Caco‐2 Cells and Mouse Small Intestinal Segments to the Same Degree by Nanogels Prepared in Inverse Miniemulsion and by Inverse Nanoprecipitation
In previous studies inhibition of SGLT1 mediated glucose uptake in small intestine of RS1 knockout mice (RS1−/−) was demonstrated by gavage of RS1−/− mice with nanohydrogels prepared in inverse miniemulsions, containing the cell‐penetrating TAT‐peptide with or without (control) mRS1‐reg(S19E). Three hours later phlorizin‐inhibited AMG uptake was measured into segments of everted jejunum, that was downregulated by mRS1‐reg(S19E).[ 25 ] In this work, we studied in more detail the influence on glucose uptake of nanohydrogels containing hRS1‐reg(S20E) and mRS1‐reg(S19E) with or without TAT‐peptide, prepared in inverse miniemulsion versus inverse nanoprecipitation, into segments of everted RS1−/− mouse small intestine (mRS1‐reg(S19E)) and differentiated Caco‐2 cells (hRS1‐reg(S20E)), which represent an established model for human enterocytes for activity assays and express SGLT1.[ 38 ] For that mouse jejunal segments or Caco‐2 cells were preincubated with the nanohydrogels or with the RS1‐reg‐peptides alone (as controls) and then phlorizin‐inhibited uptake of 10 × 10−6 m radiolabeled AMG was measured. AMG uptake was not altered after incubation with hRS1‐reg(S20E) or mRS1‐reg(S19E) peptides alone or by nanogels without those peptides, but it was downregulated to the same degree, respectively, by about 50% after treatment with mRS1‐reg(S19E) or hRS1‐reg(S20E), transported in nanohydrogels prepared in inverse miniemulsions and by nanoprecipitation, in presence or absence of TAT peptide (Figure 2A). Using a dilution series (500, 100, and 10 µg mL−1) of nanogels containing hRS1‐reg(S20E) out of nanoprecipitation without TAT for preincubation with Caco‐2 cells, we demonstrated dose‐dependency of the inhibitory effect on AMG uptake (Figure 2B). These data suggest that the way of manufacturing the nanogels as well as the presence or absence of the cell‐penetrating TAT peptide did not influence the inhibitory effect of the RS1‐reg peptides on glucose uptake in mouse small intestine and Caco‐2 cells. Furthermore, the results also indicate, that Caco‐2 cells might be a good model for peptide uptake via nanohydrogels in small intestinal mucosa in the subsequent fluorescence experiments. Besides the inhibitory effect of the RS1‐reg loaded nanogel on AMG uptake in Caco2 cells and ex vivo in mouse small intestine presented here, this inhibition has also already been shown in vivo in mice.[ 25 ]
Figure 2.
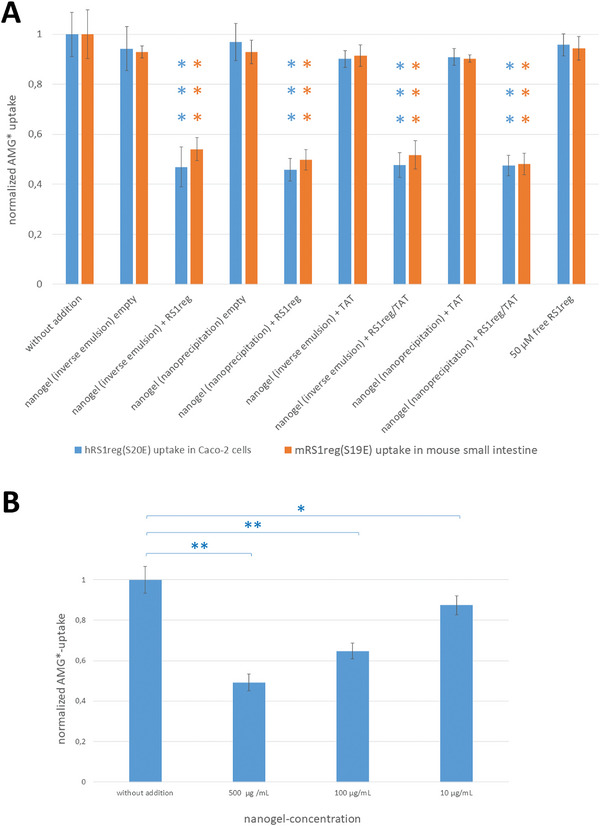
A) Downregulation of SGLT1‐mediated AMG uptake expressed in Caco‐2 cells or mouse small intestinal segments after preincubation with polyglycidol‐based nanohydrogels containing hRS1‐reg(S20E) (for experiments with Caco‐2 cells) or mRS1‐reg(S19E) (for measurements with mouse small intestinal segments), formed in inverse miniemulsion or by inverse nanoprecipitation either with or without TAT peptide. Independent of the way of preparation of nanogels containing RS1‐reg, AMG uptake was reduced to the same extent, by about 50 percent, in Caco‐2 cells and segments of mouse small intestine. No inhibition of AMG uptake was observed in approaches with empty nanogels or with free RS1reg peptides. Presence or absence of the cell‐penetrating TAT peptide did not change the results. B) Dose‐dependent inhibition of AMG uptake into Caco‐2 cells by a dilution series of hRS1‐reg(S20E) containing nanohydrogels out of nanoprecipitation without TAT peptide. Mean values ± S.E. of 9 wells with confluent Caco‐2 cells or rather mouse small intestinal segments from three independent experiments are shown, respectively. *P < 0.05; **P < 0.01; ***P < 0.001 for significance of difference from control tested by ANOVA with post hoc Tukey comparison.
2.4. Different Ways of Preparing Nanohydrogels out of the Same Chemical Compounds Result in Different Intracellular Localization of the Cargo RS1‐Reg
For investigating the influence of the nanogel preparation method and the presence or absence of the cell penetrating TAT peptide as transfection reagent on the intracellular targeting and localization of the example cargo RS1‐reg, Caco‐2 cells were preincubated with nanogels. The latter were formed either in inverse miniemulsions or by inverse nanoprecipitation and contained ATTO488 labeled hRS1‐reg(S20E) with or without TAT. The corresponding experiments have been performed by confocal laser scanning microscopy analysis (Leica TCS SP8). Nuclei were stained with Hoechst 33342 dye and the green fluorescence of the RS1‐reg peptide was detected (Figure 3 ). In case of the nanogel, which has been produced in inverse miniemulsion without TAT, a diffuse, homogenous distribution of the RS1‐reg signal in the cytosol of Caco‐2 cells could be observed. No signal was detected in the nuclei (Figure 3A). These data suggest endosomal escape of ATTO488‐hRS1‐reg(S20E) following uptake by endocytosis of the nanogel into Caco‐2 cells. The peptide distributes evenly in the cytosol and finally also reaches its site of action at the trans‐Golgi network and inhibits the release of SGLT1 containing vesicles, resulting in the observed inhibition of glucose uptake (Figure 2). In the approach of the nanogel out of inverse emulsion containing the TAT‐peptide, only a minor part of RS1‐reg signal was found in the cytosol. The majority of hRS1‐reg was localized in the nuclei of Caco‐2 cells, suggesting that TAT carried also hRS1‐reg with it into the cell nuclei (Figure 3B). This steering characteristic of TAT was also described for other compounds.[ 39 ] Figure 3C,D show a similar, selective accumulation of ATTO488‐hRS1‐reg(S20E), taken up by nanogels formed by inverse nanoprecipitation with or without TAT‐peptide, in defined cytosolic vesicles. These findings could indicate an impaired endosomal escape of hRS1‐reg, which was not influenced by TAT. A direct comparison between CLSM measurements of the uptake of Alexa647‐labeled RS1‐reg via nanogels out of inverse emulsion versus nanoprecipitation without TAT is shown in Figure 3E. In case of the nanogel formed in inverse miniemulsions the Alexa647‐RS1‐reg signal is evenly distributed in the cytosol of Caco‐2 cells, whereas the RS1‐reg peptide transported by the nanohydrogel out of nanoprecipitation accumulates selectively in distinct vesicular structures within the cytosol. These results indicate that different ways of preparing nanohydrogels out of the same chemical compounds result in different intracellular localization of the cargo RS1‐reg.
Figure 3.

Confocal laser scanning microscopy (CLSM) analysis of the localization of ATTO488‐hRS1‐reg(S20E) in fixed Caco‐2 cells after uptake via nanohydrogels formed in A,B) inverse miniemulsion or C,D) by inverse nanoprecipitation either with B,D) or without A,C) cell‐penetrating TAT peptide. While in case of nanogel out of inverse miniemulsion without A) TAT an even distribution of the green fluorescent peptide in the cytosol can be detected, suggesting successful endosomal escape, the presence of B) TAT directs the majority of RS1‐reg signal with it into the cell nuclei. Uptake via nanoprecipitation nanogels in presence or absence of C,D) TAT results in a selective accumulation of ATTO488‐RS1‐reg in defined vesicular cytosolic compartments, suggesting impaired endosomal escape. A–D) The left column displays the staining of nuclei with Hoechst 33 342, in the middle column the signal of ATTO488‐labeled hRS1‐reg(S20E) can be seen and the right column shows merged signals. In E) a direct comparison of the localization of Alexa647‐labeled hRS1‐reg following uptake via nanohydrogels out of inverse miniemulsion (even distribution in the cytosol) versus nanoprecipitation (selective enrichment in cytosolic vesicles) without TAT is depicted, indicating that different ways of preparing nanohydrogels out of the same chemical compounds result in different intracellular localization of the cargo RS1‐reg. Bars = 15 µm.
2.5. hRS1‐Reg Follows the Endosomal Pathway after Uptake via Endocytosis Using Nanohydrogels from Precipitation Polymerization and Thus Reaches Its Site of Action at the TGN via Targeted Intracellular Transport
To identify the cytosolic vesicular structures, in which hRS1‐reg(S20E) selectively accumulates, we performed structured illumination microscopy (SIM) measurements, that offer a higher resolution than CLSM analysis. Caco‐2 cells were grown on coverslides or 8 Well Lab‐Tek chamber slides, pretreated with nanogels harboring Alexa488‐labeled hRS1‐reg(S20E) peptide without TAT, fixed with paraformaldehyde and permeabilized. After incubation with primary antibodies against marker proteins of endosomes (EEA1), lysosomes (Lamp‐1), endoplasmatic reticulum (Calnexin), the trans‐Golgi network (TGN46) and against the enzyme ODC1 the cells were treated with ATTO643‐labeled secondary antibodies. Cell nuclei were stained with Hoechst 34580 and SIM analysis (Zeiss Elyra S.1 SIM, Carl Zeiss Microscopy GmbH, Jena) was performed. In case of the markers EEA1, Lamp‐1, TGN46 and ODC1 a partial colocalization with the signal of Alexa488‐labeled RS1‐reg peptide were observed. No colocalization was detected with the marker for endoplasmatic reticulum, calnexin (Figure 4 ). To validate the data with higher accuracy, the SIM measurements were repeated with embedded samples for the approaches with the markers EEA1, Lamp‐1, TGN46 and ODC1, enabling a precise channel alignment. SIM‐analysis revealed the same result, a partial colocalization with Alexa488‐hRS1‐reg(S20E), respectively (Figure 5 ). Quantitative colocalization analysis was performed, employing all pictures of 4 stacks regarding the green signals of Alexa488‐RS1‐reg (Ch2) and red fluorescence of marker proteins of endosomes (EEA1), lysosomes (Lamp‐1), trans‐Golgi network (TGN46) and the enzyme ODC1 (Ch1, respectively). Pearson's coefficients were calculated using a custom‐written ImageJ macro (see Supporting Information). A high positive linear correlation between the respective characteristics under consideration is indicated by Pearson coefficients > 0.7. As a control, the colocalization between the signals of the marker proteins (Ch1) and nuclei (Ch3) was investigated. The resulting Pearson coefficients of approximately 0 indicate no dependence of the two characteristics on each other, as expected.
Figure 4.
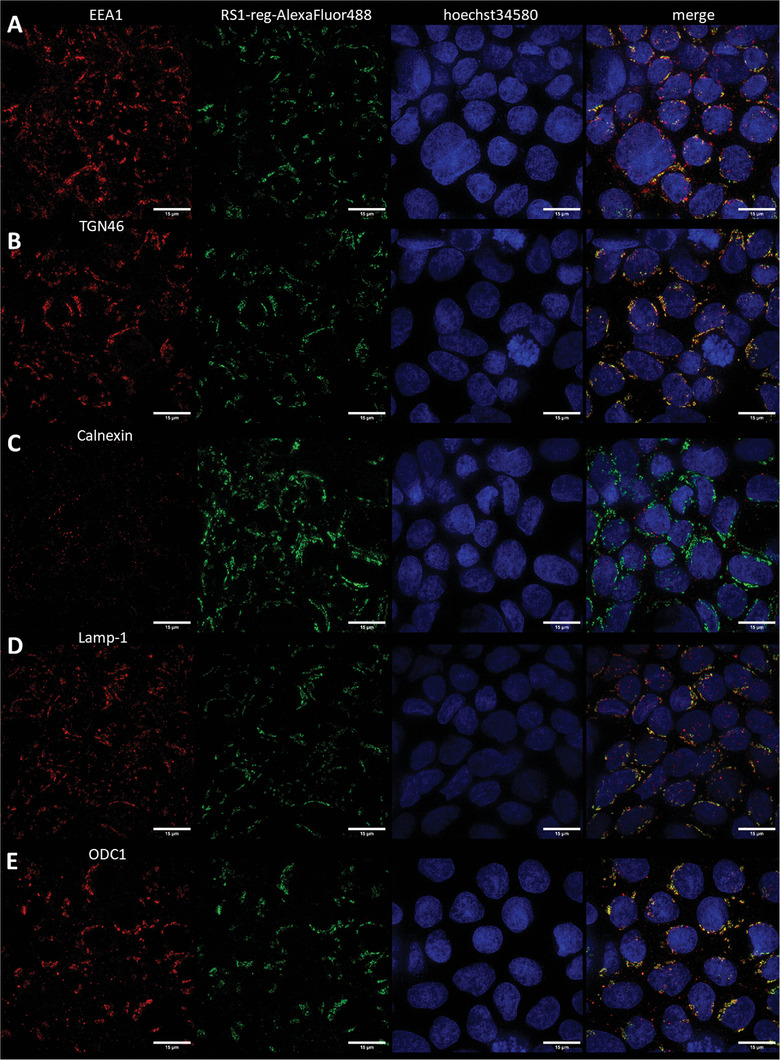
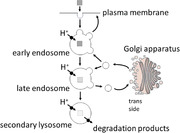
Colocalization analysis in fixed, permeabilized, nonembedded Caco‐2 cells via structured illumination microscopy (SIM) of the distribution of Alexa488‐labeled hRS1‐reg(S20E) after uptake with nanogels formed by inverse nanoprecipitation. Colocalization was tested with marker proteins for endosomes (EEA1, row A), trans‐Golgi network (TGN46, row B), endoplasmatic reticulum (Calnexin, row C), lysosomes (Lamp‐1, row D) and with the enzyme ODC1 (row E). A partial colocalization was detected with EEA1, TGN46, Lamp‐1 and ODC1, respectively, but no colocalization with calnexin. In the left column the signal of the respective marker protein is visible, in the neighboring column the Alexa488‐RS1‐reg signal is displayed, followed by the staining of cell nuclei with Hoechst 34580 in the next column and merged signals in the right column. The results suggest impaired endosomal escape of RS1‐reg, which follows the endosomal way from early to late endosomes, where partial transport to the trans‐Golgi networks occurs via vesicle exchange. Residual RS1reg is finally degraded after fusion with lysosomes in the resulting endolysosomes. Bars = 15 µm.
Figure 5.
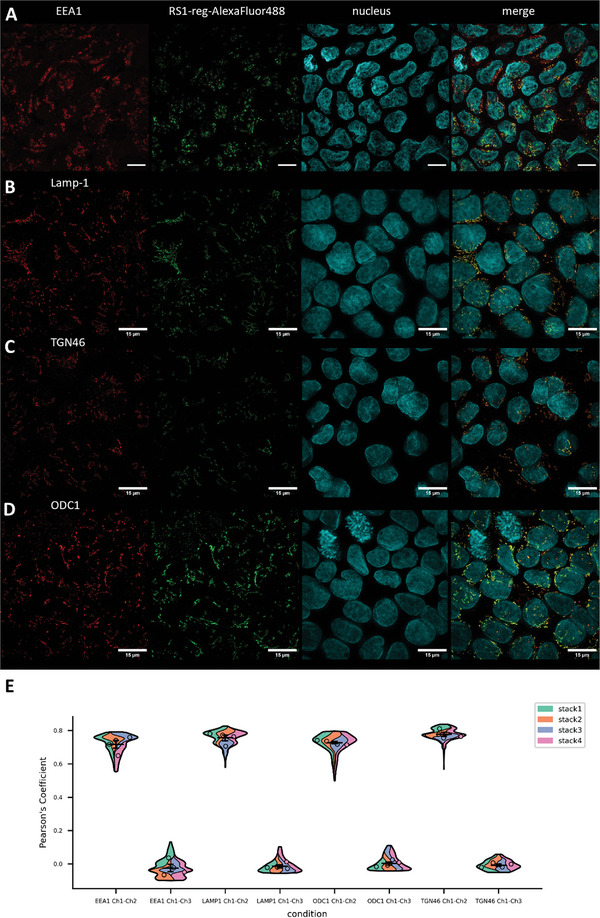
Colocalization analysis in fixed, permeabilized Caco‐2 cells via structured illumination microscopy (SIM) of the distribution of Alexa488‐labeled hRS1‐reg(S20E) after uptake with nanogels formed by inverse nanoprecipitation. Caco‐2 cells were embedded to allow for a precise channel alignment, resulting in a more precise localization of the RS1‐reg peptide and the marker proteins for different cellular structures. Marker proteins of endosomes (EEA1, row A), lysosomes (Lamp‐1, row B), trans‐Golgi network (TGN46, row C) and the enzyme ODC1 (row D) were tested for colocalization with Alexa488‐RS1‐reg. A partial colocalization was detected in all approaches. The left column shows the signal of the respective marker protein, in the neighboring column the Alexa488‐RS1‐reg signal can be seen, followed by the staining of cell nuclei with Hoechst 34580 in the next column and merged signals in the right column. The results indicate no endosomal escape of RS1‐reg. The peptide appears to pursue the endosomal route, by passing from early to late endosomes, where it can be translocated in part to the TGN by vesicle exchange. Remaining hRS1‐reg(S20E) is finally cleaved proteolytically in secondary lysosomes. Bars = 15 µm. E) Violin‐Plot of quantitative colocalization analysis employing all pictures of 4 stacks regarding the green signals of Alexa488‐RS1‐reg (Ch2) and red fluorescence of marker proteins of endosomes (EEA1), lysosomes (Lamp‐1), trans‐Golgi network (TGN46) and the enzyme ODC1 (Ch1, respectively). Pearson's coefficients were calculated using a custom‐written ImageJ macro (see Supporting Information). Pearson coefficients > 0.7 indicate a high positive linear correlation between the respective characteristics under consideration. As a control the colocalization between the signals of the marker proteins (Ch1) and nuclei (Ch3) was investigated, resulting in Pearson coefficients of approximately 0, indicating no dependence of the two characteristics on each other, as expected.
In summary, the results suggest (Figure 6 ) that in case of nanogels, formed in inverse miniemulsions, after cellular uptake by endocytosis the endosomal escape of RS1‐reg succeeds, resulting in an even distribution in the cytosol and also in reaching its site of action at the TGN. On the other hand, the endosomal escape of RS1‐reg, taken up via nanogels from nanoprecipitation, is impaired. Due to this process, we detected the colocalization with the endosomal marker EEA1. RS1‐reg passes from the early endosomes to the late endosomes, where it can be transported via vesicle exchange to the trans‐Golgi network. The inhibition of glucose uptake by RS1‐reg via the enzyme ODC1 by blocking the release of SGLT1‐containing vesicles from the TGN could be detected in activity assays (see Figure 2). Furthermore, we observed both partial colocalization with the TGN marker TGN46 and with ODC1. The residual hRS1‐reg in late endosomes is degraded after fusion with lysosomes in the resulting endolysosomes. In this context, a partial colocalization with the lysosomal maker Lamp‐1 was detected. After cellular uptake via nanohydrogels, formed by nanoprecipitation, RS1‐reg follows the endosomal pathway and reaches its site of action at the TGN. It is an example of a targeted intracellular drug transport, where endosomal escape is not required. Thus we developed a bespoken nanogel for specific organelle targeting of a drug to the TGN.
Figure 6.
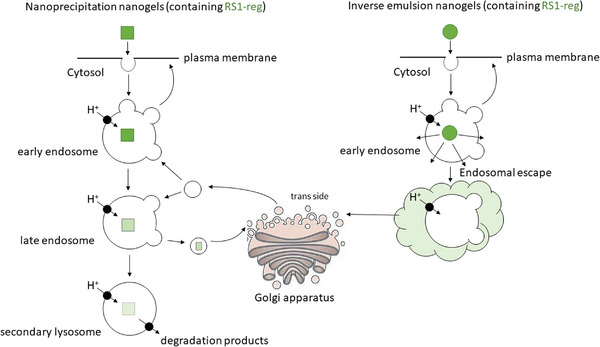
Model scheme for the uptake of the cargo RS1‐reg via nanohydrogels out of inverse miniemulsion (right side) or inverse nanoprecipitation (left side). Following cellular uptake with nanogels formed in inverse emulsion, that are softer probably due to a lower crosslinking degree, endosomal escape of RS1‐reg is possible, leading to an even distribution in the cytosol. Thus the antidiabetic peptide also reaches the trans‐Golgi network, where it can exert its biological function of inhibiting glucose uptake by blockage of the release of SGLT1‐containing vesicles. On the other hand after uptake via nanoprecipitation nanohydrogels the endosomal escape of RS1‐reg is impaired, because of the higher stability probably resulting from a higher crosslinking degree. The peptide follows the endosomal pathway from early to late endosomes, where it is partially transported to its site of action at the TGN by vesicle exchange. Residual RS1‐reg is finally degraded after fusion with lysosomes in the resulting endolysosomes.
2.6. Nanohydrogels out of Nanoprecipitation Exhibit a Higher Stability in Terms of Form and Structure
To further corroborate the assumption that the impaired endosomal escape of hRS1‐reg after endocytosis of nanohydrogels formed by nanoprecipitation may be due to a higher stability of these nanogels as a result of a higher degree of cross‐linking compared to nanogels prepared from inverse miniemulsion, we studied adsorbed nanogel colloids by PeakForce Tapping mode AFM in air. The spin‐coated miniemulsion nanogels formed film‐like structures on mica. No distinct nanogel particles could be observed within this structure. By contrast, for the precipitation method, distinct nanogel particles could be clearly resolved within in the larger aggregates. Cryo‐SEM analysis as well as DLS measurements demonstrated that nanogels from both manufacturing methods were initially present as individual colloidal particles in the suspensions (Figure S2, Supporting Information). The AFM measurements revealed the different stability of the two types of nanogels. Nanogels prepared by the inverse precipitation method are more stable due to the higher degree of cross‐linking.
3. Discussion
In this work, we provide evidence, that endosomal escape is not always necessary for a targeted, intracellular drug transport. The reason for the observed targeted, retrograde transport of the cargo RS1reg to the TGN in case of nanogels out of nanoprecipitation appears to be an impaired endosomal escape as a result of a higher disulfide bond cross‐linking degree of the nanoprecipitation nanogels in comparison to the miniemulsion nanogels. The antidiabetic peptide RS1‐reg, packed in polygycidol‐based nanohydrogels out of nanoprecipitation, remains in endosomes after cellular uptake by endocytosis as colocalization with the endosomal marker EEA1 (Figures 4 and 5) suggests. The endosomal escape is impaired probably due to a higher cross‐linking density of disulfide bridges in these nanogels in comparison to nanogels formed by inverse miniemulsions. AFM analysis supports this assumption, because the less cross‐linked nanogel particles as obtained by the miniemulsion method were hard to recognize, not dimensionally stable and flat. They dissolved on the surface under formation of a polymer film. On the other hand, nanoprecipitation nanogel particles with a presumably higher degree of cross linking were easily visible as dimensionally stable beads in nanogel aggregates ( Figure S2, Supporting Information). Turbidity measurements of the time‐dependent degradation of precipitation nanogels versus miniemulsion nanogels in presence of 10 × 10−3 m DTT delivered further proof for the assumption that the degree of disulfide cross‐linking is lower in the nanogels from inverse emulsion. The particles are therefore less stable and decompose more easily or rather more quickly in the presence of DTT than in case of nanoprecipitation nanogels (Figure S7, Supporting Information). The degradation of the nanogel under simultaneous release of the covalently bound RS1‐reg by disulfide bond cleavage seems to be an important prerequisite for the endosomal escape of the peptide. Cleavage of disulfide bridges in endosomes after receptor‐mediated endocytosis is possible and described in literature.[ 40 , 41 ] For instance after endocytosis also the activation of diphtheria toxin requires the acid‐triggered exposure of an interchain disulfide bond that has to be cleaved by reduction before endosomal escape and cell killing can take place.[ 42 , 43 , 44 ] In case of Cholera toxin and Pseudomonas exotoxin the intracellular reduction of disulfide bridges following uptake also seems to be necessary for their cytotoxic activity.[ 45 ] The RS1‐reg peptide follows the endosomal pathway, moving from early endosomes to late endosomes, where it can be transported to its site of action at the trans‐Golgi network via vesicle exchange. Based on these findings, we designed a bespoken nanogel for specific organelle targeting of a drug to the trans‐Golgi network and demonstrated a partial colocalization both with the TGN marker TGN46 and with ODC1 in SIM analysis (Figures 4 and 5). At the TGN, using a nanogel concentration of 100 µg mL−1, hRS1‐reg blocks via ODC1 the release of SGLT1‐containing vesicles, resulting in inhibition of glucose uptake that we measured in activity assays (Figure 2).[ 26 ] On the other hand after preincubation of Caco‐2 Cells with the same nanogel concentration of 100 µg/mL no hRS1‐reg(S20E) was detected in the cytosol in CLSM‐ and SIM‐measurements (Figures 3, 4, 5). These findings might indicate that after fusion of the exchange vesicles with the TGN, hRS1‐reg accumulates in the interior of the TGN and a part of the peptide passes the TGN membrane by a yet undefined mechanism, maybe mediated by a hitherto unknown transport protein or by diffusion during the release of vesicles at the very active TGN membrane, yielding a local concentration that is sufficient to exert its inhibitory effect on the release of SGLT1‐containing vesicles at the cytosolic side of the TGN membrane by the postulated mechanism via ODC1 (Figure S1, Supporting Information). Alternatively the high concentration of accumulated hRS1‐reg might act by another unknown, less specific mechanism from the interior of the TGN. Future experiments are required to clarify the underlying mechanism. After fusion with lysosomes, the remaining RS1‐reg is degraded in the formed endolysosomes (secondary lysosomes). A partial colocalization of Alexa488‐hRS1‐reg(S20E) and the lysosomal marker Lamp‐1 was found (Figures 4 and 5). Presence or absence of TAT in the nanogels had no influence on these results (Figures 2 and 3). This kind of targeted intracellular drug transport to the site of action, without necessity for endosomal escape, might also be important for other agents targeting Golgi apparatus or trans‐Golgi network. In future organelle targeting will become an important aspect for drug targeting to its active site, resulting in higher drug efficacy and reduced systemic toxicity and immunological side effects. Nanoparticles as drug delivery vehicles are an emerging field and are believed to play a crucial role for delivering cancer chemotherapeutic drugs exclusively to target sites inside tumor cells, while sparing normal proliferating cells.[ 46 ] The ER‐Golgi network is a future target for anticancer therapy and development of targeted delivery strategies of agents to this network will become important.[ 47 , 48 ] Examples are the effort to target the anticancer drugs rapamycin, the central regulator of mammalian cell growth and proliferation, in response to nutritional and environmental conditions, bortezombib and Eeyarestatin I.[ 49 , 50 , 51 ] Alterations in the neuronal Golgi apparatus also give rise to a variety of neurodegenerative disorders which include Alzheimer's, Parkinson's, Niemann‐Pick disease, and others.[ 52 ] In Alzheimer's disease (AD), cleavage of the amyloid precursor protein (APP) during its trafficking inside the nerve cells results in formation of Aβ aggregates. The Golgi apparatus plays a critical role in APP trafficking. In nerve cells of AD patients, the fragmentation of the normally highly ordered Golgi structure can be seen. Due to that the Golgi apparatus is a potential drug target for treatment of AD and targeted delivery of agents to this site of action will become important.[ 53 ] In general, the retrograde transport of proteins after endocytoses via endosomes to the TGN or rather Golgi apparatus is known and described, e.g., for cellular proteins like TGN38, bacterial and plant toxins like ricin (regulated by cholesterol, Rab6A, and Rab6A'), cholera toxin, pertussis toxin or shiga toxin via distinct retromer components that regulate the transport of these cargos.[ 54 , 55 , 56 , 57 , 58 , 59 ] Retrograde transfer from endosomes to the TGN is important for the recycling of membrane proteins, that are involved in a range of biological processes, endogenous proteins including the sorting receptors mannose‐6‐phosphate receptor (M6P‐R), sortilin and wntless, transmembrane peptidases such as furin, SNAREs, and ion and glucose transporters. Several retrograde transport pathways from the endosomal compartments to the TGN have been identified, including routes from the early/recycling endosomes to the TGN as well as trafficking from late endosomes to the TGN. Many factors were found at the TGN that regulate these retrograde endosome‐to‐TGN transport pathways, including small G proteins, SNAREs, tethering factors and scaffold molecules.[ 55 , 60 , 61 , 62 , 63 , 64 , 65 , 66 , 67 , 68 ] Qui and coworkers developed endoplasmic reticulum membrane‐decorated hybrid nanoplexes to effectively transport siRNA through the endosome‐Golgi‐ER pathway, avoiding lysosomal degradation and enhancing the silencing effects of siRNA.[ 69 ] Ross et al. report in the context of non‐viral gene delivery, after uptake of histone‐targeted polyplexes via caveolin‐dependent endocytosis, avoiding an endosomal escape, a directed, retrograde transport of these polyplexes via early and late endosomes, by means of vesicles to the Golgi, then to the ER up to the nucleus.[ 70 ] On the other hand, in case of polyglycidol‐based nanogels out of inverse miniemulsion without TAT peptide, that are smoother probably due to a lower degree of crosslinking, after cellular uptake the endosomal escape of RS1‐reg succeeds, resulting in its even cytosolic distribution (Figure 3A) and thereby in reaching the TGN, where the antidiabetic peptide exerts its biological activity (Figure 2). In the corresponding approach with TAT, certainly the cell‐penetrating peptide accelerates the endosomal escape of the RS1‐reg peptide, but it takes the majority of RS1‐reg with it into the cell nuclei, leaving only a minor part in the cytosol of Caco‐2 cells (Figure 3B) that is nevertheless sufficient for its biological activity at the TGN (Figure 2). This steering property of TAT was also described for other cargos.[ 39 ] An interesting aspect is, that on the one hand in case of miniemulsion nanogels with TAT the majority of hRS1‐reg was localized in the nuclei of Caco‐2 cells, suggesting that TAT carried also hRS1‐reg with it into the cell nuclei (Figure 3B). On the other hand, there was no significant difference in downregulation of AMG uptake compared to the nanogels without TAT (Figure 2A). Another interesting observation is that the two different intracellular trafficking processes associated with nanoprecipitation nanogels versus miniemulsion nanogels led to no significant difference in downregulation of AMG uptake (Figure 2A). The explanation for these two aspects is that it is a high‐affinity inhibition of SGLT1 via ODC1 by even small amounts of RS1reg in the cytosol. EC50 (RS1reg(S20E)) = 19 ± 0.23 × 10−12 m.[ 25 ] The low amount of RS1reg in the cytosol in case of miniemulsion gels with TAT is sufficient to exert the same maximal inhibitory effect on SGLT1 as the higher amount of RS1reg in the cytosol in case of miniemulsion gels without TAT. The same is true when comparing the two different intracellular trafficking processes. A maximum reduction of AMG uptake by RS1 or RS1reg of about 50% was observed under all conditions considered so far in all, different measurement systems (not only in mouse small intestine with RS1reg‐containing nanogels with/without TAT but also, for example, in oocytes without nanogels, but only with RS1 or RS1reg).[ 25 ] AMG uptake is determined by regulation of the amount of SGLT1 in the plasma membrane, which, however, does not fall below a certain minimum value. Therefore, AMG uptake is never zero, but can be inhibited to a maximum of about 50%.
Our experiments demonstrate that our polyglycidol‐based nanogels develop mucoadhesive properties, stick to the proximal part of small intestine and are taken up there into enterocytes (Figure S4, Supporting Information). Here the released payload RS1reg also exerts its biological function by blocking the release of SGLT1 containing vesicles to the plasma membrane, resulting in an inhibition of glucose uptake (Figure 2). Development of mucoadhesive properties according to our working model described in the introduction alone is not a sufficient explanation for our nanoparticles to traverse the mucus layer. Mucus is composed of highly cross‐linked mucin fibers by hydrophobic interactions and disulfides link, creating a dense porous structure. Thereby, to penetrate mucus, nanocarriers must be small enough to avoid steric obstruction in spite of nanoparticles (NPs) with larger size are preferred to improve drug loading and release kinetics.[ 71 ] As reported, a twofold increase in particle size, from 510 to 1190 nm for PS‐PEG NP, would lead to a 30‐fold decrease in the ensemble‐averaged mean squared displacement.[ 72 ] It also has been reported that nanospheres with a size approaching 560 nm were almost completely blocked by the sputum.[ 73 ] In case of our nanoparticles, the average size of 200–300 nm seems to be small enough to favor efficient penetrating of the mucus layer in small intestine. Mucus is a hydrogel complex composed of carbohydrates, protein, lipids, antibody, cellular debris bacteria and inorganic salts.[ 74 ] The barrier properties of mucus are rooted in its dense network of mucin fibers, which contain highly glycosylated (negatively charged) segments,[ 71 ] thus show high affinity with positively charged particles. For example, Laffleur et al. reported that the diffusion rate of neutral polyacrylic acid (PAA)‐polypropylene amide (PAM) nanoparticles is 2.5‐fold higher than positively charged PAM NPs.[ 75 ] Additionally, there exist periodic hydrophobic domains along the mucin strains, which can bind hydrophobic particles with high avidity.[ 76 ] However, after coating hydrophobic nanoparticles with a hydrophilic layer, the average transport rates can be improved. NPs properties including charge and hydrophobicity have a great influence on their behavior of penetrating through mucus. As a consequence, to prepare nanoparticles with a sufficiently hydrophilic and uncharged surface to effectively minimize the adhesive interactions between mucin and NPs by reducing hydrophobic or electrostatic interactions, show promise prospect on mucus penetrating. Coating NPs with PEG is the most widely studied mucus penetrating strategy.[ 77 ] PEG is an uncharged hydrophilic polymer and coating NPs with a high density of low MW PEG can reduce the interactions between particle and mucus. PEG coating can facilitate the NPs to pass through mucus, when the PEG density is sufficient to shield the hydrophobic core effectively.[ 78 ] In case of our nanogels there is no hydrophobic core, but they are made of polyglycidol, exhibiting similar non‐ionic, hydrophilic properties like PEG, exhibiting low hydrophobic or electrostatic interaction with mucus compounds, thereby favoring a fast and efficient penetration of the mucus layer.
Taken together, the results demonstrate that different ways of preparing nanohydrogels out of the same chemical compounds result in different intracellular localization of the antidiabetic peptide. The selected example cargo RS1‐reg can reach its site of action at the trans‐Golgi network by two ways, where its biological activity of inhibiting glucose uptake into Caco‐2 cells can be measured: either mainly via the cytosol after cellular uptake of nanohydrogels out of inverse miniemulsion and successful endosomal release or in form of a selective organelle targeting mainly via the endosomal pathway under vesicle exchange with the TGN following uptake of nanogels formed by inverse nanoprecipitation (Figure 6). This selective way of intracellular targeting might also be important for other agents with the Golgi apparatus as drug target.
4. Conclusion
We report the development of a bespoken nanogel system for specific organelle targeting of the antidiabetic peptide RS1‐reg to its site of action at the trans‐Golgi network, that might also be relevant for other compounds for treatment of certain types of cancer or neurodegenerative disorders (Alzheimer, Parkinson) with the Golgi apparatus as drug target. This might enable the future development of new agents for treatment of diabetes, cancer and neurodegenerative diseases, exhibiting a higher efficacy and less side effects.
5. Experimental Section
Animals
C57BL/6 wild‐type mice were kept in temperature controlled environment with 12 h light/dark cycles. The mice were provided with standard chow and water ad libitum (Altromin 1324; Altromin Spezialfutter GmbH, Lage, Germany). Animals were handled in compliance with the guidelines of the University of Würzburg and German laws and ethical approval was given by the government of Upper Bavaria (file number: 55.2.2‐2532‐2‐704).
Materials
α‐Methyl‐D‐[14C]glucopyranoside ([14C]AMG) (11.1 GBq per mmol) was obtained from American Labeled Chemical Inc. (St. Louis, MO). Alloxan‐monohydrate, Saponin (from Quillaja bark), Span 80 and Tween 80 were obtained from Sigma‐Aldrich GmbH (Taufkirchen, Germany). DTT was from Applichem (Darmstadt, Germany). Soluene‐350 was obtained from PerkinElmer Inc. (Waltham, MA, USA). TAT‐peptide (CGRLLRRQRRR) was delivered by Peptides International (Louisville, Kentucky, USA). Nickel(II)‐charged nitrilotriacetic acid‐agarose (Ni2+‐NTA‐agarose) was purchased from QIAGEN (Hilden, Germany). Escherichia coli strain BL21 Star(D3) (cat. no. C6010‐03) was purchased from Invitrogen/Thermo Fisher Scientific (Carlsbad, CA, USA) and plasmid pET21a (cat. no. 69740‐3) from Merck Life Science GmbH (Eppelheim, Germany). Other chemicals were purchased as described.[ 16 , 25 , 26 ]
Antibodies
Rabbit polyclonal antibody against TGN46 was obtained from Novus Biologicals (Centennial, Colorado, USA) and rabbit monoclonal antibody against EEA1 was from Cell Signaling Technology (Danvers, MA, USA). Rabbit polyclonal antibody against ODC1 and mouse monoclonal antibody against Calnexin were purchased from ThermoFisher (Waltham, MA, USA). Rabbit polyclonal anti‐human‐LAMP‐1 antibody was delivered by Abcam (Cambridge, UK). Goat‐anti‐mouse‐ATTO643 antibody was from Sigma‐Aldrich GmbH (Taufkirchen, Germany). Polyclonal donkey‐anti‐Rabbit antibody was obtained from Jackson ImmunoResearch Europe Ltd (Ely, Cambridgeshire, UK) and fluorescently labeled with NHS‐ATTO643 conjugate (ATTO‐TEC GmbH, Siegen, Germany) according to manufacturer´s protocol.
Cloning
Cloning of hRS1‐reg(S20E) and mRS1‐reg(S19E) into the plasmid pET21a was performed as described earlier.[ 25 ]
Expression and Purification of hRS1‐Reg(S20E) and mRS1‐Reg(S19E)
E. coli bacteria (strain BL21 Star) were transformed with pET21a plasmids containing genes for His‐tagged hRS1‐reg(S20E) or mRS1‐reg(S19E) and grown at 30 °C to mid‐log phase. Protein expression was induced by adding 1 × 10−3 m isopropyl‐1‐thio‐b‐D‐galactopyranoside, and bacteria were subsequently grown for 3 h at 30 °C. After 15 min centrifugation at 6000 × g, bacteria were washed and suspended in 20 × 10−3 m Tris‐HCl (pH 8.0) containing 500 × 10−3 m NaCl, 50 × 10−3 m imidazole and an EDTA‐free protease inhibitor cocktail (Roche, Basel, Switzerland). The cells were lysed by sonication at 4 °C, and cellular debris was removed by 1 h centrifugation at 100 000 × g. For protein purification, the supernatants were mixed with Ni2+‐NTA‐Agarose (Qiagen, Hilden, Germany), and the beads were incubated for 1 h under rotation and poured into an empty gravity flow column. After washing with 20 × 10−3 m Tris (pH 8.0) containing 500 × 10−3 m NaCl and 50 × 10−3 m imidazole, protein was eluted with 20 × 10−3 m Tris (pH 8.0) containing 500 × 10−3 m NaCl and 500 × 10−3 m imidazole. Fractions containing purified protein were pooled and dialyzed against PBS. For some experiments RS1‐reg peptides were labeled with NHS‐ATTO488 (ATTO‐TEC GmbH, Siegen, Germany), NHS‐Alexa488 (ThermoFisher Scientific, Waltham, MA, USA) or NHS‐Alexa647 (ThermoFisher Scientific, Waltham, MA, USA) conjugates according to manufacturer´s protocol.
SDS‐Polyacrylamide Gel Electrophoresis (SDS‐PAGE)
SDS polyacrylamide gel electrophoresis (5% stacking gel and 12% separating gel) was performed as described by Laemmli in a Mini‐PROTEAN II slab gel apparatus (Bio‐Rad, Hercules, California, USA) for 60 min at 200 volt.[ 79 ] For standard sample preparation the protein samples were pretreated for 30 min at 37 °C in 60 × 10−3 m Tris‐HCl, pH6.8, 100 × 10−3 m dithiothreitol, 2% (w/v) SDS, and 7% (v/v) glycerol and then separated by SDS‐PAGE. The gels were stained with a colloidal coomassie brilliant blue staining solution according to Candiano et al.[ 80 ]
Preparation of Nanohydrogels in Inverse Miniemulsion
The preparation of nanohydrogels in inverse miniemulsion was carried out on the basis of a previously published protocol.[ 25 ] Inverse miniemulsions were formed from an organic phase consisting of 625 µL hexane, which contained 14.1 mg Span 80 and 4.7 mg Tween 80, and an aqueous phase of 62.5 µL PBS with 25 mg thiol‐functionalized linear polyglycidol prepolymer, 44 µg RS1‐reg polypeptide with N‐terminal cysteine and in some cases 125 µg of the cell‐penetrating TAT peptide (CGRLLRRQRRR) or without cell‐penetrating peptide.[ 29 ] For the preparation of empty control nanohydrogels, the RS1‐reg polypeptide was omitted. To form the inverse miniemulsion, the organic and aqueous phases were combined, stirred and treated by ultrasonication for 60 s with a Branson sonifier 250 (duty cycle of 40% and output control of 10%). During the sonication, the reaction vessel was cooled with an ice bath. By adding 30 µL of 50 × 10−3 m alloxan, the formation of the nanohydrogel and the covalent attachment of the cell‐penetrating peptide and the RS1‐reg peptide, respectively, were achieved by oxidative coupling under formation of a disulfide bridge network. For this purpose, the mixture was immediately treated with ultrasonification for 60 s after alloxan addition and then incubated for 25 min at room temperature with stirring. The oxidation was then stopped by acidification and the formed nanohydrogels were separated by centrifugation. The organic phase was removed, the aqueous phase containing the nanohydrogels was washed twice with n‐hexane and twice with tetrahydrofuran to remove detergents and unreacted prepolymer. Remaining organic solvents and acid were finally removed by multiple dialysis against water. Suspensions of the purified, loaded nanohydrogels were stored in millipore water at 4 °C for further use for up to 4 weeks.
Preparation of Nanohydrogels via Inverse Nanoprecipitation Polymerization
Nanogels were produced by nanoprecipitation polymerization of the polymer solution in acetone.[ 13 ] In the standard procedure, 6 mg thiol‐functionalized, linear polyglycidol was dissolved in 90 µL PBS with 44 µg RS1‐reg polypeptide with N‐terminal cysteine and in some cases with 125 µg TAT‐peptide or without cell‐penetrating peptide and precipitated in 45 mL acetone by rapid addition. For the preparation of empty control nanohydrogels, RS1‐reg polypeptide was omitted. After 30 min, 30 µL of alloxane solution (32 mg mL−1 in water) was added and oxidation was carried out for another 30 min, followed by the addition of 4 mL water. The acetone still contained in the sample was evaporated overnight in a fume hood and the formed nanogels were then washed three times with water by centrifugation (16.100 x g, 15 min). Suspensions of the loaded nanohydrogels were stored at 4 °C for up to 4 weeks.
Determination of Particle Size Distribution, Particle Concentration, and ζ Potential
Measurements were performed using the nanoparticle tracking analysis (NTA) device (NS500, Nanosight). Samples were diluted in a ratio of 1:1000 and 1:10 000 in a total volume of 1 mL of Millipore water. Each sample was measured three to five times (camera level 16, threshold 4−5 with automated settings) at 23 °C. The particle concentration and size distribution of the sample were calculated as a mean value overall measurements. ζ potential measurements were performed by electrophoretic light scattering and size determination by dynamic light scattering (DLS) using a Zetasizer Nano ZSP (Malvern Instruments) instrument. Measurements were performed at 25 °C in water.
Stability Analysis of Nanogels out of Precipitation Polymerization and Inverse Miniemulsion by DLS and Fluorescence Measurements
DLS measurements of nanohydrogel stability after 1:100 dilution in 100 × 10−3 m HCl (pH 1), PBS (pH 7.4) and 20 × 10−3 m Tris/HCl (pH 8.5) were performed for 0–60 min, mimicking pH conditions in stomach (pH 1), in small intestinal lumen and mucosa after fasting (neutral pH range, exact value varies from patient to patient, animal to animal, study to study and position in small intestine) and in presence of digestive juices within small intestine (pH 7.5–8.5) after a meal.[ 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 ] DLS analysis of nanohydrogel stability was carried out for 0, 10, 20, and 30 min after 1:100 dilution in PBS in absence or presence of 10 × 10−3 m DTT, mimicking the reducing conditions in the cytoplasm. Fluorescence measurements of the stability of precipitation polymerization based nanohydrogels loaded with ATTO488‐RS1reg for 10, 20, and 30 min after 1:100 dilution in PBS with or without DTT under liberation of the labeled peptide were done with a SPARK Multimode Microplate Reader (TECAN Trading AG, Switzerland), Excitation wavelength: 500 nm, Emission wavelength: 520 nm.
AFM Imaging
AFM imaging of miniemulsion and nanoprecipitation nanogels was performed using a Dimension Icon equipped with a Nanoscope V controller (Bruker, Santa Barbara, CA) by means of PeakForce Tapping mode in air. Prior to each AFM measurement the aqueous nanogel suspension was vortexed and treated in an ultrasonic bath (120 W) each for 180 s and subsequently spin‐coated (120 s; 2100 rpm, EC101DT, Headway Research Inc., Garland, TX) onto a freshly cleaved mica sheet (#71856‐01; Electron Microscopy Sciences, Hatfield, PA). The surface of the dry mica sheet was imaged using silicon nitride cantilevers (ScanAsyst‐Air, Bruker) with a nominal spring constant of 0.4 N m−1. A PeakForce Tapping amplitude of 150 nm and a frequency of 2 kHz were used. Processing of the imaging data was performed with NanoScope Analysis 1.50 (Bruker).
Turbidity Measurements Regarding Nanogel Stability
To investigate differences concerning stability and cross‐linking density of miniemulsion nanogels versus nanoprecipitation nanogels, time‐dependent turbidity measurements of samples of both nanogel types with equal particle density (1 × 1011 particles mL−1) following supplementation with 10 × 10−3 m DTT (Figure S7, Supporting Information) in a Tecan Spark 20 M multimode microplate reader (Tecan Group Ltd., Männedorf, Switzerland) at a wavelength of 500 nm at 25 °C for 1 h. Nanogels without DTT and a 10 × 10−3 m DTT solution served as controls.
Cell Culture
Caco‐2 cells were grown and routinely maintained at 37 °C in EMEM containing 10% (v/v) FBS and penicillin (100 U mL−1)–streptomycin (100 µg mL−1) in an atmosphere of 5% CO2 and 90% relative humidity. The cells were harvested with Accutase and seeded out on 8 Well Lab‐Tek chamber slides or coverslips for immunostaining or on 12 well plates for uptake measurements. The culture medium was replaced every 2–3 d. Caco‐2 cells were used between the passage 43 and 70.
Uptake Measurements with Caco‐2 Cells
The non‐metabolizable glucose analog α‐methylglucose (AMG), a specific substrate for Na+/glucose cotransporters was used to measure cotransport activity in Caco‐2‐cell monolayers.[ 81 ] Cells were seeded in 12 well plates, grown to confluence and further cultivated for 18 d, at which time they were fully differentiated. Transport assays were performed at 37 °C in PBS as transport medium, supplemented with 10 × 10−6 m α‐methyl‐D‐glucopyranoside (AMG) containing tracer amounts of [14C]AMG (11.1 GBq per mmol, American Labeled Chemical, St. Louis, MO, USA). To evaluate nonspecific uptake of AMG SGLT1 was selectively inhibited by 1 × 10−3 m phlorizin in controls. Cells were preincubated without or with 0.5 mg mL−1 nanogel for 30 min (nanogels with TAT‐peptide) or 120 min (nanogels without TAT). Thereafter AMG‐uptake was performed for 10 min and then stopped by adding ice‐cold transport medium containing 1 × 10−3 m phlorizin and incubation for 5 min. The monolayers were washed twice with the same buffer and solubilized in 2% (w/v) sodium dodecyl sulfate (SDS). Triplicate cultures were assayed for radioactivity by liquid scintillation counting. Counts were normalized in relation to 1 for the approach with Caco‐2 cells without any addition.
Uptake Measurements with Everted Small Intestinal Segments of Mice
For preparation of small intestinal segments, mice were killed by cervical dislocation, and the small intestine was removed. After perfusion with PBS, the small intestine was everted using a steel rod. Eight 1‐cm long segments of the proximal jejunum were isolated and washed with PBS. The small intestinal segments and mucosal pieces were preincubated at 37 °C with PBS in the absence (control) and presence of 0.5 mg mL−1 nanogel for 30 min (nanogels with TAT‐peptide) or 120 min (nanogels without TAT). Thereafter the intestinal samples were washed thoroughly with PBS. For measurements of α‐methyl‐D‐glucopyranoside (AMG) uptake, the samples were incubated for 2 min at 37 °C with PBS containing 10 × 10−6 m AMG traced with [14C]AMG. The incubations were performed in the absence of phlorizin or in the presence of 1 × 10−3 m phlorizin. Uptake was stopped with ice‐cold PBS containing 1 × 10−3 m phlorizin. The segments were washed twice with the same buffer, solubilized with Tissue Solubilizer, Soluene‐350 (PerkinElmer, Waltham, MA), and their radioactivity was determined by liquid scintillation counting. Counts were normalized in relation to 1 for the approach with a small intestinal segment without any addition.
Staining of Small Intestinal Segments of Mice, Pig and Human with ATTO488‐Labeled RS1‐reg for Confocal Laser Scanning Microscopy (CLSM) Analysis
After killing of mice by cervical dislocation, removal and perfusion of small intestine 1 cm long segments of the proximal jejunum were isolated. Furthermore, 1 cm diameter slices were punched out of pig and human small intestinal segments and washed with PBS. Human small intestine segments were obtained as waste material after bariatric surgery at the Surgical Clinic of the University Hospital of Würzburg. Written informed consent was obtained from all patients. The small intestinal samples were preincubated with 0.5 mg mL−1 nanogel in PBS containing ATTO488‐RS1‐reg and TAT‐peptide for 30 min. After washing with PBS the samples were fixed for 24 h with 4% (v/v) p‐formaldehyde and embedded in Tissue Tek (Sakura), snap frozen in liquid nitrogen and histological slices produced with a Leica CM3050 S cryotome that were analyzed by CLSM (Leica TCS SP8).
Localization of ATTO488‐RS1‐reg Uptake via Nanohydrogel in Mouse Gastrointestinal Tract
One hour after administration of 30 µL 0.5 mg mL−1 nanogel in PBS containing ATTO488‐RS1‐reg and TAT peptide via gavage, mice were killed by cervical dislocation. After removal and washing of small intestine 1 cm long segments at different positions (3, 5, 10, 15, 20, 25 cm) after stomach outlet were isolated. Furthermore, stomach segments were excised and washed with PBS. Samples were fixed for 24 h with 4% (v/v) p‐formaldehyde, embedded in Tissue Tek (Sakura), snap frozen in liquid nitrogen, and histological slices produced with a Leica CM3050 S cryotome that were analyzed by CLSM (Leica TCS SP8). A corresponding animal welfare application was approved (file number: 55.2.2‐2532‐2‐704, Government of Upper Bavaria).
Immunostaining of Caco‐2 Cells for Confocal Laser Scanning Microscopy (CLSM) and Structured Illumination Microscopy (SIM) Analysis
For immunostaining, Caco‐2 cells were grown on 8 Well Lab‐Tek chamber slides (0.7 cm2 cultivated area per well) or coverslips (12 mm diameter) to 90% confluence. The cells were preincubated with 10 µg mL−1 nanogel out of inverse miniemulsion or 100 µg mL−1 nanoprecipitation nanohydrogel containing fluorescently labeled hRS1‐reg(S20E) for 30 min (nanogels with TAT) or 120 min (nanogels without TAT), washed twice with PBS, fixed for 15 min with 4% (w/v) paraformaldehyde diluted in PBS, and washed twice again. Free aldehyde groups were quenched by 10 min incubation with 100 × 10−3 m glycine (pH 9). For immunoreactions, washed cells were permeabilized and unspecific binding sites were blocked by a 60 min incubation with PBS containing 2.5% (w/v) BSA and 0.1% (w/v) saponin and incubated for 1 h with primary antibodies diluted in PBS. The dilutions of primary antibodies were as follows: rabbit‐anti‐EEA1‐Ab, 1:100; rabbit‐anti‐TGN46‐Ab, 1:50; mouse‐anti‐Calnexin, 1:50; rabbit anti‐ODC1, 1:50; and rabbit‐anti‐LAMP‐1, 1:200. After incubation with primary antibodies, cells were washed three times with PBS and incubated for 1 h at room temperature with fluorochrome‐linked secondary antibodies donkey‐anti‐rabbit‐ATTO643 or goat‐anti‐mouse‐ATTO643, diluted 1:200 each. The cells were washed five times with PBS, rinsed shortly with double‐distilled water, and for some experiments embedded in Pro Long Glass Anti Fade Mounting Medium from Invitrogen/Thermo Fisher Scientific (Carlsbad, CA, USA). Nuclei were stained with Hoechst 33342 or Hoechst 34580 (Sigma‐Aldrich GmbH, Taufkirchen, Germany).
Quantitative Colocalization Analysis
To estimate colocalization between two channels of each slice from SIM z‐stacks Pearson's coefficient was calculated using a custom‐written ImageJ macro (see Supplementary). The macro is based on the JACoP plugin (https://imagej.nih.gov/ij/plugins/track/jacop2.html) to calculate Pearson's coefficients for each individual slice of all slices in a z‐stack between two channels.[ 82 ] The results for each slice are written into a single results table for each z‐stack, respectively. Individual Pearson's coefficients of each stack, with at least 39 slices/data points, are depicted as superviolin plot using the python package superviolin 1.0.6 (https://pypi.org/project/superviolin/).[ 83 ]
Airyscan Analysis of TAT Localization
To analyze whether TAT was bound merely in the peripherie of nanogel particles or coupled to the matrix throughout the entire particle volume, the TAT peptide was fluorescently labeled with Alexa 647 and incorporated into nanogels out of nanoprecipitation. Especially particles in the sample with a much higher diameter than the mean were analyzed to get a clear result using a ZEISS LSM 900 confocal microscope with Airyscan 2 (Leica Microsystems, Wetzlar, Germany) in Airyscan mode with a laser at a wavelength of 640 nm.
Cryo Scanning Electron Microscopy (Cryo‐SEM)
The hydrogel nanoparticles were imaged using a Crossbeam 340 field emission scanning electron microscope (SEM) (Carl Zeiss Microscopy, Oberkochen, Germany). To visualize the nanoparticles in a swollen state, nanogels in a concentrated form were placed between two alumina holders (d = 3 mm), both containing a notch with a diameter of 2 mm, inclosing the sample and rapidly frozen in nitrogen slush (SN) at ‐210°C. The samples were subsequently transferred with an EM VCT100 cryo‐shuttle into an EM ACE600 sputter coater (both Leica Microsystems, Wetzlar, Germany) at ‐140°C. Here, under high vacuum (<1 × 10−3 mbar) the upper half of the sample was knocked off, creating a freshly fractured surface, and freeze‐etched at ‐85 °C for 15 min. After etching, the samples were sputtered with 2.5 nm of platinum and again transferred with the cryo‐shuttle into the SEM. Here the morphology of the fractured surface with the uncovered nanoparticles was imaged at ‐140 °C, by setting an acceleration voltage of 8 kV and detection of secondary electrons with an Everhart Thornley detector. Supplemental measurements were taken by energy dispersive X‐ray spectroscopy (EDX), to verify the positions of the nanogel on the surface. The EDX measurements were taken with an X‐MaxN 50 Silicon Drift Detector (Oxford Instruments, Abington, England), also setting the acceleration voltage to 8 kV.
Statistics
Uptake measurements indicated in Figure 2 are presented as mean ± S.E. Uptake rates of phlorizin‐inhibited AMG uptake into Caco‐2 cells were calculated from three independent experiments. In each experiment, three uptake measurements were performed in the absence of phlorizin and corrected for uptake measured in the presence of phlorizin. Uptake rates measured in segments of intestinal mucosa were calculated from measurements in three animals. For each animal, the uptake rate was calculated from three measurements using two experimental conditions (AMG uptake without and with phlorizin). When three or more groups of data were compared, the significance of differences was determined by analysis of variance (ANOVA) using post hoc Tukey comparison. The significance of the differences between two groups was determined by Student's t test. P < 0.05 was considered significant.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
This work was supported by the European Regional Development Fund (ERDF Bavaria 2014‐2020).
Open access funding enabled and organized by Projekt DEAL.
Keller T., Trinks N., Brand J., Trippmacher S., Stahlhut P., Albrecht K., Papastavrou G., Koepsell H., Sauer M., Groll J., Design of Nanohydrogels for Targeted Intracellular Drug Transport to the Trans‐Golgi Network. Adv. Healthcare Mater. 2023, 12, 2201794. 10.1002/adhm.202201794
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Peppas N. A., Bures P., Leobandung W., Ichikawa H., Eur. J. Pharm. Biopharm. 2000, 50, 27. [DOI] [PubMed] [Google Scholar]
- 2. Kamath K. R., Park K., Adv. Drug Delivery Rev. 1993, 11, 59. [Google Scholar]
- 3. Kim S. W., Bae Y. H., Okano T., Pharm Res 1992, 9, 283. [DOI] [PubMed] [Google Scholar]
- 4. Schwall C. T., Banerjee I. A., Materials 2009, 2, 577. [Google Scholar]
- 5. Cuggino J. C., Blanco E. R. O., Gugliotta L. M., Igarzabal C. I. A., Calderón M., J. Controlled Release 2019, 307, 221. [DOI] [PubMed] [Google Scholar]
- 6. Schexnailder P., Schmidt G., Colloid Polym. Sci. 2009, 287, 1. [Google Scholar]
- 7. Gao D., Xu H., Martin A. P., Kopelman R., Adv. Drug Delivery Rev. 2008, 8, 3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hamidi M., Azadi A., Rafiei P., Adv. Drug Delivery Rev. 2008, 60, 1638. [DOI] [PubMed] [Google Scholar]
- 9. Qiu Y., Park K., Adv. Drug Delivery Rev. 2012, 64, 49. [DOI] [PubMed] [Google Scholar]
- 10. Fleige E., Quadir M. A., Adv. Drug Delivery Rev. 2012, 64, 866. [DOI] [PubMed] [Google Scholar]
- 11. Klinger D., Landfester K., Polymer 2012, 53, 5209. [Google Scholar]
- 12. Groll J., Singh S., Albrecht K., Moeller M., J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 5543. [Google Scholar]
- 13. Horvat S., Yu Y., Manz H., Keller T., Beilhack A., Groll J., Albrecht K., Adv Nanobiomed Res 2021, 1, 2000060. [Google Scholar]
- 14. Zilkowski I., Ziouti F., Schulze A., Hauck S., Schmidt S., Mainz L., Sauer M., Albrecht K., Jundt F., Groll J., Biomacromolecules 2019, 20, 916. [DOI] [PubMed] [Google Scholar]
- 15. Korn T., Kühlkamp T., Track C., Schatz I., Baumgarten K., Gorboulev V., Koepsell H., J. Biol. Chem. 2001, 276, 45330. [DOI] [PubMed] [Google Scholar]
- 16. Veyhl M., Keller T., Gorboulev V., Vernaleken A., Koepsell H., Am. J. Physiol. 2006, 291, F1213. [DOI] [PubMed] [Google Scholar]
- 17. Kroiss M., Leyerer M., Gorboulev V., Kühlkamp T., Kipp H., Koepsell H., Am. J. Physiol. 2006, 291, F1201. [DOI] [PubMed] [Google Scholar]
- 18. Veyhl M., Wagner C. A., Gorboulev V., Schmitt B. M., Lang F., Koepsell H., J. Membr. Biol. 2003, 196, 71. [DOI] [PubMed] [Google Scholar]
- 19. Filatova A., Leyerer M., Gorboulev V., Chintalapathi C., Reinders Y., Mueller T. D., Srinivasan A., Hübner S., Koepsell H., Traffic 2009, 10, 1599. [DOI] [PubMed] [Google Scholar]
- 20. Wright E. M., Loo D. D. F., Hirayama B. A., Physiol. Rev. 2011, 91, 733. [DOI] [PubMed] [Google Scholar]
- 21. Vrhovac I., Balen Eror D., Klessen D., Burger C., Breljak D., Kraus O., Radović N., Jadrijević S., Aleksic I., Walles T., Sauvant C., Sabolić l., Koepsell H., Pflugers Arch. ‐ Eur. J. Physiol. 2015, 467, 1881. [DOI] [PubMed] [Google Scholar]
- 22. Gorboulev V., Schürmann A., Vallon V., Kipp H., Jaschke A., Klessen D., Friedrich A., Scherneck S., Rieg T., Cunard R., Veyhl‐Wichmann M., Srinivasan A., Balen D., Breljak D., Rexhepaj R., Parker H. E., Gribble F. M., Reimann F., Lang F., Wiese S., Sabolić I., Sendtner M., Koepsell H., Diabetes 2012, 61, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Koepsell H., Gorboulev V., Diabetes 2012, 61, e5.22618779 [Google Scholar]
- 24. Vernaleken A., Veyhl M., Gorboulev V., Kottra G., Palm D., Burckhardt B. C., Burckhardt G., Pipkorn R., Beier N., van Amsterdam C., Koepsell H., J. Biol. Chem. 2007, 282, 28501. [DOI] [PubMed] [Google Scholar]
- 25. Veyhl‐Wichmann M., Friedrich A., Vernaleken A., Singh S., Kipp H., Gorboulev V., Keller T., Chintalapati C., Pipkorn R., Pastor‐Anglada M., Groll J., Koepsell H., Mol. Pharmacol. 2016, 89, 118. [DOI] [PubMed] [Google Scholar]
- 26. Chintalapati C., Keller T., Mueller T. D., Gorboulev V., Schäfer N., Zilkowski I., Veyhl‐Wichmann M., Geiger D., Groll J., Koepsell H., Mol. Pharmacol. 2016, 90, 508. [DOI] [PubMed] [Google Scholar]
- 27. Koepsell H., Pflugers Arch 2020, 472, 1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Horvat S., Adv Nanobiomed Res. 2021, 1, 2000060. [Google Scholar]
- 29. Torchilin V. P., Adv. Drug Delivery Rev. 2008, 60, 548. [DOI] [PubMed] [Google Scholar]
- 30. Russell T. L., Berardi R. R., Barnett J. L., Dermentzoglou L. C., Jarvenpaa K. M., Schmaltz S. P., Dressman J. B., Pharm. Res. 1993, 10, 187. [DOI] [PubMed] [Google Scholar]
- 31. Koziolek M., Grimm M., Becker D., Iordanov V., Zou H., Shimizu J., Wanke C., Garbacz G., Weitschies W., J. Pharm. Sci. 2015, 104, 2855. [DOI] [PubMed] [Google Scholar]
- 32. Fallingborg J., Dan Med Bull 1999, 46, 183. [PubMed] [Google Scholar]
- 33. Barmpatsalou V., Dubbelboer I. R., Rodler A., Jacobson M., Karlsson E., Pedersen B. L., Bergström C. A. S., Eur. J. Pharm. Biopharm. 2021, 169, 156. [DOI] [PubMed] [Google Scholar]
- 34. Itoyama S., Noda E., Takamatsu S., Kondo J., Kawaguchi R., Shimosaka M., Fukuoka T., Motooka D., Nakamura S., Tanemura M., Mitsufuji S., Iwagami Y., Akita H., Tobe T., Kamada Y., Eguchi H., Miyoshi E., JGH Open 2022, 6, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jones K. K., Proc. Soc. Exp. Biol. Med. 1931, 28, 567. [Google Scholar]
- 36. Hoerner M. T., Am. J. Dig. Dis. Nutr. 1935, 2, 300. [Google Scholar]
- 37. Takeshima T., Adler M., Nacchiero M., Rudick J., Dreiling D. A., Am. J. Gastroenterol. 1977, 67, 54. [PubMed] [Google Scholar]
- 38. Kipp H., Khoursandi S., Scharlau D., Kinne R. K., Am. J. Physiol.: Cell Physiol. 2003, 284, C737. [DOI] [PubMed] [Google Scholar]
- 39. Vivès E., Brodin P., Lebleu B., J. Biol. Chem. 1997, 272, 16010. [DOI] [PubMed] [Google Scholar]
- 40. Yang J., Chen H., Vlahov I. R., Cheng J.‐X., Low P. S., Proc. Natl. Acad. Sci. USA 2006, 103, 13872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shen w.‐c., Ryser H., LaManna L., J. Biol. Chem. 1985, 260, 10905. [PubMed] [Google Scholar]
- 42. Blewitt M. G., Chung L. A., London E., Biochemistry 1985, 24, 5458. [DOI] [PubMed] [Google Scholar]
- 43. Falnes P. Ø., Olsnes S., J. Biol. Chem. 1995, 270, 20787. [DOI] [PubMed] [Google Scholar]
- 44. Ryser H., Mandel R., Ghani F., J. Biol. Chem. 1991, 266, 18439. [PubMed] [Google Scholar]
- 45. Falnes P. Ø., Sandvig K., Curr. Opin. Cell Biol. 2000, 12, 407. [DOI] [PubMed] [Google Scholar]
- 46. Sakhrani N. M., Padh H., Drug Des., Dev. Ther. 2013, 7, 585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wlodkowic D., Skommer J., McGuinness D., Hillier C., Darzynkiewicz Z., Leuk Res 2009, 33, 1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Migita T., Inoue S., Int. J. Biochem. Cell Biol. 2012, 44, 1872. [DOI] [PubMed] [Google Scholar]
- 49. Pópulo H., Lopes J. M., Soares P., Int. J. Mol. Sci. 2012, 13, 1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Drenan R. M., Liu X., Bertram P. G., Zheng X. F., J. Biol. Chem. 2004, 279, 772. [DOI] [PubMed] [Google Scholar]
- 51. Wang Q., Mora‐Jensen H., Weniger M. A., Perez‐Galan P., Wolford C., Hai T., Ron D., Chen W., Trenkle W., Wiestner A., Ye Y., Proc. Natl. Acad. Sci. USA 2009, 106, 2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Aridor M., Hannan L. A., Traffic 2000, 1, 836. [DOI] [PubMed] [Google Scholar]
- 53. Joshi G., Chi Y., Huang Z., Wang Y., Proc. Natl. Acad. Sci. USA 2014, 111, E1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mallard F., Antony C., Tenza D., Salamero J., Goud B., Johannes L., J. Cell Biol. 1998, 143, 973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lieu Z. Z., Gleeson P. A., Eur. J. Cell Biol. 2010, 89, 379. [DOI] [PubMed] [Google Scholar]
- 56. Utskarpen A., Slagsvold H. H., Iversen T.‐G., Wälchli S., Sandvig K., Traffic 2006, 7, 663. [DOI] [PubMed] [Google Scholar]
- 57. Grimmer S., Iversen T. G., van Deurs B., Sandvig K., Mol. Biol. Cell 2000, 11, 4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sandvig K., van Deurs B., EMBO J. 2000, 19, 5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Plaut R. D., Carbonetti N. H., Cell. Microbiol. 2008, 10, 1130. [DOI] [PubMed] [Google Scholar]
- 60. Chia P. Z. C., Gleeson P. A., Traffic 2011, 12, 939. [DOI] [PubMed] [Google Scholar]
- 61. Bonifacino J. S., Rojas R., Nat. Rev. Mol. Cell Biol. 2006, 7, 568. [DOI] [PubMed] [Google Scholar]
- 62. Johannes L., Popoff V., Cell 2008, 135, 1175. [DOI] [PubMed] [Google Scholar]
- 63. Sannerud R., Saraste J., Goud B., Curr. Opin. Cell Biol. 2003, 15, 438. [DOI] [PubMed] [Google Scholar]
- 64. Ghosh R. N., Mallet W. G., Soe T. T., McGraw T. E., Maxfield F. R., J. Cell Biol. 1998, 142, 923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lewis M. J., Nichols B. J., Prescianotto‐Baschong C., Riezman H., Pelham H. R., Mol. Biol. Cell 2000, 11, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ghosh P., Dahms N. M., Kornfeld S., Nat. Rev. Mol. Cell Biol. 2003, 4, 202. [DOI] [PubMed] [Google Scholar]
- 67. Shewan A. M., van Dam E. M., Martin S., Luen T. B., Hong W., Bryant N. J., James D. E., Mol. Biol. Cell 2003, 14, 973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sandvig K., van Deurs B., Gene Ther. 2005, 12, 865. [DOI] [PubMed] [Google Scholar]
- 69. Qiu C., Han H.‐H., Sun J., Zhang H.‐T., Wei W., Cui S.‐H., Chen X., Wang J.‐C., Zhang Q., Nat. Commun. 2019, 10, 2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ross N. L., Munsell E. V., Sabanayagam C., Sullivan M. O., Mol Ther Nucleic Acids 2015, 4, e226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lai S. K., Wang Y. Y., Hanes J., Adv. Drug Delivery Rev. 2009, 61, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Xu Q., Boylan N. J., Suk J. S., Wang Y. Y., Nance E. A., Yang J. C., McDonnell P. J., Cone R. A., Duh E. J., Hanes J., J. Controlled Release 2013, 167, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sanders N. N., De Smedt S. C., Van Rompaey E., Simoens P., De Baets F., Demeester J., Am J. Respir. Crit. Care Med. 2000, 162, 1905. [DOI] [PubMed] [Google Scholar]
- 74. Ensign L. M., Cone R., Hanes J., Adv. Drug Delivery Rev. 2012, 64, 557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Laffleur F., Hintzen F., Shahnaz G., Rahmat D., Leithner K., Bernkop‐Schnürch A., Nanomedicine 2014, 9, 387. [DOI] [PubMed] [Google Scholar]
- 76. Cone R. A., Adv. Drug Delivery Rev. 2009, 61, 75. [DOI] [PubMed] [Google Scholar]
- 77. Nance E., Zhang C., Shih T. Y., Xu Q., Schuster B. S., Hanes J., ACS Nano 2014, 8, 10655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wang Y. Y., Lai S. K., Suk J. S., Pace A., Cone R., Hanes J., Angew Chem Int Ed Engl 2008, 47, 9726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Laemmli U. K., Nature 1970, 227, 680. [DOI] [PubMed] [Google Scholar]
- 80. Candiano G., Bruschi M., Musante L., Santucci L., Ghiggeri G. M., Carnemolla B., Orecchia P., Zardi L., Righetti P. G., Electrophoresis 2004, 25, 1327. [DOI] [PubMed] [Google Scholar]
- 81. Matosin‐Matekalo M., Mesonero J. E., Delezay O., Poiree J. C., Ilundain A. A., Brot‐Laroche E., Biochem. J. 1998, 334, 633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bolte S., Cordelières F. P., J Microsc 2006, 224, 213. [DOI] [PubMed] [Google Scholar]
- 83. Kenny M., Schoen I., Mol. Biol. Cell 2021, 32, 1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.