

Abstract
Type 2 diabetes mellitus (T2D), a major cause of worldwide morbidity and mortality, is characterized by dysfunction of insulin-producing pancreatic islet β cells1,2. T2D genome-wide association studies (GWAS) have identified hundreds of signals in non-coding and β cell regulatory genomic regions, but deciphering their biological mechanisms remains challenging3–5. Here, to identify early disease-driving events, we performed traditional and multiplexed pancreatic tissue imaging, sorted-islet cell transcriptomics and islet functional analysis of early-stage T2D and control donors. By integrating diverse modalities, we show that early-stage T2D is characterized by β cell-intrinsic defects that can be proportioned into gene regulatory modules with enrichment in signals of genetic risk. After identifying the β cell hub gene and transcription factor RFX6 within one such module, we demonstrated multiple layers of genetic risk that converge on an RFX6-mediated network to reduce insulin secretion by β cells. RFX6 perturbation in primary human islet cells alters β cell chromatin architecture at regions enriched for T2D GWAS signals, and population-scale genetic analyses causally link genetically predicted reduced RFX6 expression with increased T2D risk. Understanding the molecular mechanisms of complex, systemic diseases necessitates integration of signals from multiple molecules, cells, organs and individuals, and thus we anticipate that this approach will be a useful template to identify and validate key regulatory networks and master hub genes for other diseases or traits using GWAS data.
T2D is a major cause of macro and microvascular morbidity and mortality worldwide. Clinically heterogenous, T2D involves genetic, environmental and pathophysiologic components that affect multiple molecular pathways and tissues6,7. Initial management of the disease frequently involves lifestyle modifications but usually escalates to medication and, often, exogenous insulin to control blood glucose. T2D prevalence increases with obesity and age, both of which reduce peripheral insulin sensitivity; however, most insulin-resistant individuals do not develop T2D. Instead, a defining feature of T2D is impaired insulin secretion1,2. Insulin is secreted by the β cell of the pancreatic islet, a mini-organ composed of endocrine cells (β, α, δ, γ and ε cells) and other cell types (for example, endothelial, immune and pericytes), which coordinate to control glucose homeostasis8. Although islet dysfunction is a hallmark of T2D, it remains unclear whether this is caused by an intrinsic defect in β cells, a reduction in β cell number, systemic signals, or some combination of these factors.
T2D has a strong genetic component; more than 400 signals have been identified through genome-wide association studies3,9,10. However, 90% of these single nucleotide polymorphisms (SNPs) are located in non-coding genomic enhancer regions, where they are expected to modulate β cell- and islet cell-specific gene expression, implicating β cell processes as a key determinant for T2D pathophysiology4,5,11–13. How population-level, disease-associated genetic variation relates to molecular changes in gene expression, tissue architecture and cellular physiology in T2D islets is not well understood. Postulated T2D disease processes, studied primarily in rodent models, include β cell loss and/or dedifferentiation, endoplasmic reticulum stress, amyloid deposition, oxidative stress, glucotoxicity, lipotoxicity and islet inflammation2,14. Notably, human islets differ from mouse islets in key features including cellular architecture, basal and stimulated insulin secretion, response to dyslipidemia and hyperglycaemia, and expression of islet-enriched transcription factors15,16, highlighting the need for studies to define initiating and sustaining mechanisms of islet dysfunction in primary human islets.
Recent advances in pancreas procurement from organ donors with diabetes have increased the availability of human tissue and isolated islets for molecular and functional profiling17–19. However, many studies utilize only tissue or islets, and further, do not differentiate outcomes based on T2D duration. Since different stages of T2D may involve different processes, such approaches make it difficult to discern cellular and molecular causes from disease consequences. The association of physiological measurements with transcriptomic profiles of islet cells have begun to identify critical pathways for β cell function17,18, but integration with disease stage, tissue-based analyses and genetic risk remains a challenge. Exacerbating this challenge is the likelihood that heterogenous pathways are involved in disease predisposition: it is not yet clear whether T2D GWAS variants are uniformly distributed across or focally concentrated in pathways.
Here we used an integrated approach to study the pancreas and islets from donors with early-stage T2D (referred to here as early T2D) and controls to identify disease-driving molecular mechanisms of T2D. We analysed islet function both ex vivo and in vivo using a transplant system and performed comprehensive transcriptional analysis by bulk RNA sequencing of whole islets and purified β and α cells, correlating these profiles to functional parameters and GWAS variants. Concurrently, we assessed changes in the pancreatic islet microenvironment via traditional and multiplexed imaging approaches, including spatial cellular relationships. We found that early T2D is defined primarily by β cell-intrinsic defects that can be proportioned into gene regulatory modules, including an RFX6-governed and GWAS-enriched transcriptional regulatory network. We validated this network at the molecular level through targeted gene perturbation and at the population scale using Mendelian randomization (MR), thus demonstrating convergence of multiple layers of genetic risk.
Dynamic functional analysis
Focusing on early T2D as defined by a combination of disease duration and treatment approach, we collected pancreata and isolated islets for multimodal analysis from individuals with T2D (n = 20; mean disease duration 3.5 years) as well as nondiabetic (ND) donors (n = 36) (Fig. 1a,b and Supplementary Table 1). Compared with age- and body mass index (BMI)-matched controls, basal insulin secretion in T2D islets was similar, but stimulated secretion was substantially reduced in response to high glucose, cyclic AMP (cAMP)-evoked potentiation and potassium chloride (KCl)-mediated depolarization when normalized by islet volume (Fig. 1c–f and Extended Data Fig. 1a–e). Inhibition of insulin secretion by low glucose and adrenaline was similar between ND and T2D islets, as was insulin content (Fig. 1g and Extended Data Fig. 1f); thus, normalization of response by islet insulin content showed similar reductions in stimulated insulin secretion but also showed reduced basal insulin secretion (Supplementary Fig. 1a–f). Together, these data suggest that early T2D islets ex vivo maintain insulin production and storage but have defects at multiple steps of the insulin secretory pathway, including those distal to glucose metabolism, which persist after islet isolation from the in vivo environment.
Fig. 1 |. Integrated analysis of islet function, gene expression, and tissue architecture in a cohort of donors with early T2D reveals reduced stimulated insulin secretion ex vivo and in vivo and highlights dysregulated pathways in purified β and α cells as well as whole islets.
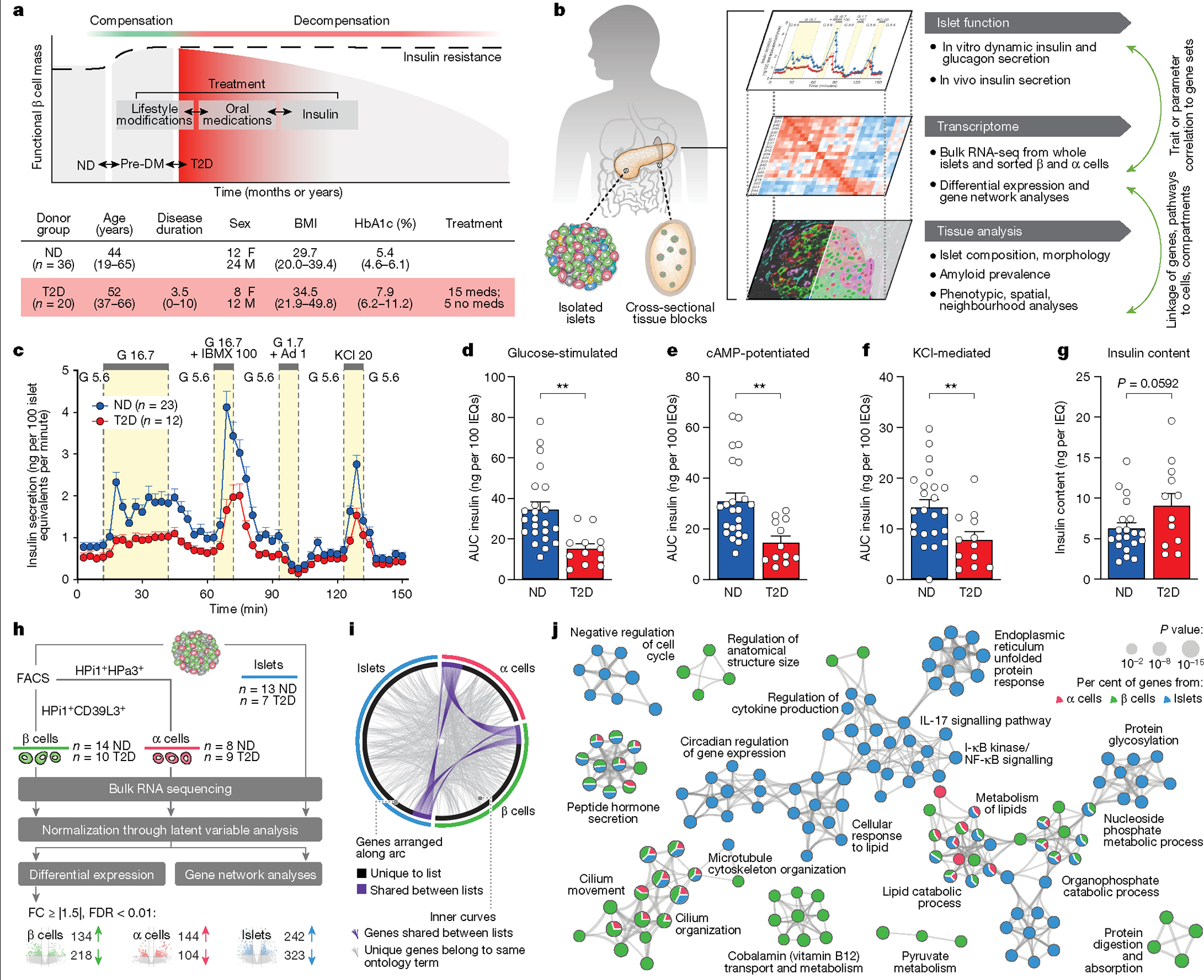
a, Top, schematic of disease progression from ND to pre-diabetes (pre-DM) and T2D, highlighting the progressive loss of functional β cell mass (red shading represents the disease stage targeted with this cohort); bottom, clinical cohort profile. See also Supplementary Table 1. HbA1c, haemoglobin A1C test. b, Application of multiple modalities, including ex vivo and in vivo islet function, tissue architecture and microenvironment, and cell type-specific gene expression in the study. Coordinated study on islets and tissue from same donor enabled integration between analyses (green arrows). RNA-seq, RNA sequencing. c, The dynamic insulin secretory response (P = 0.0005) of islets from ND (n = 23) and T2D (n = 12) independent donors measured by islet perifusion. Treatments (and concentrations) are shown along the top of the graph: Ad, adrenaline (μM); G, glucose (mM); IBMX, isobutylmethylxanthine (μM); KCl, potassium chloride (mM). d–f, Secretory response, quantified as area under the curve (AUC), to glucose (d), cAMP (e), and KCl (f). IEQs, islet equivalents. g, Islet insulin content normalized to islet volume. h, Schematic of RNA sample collection and analysis. A latent variable analysis separated biological from technical variation, followed by examination by both differential gene expression and gene network analyses. FC, fold change; FDR, false discovery rate. i, Common differentially expressed genes in T2D β cell, α cell and islet samples at the level of gene ID (purple curves) or ontology term enrichment (grey curves; P < 0.01). j, Metascape network showing a subset of enriched terms from differentially expressed genes (two-tailed hypergeometric test). Edges denote similarity and node colours reflect the contribution of sample(s). Of note, endoplasmic reticulum processing and unfolded protein pathways were more enriched in islets than in isolated α or β cells. Data in c–g are mean + s.e.m. c, Two-tailed linear mixed-effect model. d–g, Two-tailed t-test indicated as follows. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
In contrast to insulin secretion, glucagon secretion under basal, stimulatory and inhibitory conditions, as well as glucagon content was similar in T2D islets (Extended Data Fig. 1g–m and Supplementary Fig. 1g–l). Although there is substantial evidence of dysregulated glucagon secretion in T2D20, these data suggest either that α cell dysfunction is not present in early T2D, or that defects are present in vivo but not maintained after islet isolation. Stimulated insulin secretion correlated to donor HbA1c (Extended Data Fig. 1n). Therefore, to test whether the systemic environment contributed to β cell dysfunction in T2D islets, we transplanted T2D or ND islets from a subset of donors into normoglycaemic, non-insulin-resistant immunodeficient NOD-scid-Il2rgnull (NSG) mice (Extended Data Fig. 1o). After six weeks in this environment, T2D islets secreted less human insulin than ND islets despite similar engraftment, consistent with ex vivo findings of impaired stimulated insulin secretion (Extended Data Fig. 1p–s). In sum, these experiments highlight that β cell dysfunction in early T2D persists in a normoglycaemic, non-insulin-resistant environment and suggest that intrinsic β cell dysregulation and/or cellular and molecular alterations within the islet microenvironment are key features driving reduced insulin secretion.
Islet, β cell and α cell transcriptomes
Studying β and α cells purified by fluorescence-activated cell sorting (FACS) together with whole islets enabled detailed appreciation of both cell type-specific and islet-wide transcriptional changes in T2D (Fig. 1h, Extended Data Fig. 2, Supplementary Fig. 2 and Supplementary Table 2). Differential expression analysis highlighted that β cell genes involved in stimulated insulin secretion (G6PC2 and GLP1R) and mitochondrial, exocytosis, ion transport and protein secretion pathways were enriched in T2D β cells (Extended Data Fig. 2a,d). Despite diverse differentially expressed genes across sample types (Fig. 1i), there was considerable overlap at the level of biological pathways in which these genes are involved; hormone secretion, lipid metabolism and cilia organization were among the most enriched biological pathways across samples (Fig. 1j). In sum, parallel analyses emphasize common dysregulated pathways among sample types as well as cell-specific transcriptomic changes.
Endocrine cell mass
To understand the context in which functional and transcriptomic changes occur, we comprehensively evaluated the islet architecture in pancreatic tissue from T2D donors by immunohistochemistry and co-detection by indexing (CODEX), a multiplexed technique that enables simultaneous visualization of multiple tissue compartments and identification of cellular phenotypes (Supplementary Tables 3 and 4 and Extended Data Fig. 3a). Images are available in Pancreatlas (https://pancreatlas.org/datasets/904/explore) for reader exploration. Since changes in endocrine cell number or ratio could explain the reduced insulin secretion in T2D islets, we first analysed β, α, and δ cell populations across the pancreas head, body and tail. Islet cell area and islet cell count from sagittal tissue cross-sections revealed that β and α cell mass in early T2D were similar to controls (Fig. 2a and Extended Data Fig. 3b–g). The abundance of endocrine cells quantified by CODEX was also similar (Fig. 2b,c and Extended Data Fig. 3h–j). Apoptotic and/or necrotic cells were exceedingly rare in both ND and T2D islets (data not shown), and cells positive for chromogranin A (CHGA) but negative for all hormones—occasionally considered ‘dedifferentiated’ β cells—were rare but present in similar proportions (Extended Data Fig. 3k). Evidence of amyloid deposits, the abnormal buildup of β cell-produced islet amyloid polypeptide (IAPP) that manifests in T2D, was detectable in 75% of donors in this cohort but did not correlate to endocrine cell abundance or area (Extended Data Fig. 3l–m). Thus, these data suggest that changes in endocrine cell populations and β cell mass are not a substantial component of early T2D and instead point to reduction in β cell function as the predominant feature of this disease stage.
Fig. 2 |. Integrated tissue analysis reveals no change to endocrine cell mass or number and modest changes in intraislet capillaries, T cells and cellular neighbourhoods in the early T2D cohort.
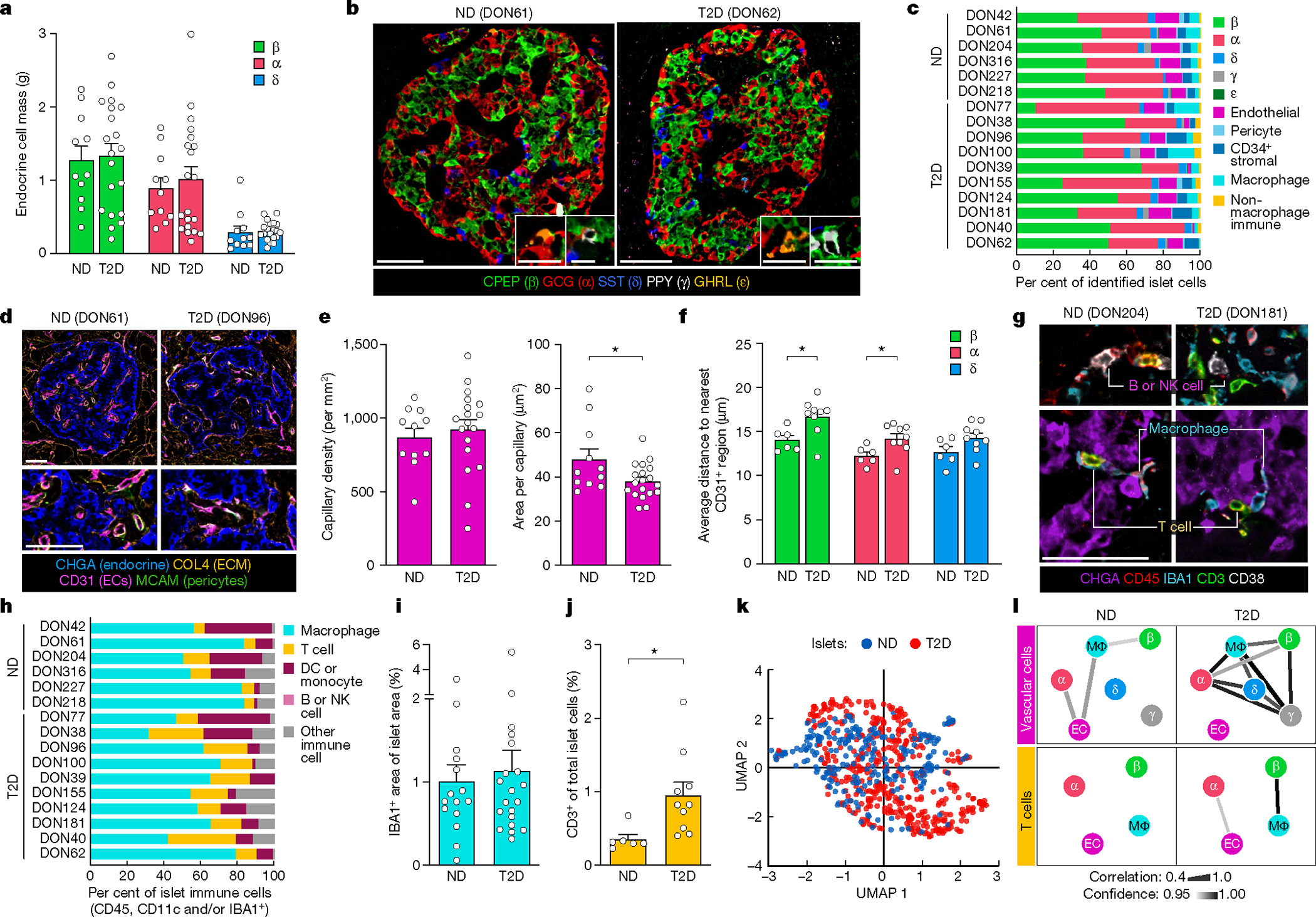
a, The mass of β, α and δ cells in 11 independent ND donors and 20 independent T2D donors. b, Representative images of islets from CODEX imaging; insets show γ and ε cells. c, Relative proportions of islet endocrine, vascular, stromal and immune cells. d, Representative images of islet capillaries, pericytes and extracellular matrix (ECM). EC, endothelial cell. e, Islet capillary density and area per capillary in n = 11 ND and n = 17 T2D donors. P = 0.0268. f, Spatial analysis of endocrine cells and islet capillaries in n = 6 ND and n = 9 T2D donors. g,h, Islet immune cell phenotypes and composition. β cells: P = 0.0247; α cells: P = 0.0479. DC, dendritic cell; NK, natural killer. i,j, Islet macrophage (i; n = 11 ND and n = 20 T2D) and T cell (j; n = 6 ND and n = 10 T2D; P = 0.0246) abundance. k, High-dimensional component analysis by uniform manifold approximation and projection (UMAP) of islet cell composition per islet (n = 255 ND, n = 426 T2D). l, Cellular neighbourhood changes in T2D versus ND islets. Line thickness and colour indicate correlation and confidence, respectively. a,e,i, Traditional immunohistochemistry data. b–d,f–h,j–l, CODEX data. Scale bars, 50 μm (inset in b, 25 μm). Bar graphs show mean + s.e.m. Two-way ANOVA with Šídák’s multiple comparisons test (a,f); two-tailed t-test (e,i,j). None of the variables shown had a statistically significant association with disease duration (Pearson correlation, threshold P < 0.05). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Islet microenvironment
Adequate islet vascularization and blood flow are critical for sensing and delivery of hormones to systemic circulation, and RNA-seq analysis highlighted enrichment in T2D samples for processes controlling capillary maintenance (Extended Data Fig. 4a–c). Morphometric analysis of islet capillary endothelial cells demonstrated that capillary size, but not density, was reduced in T2D islets (Fig. 2d,e), resulting in a greater distance of α and β cells to the nearest capillary in T2D islets (Fig. 2f and Extended Data Fig. 4d). The phenotypic markers CD34, a cell adhesion molecule, and HLA-DR, a major histocompatibility class II (MHCII) receptor, were unchanged in T2D endothelial cells (Extended Data Fig. 4e). Similar to vascular signatures, immune activity was also detectable by transcriptional profiling (Extended Data Fig. 4f,g). Macrophages, the largest population of intraislet immune cells, did not differ between ND and T2D on the basis of abundance or phenotypic classification (Fig. 2g,i and Extended Data Fig. 4h). T cells, the second most prevalent intraislet immune population, were greater in T2D islets across all phenotypes (Fig. 2j and Extended Data Fig. 4i). HLA-DR+ T cells, previously observed in T2D islets21, were not increased, though they were more abundant in a subset of T2D donors (Extended Data Fig. 4j). High-dimensional data analysis using all identified cell types within individually annotated islets revealed a high degree of overlap between islets from ND and T2D donors, emphasizing that although there are subtle differences, the overall islet composition is similar (Fig. 2k and Extended Data Fig. 4k).
We next performed cellular neighbourhood studies to identify differential cellular spatial architecture not detected by compositional analyses. A community detection algorithm tailored to islet cell frequencies, termed CF-IDF, categorized six different cellular neighbourhoods (CNs), clusters of cells with distinct cell-type compositions that were defined by the most enriched cell type (CN0–CN5; Extended Data Fig. 5a). A parallel k-means approach corroborated cellular neighbourhood classifications and similar cellular neighbourhood distribution between ND and T2D islets (Extended Data Fig. 5b,c). Endothelial cells and pericytes were depleted in β cellular neighbourhoods (CN1) of T2D islets, whereas these cellular neighbourhoods had higher β cell enrichment than ND cellular neighbourhoods (Extended Data Fig. 5d,e). Vascular cell frequencies were correlated between more cellular neighbourhoods in T2D compared with ND islets, whereas T cell frequencies were specifically correlated between endothelial cells and α cellular neighbourhoods as well as β cell and macrophage cellular neighbourhoods in T2D (Fig. 2l and Extended Data Fig. 5f,g), congruent with findings from islet RNA-seq showing that endothelial cell-specific and immune signals were upregulated in T2D. Together, these results demonstrate modest changes in islet organization by vascular and immune cells in early T2D.
Co-expression network analyses
To understand the key gene networks that were contributing to β cell dysfunction in early T2D, we performed network analysis on α cell, β cell and islet samples using weighted gene co-expression network analysis (WGCNA) (Supplementary Fig. 3). This approach created modules (eigengenes) of up to 2,000 genes each, labelled by sample type and numbered consecutively (for example, β cells constituted modules β00–β48), that enabled association of transcriptomic profiles with curated gene lists, donor traits, islet functional parameters measured by perifusion and enrichment of open chromatin peaks to overlap GWAS variants (Fig. 3, Extended Data Fig. 6 and Supplementary Table 5). Modules with significant correlations were then queried on the basis of their member genes for ontology terms to determine biological processes related to significant associations (Supplementary Fig. 4). We highlight noteworthy observations below, with results available for further exploration online (https://theparkerlab.shinyapps.io/Islet-RNAseq-WGCNA/).
Fig. 3 |. WGCNA distinguishes β cell gene modules associated with donor and islet traits as well as those enriched in GWAS loci.
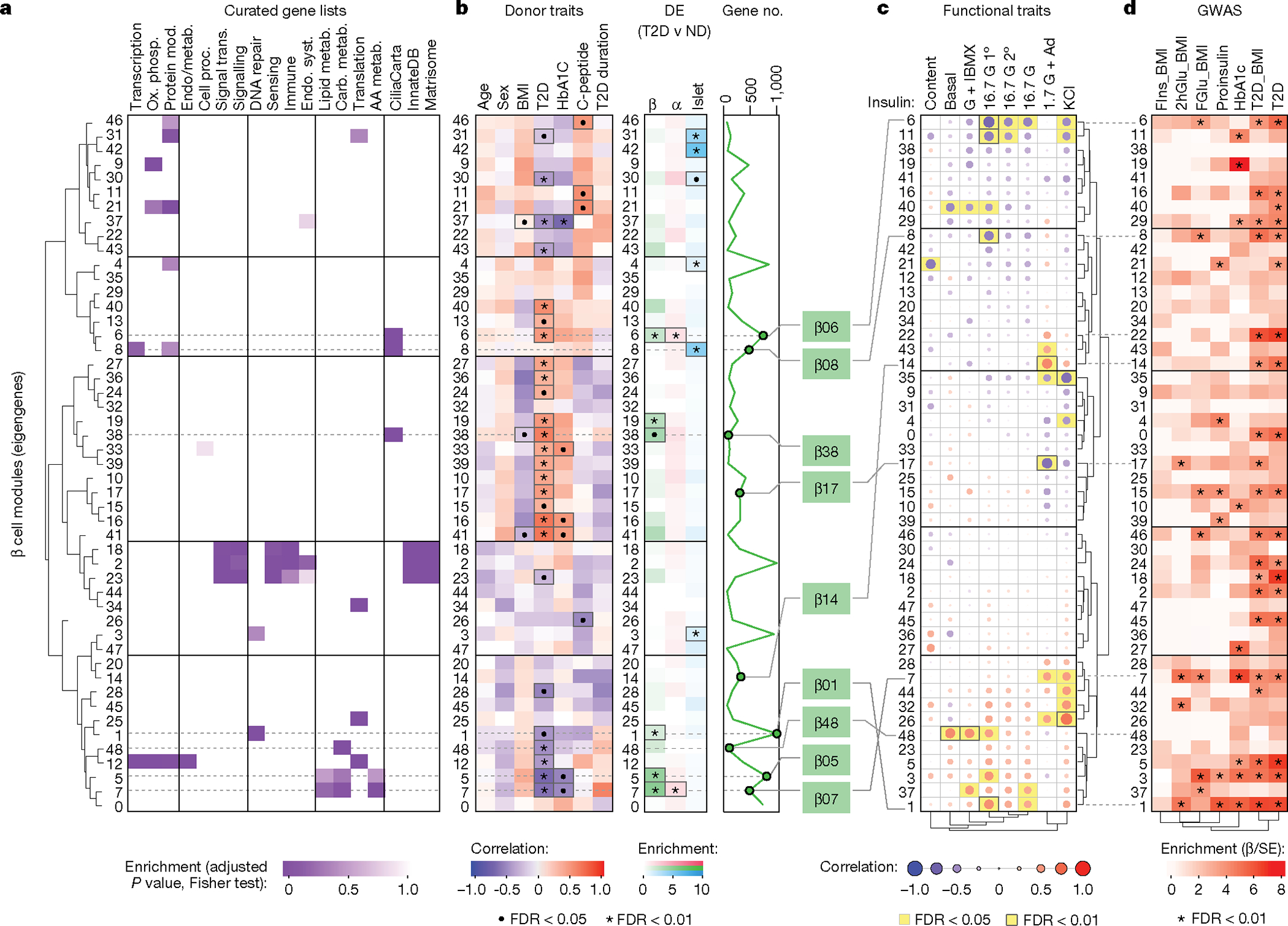
a, Relative enrichment of β cell modules (eigengenes) for curated gene lists, based on genes present in each module (two-tailed Fisher test, adjusted for multiple comparisons). See also Supplementary Table 5. AA, amino acid; carb., carbohydrate; endo., endocrine; metab., metabolism; mod, modification; ox. phosph., oxidative phosphorylation; proc. processes; Syst., system; trans., transduction. b, Module correlation to donor characteristics, enrichment of differentially expressed (DE) genes (two-tailed Fisher test, adjusted for multiple comparisons; •P < 0.05, *P < 0.01) and total number of genes per module. Modules of interest are highlighted in green. c, Module correlation to β cell functional traits described in Fig. 1; significant associations are highlighted in yellow. G + IBMX, 16.7 mM glucose with 100 μM isobutylmethylxanthine; 16.7 G, 16.7 mM glucose; 16.7 G 1°, first phase; 16.7 G 2°, second phase; 1.7 G + Ad, 1.7 mM glucose and 1 μM adrenaline; KCl, 20 mM potassium chloride. d, Module enrichment for GWAS traits using GARFIELD37. β is the regression coefficient indicating effect size and direction; SE quantifies the estimate variability. FIns, fasting insulin; 2hGlu, 2-hour glucose; FGlu, fasting glucose. See also Supplementary Fig. 4. Correlations (donor and functional characteristics) were run with a t-test, unadjusted, using Spearman correlations; see Methods for additional details.
Several β cell modules were significantly associated (FDR < 5%) with whole-body glucose homeostasis (HbA1c), and some of these (for example, β05 and β07) were also significantly enriched for genes differentially expressed in T2D β cells (Fig. 3b). Both β05 and β07 contained genes related to carbohydrate, lipid, and amino acid metabolism (Fig. 3a and Supplementary Fig. 4a), with β07 significantly correlating with KCl-mediated insulin secretion (r = 0.49, P = 0.027; Fig. 3c). Modules significantly positively correlated with glucose-stimulated insulin secretion included β01 and β48 (all enriched for metabolism-related processes), whereas β06 and β08 (both enriched for cilium movement and motility) were significantly negatively correlated with glucose-stimulated insulin secretion (Fig. 3c and Supplementary Fig. 4a). Notably, aligning functional correlations with enrichment for GWAS loci (Fig. 3d) enabled identification of modules that are more likely to be disease-causing (for example, β01) as opposed to those without GWAS enrichment (for example, β48) that may instead represent disease-induced transcriptional changes. Analyses of α cell and islet modules (Extended Data Fig. 6 and Supplementary Fig. 4b,c) further suggest that β cell function may be influenced by α and other non-endocrine cells residing within the islet.
Thus, this approach enables linking of transcriptional profiles to islet physiological parameters and facilitates prioritization of signatures based on T2D genetic risk. For example, cilia-related processes defined functionally correlated modules in every sample type and were represented proportionately on the basis of differential expression of validated cilia-related genes22 (Extended Data Fig. 7a,b). Further β06, β08 and α08 were enriched for T2D and related trait GWAS loci, suggesting a potential casual role (Fig. 3d and Extended Data Fig. 6d). We explored this finding briefly using tissue sections from the same donors and found that total cilia area within the islet was greater in T2D tissue, which was attributable to a higher cilia density with unchanged cilia size (Extended Data Fig. 7c,d). Such initial findings demonstrate how our dataset of functional, transcriptional and genetic linkages provides a practical foundation to design and prioritize studies of early T2D.
Targeted molecular perturbation of RFX6
To understand central transcriptional regulators that may be driving β cell dysfunction, we used the network analysis framework to identify ‘hub’ genes that are highly connected—that is, whose expression highly correlates with many other genes both within and across modules (Fig. 4a,b and Supplementary Fig. 5). RFX6, which has been linked to both monogenic and polygenic forms of diabetes23–25, was more highly connected in β cells than other islet-enriched transcription factors; further, RFX6 was reduced in β cells at the transcript level in T2D (Fig. 4c). Of note, RFX6 is a member of module β01, which had the strongest positive association with high glucose-stimulated insulin secretion and was among the most significantly enriched for both GWAS variants and RFX binding motifs (Fig. 3c,d and Extended Data Fig. 8a). Immunohistochemistry analysis revealed a reduction in number of β cells expressing RFX6 in T2D (Fig. 4d,e). Together, these data support RFX6 as a critical β cell hub gene that may contribute to the functional deficits observed in early T2D.
Fig. 4 |. Expression of RFX6, a central regulator of transcript changes in early T2D, is reduced in T2D β cells.
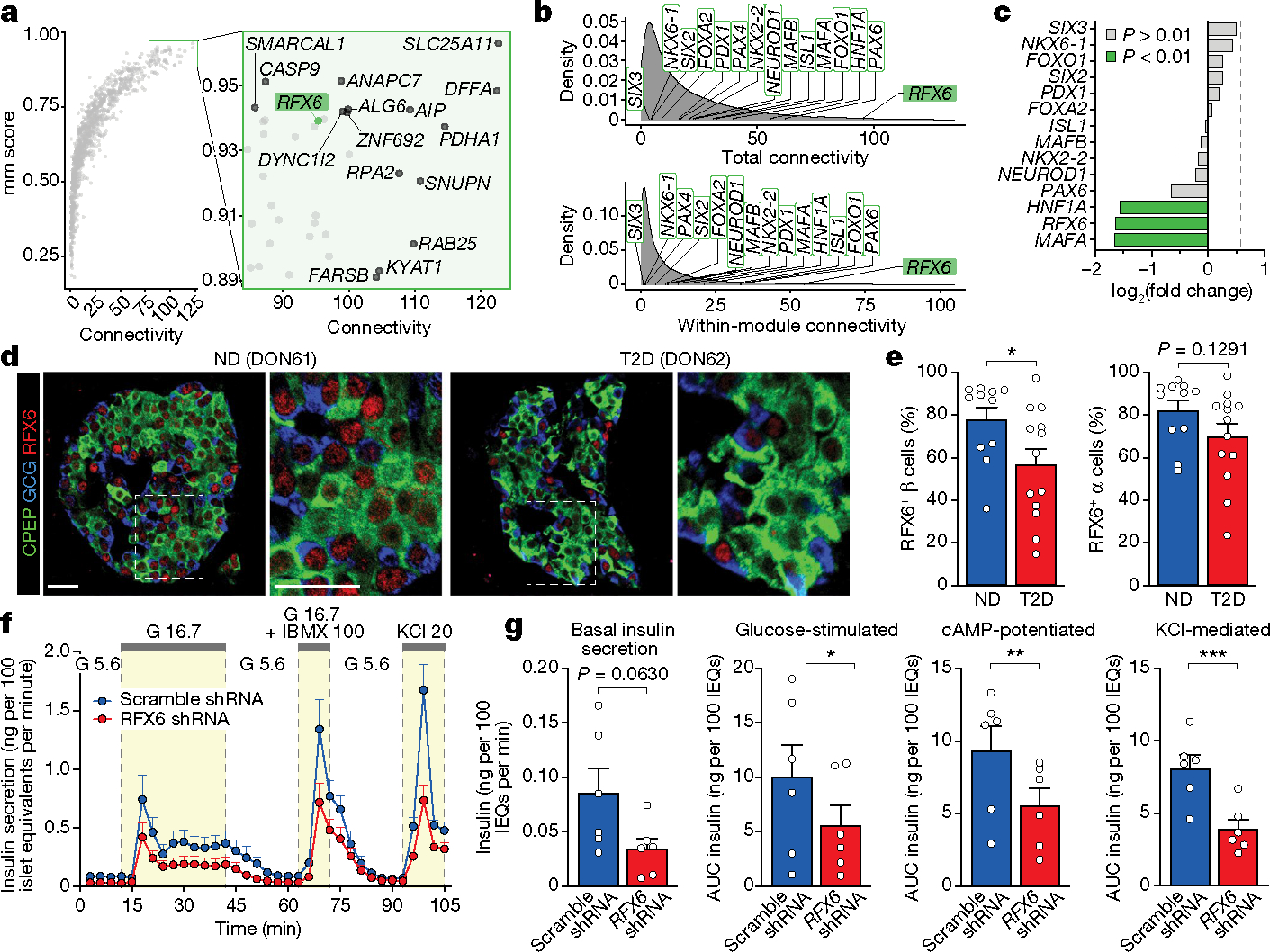
a, Overall connectivity of individual genes based on β cell WGCNA; selected genes with high connectivity scores are labelled. b, Cross-module and within-module connectivity of individual genes based on β cell WGCNA; selected transcription factor genes are labelled. c, Fold change in T2D β cell RNA expression of transcription factor genes highlighted in a. Vertical lines denote fold change of ±1.5. d,e, Images (d) and quantification (e) of expression of RFX6 in β cells and α cells of n = 11 ND and n = 13 T2D donors. Two-tailed t-test. Scale bars, 50 μm. f, Pseudoislet insulin secretion assessed by perifusion; n = 6 independent islet preparations. g, Basal insulin secretion and area under the curve (AUC) for secretory response to each stimulus. Two-tailed t-test. Data in e–g are mean + s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To determine the role of RFX6 in adult human β cell function, we used short hairpin RNA (shRNA) knockdown in a pseudoislet system26 that enables robust phenotypic analyses of genetically altered primary cells in an islet-like context (Extended Data Fig. 8b). RFX6 shRNA (shRFX6) pseudoislets showed β cell knockdown of RFX6 compared with scramble shRNA controls, but were similar in morphology and composition (Extended Data Fig. 8c–e), suggesting that acute RFX6 reduction does not cause β cell loss. Like T2D islets, dynamic insulin secretion of shRFX6 pseudoislets was significantly blunted in the presence of secretagogues (Fig. 4f,g and Extended Data Fig. 8f,g), and although shRFX6 pseudoislets had a greater insulin content, proinsulin processing was similar (Extended Data Fig. 8h,i). In sum, not only is RFX6 decreased in T2D β cells, but the results of targeted knockdown are consistent with the RFX6-containing module β01 association with glucose-stimulated insulin secretion and strongly implicate RFX6 as a major regulator of human β cell gene expression required for stimulated insulin secretion.
To determine the molecular mechanism by which RFX6 knockdown affects insulin secretion, we processed shRFX6 and control pseudoislets (n = 7 matched donors) for single-nucleus multiome profiling (Fig. 5a) to yield 15,825 (RNA) and 5,706 (assay for transposase-accessible chromatin (ATAC)) high-quality nuclei for downstream analysis (Supplementary Fig. 6a). Major islet cell types were represented across all donors and constructs (Fig. 5b,c, Extended Data Fig. 9a,b and Supplementary Fig. 6b,c). Data are available via the UCSC Cell Browser at https://theparkerlab.med.umich.edu/data/public/cellbrowser/?ds=Pseudoislet10XMultiome for further exploration. Supporting the role of RFX6 as a major β cell regulator, 13% of total detected genes were differentially expressed in β cell nuclei compared with less than 3% in other cell types (Extended Data Fig. 9c). Upregulated genes were enriched for actin filament-based movement and synaptic signalling, whereas downregulated genes were enriched for membrane trafficking, autophagy and ciliary pathways (Extended Data Fig. 9d). We identified β cell-specific regulons that were differentially active in shRFX6 nuclei, with regulon genes being significantly enriched (P < 0.001) for maturity-onset diabetes of the young and insulin secretion pathways (Fig. 5d, Extended Data Fig. 9e,f and Supplementary Fig. 6d). Of note, there was common pathway overlap between shRFX6 β cell nuclei and purified T2D β cells (Extended Data Fig. 9g), particularly in cellular membrane and vesicle components linked to T2D GWAS variants and RFX6 binding motifs by module analysis (Supplementary Fig. 6e). Together, these data highlight how reduction in RFX6 disrupts networks fundamental to β cell function and identifies exocytosis as a target of RFX6-mediated dysfunction in T2D β cells.
Fig. 5 |. Molecular perturbation and population analyses demonstrate that regulatory GWAS variants are enriched in the RFX6 network.
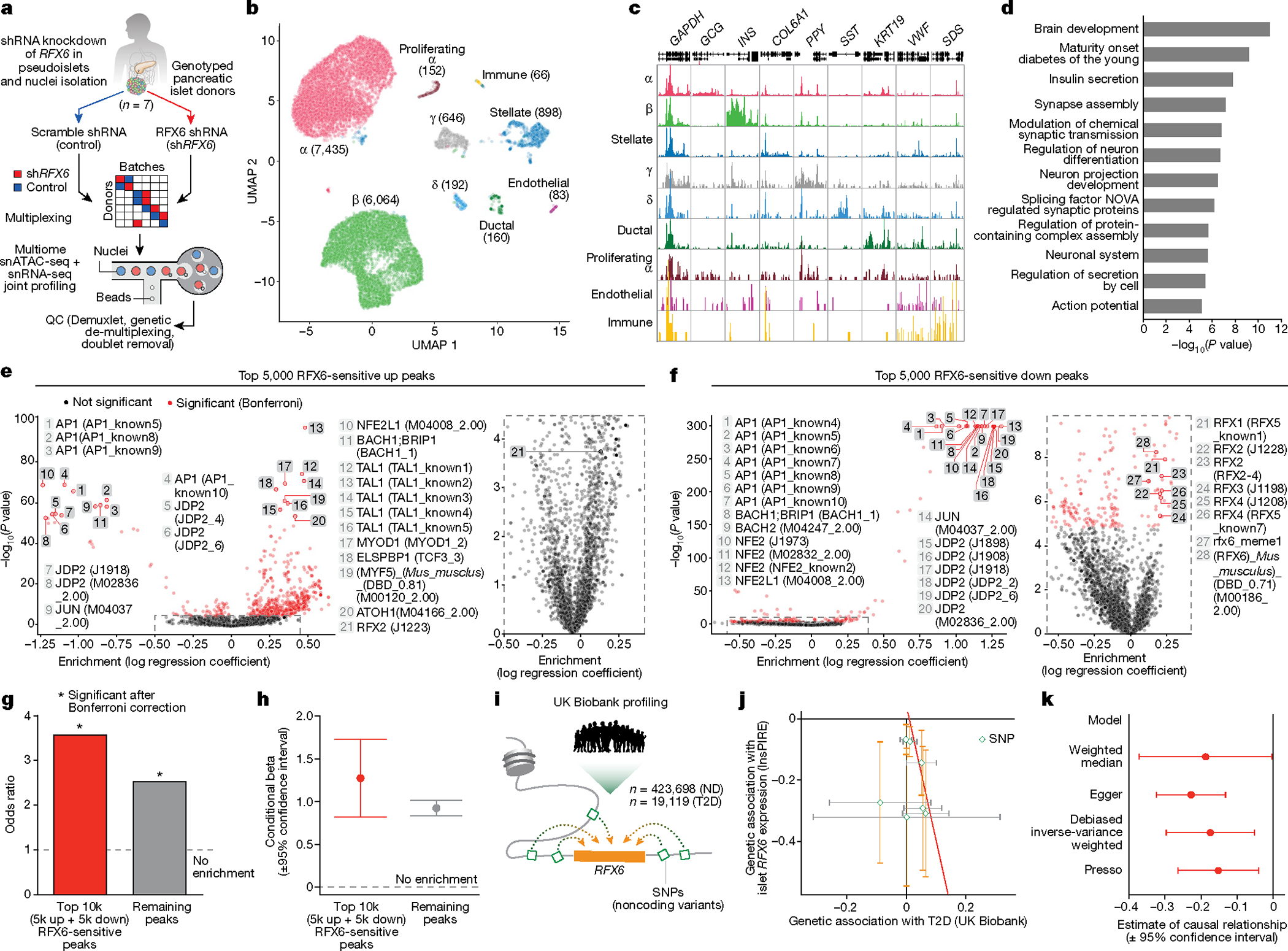
a, Scramble shRNA (control) and RFX6 shRNA (shRFX6) pseudoislets (n = 7 matched donors) were multiplexed for single-nucleus RNA-sequencing (snRNA-seq) and single-nucleus ATAC-seq (snATAC-seq) multiome profiling using a randomized block study design. n = 15,825 (snRNA-seq); n = 5,706 (snATAC-seq) nuclei. b, Cell-type assignment by clustering on snRNA-seq data. c, Pseudobulk ATAC-seq signal at marker genes. d, Pathway enrichment of genes (n = 220; g:Profiler, two-tailed hypergeometric test) in top 10 differential β cell regulons (shRFX6 versus control). Significantly enriched pathways highlight a broad similarity of β cells to neurons as electrically active and secretory cells. e,f, Motif enrichment for top 5,000 RFX6-sensitive upregulated (e) and downregulated (f) ATAC-seq peaks in shRFX6 β cell nuclei. Right panels show enlarged views of the outlined area. g,h, Single-value odds ratio of T2D GWAS enrichment (g; nominal P values 0.00478 (RFX6-sensitive peaks) and 3.613 × 10−25 (remaining peaks); Bonferroni correction) and model estimate from conditional analysis (h) of RFX6-sensitive peaks. Enrichment remained significant after conditional analysis controlled for remaining (not RFX6-sensitive) peaks. i, Interrogation of UK Biobank data to analyse RFX6 non-coding regulatory SNPs. j, Genetic association (causal effect, beta) of RFX6 regulatory SNPs (n = 7) with islet RFX6 expression (n = 420 independent samples) and T2D trait (n = 442,817 independent samples). Each symbol represents one SNP and error bars show standard errors. All eQTL effects are oriented in the same direction (effect allele leads to reduced expression). Best fit line from MR-Egger is shown in red, indicating that decreased islet RFX6 expression is associated with increased risk for T2D. k, MR analysis on UK Biobank European ancestry reveals significant causal association of RFX6 expression with T2D risk (exposure, RFX6 expression; outcome, T2D). Negative estimate values indicate an inverse causal relationship between exposure and outcome (decreased RFX6 expression leads to increased T2D risk). h,k, Data are mean ± 95% confidence interval.
We observed global changes to the chromatin landscape in shRFX6 β cells (Supplementary Fig. 6f–h), taking peaks with smallest P values in either direction (top RFX6-sensitive peaks) for use in downstream analyses. These peaks were significantly enriched for motifs corresponding to the known chromatin modifier activator protein 1 (AP1) as well as RFX6 and related family member motifs (Fig. 5e,f and Extended Data Fig. 9h,i) and were significantly enriched to occur near differentially expressed genes (Extended Data Fig. 9j), indicating concordance between the snATAC-seq and snRNA-seq modalities. β cell ATAC peaks are enriched for T2D GWAS variants4,5, and indeed, top RFX6-sensitive peaks were also significantly enriched to overlap with these variants (Fig. 5g,h and Extended Data Fig. 9k,l), emphasizing their importance in the genetic predisposition to T2D. Overall, these results show that RFX6 knockdown in β cells results in widespread transcriptional and chromatin changes that are associated with downregulated vesicle transport and coordinated disruption of genome-wide regulatory elements that overlap T2D GWAS variants, highlighting how genetic risk throughout the transcriptional regulatory network may mediate β cell dysfunction.
Large-scale population genetics of RFX6
To further investigate the role of RFX6 in T2D, we performed MR to test the causal relationship between RFX6 expression in human islets and T2D status (Extended Data Fig. 9m). Seven islet RFX6 expression quantitative trait loci (eQTLs) with low pairwise linkage disequilibrium (r2 < 0.2) from the Integrated Network for Systemic analysis of Pancreatic Islet RNA Expression (InsPIRE) study27 (n = 420 donors; Supplementary Fig. 6i), a European ancestry cohort, were selected as instrumental variables. Their association with T2D was evaluated in a large population dataset, UK Biobank3 (n = 423,698 ND, n = 19,119 T2D; Fig. 5i,j and Supplementary Table 6), selected to match the European ancestry of the InsPIRE cohort. Robust analysis by four complementary MR approaches revealed that genetically predicted decreased islet RFX6 expression is causally associated with increased risk of T2D (causal effect = −0.228, P = 3.98 × 10−6 from Egger; causal effect = −0.187, P = 0.048 from weighted median; causal effect = −0.174, P = 0.006 from debiased inverse-variance weighted method (dIVW); causal effect = −0.152, P = 0.038 from pleiotropy residual sum and outlier (PRESSO); Fig. 5k). Both MR-Egger and MR-PRESSO approaches suggest non-significant horizontal pleiotropy (effect = 0.01, P = 0.122 from Egger; test statistic = 6.59, P = 0.615 from PRESSO). MR of the same instrumental variables in a larger and more heterogeneous European T2D meta-analysis9 (n = 965,732 ND, n = 148,726 T2D) showed similar point estimates of the causal effect of RFX6 expression on T2D, however, perhaps owing to weak instrument bias (Supplementary Table 6) and/or the increased heterogeneity across contributing studies, these effects did not reach statistical significance (Extended Data Fig. 9n; for example, dIVW causal effect = −0.084, P = 0.065). Non-coding functional variation predicted to reduce RFX6 expression in islets increases T2D risk in the UK Biobank, a population-based sample of European ancestry, demonstrating an additional level of genetic risk converging on RFX6 in diabetes.
Discussion
The pancreatic β cell resides in the multicellular pancreatic islet mini-organ, where there are complex interactions between various cell types. In T2D, as in other chronic, complex, multi-organ diseases, teasing apart the causes, correlates and consequences of cellular and tissue dysfunction is challenging owing to the limited availability of primary tissue, constraints of sample processing at different disease stages, and in many cases, removal of cells from their native environment. Here, to address these challenges and identify early disease-driving events, we applied a comprehensive, multimodal, integrated approach to isolated islets and pancreatic tissue from a unique cohort of early T2D and control donors that included analyses of islet physiology, transcriptome and pancreas tissue cellular architecture. Furthermore, we integrated donor and islet functional traits with gene network analysis and GWAS to understand central transcriptional and genetic regulators driving β cell dysfunction in early T2D. Co-registration of multimodal data and clinical information yielded several important findings (summarized in Extended Data Fig. 10a): (1) impaired β cell function, a hallmark of early T2D, persisted ex vivo and in a nondiabetic environment; by contrast, α cell function was not changed; (2) islet endocrine cell composition was unchanged, although there were modest changes in the islet microenvironment including endothelial and immune cells; (3) transcriptional network analysis proportioned genetic risk into gene modules with specific functional properties; and (4) RFX6 emerged as a highly connected hub transcription factor that was reduced in T2D β cells and associated with reduced glucose-stimulated insulin secretion. We used two approaches to investigate the critical role for RFX6 and its regulatory network (summarized in Extended Data Fig. 10b): (1) molecular perturbation of RFX6 in β cells of primary human pseudoislets enabled functional, transcriptomic and epigenomic analyses; and (2) integration of UK Biobank data enabled population-scale genetic relationships to be examined. Reduction of RFX6 levels led to reduced insulin secretion defined by transcriptional dysregulation of vesicle trafficking, exocytosis and ion transport pathways, mediated by genome-wide chromatin architectural changes overlapping with T2D GWAS variants. Furthermore, MR analysis revealed a significant causal association between genetically predicted decreased islet RFX6 expression and increased risk of T2D in the UK Biobank. We also found directionally consistent point estimates of effect in a larger but more heterogeneous dataset from Vujkovic et al.9, which did not reach statistical significance. Thus, our integrated, multimodal studies identify β cell dysfunction that results from cell-intrinsic defects, including an RFX6-mediated, T2D GWAS-enriched transcriptional network as a key event in early T2D pathogenesis. This study serves as a blueprint for investigating complex diseases, taking a finding arising from unbiased genome-wide approaches and validating it at both single-cell and population scales.
Integration of functional, transcriptional and spatial analyses
This study demonstrates β cell functional defects ex vivo—which persist in culture and following transplantation into a normoglycaemic environment—but no change to insulin content or β cell mass. The relative contributions of impaired β cell function and/or reduced β cell mass have long been debated in T2D28–30. By integrating studies of pancreatic tissue and isolated islets from the same donors, our data indicate that β cell loss is not a major component in disease pathogenesis early in T2D. Further, the continued dysfunction of islets in a transplant setting underscores the persistence of the initial β cell defect.
Overall, the islet microenvironment in T2D was largely similar to that in ND donors. We identified transcriptional changes in vascular and immune signalling as features in sorted α and β cells as well as in whole islets and demonstrated that in situ T2D islets had subtle reductions in islet capillary size, increased intraislet T cells, and altered communication between cellular neighbourhoods. Although most T2D donors showed some evidence of amyloid deposits as a unique feature in the T2D islet microenvironment, only a minority of islets demonstrated detectable amyloid at this stage of disease. Together, these observations are unlikely to explain the degree of β cell dysfunction in this cohort, but given that they are present without any associated changes in endocrine cell composition, they may represent early consequences of β cell dysfunction or may act to exacerbate initial β cell-intrinsic defects. Further study is needed to determine whether changes to the microenvironment are an independent disease process or whether there is bidirectional signalling between dysfunctional β cells, α cells and/or other islet cell types.
Gene modules of genetic risk in early T2D
Co-expression network analysis and association with GWAS variants and physiological parameters enabled us to prioritize processes with physiological relevance that were more likely to be disease-causing rather than disease-induced. For instance, both β01 (metabolism-enriched) and β06 (cilia-enriched) modules are associated with T2D GWAS variants, indicating that the regulatory circuitry related to metabolism and cilia function may have causative roles in the development of T2D. Notably, insulin secretion was positively correlated to β01, whose constituent genes were decreased in T2D β cells, but negatively correlated to β06, whose genes were increased in T2D β cells. These results suggest that β01 genes enhance insulin secretion, whereas β06 genes decrease it; thus, T2D risk alleles are likely to decrease β01 gene expression and activate β06 genes, both of which would negatively influence β cell function. Future work directly testing key candidate genes from this dataset and these modules, analogous to the studies of RFX6 described here, will be important.
Genetic risk for complex metabolic diseases such as T2D results from the combined influence of many variants with small effects, with at-risk individuals likely having multiple parallel processes affected. This concept has been described as a ‘palette’ model31, and our work aids in deciphering components of the palette by proportioning genetic risk into cell-specific functional modules derived from transcriptome signatures across early stages of disease. Since this work utilizes eQTL and genetic datasets of primarily European ancestry, the extension of these findings as new multi-ancestry datasets emerge9,10 will be an important future direction. Such studies provide the opportunity to assess downstream consequences of an individual’s innate genetic risk by identifying specific molecular and functional processes that would be most affected and hopefully enabling precise targeting of those to achieve precision medicine in diabetes.
Role of RFX6 in early T2D β cell dysfunction
Although we did not set out to target RFX6 in this study, our unbiased multimodal analysis identified an RFX6 regulatory network strongly correlating with insulin secretion and T2D genetic risk, which provides new insight into RFX6 biology and how regulatory variants can influence T2D risk through key pathways. Our single-cell to population-scale results suggest that RFX6 exerts a disproportionate transcriptional influence on β cell state and that its dysregulation is a key molecular event in early T2D pathogenesis. RFX6 perturbation in primary human pseudoislets points to two major drivers of impaired β cell insulin secretion: (1) defective ion transport processes32,33 and (2) dysregulated vesicle trafficking and exocytosis pathways mediated by changes in chromatin accessibility. Dysregulated cilia-related genes were in line with evidence that the RFX family of transcription factors control ciliogenesis34,35. Given their role in environment sensing, cell–cell communication and signal transduction, cilia represent a potential link between β cell-intrinsic, RFX6-mediated dysregulation and changes within the islet microenvironment seen in early T2D and warrant future study. Additional dissection of individual-level RFX6 transcriptional heterogeneity is an exciting topic that will be enabled by large-scale single-cell studies.
Overall, our results and prior information reveal multiple layers of genetic convergence on RFX6 and its regulatory network: (1) it has been known that rare coding variants in RFX6 are associated with early-onset monogenic forms of diabetes23,24 and a recently identified rare frameshift variant in RFX6 is associated with T2D36; (2) we show that RFX6-altered binding regions genome wide are enriched to overlap common variants associated with T2D; and (3) we show that genetically predicted reduced expression of RFX6 from nearby non-coding variants is causally associated with increased T2D risk in the UK Biobank population dataset. Thus, these three different classes of genetic variations all converge on RFX6 biology in diabetes. Such a genetic convergence may be possible for other diseases and genes, especially transcription factors with hub properties as reported here.
This work raises important questions about the factors or events initially dysregulate RFX6 to begin this cascade. Given the conserved role of RFX6 in islet cell development and maintenance of functional β cell identity23,32,33, it may be that early defects driven by RFX6 dysregulation only become apparent after superimposed environmental, nutritional and/or age-related stressors. Alternatively, the strong enrichment of T2D GWAS variants in β01 (the RFX6-containing module) and the position of RFX6 as a hub gene may point to cumulative genetic effects compounding over time in a cascade that disrupts β cell homeostasis. Thus, precisely what underlies the initial RFX6 dysregulation and whether it can be targeted to prevent or reverse early-stage molecular and functional defects in the β cell should be an active area of investigation.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-023-06693-2.
Methods
Human participants
A total of 20 organ donors with T2D and 50 ND organ donors were used in this study; 36 ND samples were used as controls in molecular phenotyping experiments and 14 NDs were used in follow up pseudoislet experiments. T2D donors were identified using a national network including partnerships with the International Institute for Advancement of Medicine (IIAM), National Disease Research Interchange (NDRI), and local Organ Procurement Organizations to ensure organs met inclusion and exclusion criteria19,38 and had minimal ischaemic time. Twenty pancreata were obtained from individuals with T2D aged 37–66 years (mean 52 years) with T2D duration of 0–10 years (mean 3.5 years). Of these donors, 25% were without pharmaceutical treatment (HbA1c range 6.2–9.9; mean 7.6) and 75% were on diabetes medication, mostly oral agents (HbA1c range 6.3–11.2; mean 8.0) (Fig. 1a). Pancreata from ND donors (n = 25) were identified in a similar fashion. Additional ND donors (n = 25) were identified and studied through partnerships with the Integrated Islet Distribution Program (IIDP) and the Alberta Diabetes IsletCore. Detailed information for each donor, including donor information, sample types, and experimental usage for each case, is available in Supplementary Table 1. De-identified medical histories provided both information for T2D staging as well as clinical characteristics to correlate with generated data. The Vanderbilt University Institutional Review Board declared that studies on de-identified human pancreatic specimens do not qualify as human subject research.
Some human islets used in this research study were provided by the ADI IsletCore at the University of Alberta in Edmonton (http://www.bcell.org/adi-isletcore.html) with the assistance of the Human Organ Procurement and Exchange (HOPE) program, Trillium Gift of Life Network (TGLN), and other Canadian organ procurement organizations. Islet isolation was approved by the Human Research Ethics Board at the University of Alberta (Pro00013094). All donors’ families gave informed consent for the use of pancreatic tissue in research. This study also used data from the Organ Procurement and Transplantation Network (OPTN) that was in part compiled from the Data Hub accessible to IIDP-affiliated investigators through IIDP portal (https://iidp.coh.org/secure/isletavail). The OPTN data system includes data on all donors, wait-listed candidates, and transplant recipients in the US, submitted by the members of the OPTN. The Health Resources and Services Administration (HRSA), US Department of Health and Human Services provides oversight to the activities of the OPTN contractor. The data reported here have been supplied by UNOS as the contractor for the Organ Procurement and Transplantation Network (OPTN). The interpretation and reporting of these data are the responsibility of the authors and in no way should be seen as an official policy of or interpretation by the OPTN or the US Government.
Pancreas procurement and processing
Pancreata were processed in Pittsburgh by R. Bottino in a consistent manner that included multiple tissue processing and fixation methods and simultaneous isolation of islets and collection of pancreatic tissue from the same pancreas when possible, as previously described39–41. Pancreata were received within 18 h from cross clamp and maintained in cold preservation solution on ice until processing, as described previously41. Pancreas was then cleaned from connective tissue and fat, measured and weighed. Prior to islet isolation, multiple cross-sectional slices of pancreas with 2–3 mm thickness were obtained from the head, body and distal tail, further divided into quadrants, and processed into paraformaldehyde (PFA)-fixed cryosections as described previously41. Islet isolation was performed via ductal collagenase infusion and purification by density gradient as described previously39,41, then shipped to Vanderbilt for further analysis following shipping protocols developed by the IIDP. Islets were cultured in CMRL 1066 medium (5.5 mM glucose, 10% FBS, 1% penicillin-streptomycin and 2 mM l-glutamine) in 5% CO2 at 37 °C for 24–48 h prior to reported studies16,41,42. Pseudoislets were cultured in Vanderbilt pseudoislet medium26. Limitations of tissue availability and processing dictated that not all assays could be performed on each donor.
Assessment of native pancreatic islet and pseudoislet function by macroperifusion
Dynamic hormone secretion from ND and T2D islets and pseudoislets was assessed by a standardized perifusion approach that interrogates multiple steps of the insulin secretory pathway, a protocol that has been adopted by the Human Islet Phenotyping Program of the IIDP to assess over 400 human islet preparations43. The experiment was performed at a perifusate flow rate of 1 ml min−1 and the effluent was collected at 3-minute intervals using an automatic fraction collector26,44, then islets were retrieved and lysed with acid-ethanol solution to extract. Insulin and/or glucagon concentrations in each perifusion fraction, as well as total hormone content, were measured by radioimmunoassay (RIA) (human insulin, RI-13K, Millipore; glucagon, GL-32K, Millipore), enzyme-linked immunosorbent assay (ELISA) (Human insulin, 10-1132-01, Mercodia; glucagon, 10-1281-01, Mercodia), or homogeneous time-resolved fluorescence assay (glucagon, 62CGLPEH, Cisbio). Pseudoislet proinsulin content was measured by ELISA (10-1118-01, Mercodia). AUC above baseline hormone release was calculated with the trapezoidal method in GraphPad Prism 8.0 as previously described26, using the following time point boundaries: insulin 16.7 mM glucose, 9–60 min (1st phase 9–24 min); insulin 16.7 mM glucose + IBMX, 63–90 min; insulin 1.7 mM glucose + adrenaline, 90–114 min; insulin KCl, 120–150 min; glucagon 16.7 mM glucose, 12–51 min; glucagon 16.7 mM glucose + IBMX, 69–90 min; glucagon 1.7 mM glucose + adrenaline, 93–117 min; glucagon KCl, 120–144 min. In addition, we compared hormone secretory trajectories between ND and T2D using linear mixed-effect model45 that takes into consideration the underlying temporal correlation. In each model, natural splines were employed to capture nonlinear relationships between hormone secretory trajectory and time. The statistical significance (P < 0.05) of the interaction between the splines and group indicator (1 for T2D and 0 for ND), assessed by a likelihood ratio test, confirmed the difference in hormone secretory trajectory between the two groups.
Human islet transplantation
Immunodeficient NOD.Cg-PrkdcscidIl2rgtm1Wjl/Sz (NSG)46 10- to 12-week-old male mice were maintained by Vanderbilt Division of Animal Care in group housing in sterile containers within a pathogen-free barrier facility housed with a 12 h light:12 h dark cycle and access to free water and standard rodent chow. All animal procedures were approved by the Vanderbilt Institutional Animal Care and Use Committees. Between 1,000 and 2,000 islet equivalents per mouse (n = 4–8 mice per islet preparation) were transplanted beneath the kidney capsule. Randomization and blinding were not applicable. After 6 weeks, mice were fasted for 6 h and then injected with glucose + arginine (2 g per kg body weight) intraperitoneally as previously described16,41,42,47. Blood samples were obtained before (0 min) and after (15 min) injection and human-specific insulin was analysed by ELISA (Alpco, 80-ISNHU-E01.1) or radioimmunoassay (Millipore, RI-13K). Animals were euthanized after glucose + arginine stimulation and grafts were removed, fixed, and stained as previously described16,40,47.
Purification of α and β cells by FACS
To assess both the β and α cell-specific transcriptional landscapes as well as global islet dysregulation in the short-duration T2D cohort, we purified β and α cells by fluorescence-activated cell sorting (FACS) using well-characterized cell surface antibodies and hand-picked isolated islets for RNA-sequencing as described previously41,48,49. In brief, 0.025% trypsin was used to disperse islet cells by manual pipetting and subsequently quenched with RPMI containing 10% FBS. Cells were washed in the same medium and counted on a haemocytometer, then transferred to FACS buffer (2 mM EDTA, 2% FBS, 1× PBS). Indirect antibody labelling was completed via two sequential incubation periods at 4 °C, with one wash in the FACS buffer following each incubation. Primary and secondary antibodies, listed in Supplementary Table 2, have been characterized previously and used to isolate high-quality RNA41,48–50. Of note, there is no detectable change in expression of β cell surface marker NTPDase3 in T2D islets49, making this primary antibody suitable for the present study. Appropriate single-colour compensation controls were run alongside samples. For sorting of β cells for use in pseudoislets, quenching step post-dispersion was performed with 100% FBS at 1/3 volume trypsin. Cells then underwent an additional filtration step using a 40-μl strainer prior to staining. For all preparations, propidium iodide (0.05 μg per 100,000 cells; BD Biosciences) was added to samples prior to sorting for non-viable cell exclusion. Flow analysis was performed using an LSRFortessa cell analyser (BD Biosciences), and a FACSAria III cell sorter (BD Biosciences) was used for FACS. Cells for RNA were collected into FACS buffer, washed once in 1× PBS, and stored in RNA lysis buffer for RNA extraction. Cells for pseudoislets were washed once in 1× PBS, resuspended in Vanderbilt pseudoislet medium, and processed as described in ‘Pseudoislet’. Analysis of flow cytometry data was completed using FlowJo 10.1.5 (Tree Star).
Traditional and multiplexed immunohistochemical imaging and analysis
Traditional immunohistochemistry.
Multiple sections from pancreatic head, body, and tail regions of 20 T2D and 11 age-matched ND donors were lightly PFA-fixed and prepared for immunohistochemistry and stained as described previously41,49,51. Primary and secondary antibodies and their dilutions are listed in Supplementary Table 2. Amyloid, a marker of islet pathology in T2D52,53, was visualized using a 2-min incubation in Thioflavin S (0.5% w/v; T-1892, Sigma) followed by a brief wash in 70% ethanol as described previously16,47,54. Images were acquired at 20× with 2× digital zoom using a FV3000 confocal laser scanning microscope (Olympus) or a ScanScope FL (Leica/Aperio) and processed using cytonuclear algorithms (HighPlex FL v3.2.1) or tissue classifiers via HALO software (Indica Labs) or morphometric measurement via Metamorph software v7.10 (Molecular Devices). Analyses were run on the entire tissue section or manually annotated islets as indicated in figure legends. Endocrine cell mass was quantified by using pancreas weight and the ratio of hormone positive cells as identified by cytonuclear logarithm within the entire pancreatic section from multiple blocks representing the head, body, and tail regions. To obtain islet capillary measurements, caveolin-1 channel was isolated and colour thresholding was used on a per-image basis to gather object data using the Integrated Morphometry Analysis function (Metamorph). The following analysis metrics represent mean ± standard error: endocrine cells (Fig. 2a and Extended Data Fig. 3e–g) 16,151 ± 1,715 islet cells per donor and 570,508 ± 51,866 total cells per donor; endocrine cell area (Extended Data Fig. 3c,d) 2.34 ± 0.24 mm2 per donor; capillary morphology (Fig. 2e) 48 ± 4 islets per donor; macrophage area (Fig. 2i) 0.64 ± 0.07 mm2 per donor; amyloid (Extended Data Fig. 3l) 108 ± 19 islets per donor; cilia (Extended Data Fig. 7d) 0.32 ± 0.05 mm2 per donor; RFX6 (Fig. 4e) 1,863 ± 362 cells per donor; pseudoislets (Extended Data Fig. 8d) 2,797 ± 508 cells per sample. Islet grafts were stained, imaged, and analysed using the same technique described for tissue above, with graft area annotated by hand to exclude quantification of mouse kidney tissue. The following analysis metrics represent mean ± standard error: endocrine cells (Extended Data Fig. 1r) 2,109 ± 347 cells per donor; endocrine cell area (Extended Data Fig. 1r) 0.1606 ± 0.026 mm2 per donor; vascularization (Extended Data Fig. 1s) 0.1728 ± 0.031 mm2 per donor.
CODEX multiplexed imaging.
To visualize additional cell types modulating the islet microenvironment55, a subset of samples was analysed using CODEX56. Antibodies were purchased pre-conjugated from Akoya Biosciences or sourced from other vendors and conjugated in-house using the CODEX Conjugation Kit (Akoya Biosciences) or by Leinco Technologies (Supplementary Table 3). Ten-micrometre lightly fixed41 pancreas sections were mounted onto 22 × 22 mm glass coverslips (Electron Microscopy Sciences) coated in 0.1% poly-l-lysine (Sigma) and stained with the CODEX Staining Kit (Akoya Biosciences) in uncoated 6-well tissue culture plates (VWR) per manufacturer instructions. Fluorescent oligonucleotide-conjugated reporters were combined with Nuclear Stain and CODEX Assay Reagent (Akoya Biosciences) in light-protected 96-well plates sealed with foil (Akoya Biosciences) and automated image acquisition and fluidics exchange were performed using the Akoya CODEX instrument and CODEX Instrument Manager (CIM) v1.29 driver software (Akoya Biosciences) integrated with a BZ-X810 epifluorescence microscope (Keyence). Tissue was hydrated in 1× CODEX buffer (10× CODEX Buffer diluted in Milli-Q water) and hybridization/stripping of the fluorescent oligonucleotides was performed using dimethyl sulfoxide (Sigma). After loading of coverslip into stage insert, tissue was visualized with Nuclear Stain diluted 1:1,000 in PBS and imaging area was set by centre point and tile number using BZ-X810 viewing software (Keyence). All images were acquired using a CFI plan Apo I 20×/0.75 objective (Nikon) with 30% tile overlap and 5 z-planes (1.5 μm/z).
Processing and annotation of CODEX images.
A total of 16 tissue regions were captured from 6 ND and 10 T2D donors (mean 50 mm2 tissue per donor). Image alignment, stitching, background subtraction, and deconvolution were performed using the CODEX Processor v1.7.0.6 (Akoya Biosciences; see https://help.codex.bio/codex/processor/technical-notes for details). Individual channel images (TIFF files) were imported into HALO software v3.1 (Indica Labs) for all analyses as described below. Tissue and islet areas were annotated by hand to exclude out-of-focus regions and poor tissue quality. Islets (estimated diameter ≥50 μm; mean 42 islets per donor) were annotated based on DAPI and CHGA channels. Cell segmentation and cell-type annotations were performed using the HALO HighPlex FL v3.2.1 module with consistent cytonuclear parameters (nuclear contrast threshold 0.456, maximum cytoplasm radius 0.48). Due to marker intensity variability among samples, thresholds were manually set for each marker and donor. Unless otherwise noted, cells were counted positive for a given marker if minimum intensity was reached in 50% of cytoplasm area (see Supplementary Tables 3 and 4 for a complete list of markers and cell types). For cells with more variable morphology, positivity was also counted for nuclear area (30%: ARG1, CD11c, CD14, CD163, CD206, CD31, CD34, CD45, HLA-DR, IBA1, KRT, MCAM). Proliferating cells were counted only if minimum 60% of nuclear area met KI67 intensity threshold. Vascular structures (CD31) were also measured by random forest classification algorithm (HALO Tissue Classifier module).
The following analysis metrics represent mean ± standard error: endocrine cell area (Extended Data Fig. 3h) 0.88 ± 0.10 mm2 per donor; islet cell composition (Fig. 2c and Extended Data Fig. 3j) 7,322 ± 852 cells per donor; immune cells (Fig. 2h,j) 309 ± 43 cells per donor; endothelial cell phenotypes (Extended Data Fig. 4e) 460 ± 92 cells per donor; macrophage phenotypes (Extended Data Fig. 4h) 191 ± 29 cells per donor; T cell phenotypes (Extended Data Fig. 4i,j) 40 ± 17 cells per donor.
High-dimensional, spatial and neighbourhood analyses.
The R implementation of the UMAP algorithm (https://CRAN.R-project.org/package=umap) was used for dimensionality reduction. Cell marker percentages obtained through HALO were standardized across islets (n = 255 ND islets and 426 T2D islets; mean 172 cells per islet), and default parameters were used for UMAP reduction (Fig. 2k and Extended Data Fig. 4k) except for nearest neighbours (80) and minimum distance (0.05). For spatial analyses, CD31 area classifications were converted to an annotation layer. A nearest neighbour algorithm (HALO Spatial Analysis module) was applied to obtain average distance of endocrine cells (n = 4,830 ± 692 cells per donor) to islet capillaries (CD31+ region) (Fig. 2f and Extended Data Fig. 4d).
For CN analysis, two methods were applied in parallel to CODEX data from annotated islets. In the community detection method, termed Dynamic CF-IDF (Fig. 2l and Extended Data Fig. 5a,d), a weighted undirected heterogeneous graph for each islet was constructed based on the cell types and normalized distance between cells. A greedy-based graph community detection method57 was applied to segment the graph into a set of cell communities, then cell communities were stratified into 6 cellular neighbourhoods (n = 5,582 total cellular neighbourhoods with median 11 cells per cellular neighbourhood). Cell-type enrichment was determined by a new proposed scoring function CF-IDF, which is a modification of the widely used text sequence analysis method term frequency–inverse document frequency scoring58. Our cell frequency (CF)-inverse dataset frequency (IDF) score emphasizes the cell type that is not only prevailing, but also uniquely representative in a group of target islets. Therefore, it will deemphasize the most dominant cell types (for example, α and β) throughout all the islets while paying more attention to the relative enrichment of less abundant cell types (for example, vascular and immune cells) in the local regions. The downstream analysis not only introduces insightful results on T2D feature analysis but also shows a robust performance across different resolution levels.
The second cellular neighbourhood analysis method, a k-means approach (Extended Data Fig. 5b,e,g), built on a previously published algorithm used to identify cellular neighbourhoods in the tumour microenvironment59. For each cell, we first found its 10 nearest neighbours in the islet and assigned the ith nearest neighbour that was an α cell, β cell, macrophage, endothelial cell or γ cell, a score cos(iπ/20). Then we calculated the total score for each cell type, applied L1 normalization to the scores, and standardized them across all cells. The resulting representations of cells were finally used for k-means clustering to form 5 cellular neighbourhoods (n = 5,021 total cellular neighbourhoods with median 5 cells per cellular neighbourhood).
Transcriptional analysis of α and β cells and islets from ND and T2D donors
RNA isolation and bulk RNA sequencing.
RNA was extracted from sorted α and β cells (see ‘Purification of α and β cells by FACS’) or from pelleted whole islets using the Invitrogen RNAqueous-Micro Total RNA Isolation kit (Thermo Fisher AM1931). TURBO DNA-free (Ambion) was used to treat any trace DNA contamination. RNA was quantified by Qubit Fluorometer 2.0 and RNA integrity (RIN) was confirmed (RIN > 7) by 2100 Bioanalyzer (Agilent). Amplified cDNA libraries were constructed using SMART-seq v4 Ultra Low Input RNA-kit (Takara) and sequencing was performed on an NovaSeq platform (Illumina) using paired-end reads (100 bp) and 25 million reads per sample.
We processed the raw RNA-seq reads using FastQC (v0.11.8) for broad quality assessment. In brief, we examined the following parameters: (1) base quality score distribution, (2) sequence quality score distribution, (3) average base content per read, (4) GC distribution in thereads, (5) PCR amplification issue, (6) overrepresented sequences, (7) adapter content. Based on the quality report of fastq files, we trimmed sequence reads using fastq-mcf (v1.05) and cutadapt (v2.5) to only retain high-quality sequence for further analysis. The paired-end reads were aligned to the GRCh37/hg19 human reference with GENCODE v19 gene annotation using STAR splice-aware aligner (v2.5.4b;–outSAMUnmapped Within KeepPairs)60.
We counted fragments mapping to features type in GENCODE v19 gene annotation using featureCounts from Subread package61. The gene list was pruned to contain only protein-coding genes mapping to autosome and chrX, resulting in a total of 20,260 genes. We assessed libraries using comprehensive quality metrics generated by QoRTs62 as well as computed derived metrics. In brief, on the top of QoRTs reported metrics, we computed: (1) 5′–3′ gene coverage bias (as the ratio of coverage values at the 90th percentile and 10th percentile of the coverage distribution); (2) Kolmogorov–Smirov test statistic between cumulative gene diversity of each library relative to median distribution of all libraries within each cell type and standardized to a mean of 0 and s.d. of 1 to yield a z-score; (3) number of reads mapped mapped to Xist and SRY genes; (4) average number of reads mapped to chrM; and (5) transcript integrity number (TIN)63 for each library. The labelled sex of donors was matched against the gene expression quantified for sex genes to rule out any sample swaps or mislabelling. We also computed principal components for TPM (transcript per million) normalized count matrix for each cell type in order to detect potential outliers.
Differential gene expression analysis.
As collection of these rare tissues spanned more than five years, we used a latent variable analysis to discern biological variation from technical variation and then examined the datasets by both differential gene expression and gene network analyses. We performed differential gene expression analysis between T2D and ND samples for each cell type individually using DESeq264. In order to minimize potential effects of known and unknown confounding factors, we included known covariates in the DESeq2 model as well accounted for unknown covariates using the RUVseq latent variable approach65. In brief, we used the following multi-step process: (1) we first removed genes from the raw count matrix that had less than 10 reads in fewer than 25% of the samples for that cell type. (2) We then ran a first-pass differential expression analysis using DESeq2 with age, sex, BMI and batch as known covariates. The output result was filtered for genes that were non-significant (that is, not differentially expressed between T2D and ND samples) and had P values > 0.5. These genes were used as ‘control’ or ‘empirical’ genes for the RUVSeq::RUVg function to estimate latent variables accounting for variation in the data not attributed to disease status. (3) The latent variables estimated from the RUVseq run were then used as additional covariates (on the top of age, sex, BMI and batch where applicable) for the second run of DESeq2. We selected the number of latent variables to provide the most reasonable separation between T2D and ND samples and minimal deviation from mean in the relative log expression plots. The output results from DESeq2 were filtered for 1% FDR to generate the final list of genes that were differentially expressed between T2D and ND for each cell type. We performed functional enrichment analysis using RNA-Enrich66 and retained terms with an FDR threshold of 5%. Terms were condensed using the RelSim function in REVIGO67 with similarity parameter set to 0.5 and visualized in semantic space using an.xgmml file imported into Cytoscape software68 v3.8.2.
Combined analysis of differentially expressed genes (fold change ≥1.5 or ≤−1.5; P < 0.01) was performed using Metascape69. For visualization of overlap between samples (Fig. 1i), each gene list (α, β and islet) was independently analysed to calculate all statistically significant (P < 0.01) ontology terms enriched (two-tailed hypergeometric test). Grey curves on the diagram that connect two lists indicate that an ontology term containing ≤100 genes was statistically enriched in both lists (that is, the genes in the two lists are not identical, but they are both members of terms that are statistically enriched in their respective list). For visualization of enrichment across samples (Fig. 1j), all three gene lists (α, β and islet) were provided and Metascape’s heuristic algorithm sampled the 20 top-score clusters, selected up to the 10 best scoring terms (lowest P values) within each cluster, and connected terms pairs with Kappa similarity above 0.3. The resulting network was exported as a.cys file and visualized using Cytoscape, with the most representative term name in each cluster selected manually.
Gene network analysis.
We adopted the WGCNA70 approach to create networks from the gene expression data. This approach created modules (eigengenes) of up to 2,000 genes each, labelled by sample type and numbered consecutively (β cells, modules β00–β48; α cells, modules α00–α54; and islets, modules i00-i67). Importantly, collapsing the expression patterns across >14,000 genes into a smaller number of modules reduced gene-level multiple testing burden and enabled association of transcriptomic profiles with sample features including donor traits71, islet functional parameters from the same donors defined by dynamic islet perifusion, and enrichment of open chromatin peaks to overlap GWAS variants. In brief, we first filtered genes following the same rule established in ‘Differential gene expression’, where we only kept genes that had at least 10 reads in at least 25% of the samples for each cell type. We then processed raw counts using the varianceStabilizedTransformation function in DESeq2 package and used removeBatchEffect from the limma R package72 to adjust for effects of age, sex and BMI while protecting for disease status in the design matrix. The normalized and batch-corrected count matrix was then used as input to blockwiseModules to create a ‘signed hybrid’ network with ‘bicor’ as the correlation function. The power (k) parameter was selected such that the scale free topology fit reached at least 80% fit. To examine cell-type modules associated with quantitative traits of interest, we utilized a linear regression-based framework. We: (1) inverse-normalized the raw quantitative trait; (2) adjusted for age, sex and BMI by linear regression; and (3) computed the spearman rank correlation between residuals and eigengene of all modules. Within each network, we also computed the module membership score and network connectivity for each gene. Estimated enrichment of curated gene lists22,73,74 (Supplementary Table 5) was calculated using Fisher’s exact test. Functional enrichment of genes in each module was performed using gprofiler275, and the results were visualized as a dotplot.
Integration of network analysis with chromatin accessibility.
We integrated chromatin accessibility information with gene network analysis using single-cell combinatorial indexing on ATAC-seq (sci-ATAC-seq) data for α and β cells derived from our previously published study4. For each module within each cell type, we selected: (1) accessible sites that were present within a specified distance of the transcription start site (TSS) of the genes within that module; and (2) the distal chromatin peaks that were linked to the peaks within this set based on the Cicero peak interaction results from the same study. This set of TSS proximal and distal peaks for all of the genes within each module and for each cell type were then used for downstream enrichment analyses.
For variant enrichment analysis in the module linked peaks, we collected the latest published summary statistics for selected traits3,76. Using a threshold of ±10 kb to define our gene TSS boundary for linking peaks with modules, we created a set of accessible sites for each module. The union of peaks across all modules was used as a ‘bulk’ positive enrichment control. We then tested the enrichment of trait-associated variants from multiple GWAS across module peaks using GARFIELD37 and used a P value threshold of 5 × 10−8 as input parameter for selecting trait-associated variants.
Next, we considered whether specific transcription factor binding motifs (TFBMs) are enriched to occur in certain modules. To test this, we defined module linked peaks for each module as described before but using a threshold of ±1 kb from gene TSS. For each peak within a module, we then identified the peak summit and extended the summit by 50 bp in each direction. Using genomic sequence in this region as our test sequence, we used the Analysis of Motif Enrichment (AME, v5.3.2) tool from MEME suite77 (using default parameters) to identify enriched TFBMs represented in cisBP v.2.078. The control set of sequence was generated using –shuffle–parameter in AME that generates a control sequence by shuffling the test sequence but preserving the 2-mer frequency. The enrichment score was computed as scaled log2-transformed (true positives + 1)/(false positives + 1) for each TFBM.
Pseudoislet formation and assessment of RFX6 knockdown
Pseudoislets were formed as previously described26. In brief, nondiabetic human islets were hand-picked to purity and then dispersed with 0.025% HyClone trypsin (Thermo Scientific) for 7 min at room temperature before counting with an automated Countess II cell counter or manually by haemacytometer. Dispersed human islets or purified β cells (see ‘Purification of α and β cells by FACS’) were incubated in adenovirus at a multiplicity of infection of 500 for 2 h in Vanderbilt pseudoislet medium before being spun and washed. Adenovirus containing U6 driven scramble or RFX6-targeted shRNA as well as CMV driven mCherry or mKate2 red fluorescent tag were prepared, amplified, and purified by Welgen. Cells were then resuspended in appropriate volume of Vanderbilt pseudoislet medium to allow for seeding into wells at 2,000 cells per 200 μl in each well of CellCarrier Spheroid Ultra Low Attachment microplates (PerkinElmer). Pseudoislets were allowed to reaggregate for six days before being collected and studied.
To assess knockdown, RNA was extracted from pseudoislets containing only β cells using an RNAqueous RNA isolation kit (Ambion). cDNA synthesis and quantitative reverse transcriptase PCR were performed as previously described40; in brief, cDNA was synthesized using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems 4368814) according to the manufacturer’s instructions. Quantitative PCR (qPCR) was performed using TaqMan probes for ACTB (Hs99999903_m1) as endogenous control and RFX6 (Hs00941591_m1). Relative changes in mRNA expression were calculated by the comparative ΔCt method.
Multiome snRNA-seq and snATAC-seq
Isolation of nuclei.
Pseudoislet samples treated with RFX6 shRNA or scramble RNA were pooled together using a randomized study design, so the targeting and scramble conditions were not confounded by batch (Fig. 5a). To accomplish this, samples were allocated into 6 groups (batches) of n = 490–494 pseudoislets for nuclei isolation. A customized protocol was developed based on recommendations by 10x Genomics (https://www.10xgenomics.com/resources/demonstrated-protocols/), which included optimization steps described below. In brief, the samples were suspended in 1× PBS and pelleted at 2,000g for 3 min at 4 °C. The pellet was resuspended in lysis buffer (10 mM Tris-HCl 7.4 pH, 10 mM NaCl, 3 mM MgCl2, 0.1% Tween-20, 0.1% NP40, 0.01% digitonin, 1% BSA, 1 mM DTT, and 2 U μl−1 RNAse inhibitor) and rocked in an Eppendorf thermomixer C (EP 5382000015) at 300g for 5 min at 4 °C. Keeping the samples on ice as much as possible, tubes were then transferred to a pre-chilled 2-ml glass dounce homogenizer and homogenized with 15 strokes of tight pestle B before being transferred to a 1.5-ml tube and centrifuged at 500g for 5 min at 4 °C. The resulting pellet was then resuspended in 1 ml wash buffer (10 mM Tris-HCL 7.4 pH, 10 mM NaCl, 3 mM MgCl2, 1% BSA, 0.1% Tween-20, 1 mM DTT and 2 U μl−1 RNAse inhibitor) and centrifuged at 100g for 1 min at 4 °C. The supernatant was collected, filtered through a pre-wetted 30-μm filter, and centrifuged at 500g for 5 min at 4 °C. Nuclei were resuspended in 300 μl of wash buffer, then 300 μl of sucrose cushion (0.88 M sucrose, 1 mM DTT, 1 mM RNAse inhibitor, and 10% wash buffer) was added to the bottom of the tube and the resulting layered solution was centrifuged at 1,000g for 10 min at 4 °C. Both layers of supernatant were removed, and pellet was resuspended in 1 ml wash buffer and centrifuged at 500g for 5 min at 4 °C. Nuclei were then resuspended in 30 μl of nuclei resuspension buffer before counting and quality assessment. The desired concentration of nuclei was achieved by resuspending the appropriate number of nuclei in 1× diluted nuclei buffer for joint (on the same nucleus) snATAC-seq and snRNA-seq multiome profiling. Nuclei were processed by the University of Michigan Advanced Genomics Core using the 10x Genomics Chromium platform at 20,000 nuclei per well.
Multiome sample genotyping and imputation.
Samples were genotyped with the Infinium Multi-Ethnic Global-8 v1.0 kit using 50 ng I−1 DNA samples in two batches. Probes were mapped to Build 37. We merged the.ped files for the two batches along with samples from other projects that were genotyped on the same chip (resulting in a combined 68 samples). We removed variants with multi mapping probes and updated the variant rsIDs using Illumina support files Multi-EthnicGlobal_D1_MappingComment.txt and Multi-EthnicGlobal_D1.annotated.txt (downloaded from https://support.illumina.com/downloads/infinium-multi-ethnic-global-8-v1-support-files.html). We performed pre-imputation QC using the HRC-1000G-check-bim. pl script (version 4.2.9) obtained from https://www.well.ox.ac.uk/~wrayner/tools/ to check for strand, alleles, position, Ref/Alt assignments and update the same based on the 1000 Genomes reference (https://www.well.ox.ac.uk/~wrayner/tools/1000GP_Phase3_combined.legend.gz). We did not conduct allele frequency checks at this step (that is, used the–noexclude flag) since we had 68 samples from mixed ancestries. These filters resulted in 958,427 variants. We performed pre-phasing and imputation using the Michigan Imputation Server79. The standard pipeline (https://imputationserver.readthedocs.io/en/latest/pipeline/) included pre-phasing using Eagle280 and genotype dosage imputation using Minimac4 (https://github.com/statgen/Minimac4) and the 1000 Genomes phase 3 v5 (build GRCh37/hg19) reference panel81. Post-imputation, we selected biallelic variants with estimated imputation accuracy (r2) > 0.3, variants not significantly deviating from Hardy Weinberg equilibrium (P > 10−6), minor allele frequency (MAF) in 1000 Genomes European individuals > 0.05 and minor allele count (MAC) > 1 in our 12 samples, resulting in 6,665,607 variants.
Data processing for RNA component.
The RNA component of the multiome data was processed using starSOLO (STAR v. 2.7.3a, with GENCODE v19 annotation; options–soloUMIfiltering MultiGeneUMI–soloCBmatchWLtype 1MM_multi_pseudocounts–soloCellFilter None), which outputs the count matrices needed for most of the analyses60. Quality control metrics were gathered on a per-nucleus basis using a custom Python script on the corrected gene counts and aligned BAM file.
Following processing with STAR, we constructed a custom count matrix by combining information from the GeneFull and Gene matrices output by STAR. The GeneFull matrix contains per-gene counts based on intronic and exonic reads, while the Gene matrix contains per-gene counts based on exonic reads only. As nuclear RNA may contain introns, the GeneFull matrix should be preferred. However, due to overlapping transcript annotations that render some read gene assignments ambiguous, some genes may receive fewer counts in the GeneFull matrix than in the Gene matrix. The INS gene was an extreme example of this, receiving very low counts in the GeneFull matrix but high counts in the Gene matrix. To salvage counts for such genes, our custom matrix utilized the GeneFull counts for most genes but utilized the Gene counts for the subset of genes that had greater counts in the Gene matrix than in the GeneFull matrix.
Data processing for the ATAC component.
Adapters were trimmed using cta (https://github.com/ParkerLab/cta). We used a custom Python script, available in the Parker laboratory Github repository, for barcode correction. Barcodes were corrected in a similar manner as in the 10x Genomics Cell Ranger ATAC v. 1.0 software. In brief, barcodes were checked against the 10x Genomics whitelist. If a barcode was not on the whitelist, then we found all whitelisted barcodes within a hamming distance of two from the bad barcode. For each of these whitelisted barcodes, we calculated the probability that the bad barcode should be assigned to the whitelisted barcode using the Phred scores of the mismatched base(s) and the prior probability of a read coming from the whitelisted barcode (based on the whitelisted barcode’s abundance in the rest of the data). If there was at least a 97.5% probability that the bad barcode was derived from one specific whitelisted barcode, it was corrected to the whitelisted barcode.
Reads were mapped using BWA-MEM82 with flags ‘-I 200,200, 5000 -M’ (v. 0.7.15-r1140). We used Picard MarkDuplicates (v. 2.25.1; https://broadinstitute.github.io/picard/) to mark duplicates, and filtered to high-quality, non-duplicate autosomal read pairs using SAMtools view83 with flags ‘-f 3 -F 4 -F 8 -F 256 -F 1024 -F 2048 -q 30’ (v. 1.10). Quality control metrics were gathered on a per-nucleus basis using ataqv84 (v. 1.2.1) on the BAM file with duplicates marked.
Selection of quality nuclei for downstream analysis.
We performed rigorous QC of all RNA nuclei and only included those deemed as high-quality based on the following four definitions: (1) number of unique molecular identifiers (UMIs) >1,000; (2) mitochondrial fraction <0.2; (3) nuclei where the RNA profile was statistically different from the background or ambient RNA signal; and (4) nuclei were identifiable as a singlet and assignable to a sample using genotypes. We considered droplets with UMIs <10 to be empty and therefore representative of the background or ambient RNA profile. Top genes in the ambient RNA included highly expressed genes across prominent islet cell types such as INS, GCG and SST, along with several mitochondrial genes. We used the testEmptyDrops function from DropletUtils (v 1.6.1)85, specifying the lower parameter as 10 and selecting droplets with P < 0.05 as droplets significantly different from the ambient RNA profile. To identify singlets and assign to samples, we ran Demuxlet86 using using the BAM files and the genotype VCF file considering all post-QC variants in gene bodies with minor allele count (MAC) > 1. We used the command “demuxlet–sam $bam–tag-group CB–tag-UMI UB–vcf ${vcf}–alpha 0–alpha 0.5–field GT”, and selected singlets. To account for ambient RNA contamination while identifying singlets, we also masked the top 1% genes expressed in the ambient RNA and re-ran Demuxlet with the same parameters; nuclei were considered singlets and kept for downstream analysis if they were called as singlets in either Demuxlet run.
We also performed QC of the ATAC component of the multiome data. For ATAC, we required nuclei to have a minimum TSS enrichment (as calculated by ataqv) of 2, minimum filtered read count of 1.000 (ataqv ‘HQAA’ metric), and maximum mitochondrial fraction of 0.5. We also ran Demuxlet on the ATAC component (command: “demuxlet–sam $bam–tag-group CB–vcf ${vcf}–field GT”) and required that a prospective nucleus be called as a singlet. The ATAC component of nuclei in two wells showed low TSS enrichment and all nuclei from these two wells were therefore excluded from analysis.
If the RNA and the ATAC component of a barcode both passed QC and the Demuxlet sample assignment was the same, both modalities were utilized for downstream analysis. If only the RNA component passed QC, only the RNA component was used in downstream analysis. As we performed clustering on the RNA component, we excluded the few (twelve) barcodes that passed ATAC QC and failed RNA QC. The final count was 15,825 (RNA) and 5,706 (ATAC) high-quality nuclei for downstream analysis.
Removal of ambient RNA counts from single-nucleus gene expression UMI matrices.
Prior to clustering and downstream analysis, we used DecontX87 (celda v. 1.8.1, in R v. 4.1.1)88 to adjust the nucleus × gene expression count matrices for ambient RNA. DecontX was run on a per-batch basis, as the amount of ambient contamination may vary across batches. Decontaminated counts were generated via the decontX() function, passing barcodes with total UMI count ≤10 to the background argument. Rounded decontaminated counts were used for clustering and all downstream analyses. Nuclei with estimated contamination level >0.2 were excluded from downstream analysis.
Clustering of multiome data.
Nuclei were clustered on the RNA component using Seurat89–91 (v. 3.9.9.9010, in R v. 3.6.3). After normalizing counts with the NormalizeData function, we identified the top 2,000 variable features (FindVariableFeatures function, with selection. method=‘vst’) and scaled with the ScaleData function. We identified neighbours using the top 20 principal components and k.param = 20, and called clusters using resolution = 0.1 with n.start = 100. We used the top 20 principal components for generating the UMAP.
This clustering protocol identified 10 clusters. One of the smaller clusters shows expression of both INS and GCG, suggesting it may consist of doublets that were not caught by demuxlet. To verify this was a doublet cluster, we ran a different, genotype-independent, ATAC-based doublet detection method (AMULET; v. 1.0-beta, run with default parameters separately on data from each multiome well)92 on the ATAC nuclei that otherwise passed QC. This method tagged ~40% of the nuclei in the suspected doublet cluster as doublets, while only ~5% of nuclei in any other cluster were tagged as doublets. We therefore removed the small doublet cluster from the clustering and downstream analysis. Data are available via the UCSC Cell Browser93 at https://theparkerlab.med.umich.edu/data/public/cellbrowser/?ds=Pseudoislet10XMultiome for further exploration.
Differential gene expression analysis.
Differential gene expression was performed within each cluster using DESeq2 (v. 1.28.0)64 on pseudobulk counts. UMI counts were summed across nuclei within a donor + construct + cluster. Only donors with paired data (RFX6–2896 and scrambled–mCherry constructs) were used, and the analysis was performed in a paired fashion (DESeq2 model: ~donor + construct). We used an FDR threshold of 5% for considering genes differentially expressed. To compare differential gene expression between multiome data and data from sorted β cells (Extended Data Fig. 9g), Metascape was utilized as described above (‘Transcriptional analysis of α and β cells and islets from ND and T2D donors’, ‘Differential gene expression analysis’).
Gene network exploration for the RNA component.
Single-cell regulatory network inference and clustering (SCENIC)94 was applied to the RNA modality (~15,300 nuclei) to identify cell-specific regulons (small gene regulatory networks of transcription factors and their target genes) and discern changes induced by RFX6 knockdown. Soft gene filtering was applied to post-QC nuclei to remove genes present in <1% of all the nuclei and used as the input for SCENIC. In brief, the following steps were performed: network inference based on GRNBoost2, regulon prediction (cisTarget), and cellular enrichment (AUCell). Adjacency matrices, listing interactions of transcription factors and a numerical score, were generated, as were lists of regulons, their regulon specificity scores (RSS), and UMAP of nuclei based on RSS. Using previously identified cluster labels for each nucleus, average RSS was calculated for each regulon to generate a ranked list for each cluster. For comparison between shRFX6 and control nuclei in the β cluster, absolute difference of the RSS score for each regulon was computed between the two groups to identify the most differential regulons. Pathway enrichment was performed using Metascape as described above, using the input of genes (n = 220) contained in top 10 differential β regulons.
Per-cluster processing of ATAC component.
All ATAC reads from pass-QC, clustered nuclei were merged within each cluster. To generate per-cluster peaks, these BAM files were converted to single-ended BED format using bedtools bamtobed95 before calling ATAC-seq peak summits with MACS296 (flags -g hs–nomodel–shift −37–extsize 73 -B–keep-dup all–call-summits). We removed summits in blacklist regions, filtered to FDR 0.1% summits, and then generated a peak list from the summits by extending the ATAC-seq peak summits for each cluster +/− 150 bps to get 300-bp peaks (within each cluster, if two 300-bp peaks overlapped the one with the greater MACS2 score was kept). We then removed peaks in blacklist regions. To get the ATAC peak counts used in the ATAC principal components analysis (PCA) and differential chromatin accessibility analyses, we determined the number of ATAC fragments overlapping each of these peaks in each of the per-cluster, per-donor, per-construct pseudobulk samples.
For visualization of ATAC signal, we generated a normalized bedGraph file using MACS2 on the single-end BED file (macs2 callpeak command, with options–SPMR–nomodel–shift −100–extsize 200 -B–broad–keep-dup all) and then converted to bigWig format using the UCSC bedGraphToBigWig97. For PCA on the pseudobulk ATAC counts, we first removed any peaks on the mCherry or mKate2 contigs. We then converted peak counts to counts per million and removed the bottom 10% of features with the lowest average CPM across samples. For each peak, we filled any zeros with a value equal to half of the minimum non-zero CPM for that peak across samples. We then log transformed prior to performing the PCA.
Differential chromatin accessibility analysis.
Differential chromatin accessibility was performed within each cluster using DESeq2 (v. 1.28.0)64 on pseudobulk ATAC peak counts. Only donors with paired ATAC data (RFX6–2896 and scrambled–mCherry constructs) were used, and we additionally excluded donor 17277513 due to very low read counts. The DESeq2 analysis was performed in a paired fashion, with model: ~donor + tss_enrichment + construct. To compute TSS enrichment for each pseudobulk sample, we merged all ATAC nuclei (regardless of cluster) from each donor and computed TSS enrichment with ataqv.
Testing for enrichment of peak subsets near differential genes.
We used a permutation test to determine whether the most significant peaks (‘top peaks’) from the beta cell differential peak analysis were enriched near beta cell differentially expressed genes. First, we assigned each peak to the gene with the nearest TSS (if multiple TSS were equally close, we took the TSS with the smallest chromosomal coordinate). We then calculated the fraction of top peaks whose nearest gene was differentially expressed. To get the null expectation for this value, we permuted the ‘DE/not DE’ gene labels, such that the same number of genes were always labelled as ‘DE’ but the identity of these differentially expressed genes changed in each permutation. While permuting, we split genes into deciles based on the expression of each gene and permuted the labels only within each decile (this controls for the fact that highly expressed genes are more likely to be differentially expressed than lowly expressed genes due to statistical power in the differentially expressed analysis). We performed 10,000 permutations, in each permutation re-calculating the fraction of top peaks whose nearest gene was differentially expressed to build up the null distribution. We then calculated an empirical P value based on our observed value and the null distribution, adding a pseudocount to avoid a P value of 0 (P = (1 + no. of permutations where the test statistic was greater than or equal to our observed value)/10,001).
Motif scanning for multiome motif enrichment analyses.
The motif scans were performed using FIMO (v. 5.0.4) with a background model calculated from the hg19 reference genome98 and otherwise default parameters. We used the motifs from Kheradpour and Kellis 201499, excluding “*_disc” motifs; motifs from cisBP v. 2.078; motifs from Jolma et al. (2013)100; and custom RFX6 motifs generated using mouse Rfx6 ChIP-seq data from Piccand et al. (2014)33.
The custom RFX6 motifs were generated during a previous project25. Sequencing reads from Piccand et al. (2014)33 were mapped to the mouse mm9 genome101 using bwa (v. 0.7.12-r1039) and peaks were called using MACS2 (flags: -t MIN6_Rfx6-HA_IP.bam -c MIN6_Control-HA.bam -B–nomodel -g mm–keep-dup 1 -q 1.00e-4). The MEME (v. 4.11.0)102 and DREME (v. 4.9.1)103 tools from the MEME suite104 were used to discover novel motifs in the resulting peaks. One non-repetitive motif from the MEME tool and two motifs from the DREME tool, bearing similarity to known RFX family motifs, were selected for use in downstream analysis.
Motif enrichment in most significant peaks.
We used logistic regression to measure enrichment of motifs in subsets of ATAC-seq peaks. We ran one model per peak category and motif. For testing for enrichment in the peaks that had the smallest P values and leaned towards higher signal in shRFX6 samples, we modelled:
Where ‘peak_leans_higher_in_shRFX6’ is 1 if the peak was one of the most significant peaks in the ‘up in RFX6 KD condition’ direction and 0 otherwise; peak_gc_content was the GC content of the sequence within the peak; peak_size was the mean DESeq2-normalized count for the peak across the samples in the DESeq2 analysis; and n_motif_hits_in_peak was the number of motif hits in the peak as determined by the FIMO motif scans. The coefficient of the n_motif_hits_in_peak term was taken as the measure of motif enrichment. For testing for enrichment in the peaks that had the smallest P values and leaned towards lower signal in shRFX6 samples, we used the same model except the outcome variable was ‘peak_leans_lower_in_shRFX6’.
Generation of ATAC footprint plots.
To generate the ATAC footprint plots, we first separated the motif occurrences into those within the beta cells ATAC peaks and those outside of peaks. For each of these two groups, we computed an aggregate Tn5 cut matrix for the 500 bps on either side of the motifs, using beta cell ATAC reads from each individual donor plus construct (using the make_cut_matrix script within the atactk package (https://github.com/ParkerLab/atactk); options -a -r 500). The cut matrices were generated separately for each donor+construct, utilizing only donors with paired ATAC data (RFX6–2896 and scrambled–mCherry constructs) and additionally excluding donor 17277513 due to very low ATAC read counts. To reduce the impact of Tn5 insertion sequence bias, we normalized the Tn5 cut frequency at each position for the motifs in peaks by the corresponding frequencies for the motifs outside of peaks. To adjust for technical differences (for example, TSS enrichment) between the donors plus constructs, we then divided these normalized cut frequencies by the average normalized cut frequency between the −500 and −400 bp positions.
GWAS enrichment in most significant peaks.
We considered if β cell ATAC-seq peaks that score highly for differential accessibility, as measured by P value, are specifically enriched to overlap T2D GWAS variants. We compared the enrichment of T2D (adjusted BMI) GWAS variants (n = 3,062,361) to overlap top 5,000 ATAC-seq differential peaks leaning up and down with the remaining peaks for β cell using GARFIELD37. Using a P value threshold of 10−5, we also performed a conditional analysis where GARFIELD evaluates if both annotations are conditionally independent of each other in the enrichment model. The coefficients corresponding to each annotation from the conditional enrichment model were shown along with the 95% confidence interval. To ensure robustness of our results, we repeated the analysis for top 2,000 (up and down each) and top 10,000 (up and down each) differential peaks.
Biobank interrogation and MR analysis
MR was performed to explore causal effects of RFX6 expression in human islets on T2D. Seven independent instrumental variables were selected from the islet eQTL analysis of InsPIRE27 (P value < 0.01; linkage disequilibrium (LD) pruning with 50 SNP windows, 5 SNPs each step, and LD < 0.2 by PLINK and based on an LD panel from the 1000 Genomes Project European population81). Data from the UK Biobank3 and the European T2D meta-analysis from Vujkovic et al.9 were used to determine the association between instrumental variables and T2D status. Four MR approaches were performed: Egger, weighted median, dIVW and PRESSO. These approaches offer different strategies for estimating the causal effects of the exposure (RFX6 expression) on outcome (T2D). MR-Egger uses the intercept to correct for pleiotropic effects of instrumental variables105, while the more precise weighted median approach offers a robust effect-size estimation from median values even in the presence of invalid instrumental variables106. The dIVW method removes the weak instrument bias of inverse-variance weighted estimator and is more robust under many weak instruments, formulated using a bias correction factor107. Similar to MR-Egger, MR-PRESSO quantifies the horizontal pleiotropy and estimates the causal effect after outlier removal108. MR-Egger, weighted median, and dIVW approach were performed using the R package Mendelian-Randomization109, and robust regression was applied for MR-Egger. MR-PRESSO was conducted with its R package, MR-PRESSO.
The Integrated Network for Systematic analysis of Pancreatic Islet RNA Expression (InsPIRE) consortium data comprise collected human islet tissue and genetic data from 420 participants. The islet transcriptome was profiled by Illumina HiSeq2000 platform. The sequencing reads were aligned to GRCh37 reference genome by GEM, raw read counts were scaled to ten million reads to address inconsistent library size and scaled read counts were normalized via quantile normalization. All individuals were genotyped on the OmniExpress and Omni2.5 genotype arrays and imputed with 1000 Genomes phase I as reference panel. Cis-eQTL analysis was restricted to the 1-Mb region upstream and downstream of the TSS, and fastQTL was conducted for the eQTL analyses with the adjustment of gender, 4 genetic principal components, 25 transcriptome principal components, and a variable indicating experimental batch. Additional details regarding the data and eQTL analyses have been previously published27. The human islet eQTL results were downloaded from https://zenodo.org/record/3408356#.Yo0B-5PMKCc.
UK Biobank is a large nation-wide biobank study including genetic and medical data for over 500,000 participants aged110 40–69. All individuals were genotyped by UK Biobank Axiom Array and imputed to the Haplotype Reference Consortium (HRC) reference panel111. T2D status was determined via self-reported medical history and medication, as previously described3. T2D GWAS was performed in 19,119 cases and 423,698 controls of European ancestry3. BOLT-LMM, a mixed-effect model, was applied to adjust for population structure and relatedness112. Additional details of this analysis have been previously published3. Summary statistics from this T2D GWAS in UK Biobank were downloaded from http://diagram-consortium.org/. Summary statistics from the European T2D meta-analysis9 were downloaded from dbGAP (study phs001672.v4.p1, analysis pha004945.v1.p1).
Statistical information
Specific statistical tests used for each dataset are described in the figure legends and text where appropriate. Data are represented as mean ± s.e.m. unless otherwise noted. A P value of 0.05 was considered significant except for bulk RNA-seq differential expression where we used a more stringent cut-off of 0.01. Statistical comparisons were performed using GraphPad Prism software 8.0 or using R and are specified in the figure legends.
Extended Data
Extended Data Fig. 1 |. (related to Fig. 1). Ex vivo and in vivo functional profiling of islets from donors with early T2D demonstrates reduced stimulated insulin secretion.
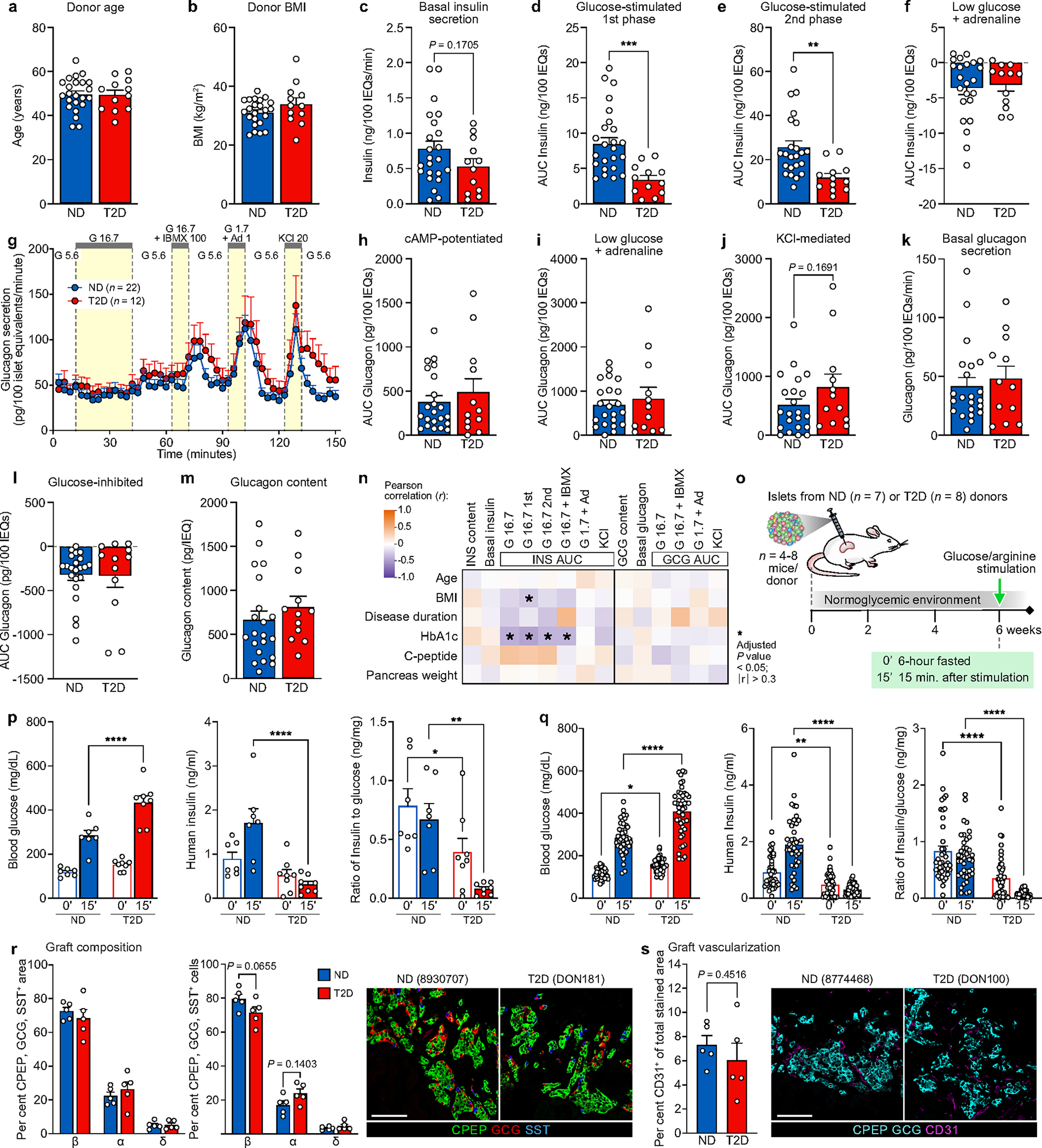
(a-b) Matching of BMI (a) and age (b) in n = 23 ND and n = 12 T2D independent donors for perifusion experiments. (c-m) Perifusion metrics for islets from n = 23 ND and n = 12 T2D independent donors. (c) Basal insulin secretion calculated as the average of the first three points of perifusion trace. (d-e) Integrated area under the curve (AUC) breaking down the total 16.7 mM glucose response into the first phase (d; through minute 24) and second phase (e; remainder of stimulation). Both first and second phases of insulin secretion were reduced, with the first phase showing a more significant reduction. (f) Area “under” the curve calculated from trace baseline for inhibition with low glucose and epinephrine. (g-k) Dynamic glucagon secretory response (g) measured by islet perifusion and secretory response as area under the curve (AUC) (h-j) normalized to islet volume. Ad, adrenaline (μM); G, glucose (mM); IBMX, isobutylmethylxanthine (μM); KCl, potassium chloride (mM). (k) Basal glucagon secretion calculated as average of first three points of perifusion trace. (l) Area “under” the curve calculated from trace baseline for inhibition with high glucose; both ND and T2D islets showed glucose-mediated suppression of glucagon secretion. (m) Glucagon content normalized to islet volume. (n) Pearson correlation of donor attributes to insulin and glucagon secretory metrics highlighted a significant negative correlation between donor HbA1c and stimulated insulin secretion (r < −0.30, P < 0.05). (o) Schematic of human islet transplantation and in vivo assessment of function. (p-q) After six weeks T2D islets secreted less human insulin than ND islets, especially after stimulation with glucose/arginine (p: average per donor of n = 7 ND and n = 8 T2D independent islet preparations; q: n = 41 individual mice with engrafted ND islets and n = 45 individual mice with engrafted T2D islets). Blood glucose, human insulin levels, and human insulin:blood glucose ratio were measured at 0’ (six-hour fasted) and 15’ after glucose and arginine administration in mice with human islet grafts. (r-s) Endocrine composition (r) and vascularization (s) of islet grafts from n = 5 individual mice representing n = 5 ND donor islet preparations and n = 5 individual mice representing n = 5 T2D donor islet preparations. Engraftment was similar between ND and T2D islets. Scale bars, 100 μm. Data in panels a-m and p-s show mean + SEM; statistical results (e: two-tailed linear mixed-effect model, P = 0.518; a-d, f-k, s: two-tailed t-test; p-r: two-way ANOVA with Šídák’s multiple comparisons test) indicated as follows: *P < 0.05, **P < 0.01, ***P < 0.001, **** P < 0.0001.
Extended Data Fig. 2 |. (related to Fig. 1). Transcriptional analysis of islets and sorted α and β cells reveals dysregulation of metabolic pathways in T2D β cells and immune signaling in T2D islets.
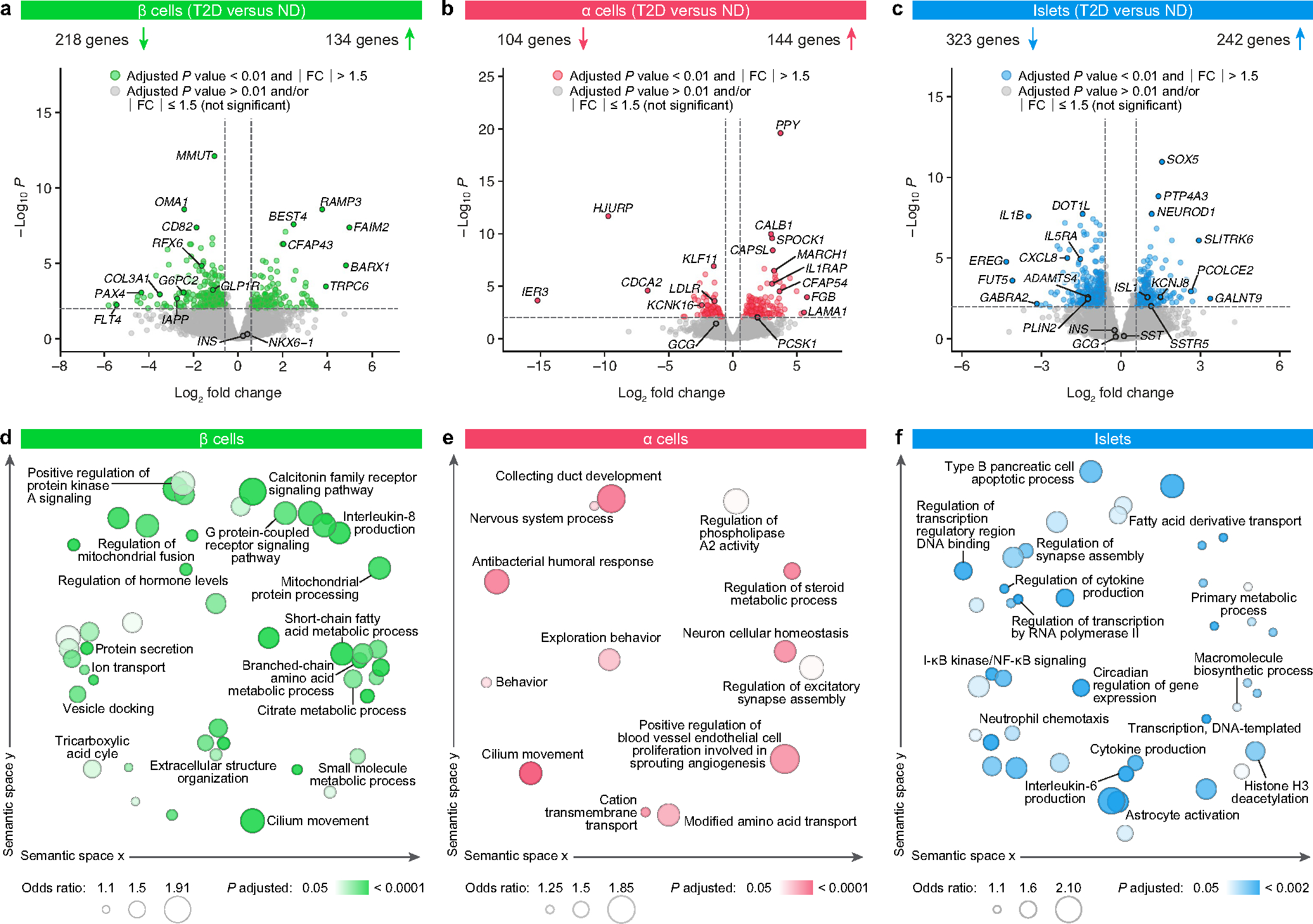
(a-c) Volcano plots illustrating differentially expressed genes between ND and T2D β cells (a; 352 genes), α cells (b; 248 genes), and islets (c; 565 genes) as obtained by DESeq264 (two-sided; multiple hypothesis corrected at FDR < 0.01). Lines denote cutoffs for fold-change (±1.5) and significance (FDR < 0.01); genes passing both thresholds are colored and select genes are labeled. (d-f) Enriched gene ontology terms (two-sided; multiple hypothesis corrected at FDR < 0.05) obtained from RNA-Enrich66 were condensed using the RelSim function of Revigo (similarity = 0.5) and plotted in semantic space to emphasize relatedness. Dot size represents odds ratio and color represents p-value. Select terms are labeled. For α cells gene changes were most evident in amino acid and steroid signaling pathways and regulation of blood vessel morphology and for islets in cytokine signaling and other immune terms.
Extended Data Fig. 3 |. (related to Fig. 2). Parallel approaches of multiplexed imaging and high-throughput traditional immunohistochemistry enable detailed profiling of the endocrine compartment of the islet.
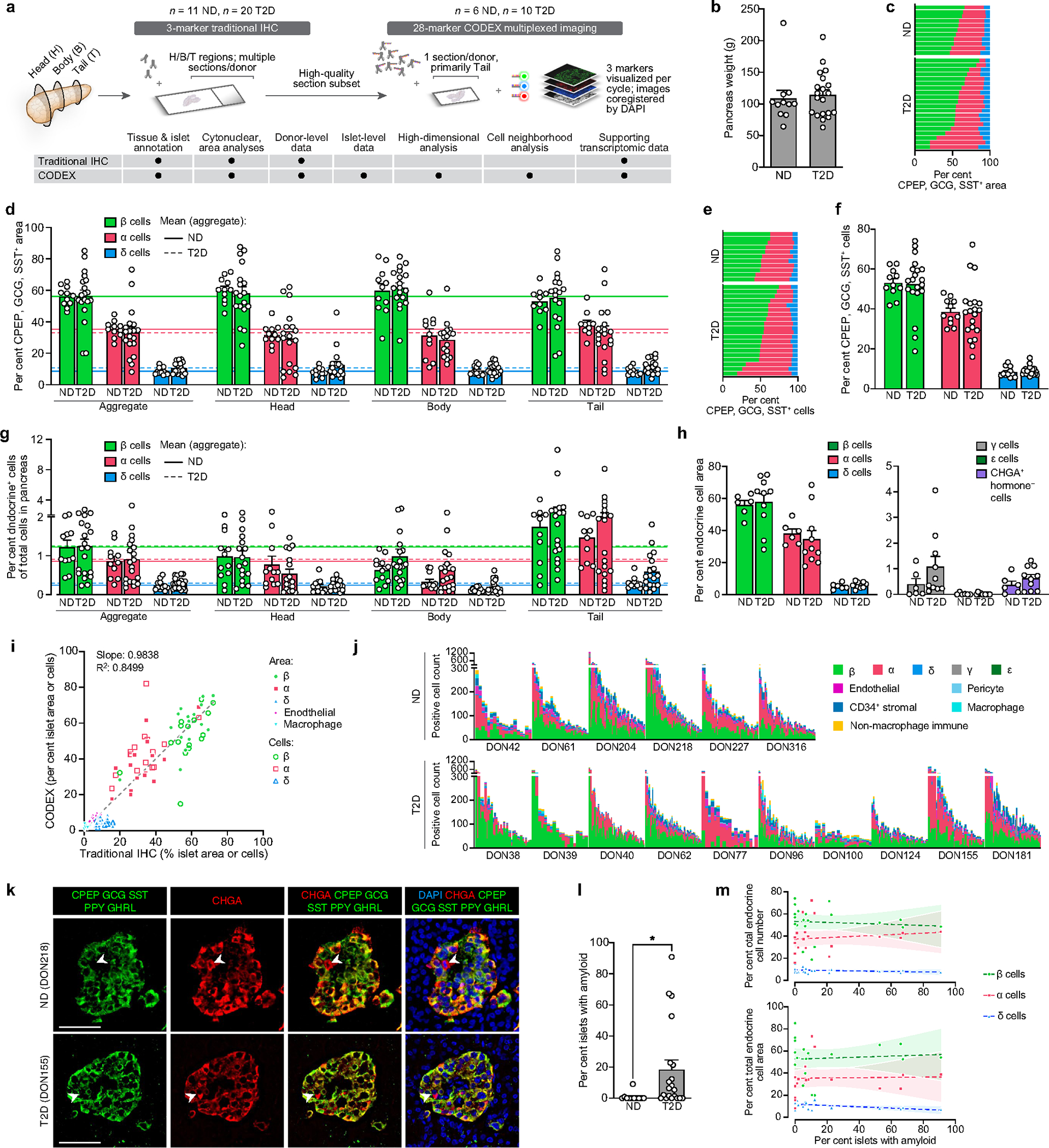
(a) High-throughput traditional immunohistochemistry (IHC) with whole-slide imaging was applied across pancreas head, body, and tail regions for the entire donor cohort, and in parallel, a subset of samples was analyzed with a 28-marker panel using co-detection by indexing (CODEX), a multiplexed technique enabling fluorescence-based imaging of large tissue sections without tissue destruction to spatially resolve many cellular phenotypes. A full list of inclusionary and exclusionary markers for cellular phenotypes analyzed is provided in Supplementary Table 4. (b-g) Analysis by traditional IHC of tissue from n = 11 ND and n = 20 T2D independent donors. (b) Pancreas weight measured during organ procurement was used to calculate endocrine cell mass in Fig. 2a. (c-g) Cross-sectional area (c-d) and cytonuclear quantification (e-g) of β cells (CPEP; green), α cells (GCG; red), and δ cells (SST; blue). Individual donor data shown in stacked bar graphs (c, e); stratification by pancreas region (d, g) includes horizontal lines (solid, ND; dotted, T2D) for mean values from combined analysis (‘Aggregate’). Donor-to-donor variability in β and α cell ratio was notable, underscoring the challenge in working with heterogeneous human tissues113, but no differences were detected between groups in this cohort30,114–116. (h-j) Analysis by CODEX of tissue from n = 6 ND and n = 10 T2D independent donors. (h) Cross-sectional area of endocrine cell types as measured by CODEX, including rarer γ and ε cell populations. (i) Correlation of paired cell type measurements from traditional IHC (x-axis) and CODEX (y-axis); the two methods were highly concordant (R2 = 0.8499, slope=0.9828, P < 0.0001; two-tailed linear regression). (j) Abundance of endocrine and non-endocrine cells in ND and T2D islets; one vertical bar per islet and colored by cell type. Islets are grouped by donor and ordered from largest (highest total cell number) to smallest. See also Fig. 2c. (k) Rare cells positive for chromogranin A (CHGA; red) but negative for all hormones (green), postulated to represent dedifferentiated endocrine cells117, were present in both ND and T2D at similar proportions. Scale bars, 50 μm; arrowheads denote CHGA+ hormone− cells. (l) Amyloid prevalence (% total islets with amyloid, averaged over multiple regions) for n = 11 ND and n = 20 T2D independent donors. (m) Correlation of amyloid prevalence with β, α, and δ cell populations as percentage of total endocrine cell number or cross-sectional area; one symbol per donor with 95% confidence interval of linear regression (shading). No slopes were significantly nonzero at P < 0.01 threshold. Traditional IHC data: panels c-h, l-m; CODEX data: panels i-k. Data in panels b, d, f-h, and l show mean + SEM; statistical results (b, l: two-tailed t-test; d, f-h: two-way ANOVA with Šídák’s multiple comparisons test) indicated as follows: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. None of the variables shown had a statistically significant association with disease duration (Pearson correlation, threshold P < 0.05).
Extended Data Fig. 4 |. (related to Fig. 2). Integration of multiplexed imaging and transcriptional profiling highlight disrupted capillaries and immune cells within T2D islets.
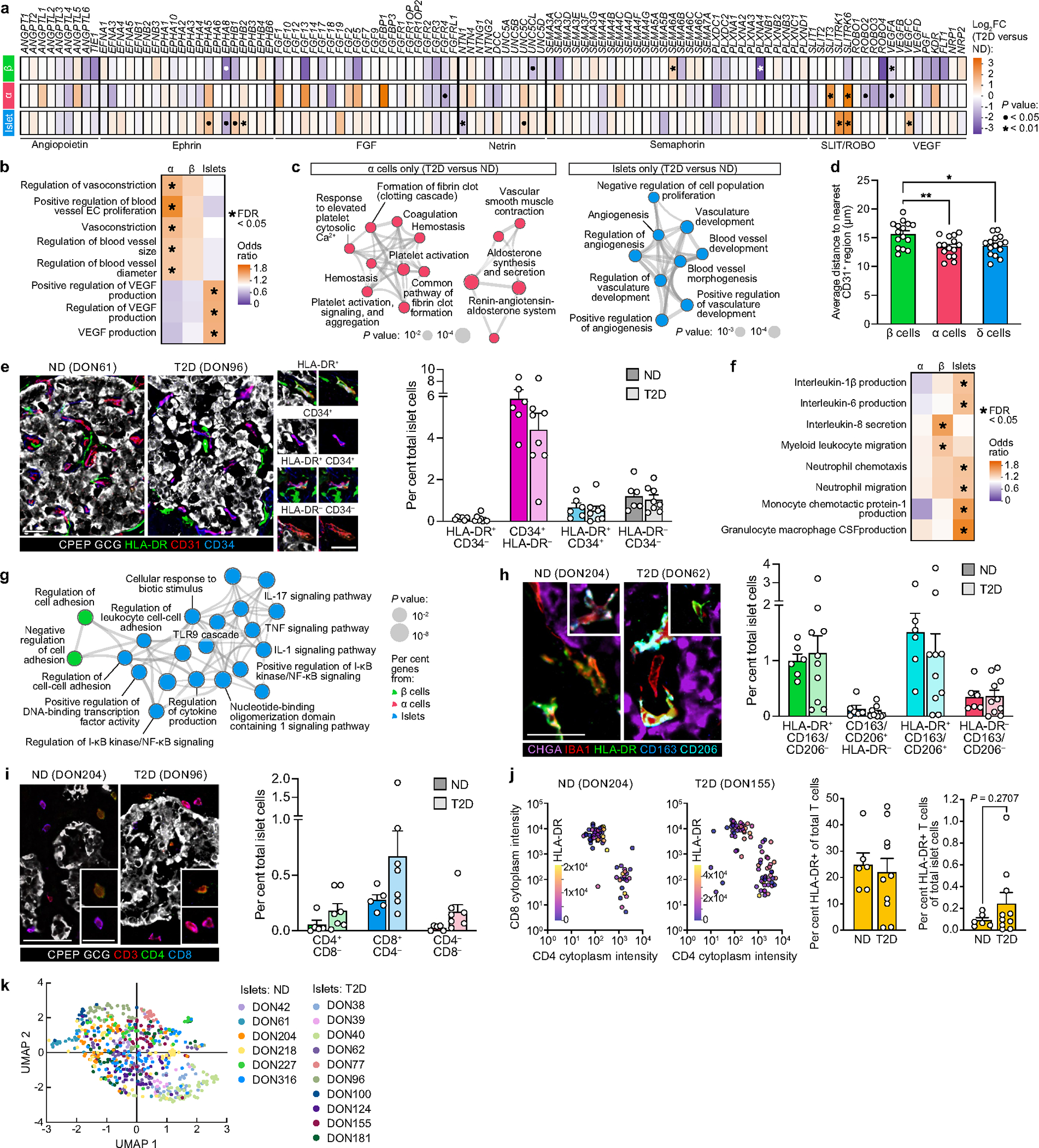
(a) Gene expression fold-change (DESeq264; two-sided and multiple hypothesis corrected at FDR < 0.05) of selected vascular and neuronal ligands and their receptors in β cells, α cells, and islets; • FDR < 0.05; * FDR < 0.01. Alpha cells expressed more angiogenic ligands and receptors than β cells. (b-c) RNA-sequencing analysis highlighted enrichment in T2D samples for processes controlling blood vessel size, particularly in α cells, as well as regulation of growth factors critical to islet capillary maintenance. Panel (b) shows select vascular-related ontology terms (RNA-Enrich66; two-sided and multiple hypothesis corrected at FDR < 0.05); panel (c) shows Metascape visualization of select terms enriched for differentially expressed genes (two-tailed hypergeometric test)69 in T2D α cells (left) and islets (right). (d) Average distance of each endocrine cell type to nearest capillary (n = 6 ND and n = 9 T2D independent donors). Interestingly, α and δ cells were slightly closer to capillaries than β cells in both ND and T2D islets. (e) Phenotypes of endothelial cells (CD31; red) defined by single or dual positivity for HLA-DR (green) and CD34 (blue). Examples of each combination (HLA-DR+ CD34−, CD34+ HLA-DR−, HLA-DR+ CD34+, and HLA-DR− CD34−) are shown to right. Data from n = 6 ND and n = 10 T2D independent donors. Scale bars, 25 μm. (f-g) Enrichment in T2D β cells and islets for cytokine signaling and immune cell recruitment pathways118,119. Panel (f) shows select immune-related ontology terms (RNA-Enrich66; two-sided and multiple hypothesis corrected at FDR < 0.05); panel (g) shows magnification of select clusters depicted in Fig. 1j (terms enriched across β, α, and islet samples). (h-i) Macrophages (IBA1+) and T cells (CD3+) phenotyped by various cell surface markers; insets show additional cells to illustrate phenotypic variety. Proinflammatory (HLA-DR+), anti-inflammatory (CD163 and/or CD206+), helper (CD4+), and cytotoxic (CD8+) phenotypes are detailed in Supplementary Table 4. Scale bars, 25 μm (h, i inset) and 50 μm (i). Data from n = 6 ND and n = 10 T2D (h) or n = 5 ND and n = 8 T2D (i-j) independent donors. (j) Expression of HLA-DR in CD4+ and CD8+ T cell populations. (k) High-dimensional component analysis of islet cell composition per islet (n = 681), shown by donor; corresponds to Fig. 2k. RNA data: panels a-c, f-g; * FDR < 0.05; CODEX data: panels d, h-j. Bar graphs in panels d-e and h-j show mean + SEM with symbols representing individual donors; statistical results (d: one-way ANOVA with Tukey’s multiple comparisons test; e, h-i: two-way ANOVA with Šídák’s multiple comparisons test; j: two-tailed t-test) indicated as follows: * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001. None of the variables shown had a statistically significant association with disease duration (Pearson correlation, threshold P < 0.05).
Extended Data Fig. 5 |. (related to Fig. 2). Cellular neighborhood assignment and (d-e) corresponding cell composition changes in T2D and ND islets.
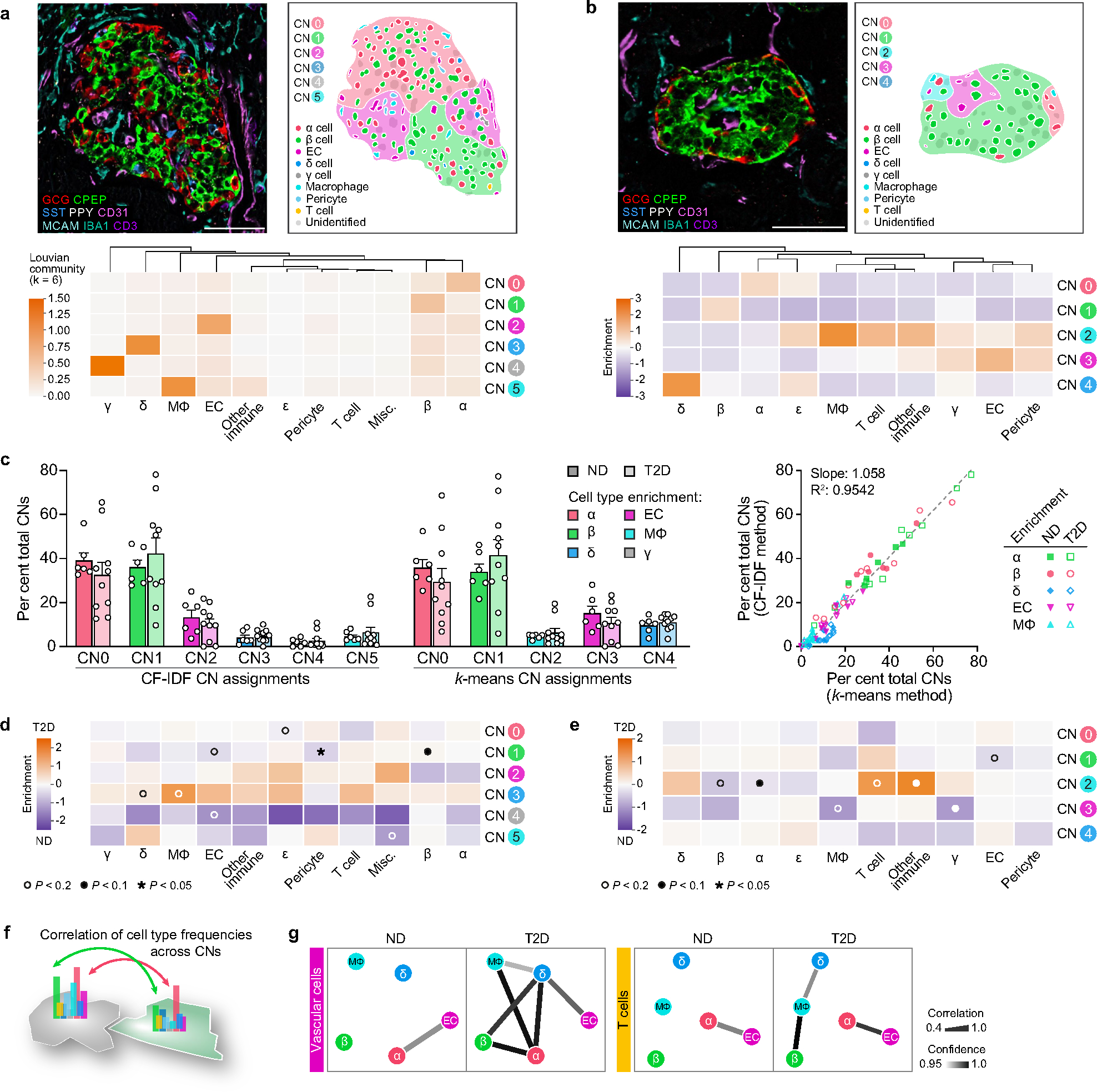
(a) Heat map showing cellular neighborhoods (CNs) identified by CF-IDF method and representative islet image with cell and CN annotation overlay. See Methods for more details. Scale bar, 50 μm. (b-c) A k-means cellular neighborhood analysis (b) was applied in parallel, and results were highly concordant between the two methods (c; R2 = 0.9542, slope = 1.058, P < 0.0001; two-tailed linear regression). Scale bar (b), 50 μm. Bar graphs show mean + SEM with symbols representing individual donors. (d-e) Differential cell type enrichment (two-tailed t-test) in T2D vs. ND islets by CF-IDF (d) and k-means (e) analyses. ECs and pericytes were depleted in β CNs (CN1) of T2D islets, consistent with our findings of decreased proximity between β cells and ECs in T2D. Symbols indicate unadjusted P-values as follows: ◦ P < 0.2, • P < 0.1, *P < 0.05. (f-g) Correlation of cell compositions across CNs; these analyses ask whether cell type frequencies are correlated between CNs, i.e., if there was evidence for connectivity between spatially distinct regions. Panel (g) shows results from k-means method; see also Fig. 2l (CF-IDF method). All cellular neighborhood data is derived from CODEX imaging of tissue from n = 6 ND and n = 10 T2D independent donors.
Extended Data Fig. 6 |. (related to Fig. 3). WGCNA emphasizes α and islet cell gene modules associated with donor and islet traits as well as those enriched in GWAS loci.
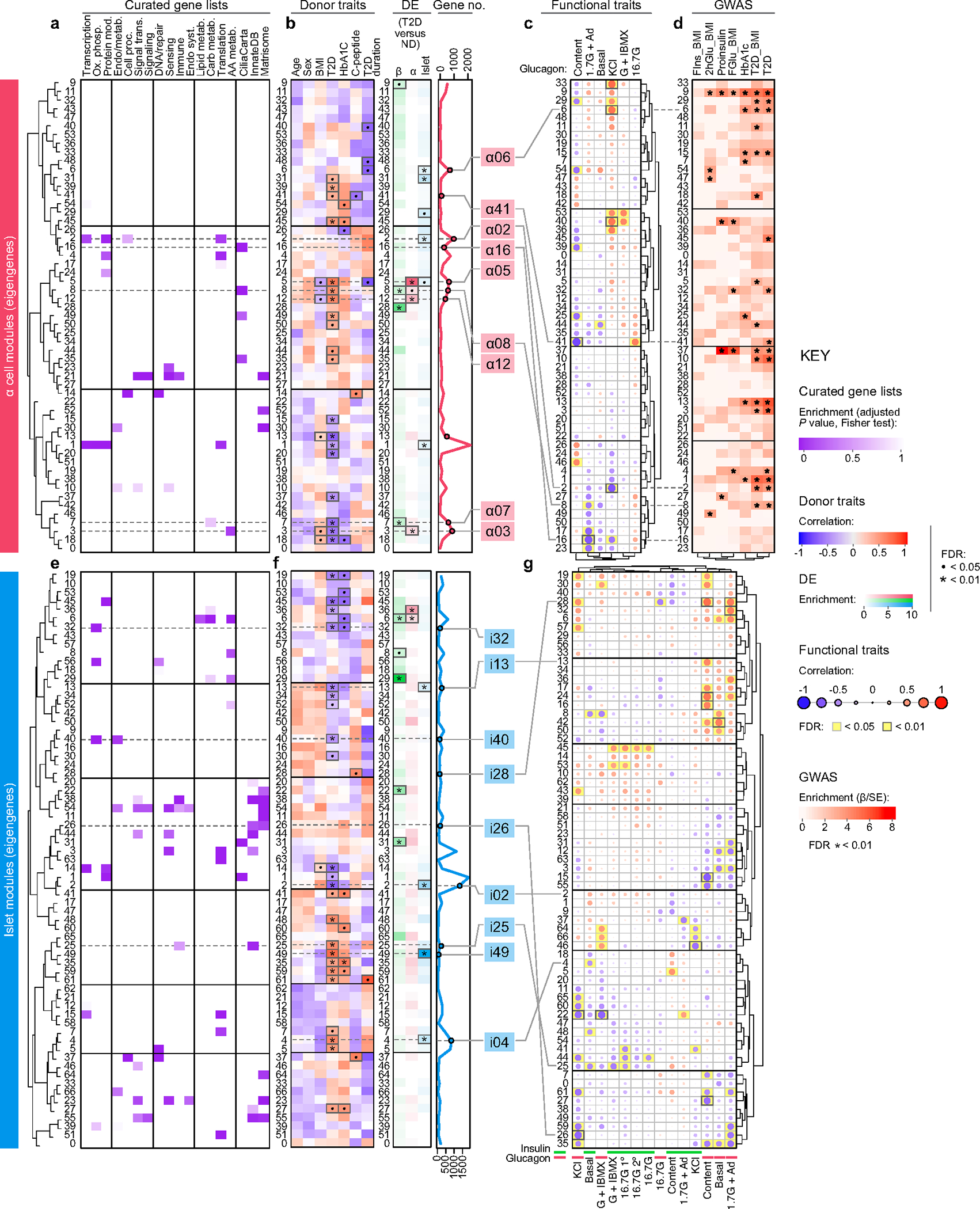
(a-d) Though α cell modules showed weaker correlations to donor and functional traits than did β cell modules, several modules were significantly enriched for cilia-related genes (a) and α08 was also enriched for α cell genes differentially expressed in T2D α cells (b). Both α08 and α16 significantly inversely correlated with epinephrine-mediated glucagon secretion and were closely related across functional parameters (c). Module α08 showed significant enrichment for T2D GWAS variants (d). See also Supplementary Fig. 4b. (e-g) Several islet modules showed notable enrichment for immune- and matrisome-related genes (e). Module i25 correlated positively with T2D status (f) and inversely with basal insulin secretion and GSIS, while i26 correlated inversely with KCl-mediated insulin secretion (g). See also Supplementary Fig. 4c. Panels a and e show module eigengenes clustered by similarity and relative enrichment of curated gene lists (see also Supplementary Table 5). Panels b and f provide correlation to donor characteristics, enrichment of differentially expressed (DE) genes, and total number of genes per module (• P < 0.05; * P < 0.01). Modules of interest highlighted (b: red, f: blue). Panels c and g show module correlation to α and β cell function (Fig. 1); significant associations highlighted (yellow). For islets (g), modules were correlated to both insulin and glucagon secretion (G + IBMX, 16.7 mM glucose with 100 μM isobutylmethylxanthine; 16.7 G, 16.7 mM glucose; 16.7 G 1°, first phase; 16.7 G 2°, second phase; 1.7 G+Epi, 1.7 mM glucose and 1 μM epinephrine; KCl, 20 mM potassium chloride). Panel d shows α cell module enrichment for GWAS traits (FIns, fasting insulin; 2hGlu, 2-hour glucose; FGlu, fasting glucose; * FDR < 0.01). Panels a, e, b and f (DE genes): two-tailed Fisher test adjusted for multiple comparisons; panels c, g, b and f (donor traits): t-test, unadjusted, using Spearman correlations (see Methods for additional details); panel d: enrichment using GARFIELD37.
Extended Data Fig. 7 |. (related to Fig. 3). Both transcriptomic and histologic data suggest changes in cilia processes in early T2D.
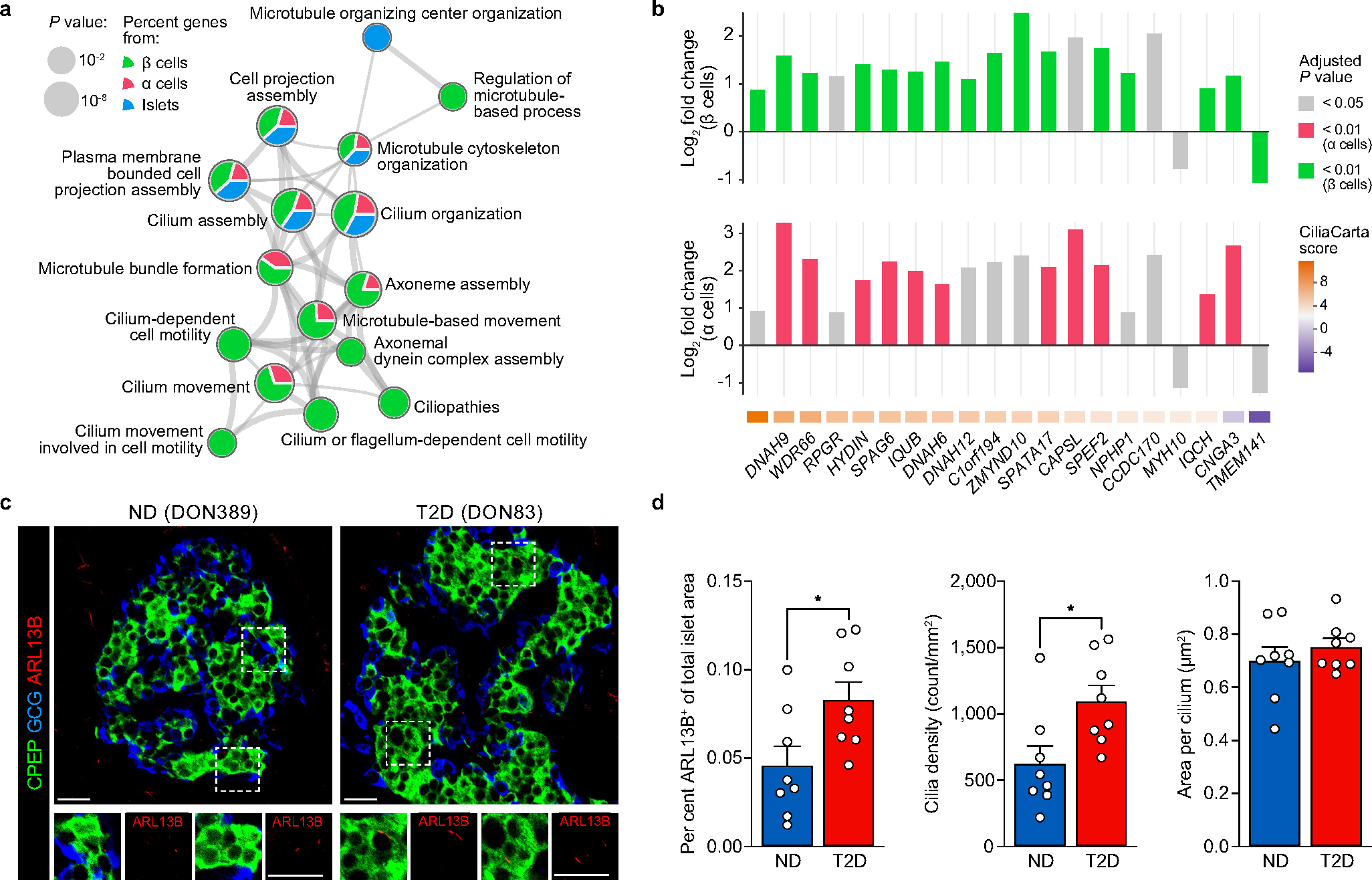
(a) Magnification of select clusters depicted in Fig. 1j (terms enriched across β, α, and islet samples). (b) Fold change of validated cilia-related genes (shown here, those ≥ |1.5| in both α and β cells); the majority were expressed at higher levels in T2D compared to ND. (c-d) Visualization by immunohistochemistry of cilia (ARL13B; red) and quantification of abundance, density, and size in tissue from n = 8 ND and n = 8 T2D independent donors. Tissue sections are from the same donors shown in panel b. Scale bars, 50 μm. Bar graphs show mean + SEM with symbols representing individual donors; statistical results (d: two-tailed t-test) indicated as follows: * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
Extended Data Fig. 8 |. (related to Fig. 4). RFX motif enrichment across β cell modules and β cell RFX6 reduction in pseudoislets impairs insulin secretion.
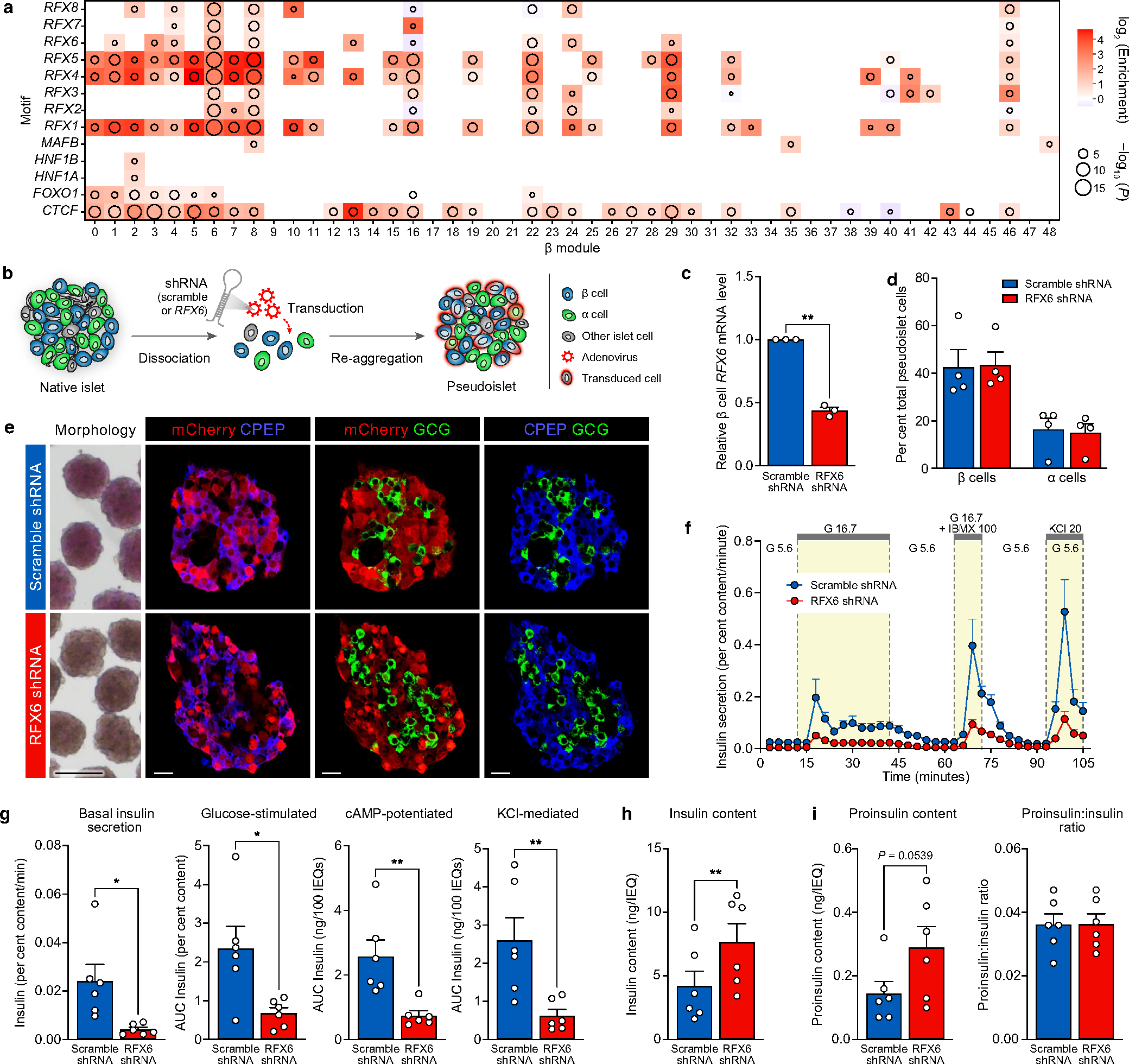
(a) Enrichment of selected transcription factor120 motifs in β cell modules, calculated using the AME tool from MEME-Suite with Bonferroni correction77; log10(P-values) are unadjusted. (b) Schematic of adenoviral shRNA delivery and formation of pseudoislets, which mirror native human islet architecture26,121. (c) Relative RFX6 mRNA expression in β cells treated with scramble shRNA (‘control’) or RFX6 shRNA (‘shRFX6’). Data represents n = 3 independent islet preparations. (d) RFX6 knockdown did not change β or α cell proportion (n = 4 independent islet preparations); acute (6-day) reduction of RFX6 expression does not lead to β cell loss. (e) Control and shRFX6 pseudoislets exhibited similar size and morphology. Transduced cells marked by mCherry; distribution of β cells shown by C-peptide (CPEP; blue) and α cells by glucagon (GCG; green). Scale bars, 200 μm (morphology) and 50 μm (immunostaining). (f-i) Dynamic insulin secretion and metrics equivalent to Fig. 4f, g but normalized by total insulin content (h) in control and shRFX6 pseudoislets. (i) Proinsulin content and proinsulin:insulin ratio in control (scramble) and shRFX6 pseudoislets. Proinsulin content was not significantly greater in shRFX6 pseudoislets and the proinsulin:insulin ratio was comparable to controls, suggesting secretory defects were not due to inappropriate insulin processing. Functional data (f-i) represents n = 6 independent islet preparations. Data in panels c-d and f-i show mean + SEM; statistical results (c-d, g-i: two-tailed t-test; f: linear mixed-effect model) indicated as follows: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Extended Data Fig. 9 |. (related to Fig. 5). RFX6 controls stimulated insulin secretion through genome-wide chromatin alterations disrupting transcripts controlling exocytotic pathways.
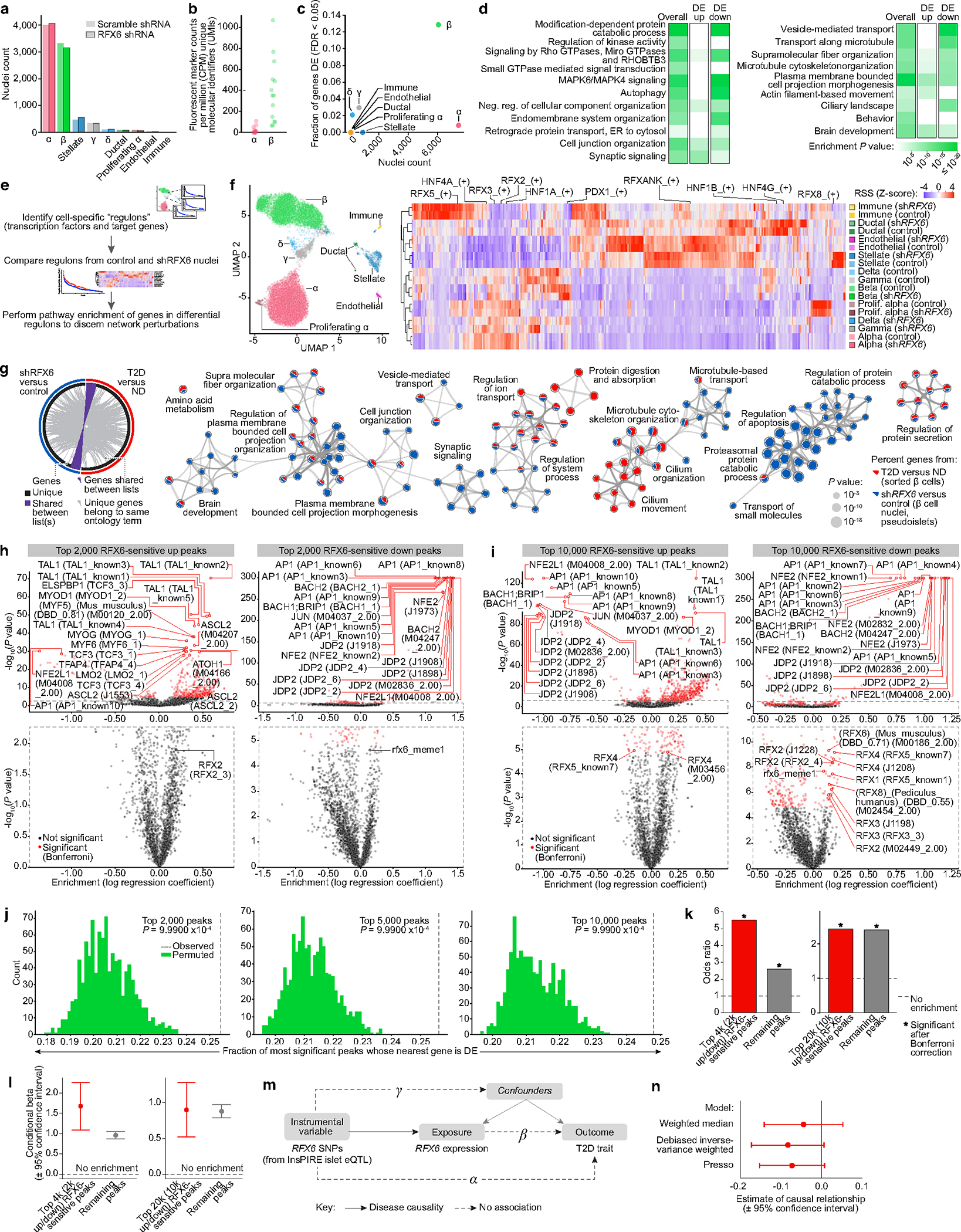
(a) Post-QC nuclei counts from control and shRFX6 pseudoislets. (b) Abundance of fluorescent marker gene expression (mCherry/mKate2) in α and β cell nuclei. Reporter expression was much higher in β cell nuclei than in α cell nuclei, consistent with the previously observed preferential adenoviral targeting of β relative to α cells26. (c) Proportion of differentially expressed (DE) genes per cell type. Nuclear RFX6 was not among those reduced in β cell nuclei, consistent with shRNA silencing occurring in the cytoplasm. (d) Pathway enrichment (g:Profiler75, two-tailed hypergeometric test) for DE genes (FDR < 0.01); second two columns separate genes up- or downregulated in shRFX6. (e-f) To identify gene regulatory networks driving cell states, we employed Single-Cell rEgulatory Network Inference and Clustering (SCENIC) and discovered cell type specific regulons governing cell identity. (f) UMAP clustering of nuclei (RNA component) by SCENIC using regulon activity (left) and average regulon activity across cell types (right). See also Fig. 5d. (g) To investigate overlap in differentially expressed genes between shRFX6 β cell nuclei and sorted T2D β cells, we compared the top 1,000 most significant DE genes in shRFX6 vs. control β cell nuclei (blue) and T2D vs. ND sorted β cells (red). Circos plot illustrates commonality at the level of gene IDs (purple) or ontology term enrichment (grey; p < 0.01). Metascape network analsyis69 displays a subset of enriched terms, where edges denote term similarity and node colors represent contribution of each gene list. Common pathway enrichment related to microtubule cytoskeleton organization, ion transport, and regulation of protein secretion. (h-i) Motif enrichment for top 2,000 (h) or 10,000 (i) RFX6-sensitive up- and downregulated ATAC peaks in shRFX6 β cell nuclei. Motifs with highest significance are labeled in top panels; significant RFX motifs (or the single RFX motif closest to significance, in the case that no RFX motifs reach significance) are labeled in bottom panels. (j) Enrichment of top RFX6-sensitive up- and downregulated ATAC peaks (n = 2,000, 5,000, or 10,000) in shRFX6 β cell nuclei near shRFX6 β cell differentially expressed genes. (k-l) Single value odds ratio of T2D GWAS enrichment (k) and model estimate from conditional analysis (l) of top 2,000 or 10,000 RFX6-sensitive peaks; see Methods for details. Nominal P-values for panel k, left to right: 0.00336, 5.53e-28, 0.0194, 9.28e-22. (m) Model of Mendelian randomization (MR) depicting the relationship of an instrumental variable (here, RFX6 SNPs) related to exposure (RFX6 expression) and outcome (T2D). Dotted lines denote assumptions of no association while solid and dashed lines indicate disease causality. (n) MR analyses using the European ancestry cohort from Vujkovic et al. meta-analysis9 (n = 1,114,458 independent samples) resulted in point estimates that were directionally consistent with the findings for the UK Biobank (see Fig. 5k). The heterogeneity across contributing studies (for example Million Veterans Program data reflect an older T2D case set, more comorbidities, and a strong male bias compared to UK Biobank) likely reduces our power. We leveraged several MR approaches to determine whether results were robust to their varying assumptions. Debiased inverse variant weighted method (robust to weak instrument bias): causal effect = −0.084, P = 0.065; Pleiotropy RESidual Sum and Outlier (PRESSO) method (removes outlier instrumental variable effects): causal effect = −0.073, P = 0.251; weighted median method (robust when up to 50% of instrumental variables are invalid): causal effect = −0.045, P = 0.372. Results from MR-Egger are not shown due to strong weak instrument bias (I2_GX = 0). Error bars in panels l and n represent 95% confidence intervals.
Extended Data Fig. 10 |. (related to Fig. 5). Summary schematic of key molecular alterations in short-duration T2D cohort and of the convergence of genetic risk on an RFX6-mediated gene regulatory network.
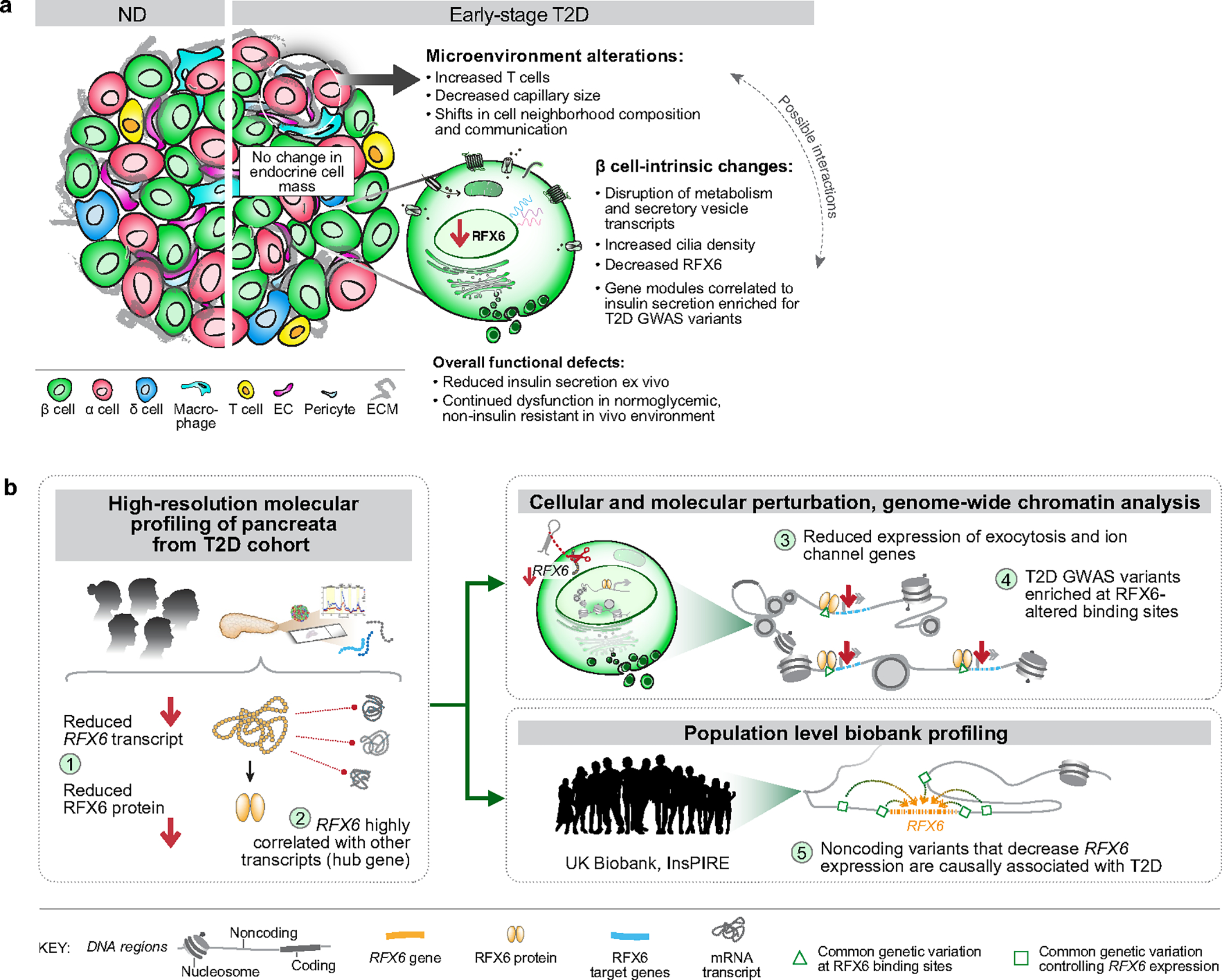
(a) Major β cell-intrinsic and islet microenvironment alterations that define islet dysfunction in early T2D. Observations from transcriptomic and histologic studies revealed no change to endocrine cell composition but evidence of dysregulated β cell processes and modest changes to intraislet vascular and immune cell populations. Insulin secretion was reduced and persisted in a nondiabetic environment; glucagon hypersecretion was not observed122. (b) After identifying RFX6 as a candidate disease-associated gene through unbiased analysis of a small cohort (left panel; labels 1–2), we used two approaches for validation: first, molecular perturbation of RFX6 in β cells of primary human pseudoislets allowed functional, transcriptomic and epigenomic analyses (top right panel; labels 3–4) and second, integration of UK Biobank data allowed population-scale genetic relationship to be examined (bottom right panel; label 5). Reduction of RFX6 led to reduced insulin secretion defined by transcriptional dysregulation of vesicle trafficking, exocytosis, and ion transport pathways that was mediated by genome-wide chromatin architectural changes overlapping with T2D GWAS variants. Furthermore, Mendelian randomization analysis revealed that reduced islet RFX6 expression is causally associated with T2D.
Supplementary Material
Acknowledgements
The authors thank the organ donors and their families for their invaluable donations and the International Institute for Advancement of Medicine (IIAM), Organ Procurement Organizations, National Disease Research Exchange (NDRI), and the Alberta Diabetes Institute IsletCore together with the Human Organ Procurement and Exchange (HOPE) program and Trillium Gift of Life Network (TGLN) for their partnership in studies of human pancreatic tissue for research. We thank J. Hughes, J. H. Jo, S. Kim, Y. Hang, J. Almaça, R. Stein and R. Haliyur for their valuable scientific insight regarding experimental design and methods. This study used human pancreatic islets that were provided by the NIDDK-funded Integrated Islet Distribution Program at the City of Hope (DK098085). This work was supported by the Human Islet Research Network (RRID:SCR_014393), the Human Pancreas Analysis Program (RRID:SCR_016202), DK106755, DK123716, DK123743, DK120456, DK104211, DK108120, DK104218, DK112232, DK112217, DK117960, DK126185, DK117147, HL142302, DK127084, HL163262, DK129469, DK135017, EY032442, T32GM007347, F30DK118830, DK020593 (Vanderbilt Diabetes Research and Training Center), The Leona M. and Harry B. Helmsley Charitable Trust, JDRF, Doris Duke Charitable Foundation, and the Department of Veterans Affairs (BX000666). Cell sorting was performed in the Vanderbilt Flow Cytometry Shared Resource (P30 CA068485, DK058404) and whole-slide imaging was performed in the Islet and Pancreas Analysis Core of the Vanderbilt DRTC (DK020593). Authors A.C.P. and M.B. are principal investigators and D.C.S. and C.D. are co-investigators under the Human Pancreas Analysis Program. S.C.J.P. is a principal investigator in the Accelerating Medicines Partnership for Common Metabolic Diseases (AMP-CMD).
The HPAP Consortium
Marcela Brissova2, Chunhua Dai2, Alvin C. Powers1,2,12 & Diane C. Saunders2
A full list of members and their affiliations appears in the Supplementary Information.
Footnotes
Competing interests The authors declare no competing interests.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Code availability
Packages and algorithms used for transcriptomic and cell neighbourhood analyses are provided in the Github repositories https://github.com/ParkerLab and https://github.com/liu-bioinfo-lab.
Additional information
Peer review information Nature thanks Joanna Howson, Peter Tessarz and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Reprints and permissions information is available at http://www.nature.com/reprints.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-023-06693-2.
Data availability
Raw genotyping, bulk RNA-seq and single-nucleus multiome data are available via the European Genome-Phenome Archive (EGA; RRID SCR_004944) as study EGAS00001006273. Raw imaging datasets are available from the authors upon request. Processed imaging data is available via Pancreatlas (RRID SCR_018567; https://pancreatlas.org) and at https://zenodo.org/doi/10.5281/zenodo.8125025; weighted gene co-expression network analysis is available at https://theparkerlab.shinyapps.io/Islet-RNAseq-WGCNA/; and processed pseudoislet multiome data are available at https://theparkerlab.med.umich.edu/data/public/cellbrowser/?ds=Pseudoislet10XMultiome. In addition, intermediate processed results (expression matrices and others) are available via Zenodo (https://zenodo.org/doi/10.5281/zenodo.6515986). All datasets are linked from http://theparkerlab.org/manuscripts/2021_islet-rfx6/. Source data are provided with this paper.
References
- 1.Kahn SE, Hull RL & Utzschneider KM Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444, 840–846 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Halban PA et al. β-cell failure in type 2 diabetes: postulated mechanisms and prospects for prevention and treatment. Diabetes Care 37, 1751–1758 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mahajan A et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 50, 1505–1513 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rai V et al. Single-cell ATAC-seq in human pancreatic islets and deep learning upscaling of rare cells reveals cell-specific type 2 diabetes regulatory signatures. Mol. Metab. 32, 109–121 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiou J et al. Single-cell chromatin accessibility identifies pancreatic islet cell type- and state-specific regulatory programs of diabetes risk. Nat. Genet. 53, 455–466 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahlqvist E, Prasad RB & Groop L Subtypes of type 2 diabetes determined from clinical parameters. Diabetes 69, 2086–2093 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Redondo MJ et al. The clinical consequences of heterogeneity within and between different diabetes types. Diabetologia 63, 2040–2048 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weitz J, Menegaz D & Caicedo A Deciphering the complex communication networks that orchestrate pancreatic islet function. Diabetes 70, 17–26 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vujkovic M et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat. Genet. 52, 680–691 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mahajan A et al. Multi-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation. Nat. Genet. 54, 560–572 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parker SCJ et al. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc. Natl Acad. Sci. USA 110, 17921–17926 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trynka G et al. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat. Genet. 45, 124–130 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pasquali L et al. Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat. Genet. 46, 136–43 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walker JT, Saunders DC, Brissova M & Powers AC The human islet: mini-organ with mega-impact. Endocr. Rev. 42, bnab010 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brissova M et al. Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J. Histochem. Cytochem. 53, 1087–1097 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Dai C et al. Stress-impaired transcription factor expression and insulin secretion in transplanted human islets. J. Clin. Invest. 126, 1857–1870 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wigger L et al. Multi-omics profiling of living human pancreatic islet donors reveals heterogeneous beta cell trajectories towards type 2 diabetes. Nat. Metab. 3, 1017–1031 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Camunas-Soler J et al. Patch-seq links single-cell transcriptomes to human islet dysfunction in diabetes. Cell Metab. 31, 1017–1031.e4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shapira SN, Naji A, Atkinson MA, Powers AC & Kaestner KH Understanding islet dysfunction in type 2 diabetes through multidimensional pancreatic phenotyping: The Human Pancreas Analysis Program. Cell Metab. 34, 1906–1913 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Albrechtsen NJW et al. The liver–α-cell axis and type 2 diabetes. Endocr. Rev. 40, 1353–1366 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Wu M et al. Single-cell analysis of the human pancreas in type 2 diabetes using multi-spectral imaging mass cytometry. Cell Rep. 37, 109919 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dam TJ Pvan et al. CiliaCarta: an integrated and validated compendium of ciliary genes. PLoS ONE 14, e0216705 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith SB et al. Rfx6 directs islet formation and insulin production in mice and humans. Nature 463, 775–780 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patel KA et al. Heterozygous RFX6 protein truncating variants are associated with MODY with reduced penetrance. Nat. Commun. 8, 888 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Varshney A et al. Genetic regulatory signatures underlying islet gene expression and type 2 diabetes. Proc. Natl Acad. Sci. USA 114, 2301–2306 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walker JT et al. Integrated human pseudoislet system and microfluidic platform demonstrates differences in G-protein-coupled-receptor signaling in islet cells. JCI Insight 5, e137017 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Viñuela A et al. Genetic variant effects on gene expression in human pancreatic islets and their implications for T2D. Nat. Commun. 11, 4912 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kahn SE, Zraika S, Utzschneider KM & Hull RL The beta cell lesion in type 2 diabetes: there has to be a primary functional abnormality. Diabetologia 52, 1003–1012 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meier JJ & Bonadonna RC Role of reduced β-cell mass versus impaired β-cell function in the pathogenesis of type 2 diabetes. Diabetes Care 36, S113–S119 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cohrs CM et al. Dysfunction of persisting β cells is a key feature of early type 2 diabetes pathogenesis. Cell Rep. 31, 107469 (2020). [DOI] [PubMed] [Google Scholar]
- 31.McCarthy MI Painting a new picture of personalised medicine for diabetes. Diabetologia 60, 793–799 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chandra V et al. RFX6 regulates insulin secretion by modulating Ca2+ homeostasis in human β cells. Cell Rep. 9, 2206–2218 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Piccand J et al. Rfx6 maintains the functional identity of adult pancreatic β cells. Cell Rep. 9, 2219–2232 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choksi SP, Lauter G, Swoboda P & Roy S Switching on cilia: transcriptional networks regulating ciliogenesis. Development 141, 1427–1441 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Piasecki BP, Burghoorn J & Swoboda P Regulatory factor X (RFX)-mediated transcriptional rewiring of ciliary genes in animals. Proc. Natl Acad. Sci. USA 107, 12969–12974 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kurki MI et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 613, 508–518 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iotchkova V et al. GARFIELD classifies disease-relevant genomic features through integration of functional annotations with association signals. Nat. Genet. 51, 343–353 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gloyn AL et al. Every islet matters: improving the impact of human islet research. Nat. Metab. 4, 970–977 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Balamurugan AN, Chang Y, Fung JJ, Trucco M & Bottino R Flexible management of enzymatic digestion improves human islet isolation outcome from sub-optimal donor pancreata. Am. J. Transplant. 3, 1135–1142 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Dai C et al. Age-dependent human β cell proliferation induced by glucagon-like peptide 1 and calcineurin signaling. J. Clin. Invest. 127, 3835–3844 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brissova M et al. α cell function and gene expression are compromised in type 1 diabetes. Cell Rep. 22, 2667–2676 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brissova M et al. Islet microenvironment, modulated by vascular endothelial growth factor-A signaling, promotes β cell regeneration. Cell Metab. 19, 498–511 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brissova M et al. The Integrated Islet Distribution Program answers the call for improved human islet phenotyping and reporting of human islet characteristics in research articles. Diabetologia 62, 1312–1314 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kayton NS et al. Human islet preparations distributed for research exhibit a variety of insulin-secretory profiles. Am. J. Physiol. Endocrinol. Metab. 308, E592–E602 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fitzmaurice GM, Laird NM & Ware JH Applied Longitudinal Analysis (Wiley, 2011). [Google Scholar]
- 46.Shultz LD et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2Rγnull mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 174, 6477–6489 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Dai C et al. Tacrolimus- and sirolimus-induced human β cell dysfunction is reversible and preventable. JCI Insight 5, e130770 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dorrell C et al. Transcriptomes of the major human pancreatic cell types. Diabetologia 54, 2832 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saunders DC et al. Ectonucleoside triphosphate diphosphohydrolase-3 antibody targets adult human pancreatic β cells for in vitro and in vivo analysis. Cell Metab. 29, 745–754.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dorrell C et al. Human islets contain four distinct subtypes of β cells. Nat. Commun. 7, 11756 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haliyur R et al. Human islets expressing HNF1A variant have defective β cell transcriptional regulatory networks. J. Clin. Invest. 129, 246–251 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marzban L, Park K & Verchere CB Islet amyloid polypeptide and type 2 diabetes. Exp. Gerontol. 38, 347–351 (2003). [DOI] [PubMed] [Google Scholar]
- 53.Westermark P, Andersson A & Westermark GT Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol. Rev. 91, 795–826 (2011). [DOI] [PubMed] [Google Scholar]
- 54.Hart NJ et al. Cystic fibrosis–related diabetes is caused by islet loss and inflammation. JCI Insight 3, e98240 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Noguchi GM & Huising MO Integrating the inputs that shape pancreatic islet hormone release. Nat. Metab. 1, 1189–1201 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Black S et al. CODEX multiplexed tissue imaging with DNA-conjugated antibodies. Nat. Protoc. 16, 3802–3835 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Blondel VD, Guillaume J-L, Lambiotte R & Lefebvre E Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, P10008 (2008). [Google Scholar]
- 58.Luhn HP The automatic creation of literature abstracts. IBM J. Res. Dev. 2, 159–165 (1958). [Google Scholar]
- 59.Schürch CM et al. Coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive front. Cell 182, 1341–1359.e19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liao Y, Smyth GK & Shi W featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 62.Hartley SW & Mullikin JC QoRTs: a comprehensive toolset for quality control and data processing of RNA-Seq experiments. BMC Bioinformatics 16, 224 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang L et al. Measure transcript integrity using RNA-seq data. BMC Bioinformatics 17, 58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Risso D, Ngai J, Speed TP & Dudoit S Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 32, 896–902 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee C, Patil S & Sartor MA RNA-Enrich: a cut-off free functional enrichment testing method for RNA-seq with improved detection power. Bioinformatics 32, 1100–1102 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Supek F, Bošnjak M, Škunca N & Šmuc T REVIGO summarizes and visualizes long lists of Gene Ontology terms. PLoS ONE 6, e21800 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shannon P et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou Y et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Langfelder P & Horvath S WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saeedi P et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 157, 107843 (2019). [DOI] [PubMed] [Google Scholar]
- 72.Ritchie ME et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Naba A et al. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell Proteomics 11, M111.014647 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Breuer K et al. InnateDB: systems biology of innate immunity and beyond—recent updates and continuing curation. Nucleic Acids Res. 41, D1228–D1233 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kolberg L, Raudvere U, Kuzmin I, Vilo J & Peterson H gprofiler2–an R package for gene list functional enrichment analysis and namespace conversion toolset g:Profiler. F1000research 9, ELIXIR–709 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen J et al. The trans-ancestral genomic architecture of glycemic traits. Nat. Genet. 53, 840–860 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bailey TL et al. MEME Suite: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Weirauch MT et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Das S et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Loh P-R et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 48, 1443–1448 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Auton A et al. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li H & Durbin R Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Orchard P, Kyono Y, Hensley J, Kitzman JO & Parker SCJ Quantification, dynamic visualization, and validation of bias in ATAC-seq data with ataqv. Cell Syst. 10, 298–306.e4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lun ATL et al. EmptyDrops: distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biol. 20, 63 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kang HM et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol. 36, 89–94 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yang S et al. Decontamination of ambient RNA in single-cell RNA-seq with DecontX. Genome Biol. 21, 57 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.R Core Team. R: A Language and Environment for Statistical Computing. http://www.R-project.org/ (R Foundation for Statistical Computing, 2020). [Google Scholar]
- 89.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stuart T et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hao Y et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Thibodeau A et al. AMULET: a novel read count-based method for effective multiplet detection from single nucleus ATAC-seq data. Genome Biol. 22, 252 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Speir ML et al. UCSC Cell Browser: visualize your single-cell data. Bioinformatics 37, 4578–4580 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sande B Vde et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 15, 2247–2276 (2020). [DOI] [PubMed] [Google Scholar]
- 95.Quinlan AR BEDTools: the Swiss-army tool for genome feature analysis. Curr. Protoc. Bioinform. 47, 11.12.1–11.12.34 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang Y et al. Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, R137–R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kent WJ, Zweig AS, Barber G, Hinrichs AS & Karolchik D BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Grant CE, Bailey TL & Noble WS FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kheradpour P & Kellis M Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic Acids Res. 42, 2976–2987 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jolma A et al. DNA-binding specificities of human transcription factors. Cell 152, 327–339 (2013). [DOI] [PubMed] [Google Scholar]
- 101.Chinwalla AT et al. Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562 (2002). [DOI] [PubMed] [Google Scholar]
- 102.Bailey TL & Elkan C Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2, 28–36 (1994). [PubMed] [Google Scholar]
- 103.Bailey TL DREME: motif discovery in transcription factor ChIP–seq data. Bioinformatics 27, 1653–1659 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bailey TL, Johnson J, Grant CE & Noble WS The MEME suite. Nucleic Acids Res. 43, W39–W49 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bowden J, Smith GD & Burgess S Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 44, 512–525 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bowden J, Smith GD, Haycock PC & Burgess S Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 40, 304–314 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ye T, Shao J & Kang H Debiased inverse-variance weighted estimator in two-sample summary-data Mendelian randomization. Ann. Stat. 49, 2079–2100 (2021). [Google Scholar]
- 108.Verbanck M, Chen C-Y, Neale B & Do R Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 50, 693 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yavorska OO & Burgess S MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int. J. Epidemiol. 46, 1734–1739 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sudlow C et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.McCarthy S et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Loh P-R et al. Efficient Bayesian mixed model analysis increases association power in large cohorts. Nat. Genet. 47, 284–290 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bonner-Weir S & O’Brien TD Islets in type 2 diabetes: in honor of Dr. Robert C. Turner. Diabetes 57, 2899–2904 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sakuraba H et al. Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese type II diabetic patients. Diabetologia 45, 85–96 (2002). [DOI] [PubMed] [Google Scholar]
- 115.Butler AE et al. β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes 52, 102–110 (2003). [DOI] [PubMed] [Google Scholar]
- 116.Rahier J, Guiot Y, Goebbels RM, Sempoux C & Henquin JC Pancreatic β-cell mass in European subjects with type 2 diabetes. Diabetes Obes. Metab. 10, 32–42 (2008). [DOI] [PubMed] [Google Scholar]
- 117.Talchai C, Xuan S, Lin HV, Sussel L & Accili D Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 150, 1223–1234 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Masters SL et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat. Immunol. 11, 897–904 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Westwell-Roper CY, Ehses JA & Verchere CB Resident macrophages mediate islet amyloid polypeptide–induced islet IL-1β production and β-cell dysfunction. Diabetes 63, 1698–1711 (2014). [DOI] [PubMed] [Google Scholar]
- 120.Nair G & Hebrok M Islet formation in mice and men: lessons for the generation of functional insulin-producing β-cells from human pluripotent stem cells. Curr. Opin. Genet. Dev. 32, 171–180 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Arrojo e Drigo R et al. New insights into the architecture of the islet of Langerhans: a focused cross-species assessment. Diabetologia 58, 2218–2228 (2015). [DOI] [PubMed] [Google Scholar]
- 122.Unger RH & Cherrington AD Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J. Clin. Invest. 122, 4–12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw genotyping, bulk RNA-seq and single-nucleus multiome data are available via the European Genome-Phenome Archive (EGA; RRID SCR_004944) as study EGAS00001006273. Raw imaging datasets are available from the authors upon request. Processed imaging data is available via Pancreatlas (RRID SCR_018567; https://pancreatlas.org) and at https://zenodo.org/doi/10.5281/zenodo.8125025; weighted gene co-expression network analysis is available at https://theparkerlab.shinyapps.io/Islet-RNAseq-WGCNA/; and processed pseudoislet multiome data are available at https://theparkerlab.med.umich.edu/data/public/cellbrowser/?ds=Pseudoislet10XMultiome. In addition, intermediate processed results (expression matrices and others) are available via Zenodo (https://zenodo.org/doi/10.5281/zenodo.6515986). All datasets are linked from http://theparkerlab.org/manuscripts/2021_islet-rfx6/. Source data are provided with this paper.