

Abstract
γ-Secretase (GS) is a multi-subunit membrane-embedded aspartyl protease that cleaves more than 80 integral membrane proteins, including the amyloid precursor protein (APP) to produce the amyloid-β (Aβ) peptide. Oligomerization and aggregation of the 42-amino acid length Aβ isoform in the brain has been associated with the development and progression of Alzheimer’s disease (AD). Based on recent experimental structural studies and using multiscale computational modeling approaches, the conformational states and protein-membrane interactions of the GS complex embedded in six homogeneous and six heterogeneous lipid bilayers were characterized. In order to identify potential lipid and cholesterol binding sites, GS regions with high lipid/cholesterol occupancy values were analyzed using atomistic and coarse-grained simulations. Long lipid residence times were observed to be correlated with a large number of hydrogen bonds between the charged headgroups and key GS amino acids. This observation provides a plausible explanation for the inhibition of GS by charged lipids observed in previous experimental studies. Computed lateral pressure profiles suggest that higher trans-membrane pressures favor active state conformations of the catalytic subunit. A probable mechanism for the regulation of the local stress response in cholesterol-rich multicomponent lipid bilayers is identified. Finally, it is demonstrated that interactions between the nicastrin extracellular domain and lipid headgroups leads to a compact structural conformation of the GS complex. Overall, this study provides valuable insight into the effect of bilayer lipid composition on the GS structural ensemble and its function.
Table of contents
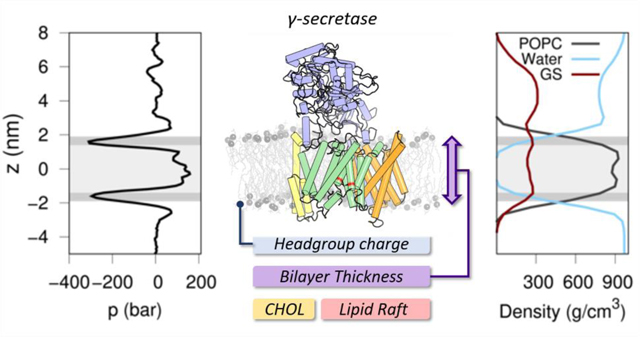
Insight into the effect of bilayer lipid composition on the GS structural ensemble and function using multiscale computational modeling approaches.
Introduction
γ-secretase (GS) is an intramembrane aspartyl protease involved in the ultimate step in the cleavage of amyloid precursor protein (APP) to generate amyloid-β (Aβ) peptides.1 In the penultimate step in this process, APP is cleaved by the membrane-associated β-site APP-cleaving enzyme 1 (BACE1, β-secretase) to produce an intramembrane 99-residue C-terminal fragment (C99).2 The intramembrane cleavage of the C99 fragment by GS leads to the formation of the APP intracellular domain (AICD), which mediates nuclear signaling, and Aβ peptides of varying length.3 Normal variation in the cleavage of C99 leads to the production of a 40-amino acid length isoform of Aβ (Aβ40). However, aberrant processing of C99 by GS can increase the production of the toxic and more amyloidogenic 42-amino acid species (Aβ42).4 Oligomerization of the Aβ42 isoform in the brain has been proposed to be the causative agent of Alzheimer’s disease (AD).5
GS is a multi-subunit integral membrane protein complex consisting of four components: presenilin 1 (PS1), presenilin enhancer 2 (PEN-2), anterior pharynx-defective 1A (APH-1A) and nicastrin (NCT) (Figure 1A).6 PS1 is the catalytic component comprised of nine transmembrane helices (TM1–9) in which the two catalytic aspartate residues are located at TM6 (D257) and TM7 (D385).7 GS maturation involves the autoproteolytic processing of the intracellular loop connecting TM6 and TM7 of this subunit leading to the formation of N-terminal (NTF, TM1–6) and C-terminal (CTF, TM7–9) fragments.8 These fragments are stabilized by PEN-2 and APH-1A, which have key structural roles during the maturation and assembly of the complex.9 NCT is a type-1 transmembrane glycoprotein formed by a single TM helix and a large extracellular domain (ECD). It has been proposed that NCT plays a critical role in the recruitment of GS substrates.10 To date, only six cryo-electron microscopy structures of the complete human GS complex have been deposited in the Protein Data Bank (PDB): 4UI2, 5A63, 5FN2, 5FN3, 5FN4 and 5FN5.11–14
Figure 1.

Overview of the GS structure. (A) Depiction of γ-secretase structure derived from 5FN2 PDB structure colored by its four components: NCT (violet), PS1 (green), APH-1A (orange) and PEN-2 (yellow), highlighting the catalytic aspartic residues (red). (B) Hydrophobic (yellow) and hydrophilic (teal) residue distribution in the GS surface. (C) Depiction of the hydropathy index distribution ranging from the most (blue) to the least hydrophilic (red) regions. (D) Distribution of the negatively (red) and positively (blue) charged amino acids in the GS.
The production of different Aβ isoforms by GS is regulated by structural and external factors that alter the protease activity of the enzyme.15 Some of the factors that regulate GS activity are (1) autosomal-dominant mutations in the substrate and/or the PS1 component, (2) external agents that inhibit or modulate the proteolytic cleavage, and (3) variations in the bilayer lipid composition. In the first, certain congenital mutations in APP and PS1 have been found to increase the Aβ42/Aβ40 ratio, causing early-onset familial Alzheimer’s disease (FAD).16 GS inhibitors (GSIs) and modulators (GSMs) seek to regulate the Aβ42 production caused by these FAD mutations through binding to the PS1 active site or to an allosteric site, respectively.17 These two factors regulate cleavage processing during the active state of the GS complex; however, this activation is also directly regulated by the lipid environment in which this enzyme is found.18 Since cleavage of substrates by GS is carried out in the middle of the membrane, it is expected that the lipid composition will play an important role in the proteolytic activity.19
The GS complex was first observed in lipid raft domains in human brain derived reconstituted lipid membranes.20 Changes in the composition of the lipid rafts were associated with the expression of the PS1 gene (PSEN1).21 A lipidomic analysis showed that these membrane rafts are enriched with 11 different major lipid classes including sphyngolipids, cholesterol and phosphatidylcholine.22 Importantly, the lipid composition was consistent with the lipid characterization in mice and the brains of humans diagnosed with Alzheimer’s disease.23 The membrane lipid composition regulates the activity of GS and is essential to GS integrity and signaling in nerve cells.22,24,25
It is critical to investigate and characterize the importance of lipid composition on the conformational ensemble and stability of the GS complex in order to develop a complete understanding of the cleavage process and mechanism of activity of GS. In this study, we employed a multiscale methodology, combining coarse-grained (CG) and all-atom (AA) models, to perform molecular dynamics (MD) simulations and explore the dynamic structural ensemble of GS embedded in a variety of bilayer lipid compositions. A total of twelve different lipid membranes were constructed for this study, in which we evaluated the effect of (1) the headgroup charge, (2) the bilayer thickness, (3) the cholesterol concentration, and (4) the presence of three lipid mixtures mimicking membrane rafts on the structural conformational ensemble and the dynamic properties of GS. Detailed analysis was used to characterize the simulated GS structural ensemble and the surrounding lipid bilayer environment. Key lipid-protein interactions involving residues known to play a role in C99 substrate recognition and cleavage were identified, providing insight into the role of membrane and protein-lipid specific interactions in GS activation and inhibition. Overall, the simulation results support the validity of the simulation models and address a number of outstanding questions related to the role of membrane lipid composition in modulating the structure and activity of GS.
METHODS
Model preparation
The cryo-EM structure of GS resolved at 4.2 Å resolution (PDB ID: 5FN2) was employed as the initial 3D coordinate model to perform our multiscale MD simulations in a variety of lipid compositions.14 The missing side-chains of the structure were completed using WHAT IF server.26 The intracellular loop connecting TM6 and TM7 of the PS1 component (amino acids 264 to 377) was not modeled due to its extended length and the lack of available structural information to inform its structure. Modeling PS1 absent the loop allowed us to mimic the mature (auto-protolyzed) form of PS1 in the complex. Simulations employing a model of the complete mature and in-mature forms have been reviewed by Kepp’s group.27 The arrangement of our GS models with respect to a lipid membrane was obtained from the Orientation of Proteins in Membranes (OPM) server.28 The average hydropathy index of each amino acid was calculated with the hydropathy values reported by Kyte and Doolittle using a nine-residue window of the GS sequence.29
Molecular dynamics simulations
We used a multiscale computational modeling approach, combining all-atom and coarse-grained representations, to characterize the structural ensemble and conformational behavior of GS in twelve different lipid bilayers (see Table 1). For the CG models, we simulated GS in the (1) unprotonated and (2) protonated states of Asp385 residue.30
Table 1.
Membrane lipid compositions
| Homogeneous systems | MD replicas | |||
|---|---|---|---|---|
|
| ||||
| ID | Name | CG (1.5 μs) | AA (200 ns) | |
|
| ||||
| Unprotonated Asp385 | Protonated Asp385 | Unprotonated Asp385 | ||
|
| ||||
| POPC | 1-Palmitoyl-2-oleoyl-phosphatidylcholine | 20 | 20 | 1 |
| POPE | 1-Palmitoyl-2-oleoyl-phosphatidylethanolamine | 20 | 20 | 1 |
| POPA | 1-Palmitoyl-2-oleoyl-phosphatidic acid | 20 | 20 | 1 |
| DLPC | Dilauroyl-phosphatidylcholine | 20 | 20 | - |
| DPPC | Dipalmitoyl-phosphatidylcholine | 20 | 20 | - |
| DGPC | Dieicosenoyl-phosphatidylcholine | 20 | 20 | - |
|
| ||||
| Heterogeneous systems | MD replicas | |||
|
| ||||
| ID | Type (%) | CG (1.5 μs) | AA (200 ns) | |
|
| ||||
| Unprotonated Asp385 | Protonated Asp385 | Unprotonated Asp385 | ||
|
| ||||
| POPC(80):CHOL(20) | POPC(80) and CHOL(20) | 20 | 20 | - |
| POPC(60):CHOL(40) | POPC(60) and CHOL(40) | 20 | 20 | 2 |
| POPC(40):CHOL(60) | POPC(40) and CHOL(60) | 20 | 20 | - |
| Lipid Raft45 (Raft 1) |
DOPC(25), POPC(15), DOPE(10), POPI(10), POPS(5), LPPC(5), DOPS(3), DPSM(4), POPG(2), and CHOL(20) | 20 | 20 | 1 |
| Lipid Raft46 (Raft 2) |
DPPC(40), DUPC(30), and CHOL(30) | 20 | 20 | - |
| Lipid Raft23,47 (Raft 3) |
POPC(7), PRPE(6), DPSM(4), PGPC(4), PAPC(3), DPPC(3), PUPE(2), PUPC(2), PRPC(2), PRPS(2), PEPC(2), PNSM(1), PGPE (1), PGPS(1), PIPC(1), POPE(1), POPA(1), POSM(1), PVSM(1), and CHOL(55) | 20 | 20 | - |
Coarse-grained simulations.
The unprotonated and protonated Asp385 CG models were embedded in the different lipid membranes using the CHARMM-GUI Martini bilayer maker31 and the MARTINI v2.2 forcefield.32 The pressure was set at 1.0 bar with a semi-isotropic Berendsen coupling and the temperature was set to 315 K using V-rescale coupling. This temperature was used for all simulated systems since it corresponds to the highest phase transition temperature within the lipids used in this work.33 Each system was neutralized and, except for POPA system, brought to a concentration of 0.15 M with randomly placed sodium and chloride ions. We performed 20 NPT replicas of the protonated and unprotonated states of GS-membrane models for 1.5 μs, for a total of 60 μs of dynamics for each complex, using GROMACS 5.0.6 software.34 The MD simulations were analyzed using the inbuilt GROMACS tools, MDAnalysis libraries35,36 for python, g_lomepro37, GridMAT-MD38 and FATSLiM39 tools. An extra simulation of 1.5 μs was performed for each GS model using the customized version of GROMACS 4.5 to calculate the 3D-resolved local pressure of the last 500 ns (10,000 frames).40,41 Plots were generated with Gnuplot 5.042 and Matplotlib v2.043 and protein figures with PyMOL v0.944.
All-atom simulations.
Five atomistic GS models were constructed with the CHARMM-GUI membrane builder48 module in POPC, POPE, POPA, POPC(60):CHOL(40) and a lipid raft (Raft 1) bilayer compositions. The systems were energy minimized and equilibrated under standardized NVT and NPT conditions. Equilibration of each simulation was followed by 200 ns production run with a 2 fs time step. Temperature was set to 315 K using the Nosé–Hoover coupling thermostat algorithm. The pressure was set to 1.0 bar using the semi-isotropic Parrinello–Rahman barostat algorithm. The Lennard-Jones potential was set using a shift function between 0.9 and 1.2 nm, and the electrostatic interactions were calculated between 0 and 1.2 nm after which were calculated using the Particle Mesh Ewald (PME) approach. Neighbor lists were updated every 20 steps and bonds involving hydrogens were constrained using the Linear Constraint Solver (LINCS) algorithm.49 The simulations were performed using GROMACS 5.0.6 with the CHARMM36 force field50 and the flexible TIP3P water model. Each system was neutralized and, except for POPA system, brought to a concentration of 0.15 M with randomly placed sodium and chloride ions.
Results and discussion
The structure of a membrane protein, including its insertion and orientation in a lipid membrane, is dependent on the protein’s topology and amino acid distribution, as well as the lipid composition of the membrane in which it is embedded.28 The amino acid composition of a protein defines the way it interacts with the membrane lipids, altering its folding, thermodynamic stability, and biological activity.28,51,52 Key factors impacting protein structure and activity are the nature of the lipid headgroup, the membrane thickness, and the lipid mixture composition.53–55 This also occurs with GS, since different lipid environments have been shown to affect the biological activity of the PS1 component of the GS complex. Selkoe and coworkers reported that GS is fully active in a membrane composed solely of phosphatidylcholine (PC) lipids, while other homogeneous membranes reduce GS cleavage activity almost entirely.18 In addition, it has been demonstrated that high cholesterol concentrations and lipid raft membrane compositions enhance GS activity.23,56
To evaluate the influence of the lipid environment on the structural ensemble and dynamics of GS, we performed CG MD simulations of the initial GS complex (PDB ID: 5FN2) embedded in six homogeneous and six heterogeneous membranes, comprising 20 independent replicas each of 1.5 μs. In the first set of simulations, we evaluated the effect of (1) lipid headgroup charge and (2) lipid length in homogeneous lipid bilayers, followed by a second set of simulations of heterogeneous systems in which we analyzed the influence of (3) cholesterol concentration and (4) lipid raft mixtures (Figures S1-S7).
To develop a detailed analysis of how the membrane lipid composition influences the GS structural ensemble and dynamics, we evaluated the (1) lateral pressure profile (stress profile) of the membrane, (2) the transition between the active/inactive states of the PS1 component and (3) the general behavior of the lipids surrounding the GS complex. The local stress profile of a membrane is useful to describe alterations in protein activation or association due to pressure variations in the headgroup, interfacial region and/or chain of the lipids.40,57 Similar to other intramembrane proteases, the PS1 component of GS needs the catalytic aspartic acid residues in close proximity to form an intramolecular hydrogen-bond required for the formation of its active state.58 Alterations in the lateral pressure profile embedding the GS-membrane complex could interfere with the active/inactive transition of PS1. In a previous study, we defined inactive state (state 1) and active state (state 2) structural ensembles of the GS complex based on PS1 experimental conformations. Those structural ensembles were analyzed using two principal order parameters (1) the distance between its catalytic aspartates (ddAsp) and (2) the tilt angle distribution of PS1 TM6 (TTM6).30 In this work we have employed a similar approach to analyze the impact of the protonation state of Asp385 on the inactive (state 1) and active (state 2) structural ensembles. Finally, we characterized the protein-lipid interactions and the general behavior of the surrounding lipids with GS (e.g. lipid displacements, membrane thickness, and residence times).
GS topology defines its orientation in the lipid membrane
To understand the effect of the membrane lipid composition on the structure and dynamics of GS, it is first necessary to characterize the amino acid properties and distribution of the complex.53 Like most membrane proteins, the majority of surface exposed residues in the TM region of GS are hydrophobic, while the majority of hydrophilic residues are surface oriented in the extra-membrane domains (Figure 1B).29 Using the hydropathic profile proposed by Kyte and Doolittle, GS regions were defined so as to optimize the number of protein-membrane interactions (Figure 1C). Except for TM3 of APH-1A, all GS TMs have a high index of hydropathy at the center region of the helices (Figure S1). Charged amino acids located close to the bilayer-water interface are usually involved in protein stability and conformational state transitions due to their interaction with surrounding water molecules and lipid headgroups.59 A large number of charged amino acids can be found in these GS regions, where a majority of positively charged amino acids (Lys and Arg) in the TMs are exposed to the cytosol rather than the extracellular space (“positive-inside” rule) (Figure 1D).60 This initial structural analysis was used to organize our observations of membrane and lipid-specific interactions with GS domains and residues.
Lipid headgroup charge modulates the structure and activity of GS
We built three different protein-membrane systems, each consisting of GS in a lipid bilayer composed of (1) zwitterion phosphatidylcholine (PC) lipids, (2) hydrogen-bond donor phosphatidylethanolamine (PE) lipids, and (3) negatively charged phosphatidic acid (PA) lipids, leaving the acyl chains 1-palmitoyl-2-oleoyl (PO) constant. The local stress profile of our reference POPC bilayer system presents a homogeneous lateral pressure profile in the transmembrane region, with lower pressure values characteristic of the interfacial region (z ≈ 1.8 nm and z ≈ −1.8 nm) and slightly increased pressure values in the lipid acyl tail (1.5 > z > −1.5 nm) and headgroup regions (z ≈ 2.2 nm and z ≈ −2.2 nm) due to repulsive interactions (Figure 2A). Pressure fluctuations observed in the upper part of the lateral pressure plots (z > 2.0 nm) are derived from the collision of water molecules with the ECD of NCT. Figure 2B shows the probability distribution of our previously described order parameters: ddAsp distance and TTM6 angle obtain from our unprotonated and protonated Asp385 GS systems. As in our previous study, we found that the protonated Asp385 form facilitates the transition of PS1 to the active state (state 2) conformation (characterized by short ddAsp and proper TTM6 angles), while the unprotonated systems were found predominantly in the inactive state (state 1) conformation (long ddAsp and proper TTM6 angle). It is worth mentioning that the experimental GS structures were transformed to a CG resolution and minimized using backbone-restrains in order to compare our CG model simulations.
Figure 2.

Effect of the headgroup charge (POPC, POPE, and POPA) and membrane thickness (DLPC, DPPC, and DGPC) in the structural ensemble of 5FN2 derived CG models. (A) System density and lateral pressure (LP) profiles as a function of lipid composition. The dashed black line shows the POPC lateral pressure profile and the gray background depicts the headgroups (dark gray) and tails (light gray) of the lipids in the bilayer. (B) Distribution of our 20 replicas CG simulations projected onto the distances between the catalytic residues (D257 and D385) and the calculated TM6 tilt angles in the unprotonated and protonated states of Asp385. The colored scale on the right defines the relative populations. The black circles mark the locations of the experimental GS structures at CG resolution after backbone-restrained minimization (PDB IDs: 5A63, 4UIS, 5FN2, 5FN3, 5FN4 and 5FN5).12–14 (C) Representation of lipid displacement in the upper (UL) and lower (LL) leaflets of the lipid bilayer relative to GS (purple). The intensity of the color in both leaflets indicates the smaller (white) and greater (colored) displacement of the lipids in the three simulated membranes. The figure on the left depicts the perspective of the GS complex used for the graphs of lipid displacement and membrane thickness (C and D). Also shown are the proposed initial substrate-binding sites (SBS1 and SBS2). (D) Local membrane thickness analysis of DLPC, DPPC and DGPC lipid bilayers.
The local stress profile obtained for our POPE bilayer simulations showed a slight increment in the transmembrane pressure profile values with respect to our POPC system, in agreement with previously reported values from similar CG bilayer systems.48 Interestingly, the higher lateral pressure values favoured the active state (state 2) PS1 conformations in our CG models with unprotonated Asp385. In order to corroborate the impact of higher lateral pressures in our structural ensemble of GS systems, we performed 1.5 μs CG MD simulations for three model systems and replicas at higher pressures with the unprotonated Asp385 CG model embedded in a POPC bilayer (Figure S8). The resulting probability distribution of these simulations confirmed that an increased lateral pressure of the system promotes the active state (state 2) conformation. Despite the behavior of the high-pressure profile of the system with POPE lipids, no significant changes were observed in the protonated Asp385 CG models with respect to GS in the POPC bilayer.
The local stress profile in our POPA bilayer system showed higher interfacial pressure values in the upper leaflet, suggesting structural modifications in the lipid bilayer due to its interaction with the dynamic GS complex. Additionally, the decreased pressure in the lower leaflet headgroup region confirms the presence of attractive electrostatic interactions between the lipid headgroups and the positively charged amino acids located in the intracellular region of GS. The repulsive interactions between lipids and the GS complex in the upper leaflet and relatively attractive lipid-GS interactions in the lower leaflet are consistent with the small lipid displacement observed in the POPA bilayer relative to the POPC and POPE bilayers (Figure 2C). Nevertheless, we observed no differences in the structural ensemble of GS obtained with the protonated Asp385 embedded in a POPA bilayer compared to the equivalent system simulated in a POPC or POPE bilayer.
The CG simulations allowed us to obtain a general overview of the GS behavior when varying the headgroup charge of the bilayer. In order to obtain an atomistic description of the GS-lipid interactions and explore the origin of the low observed GS activity in the presence of POPE or POPA lipid bilayers,18 we performed 200 ns all-atom MD simulations (Figure 3) of the GS complex employing the CHARMM36 force field in three single component lipid bilayers (i.e., POPC, POPE and POPA). We evaluated the residence times of lipids located close to GS in order to identify those lipids in contact with the protein more than 70% of the time during the last 100 ns in our all-atom MD simulations. For those identified lipids, we further evaluated the spatial distribution and occupancies in upper and lower leaflets. Figure 3 (A and B) show occupancy plots for POPC, POPE and POPA lipids, located in the upper and lower leaflets, in contact with GS. A greater population of lipids was found in the lower leaflets due to non-bonded interactions of the headgroups (phosphate or phosphodiester groups) with the inner positive amino acids of the complex (Figure S9).54 These interactions impact the membrane thickness of the charged headgroup systems (Figure 3C). In addition, we found that none of the lipid headgroups interact with the catalytic aspartates, suggesting that the inhibitory mechanism of POPE and POPA does not require direct contact between the lipids and the catalytic residues as was previously proposed.61 However, the high POPA lipid occupancy values obtained in the lower leaflet in our all-atom simulations, together with the high lateral pressure values in the upper leaflet and the significant lipid displacement alterations, suggest an alternative mechanism in which GS activity is inhibited in a POPA lipid bilayer due to high PS1 structural restrictions, possibly affecting the substrate recruitment and/or entry into the GS active site.
Figure 3.

Atomistic simulations of GS embedded in charged lipid bilayers. (A) Occupancy plots of POPC, POPE and POPA lipids located in the upper and lower leaflets in contact with GS for more than 70% of the time during the last 100 ns of the all atom simulations. The colored scale on the right defines occupancy values of lipids with higher residence times during the last 100ns of simulation. (B) Three-dimensional density distribution of selected POPC lipids with high residence times around GS shown from forward (left) and rear (right) views. The substrate (SBS) and lipid binding sites (LBS) are highlighted in red and blue, respectively. (C) Time evolution of POPC, POPE and POPA membrane thickness (Top) and the minimum distance between the catalytic aspartates and the lipid headgroup (Bottom) through the last 100 ns of our all-atom MD simulations. (D) Depiction of POPE (purple) lipid interactions with PS1 (green) at LBS1 and LBS2. The dotted lines represent proper hydrogen bond distances.
The lipid residence time analysis of our all-atom MD simulations led to the identification of four key lipid binding sites (LBS) with high occupancy values consistent across the three GS-bilayer systems (Figure 3A and B). LBS1 and LBS2 were assigned to density distributions located respectively in the TM2-TM3 and TM2-TM6 interfaces of PS1 component (Figure 3D). The sites LBS3 and LBS4 were assigned to density distributions located on the internal side of the GS horseshoe and near the juxtamembrane helix of APH-1A (Figure S10). Interestingly, two distearoyl-phosphatidylcholine lipids were also found at these LBS in the 5A63 PDB structure of GS, which confirms the high lipid occupancies in these regions.13 In addition to the aforementioned sites, a high prevalence of lower leaflet lipids was observed in regions previously identified as initial substrate-binding sites.7,62 This observation suggests that displacement of the resident lipids is required in the process of ligand or substrate binding.
Hydrogen bond analysis of our all-atom MD simulations indicate that interactions between the lipid headgroups and the hydrophilic loop (HL1) that connects TM1 and TM2 are responsible for the longer lipid residence times observed at LBS1 and LBS2 (Figures S9 and S11). POPE lipids in our all-atom bilayer system showed the highest number of hydrogen bonds with HL1. The increase in membrane thickness facilitates the residence of POPE lipids in LBS1 and LBS2 (Figure 3D). Consequently, when comparing RMSF values of AA and CG simulations of GS in POPE and POPC lipid bilayers (Figures S4, S5 and S7) significant alterations in HL1 flexibility are observed. The region with the highest ΔRMSFPOPC-POPE values is related to hydrogen bond formation between POPE lipids and Tyr115. It is well known that tyrosine residues are suitable polar ligands for the interaction with phosphodiester groups or positively charged molecules,54 and Tyr115 could play an important role in the GS cleavage mechanism due to its close proximity to the active site.63,64 In addition, alterations in the HL1 movement could explain the experimentally observed POPE inhibition of GS activity.18
Bilayer thickness and degree and location of lipid unsaturation impacts the PS1 conformational ensemble
Selkoe and coworkers demonstrated that lipid membrane thickness affects the C99 cleavage pattern leading to the formation of different Aβ isoforms.18 The authors found that thinner membranes favor Aβ42 production, whereas thicker membranes favor the production of Aβ40. In a later study, the same research group65 confirmed that increasing the number of carbons in the lipid chain also increase GS activity, reducing the Aβ42/Aβ40 ratio. Despite the membrane composition and PS1 conformational studies, the mechanism by which the different Aβ isoforms are produced remains controversial.66 Herein, we focused this part of our study to identify the general behavior of three lipid types containing a phosphatidylcholine (PC) headgroup and different chain lengths: DLPC (12:0/12:0), DPPC (16:0/16:0) and DGPC (20:1/20:1). Figure 2 presents lateral pressure profiles, together with the PS1 structural ensemble, and the local membrane thickness analysis obtained for our CG simulated systems of GS embedded in DLPC (thinnest), DPPC and DGPC (thickest) lipid bilayers. Without considering the thickness distributions, the three systems show similar local stress profiles to that of POPC. The DPPC bilayer has the greatest similarities with POPC because both lipid acyl tails have the same length, differing only by the single unsaturation in the oleyl chain of POPC. Interestingly, the local membrane thickness analysis (Figure 2D) shows a thicker bilayer region close to the SBS1 of GS, in agreement with the lower leaflet density distribution observed in the POPC systems.
Hydrophobic mismatch between a protein and lipid membrane can induce conformational changes in the tilt and rotation of the helices in a way that enhances protein-lipid interactions.53 The hydrophobic mismatches observed in the simulated GS systems appear to modulate the conformational states explored by PS1. From these three systems, the thinner (DLPC) bilayer is observed to increase the population of the active state (state 2) whereas the thickest (DGPC) bilayer exhibits a broader distribution of PS1 conformations with ddAsp values greater than 1.1 nm. In general, GS with protonated Asp385 simulated in the three lipid bilayers were observed to have a smaller population of active state (state 2) conformations when compared to previous simulations of GS in monounsaturated lipid bilayer systems. Therefore, the absence of unsaturated chains in DLPC and DPPC lipids appears to alter the spatial arrangement and packing of the protein, hindering the transition between the inactive state (state 1) and active state (state 2) conformations.57 This result is in agreement with the study of Holmes et al. in which they demonstrated that the trans isomer of a monounsaturated fatty acid chain in phosphatidylcholine lipids increases the activity of the enzyme.65 Meanwhile, the protein conformational freedom allowed by the diunsaturated DGPC bilayer thickness results in a broad probability distribution for the inactive state (state 1), reducing the population of active state (state 2) conformations. Moreover, it has been observed that the position of the unsaturation is also crucial for GS activity, with the double bond in the omega-9 position of the oleic acid chain promoting the greatest activity.65 These results suggest that GS activity may not be entirely associated with membrane thickness alone but also depends critically on the degree and location of unsaturation. The lipid composition and site-specific protein-lipid interactions may also play a critical role. It is therefore critical to study the effect of thickness variation for those lipid mixtures in which GS has shown increased cleavage activity.18
Site-specific interaction of cholesterol with GS modulates the PS1 structural ensemble
Cholesterol (CHOL) is an essential component of lipid rafts. Its high concentration in the brains of patients suffering from AD has been related to enhanced GS activity.56,67 Osenkowski et. al. observed that Aβ production is directly proportional to the abundance of CHOL in various lipid membranes.18 The relative abundance of CHOL in lipid rafts has been observed to vary from 20 to 25% in the plasma membrane and from 40 to 55% in AD patient brain tissues and Chinese hamster ovary (CHO) cells.18,23,45
To evaluate the influence of CHOL concentration on the structural ensemble of GS, we conducted CG MD simulations of three POPC:CHOL systems with 20, 40, and 60% CHOL as a function of the Asp385 protonation state. Figure 4 shows the lateral pressure profiles, the probability distributions of ddAsp distance and TTM6 angle, and the local bilayer thickness analysis of the POPC:CHOL bilayer mixtures. As expected, increasing CHOL concentration results in an increase in (1) bilayer lateral pressure and (2) bilayer thickness.68–71 As mentioned earlier, higher lateral pressures promote the population of the PS1 active state (state 2), even in the unprotonated Asp385 CG models of POPC(40):CHOL(60) (Figure S12). This result suggests that lateral pressure at high cholesterol concentrations could facilitate the transition from an inactive state (state 1) to an active state (state 2) of GS. However, such high CHOL concentrations also reduce the flexibility of GS (Figures S4 and S5), which could hinder TM2 movement and blocking the substrate entry to the active site.62
Figure 4.

Changes in GS dynamics in POPC bilayers containing 20%, 40%, and 60% of cholesterol (CHOL). (A) Lateral pressure profiles as a function of cholesterol concentration. The dashed black line shows the POPC lateral pressure profile without CHOL and the gray background marks the location of the headgroups (dark gray) and tails (light gray) of the membrane. (B) Distribution of the 20 replicas CG simulations projected onto the distances between catalytic residues (Asp257 and Asp385) and the calculated TM6 tilt angles in the protonated state of Asp385. The colored scale defines the relative populations. The black circles depict the values obtained from experimental structures of GS at CG resolution after backbone-restrained minimization (PDB IDs: 5A63, 4UIS, 5FN2, 5FN3, 5FN4 and 5FN5). (C) Local membrane thickness analysis of the CG membranes with different POPC:CHOL mixtures. (D) Lipid occupancy plots for CHOL in the AA MD simulation located in the upper and lower leaflets of our POPC lipid bilayer systems. The colored scale defines the occupancy of the CHOL molecules with higher residence times. (Right) Structure of CHOL molecule interacting with NCT and APH-1A components. (E) Per amino acid occupancy contacts of CHOL with each GS subunit in our AA simulated systems. (F) Depiction of amino acids that achieved the greatest number of contacts with CHOL molecules, constituting the CHOL binding sites (CBS).
A recent experimental study reported that only 8% of CHOL molecules bind to the GS complex in a membrane with approximately 43% relative abundance of CHOL.45 To characterize CHOL binding sites in GS, we performed 200 ns all-atom MD simulations of GS in a POPC(60):CHOL(40) bilayer system (R1). The CHOL molecules with higher lifetimes (residence time > 70%) located close to GS during the last 100 ns of the all-atom MD simulations display seven potential CHOL binding sites (CBS) (Figure 4D). CHOL molecules have longer residence times in the upper leaflet binding sites than the other simulated lipids. Key residues involved in the GS-CHOL contacts were identified using per amino acid occupancy analysis of the all-atom simulation (Figure 4E). To corroborate the occupancy analysis results for the all-atom MD simulation, a second 200 ns simulation (R2) was performed with a different initial CHOL spatial distribution. These AA simulations were analyzed and compared with the CG simulations (Figure S13). In general, we observed high CHOL preference for amino acids located in TM2, TM3, TM5, TM6 and the PAL motif (TM8-TM9) of PS1; TM1 of NCT; TM3, TM6, TM7 and the juxtamembrane helix of APH-1A; and TM3 of PEN2 (Figure 4F). Table S1 of the supplementary material lists the amino acids that constitute the lipids and cholesterol binding sites. Interestingly, the cholesterol binding sites identified as CBS1, CBS3 and CBS5 involve the same amino acids as the lipid binding sites LBS1, LBS4 and SBS1. Moreover, the local membrane thickness of AA simulations shows a thicker region close to the CBS3 site, as observed in the DLPC, DPPC and DGPC CG simulated systems (Figure 4C). Nevertheless, the presence of a CHOL molecule in CBS3 was not observed in the all-atom MD simulation replica (R2), suggesting a low turnover rate with other lipids at this site. Similarly, low occupancy values in amino acids comprising CBS2 were also observed in our R2 atomistic simulation. However, increased CHOL lifetimes in the N-terminal regions of TM2 and TM6 suggest that this molecule prefers the TM interfaces rather than the interaction with the lipid-exposed surface of these TMs observed in the R1 simulation. Note that the separation between TM2 and TM6 facilitating interaction with CHOL could be related to the possible “gate-open” movement essential for the substrate entry mechanism. The CBS2 in our second replica (R2) agrees with the POPC binding site LBS2. The binding of CHOL to this site and to the CBS1 would explain the reduction of HL1 flexibility observed in these simulations (Figures S4 to S7). This suggests the existence of a specific site for CHOL binding that results in the modulation of Aβ production.72 Finally, the proximity of CBS4 and CBS7 to the proposed substrate binding sites (SBSs) suggests that these sites could intervene in the initial recruitment of C99 through the recognition of a bound CHOL molecule.73,74
To validate the observations drawn from simulations of the POPC(40):CHOL(60) CG models, we performed CG MD simulations of GS in three different lipid mixtures consistent with experimental and theoretical abundances in lipid rafts (see Figure 5 and Table 1).23,45,46 Raft 1 was constructed from the information derived from the lipidome associated with GS, reported by Ayciriex and coworkers.45 For the Raft 2 system, the composition of the lipid rafts studied by Risselada and Marrink with CG simulations was used.46 Finally, the lipidomic analysis performed by Di Paolo and coworkers on the tissue derived from the brain of an Alzheimer’s disease patient was used for the construction of Raft 3, following Dal Peraro and coworkers.23,47 The lateral pressure profile analysis obtained for Raft 1 (Figure 5A) results in a similar profile to that observed in a pure-POPC lipid bilayer, while Rafts 2 and 3 displayed alterations in headgroup and interfacial region pressures, while no significant pressure changes were observed in the transmembrane region. The latter suggests that the use of a more complex lipid mixture reduces the effect of transmembrane lateral pressures compared with the POPC:CHOL systems. In addition, Raft 1 (with protonated Asp385) exhibited ddAsp distance and TTM6 angle probability distributions similar to those observed in the POPC system with a uniform membrane thickness and a high flexibility of the PS1 TMs (Figure S14A).
Figure 5.

(A) Lateral pressure profiles of three different lipid raft systems. The dashed black line shows the POPC lateral pressure profile without CHOL and the gray background depicts the headgroups (dark gray) and tails (light gray) of the membrane. (B) Distribution of the 20 replicas CG simulations of Raft 1 projected onto the distances between the catalytic residues (Asp257 and Asp385) and the calculated TM6 tilt angles in the protonated state of Asp385. The colored scale defines the relative populations. The black circles depict the values obtained from experimental structures of GS at CG resolution after backbone-restrained minimization (PDB IDs: 5A63, 4UIS, 5FN2, 5FN3, 5FN4 and 5FN5). (C) Local membrane thickness analysis of Raft 1. (D) Distribution of major axis length of the GS complex embedded in the twelve different membrane systems from the CG replica simulations (see Table 1). The gray line indicates the GS major axis lengths employed to classify our GS structures in an intermediate (In) conformation, while the upper and lower white zones display GS lengths in extended (Ex) and compact (Co) forms, respectively.
NCT ECD adopts a compact conformation in the presence of lipid rafts
In our previous study, we characterized three unique conformational states of NCT ECD: compact, intermediate and extended in agreement with experimental single-particle EM results reported by Chávez-Gutiérrez and coworkers.30,75 As in the previous study, the major axis length of the GS structures was measured using the distance between the furthest amino acids located at the complex. Figure 5D shows the major axis length distribution of ten GS-membrane CG models with a gray shadow that divides the compact (< 10.3 nm), intermediate (10.3 to 11.3 nm) and extended (> 11.3 nm) conformations. In agreement with our previous study, we observed a dominant preference for the intermediate conformation. This analysis also showed that as the thickness of the membrane increases, the preference for a more extended GS form is preferred. However, Raft 1 and Raft 3 display a wide up/down movement of NCT ECD with a high population of structures close to the compact conformation. This proximity of the NCT ECD to the outer monolayer was also observed in the CG MD simulations of GS performed by Audagnotto et. al. in a lipid raft of similar composition to Raft 3.47 The latter behavior of GS embedded in lipid rafts may be associated with the bilayer thickness that facilitates a close interaction between NCT ECD and the lipid headgroups surrounding GS (Figure S14B). Further studies are needed to evaluate the impact of glycosylated NCT ECD in the distribution of compact, intermediate and extended GS conformational states.76,77
Conclusions
The interaction of a membrane protein with the surrounding lipid environment plays a critical role in defining the protein’s conformation and its orientation in the membrane. The principal characteristics of the membrane environment that play a role defining the membrane protein structure include the lipid headgroup type, membrane thickness, and the composition of the lipid mixture. In this study, we employed a multiscale computational modeling approach to characterize conformational states the GS protein complex as defined by its interaction with water, lipid and cholesterol molecules composing the bilayer environment. To gain insight into the GS ensemble in various lipid environments, we embedded the entire GS complex in six homogeneous and six heterogenous lipid bilayers. Homogenous systems were employed to understand the effect of the lipid headgroup charge and the acyl chain length embedding GS, while with heterogeneous membranes were employed to evaluate the influence of cholesterol concentration and lipid raft mixtures on the GS structural ensemble.
An initial structural analysis was performed to evaluate the environmental dependence of the GS structural ensemble and to further investigate protein-lipid interactions in a variety of lipid environments. The membranes containing charged lipid headgroups (i.e. POPE and POPA) displayed long lipid residence times due to a large number of hydrogen bonds between lipid headgroups and GS. Since these interactions mainly affect the flexibility of HL1 and TM2, we believe that the observed long lifetimes could be related to the lipid inhibition mechanism observed in previous experimental studies.18 Moreover, this observations could explain the low stability of GS induced by PS1 mutations with high polarity.78 Characterization of lipid residence times and interactions allowed us to identify 6 potential lipid binding sites, among which two were found in the possible initial substrate binding sites (SBS), one adjacent to the active site (LBS1) and another at the entry gateway (LBS2).
A bilayer thickness analysis confirmed that PS1 adapts its conformational state in response to the hydrophobic mismatch with the lipid bilayer; a greater population of active state (state 2) conformation was observed in the thinnest bilayer (DLPC) compared to the thickest one (DPPC). Moreover, we observed that the absence of unsaturated chains in DLPC and DPPC lipids alters the spatial arrangement and packing of the protein, hindering the transition between the active and inactive conformational states.
As expected, rising CHOL concentration in a POPC bilayer resulted in an increased lateral pressure and thickness. Interestingly, the high intramembrane pressure observed in the POPC(40):CHOL(60) system favors the active state (state 2) over the inactive state (state 1) conformations. Nevertheless, simulations of GS in the presence of lipid mixtures designed to model lipid rafts resulted in reduced lateral pressure induced by CHOL, thus favoring the transition between active and inactive states of the GS complex. Remarkably, interaction of NCT ECD with lipid headgroups in the lipid raft mixtures leads to more compact conformations of the GS complex, as observed in CG simulations of Dal Peraro and coworkers.47 Finally, we identified CHOL binding sites in GS located at the proposed substrate-binding sites (CBS3 and CBS7), nearby the active site (CBS1) and at the proposed entry gateway (CBS2). It is worth noting that the proximity of CBS4 and CBS7 to the proposed SBSs suggests that these sites could intervene in the initial recruitment of C99 through the recognition of a bound CHOL molecule.
In general, we observed that our CG models with protonated Asp385 showed a greater number of active state (state 2) conformations than the unprotonated systems, in agreement with the results of our previous study.30 Furthermore, lateral pressure profiles with high pressures in the transmembrane region were observed to facilitate the transition of the PS1 subunit to the active state (state 2) conformation. However, this high intramembrane pressure also affects the TMs flexibility, which could interfere with the substrate entry or trimming mechanisms. Overall, our study provides valuable insight into the effect of the bilayer lipid composition on the structural ensemble and activity of the GS complex.
Supplementary Material
Acknowledgements
R. Aguayo-Ortiz (No. 510728/288862) is very grateful to CONACyT for the fellowship granted. The authors gratefully acknowledge the support of the Programa de Apoyo a la Investigación y el Posgrado (PAIP 5000–9155) and Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (PAPIIT IA201118). LD is very grateful to CONACyT for financial support in project A1-S-8866. JES acknowledges the generous support of the National Institutes of Health (R01GM107703). LD acknowledges the support of the Dirección General de Cómputo y de Tecnologías de Información (LANCAD-UNAM-DGTIC-306).
Footnotes
Conflicts of interest
There are no conflicts to declare.
References
- 1.Zhang H, Ma Q, Zhang Y and Xu H, J. Neurochem, 2012, 120, 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL and Wong PC, Nat. Neurosci, 2001, 4, 233–234. [DOI] [PubMed] [Google Scholar]
- 3.Zhang X and Song W, Alzheimers. Res. Ther, 2013, 5, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haass C, Kaether C, Thinakaran G and Sisodia S, Cold Spring Harb. Perspect. Med, 2012, 1–26. [DOI] [PMC free article] [PubMed]
- 5.Jang H, Connelly L, Teran Arce F, Ramachandran S, Kagan BL, Lal R and Nussinov R, J. Chem. Theory Comput, 2013, 9, 822–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Strooper B, Neuron, 2003, 38, 9–12. [DOI] [PubMed] [Google Scholar]
- 7.Watanabe N, Takagi S, Tominaga A, Tomita T and Iwatsubo T, J. Biol. Chem, 2010, 285, 19738–19746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Strooper B, Iwatsubo T and Wolfe MS, Cold Spring Harb. Perspect. Med, 2012, 2, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, Nicoll M, Maxwell M, Hai B, Ellis MC, Parks AL, Xu W, Li J, Gurney M, Myers RL, Himes CS, Hiebsch R, Ruble C, Nye JS and Curtis D, Dev. Cell, 2002, 3, 85–97. [DOI] [PubMed] [Google Scholar]
- 10.Bolduc DM, Montagna DR, Gu Y, Selkoe DJ and Wolfe MS, Proc. Natl. Acad. Sci, 2016, 113, E509–E518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu P, Bai XC, Ma D, Xie T, Yan C, Sun L, Yang G, Zhao Y, Zhou R, Scheres SH and Shi Y, Nature, 2014, 512, 166–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun L, Zhao L, Yang G, Yan C, Zhou R, Zhou X, Xie T and Zhao Y, Proc. Natl. Acad. Sci. U. S. A, 2015, 112, 6003–6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bai X, Yan C, Yang G, Lu P, Ma D, Sun L, Zhou R, Scheres SHW and Shi Y, Nature, 2015, 525, 212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bai X-C, Rajendra E, Yang G, Shi Y and Scheres SH, Elife, 2015, 4, e11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aguayo-Ortiz R and Dominguez L, Isr. J. Chem, 2017, 57, 574–585. [Google Scholar]
- 16.Goate A and Hardy J, J. Neurochem, 2012, 120, 3–8. [DOI] [PubMed] [Google Scholar]
- 17.Wolfe MS, Neurochem J., 2012, 120, 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Osenkowski P, Ye W, Wang R, Wolfe MS and Selkoe DJ, J. Biol. Chem, 2008, 283, 22529–22540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hartmann T, Kuchenbecker J and Grimm MOW, J. Neurochem, 2007, 103, 159–170. [DOI] [PubMed] [Google Scholar]
- 20.Hur JY, Welander H, Behbahani H, Aoki M, Frånberg J, Winblad B, Frykman S and Tjernberg LO, FEBS J, 2008, 275, 1174–1187. [DOI] [PubMed] [Google Scholar]
- 21.Eckert GP and Müller WE, Biochem. Biophys. Res. Commun, 2009, 382, 673–677. [DOI] [PubMed] [Google Scholar]
- 22.Ayciriex S, Gerber H, Osuna GMG, Chami M, Stahlberg H, Shevchenko A and Fraering PC, Biochem. J, 2016, 473, 321–334. [DOI] [PubMed] [Google Scholar]
- 23.Chan RB, Oliveira TG, Cortes EP, Honig LS, Duff KE, Small SA, Wenk MR, Shui G and Di Paolo G, J. Biol. Chem, 2012, 287, 2678–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hicks DA, Nalivaeva NN and Turner AJ, Front. Physiol, 2012, 3 JUN, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ehehalt R, Keller P, Haass C, Thiele C and Simons K, J. Cell Biol, 2003, 160, 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vriend G, J. Mol. Graph. Model, 1990, 8, 52–56. [DOI] [PubMed] [Google Scholar]
- 27.Somavarapu AK and Kepp KP, ACS Chem. Neurosci, 2017, 8, 2424–2436. [DOI] [PubMed] [Google Scholar]
- 28.Lomize AL, Pogozheva ID, Lomize MA and Mosberg HI, Protein Sci, 2006, 15, 1318–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kyte J and Doolittle RF, J. Mol. Biol, 1982, 157, 105–132. [DOI] [PubMed] [Google Scholar]
- 30.Aguayo-Ortiz R, Chávez-García C, Straub JE and Dominguez L, Chem. Sci, 2017, 8, 5576–5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qi Y, Ingólfsson HI, Cheng X, Lee J, Marrink SJ and Im W, J. Chem. Theory Comput, 2015, 11, 4486–4494. [DOI] [PubMed] [Google Scholar]
- 32.Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP and De Vries AH, J. Phys. Chem. B, 2007, 111, 7812–7824. [DOI] [PubMed] [Google Scholar]
- 33.Nagle JF, Biophys J, 1993, 64, 1476–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abraham MJ, Murtola T, Schulz R, Páall S, Smith JC, Hess B and Lindah E, SoftwareX, 2015, 1–2, 19–25.
- 35.Michaud-Agrawal N, Denning E, Woolf TB and Beckstein O, J. Comput. Chem, 2011, 32, 2319–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chavent M, Reddy T, Goose J, Dahl ACE, Stone JE, Jobard B and Sansom MSP, Faraday Discuss, 2014, 169, 455–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gapsys V, De Groot BL and Briones R, J. Comput. Aided. Mol. Des, 2013, 27, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allen WJ, Lemkul JA and Bevan DR, J. Comput. Chem, 2009, 30, 1952–1958. [DOI] [PubMed] [Google Scholar]
- 39.Buchoux S, Bioinformatics, 2017, 33, 133–134. [DOI] [PubMed] [Google Scholar]
- 40.Ollila OHS, Risselada HJ, Louhivuori M, Lindahl E, Vattulainen I and Marrink SJ, Phys. Rev. Lett, 2009, 102, 1–4. [DOI] [PubMed] [Google Scholar]
- 41.Kasson PM, Hess B and Lindahl E, Chem. Phys. Lipids, 2013, 169, 106–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williams T and Kelley C, 2016.
- 43.Hunter JD, Comput. Sci. Eng, 2007, 9, 99–104. [Google Scholar]
- 44.W. L. DeLano, 2007, http://www.pymol.org.
- 45.Ayciriex S, Gerber H, Osuna GMG, Chami M, Stahlberg H, Shevchenko A and Fraering PC, Biochem. J, 2016, 473, 321–334. [DOI] [PubMed] [Google Scholar]
- 46.Risselada HJ and Marrink SJ, Proc. Natl. Acad. Sci, 2008, 105, 17367–17372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Audagnotto M, Kengo Lorkowski A and Dal Peraro M, Biochem. Biophys. Res. Commun, 2017, 1–8. [DOI] [PubMed]
- 48.Qi Y, Cheng X, Lee J, Vermaas JV, Pogorelov TV, Tajkhorshid E, Park S, Klauda JB and Im W, Biophys. J, 2015, 109, 2012–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hess B, Bekker H, Berendsen HJC and Fraaije JGEM, J. Comput. Chem, 1997, 18, 1463–1472. [Google Scholar]
- 50.Best RB, Zhu X, Shim J, Lopes PEM, Mittal J, Feig M and MacKerell J, Alexander D, J. Chem. Theory Comput, 2012, 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bondar A-N and White SH, Biochim. Biophys. Acta - Biomembr, 2012, 1818, 942–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ng HW, Laughton CA and Doughty SW, J. Chem. Inf. Model, 2014, 54, 573–581. [DOI] [PubMed] [Google Scholar]
- 53.Lee AG, Biochim. Biophys. Acta - Biomembr, 2003, 1612, 1–40. [DOI] [PubMed] [Google Scholar]
- 54.Palsdottir H and Hunte C, Biochim. Biophys. Acta - Biomembr, 2004, 1666, 2–18. [DOI] [PubMed] [Google Scholar]
- 55.Dickey A and Faller R, Biophys. J, 2008, 95, 2636–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xiong H, Callaghan D, Jones A, Walker DG, Lue LF, Beach TG, Sue LI, Woulfe J, Xu H, Stanimirovic DB and Zhang W, Neurobiol. Dis, 2008, 29, 422–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vanegas JM, Torres-Sánchez A and Arroyo M, J. Chem. Theory Comput, 2013, 10, 691–702. [DOI] [PubMed] [Google Scholar]
- 58.Li X, Dang S, Yan C, Gong X, Wang J and Shi Y, Nature, 2012, 493, 56–61. [DOI] [PubMed] [Google Scholar]
- 59.Deol SS, Bond PJ, Domene C and Sansom MSP, Biophys. J, 2004, 87, 3737–3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van Heijne G and Gavel Y, Eur.J.Biochem, 1988, 174, 671–678. [DOI] [PubMed] [Google Scholar]
- 61.Kong R, Chang S, Xia W and Wong STC, J. Struct. Biol, 2015, 191, 120–129. [DOI] [PubMed] [Google Scholar]
- 62.Fukumori A and Steiner H, EMBO J, 2016, 35, 1628–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takeo K, Tanimura S, Shinoda T, Osawa S, Zahariev IK, Takegami N, Ishizuka-Katsura Y, Shinya N, Takagi-Niidome S, Tominaga A, Ohsawa N, Kimura-Someya T, Shirouzu M, Yokoshima S, Yokoyama S, Fukuyama T, Tomita T and Iwatsubo T, Proc. Natl. Acad. Sci. U. S. A, 2014, 111, 10544–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee JY, Feng Z, Xie X-Q and Bahar I, Biophys. J, 2017, 113, 2634–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Holmes O, Paturi S, Ye W, Wolfe MS and Selkoe DJ, Biochemistry, 2012, 51, 3565–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tominaga A, Cai T, Takagi-Niidome S, Iwatsubo T and Tomita T, J. Neurosci, 2016, 36, 1362–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sun J-H, Yu J-T and Tan L, Mol. Neurobiol, 2015, 51, 947–965. [DOI] [PubMed] [Google Scholar]
- 68.Cantor RS, Biophys J, 1999, 76, 2625–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patra M, Eur. Biophys. J, 2005, 35, 79–88. [DOI] [PubMed] [Google Scholar]
- 70.Samuli Ollila OH, Róg T, Karttunen M and Vattulainen I, J. Struct. Biol, 2007, 159, 311–323. [DOI] [PubMed] [Google Scholar]
- 71.De Jong DH, Singh G, Bennett WFD, Arnarez C, Wassenaar TA, Schäfer LV, Periole X, Tieleman DP and Marrink SJ, J. Chem. Theory Comput, 2013, 9, 687–697. [DOI] [PubMed] [Google Scholar]
- 72.Jung JI, Price AR, Ladd TB, Ran Y, Park HJ, Ceballos-Diaz C, Smithson LA, Hochhaus G, Tang Y, Akula R, Ba S, Koo EH, Shapiro G, Felsenstein KM and Golde TE, Mol. Neurodegener, 2015, 10, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Beel AJ and Sanders CR, Cell. Mol. Life Sci, 2008, 65, 1311–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Panahi A, Bandara A, Pantelopulos GA, Dominguez L and Straub JE, J. Phys. Chem. Lett, 2016, 7, 3535–3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Elad N, De Strooper B, Lismont S, Hagen W, Veugelen S, Arimon M, Horré K, Berezovska O, Sachse C and Chávez-Gutiérrez L, J. Cell Sci, 2015, 128, 589–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Taylor Kimberly W, Lavoie MJ, Ostaszewski BL, Ye W, Wolfe MS and Selkoe DJ, J. Biol. Chem, 2002, 277, 35113–35117. [DOI] [PubMed] [Google Scholar]
- 77.Yang DS, Tandon A, Chen F, Yu G, Yu H, Arawaka S, Hasegawa H, Duthie M, Schmidt SD, Ramabhadran TV, Nixon RA, Mathews PM, Gandy SE, Mount HTJ, George-Hyslop PS and Fraser PE, J. Biol. Chem, 2002, 277, 28135–28142. [DOI] [PubMed] [Google Scholar]
- 78.Somavarapu AK and Kepp KP, J. Neurochem, 2016, 137, 101–111. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.