

Abstract
Laminin, one of the most widely expressed extracellular matrix proteins, exerts many important functions in multiple organs/systems and at various developmental stages. Although its critical roles in embryonic development have been demonstrated, laminin’s functions at later stages remain largely unknown, mainly due to its intrinsic complexity and lack of research tools (most laminin mutants are embryonic lethal). With the advance of genetic and molecular techniques, many new laminin mutants have been generated recently. These new mutants usually have a longer lifespan and show previously unidentified phenotypes. Not only do these studies suggest novel functions of laminin, but also they provide invaluable animal models that allow investigation of laminin’s functions at late stages. Here, I first briefly introduce the nomenclature, structure, and biochemistry of laminin in general. Next, all the loss-of-function mutants/models for each laminin chain are discussed and their phenotypes compared. I hope to provide a comprehensive review on laminin functions and its loss-of-function models, which could serve as a reference for future research in this understudied field.
Keywords: Laminin, Knockout, Loss-of-function, Animal model, Basement membrane
Introduction
Laminin, a major constituent of the basement membrane, is a group of cross- or T-shaped heterotrimeric glycoproteins. Each laminin molecule is composed of one α, one β, and one γ polypeptide chains, and has a molecular weight ranging from 400 to 900 kD [1, 2] (Fig. 1). So far, five α, four β, and three γ variants have been identified [2]. The genes coding these chains, their chromosome locations and structures are summarized in Table 1. Different combinations of these chains give rise to a large number of laminin molecules [1, 3]. It is worth noting that not all possible combinations have been experimentally isolated or demonstrated. So far, 16 laminin molecules have been identified in mammals and four novel combinations of these chains have been proposed based on in vivo and in vitro studies [3]. All these laminin molecules are summarized in Table 2.
Fig. 1.

General structure of laminins. Laminin-111 is used as an example in this figure
Table 1.
The gene, chromosomal location, and structure of each laminin chain
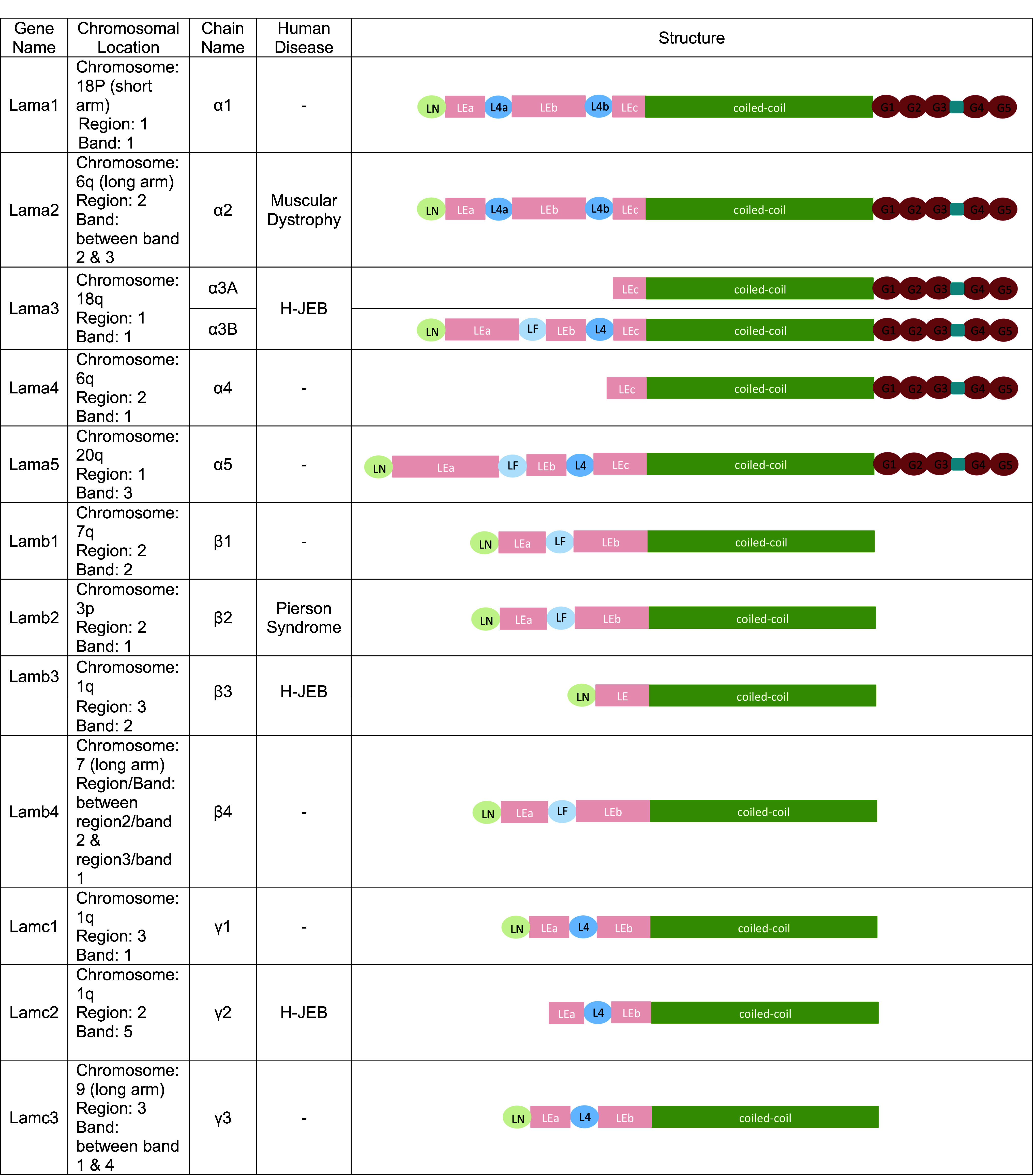
Table 2.
Laminin molecules and their names
| Old nomenclature | New nomenclature | |
|---|---|---|
| Standard | Abbreviation | |
| Laminin-1 | Laminin-α1β1γ1 | Laminin-111 |
| Laminin-2 | Laminin-α2β1γ1 | Laminin-211 |
| Laminin-3 | Laminin-α1β2γ1 | Laminin-121 |
| Laminin-4 | Laminin-α2β2γ1 | Laminin-221 |
| Laminin-5 | Laminin-α3(A)β3γ2 | Laminin-3(A)32 |
| Laminin-5B | Laminin-α3Bβ3γ2 | Laminin-3B32 |
| Laminin-6 | Laminin-α3β1γ1 | Laminin-311 |
| Laminin-7 | Laminin-α3β2γ1 | Laminin-321 |
| Laminin-8 | Laminin-α4β1γ1 | Laminin-411 |
| Laminin-9 | Laminin-α4β2γ1 | Laminin-421 |
| Laminin-10 | Laminin-α5β1γ1 | Laminin-511 |
| Laminin-11 | Laminin-α5β2γ1 | Laminin-521 |
| Laminin-12 | Laminin-α2β1γ3 | Laminin-213 |
| Laminin-13 | Laminin-α3β2γ3 | Laminin-323 |
| Laminin-14 | Laminin-α4β2γ3 | Laminin-423 |
| Laminin-15 | Laminin-α5β2γ3 | Laminin-523 |
| – | Laminin-α2β1γ2 | Laminin-212 |
| – | Laminin-α2β2γ2 | Laminin-222 |
| – | Laminin-α3β3γ3 | Laminin-333 |
| – | Laminin-α5β2γ2 | Laminin-522 |
Accumulating evidence shows that laminin plays multiple important roles at various developmental stages. However, due to its critical roles in embryonic development, most laminin mutants are embryonic lethal (see Table 3), which prevents the investigation of laminin’s functions at later stages. The intrinsic complexity of laminin, together with limited research tools (genetic models), makes research in this field understudied and laminin’s functions at later developmental stages elusive.
Table 3.
Laminin single knockout/mutant lines
| Genes | Mutation | Types | Life span | Phenotypes | References |
|---|---|---|---|---|---|
| Lama1 | Deletion of proximal promoter region and exon 1: Lama1−/− | Global KO | Die at E6.5 | Lack of Reichert’s membrane; defects in endoderm differentiation | [48] |
| Gene trap-βgeo fusion: Lama1 trap | Nonfunctional mutation | Die at E7 | Lack of Reichert’s membrane | [42] | |
| Deletion of globular domains 4–5: LG4–5−/− | Global KO | Die at E6.5 | Defects in epiblast polarization and cavity formation | [49] | |
| Floxed exon 1: Lama1fl/fl | Conditional KO in epiblasts | Normal | Loss of inner limiting membrane; defects in retina | [48, 50] | |
| Lama2 | Unidentified: dy/dy | Unidentified mutation | Die around 3–6 months | Muscular dystrophy | [59, 62, 63] |
| G-to-A mutation: dy2J/dy2J | Point mutation | Normal | Muscular dystrophy | [60] | |
| Deletion of amino acids 128–209: dy3K/dy3K | Global KO | Die at 5 weeks | Severe muscle pathology; disrupted myelination; abnormal nerve electrophysiology; compromised BBB | [65–67] | |
| Lama3 | Removal of exon A3: Lama3−/− | Global KO | Die at P2–3 | H-JEB; defects in epithelial survival and ameloblast differentiation | [96] |
| Floxed exon 42: Lama3fl/fl | Conditional KO in lung | Normal | Resistance to mechanical ventilation; mild inflammation | [98] | |
| Lama4 | Deletion of promoter and exons 1 and 2: Lama4−/− | Global KO | Normal, if not die at perinatal stage | Embryonic & perinatal hemorrhage; defect in neuromuscular junction formation; fibrotic kidney; abnormal adipose tissue function; reduced disease susceptibility, severity, and T cell infiltration during multiple sclerosis | [103–107] |
| Lama5 | Deletion of exons encoding 113 amino acids: Lama5−/− | Global KO | Die around E17 | Exencephaly; syndactyly; dysmorphogenesis of the placental labyrinth; defects in kidney, lung, hair, intestine, tooth bud, and submandibular gland development | [37, 110–117] |
| Insertion of a PGKneo cassette in intron 21: Lama5neo/neo | Hypomorphic mutation | Die at 4 weeks | Polycystic kidney disease; proteinuria; renal failure | [120] | |
| Gene trap-βgeo fusion: Lama5 trap | Nonfunctional mutation | Die around E17 | Defects in tooth bud and submandibular gland development | [122, 123] | |
| Floxed exons 15-21: Lama5fl/fl | Inducible (at E6.5) conditional KO in lung epithelium | Die a few hours after birth | Abnormal alveolar epithelial cell differentiation and lung development | [124] | |
| Floxed exons 15-21: Lama5fl/fl | Conditional KO in podocytes | A few weeks to >8 months | Proteinuria; nephrotic syndrome | [121] | |
| Floxed exons 15-21: Lama5fl/fl | Conditional KO in skeletal muscles | Normal | Delayed synaptic maturation at the neuromuscular junction | [119] | |
| Lamb1 | Gene trap-βgeo fusion: Lamb1-trap | Nonfunctional mutation | Die at E5.5 | Lack of basement membranes (Reichert’s membrane and embryonic basement membrane); defect in cavitation; reduced trophoblast invasion | [42] |
| Point mutation: T5460A | Point mutation | Normal | Dystonia-like movements when awake and hyperextension when asleep | [125] | |
| Lamb2 | Deletion of exon 2: Lamb2−/− | Global KO | Die at P15–P30 | Abnormal neuromuscular junctions; increased glomerular filtration; retinal defects | [126–131] |
| Lamb3 | Insertion at an exon/intron junction: Lamb3−/− | Global KO | Die within 24 h of birth | H-JEB | [135] |
| Lamb4 | N/A | ||||
| Lamc1 | Deletion of exon 1: Lamc1−/− | Global KO | Die at E5.5 | Lack of basement membranes; failure of endoderm differentiation; abnormal epiblast cell development | [142, 143] |
| Gene trap-βgeo fusion: Lamc1-trap | Nonfunctional mutation | Die at E5.5 | Severe defects in embryonic development | [43] | |
| Deletion of nidogen-binding site: lamc1III4−/− | Global KO | Die before E11.5 or soon after birth | Renal agenesis and impaired lung development; ruptures of basement membranes in the elongating Wolffian duct and alveolar sacculi | [29] | |
| Floxed exon 2: Lamc1fl/fl | Conditional KO in hippocampi, spinal cord and peripheral nerves | More than 4 months | Defects in neurite growth and neuronal migration; hindlimb paralysis; tremor | [144, 145] | |
| Floxed exon 2: Lamc1fl/fl | Conditional KO in Schwann cells | Die within 2 months | Tremor; progressive hindlimb paralysis | [146] | |
| Floxed exon 2: Lamc1fl/fl | Conditional KO in ureteric bud | Shortly after birth to >10 weeks | Lack of kidneys; small kidneys; abnormal renal vesicle formation | [147] | |
| Floxed exon 2: Lamc1fl/fl | Conditional KO in neural progenitors | >6 months | Age-dependent and region-specific intracerebral hemorrhage; breakdown of blood brain barrier; unable to respond to angiotensin II | [148–150] | |
| Floxed exon 2: Lamc1fl/fl | Conditional KO in vascular smooth muscles | Normal | Lower baseline blood pressure; reduced response to angiotensin II | [150] | |
| Floxed exon 2: Lamc1fl/fl | Conditional KO in PDGFRβ+ Cells | Die within 4–6 months | Progressive muscular dystrophy | [79] | |
| Lamc2 | Deletion of exon 8: Lamc2−/− | Global KO | Die before P5 | H-JEB; abnormal tracheal hemidesmosomes | [152, 153] |
| Lamc3 | Replacement of exon1 and part of intron 1 with βgeo: Lamc3−/− | Global KO | Normal | Ectopic granule cells in cerebellum; enhanced capillary branching in the outer retina | [131, 155] |
With the advance of genetic and molecular techniques, many new laminin mutants have been generated. These new mutants usually have a longer lifespan, enabling the study of laminin’s functions at later stages; and show previously unidentified phenotypes, indicating innovative functions. Here, I first briefly introduce the nomenclature, structure, and biochemistry of laminin in general. Then, I discuss all the loss-of-function mutants/models for each laminin chain and compare their phenotypes. In addition, all loss-of-function models, their mutations, and phenotypes are summarized in Table 3 (single knockout), Table 4 (knockout–knockin) and Table 5 (double knockout). The goal of this study is to provide a comprehensive review on laminin functions and its loss-of-function models, which could serve as a reference for future laminin studies.
Table 4.
Laminin knockout–knockin transgenic lines
| Background | Transgenes | Targeted Tissues | Life Span | Phenotypes | References |
|---|---|---|---|---|---|
| Lama2−/− | lacZ | Almost all tissues | Die at 2–4 weeks | Muscular dystrophy | [64] |
| Lama2−/− | Mouse lama1 (line 12) | Almost all tissues but not the PNS | Normal | Almost normal; defects observed in lama2−/− mice substantially improved | [69–72] |
| Lama2−/− | Mouse lama1 (line 8) | Almost all tissues | Normal | Almost normal; defects observed in lama2−/− mice substantially improved | [73] |
| Lama2−/− | Mouse lama1 lacking globular domains 4–5 | Almost all tissues | ~75% survive up to 3 months | Muscular dystrophy in limb muscles, but not in diaphragm or heart muscles; lack of myelination in peripheral nerves | [74] |
| Lama5−/− | Murine lama5 | Almost all tissues | Normal | Rescue of all embryonic developmental defects in Lama5−/− mice; architecture transformation from small intestine to colonic mucosa | [115, 118] |
| Lama5−/− | Chimeric lama5 with all 5 LG domains replaced by homologous sequences from lama1 | Almost all tissues | Die before or shortly after birth | Similar to Lama5−/− mice with reduced incidence of exencephaly | [118] |
| Lama5−/− | Chimeric lama5 with its last 3 LG domains replaced by homologous sequences from lama1 | Almost all tissues | Several months | Rescue of the embryonic lethality in Lama5−/− mice; rescue of presynaptic defects in Lama5M/M mice | [118, 119] |
| Lama5neo/neo | Human lama5 | Podocytes | >6 months | Rescue of kidney phenotype in Lama5neo/neo mice | [121] |
| Lamb2−/− | Rat lamb2 | Muscles | Die at about 1 month | Rescue of abnormal synaptic structure and failure to thrive in Lamb2−/− mice | [128] |
| Lamb2−/− | Rat lamb2 | Podocytes | Die at 3–4 weeks | Rescue of kidney defects in Lamb2−/− mice | [128] |
| Lamc2−/− | Human lamc2 | Keratinocytes | >1.5 years | Rescue of the H-JEB phenotype in Lamc2−/− mice | [154] |
Table 5.
Laminin double knockout lines
| Genes | Mutation | Types | Life span | Phenotypes | References |
|---|---|---|---|---|---|
|
Lama2 Lama4 |
Dy2J/dy2J Lama4−/− |
Lama2 mutation and Lama4 global KO | Up to 3 months | Lack of myelination in peripheral nerves; altered basement membrane composition | [108, 109] |
|
Lama2 Lama4 |
Dy3k/dy3k Lama4−/− |
Global KO | Die within 2 weeks | Almost complete absence of sorting in spinal roots and peripheral nerves; altered basement membrane composition | [108, 109] |
|
Lama4 Lama5 |
Lama4−/−Lama5M/M | Lama4 global KO and Lama5 conditional KO in skeletal muscles | Die around 3 months | Smaller and weaker; arrested synaptic maturation | [119] |
|
Lamb2 Lamc3 |
Lamb2−/− Lamc3−/− | Global KO | Die within 4 weeks | Fractured pia basement membrane; cobblestone lissencephaly; retinal defects | [131, 156–159] |
Laminin nomenclature, structure and biochemistry
Laminin nomenclature
Laminin is a large family of heterotrimeric proteins composed of α, β, and γ chains. In mouse and human, 5 α, 4 β, and 3 γ variants have been identified [2]. Different combination of these variants generates a large variety of laminin molecules. Two systems have been developed to name different laminin molecules. The first (old) one involves assigning Arabic numerals to laminins based on the order of their discovery, such as Laminin-1, Laminin-2, Laminin-3, et al. [4]. Although this nomenclature is simple to use, it fails to provide information regarding the trimeric composition of the laminins. Thus, an alternative (new) nomenclature system that indicates each individual polypeptide chain was developed. Specifically, laminins are named by Greek letters representing polypeptide chains followed by numbers indicating the variants [1]. For example: laminin-1 is written as laminin-α1β1γ1 or simply abbreviated as laminin-111. Sometimes, one Greek letter and one number are used to define all laminin isoforms that contain this subunit. For example, “α3 laminins” means all α3-containing laminins.
Some genes encoding laminins may produce more than one peptide due to alternative splicing [5]. For these chains, letters A and B are added after the numbers to indicate the low-molecular-weight and high-molecular-weight peptides, respectively [1]. For example, the gene encoding α3 chain can produce a short laminin α3 variant named α3A and a long laminin α3 variant named α3B [5]. It should be noted that the short α chain variants can be named without mentioning the A letter. Thus, laminin-332 implies laminin-3A32 but not laminin-3B32. A side-by-side comparison of the old and new nomenclature systems is summarized in Table 2.
Laminin structure
Laminin consists of three polypeptide chains (α, β, and γ). Each laminin chain is composed of various globular domains, rod-like tandems or repeats called Epidermal Growth Factor-like “LE” domains, and a coiled-coil domain [1]. The globular domains include laminin N-terminal globular domain (LN), laminin four globular domain (LF), and L4. For example, laminin α1 chain contains a globular LN domain at the N-terminus followed by alternating globular “L4” and LE domains, a coiled coil domain, and five homologous globular “LG” domains at the C-terminus. Compared to the α chain, β and γ chains have similar domains in the N-terminus, but do not have the LG domains in the C-terminus. These chains join together via their coiled-coil domains forming a molecule with one long arm and up to three short arms [1]. The structure of laminin-111 is shown in Fig. 1. The structure of each individual laminin chain is summarized in Table 1. The nomenclature system for these laminin polypeptide chains is well reviewed in other articles [1, 6, 7] and thus not discussed here.
Laminin biochemistry
As a major component of the basement membrane, laminin not only interacts with other extracellular matrix proteins (e.g., nidogen) contributing to basement membrane assembly, but also binds to and activates cell-surface proteins (e.g., receptors) transducing molecular signals. These activities have been mapped to specific domains/regions. In general, sites that mediate laminin-extracellular matrix protein interaction are located in the short arms of all three chains [8]. Laminin-receptor interaction, however, is confined to the N-terminal and C-terminal domains of the α chain [8]. Laminins are usually glycosylated, which increases their molecular masses and stabilizes them against degradation [9]. It should be noted that distinct laminins are not glycosylated to the same extent due to different glycosylation sites [10].
It has been shown that laminin trimers are assembled before being secreted to the extracellular matrix [11, 12]. The assembly starts by combining β and γ chains together, followed by the association of α chain to the β/γ dimer, which is the rate-limiting step [12, 13]. Once secreted, laminins together with other extracellular matrix proteins, including collagen IV and nidogen, form a dense protein network called basement membrane. Although the precise mechanism underlying basement membrane assembly is not fully understood, laminin–nidogen interaction is believed to play an important role in this process [14–17]. The nidogen-binding site on laminin is mapped to a single epidermal growth factor-like domain of the laminin γ1 chain, named γ1III4 [18–23]. It is proposed that nidogen interconnects the two networks formed by laminins and type IV collagen [24], forming a unique 3D structure with high stability. Consistent with this hypothesis, inhibiting laminin γ1-nidogen interaction with antibodies or a recombinant laminin γ1 fragment compromised basement membrane formation in many tissues and organ cultures [25–28]. To further investigate the role of laminin–nidogen interaction in basement membrane assembly, a mouse line (lamc1III4−/−) with genetic deletion of the nidogen-binding region γ1III4 in laminin γ1 was generated [29]. Interestingly, although the basement membrane in Wolffian duct is compromised [29], skin basement membrane is not affected [30]. These results suggest that laminin–nidogen interaction may play different roles in basement membrane formation/assembly depending on the tissues.
The basement membrane exerts many important functions, including sequestering growth factors and cytokines, establishing concentration gradients and regulating their spatial and temporal distribution [31]. For example, fibroblast growth factors, Wnt factors, and hedgehog are able to strongly bind to the basement membrane. In addition, the basement membrane can also serve as a reservoir for many bioactive fragments usually derived from proteolysis [31]. Furthermore, the basement membrane also participates in ligand maturation/activation. For instance, TGF-β is secreted in its latent inactive form, which is activated by matrix metalloproteinases at the basement membrane [32].
To accurately and timely regulate a variety of critical biological processes, including angiogenesis, the basement membrane undergoes active remodeling via proteolysis [32]. Some laminin chains could be truncated into smaller parts by the action of proteolytic enzymes [7, 11, 33]. The fragments generated by each single enzymatic action are usually named after the acting enzyme. For example: pepsin generates P fragments, trypsin generates T fragments, and elastase generates E fragments [1]. Such enzymatic truncation directly affects the physiological and pathophysiological functions of laminins, and thus the basement membrane [11]. The readers are referred to other excellent reviews for other biochemical properties of the laminin family [8, 34–36].
Laminin function and loss-of-function studies
Laminin exerts many different functions, including embryogenesis, vascular maturation, and neuromuscular development [8, 34, 35, 37–41]. These functions are highly dependent on laminin isoforms, their cellular sources, and developmental stages. Here I discuss and compare loss-of-function studies on each individual laminin chain.
Laminin α1
To investigate the physiological function of laminin α1 chain, a few lama1 loss-of-function mouse lines have been generated. The first lama1 mutant was generated by Miner and colleagues using gene trap technique [42, 43]. Specifically, a gene trap vector was inserted in the third intron of lama1 to produce a fusion protein containing the first 120 amino acids of lama1 and the vector-coded sequences (trans-membrane domain and βgeo). The expression pattern of lama1 (demonstrated by X-gal) in these lama1 trap mice is consistent with previous reports [44–47]. These lama1 trap embryos usually die by embryonic day (E)7 and fail to form Reichert’s membrane [42]. Interestingly, overexpression of lama5, the other laminin α chain (in addition to lama1) expressed in early post-implantation embryos, enables the lama1 trap embryos to initiate gastrulation, but fails to rescue Reichert’s membrane [42]. These studies clearly demonstrate a pivotal role of lama1 in the integrity of basement membranes and early embryonic development.
The second lama1 mutant line was generated by targeted deletion of the proximal promoter region and exon 1 of this gene [48]. Like the lama1 trap mice, these lama1 null mutants die at approximately E6.5, lack Reichert’s membrane, and show defects in parietal and visceral endoderm differentiation [48], again suggesting an important role of lama1 in basement membrane formation and early embryonic development.
Another lama1 mutant line is the LG4–5−/− mice, which lack the lama1 globular domains 4–5 [49]. Similar to the lama1 mutants described above, these LG4–5−/− mice die at E6.5 [49]. Although Reichert’s membrane and embryonic basement membranes are grossly normal, defects in epiblast polarization and cavity formation are detected in these mutants [49]. Further studies demonstrate that the truncated lama1 protein lacking globular domains 4–5 is secreted and incorporated into the Reichert’s membrane and embryonic basement membrane in these mutants [49]. Together, these results suggest that globular domains 4–5 of lama1 are required for the conversion of stem cells to polarized epithelium, but are not essential for the assembly of Reichert’s membrane and embryonic basement membrane in vivo.
Recently, a missense mutation in mouse lama1 gene (Y265C) has been found to contribute to the retinal vasculopathy, a disorder characterized by vitreal fibroplasia and vessel tortuosity [50, 51]. To further study the role of lama1 in this disease and bypass the embryonic lethal phenotype of lama1 null mice, a conditional knockout line with lama1 deficiency in epiblasts was generated by crossing the lama1 floxed (lama1fl/fl) mice with the Sox2-Cre line [50]. These conditional knockouts are slightly lighter than the controls at birth but catch up by 8 weeks of age. They demonstrate diminished lama1 expression in the inner limiting membrane and exhibit similar but more severe retinal phenotype when compared to the lama1 Y265C mutants [50, 51], suggesting critical roles of lama1 in retinal development. In addition, these conditional knockout mice also show behavioral abnormalities and aberrant cerebellum formation [52], including decreased dendritic processes in Purkinje cells, disorganized Bergmann glial fibers and endfeet, and diminished proliferation and migration of granule cell precursors.
In addition to mice, zebrafish mutants have also been used to study the function of lama1. For example, lama1-knockdown zebrafish show degeneration of lens and reduction of eye size by 2 and 3 days postfertilization [53]. Additionally, axonal guidance defects, inner limiting membrane disruption, and shortened Muller cell endfeet are also observed in zebrafish with mutations in lama1 [54–56]. Furthermore, a zebrafish mutant lama1a69/a69, which carries a cysteine-to-serine substitution at amino acid 56, displays shortened body axis and eye/len defects [57, 58]. These data suggest a critical role of lama1 in retinal development. However, it should be noted that these zebrafish mutants are larval lethal, which prevents the analysis of lama1’s function at later developmental stages.
Together, these data support an indispensable role of lama1 in embryonic development, and an important function of Sox2+ cell-derived α1-containing laminins in retina and cerebellum development/maturation. Although the functions of lama1 in other tissues/cell types remain elusive, the lama1fl/fl line provides a valuable tool for these studies.
Laminin α2
Lama2 is predominantly expressed in striated muscles and peripheral nerves. Loss of function in this gene leads to aggressive muscle degeneration, known as muscular dystrophy [59–61]. The first studied mouse model for this disorder is dy/dy mice [62], which carries an unidentified mutation in lama2 gene [59, 63]. The dy/dy mice, which express low but detectable levels of lama2, develop muscle pathology at 3 weeks of age and usually die of unknown causes around 3–6 months. Another muscular dystrophy model involves a G-to-A mutation in the lama2 gene (named dy2J), which results in abnormal splicing and generation of a truncated and non-functional lama2 protein [60]. Homozygous dy2J (dy2J/dy2J) mice have a normal lifespan, but develop muscle weakness at about 3 weeks of age, which worsens progressively[60].
To further study the role of lama2, a lama2 null mouse line, termed dyW/dyW, was generated [64]. In these mice, under the endogenous lama2 promoter/enhancer elements, the lama2-coding sequence was replaced by lacZ sequence [64]. The dyW/dyW mice are passive, small, emaciated, and usually die between 2 and 4 weeks after birth [64]. They demonstrate muscle phenotypes similar to a disease known as congenital muscular dystrophy, including muscle fiber necrosis with regeneration, small myofibers with central nuclei, pronounced fibrosis, progressive lameness, and muscle paralysis [64]. In addition, another lama2 null mouse line (dy3k/dy3k) was generated by targeted deletion of amino acids 128–209 [65]. Like the dyW/dyW mice, lama2 expression and basement membrane structure are completely absent in the muscles of dy3k/dy3k mice [65]. These dy3k/dy3k mice show muscle pathology in early postnatal (P) stage (P9), deteriorate rapidly and aggressively, and die at 5 weeks of age [65], suggesting a more severe muscular dystrophic phenotype. All three lama2 mutant lines have been widely used as mouse models for congenital muscular dystrophy. In addition to the muscle phenotype, the dy3k/dy3k mice also demonstrate other phenotypes, including thinner and disrupted myelination [66], reduced motor conduction velocity in the sciatic nerve [66], compromise of blood brain barrier integrity [67], and smaller body size. Consistent with these reports, loss of lama2 has been found in human patients with congenital muscular dystrophy [61]; and brain abnormalities, including cortical anomaly, polymicrogyria, and abnormal white matter signals, have been reported in patients with lama2-deficient congenital muscular dystrophy [68]. Analyses of the dyW/dyW and dy3k/dy3k mice have significantly increased our understanding of the pathogenesis of congenital muscular dystrophy, and these mouse lines are two of the most widely used congenital muscular dystrophy models. These results suggest that lama2 exerts crucial functions primarily in skeletal muscles, nerves, and the CNS. It is worth noting that the smaller body size observed in dyW/dyW and dy3k/dy3k mice is not found in human patients. What causes this species-specific effect remains unclear. Answers to this question may lead to a novel congenital muscular dystrophy model that better replicates human pathology.
Due to structural similarity between lama1 and lama2, the therapeutic potential of lama1 in lama2-deficient mice was investigated extensively. Gawlik and colleagues generated a transgenic mouse line (dy3kLNα1TG) that expresses lama1 in dy3k/dy3k background [69]. One line (No. 12) of these mutants shows strong lama1 expression in skeletal muscles and testes [69–72]. Compared to the dy3k/dy3k mice, these dy3kLNα1TG mice (line No. 12) are bigger in size and have a normal lifespan [69, 72]. Muscle pathology (morphology and function) is improved to a remarkable degree in these mice. In addition, the aberrant distribution of integrin α7B and α- and β-dystroglycan in dy3k/dy3k mice is also reversed by the expression of lama1 in dy3kLNα1TG mice [71]. Furthermore, loss-of-lama2-induced defects in testes, including abnormal timing in seminiferous tubule lumen formation, are partially rescued by the overexpression of lama1 [70]. In another line (No. 8) of the dy3kLNα1TG mice, lama1 expression is detected in both skeletal muscles and the peripheral nervous system (PNS) [73]. Like line No. 12 described above, this line has a normal lifespan and body weight [73]. Additionally, the muscular dystrophy and peripheral nerve defects observed in dy3k/dy3k mice are dramatically reduced/corrected in this line (No. 8) of the dy3kLNα1TG mice [73]. These studies clearly show that lama1 is able to rescue most of the phenotypes caused by loss of lama2, suggesting great therapeutic potential of lama1 in congenital muscular dystrophy. To further investigate the function of lama1 globular domains, a transgenic line (dy3k/δE3) expressing lama1 lacking globular domains 4–5 in lama2 null background was generated [74]. Compared to the dy3k/dy3k mice, these dy3k/δE3 mutants have prolonged lifespan and improved health [74]. Interestingly, diaphragm and heart muscle deficits, but not limb muscle defects are corrected by overexpression of the truncated lama1 [74]. Additionally, many unmyelinated axons and aberrant basement membranes in neuromuscular system are still observed in dy3k/δE3 mice [74]. These results suggest that different muscles have different requirements for lama1 globular domains 4-5, and that these domains are essential for myelination in the PNS and important for basement membrane assembly.
Next, the therapeutic effect of laminin-111 protein was also tested in a variety of muscular dystrophy models. It has been shown that exogenous laminin-111 is able to reduce muscle pathology, enhance viability, improve muscle repair, and increase muscle strength in both dyW/dyW and mdx mice [75–78]. Furthermore, using a new muscular dystrophy model developed in our laboratory [79], we find that exogenous laminin is able to partially rescue muscle pathology at structural, biochemical, and functional levels [79]. Together, these data suggest that laminin-111 has therapeutic effect in muscular dystrophy and can be used to treatment this devastating disorder.
In addition to laminin, the therapeutic effect of agrin, another extracellular matrix protein, has been extensively studied. It has been shown that muscle-specific expression of a miniaturized form of agrin (miniagrin) in dyW/dyW and dy3k/dy3k mice significantly ameliorates muscle pathology and improves overall health [80, 81]. Late-onset expression of miniagrin remains beneficial, but to a lesser extent compared to early expression [82]. Similar result was reported in dyW/dyW mice administered with adeno-associated virus-expressing miniagrin [83]. Furthermore, expression of full-length agrin or a chimeric protein, in which the C-terminus of agrin is replaced by perlecan domain V, has been demonstrated to substantially slow down disease and lessen muscle pathology [82]. These data strongly suggest a therapeutic potential of agrin in lama2-deficient muscular dystrophy.
Besides extracellular matrix proteins, many other therapeutic options, including apoptosis inhibition, proteasome suppression, autophagy suppression, and fibrosis inhibition, have also been investigated. Genetic inactivation of Bax (proapoptotic protein) or overexpression of Bcl-2 (antiapoptotic protein) in dyW/dyW mice was able to partially ameliorate disease pathology [84]. Consistent with these results, pharmacological inhibition of apoptosis with Doxycycline or Omigapil in dyW/dyW mice partially amended muscle pathological changes [85, 86]. Proteasome inhibitors, MG-132 and Bortezomib, were able to elongate lifespan and diminish muscle pathology in dy3k/dy3k mice [87, 88]. Similar improvements in muscle pathology have been found in dy3k/dy3k mice after treatment with autophagy inhibitor 3-methyladenine [89]. Antifibrotic treatment with L-158809, a losartan derivative and angiotensin II receptor antagonist, led to reduced fibrosis and alleviated muscle pathology in dyW/dyW mice [90]. Together, these data suggest that apoptosis, proteasome, autophagy, and fibrosis can be targeted in the treatment of muscular dystrophy. However, it should be noted that these treatments are not as effective as targeting laminin or agrin.
Given that muscular dystrophy affects multiple signaling pathways and cellular functions, combinatorial approaches have been explored. For example, a cumulative effect was reported in dyW/dyW mice with both antiapoptosis (Bcl-2 overexpression or Omigapil application) and miniagrin treatments [91]. In addition, antiapoptosis (Bax inactivation) and proregeneration (muscle-specific IGF-1 overexpression) also demonstrated an additive beneficial effect in dyW/dyW mice [92]. The readers are referred to reference [93] for a systemic review of the treatments of lama2-deficient muscular dystrophy.
Laminin α3
Laminin-332 is expressed in various epithelial tissues [94, 95] and is linked to Herlitz junctional epidermolysis bullosa (H-JEB), a severe autosomal recessively inherited blistering skin disease that has no successful treatments so far [96, 97]. A C-to-T mutation, which results in a premature termination codon in lama3 gene, was identified in a human patient with H-JEB [97]. Next, a lama3 null mouse line was generated by removing exon A3, which causes a frame shift mutation and a premature stop codon that disrupt all possible α3-laminins [96]. The lama3 null mice are born at the Mendelian ratio, but develop progressive blistering in the skin after birth and die within 2–3 days [96]. Further studies show that these mutants develop skin phenotype similar to that in human H-JEB patients, including absence of lama3 expression in epidermal basement membrane, loss of anchorage function in the epidermis, abnormal hemidesmosomes, and defects in epithelial survival and ameloblast differentiation [96]. These results strongly suggest that the lama3 null mouse line is a valuable genetic model for H-JEB. Future research should focus on investigating the therapeutic potential of laminin-332 in H-JEB using lama3 null mice.
To further study the function of lama3, a lama3 floxed (lama3fl/fl) line was generated by targeting the exon 42 of mouse lama3 gene [98]. By intratracheally treating these mice with adenovirus expressing Cre, Urich and colleagues reported that lung-specific loss of lama3 confers resistance to mechanical injury by increasing lung collagen expression [98]. In addition, resistance to mechanical ventilation and a mild inflammation were also found in these mice [98]. Together, these studies suggest fundamental roles of lama3 in skin epithelial development and lung function. The lama3fl/fl mice will server as a valuable genetic tool and enable investigation of lama3’s roles in other tissues/organs.
Laminin α4
Lama4 is widely distributed in vascular basement membrane and basement membranes of other tissues, including skeletal muscle, nerves, heart, and kidney [99–102]. The lama4 null mice were generated by Thyboll and colleagues by targeting the promoter and exons 1 and 2 of lama4 gene [103]. The lama4 mutants are born at the expected Mendelian ratio, develop subcutaneous hemorrhage during embryonic and perinatal stages, and show anemia probably secondary to the internal bleeding [103], suggesting an important role of lama4 in vascular integrity at this stage. Interestingly, lama4 null mice surviving beyond the perinatal period have a lifespan comparable to the wild-type controls [103]. Consistent with these observations, defects in vascular basement membrane are observed in newborn but not adult (except the loss of lama4) lama4 null mice [103], suggesting compensation at later stages. Although most extracellular matrix proteins are not changed [103], lama5, which predominantly covers capillaries and postcapillary venules in wild-type brains [35], is expressed ubiquitously along the vascular tree in lama4 null mice [104]. In a multiple sclerosis model, lama4 null mice show substantially decreased disease susceptibility/severity owing to dramatically reduced T lymphocyte infiltration into the brain compared to wild-type mice [104], suggesting a permissive role of lama4 in T lymphocyte migration into the CNS. Further studies demonstrate that the ubiquitously expressed lama5 selectively inhibits integrin α6β1-mediated migration of T lymphocytes through lama4 into the brain in lama4 null mice [104]. These results suggest that compensatory expression of lama5 is also responsible for the less severe phenotype in lama4 null mice during multiple sclerosis.
Additional studies in adult lama4 null mice reveal defects in the neuromuscular junction, kidney, and adipose tissue. Patton and colleagues reported that the active zones and junctional folds are not precisely apposed to each other, although they form in normal numbers [105], suggesting that lama4 actively regulates the localization of synaptic specializations. Abrass and colleagues showed that these lama4 null mice have progressive fibrotic alterations in the kidney, including dilated microvessels, perivascular inflammation, tubulointerstitial fibrosis, and increased expression of collagen and fibronectin [106]. Moreover, the lama4 null mice also exhibit reduced weight gain in response to age and/or high fat diet, reduced epididymal adipose tissue mass, and altered lipogenesis [107], suggesting that lama4 modulates adipose tissue structure and function in a depot-specific manner. It is unclear, however, whether these phenotypes are caused by loss of lama4 directly or loss of lama4-induced lama5 expression indirectly. Elucidating this question would enrich our knowledge on laminin compensation and allow us to distinguish lama4’s function from lama5’s function.
To study the redundancy and compensation in the basal lamina of Schwann cells, which synthesize α2- and α4-containing laminins, two double mutant mouse lines (dy2J/lama4−/− and dy3k/lama4−/−) were generated. The dy2J/lama4−/− mice are smaller in size and rarely survive 3 months [108, 109]. Compared to either dy2J/dy2J or lama4−/− mice, these double mutants demonstrate more severe phenotypes, including lack of myelination in peripheral nerves but not spinal roots and altered pancreatic acinar basement membrane composition [108, 109]. The dy3k/lama4−/− mice, on the other hand, are normal at birth but usually die within 2 weeks [108, 109]. These mutants show a nearly complete absence of sorting in both spinal roots and peripheral nerves [108], indicating a more substantial pathology. Like the dy2J/lama4−/− mice, the composition of pancreatic acinar basement membrane is affected in these dy3k/lama4−/− mutants [109]. These studies suggest that lama2 and lama4 do not fully compensate each other’s loss and they together coordinately regulate axonal ensheathment and Schwann cell function.
Laminin α5
Lama5 is expressed in virtually all basement membranes at early embryonic stage and then becomes restricted to specific basement membranes as development proceeds [37]. In mature vasculature, lama5 is predominantly found in capillary and postcapillary venule basement membranes [35]. To investigate lama5’s role in embryogenesis, Miner and colleagues generated lama5 null mice by targeting domains VI and V in the N-terminus of lama5 gene. The lama5 null embryos survive well past implantation stage, show apparent defects after E9, and die before E17 [37]. These lama5 mutants show defects in neural tube closure (exencephaly), digit septation (syndactyly), and dysmorphogenesis of the placental labyrinth [37], indicating a critical role of lama5 in embryonic development. Consistent with these data, defects in neural crest migratory pathways and condensation into ganglia are also found in the lama5 null embryos [110], suggesting that lama5 is indispensable for proper migration and timely differentiation of some neural crest populations. Next, kidney defects characterized by abnormal glomerular basement membrane and glomerulogenesis are observed in these lama5 null embryos [111]. Some of mutants even lack one or two of the kidneys [111], indicating a crucial role of lama5 in kidney development. Lung defects manifested by incomplete lobar separation and absence of the visceral pleura basement membrane with normal lung branching morphogenesis and vasculogenesis are detected [112]. In addition, loss of lama5-induced defects in hair [113, 114] and intestine [115–117] are also reported. Fewer hair germs, failure of hair germ elongation, decreased length and structure of primary cilia at dermal papilla, as well as compromised expression of key morphogen/molecules are found in the skin of lama5 null mice [113, 114]. These lama5 mutants also show excessive folding of intestinal loops and delayed expression of specific markers in the intestine, suggesting an essential role of lama5 in intestinal smooth muscle development [117]. Mechanistic studies revealed abnormal Wnt and PI3Kinase signaling in the malformed intestine, linking these signaling pathways to the observed phenotype.
To further assess lama5’s function, a transgenic line that expresses a full-length lama5 transgene in lama5 null background (KO/Tg), was generated [118]. Transgene-derived lama5 successfully rescues the developmental defects of lama5 null mice and the KO/Tg mice are viable and fertile [118]. These KO/Tg mice, however, demonstrate very low level of lama5 expression in small intestine compared to wild-type controls [115]. This reduced lama5 expression in small intestine basement membrane is compensated by deposition of colonic laminin isoforms (lama1 and lama4), which leads to an architectural transformation from small intestinal mucosa to colonic mucosa [115]. To further study the specific function of lama5, two additional transgenic lines that ubiquitously express laminin α1/α5 chimeric proteins were generated. In one line, all five laminin-globular (LG) domains of lama5 are replaced by homologous sequences from lama1 in lama5 null background [118]. These transgenic mice die either before or shortly after birth, and show only limited improvement when compared to lama5 null embryos (reduced incidence of exencephaly) [118]. In the other transgenic line, the last three laminin-globular domains are replaced by homologous sequences from lama1 in lama5 null background [118, 119]. These transgenic mice are outwardly normal, live for several months, and die from nephrotic syndrome [118]. Additionally, they also demonstrate impaired postsynaptic but not presynaptic maturation at the neuromuscular junction [119]. These data suggest that lama5, especially its LG domains, has multiple functions and is essential for development.
Like the lama5 null mice, a hypomorphic mutation caused by the insertion of a PGKneo cassette in intron 21 of the lama5 gene leads to similar phenotypes [120]. This insertion causes aberrant splicing and exon skipping, resulting in dramatically reduced expression of lama5 [120]. Mice homozygous for this hypomorphic mutation show polycystic kidney disease, proteinuria, and death due to renal failure by 4 weeks of age [120], which can be rescued by expressing human lama5 transgene in podocytes [121]. These data again suggest an absolute requirement of lama5 in early kidney development.
Another lama5 mutant line that produces a nonfunctional lama5/βgeo fusion protein (lama5 trap mice), was generated by putting a trans-membrane domain and βgeo after the first 1763 amino acids of lama5 gene [43]. In both lama5 null and lama5 trap mice, discontinuous inner dental epithelial basement membrane, small tooth germ with defective cusp formation, decreased proliferation of dental epithelium, and defective enamel knot are observed [122], suggesting that lama5 is necessary for the proliferation and polarity of basal epithelial cells. In addition, severe defects in submandibular glands, including delayed epithelial clefting, reduced FGFR1b and FGFR2b expression, and disrupted epithelial cell organization and lumen formation, are found in both lama5 mutants [123], suggesting that lama5 actively regulates epithelial morphogenesis in submandibular glands.
To study the roles of lama5 at later developmental stages, a lama5 floxed (lama5fl/fl) line was generated by inserting two loxP sites in the lama5 gene: one before exon 15 and the other after exon 21 [124]. By crossing the lama5fl/fl line with lung epithelial cell-specific rtTA/tetO-Cre mice, an inducible lama5 conditional knockout mouse line was generated [124]. Mice exposed to doxycycline at E6.5 survive after birth and show severe defects in the lungs, including dilated/enlarged distal airspaces, perturbed distal epithelial cell differentiation, reduced alveolar type I and type II cells, increased apoptosis, decreased proliferation, and diminished capillary density [124], suggesting that epithelial cell-derived lama5 is necessary for alveolar epithelial cell differentiation and lung development. By utilizing podocyte-specific 2.5P-Cre, a conditional knockout mouse line with lama5 abrogation in podocytes was obtained [121]. These mutant mice are viable and show proteinuria, which deteriorates with age and eventually progresses to nephrotic syndrome [121], suggesting that podocyte-derived lama5 is crucial for the maintenance of glomerular filtration barrier integrity [121]. To study the effect of lama5 in neuromuscular junction maturation at postnatal stage, a conditional knockout line with muscle-specific deletion of lama5 was generated by crossing the lama5fl/fl mice with human skeletal actin-driven Cre line [119]. These mutants are grossly normal with a normal lifespan, but demonstrate delayed pre- and post-synaptic maturation at the neuromuscular junction [119]. Furthermore, synaptic maturation is arrested in lama4/lama5 double mutants [119], suggesting that both lama4 and lama5 contribute to synaptic maturation at the neuromuscular junction. Altogether, these results suggest that lama5 has many important biological functions and the exact roles it plays depend on various factors, including tissue type, developmental stage, and compensation by other laminin α chains.
Laminin β1
Lamb1, a β subunit found in most laminin isoforms, is ubiquitously deposited in all basement membranes [42]. To investigate its function, a lamb1 trap line was generated [42, 43]. In these lamb1 trap mice, a gene trap vector is inserted in the 23rd intron of lamb1 to produce a fusion protein containing the first 1178 amino acids of lamb1 followed by a trans-membrane domain and βgeo [42, 43]. The lamb1 mutants die at E5.5 without the formation of either Reichert’s membrane or embryonic basement membrane [42]. In addition, no signs of cavitation and dramatically reduced trophoblast invasion are observed in these lamb1 mutants [42]. These defects are much worse than those in lama1 mutants and may explain the more severe phenotype of the lamb1 mutants. Recent study has identified a point mutation (T5460A) in lamb1 gene, which introduces a premature termination codon, leading to deletion of the last 57 amino acids from lamb1 chain [125]. Mice with this mutation exhibit dystonia-like hindlimb movements when awake and hyperextension when asleep [125], suggesting a pivotal role of the last 57 amino acids of lamb1 in CNS development and neuromuscular junction formation/maturation. This mouse line is a valuable tool for researchers studying movement disorders and neurological diseases.
Laminin β2
Lamb2 is widely expressed at the neuromuscular junction, kidney, and retina. To investigate its functional importance, a lamb2 null line was generated by targeting the second exon of lamb2 gene [126]. The lamb2 null mice are born at the expected Mendelian ratio and indistinguishable from their littermate controls at early postnatal stage [126]. Starting from P7, they become progressive weaker and die between P15 and P30 [126], suggesting that lamb2 is required for postnatal but not embryonic development. As expected, these lamb2 null mice demonstrate abnormal neuromuscular junctions. Structurally, the mutants show few active zones, evenly distributed vesicles in nerve terminals, abnormal Schwann cell processes, and few junctional folds in the postsynaptic membrane [126]. Functionally, significantly reduced miniature endplate potential frequency is observed in lamb2 mutants, although the passive membrane properties of the muscle fibers are normal [126].
In addition to the neuromuscular junction phenotype, a severe kidney defect, characterized by abnormal podocytes, increased glomerular filtration, and disorganized glomerular basement membrane, is observed [127–129]. To isolate the neuromuscular and kidney phenotypes, Miner and colleagues generated two transgenic mouse lines that express rat lamb2 in either muscle or podocytes under the lamb2 null background [128]. They elegantly show that deficiency of lamb2 in podocytes leads to disrupted glomerular filtration barrier, whereas loss of lamb2 in muscle is responsible for the abnormal synaptic architecture and failure to thrive [128].
Additionally, these lamb2 null mice also demonstrate retina defects, including abnormal outer segment elongation/electroretinograms/rod photoreceptor synapses, reduced tyrosine hydroxylase (TH) expression in type I TH neurons, decreased and increased density of type I and type II TH neurons, respectively [130, 131]. Whether retina-derived lamb2 is responsible for these defects, however, is unclear. Since lamb2 null phenotypes closely mimic the pathology of Pierson syndrome, which includes congenital nephrotic syndrome, ocular abnormalities, and muscular/neurological defects [132], these mice are used as an animal model for Pierson syndrome [128]. Consistent with these data, mutations in lamb2 gene have been found in human patients with Pierson syndrome [133, 134]. Based on these results, it is logical to hypothesize that adding lamb2 back would have therapeutic effect in Pierson syndrome. Future studies should focus on investigating the therapeutic potential of lamb2 in this disorder using lamb2 null mice.
Laminin β3
Lamb3 is a subunit of laminin-332, which actively participates in the pathogenesis of H-JEB. A mutation caused by insertion of an intracisternal-A particle (IAP) at an exon/intron junction, which disrupts the coding sequence of lamb3, has been identified in blistering (lamb3IAP) mice [135]. Like lama3 null mice, these lamb3IAP mice develop sub-epithelial blisters and abnormal hemidesmosomes lacking sub-basal dense plates, representing another mouse model of H-JEB [135]. Using this model, Muhle and colleagues demonstrated that prenatal delivery of lamb3 cDNA into the amniotic cavities of lamb3IAP mice increases their lifespan slightly [136].
Recently, a point mutation in lamb3 (G628A), which is frequently observed in humans [12], was introduced into the mouse lamb3 gene. This mutation causes deletion of exon 7 and introduces a premature termination codon, most likely leading to a complete loss of lamb3 protein [137]. The homozygous G628A knockin mice show a phenotype very similar to the lamb3IAP mice [137], suggesting that they can be used as an alternative mouse model of H-JEB. Future studies should focus on generating H-JEB models with longer lifespan, which would allow examination of these laminin chains at older ages. In addition, the therapeutic potential of laminin-332 in H-JEB should also be investigated in these lamb3 mutants.
Laminin β4
Lamb4 is a recently identified β chain. It is considered as a potential tumor suppressor that predisposes to colorectal cancer [138]. Consistent with this study, mutation in lamb4 and loss of lamb4 expression are found in some human colorectal cancer samples [139]. Currently, no lamb4 null mice are available.
Laminin γ1
Lamc1 is the most used γ chain and is found in the majority of laminin molecules [3, 4, 140]. The first lamc1 null mouse line was generated by homologous recombination targeting the first exon of lamc1 gene [141, 142]. These lamc1 mutants die at E5.5 due to severe developmental defects, including lack of basement membranes and failure of endoderm differentiation and parietal yolk sac development, suggesting that laminin γ1 chain is necessary for laminin assembly, endoderm differentiation, and early embryonic development [142]. Using these lamc1 mutants, Murray and Edgar further demonstrate that lamc1 regulates epiblast development and programmed cell death necessary for cavity formation [143]. Additionally, a lamc1 trap line that synthesizes a nonfunctional fusion protein containing the first 1091 amino acids of lamc1 followed by a trans-membrane domain and βgeo was also generated [43]. Like lamc1 null mice, these lamc1 trap mice die during embryogenesis at E5.5 [43], again indicating a critical role of lamc1 in early embryonic development. Recently, a lamc1 mutant line lacking the nidogen-binding site (lamc1III4−/−) was generated by Willem and colleagues [29]. Approximately 40 % of the mutants die before E11.5, whereas 60 % of them die soon after birth [29]. These lamc1III4−/− mutants show renal agenesis and impaired lung development due to locally restricted ruptures of basement membranes in the elongating Wolffian duct and alveolar sacculi, respectively [29]. These data suggest that interaction between nidogen and lamc1 is critical for early kidney morphogenesis and the formation of air–blood interface.
The early embryonic lethality of lamc1 null mice prevents investigation of lamc1’s functions at later stages. To overcome this limitation, a lamc1 floxed (lamc1fl/fl) mouse line, in which exon 2 of the lamc1 gene was flanked by two loxP sites, was generated [144]. This lamc1fl/fl line, by crossing with different promoter-driven Cre lines, allows abrogation of lamc1 expression in specific cell types, enabling loss-of-function studies of lamc1 in a cell type-specific manner.
By crossing this line with CaMKII-Cre, a conditional knockout line (CKO) with lamc1 deficiency in hippocampi, spinal cord, and peripheral nerves was generated [144]. These CKO mice show defects in neurite outgrowth and neuronal migration, including disrupted cortical layer structures and impaired axonal pathfinding, suggesting a critical role of lamc1 in brain development [145]. In additional to the CNS phenotype, these CKO mice also exhibit hindlimb paralysis and tremor due to disrupted lamc1 expression in peripheral nerves, which leads to failure in Schwann cell differentiation, axonal sorting and myelination [144].
By crossing the lamc1fl/fl mice with P0-Cre, a Schwann cell-specific lamc1 knockout line (P0KO) was generated [146]. Similar to the CKO mice, these P0KO mice develop tremor and progressive hindlimb paralysis, and usually die within 2 months [146]. Schwann cells in the mutants show reduced proliferation, increased apoptosis, and compromised extension of processes required for axonal sorting and axon-Schwann cell interaction [146], again suggesting that lamc1 is indispensable for Schwann cell survival, differentiation, and function.
Inactivating lamc1 gene in the developing mouse ureteric bud by crossing the lamc1fl/fl mice with Hoxb7-Cre line leads to compromised renal collecting system growth and function [147]. These mice either fail to develop a ureteric bud or exhibit small kidneys with reduced proliferation/branching, abnormal renal vesicle formation, and water transport defect [147], suggesting γ1-containing laminins are required for normal kidney development and function.
By crossing the lamc1fl/fl line with Nestin-Cre mice, a conditional knockout line (NKO) with lamc1 deficiency in neural progenitor cells (both neurons and glial cells) was generated [148, 149]. These NKO mice develop age-dependent and region-specific intracerebral hemorrhage [148] and blood–brain barrier breakdown in non-hemorrhagic mice [149]. By using the CKO mice and adenovirus expressing Cre under GFAP promoter, it has been demonstrated that these phenotypes are due to loss of γ1-containing laminins in astrocytes specifically [148, 149]. Further mechanistic studies reveal that loss of astrocytic laminin impairs vascular smooth muscle cell maturation [148] and pericyte differentiation [149], leading to intracerebral hemorrhage and blood–brain barrier breakdown, respectively.
Additionally, these NKO mice are unable to respond to angiotensin II (increase blood pressure), although they maintain normal baseline blood pressure [150]. In contrast to the NKO mice, mice with lamc1 deficiency in vascular smooth muscle cells specifically (SKO), generated by crossing the lamc1fl/fl with SM22α-Cre mice, have a lower baseline blood pressure and reduced response to Angiotensin II [150]. These data suggest that nestin+ cell- and vascular smooth muscle cell-derived γ1-laminins have distinct functions, probably due to different laminin isoforms synthesized by these cells.
To assess the functional role of lamc1 in mural cells, a conditional knockout (PKO) line with lamc1 deficiency in PDGFRβ+ mural cells was generated by crossing the lamc1fl/fl line with Pdgfrβ-Cre mice. These PKO mice are born at the expected Mendelian ratio, develop progressive muscle weakness soon after birth, and usually die within 4 months [79]. Further mechanistic studies demonstrate that PDGFRβ+ mural cell-derived γ1-containing laminins inhibit their proliferation, negatively regulate their adipogenic differentiation, and are required for their myogenic differentiation [79], suggesting a critical role of lamc1 in PDGFRβ+ mural cell stemness regulation.
Altogether, these studies suggest that lamc1 exerts a large variety of important functions, which are dependent on its cellular sources. The lamc1fl/fl mouse line, which allows deletion of this gene in any cell type by crossing with cell type-specific promoter-driven Cre lines, provides a valuable tool for future studies.
Laminin γ2
Like lama3 and lamb3, mutations in lamc2 have also been identified in human patients with H-JEB [151]. To better study the functional role of lamc2, a lamc2 null mouse line was generated by targeted frameshift deletion of exon 8 of the lamc2 gene [152]. The lamc2 null mice are born at the expected Mendelian ratio, show blistering phenotype at P1–P2, and die by P5 [152], suggesting that lamc2 is not required for embryonic development. Poorly developed hemidesmosomes and induced apoptosis in the basal cells are observed in these mutants [152], replicating human H-JEB pathology. Additionally, a mild lung defect, characterized by abnormal tracheal hemidesmosomes but normal branching morphogenesis and epithelial differentiation, is found in these lamc2 mutants at P1–P2 [153], suggesting a minimal role of lamc2 in lung development through the saccular stage. Similarly, the therapeutic potential of laminin-332 in H-JEB should also be investigated in this mouse model.
To bypass the early lethality of lamc2 null mice and enable studies of lamc2’s function at later stages, a transgenic line expressing doxycycline-controllable human lamc2 gene under a keratinocyte-specific promoter on the lamc2 null background was generated [154]. These transgenic mice do not have skin blisters, are fertile, and survive over 1.5 years [154]. In these mice, human lamc2 co-localizes with mouse lama3 and lamb3, forming functional laminin-332, which rescues the alterations in the deposition of hemidesmosomal components, leading to restored formation of hemidesmosomes [154]. This transgenic line will be a valuable tool to study the functions of lamc2 in other organs and/or at later stages.
Laminin γ3
Lamc3 is widely but differentially expressed in a variety of tissues in mice. To investigate its biological function, a lamc3 null mouse line was generated by replacing exon 1 and part of the intron 1 of the lamc3 gene with a promoter-less IRES βgeo sequence [155]. Unlike other mutants, the lamc3 null mice develop a normal lifespan and show only minor abnormalities, including ectopic granule cells in the cerebellum and enhanced capillary branching in the outer retina [131, 155]. These data suggest that lamc3 may have some unique functions in the CNS and retina.
Both laminin β2- and γ3-containing laminins are present in the brain and retina during development. To further investigate their roles, a double knockout mouse line lacking both lamb2 and lamc3 (compound null) was generated by crossing lamb2 heterozygote with lamc3 null mice [131]. Like the lamb2 null mice, the compound null mice usually die within 4 weeks of age. In addition to fractured pial basement membrane, the compound null mice exhibit hallmarks of human cobblestone lissencephaly (type II), including laminar disruption, midline fusion, ectopic germinal zones, altered distribution of Cajal-Retzius cells, and abnormal radial glial cell morphology [156], suggesting that lamb2 and lamc3 are necessary for proper cortical lamination. In addition, these compound null mice also show defects in retina, the inner limiting membrane (ILM) in particular, at structural, biochemical, and functional levels. Besides the retinal defects observed in lamb2 null mice, the compound null mice also demonstrate disruptions in the laminar arrangement and ILM as well as alterations in Muller cell polarity and electrophysiology [131, 157, 158]. Many other features, including astrocyte migration and spatial distribution in the retina, retinal vascular development and maturation, and inwardly rectifying potassium channel and aquaporin-4 expression in Muller cells, are also affected in the compound null mice [158, 159]. Additionally, ERG recordings in response to flash stimuli delivered after prolonged dark adaption show that both the postsynaptic responses and photoreceptor-generated currents are severely disrupted in the compound null mice, whereas only the former is disrupted in lamb2 null mice [157]. Altogether, these data suggest that β2- and γ3-containing laminins are critical for the development of the CNS and retina.
Future directions
Although recent studies dramatically enriched our knowledge in laminin biology/function and provided various invaluable genetic tools for future studies, many key questions still need to be answered. First, what are the functions of each laminin subunit in different tissues/organs? Answers to this question would help isolate different phenotypes, which may lead to more precise treatments with few side effects. The floxed mice and the knockout–knockin transgenic lines could be used to answer this question. Future studies should focus on investigating tissue/organ-specific functions of laminin.
Second, what are the functions of each laminin subunit at different developmental stages? Understanding this question would produce a temporal profile of laminin’s functions and allow us to manipulate laminin’s function at a specific time window, which will not only generate innovative disease models that better replicate human pathology, but also promote more efficient and safer therapies. Inducible knockout models would address this key question. Unfortunately, such models are currently not available for most laminin subunits. Efforts should be put on generating such tools in the future.
Third, what are the compensatory changes in these genetic models? Specifically, does deletion of one laminin subunit affect the expression of other variants of this subunit? How about other laminin subunits? How about other extracellular matrix proteins? If yes, are the phenotypes caused by loss of the deleted target genes directly or indirectly induced by compensatory expression of other variants/subunits/extracellular matrix proteins? Answers to these questions would enhance our understanding of laminin’s compensatory mechanism and allow more accurate interpretation of the results.
Fourth, what signaling pathways regulate laminin chain expression? Understanding the molecular mechanisms and signaling pathways that control the expression of each laminin chain enables modulation of laminins at a much delicate level. With such knowledge, we can increase/decrease the expression of any laminin chain by activating/inhibiting corresponding receptor(s) and/or signaling pathway(s) specifically. This will significantly enhance the accuracy and safety of laminin-based treatments. Currently, such information remains largely unknown. Future studies should focus on dissecting the molecular mechanisms and signaling pathways that mediate/regulate laminin expression.
Fifth, how is the basement membrane assembled? Specifically, what factors regulate laminin polymerization and basement membrane assembly? Although substantial progress has been made in the biochemistry of laminin polymerization and basement membrane assembly, the exact mechanism is still elusive. Answers to these questions will enrich our knowledge in laminin biochemistry and provide scientific evidence for laminin-based therapies. More importantly, this knowledge may lead to innovative and more effective therapeutic options or formulations.
Last, how does the basement membrane participate in molecular signaling? It has been shown that the basement membrane can act as a reservoir for grow factors/cytokines/bioactive fragments and activate key signaling molecules. How exact the basement membrane (or its components) exerts these functions and how they are regulated are mostly unknown. Answers to these questions will allow a deeper understanding of basement membrane biology & function and promote the development of novel therapies for various disorders by targeting the basement membrane and/or its components.
Acknowledgments
This work was supported by Myotonic Dystrophy Foundation Fund-A-Fellow Grant (Y.Y.). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Compliance with ethical standards
Conflict of interest
The author declares no competing interests.
References
- 1.Aumailley M, Bruckner-Tuderman L, Carter WG, Deutzmann R, Edgar D, Ekblom P, Engel J, Engvall E, Hohenester E, Jones JC, Kleinman HK, Marinkovich MP, Martin GR, Mayer U, Meneguzzi G, Miner JH, Miyazaki K, Patarroyo M, Paulsson M, Quaranta V, Sanes JR, Sasaki T, Sekiguchi K, Sorokin LM, Talts JF, Tryggvason K, Uitto J, Virtanen I, von der Mark K, Wewer UM, Yamada Y, Yurchenco PD. A simplified laminin nomenclature. Matrix Biol. 2005;24(5):326–332. doi: 10.1016/j.matbio.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 2.Aumailley M. The laminin family. Cell Adhes Migration. 2013;7(1):48–55. doi: 10.4161/cam.22826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Durbeej M. Laminins. Cell Tissue Res. 2010;339(1):259–268. doi: 10.1007/s00441-009-0838-2. [DOI] [PubMed] [Google Scholar]
- 4.Burgeson RE, Chiquet M, Deutzmann R, Ekblom P, Engel J, Kleinman H, Martin GR, Meneguzzi G, Paulsson M, Sanes J, et al. A new nomenclature for the laminins. Matrix Biol. 1994;14(3):209–211. doi: 10.1016/0945-053X(94)90184-8. [DOI] [PubMed] [Google Scholar]
- 5.Ferrigno O, Virolle T, Galliano MF, Chauvin N, Ortonne JP, Meneguzzi G, Aberdam D. Murine laminin alpha3A and alpha3B isoform chains are generated by usage of two promoters and alternative splicing. J Biol Chem. 1997;272(33):20502–20507. doi: 10.1074/jbc.272.33.20502. [DOI] [PubMed] [Google Scholar]
- 6.Sasaki T, Timpl R. Laminins. In: Kries T, Vale R, editors. Guidebook to the extracellular matrix, anchor, and adhesion proteins. Oxford: Oxford University Press; 1999. pp. 434–443. [Google Scholar]
- 7.Maurer P, Engel J. Structure of laminins and their chain assembly. In: Ekblom P, Timpl R, editors. The laminins. Amsterdam: Harwood Academic Press; 1996. pp. 27–49. [Google Scholar]
- 8.Miner JH, Yurchenco PD. Laminin functions in tissue morphogenesis. Annu Rev Cell Dev Biol. 2004;20:255–284. doi: 10.1146/annurev.cellbio.20.010403.094555. [DOI] [PubMed] [Google Scholar]
- 9.Morita A, Sugimoto E, Kitagawa Y. Post-translational assembly and glycosylation of laminin subunits in parietal endoderm-like F9 cells. Biochem J . 1985;229(1):259–264. doi: 10.1042/bj2290259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Champliaud MF, Virtanen I, Tiger CF, Korhonen M, Burgeson R, Gullberg D. Posttranslational modifications and beta/gamma chain associations of human laminin alpha1 and laminin alpha5 chains: purification of laminin-3 from placenta. Exp Cell Res. 2000;259(2):326–335. doi: 10.1006/excr.2000.4980. [DOI] [PubMed] [Google Scholar]
- 11.Tzu J, Marinkovich MP. Bridging structure with function: structural, regulatory, and developmental role of laminins. Int J Biochem Cell Biol. 2008;40(2):199–214. doi: 10.1016/j.biocel.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schneider H, Muhle C, Pacho F. Biological function of laminin-5 and pathogenic impact of its deficiency. Eur J Cell Biol. 2007;86(11–12):701–717. doi: 10.1016/j.ejcb.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 13.Yurchenco PD, Wadsworth WG. Assembly and tissue functions of early embryonic laminins and netrins. Curr Opin Cell Biol. 2004;16(5):572–579. doi: 10.1016/j.ceb.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 14.Kohfeldt E, Sasaki T, Gohring W, Timpl R. Nidogen-2: a new basement membrane protein with diverse binding properties. J Mol Biol. 1998;282(1):99–109. doi: 10.1006/jmbi.1998.2004. [DOI] [PubMed] [Google Scholar]
- 15.Fox JW, Mayer U, Nischt R, Aumailley M, Reinhardt D, Wiedemann H, Mann K, Timpl R, Krieg T, Engel J, et al. Recombinant nidogen consists of three globular domains and mediates binding of laminin to collagen type IV. EMBO J. 1991;10(11):3137–3146. doi: 10.1002/j.1460-2075.1991.tb04875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aumailley M, Battaglia C, Mayer U, Reinhardt D, Nischt R, Timpl R, Fox JW. Nidogen mediates the formation of ternary complexes of basement membrane components. Kidney Int. 1993;43(1):7–12. doi: 10.1038/ki.1993.3. [DOI] [PubMed] [Google Scholar]
- 17.Salmivirta K, Talts JF, Olsson M, Sasaki T, Timpl R, Ekblom P. Binding of mouse nidogen-2 to basement membrane components and cells and its expression in embryonic and adult tissues suggest complementary functions of the two nidogens. Exp Cell Res. 2002;279(2):188–201. doi: 10.1006/excr.2002.5611. [DOI] [PubMed] [Google Scholar]
- 18.Mayer U, Nischt R, Poschl E, Mann K, Fukuda K, Gerl M, Yamada Y, Timpl R. A single EGF-like motif of laminin is responsible for high affinity nidogen binding. EMBO J. 1993;12(5):1879–1885. doi: 10.1002/j.1460-2075.1993.tb05836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poschl E, Fox JW, Block D, Mayer U, Timpl R. Two non-contiguous regions contribute to nidogen binding to a single EGF-like motif of the laminin gamma 1 chain. EMBO J. 1994;13(16):3741–3747. doi: 10.1002/j.1460-2075.1994.tb06683.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poschl E, Mayer U, Stetefeld J, Baumgartner R, Holak TA, Huber R, Timpl R. Site-directed mutagenesis and structural interpretation of the nidogen binding site of the laminin gamma1 chain. EMBO J. 1996;15(19):5154–5159. [PMC free article] [PubMed] [Google Scholar]
- 21.Stetefeld J, Mayer U, Timpl R, Huber R. Crystal structure of three consecutive laminin-type epidermal growth factor-like (LE) modules of laminin gamma1 chain harboring the nidogen binding site. J Mol Biol. 1996;257(3):644–657. doi: 10.1006/jmbi.1996.0191. [DOI] [PubMed] [Google Scholar]
- 22.Timpl R, Brown JC. Supramolecular assembly of basement membranes. BioEssays. 1996;18(2):123–132. doi: 10.1002/bies.950180208. [DOI] [PubMed] [Google Scholar]
- 23.Mayer U, Kohfeldt E, Timpl R. Structural and genetic analysis of laminin-nidogen interaction. Ann N Y Acad Sci. 1998;857:130–142. doi: 10.1111/j.1749-6632.1998.tb10113.x. [DOI] [PubMed] [Google Scholar]
- 24.Yurchenco PD, Patton BL. Developmental and pathogenic mechanisms of basement membrane assembly. Curr Pharm Des. 2009;15(12):1277–1294. doi: 10.2174/138161209787846766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ekblom P, Ekblom M, Fecker L, Klein G, Zhang HY, Kadoya Y, Chu ML, Mayer U, Timpl R. Role of mesenchymal nidogen for epithelial morphogenesis in vitro. Development. 1994;120(7):2003–2014. doi: 10.1242/dev.120.7.2003. [DOI] [PubMed] [Google Scholar]
- 26.Kadoya Y, Salmivirta K, Talts JF, Kadoya K, Mayer U, Timpl R, Ekblom P. Importance of nidogen binding to laminin gamma1 for branching epithelial morphogenesis of the submandibular gland. Development. 1997;124(3):683–691. doi: 10.1242/dev.124.3.683. [DOI] [PubMed] [Google Scholar]
- 27.Tunggal J, Wartenberg M, Paulsson M, Smyth N. Expression of the nidogen-binding site of the laminin gamma1 chain disturbs basement membrane formation and maintenance in F9 embryoid bodies. J Cell Sci. 2003;116(Pt 5):803–812. doi: 10.1242/jcs.00293. [DOI] [PubMed] [Google Scholar]
- 28.Breitkreutz D, Mirancea N, Schmidt C, Beck R, Werner U, Stark HJ, Gerl M, Fusenig NE. Inhibition of basement membrane formation by a nidogen-binding laminin gamma1-chain fragment in human skin-organotypic cocultures. J Cell Sci. 2004;117(Pt 12):2611–2622. doi: 10.1242/jcs.01127. [DOI] [PubMed] [Google Scholar]
- 29.Willem M, Miosge N, Halfter W, Smyth N, Jannetti I, Burghart E, Timpl R, Mayer U. Specific ablation of the nidogen-binding site in the laminin gamma1 chain interferes with kidney and lung development. Development. 2002;129(11):2711–2722. doi: 10.1242/dev.129.11.2711. [DOI] [PubMed] [Google Scholar]
- 30.Mokkapati S, Fleger-Weckmann A, Bechtel M, Koch M, Breitkreutz D, Mayer U, Smyth N, Nischt R. Basement membrane deposition of nidogen 1 but not nidogen 2 requires the nidogen binding module of the laminin gamma1 chain. J Biol Chem. 2011;286(3):1911–1918. doi: 10.1074/jbc.M110.149864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yue B. Biology of the extracellular matrix: an overview. J Glaucoma. 2014;23(8 Suppl 1):S20–S23. doi: 10.1097/IJG.0000000000000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu P, Takai K, Weaver VM, Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol. 2011 doi: 10.1101/cshperspect.a005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yurchenco PD, Smirnov S, Mathus T. Analysis of basement membrane self-assembly and cellular interactions with native and recombinant glycoproteins. Methods Cell Biol. 2002;69:111–144. doi: 10.1016/S0091-679X(02)69010-7. [DOI] [PubMed] [Google Scholar]
- 34.Colognato H, Yurchenco PD. Form and function: the laminin family of heterotrimers. Dev Dyn. 2000;218(2):213–234. doi: 10.1002/(SICI)1097-0177(200006)218:2<213::AID-DVDY1>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 35.Hallmann R, Horn N, Selg M, Wendler O, Pausch F, Sorokin LM. Expression and function of laminins in the embryonic and mature vasculature. Physiol Rev. 2005;85(3):979–1000. doi: 10.1152/physrev.00014.2004. [DOI] [PubMed] [Google Scholar]
- 36.Ekblom P, Lonai P, Talts JF. Expression and biological role of laminin-1. Matrix Biol. 2003;22(1):35–47. doi: 10.1016/S0945-053X(03)00015-5. [DOI] [PubMed] [Google Scholar]
- 37.Miner JH, Cunningham J, Sanes JR. Roles for laminin in embryogenesis: exencephaly, syndactyly, and placentopathy in mice lacking the laminin alpha5 chain. J Cell Biol. 1998;143(6):1713–1723. doi: 10.1083/jcb.143.6.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sasaki T, Fassler R, Hohenester E. Laminin: the crux of basement membrane assembly. J Cell Biol. 2004;164(7):959–963. doi: 10.1083/jcb.200401058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miner JH. Laminins and their roles in mammals. Microsc Res Tech. 2008;71(5):349–356. doi: 10.1002/jemt.20563. [DOI] [PubMed] [Google Scholar]
- 40.Samuel MA, Valdez G, Tapia JC, Lichtman JW, Sanes JR. Agrin and synaptic laminin are required to maintain adult neuromuscular junctions. PLoS ONE. 2012;7(10):e46663. doi: 10.1371/journal.pone.0046663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patton BL. Laminins of the neuromuscular system. Microsc Res Tech. 2000;51(3):247–261. doi: 10.1002/1097-0029(20001101)51:3<247::AID-JEMT5>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 42.Miner JH, Li C, Mudd JL, Go G, Sutherland AE. Compositional and structural requirements for laminin and basement membranes during mouse embryo implantation and gastrulation. Development. 2004;131(10):2247–2256. doi: 10.1242/dev.01112. [DOI] [PubMed] [Google Scholar]
- 43.Yin Y, Kikkawa Y, Mudd JL, Skarnes WC, Sanes JR, Miner JH. Expression of laminin chains by central neurons: analysis with gene and protein trapping techniques. Genesis. 2003;36(2):114–127. doi: 10.1002/gene.10206. [DOI] [PubMed] [Google Scholar]
- 44.Virtanen I, Gullberg D, Rissanen J, Kivilaakso E, Kiviluoto T, Laitinen LA, Lehto VP, Ekblom P. Laminin alpha1-chain shows a restricted distribution in epithelial basement membranes of fetal and adult human tissues. Exp Cell Res. 2000;257(2):298–309. doi: 10.1006/excr.2000.4883. [DOI] [PubMed] [Google Scholar]
- 45.Lentz SI, Miner JH, Sanes JR, Snider WD. Distribution of the ten known laminin chains in the pathways and targets of developing sensory axons. J Comp Neurol. 1997;378(4):547–561. doi: 10.1002/(SICI)1096-9861(19970224)378:4<547::AID-CNE9>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 46.Thomas T, Dziadek M. Genes coding for basement membrane glycoproteins laminin, nidogen, and collagen IV are differentially expressed in the nervous system and by epithelial, endothelial, and mesenchymal cells of the mouse embryo. Exp Cell Res. 1993;208(1):54–67. doi: 10.1006/excr.1993.1222. [DOI] [PubMed] [Google Scholar]
- 47.Ekblom M, Klein G, Mugrauer G, Fecker L, Deutzmann R, Timpl R, Ekblom P. Transient and locally restricted expression of laminin A chain mRNA by developing epithelial cells during kidney organogenesis. Cell. 1990;60(2):337–346. doi: 10.1016/0092-8674(90)90748-4. [DOI] [PubMed] [Google Scholar]
- 48.Alpy F, Jivkov I, Sorokin L, Klein A, Arnold C, Huss Y, Kedinger M, Simon-Assmann P, Lefebvre O. Generation of a conditionally null allele of the laminin alpha1 gene. Genesis. 2005;43(2):59–70. doi: 10.1002/gene.20154. [DOI] [PubMed] [Google Scholar]
- 49.Scheele S, Falk M, Franzen A, Ellin F, Ferletta M, Lonai P, Andersson B, Timpl R, Forsberg E, Ekblom P. Laminin alpha1 globular domains 4-5 induce fetal development but are not vital for embryonic basement membrane assembly. Proc Natl Acad Sci USA. 2005;102(5):1502–1506. doi: 10.1073/pnas.0405095102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Edwards MM, Mammadova-Bach E, Alpy F, Klein A, Hicks WL, Roux M, Simon-Assmann P, Smith RS, Orend G, Wu J, Peachey NS, Naggert JK, Lefebvre O, Nishina PM. Mutations in Lama1 disrupt retinal vascular development and inner limiting membrane formation. J Biol Chem. 2010;285(10):7697–7711. doi: 10.1074/jbc.M109.069575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Edwards MM, McLeod DS, Grebe R, Heng C, Lefebvre O, Lutty GA. Lama1 mutations lead to vitreoretinal blood vessel formation, persistence of fetal vasculature, and epiretinal membrane formation in mice. BMC Dev Biol. 2011;11:60. doi: 10.1186/1471-213X-11-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ichikawa-Tomikawa N, Ogawa J, Douet V, Xu Z, Kamikubo Y, Sakurai T, Kohsaka S, Chiba H, Hattori N, Yamada Y, Arikawa-Hirasawa E. Laminin alpha1 is essential for mouse cerebellar development. Matrix Biol. 2012;31(1):17–28. doi: 10.1016/j.matbio.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zinkevich NS, Bosenko DV, Link BA, Semina EV. laminin alpha 1 gene is essential for normal lens development in zebrafish. BMC Dev Biol. 2006;6:13. doi: 10.1186/1471-213X-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Karlstrom RO, Trowe T, Klostermann S, Baier H, Brand M, Crawford AD, Grunewald B, Haffter P, Hoffmann H, Meyer SU, Muller BK, Richter S, van Eeden FJ, Nusslein-Volhard C, Bonhoeffer F. Zebrafish mutations affecting retinotectal axon pathfinding. Development. 1996;123:427–438. doi: 10.1242/dev.123.1.427. [DOI] [PubMed] [Google Scholar]
- 55.Paulus JD, Halloran MC. Zebrafish bashful/laminin-alpha 1 mutants exhibit multiple axon guidance defects. Dev Dyn. 2006;235(1):213–224. doi: 10.1002/dvdy.20604. [DOI] [PubMed] [Google Scholar]
- 56.Biehlmaier O, Makhankov Y, Neuhauss SC. Impaired retinal differentiation and maintenance in zebrafish laminin mutants. Invest Ophthalmol Vis Sci. 2007;48(6):2887–2894. doi: 10.1167/iovs.06-1212. [DOI] [PubMed] [Google Scholar]
- 57.Semina EV, Bosenko DV, Zinkevich NC, Soules KA, Hyde DR, Vihtelic TS, Willer GB, Gregg RG, Link BA. Mutations in laminin alpha 1 result in complex, lens-independent ocular phenotypes in zebrafish. Dev Biol. 2006;299(1):63–77. doi: 10.1016/j.ydbio.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 58.Pathania M, Semina EV, Duncan MK. Lens extrusion from Laminin alpha 1 mutant zebrafish. Sci World J. 2014;2014:524929. doi: 10.1155/2014/524929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu H, Christmas P, Wu XR, Wewer UM, Engvall E. Defective muscle basement membrane and lack of M-laminin in the dystrophic dy/dy mouse. Proc Natl Acad Sci USA. 1994;91(12):5572–5576. doi: 10.1073/pnas.91.12.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu H, Wu XR, Wewer UM, Engvall E. Murine muscular dystrophy caused by a mutation in the laminin alpha 2 (Lama2) gene. Nat Genet. 1994;8(3):297–302. doi: 10.1038/ng1194-297. [DOI] [PubMed] [Google Scholar]
- 61.Tome FM, Evangelista T, Leclerc A, Sunada Y, Manole E, Estournet B, Barois A, Campbell KP, Fardeau M. Congenital muscular dystrophy with merosin deficiency. C R Acad Sci. 1994;317(4):351–357. [PubMed] [Google Scholar]
- 62.Michelson AM, Russell ES, Harman PJ. Dystrophia muscularis: a hereditary primary myopathy in the house mouse. Proc Natl Acad Sci USA. 1955;41(12):1079–1084. doi: 10.1073/pnas.41.12.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sunada Y, Bernier SM, Kozak CA, Yamada Y, Campbell KP. Deficiency of merosin in dystrophic dy mice and genetic linkage of laminin M chain gene to dy locus. J Biol Chem. 1994;269(19):13729–13732. [PubMed] [Google Scholar]
- 64.Kuang W, Xu H, Vachon PH, Liu L, Loechel F, Wewer UM, Engvall E. Merosin-deficient congenital muscular dystrophy. Partial genetic correction in two mouse models. J Clin Invest. 1998;102(4):844–852. doi: 10.1172/JCI3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miyagoe Y, Hanaoka K, Nonaka I, Hayasaka M, Nabeshima Y, Arahata K, Nabeshima Y, Takeda S. Laminin alpha2 chain-null mutant mice by targeted disruption of the Lama2 gene: a new model of merosin (laminin 2)-deficient congenital muscular dystrophy. FEBS Lett. 1997;415(1):33–39. doi: 10.1016/S0014-5793(97)01007-7. [DOI] [PubMed] [Google Scholar]
- 66.Nakagawa M, Miyagoe-Suzuki Y, Ikezoe K, Miyata Y, Nonaka I, Harii K, Takeda S. Schwann cell myelination occurred without basal lamina formation in laminin alpha2 chain-null mutant (dy3K/dy3K) mice. Glia. 2001;35(2):101–110. doi: 10.1002/glia.1075. [DOI] [PubMed] [Google Scholar]
- 67.Menezes MJ, McClenahan FK, Leiton CV, Aranmolate A, Shan X, Colognato H. The extracellular matrix protein laminin alpha2 regulates the maturation and function of the blood–brain barrier. J Neurosci. 2014;34(46):15260–15280. doi: 10.1523/JNEUROSCI.3678-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sunada Y, Edgar TS, Lotz BP, Rust RS, Campbell KP. Merosin-negative congenital muscular dystrophy associated with extensive brain abnormalities. Neurology. 1995;45(11):2084–2089. doi: 10.1212/WNL.45.11.2084. [DOI] [PubMed] [Google Scholar]
- 69.Gawlik K, Miyagoe-Suzuki Y, Ekblom P, Takeda S, Durbeej M. Laminin alpha1 chain reduces muscular dystrophy in laminin alpha2 chain deficient mice. Hum Mol Genet. 2004;13(16):1775–1784. doi: 10.1093/hmg/ddh190. [DOI] [PubMed] [Google Scholar]
- 70.Hager M, Gawlik K, Nystrom A, Sasaki T, Durbeej M. Laminin {alpha}1 chain corrects male infertility caused by absence of laminin {alpha}2 chain. Am J Pathol. 2005;167(3):823–833. doi: 10.1016/S0002-9440(10)62054-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gawlik KI, Mayer U, Blomberg K, Sonnenberg A, Ekblom P, Durbeej M. Laminin alpha1 chain mediated reduction of laminin alpha2 chain deficient muscular dystrophy involves integrin alpha7beta1 and dystroglycan. FEBS Lett. 2006;580(7):1759–1765. doi: 10.1016/j.febslet.2006.02.027. [DOI] [PubMed] [Google Scholar]
- 72.Gawlik KI, Durbeej M. Transgenic overexpression of laminin alpha1 chain in laminin alpha2 chain-deficient mice rescues the disease throughout the lifespan. Muscle Nerve. 2010;42(1):30–37. doi: 10.1002/mus.21616. [DOI] [PubMed] [Google Scholar]
- 73.Gawlik KI, Li JY, Petersen A, Durbeej M. Laminin alpha1 chain improves laminin alpha2 chain deficient peripheral neuropathy. Hum Mol Genet. 2006;15(18):2690–2700. doi: 10.1093/hmg/ddl201. [DOI] [PubMed] [Google Scholar]
- 74.Gawlik KI, Akerlund M, Carmignac V, Elamaa H, Durbeej M. Distinct roles for laminin globular domains in laminin alpha1 chain mediated rescue of murine laminin alpha2 chain deficiency. PLoS ONE. 2010;5(7):e11549. doi: 10.1371/journal.pone.0011549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rooney JE, Knapp JR, Hodges BL, Wuebbles RD, Burkin DJ. Laminin-111 protein therapy reduces muscle pathology and improves viability of a mouse model of merosin-deficient congenital muscular dystrophy. Am J Pathol. 2012;180(4):1593–1602. doi: 10.1016/j.ajpath.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Van Ry PM, Minogue P, Hodges BL, Burkin DJ. Laminin-111 improves muscle repair in a mouse model of merosin-deficient congenital muscular dystrophy. Hum Mol Genet. 2014;23(2):383–396. doi: 10.1093/hmg/ddt428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Goudenege S, Lamarre Y, Dumont N, Rousseau J, Frenette J, Skuk D, Tremblay JP. Laminin-111: a potential therapeutic agent for Duchenne muscular dystrophy. Mol Therapy. 2010;18(12):2155–2163. doi: 10.1038/mt.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rooney JE, Gurpur PB, Burkin DJ. Laminin-111 protein therapy prevents muscle disease in the mdx mouse model for Duchenne muscular dystrophy. Proc Natl Acad Sci USA. 2009;106(19):7991–7996. doi: 10.1073/pnas.0811599106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yao Y, Norris EH, Widdowson EM, Strickland S. Laminin regulates PDGFRbeta cell stemness and muscle development. Nat Commun. 2016;7:11415. doi: 10.1038/ncomms11415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moll J, Barzaghi P, Lin S, Bezakova G, Lochmuller H, Engvall E, Muller U, Ruegg MA. An agrin minigene rescues dystrophic symptoms in a mouse model for congenital muscular dystrophy. Nature. 2001;413(6853):302–307. doi: 10.1038/35095054. [DOI] [PubMed] [Google Scholar]
- 81.Bentzinger CF, Barzaghi P, Lin S, Ruegg MA. Overexpression of mini-agrin in skeletal muscle increases muscle integrity and regenerative capacity in laminin-alpha2-deficient mice. FASEB J. 2005;19(8):934–942. doi: 10.1096/fj.04-3376com. [DOI] [PubMed] [Google Scholar]
- 82.Meinen S, Barzaghi P, Lin S, Lochmuller H, Ruegg MA. Linker molecules between laminins and dystroglycan ameliorate laminin-alpha2-deficient muscular dystrophy at all disease stages. J Cell Biol. 2007;176(7):979–993. doi: 10.1083/jcb.200611152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Qiao C, Li J, Zhu T, Draviam R, Watkins S, Ye X, Chen C, Li J, Xiao X. Amelioration of laminin-alpha2-deficient congenital muscular dystrophy by somatic gene transfer of miniagrin. Proc Natl Acad Sci USA. 2005;102(34):11999–12004. doi: 10.1073/pnas.0502137102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Girgenrath M, Dominov JA, Kostek CA, Miller JB. Inhibition of apoptosis improves outcome in a model of congenital muscular dystrophy. J Clin Investig. 2004;114(11):1635–1639. doi: 10.1172/JCI22928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Girgenrath M, Beermann ML, Vishnudas VK, Homma S, Miller JB. Pathology is alleviated by doxycycline in a laminin-alpha2-null model of congenital muscular dystrophy. Ann Neurol. 2009;65(1):47–56. doi: 10.1002/ana.21523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Erb M, Meinen S, Barzaghi P, Sumanovski LT, Courdier-Fruh I, Ruegg MA, Meier T. Omigapil ameliorates the pathology of muscle dystrophy caused by laminin-alpha2 deficiency. J Pharmacol Exp Ther. 2009;331(3):787–795. doi: 10.1124/jpet.109.160754. [DOI] [PubMed] [Google Scholar]
- 87.Carmignac V, Quere R, Durbeej M. Proteasome inhibition improves the muscle of laminin alpha2 chain-deficient mice. Hum Mol Genet. 2011;20(3):541–552. doi: 10.1093/hmg/ddq499. [DOI] [PubMed] [Google Scholar]
- 88.Korner Z, Fontes-Oliveira CC, Holmberg J, Carmignac V, Durbeej M. Bortezomib partially improves laminin alpha2 chain-deficient muscular dystrophy. Am J Pathol. 2014;184(5):1518–1528. doi: 10.1016/j.ajpath.2014.01.019. [DOI] [PubMed] [Google Scholar]
- 89.Carmignac V, Svensson M, Korner Z, Elowsson L, Matsumura C, Gawlik KI, Allamand V, Durbeej M. Autophagy is increased in laminin alpha2 chain-deficient muscle and its inhibition improves muscle morphology in a mouse model of MDC1A. Hum Mol Genet. 2011;20(24):4891–4902. doi: 10.1093/hmg/ddr427. [DOI] [PubMed] [Google Scholar]
- 90.Meinen S, Lin S, Ruegg MA. Angiotensin II type 1 receptor antagonists alleviate muscle pathology in the mouse model for laminin-alpha2-deficient congenital muscular dystrophy (MDC1A) Skelet Muscle. 2012;2(1):18. doi: 10.1186/2044-5040-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Meinen S, Lin S, Thurnherr R, Erb M, Meier T, Ruegg MA. Apoptosis inhibitors and mini-agrin have additive benefits in congenital muscular dystrophy mice. EMBO Mol Med. 2011;3(8):465–479. doi: 10.1002/emmm.201100151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yamauchi J, Kumar A, Duarte L, Mehuron T, Girgenrath M. Triggering regeneration and tackling apoptosis: a combinatorial approach to treating congenital muscular dystrophy type 1 A. Hum Mol Genet. 2013;22(21):4306–4317. doi: 10.1093/hmg/ddt280. [DOI] [PubMed] [Google Scholar]
- 93.Durbeej M. Laminin-alpha2 chain-deficient congenital muscular dystrophy: pathophysiology and development of treatment. Curr Top Membr. 2015;76:31–60. doi: 10.1016/bs.ctm.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 94.Carter WG, Ryan MC, Gahr PJ. Epiligrin, a new cell adhesion ligand for integrin alpha 3 beta 1 in epithelial basement membranes. Cell. 1991;65(4):599–610. doi: 10.1016/0092-8674(91)90092-D. [DOI] [PubMed] [Google Scholar]
- 95.Aberdam D, Galliano MF, Vailly J, Pulkkinen L, Bonifas J, Christiano AM, Tryggvason K, Uitto J, Epstein EH, Jr, Ortonne JP, et al. Herlitz’s junctional epidermolysis bullosa is linked to mutations in the gene (LAMC2) for the gamma 2 subunit of nicein/kalinin (LAMININ-5) Nat Genet. 1994;6(3):299–304. doi: 10.1038/ng0394-299. [DOI] [PubMed] [Google Scholar]
- 96.Ryan MC, Lee K, Miyashita Y, Carter WG. Targeted disruption of the LAMA3 gene in mice reveals abnormalities in survival and late stage differentiation of epithelial cells. J Cell Biol. 1999;145(6):1309–1323. doi: 10.1083/jcb.145.6.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.McGrath JA, Kivirikko S, Ciatti S, Moss C, Dunnill GS, Eady RA, Rodeck CH, Christiano AM, Uitto J. A homozygous nonsense mutation in the alpha 3 chain gene of laminin 5 (LAMA3) in Herlitz junctional epidermolysis bullosa: prenatal exclusion in a fetus at risk. Genomics. 1995;29(1):282–284. doi: 10.1006/geno.1995.1246. [DOI] [PubMed] [Google Scholar]
- 98.Urich D, Eisenberg JL, Hamill KJ, Takawira D, Chiarella SE, Soberanes S, Gonzalez A, Koentgen F, Manghi T, Hopkinson SB, Misharin AV, Perlman H, Mutlu GM, Budinger GR, Jones JC. Lung-specific loss of the laminin alpha3 subunit confers resistance to mechanical injury. J Cell Sci. 2011;124(Pt 17):2927–2937. doi: 10.1242/jcs.080911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Frieser M, Nockel H, Pausch F, Roder C, Hahn A, Deutzmann R, Sorokin LM (1997) Cloning of the mouse laminin alpha 4 cDNA. Expression in a subset of endothelium. Eur J Biochem/FEBS 246 (3):727–735 [DOI] [PubMed]
- 100.Iivanainen A, Kortesmaa J, Sahlberg C, Morita T, Bergmann U, Thesleff I, Tryggvason K. Primary structure, developmental expression, and immunolocalization of the murine laminin alpha4 chain. J Biol Chem. 1997;272(44):27862–27868. doi: 10.1074/jbc.272.44.27862. [DOI] [PubMed] [Google Scholar]
- 101.Lefebvre O, Sorokin L, Kedinger M, Simon-Assmann P. Developmental expression and cellular origin of the laminin alpha2, alpha4, and alpha5 chains in the intestine. Dev Biol. 1999;210(1):135–150. doi: 10.1006/dbio.1999.9270. [DOI] [PubMed] [Google Scholar]
- 102.Patton BL, Miner JH, Chiu AY, Sanes JR. Distribution and function of laminins in the neuromuscular system of developing, adult, and mutant mice. J Cell Biol. 1997;139(6):1507–1521. doi: 10.1083/jcb.139.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Thyboll J, Kortesmaa J, Cao R, Soininen R, Wang L, Iivanainen A, Sorokin L, Risling M, Cao Y, Tryggvason K. Deletion of the laminin alpha4 chain leads to impaired microvessel maturation. Mol Cell Biol. 2002;22(4):1194–1202. doi: 10.1128/MCB.22.4.1194-1202.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wu C, Ivars F, Anderson P, Hallmann R, Vestweber D, Nilsson P, Robenek H, Tryggvason K, Song J, Korpos E, Loser K, Beissert S, Georges-Labouesse E, Sorokin LM. Endothelial basement membrane laminin alpha5 selectively inhibits T lymphocyte extravasation into the brain. Nat Med. 2009;15(5):519–527. doi: 10.1038/nm.1957. [DOI] [PubMed] [Google Scholar]
- 105.Patton BL, Cunningham JM, Thyboll J, Kortesmaa J, Westerblad H, Edstrom L, Tryggvason K, Sanes JR. Properly formed but improperly localized synaptic specializations in the absence of laminin alpha4. Nat Neurosci. 2001;4(6):597–604. doi: 10.1038/88414. [DOI] [PubMed] [Google Scholar]
- 106.Abrass CK, Hansen KM, Patton BL. Laminin alpha4-null mutant mice develop chronic kidney disease with persistent overexpression of platelet-derived growth factor. Am J Pathol. 2010;176(2):839–849. doi: 10.2353/ajpath.2010.090570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vaicik MK, Thyboll Kortesmaa J, Moverare-Skrtic S, Kortesmaa J, Soininen R, Bergstrom G, Ohlsson C, Chong LY, Rozell B, Emont M, Cohen RN, Brey EM, Tryggvason K. Laminin alpha4 deficient mice exhibit decreased capacity for adipose tissue expansion and weight gain. PLoS ONE. 2014;9(10):e109854. doi: 10.1371/journal.pone.0109854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang D, Bierman J, Tarumi YS, Zhong YP, Rangwala R, Proctor TM, Miyagoe-Suzuki Y, Takeda S, Miner JH, Sherman LS, Gold BG, Patton BL. Coordinate control of axon defasciculation and myelination by laminin-2 and -8. J Cell Biol. 2005;168(4):655–666. doi: 10.1083/jcb.200411158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Miner JH, Li C, Patton BL. Laminins alpha2 and alpha4 in pancreatic acinar basement membranes are required for basal receptor localization. J Histochem Cytochem. 2004;52(2):153–156. doi: 10.1177/002215540405200202. [DOI] [PubMed] [Google Scholar]
- 110.Coles EG, Gammill LS, Miner JH, Bronner-Fraser M. Abnormalities in neural crest cell migration in laminin alpha5 mutant mice. Dev Biol. 2006;289(1):218–228. doi: 10.1016/j.ydbio.2005.10.031. [DOI] [PubMed] [Google Scholar]
- 111.Miner JH, Li C. Defective glomerulogenesis in the absence of laminin alpha5 demonstrates a developmental role for the kidney glomerular basement membrane. Dev Biol. 2000;217(2):278–289. doi: 10.1006/dbio.1999.9546. [DOI] [PubMed] [Google Scholar]
- 112.Nguyen NM, Miner JH, Pierce RA, Senior RM. Laminin alpha 5 is required for lobar septation and visceral pleural basement membrane formation in the developing mouse lung. Dev Biol. 2002;246(2):231–244. doi: 10.1006/dbio.2002.0658. [DOI] [PubMed] [Google Scholar]
- 113.Gao J, DeRouen MC, Chen CH, Nguyen M, Nguyen NT, Ido H, Harada K, Sekiguchi K, Morgan BA, Miner JH, Oro AE, Marinkovich MP. Laminin-511 is an epithelial message promoting dermal papilla development and function during early hair morphogenesis. Genes Dev. 2008;22(15):2111–2124. doi: 10.1101/gad.1689908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Li J, Tzu J, Chen Y, Zhang YP, Nguyen NT, Gao J, Bradley M, Keene DR, Oro AE, Miner JH, Marinkovich MP. Laminin-10 is crucial for hair morphogenesis. EMBO J. 2003;22(10):2400–2410. doi: 10.1093/emboj/cdg239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mahoney ZX, Stappenbeck TS, Miner JH. Laminin alpha 5 influences the architecture of the mouse small intestine mucosa. J Cell Sci. 2008;121(Pt 15):2493–2502. doi: 10.1242/jcs.025528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ritie L, Spenle C, Lacroute J, Bolcato-Bellemin AL, Lefebvre O, Bole-Feysot C, Jost B, Klein A, Arnold C, Kedinger M, Bagnard D, Orend G, Simon-Assmann P. Abnormal Wnt and PI3Kinase signaling in the malformed intestine of lama5 deficient mice. PLoS ONE. 2012;7(5):e37710. doi: 10.1371/journal.pone.0037710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bolcato-Bellemin AL, Lefebvre O, Arnold C, Sorokin L, Miner JH, Kedinger M, Simon-Assmann P. Laminin alpha5 chain is required for intestinal smooth muscle development. Dev Biol. 2003;260(2):376–390. doi: 10.1016/S0012-1606(03)00254-9. [DOI] [PubMed] [Google Scholar]
- 118.Kikkawa Y, Miner JH. Molecular dissection of laminin alpha 5 in vivo reveals separable domain-specific roles in embryonic development and kidney function. Dev Biol. 2006;296(1):265–277. doi: 10.1016/j.ydbio.2006.04.463. [DOI] [PubMed] [Google Scholar]
- 119.Nishimune H, Valdez G, Jarad G, Moulson CL, Muller U, Miner JH, Sanes JR. Laminins promote postsynaptic maturation by an autocrine mechanism at the neuromuscular junction. J Cell Biol. 2008;182(6):1201–1215. doi: 10.1083/jcb.200805095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shannon MB, Patton BL, Harvey SJ, Miner JH. A hypomorphic mutation in the mouse laminin alpha5 gene causes polycystic kidney disease. J Am Soc Nephrol JASN. 2006;17(7):1913–1922. doi: 10.1681/ASN.2005121298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Goldberg S, Adair-Kirk TL, Senior RM, Miner JH. Maintenance of glomerular filtration barrier integrity requires laminin alpha5. J Am Soc Nephrol JASN. 2010;21(4):579–586. doi: 10.1681/ASN.2009091004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fukumoto S, Miner JH, Ida H, Fukumoto E, Yuasa K, Miyazaki H, Hoffman MP, Yamada Y. Laminin alpha5 is required for dental epithelium growth and polarity and the development of tooth bud and shape. J Biol Chem. 2006;281(8):5008–5016. doi: 10.1074/jbc.M509295200. [DOI] [PubMed] [Google Scholar]
- 123.Rebustini IT, Patel VN, Stewart JS, Layvey A, Georges-Labouesse E, Miner JH, Hoffman MP. Laminin alpha5 is necessary for submandibular gland epithelial morphogenesis and influences FGFR expression through beta1 integrin signaling. Dev Biol. 2007;308(1):15–29. doi: 10.1016/j.ydbio.2007.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nguyen NM, Kelley DG, Schlueter JA, Meyer MJ, Senior RM, Miner JH. Epithelial laminin alpha5 is necessary for distal epithelial cell maturation, VEGF production, and alveolization in the developing murine lung. Dev Biol. 2005;282(1):111–125. doi: 10.1016/j.ydbio.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 125.Liu YB, Tewari A, Salameh J, Arystarkhova E, Hampton TG, Brashear A, Ozelius LJ, Khodakhah K, Sweadner KJ (2015) A dystonia-like movement disorder with brain and spinal neuronal defects is caused by mutation of the mouse laminin beta1 subunit, Lamb1. eLife 4. doi:10.7554/eLife.11102 [DOI] [PMC free article] [PubMed]
- 126.Noakes PG, Gautam M, Mudd J, Sanes JR, Merlie JP. Aberrant differentiation of neuromuscular junctions in mice lacking s-laminin/laminin beta 2. Nature. 1995;374(6519):258–262. doi: 10.1038/374258a0. [DOI] [PubMed] [Google Scholar]
- 127.Noakes PG, Miner JH, Gautam M, Cunningham JM, Sanes JR, Merlie JP. The renal glomerulus of mice lacking s-laminin/laminin beta 2: nephrosis despite molecular compensation by laminin beta 1. Nat Genet. 1995;10(4):400–406. doi: 10.1038/ng0895-400. [DOI] [PubMed] [Google Scholar]
- 128.Miner JH, Go G, Cunningham J, Patton BL, Jarad G. Transgenic isolation of skeletal muscle and kidney defects in laminin beta2 mutant mice: implications for Pierson syndrome. Development. 2006;133(5):967–975. doi: 10.1242/dev.02270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jarad G, Cunningham J, Shaw AS, Miner JH. Proteinuria precedes podocyte abnormalities inLamb2−/− mice, implicating the glomerular basement membrane as an albumin barrier. J Clin Investig. 2006;116(8):2272–2279. doi: 10.1172/JCI28414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Libby RT, Lavallee CR, Balkema GW, Brunken WJ, Hunter DD. Disruption of laminin beta2 chain production causes alterations in morphology and function in the CNS. J Neurosci. 1999;19(21):9399–9411. doi: 10.1523/JNEUROSCI.19-21-09399.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Denes V, Witkovsky P, Koch M, Hunter DD, Pinzon-Duarte G, Brunken WJ. Laminin deficits induce alterations in the development of dopaminergic neurons in the mouse retina. Vis Neurosci. 2007;24(4):549–562. doi: 10.1017/S0952523807070514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zenker M, Tralau T, Lennert T, Pitz S, Mark K, Madlon H, Dotsch J, Reis A, Muntefering H, Neumann LM. Congenital nephrosis, mesangial sclerosis, and distinct eye abnormalities with microcoria: an autosomal recessive syndrome. Am J Med Genet Part A. 2004;130A(2):138–145. doi: 10.1002/ajmg.a.30310. [DOI] [PubMed] [Google Scholar]
- 133.Zenker M, Pierson M, Jonveaux P, Reis A. Demonstration of two novel LAMB2 mutations in the original Pierson syndrome family reported 42 years ago. Am J Med Genet Part A. 2005;138(1):73–74. doi: 10.1002/ajmg.a.30894. [DOI] [PubMed] [Google Scholar]
- 134.Zenker M, Aigner T, Wendler O, Tralau T, Muntefering H, Fenski R, Pitz S, Schumacher V, Royer-Pokora B, Wuhl E, Cochat P, Bouvier R, Kraus C, Mark K, Madlon H, Dotsch J, Rascher W, Maruniak-Chudek I, Lennert T, Neumann LM, Reis A. Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum Mol Genet. 2004;13(21):2625–2632. doi: 10.1093/hmg/ddh284. [DOI] [PubMed] [Google Scholar]
- 135.Kuster JE, Guarnieri MH, Ault JG, Flaherty L, Swiatek PJ. IAP insertion in the murine LamB3 gene results in junctional epidermolysis bullosa. Mamm Genome. 1997;8(9):673–681. doi: 10.1007/s003359900535. [DOI] [PubMed] [Google Scholar]
- 136.Muhle C, Neuner A, Park J, Pacho F, Jiang Q, Waddington SN, Schneider H. Evaluation of prenatal intra-amniotic LAMB3 gene delivery in a mouse model of Herlitz disease. Gene Ther. 2006;13(23):1665–1676. doi: 10.1038/sj.gt.3302832. [DOI] [PubMed] [Google Scholar]
- 137.Hammersen J, Hou J, Wunsche S, Brenner S, Winkler T, Schneider H. A new mouse model of junctional epidermolysis bullosa: the LAMB3 628G>A knockin mouse. J Invest Dermatol. 2015;135(3):921–924. doi: 10.1038/jid.2014.466. [DOI] [PubMed] [Google Scholar]
- 138.Smith CG, Naven M, Harris R, Colley J, West H, Li N, Liu Y, Adams R, Maughan TS, Nichols L, Kaplan R, Wagner MJ, McLeod HL, Cheadle JP. Exome resequencing identifies potential tumor-suppressor genes that predispose to colorectal cancer. Hum Mutat. 2013;34(7):1026–1034. doi: 10.1002/humu.22333. [DOI] [PubMed] [Google Scholar]
- 139.Choi MR, An CH, Yoo NJ, Lee SH. Laminin gene LAMB4 is somatically mutated and expressionally altered in gastric and colorectal cancers. APMIS. 2015;123(1):65–71. doi: 10.1111/apm.12309. [DOI] [PubMed] [Google Scholar]
- 140.Miner JH, Patton BL, Lentz SI, Gilbert DJ, Snider WD, Jenkins NA, Copeland NG, Sanes JR. The laminin alpha chains: expression, developmental transitions, and chromosomal locations of alpha1-5, identification of heterotrimeric laminins 8–11, and cloning of a novel alpha3 isoform. J Cell Biol. 1997;137(3):685–701. doi: 10.1083/jcb.137.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Smyth N, Vatansever HS, Meyer M, Frie C, Paulsson M, Edgar D. The targeted deletion of the LAMC1 gene. Ann N Y Acad Sci. 1998;857:283–286. doi: 10.1111/j.1749-6632.1998.tb10133.x. [DOI] [PubMed] [Google Scholar]
- 142.Smyth N, Vatansever HS, Murray P, Meyer M, Frie C, Paulsson M, Edgar D. Absence of basement membranes after targeting the LAMC1 gene results in embryonic lethality due to failure of endoderm differentiation. J Cell Biol. 1999;144(1):151–160. doi: 10.1083/jcb.144.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Murray P, Edgar D. Regulation of programmed cell death by basement membranes in embryonic development. J Cell Biol. 2000;150(5):1215–1221. doi: 10.1083/jcb.150.5.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chen ZL, Strickland S. Laminin gamma1 is critical for Schwann cell differentiation, axon myelination, and regeneration in the peripheral nerve. J Cell Biol. 2003;163(4):889–899. doi: 10.1083/jcb.200307068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chen ZL, Haegeli V, Yu H, Strickland S. Cortical deficiency of laminin gamma1 impairs the AKT/GSK-3beta signaling pathway and leads to defects in neurite outgrowth and neuronal migration. Dev Biol. 2009;327(1):158–168. doi: 10.1016/j.ydbio.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yu WM, Feltri ML, Wrabetz L, Strickland S, Chen ZL. Schwann cell-specific ablation of laminin gamma1 causes apoptosis and prevents proliferation. J Neurosci. 2005;25(18):4463–4472. doi: 10.1523/JNEUROSCI.5032-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yang DH, McKee KK, Chen ZL, Mernaugh G, Strickland S, Zent R, Yurchenco PD. Renal collecting system growth and function depend upon embryonic gamma1 laminin expression. Development. 2011;138(20):4535–4544. doi: 10.1242/dev.071266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chen ZL, Yao Y, Norris EH, Kruyer A, Jno-Charles O, Akhmerov A, Strickland S. Ablation of astrocytic laminin impairs vascular smooth muscle cell function and leads to hemorrhagic stroke. J Cell Biol. 2013;202(2):381–395. doi: 10.1083/jcb.201212032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yao Y, Chen ZL, Norris EH, Strickland S. Astrocytic laminin regulates pericyte differentiation and maintains blood brain barrier integrity. Nat Commun. 2014;5:3413. doi: 10.1038/ncomms4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yao Y, Norris EH, Strickland S. The cellular origin of laminin determines its role in blood pressure regulation. Cell Mol Life Sci. 2015;72(5):999–1008. doi: 10.1007/s00018-014-1732-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pulkkinen L, Christiano AM, Airenne T, Haakana H, Tryggvason K, Uitto J. Mutations in the gamma 2 chain gene (LAMC2) of kalinin/laminin 5 in the junctional forms of epidermolysis bullosa. Nat Genet. 1994;6(3):293–297. doi: 10.1038/ng0394-293. [DOI] [PubMed] [Google Scholar]
- 152.Meng X, Klement JF, Leperi DA, Birk DE, Sasaki T, Timpl R, Uitto J, Pulkkinen L. Targeted inactivation of murine laminin gamma2-chain gene recapitulates human junctional epidermolysis bullosa. J Invest Dermatol. 2003;121(4):720–731. doi: 10.1046/j.1523-1747.2003.12515.x. [DOI] [PubMed] [Google Scholar]
- 153.Nguyen NM, Pulkkinen L, Schlueter JA, Meneguzzi G, Uitto J, Senior RM. Lung development in laminin gamma2 deficiency: abnormal tracheal hemidesmosomes with normal branching morphogenesis and epithelial differentiation. Respir Res. 2006;7:28. doi: 10.1186/1465-9921-7-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Adair-Kirk TL, Griffin GL, Meyer MJ, Kelley DG, Miner JH, Keene DR, Marinkovich MP, Ruppert JM, Uitto J, Senior RM. Keratinocyte-targeted expression of human laminin gamma2 rescues skin blistering and early lethality of laminin gamma2 deficient mice. PLoS ONE. 2012;7(9):e45546. doi: 10.1371/journal.pone.0045546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Li YN, Radner S, French MM, Pinzon-Duarte G, Daly GH, Burgeson RE, Koch M, Brunken WJ. The gamma3 chain of laminin is widely but differentially expressed in murine basement membranes: expression and functional studies. Matrix Biol. 2012;31(2):120–134. doi: 10.1016/j.matbio.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Radner S, Banos C, Bachay G, Li YN, Hunter DD, Brunken WJ, Yee KT. beta2 and gamma3 laminins are critical cortical basement membrane components: ablation of Lamb2 and Lamc3 genes disrupts cortical lamination and produces dysplasia. Dev Neurobiol. 2013;73(3):209–229. doi: 10.1002/dneu.22057. [DOI] [PubMed] [Google Scholar]
- 157.Pinzon-Duarte G, Daly G, Li YN, Koch M, Brunken WJ. Defective formation of the inner limiting membrane in laminin beta2- and gamma3-null mice produces retinal dysplasia. Invest Ophthalmol Vis Sci. 2010;51(3):1773–1782. doi: 10.1167/iovs.09-4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hirrlinger PG, Pannicke T, Winkler U, Claudepierre T, Varshney S, Schulze C, Reichenbach A, Brunken WJ, Hirrlinger J. Genetic deletion of laminin isoforms beta2 and gamma3 induces a reduction in Kir4.1 and aquaporin-4 expression and function in the retina. PLoS ONE. 2011;6(1):e16106. doi: 10.1371/journal.pone.0016106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Gnanaguru G, Bachay G, Biswas S, Pinzon-Duarte G, Hunter DD, Brunken WJ. Laminins containing the beta2 and gamma3 chains regulate astrocyte migration and angiogenesis in the retina. Development. 2013;140(9):2050–2060. doi: 10.1242/dev.087817. [DOI] [PMC free article] [PubMed] [Google Scholar]