

Abstract
Broadly neutralizing antibodies (bnAbs) can protect against HIV infection but have not been induced by human vaccination. A key barrier to bnAb induction is vaccine priming of rare bnAb-precursor B cells. In a randomized, double-blind, placebo-controlled phase I clinical trial, the HIV vaccine priming candidate eOD-GT8 60mer adjuvanted with AS01B had a favorable safety profile and induced VRC01-class bnAb precursors in 97% of vaccine recipients with median frequencies reaching 0.1% among Immunoglobulin G B cells in blood. BnAb precursors shared properties with bnAbs and gained somatic hypermutation and affinity with the boost. The results establish clinical proof of concept for germline-targeting vaccine priming, support development of boosting regimens to induce bnAbs, and encourage application of the germline-targeting strategy to other targets in HIV and other pathogens.
One-Sentence Summary:
Proof of principle for germline-targeting vaccine priming in humans.
Development of a preventative HIV vaccine is needed to end the HIV/AIDS pandemic (1). Broadly neutralizing antibodies (bnAbs), Abs that bind the envelope (Env) trimer and neutralize diverse HIV isolates, have been shown to provide sterilizing protection in non-human primate models (2), and infusion of the bnAb VRC01 was shown to protect against neutralization-sensitive HIV isolates in humans (3, 4). It is widely thought that an effective preventative HIV vaccine will need to induce bnAbs.
HIV vaccine design strategies to elicit broadly neutralizing antibodies
BnAbs, like all antibodies, are produced by B cells and acquire affinity-enhancing mutations when a B cell matures from the original naive (or germline) state. The discovery that most HIV Env proteins have no detectable affinity for bnAb germline precursors greatly influenced the development of HIV vaccine strategies, by indicating that special immunogens with affinity for bnAb germline precursors would be needed to prime bnAb responses, and different booster immunogens would be needed to select for antibody maturation to produce bnAbs (5–14).
The HIV vaccine field is currently pursuing at least three strategies to elicit bnAbs, each of which involves sequential vaccination with different antigens to guide the immune response through several stages of maturation. These strategies include: (i) B cell lineage vaccine design, in which the series of immunogens derives from the series of Env variants isolated from longitudinal analysis of bnAb development in a person with natural HIV-1 infection, and the first (priming) immunogen is selected to have affinity for the unmutated common ancestor for the bnAb lineage in that case study, and is usually the transmitted-founder Env in that case study (15–20); (ii) germline-targeting vaccine design, in which the priming immunogen is engineered to bind diverse precursors within a bnAb class (spanning many lineages), and boost immunogens are successively more like native Env trimers (13, 14, 21–29); and (iii) epitope-focused vaccine design, in which the series of immunogens aims to focus responses to one or more particular structural epitopes on the trimer (30–38). In each strategy, the priming stage is critical, because if appropriate B cell precursors with potential to develop into bnAbs are not stimulated at that stage, then the rest of the sequential vaccine will likely fail. Experimental medicine (phase 1) clinical trials are now underway or planned for each strategy, to test priming immunogens or sequential combinations of immunogens for their abilities to elicit desired Ab responses.
First-in-human test of germline targeting
We conducted a first-in-human test of the germline-targeting strategy, by evaluating the safety and immune responses of a germline-targeting priming vaccine candidate, eOD-GT8 60mer adjuvanted with AS01B, in the IAVI G001 Phase 1 clinical trial. The vaccine immunogen is a self-assembling nanoparticle presenting 60 copies of an HIV gp120 engineered outer domain, germline-targeting version 8 (eOD-GT8), genetically fused to and arrayed externally on an interior lumazine synthase nanoparticle (13, 21, 39, 40). eOD-GT8 was designed to have affinity for inferred-germline precursors to VRC01-class bnAbs (13, 21, 39). VRC01-class antibodies are minimally defined as those with heavy chain (HC) V gene alleles VH1–2*02 or *04, and any light chain (LC) complementarity determining region 3 (LCDR3) with a length of five amino acids (13, 14, 41, 42). These sequence features define a broad class of antibodies with diverse LCs and HCDR3s. In pre-clinical experiments, eOD-GT8 was shown to bind to diverse VRC01-class human naive B cells at an average frequency of approximately 1 in 300,000 naive B cells, in 26 of 27 donors tested (96%), and with substantial affinity (geomean dissociation constant (KD) of 4 μM) (21, 43, 44). Adjuvanted eOD-GT8 60mer was shown capable of priming VRC01-class B cell responses in multiple different engineered mouse models (27, 39, 45–49), including stringent models mimicking two key parameters of human vaccination, precursor frequency and affinity (27, 46, 48, 49). Adjuvanted eOD-GT8 60mer was also shown to prime VRC01-class responses that can be boosted toward bnAb development in mouse models (26, 27, 29). However, adjuvanted eOD-GT8 60mer failed to elicit VRC01-class responses in NHPs despite inducing robust germinal center (GC) B and T cell responses, and serum responses (50), likely due to the lack of a suitable human VH1–2 analog (13, 41, 51, 52) and a lower rate of 5-amino acid LCDR3s in NHPs (50, 51). In the IAVI G001 trial, we sought to determine if human immunization with adjuvanted eOD-GT8 60mer is safe and effective for inducing VRC01-class IgG B cell responses. Forty-eight participants were immunized with either 20 μg eOD-GT8 60mer and 50 μg AS01B (N=18), 100 μg eOD-GT8 60mer and 50 μg AS01B (N=18), or the placebo DPBS sucrose, the buffer diluent used in the vaccine (N=12) (fig. S1 and table S1). Vaccine or placebo were administered at weeks 0 and 8 intramuscularly in the same deltoid. The full schedule of procedures for safety and immunogenicity evaluation is given in table S2.
Completeness, safety, and reactogenicity
All but one study participant received both vaccinations; one declined the second vaccination due to a medical diagnosis unrelated to the trial. Forty-five of 48 participants completed all study procedures, and only 1.0% of all 768 visits were missed (fig. S1 and table S3). No serious adverse events (AEs) were reported, and no participants acquired HIV-1 infection or developed serum positivity for HIV. Forty seven of 48 participants (97.9%) reported local and/or systemic AEs (tables S4–S6), but AEs were generally mild or moderate, resolved in most cases within 1–2 days, and were consistent with other vaccines (53). Overall, the vaccine had an acceptable safety and tolerability profile.
Serum antibody responses
After the first immunization, all vaccine recipients but no placebo recipients produced serum IgG binding antibodies to eOD-GT8 60mer and monomer, and to the eOD-GT8 CD4 binding site (CD4bs) epitope, as indicated by stronger binding to eOD-GT8 than to eOD-GT8-KO11, an epitope-knockout mutant that blocks binding by VRC01-class precursor Abs and bnAbs (43, 46–49) (figs. S2–3 and tables S7–8). Vaccine-induced responses to eOD-GT8 60mer and monomer increased after the second vaccination, while CD4bs-specific responses remained relatively constant (figs. S2–3 and tables S7–8). All participants exhibited pre-immunization reactivity to the lumazine synthase (LS) base nanoparticle, and LS responses increased with both immunizations in vaccine recipients (figs. S2–3 and tables S7–8). However, the magnitude of the baseline LS reactivity was not associated with stronger or weaker responses to the eOD-GT8 60mer, eOD-GT8 monomer, or the CD4bs epitope (fig. S4). We detected no serum neutralizing activity to any of several viruses tested (table S9), as expected, because the CD4bs in eOD-GT8 has been substantially modified to enable binding to VRC01-class precursors. Induction of binding antibodies to eOD-GT8 and its CD4bs but not neutralizing antibodies was consistent with pre-clinical experiments (27, 39, 45, 46, 48–50, 54). We concluded that the vaccine was highly immunogenic and induced class-switched, antigen-specific and CD4bs-specific serum IgG responses.
B cell sorting and receptor sequencing as the critical immunological assay
The major immunological objective of the trial was to determine if the vaccine could induce VRC01-class IgG B cells, defined as IgG B cells with VRC01-class B cell receptors (BCRs). Toward that end, we developed an analysis workflow to determine and interpret BCR sequences for eOD-GT8 CD4bs-specific IgG B cells, using single B cell sorting, reverse transcriptase-polymerase chain reaction (RT-PCR), DNA sequencing, and bioinformatic analysis (figs. S5–S10 and tables S10–S19). For each trial participant, we attempted to interrogate eight samples with this workflow, including memory B cells (MBCs) and plasmablasts (PBs) from peripheral blood mononuclear cell (PBMC) samples, and germinal center (GC) B cells from lymph node (LN) cells obtained by fine-needle aspiration (FNA) (Fig. 1A and tables S10–11). Complete data from sorting and sequencing, including quality-filtered sequences for the heavy and light chain of at least one CD4bs-specific BCR per sample, was obtained for 69.3% (266/384) of attempted samples (table S20).
Fig. 1. Frequencies of antigen-specific and epitope-specific B cells.
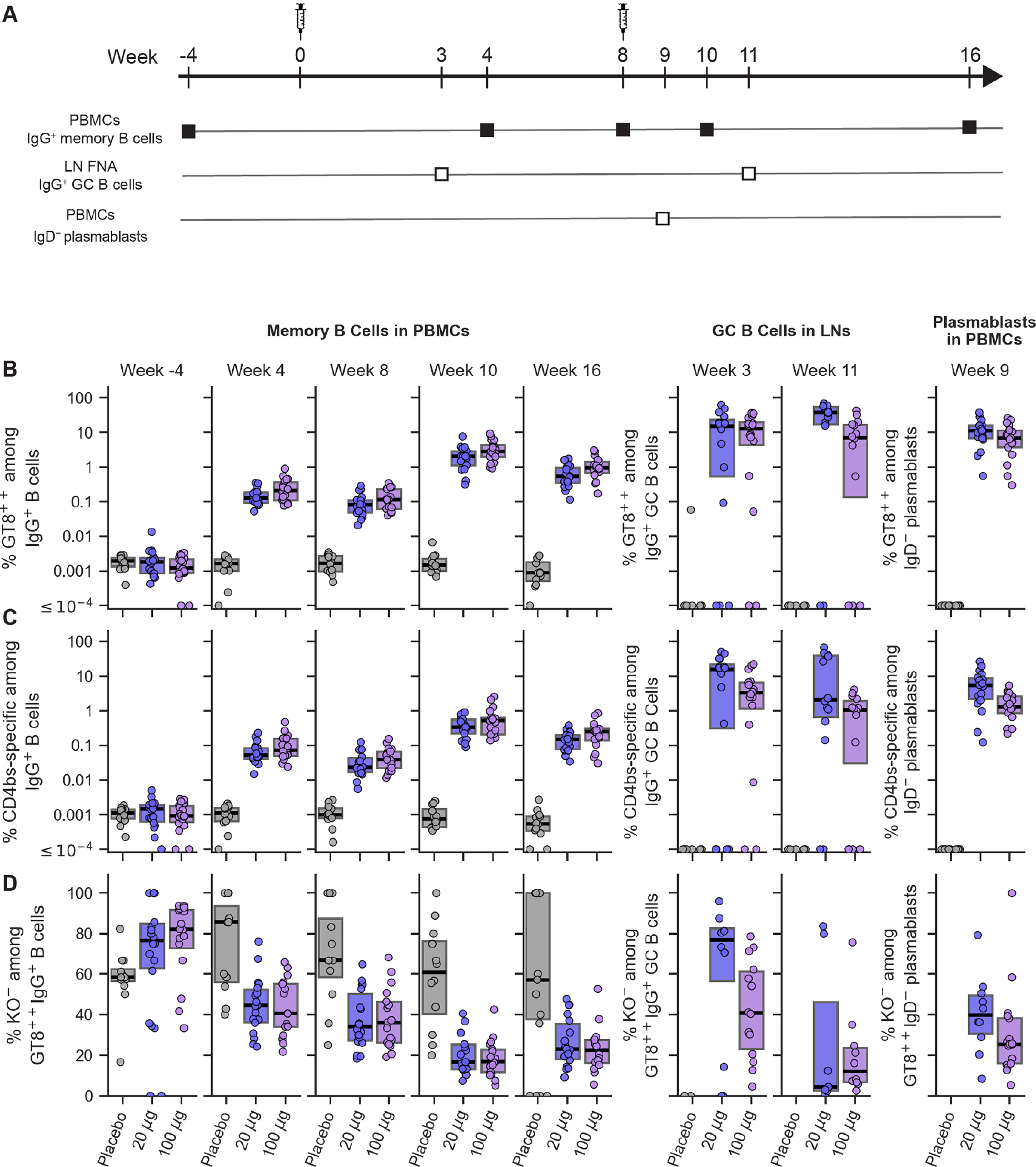
(A) Schedule for immunization and B cell sampling. (B) Frequency of eOD-GT8-specific (GT8++) IgG memory B cells (left), IgG GC B cells (middle), and IgD− plasmablasts (right) shown over time for each participant, grouped by vaccine treatment. GT8++ indicates binding to two different eOD-GT8 fluorescent probes. (C) Frequency of CD4bs-specific (KO−GT8++) IgG B cells, displayed as in (B). KO− indicates lack of binding to the eOD-GT8-KO11 probe. (D) Percent of eOD-GT8-specific IgG B cells that are CD4bs-specific (KO−), displayed as in (B). Each symbol represents frequency for one participant. Thick lines are median values; boxes indicate 25% and 75% quantiles.
Immunogenicity assessed by antigen- and epitope-specific B cell sorting
In PBMCs, all vaccine recipients produced eOD-GT8- and CD4bs-specific IgG memory B cells after the first immunization, with frequencies significantly higher than in preimmunization or placebo recipient samples, indicating vaccine-induced responses (Fig. 1B–C, figs. S11–S12, and tables S21–S28). Frequencies of eOD-GT8- and CD4bs-specific IgG memory B cells in vaccine-recipient PBMCs increased significantly following the second immunization (Fig. 1B–C, tables S29–S30), although the fraction of eOD-GT8-specific cells that were CD4bs-specific (KO−) decreased with the boost (Fig. 1D, table S31). CD4bs-specific IgG memory B cell frequencies in PBMC samples peaked two weeks after the boost, reaching median frequencies of approximately 1 in 300 and 1 in 200 IgG B cells in the low and high dose vaccine groups, respectively (Fig. 1C, tables S27 and S30). Thus, the germline-targeting immunogen induced substantial frequencies of CD4bs-specific IgG memory B cells in peripheral blood.
In lymph node and PB samples, eOD-GT8- and CD4bs-specific GC B cell or PB frequencies were significantly higher in vaccine than placebo recipients (Fig. 1B–C, tables S25 and S27), indicating vaccine-induced responses. Among vaccine recipients, frequencies of GT8- and CD4bs-specific IgG GC B cells or IgD− PBs were generally higher than for IgG memory in PBMCs (Fig. 1), reflecting the spatial and/or temporal enrichment of vaccine-specific cells among GC B cells and PBs. Overall, postimmunization class-switched (IgD−) PBMC memory and lymph node GC B cells specific for eOD-GT8 or CD4bs were predominantly IgG and were enriched for IgG compared to preimmunization memory (fig. S13 and tables S32–34), justifying our focus on IgG responses.
Detection and frequency quantification of VRC01-class IgG B cells
A total of 11,372 CD4bs-specific BCR sequences with paired HCs and LCs were available from 266 samples to assess vaccine performance (fig. S14). For each of these samples, we measured the number of VRC01-class IgG B cells and their frequencies among IgG memory B cells, GC B cells or PBs, and we grouped the results by timepoint, sample type, and vaccine treatment (Fig. 2A–B and tables S35–36. We then computed the positivity rate, defined as the percent of each group with at least one VRC01-class IgG B cell detected, at each timepoint (Fig. 2C and table S37).
Fig. 2. Detection of VRC01-class IgG B cells in blood and lymph nodes.
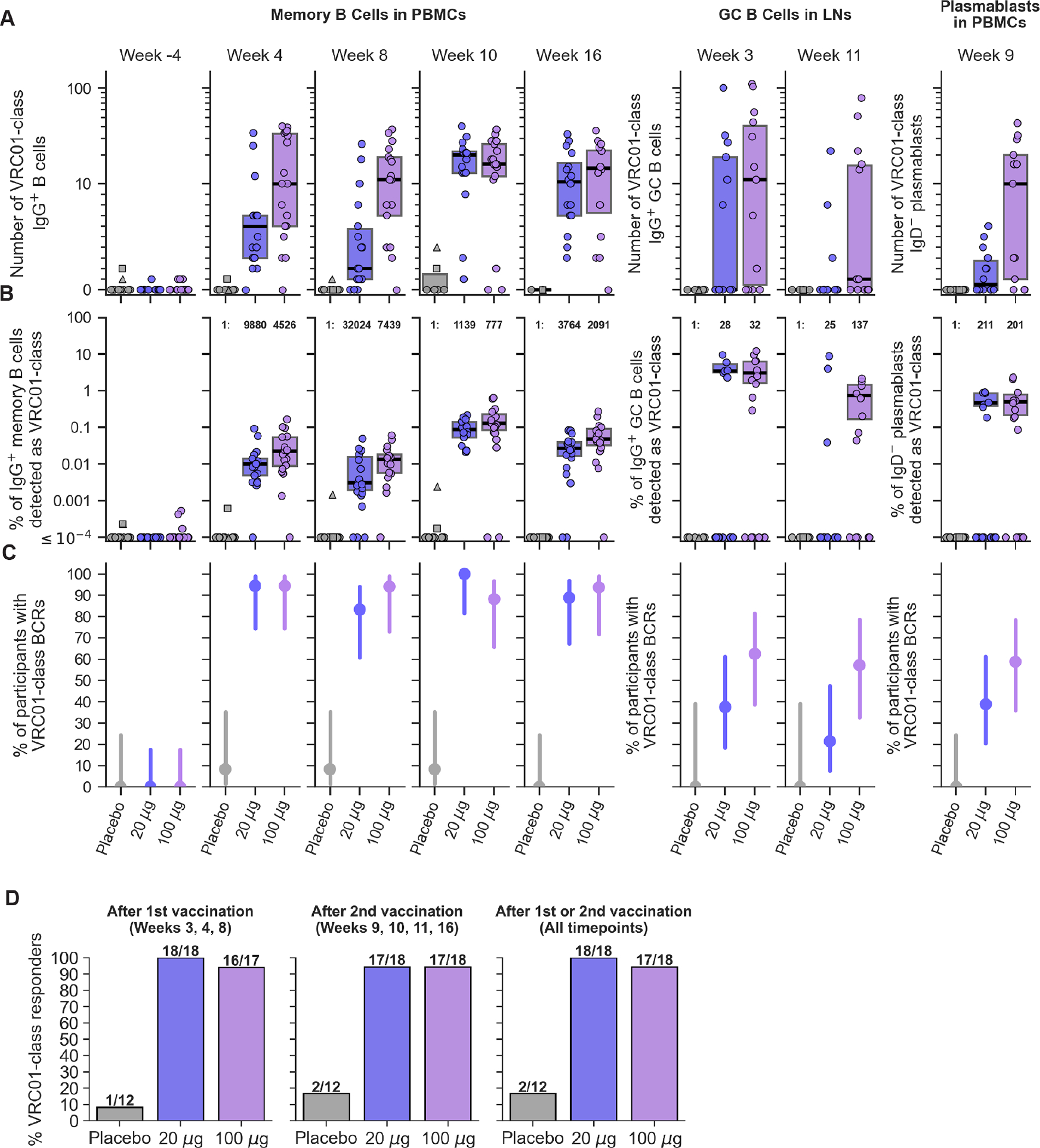
(A) Number of VRC01-class IgG B cells detected over time in each participant. (B) Frequency of VRC01-class IgG B cells as a percent of IgG B cells in each participant. Median post-vaccination frequencies are stated as 1:number of IgG B cells. In (A) and (B), symbols represent participants, and the two placebo participants with pre-existing (week -4) VRC01-class B cells are indicated as a square and a triangle. Thick lines indicate medians, and boxes indicate 25% and 75% quantiles. Medians and quantiles were computed over non-zero values only, because non-responders are accounted for in (C). (C) Positivity of VRC01-class IgG B cell detection, the percent of participants in each group with at least one VRC01-class B cell detected, for each timepoint and sample type. Circles indicate median values, and lines indicate 95% confidence intervals computed using the Wilson score method. (D) Positivity of VRC01-class responses over all timepoints or only after the first or second vaccination.
In pre-immunization (baseline) samples, we detected one or two VRC01-class IgG memory B cells in 6 of 48 participants (12.5%), with a median frequency over responders of 2.33×10−4% (one VRC01-class B cell in 429,000 IgG memory B cells) (week -4 in Fig. 2A–B). Thus, pre-existing VRC01-class IgG memory was present in at least a minority of participants. In post-immunization PBMC samples (weeks 4, 8, 10, 16), we detected VRC01-class IgG memory B cells in two placebo recipients (2/12, 16.7%), both of whom also showed pre-immunization VRC01-class IgG memory (participants 001 and 080 indicated in Fig. 2). Detection of post-placebo VRC01-class IgG memory B cells in those participants was likely due to pre-existing VRC01-class IgG memory.
In post-immunization PBMC samples, we detected VRC01-class IgG memory B cells in significantly higher fractions of vaccine recipients compared to baseline or placebo recipients (Fig. 2C and tables S38–39), and frequencies of VRC01-class IgG memory B cells among IgG B cells were significantly higher after vaccination compared to baseline (Fig. 2B and table S40). Our predetermined definition of a vaccine-induced VRC01-class IgG memory B cell response was the detection of one or more VRC01-class IgG memory B cells with a frequency higher than baseline for the same participant. All post-vaccination IgG memory samples with VRC01-class B cells detected met that definition (table S36). In week 4 PBMCs, we detected VRC01-class memory B cell responses in 17 of 18 participants in each vaccine group (94.4%; 95% confidence interval [CI], 74.2%–99.0%) (Fig. 2C and table S37), with median frequencies of approximately 1 in 1000 and 1 in 450 IgG B cells among positive responders in the low and high dose groups, respectively (Fig. 2B and table S41). These week 4 frequencies of VRC01-class memory B cells in the low and high dose groups were higher than the median pre-vaccination frequency among responders (1 in 429,000) by factors of 43 and 94, respectively, and higher than the previously reported average frequency for naive VRC01-class precursors (1 in 300,000; (21, 43)), by factors of 30 and 67, respectively. In week 8 PBMCs, VRC01-class positivity remained high, at 83.3% of the low dose group and 94.4% of the high dose group (Fig. 2C and table S37). Median VRC01-class IgG memory frequencies among responders declined modestly from week 4, by factors of 3.2 and 1.7 in the low and high dose groups, respectively (Fig. 2B and tables S41 and S42). Week 8 median frequencies remained >12-fold above the baseline VRC01-class IgG B cell frequency. We concluded that a single vaccination consistently induced strong VRC01-class IgG memory B cell responses in the peripheral blood.
Following the second vaccination, VRC01-class positivity in week 10 and 16 PBMCs remained high (88–100%) in both vaccine groups (Fig. 2C and table S37). Median VRC01-class IgG memory frequencies among responders in the low dose group increased markedly, by a factor of 28, to reach a week 10 peak frequency of 0.088% (1 in 1,140), and finally declined, by a factor of 3.3, to a week 16 frequency of 0.027% (1 in 3,768) (Fig. 2B and tables S41 and S42). Trends were similar but more favorable in the high dose group, with frequencies at weeks 10 and 16 of 0.13% (1 in 777) and 0.048% (1 in 2,092), respectively (Fig. 2B and table S41). Thus, at the peak response two weeks after the second vaccination, median frequencies of VRC01-class IgG memory B cells for >88% of vaccine recipients in the low and high dose groups were higher than the median pre-vaccination frequency by factors of 380 and 560, respectively, and higher than the previously reported average frequency for VRC01-class naive B cells by factors of 260 and 390, respectively (21, 43). Six weeks after the peak, at week 16, VRC01-class IgG memory B cell frequencies remained significantly higher than on the day of the second immunization (Fig. 2B and table S42). We concluded that the second immunization consistently increased VRC01-class IgG B cell frequencies in the peripheral blood.
In lymph node and PB samples, VRC01-class response rates were generally lower than in PBMCs (Fig. 2C). However, among positive responders, VRC01-class frequencies among IgG GC B cells and IgD− PBs were generally higher than among IgG memory B cells in PBMCs (Fig. 2B), illustrating the strong VRC01-class response in the GC reactions.
Combining all post-immunization data, we detected VRC01-class IgG B cell responses in 100% (18/18) of participants in the low dose group (median of 44 per participant) and 94.4% (17/18) of participants in the high dose group (median of 91 per participant), for an overall response rate of 97% (35/36) (Fig. 2D and table S37). After the first vaccination only (weeks 3, 4, and 8), we measured response rates of 94.4% (17/18) in both the low and high dose groups, giving an overall response rate of 94.4% (34/36) (Fig. 2D and table S37). We concluded that the eOD-GT8 60mer vaccine induced VRC01-class IgG B cell responses with high consistency across vaccine recipients and over time after one or two vaccinations, in both dose groups.
VH1–2 genotype analysis
One high dose vaccine participant contributed samples from all eight visits but had no detectable VRC01-class IgG B cells. Only two of the 540 heavy chain sequences from this individual (PubID 059) were VH1–2, and both used the *06 allele, which suggested that the individual might not possess either of the required *02 or *04 alleles. To evaluate the VH1–2 allele content in that individual and in all other participants, we carried out VH1–2 genotype analysis using IgDiscover for all 48 participants. Across all individuals, we detected five VH1–2 alleles, including *02 and a novel variant of *02 with a non-coding polymorphism, *02_S4953, both of which we will refer to as *02 here. The remaining alleles included *04, *05, and *06. Accounting for hetero- and homo-zygosity, we identified a total of 9 genotypes, among which the most common were: *02/*04 (27.1%), *02/*02 (22.9%), *04/*04 (20.8%), and *04/*06 (14.6%) (table S43). Only one of the genotypes, *05/*06, included neither of the required VRC01-class alleles, and that genotype was only carried by the participant who did not produce detectable VRC01-class IgG B cell responses. Thus, we found that vaccine induction of VRC01-class responses by eOD-GT8 60mer is limited by VH1–2 genotype. However, individuals lacking at least one of the required alleles represented only 2% (1 of 48) of this study population, consistent with prior analyses (13, 41, 44).
Polyclonality of VRC01-class responses
Despite sharing the VH1–2 gene, VRC01-class responses were highly polyclonal, with diverse HCDR3s and LCs (fig. S15). BCR sequence hierarchical clustering showed that all 2,865 post-vaccination VRC01-class BCRs were represented by 1,779 independent clusters (lineages) originating from independent germline recombination events, thus >60% of VRC01-class responses derived from distinct precursors (Fig. 3A–B). Very few clusters (0.11%) were shared between participants (Fig. 3C); relatively few clusters (9.7%) were shared over time within individuals (Fig. 3C); and the majority of clusters (82.1% and 80.9% in the low and high dose groups, respectively) contained a single member (Fig. 3D). The depth of sampling of the CD4bs-specific BCR repertoire was limited by practical constraints, and deeper sampling might have identified additional clusters or members for each cluster. Nevertheless, the number of independent VRC01-class clusters per participant was substantial (medians of 32 and 65 in the low and high dose groups, respectively) (Fig. 3E), showing that a large number of distinct precursors were primed in each individual. High VRC01-class polyclonality was observed in memory B cells at all timepoints after the prime and boost, and in plasmablasts, whereas in GCs we observed reduced levels of polyclonality concomitant with detection of larger clonal families (Fig. 3F). Thus, eOD-GT8 60mer primed responses from a diverse pool of VRC01-class precursors, and the clonal diversity was maintained after the boost.
Fig. 3. VRC01-class BCR hierarchical clustering and genetic diversity.
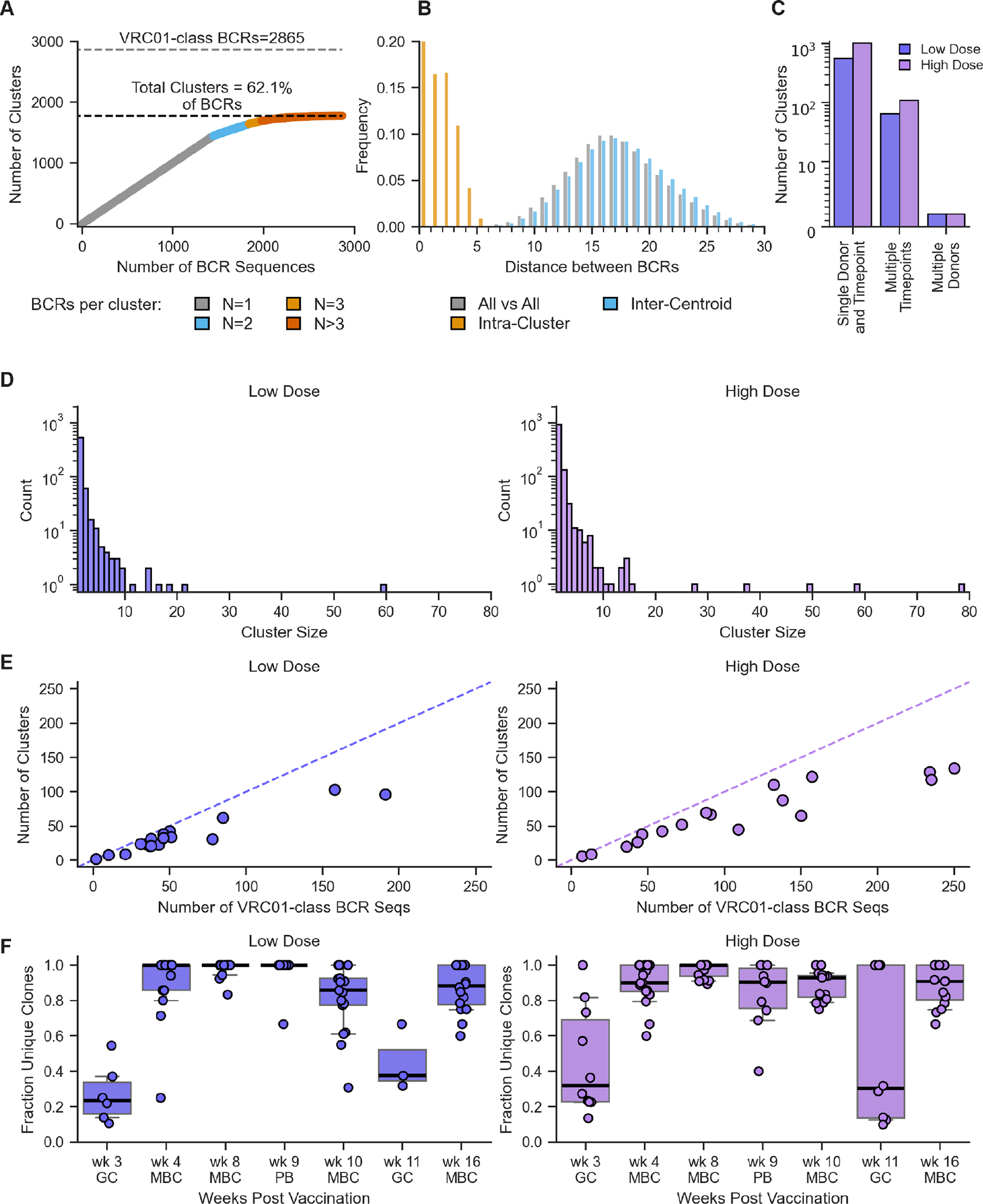
(A) Number of clusters versus number of BCR sequences, for all post-vaccination VRC01-class BCR sequences in both vaccine groups. Number of sequences per cluster indicated at bottom. (B) Distributions of distances between BCR sequences in (A), including all versus all, intra-cluster (all versus all within cluster), and inter-centroid (between cluster centroids). (C) Number of clusters involving single donor and timepoint, single donor and multiple timepoints, and multiple donors, separately for low and high dose groups. (D) Histogram of cluster size for low and high dose groups. (E) Number of clusters versus number of VRC01-class BCR sequences, for each participant in the low and high dose groups. (F) VRC01-class BCR polyclonality over time. Each symbol reports the fraction of BCR sequences that cluster as unique clones within a single donor at a single timepoint. Thick lines represent median values; boxes indicate 25% and 75% quantiles; whiskers approximate 10% and 90% quantiles.
Competitor responses — non-VRC01-class CD4bs-specific responses
VRC01-class responses were a minority of CD4bs-specific IgG B cell responses. The VRC01-class fraction of CD4bs-specific IgG BCRs had median per dose group values of 16% to 28% in PBMC memory B cells (MBCs), and 0% to 36% in lymph node GC B cells and PBMC plasmablasts (PBs), across both vaccine groups and over time (Fig. 4A). CD4bs-specific VRC01-class responses were an even smaller fraction of total eOD-GT8-specific IgG B cells, with median per dose group values of 3.5 to 8% in MBCs (Fig. 4B). These values were substantially higher than results from pre-clinical experiments with adjuvanted eOD-GT8 60mer in Kymab mice (46) and VH1–2 recombining mice (27, 54), in which only 1% and 0.3–3.2% of CD4bs-specific IgG BCRs were VRC01-class, respectively, and were similar to results from naive human B cell sorting with eOD-GT8 tetramers, in which 15–20% of CD4bs-specific naive BCRs were VRC01-class (21, 43, 44). Here, CD4bs-specific, non-VRC01-class IgG BCRs, defined as any CD4bs-specific BCR not meeting the VRC01-class definition, included non-VH1–2 BCRs as well as VH1–2 BCRs with LCDR3 lengths other than five. However, VRC01-class BCRs comprised a dominant 83% of VH1–2/kappa BCRs and 43% of VH1–2/lambda BCRs, due to strong enrichment for 5-amino acid LCDR3s (fig. S16). CD4bs-specific, non-VRC01-class IgG B cells were highly polyclonal (fig. S17), with diverse gene usage and CDR3 lengths (fig. S18), just as VRC01-class were highly polyclonal (Fig. 3). However, non-VRC01-class B cells reached higher frequencies than VRC01-class B cells, among all IgG B cells. Median frequencies of non-VRC01-class IgG B cells peaked at week 10 values of 0.27% and 0.39% in the low and high dose vaccine groups, respectively (fig. S19; table S44), compared to VRC01-class frequencies of 0.09% and 0.13%, respectively. Thus, VRC01-class B cells were induced despite a dominant competing CD4bs response by a highly diverse pool of non-VRC01-class B cells.
Fig. 4. Frequency of VRC01-class B cells among CD4bs- or eOD-GT8-specific IgG B cells and plasmablasts.
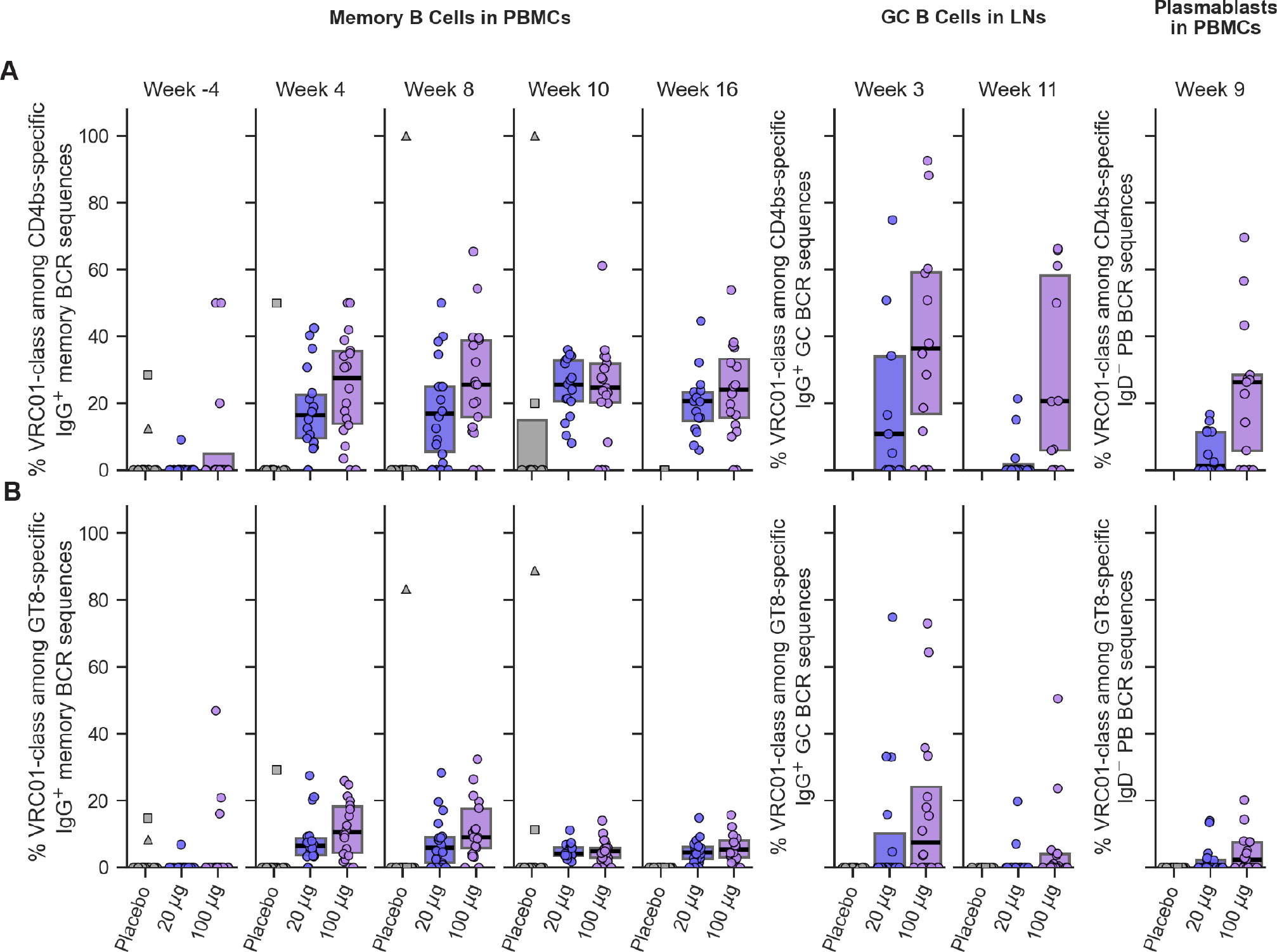
(A) VRC01-class frequencies among CD4bs-specific B cells. (B) VRC01-class frequencies among eOD-GT8-specific B cells. Each symbol represents frequency for one participant. The two placebo participants with pre-existing (week -4) VRC01-class B cells are indicated as a square and a triangle. Thick lines are median values; boxes indicate 25% and 75% quantiles.
BCR mutation levels
Changes in vaccine-induced BCR mutation levels over time can provide insight into the immune processes underlying the response. To assess changes in CD4bs-specific BCR mutation levels, we computed the median percent mutation per participant per timepoint, for heavy chain V genes (VH) and light chain kappa and lambda V genes (VK/VL), in VRC01-class and non-VRC01-class BCRs, for both nucleotide and amino acid mutations (Fig. 5A–B; fig. S20; table S45). We made statistical comparisons between timepoints using paired data in which mutation levels were available from the same individual at two timepoints (tables S46–48). We also monitored the mutation distributions across all antibodies in each group and timepoint, through violin plots (Fig. 5C–D). In the narrative below, we provide example VH amino acid mutation values as indicators for change.
Fig. 5. Amino acid mutation levels in heavy and light chain V genes over time, for VRC01-class and non-VRC01-class BCRs from vaccine recipients.
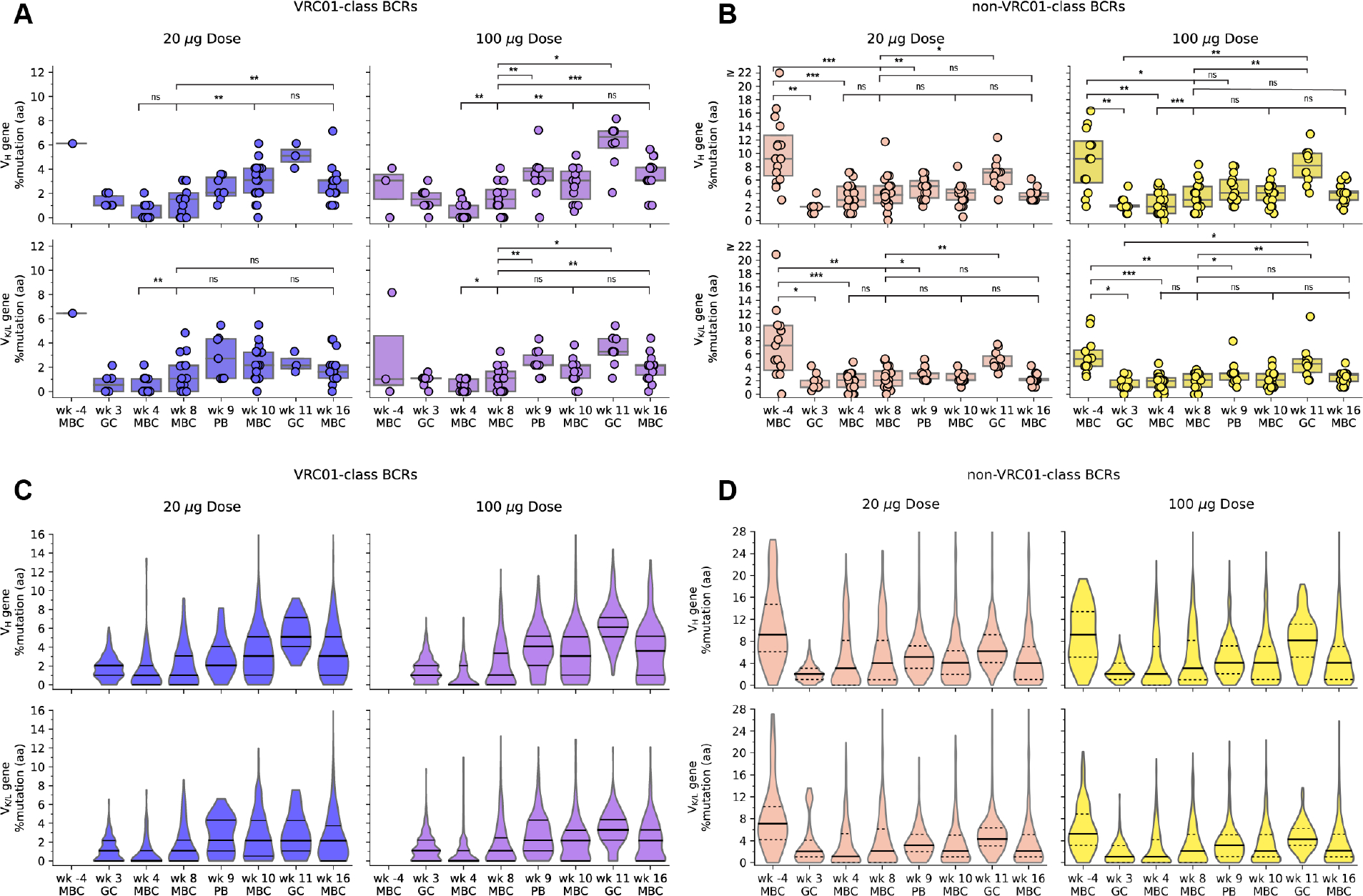
(A) and (B) VRC01-class (A) and non-VRC01-class (B) BCR VH percent amino acid (aa) mutation (top) and VK/L %aa mutation (bottom) for low dose (left) and high dose (right), with symbols representing the median per participant per timepoint; (C) and (D) VRC01-class (C) and non-VRC01-class (D) BCR VH %aa mutation (top) and VK/L %aa mutation (bottom) for low dose (left) and high dose (right), with violin plots representing the distribution of all BCRs per group per timepoint. In (A) and (B), thick lines are medians, and box plots show 25% and 75% quantiles, for each dose group at each timepoint. Statistical significance is indicated for all comparisons with pairs of measurements from at least 8 participants (tables S46 and S48). Significance testing was done using Wilcoxon signed-rank test for paired data (two-sided, α = 0.05); ns, not significant; *, P<0.05; **, P<0.01; ***, P<0.001. In (C) and (D), solid lines are medians, and dashed lines show 25% and 75% quantiles, for each dose group at each timepoint.
After the first immunization, nucleotide and amino acid mutation levels in VRC01-class memory IgG VH and VK/L genes generally increased significantly from week 4 to week 8 in both vaccine groups, the only exception being VH amino acid mutation in the low dose group, which showed an increase that barely missed significance (Figs. 5A and 5C; fig. S20; tables S46 and S47). Thus, GCs remained active and produced memory B cells with increased SHM beyond week 4. By week 8, VH amino acid mutation levels reached median values of 1.5% in both dose groups, respectively (table S45).
After the second immunization, nucleotide and amino acid mutation levels in VRC01-class memory BCR VH genes from both vaccine groups increased significantly from week 8 to week 10 but did not change significantly from week 10 to 16 (Figs. 5A and 5C; fig. S20; tables S46 and S47). Week 10 VH amino acid mutation levels in VRC01-class IgG memory BCRs reached median values of 3.0% in both vaccine groups (table S45). Thus, the week 8 boost caused a relatively rapid increase in SHM within the VRC01-class IgG memory pool. Increased mutation levels also appeared in PBs 4 to 8 days after the boost (Figs. 5A and 5C; fig. S20; tables S46 and S47). GCs remained active at week 11, reflected by the relatively high median VH amino acid mutation levels of 5.1% and 6.7% in week 11 GC BCRs in the low and high dose groups, respectively (table S45). The increased SHM in GCs indicated an ongoing physiological response to the boost in which B cells continued acquiring mutations and likely exiting to blood. Our finding that an autologous boost immunization increased mutation levels in VRC01-class GC and memory B cells provides support for a key assumption underlying the germline-targeting vaccine design strategy, namely that sequential vaccination can increase the maturation of targeted B cell classes in humans.
Comparing GC BCR mutation levels at weeks 3 and 11 provides insight into mechanism. In GC BCRs, the distributions of VH and VK/VL mutation levels computed over all VRC01-class responders were substantially higher at week 11 than week 3 (Figs. 5A and 5C; fig. S20; table S45). For example, median [interquartile range] VH amino acid mutation levels were 1.0% [1.0–1.8%] and 1.3% [1.0–2.0%] at week 3 in the low and high dose groups, respectively, compared to 5.1% [4.6–5.6%] and 6.7% [5.0–7.1%] at week 11 (Fig. 5A; fig. S20; table S45), and the week 11 violin plot distributions over all BCRs show little similarity to the lower SHM week 3 violin plots (Fig. 5C). With paired data from weeks 3 and 11 available for only a few individuals (N=1 at low dose and N=5 at high dose; [fig. S21]) we could not ascribe significance to the higher mutation levels at week 11 (tables S46 and S47). Nevertheless, the above comparisons between weeks 3 and 11, and the minimal overlap in mutation distributions for four individuals with multiple datapoints at both week 3 and 11 (fig. S21A), suggested that the second immunization did not cause substantial priming of naive B cells but instead primarily induced additional maturation of VRC01-class GC and/or memory B cells generated by the first immunization.
SHM in non-VRC01-class IgG memory BCRs increased significantly from week 4 to 8 for the high dose group only, and showed no significant increase from week 8 to week 10 or 16 in either group (Figs. 5B and 5D; fig. S20; tables S48 and S49). Thus, the germline-targeting vaccine boost succeeded to increase memory IgG BCR median mutation levels for the targeted class of B cells without causing similar increases in BCRs competing for the same epitope. Non-VRC01-class BCRs did show significant mutational increases between memory B cells at week 8 and either PBs at week 9 or GC BCRs at week 11 (Figs. 5B and 5D; fig. S20; tables S48 and S49). SHM in non-VRC01-class GC BCRs was significantly higher at week 11 than at week 3 for the high dose group, and nearly significantly higher for the low dose group (Figs. 5B and 5D, fig. S21B and tables S48 and S49), indicating that the second immunization primarily activated previously mutated GC and/or memory B cells instead of naive B cells, similar to our observation with VRC01-class responses.
Precursor origins
We detected pre-vaccination VRC01-class IgG memory B cells in only 12.5% (6/48) of participants, and 71% (5/7) of those BCRs had mutation levels substantially above the levels detected post-vaccination at weeks 3 or 4 (tables S45 and S50), indicating that the VRC01-class IgG B cells detected post-vaccination predominantly originated from naive VRC01-class B cells rather than IgG memory B cells. However, clustering analysis indicated that one week -4 VRC01-class IgG memory BCR was potentially clonally related to two post-vaccination BCRs in the same individual (fig. S22), providing evidence that at least some VRC01-class IgG BCRs detected post-vaccination may have originated from VRC01-class IgG memory B cells.
Non-VRC01-class IgG BCRs had significantly higher mutation levels at week -4 compared to weeks 3 or 4 (Figs. 5B and 5D; fig. S20; tables S45, S48 and S49), indicating that the majority of non-VRC01-class IgG B cells detected post-vaccination probably also originated from naive rather than memory B cells. However, sequence clustering analysis indicated that, among 26 non-VRC01-class memory IgG BCR lineages with members from both pre- and post-vaccination timepoints, evidence allowing for a potential pre-existing memory B cell precursor could be found in 11 (42%) of the lineages (fig. S23). Non-VRC01-class BCR mutation levels at weeks 3–8 were substantially higher than for VRC01-class (Fig. 5 and table S45), which might have been due to non-VRC01-class responses deriving in part from already mutated memory B cells, but also might have been due to low-affinity, naive precursor-derived, non-VRC01-class BCRs gaining SHM more rapidly than high-affinity VRC01-class BCRs after the first immunization.
BnAb characteristics shared by vaccine-induced VRC01-class IgG BCRs
Vaccine-induced VRC01-class BCRs shared other characteristic features of VRC01-class bnAbs in addition to the VH1–2 alleles and 5-amino acid LCDR3. VRC01-class bnAbs utilize a subset of human VK or VL genes (Data S1), and more than 90% of vaccine-induced VRC01-class IgG BCRs used known bnAb VK or VL genes, in both groups after the prime or boost (Fig. 6A and fig. S24A). The LCDR3 is an important site of affinity selection in VRC01-class bnAbs, with all known bnAbs possessing Glu or Gln at position 96, and most bnAbs exhibiting LCDR3 sequence motifs with substantially reduced diversity at several positions compared to naive VRC01-class precursors (Fig. 6B; Data S1) (21, 39, 41, 43, 44, 46, 55). LCDR3s from vaccine-induced VRC01-class BCRs showed signs of selection toward bnAb sequences, especially at position 96 (Fig. 6B–D; figs. S24B and S25). For VRC01-class BCRs with kappa LCs, the median fraction of LCDR3s perfectly matching a bnAb LCDR3 was 39% or 35% in the low or high dose groups, respectively, while lambda chain BCRs had no bnAb LCDR3s (Fig. 6E and fig. S24C–D). Among non-VRC01-class BCRs, a small fraction (1.8%) had 5-amino acid LCDR3s, many of which had high similarity to VRC01-class bnAb LCDR3s, whereas control sequences did not (Fig. 6C–E). This suggested the possibility that eOD-GT8 60mer vaccination might have selected for BCRs using a VRC01-class binding mode with VH genes other than VH1–2, which will be investigated further. Most VRC01-class bnAbs with K3–20 LCs have deletions in LCDR1 important for accommodating the N276 glycan conserved on the HIV spike (42, 55, 56). We observed K3–20 LCDR1 deletions at rates of 5–10% in both VRC01-class and non-VRC01-class BCRs from only a few participants each (fig. S26), indicating that such deletions were not common and were not specifically selected by eOD-GT8, which was not surprising since eOD-GT8 lacks the N276 glycan. VRC01-class bnAbs exhibit a wide range of HCDR3 lengths, from 12 to 18, and nearly all encode a Trp five residues before the end of the HCDR3, a position that we and others have previously inaccurately referred to as “100B” that we here term “103–5” to count backward from position 103 at the end of the HCDR3 (Data S1). Trp103–5 was previously found in approximately 31% of eOD-GT8-specific human naive VRC01-class BCRs (21, 43, 44) (fig. S27). HCDR3 lengths for vaccine-induced VRC01-class BCRs spanned those of VRC01-class bnAbs (fig. S15C), and the fraction of VRC01-class BCR HCDR3s with Trp103–5 had median values over all participants of >47% in both dose groups and after the prime or boost (Fig. 6F and figs. S24E and S27), suggesting enrichment of Trp103–5 due to vaccination. Accounting for combinations of bnAb features, we found that the median fraction of VRC01-class BCRs with four of the above described bnAb features was >25% in both dose groups and after the prime or boost (fig. S28). Finally, we considered the acquisition of key amino acid mutations in the HC, an essential aspect of vaccination to induce bnAbs. From a representative set of 19 potent VRC01-class bnAbs that included all known VRC01-class VK and VL genes but also minimized the inclusion of bnAbs with insertions or deletions, we identified a set of 20 positions (19 within VH1–2, plus Trp103–5) at which key VRC01-class residues are observed, four of which are germline-encoded in the VH1–2*02 and *04 alleles compatible with a VRC01-class antibody (Data S1). We counted these key VRC01-class residues on a scale ranging from -4 to +16, to allow for all possibilities from losing all germline-encoded key residues, to gaining key residues at all 16 positions not containing a germline-encoded key residue. The representative bnAbs had a median of 13 key residues, and a range of +8 to +16 (Fig. 6G). We computed 90th percentile values among VRC01-class BCRs in each study participant as representative for the best 20% of BCRs in that individual. The median of 90th percentile values for key HC residues was ≥+2 at nearly all timepoints at week 8 or later in both the low and high dose groups (Fig. 6G). Thus, vaccination selected for the acquisition of important HC residues in VRC01-class BCRs, suggesting that they could be guided toward bnAb activity with further boosting.
Fig. 6. Properties of post-vaccination BCRs shared with VRC01-class bnAbs.
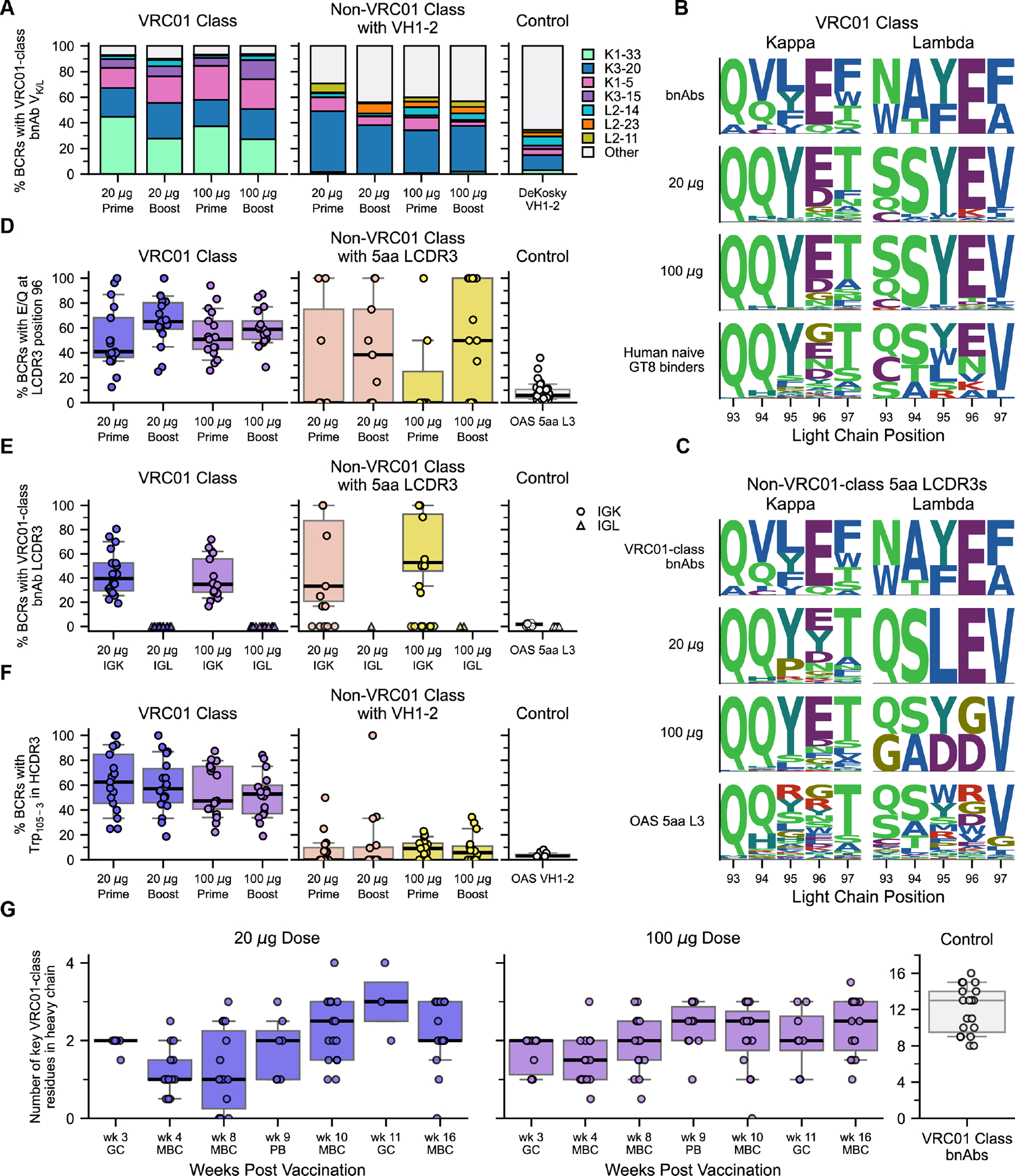
(A) Percent of BCRs using VRC01-class bnAb VK/L genes, for VRC01-class and VH1–2-using non-VRC01-class BCRs, and for control VH1–2 BCRs from HIV-unexposed individuals from DeKosky et al. (99); VRC01-class bnAb VK/L are indicated in the color key. (B) Sequence logos for 5-amino acid LCDR3s from VRC01-class BCRs, for bnAbs (top row), low dose (second row) and high dose (third row) groups, and human naive precursors from prior studies (39, 43, 44) (bottom row), distinguishing kappa (left) and lambda (right) LCs. (C) Sequence logos for 5-amino acid LCDR3s from non-VRC01-class BCRs from the low dose (second row) and high dose (third row) groups, and control data human LCs from HIV-unexposed individuals from the Observed Antibody Space (OAS) (92, 93) (bottom row), with VRC01-class bnAbs (top row) shown for reference, distinguishing kappa (left) and lambda (right) LCs. (D) Percent of BCRs using Glu or Gln at LC position 96, for 5-amino acid LCDR3s from VRC01-class BCRs, non-VRC01-class BCRs, and OAS control data LCs (92, 93). (E) Percent of BCRs with LCDR3 matching a VRC01-class bnAb sequence, for 5-amino acid LCDR3s from VRC01-class BCRs, non-VRC01-class BCRs, and OAS control data LCs (92, 93), distinguishing IGK and IGL LCs. (F) Percent of BCRs with Trp105–3, for VRC01-class and VH1–2-using non-VRC01-class BCRs, and for OAS (92, 93) control data VH1–2 HCs. (G) Number of key VRC01-class residues in VRC01-class HCs, for all timepoints in low dose (left) and high dose (middle) groups, with symbols indicating the 90% quantile per participant per timepoint, and for VRC01-class bnAbs (right, different y-axis scale), with symbols denoting bnAbs. 20 μg, low dose; 100 μg, high dose. Prime, data from weeks 3, 4, and 8; Boost, data from weeks 9, 10, 11, and 16. In (D)-(G), symbols represent individual participants [except the bnAb controls in (G)]; thick lines are medians; boxes show 25% and 75% quantiles; whiskers approximate 10% and 90% quantiles.
BCR affinity dynamics
BCR-antigen affinity influences B cell fate throughout an immune response. To understand how VRC01-class and non-VRC01-class BCR affinities for eOD-GT8 evolved over time, we carried out surface plasmon resonance (SPR) analyses of the interactions between eOD-GT8 monomer and recombinant IgGs corresponding to BCRs from the low dose vaccine group, including post-vaccination BCRs and their inferred-germline (iGL) variants representing naive precursors (Fig. 7, fig. S29, and Data S2).
Fig. 7. SPR analysis of BCR affinities for eOD-GT8.
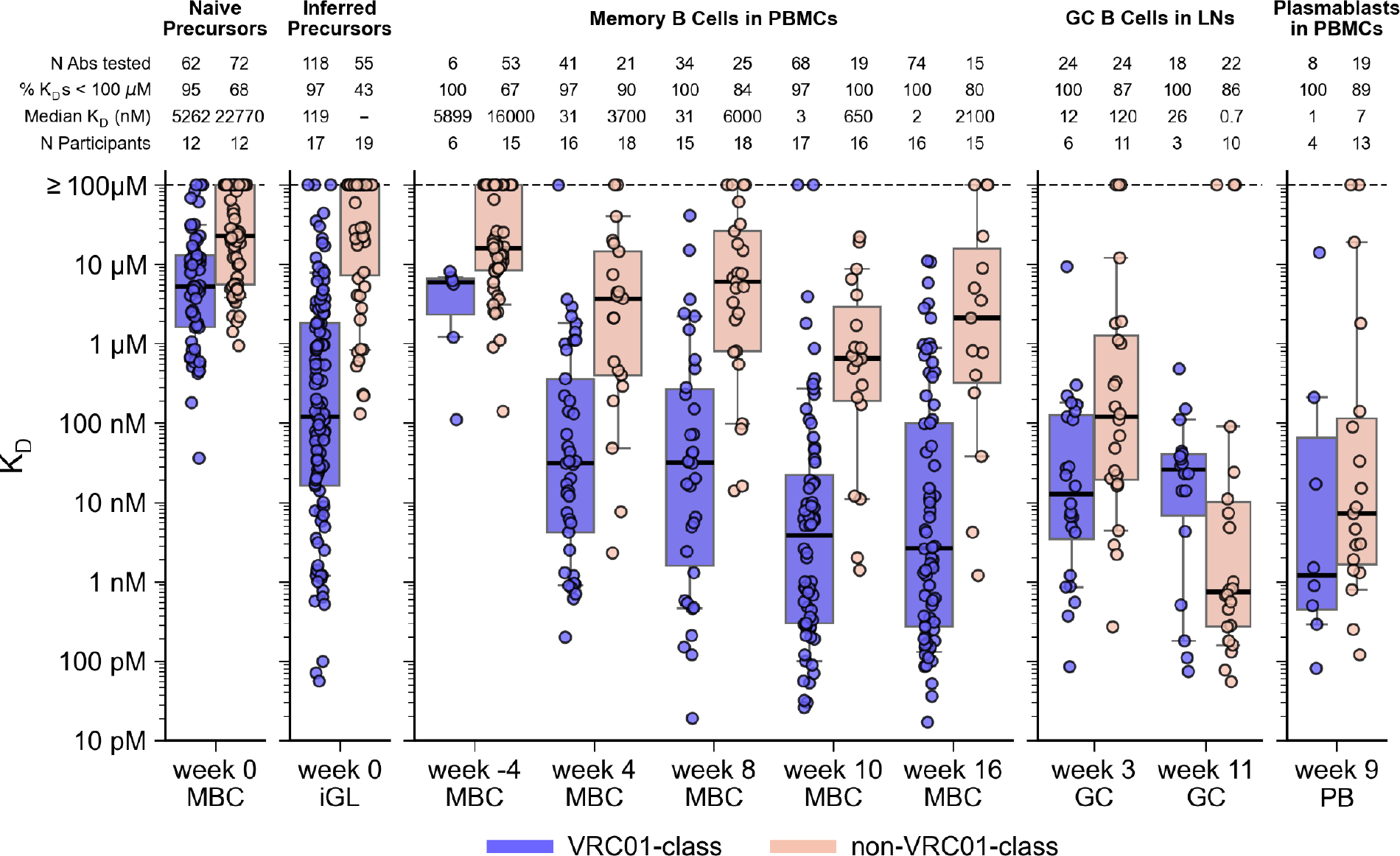
Monovalent KD values for antibody ligands binding to eOD-GT8 monomer analyte, for VRC01-class and non-VRC01-class BCRs from the low dose group post-vaccination (weeks 4–16) and pre-vaccination (week -4 and week 0 iGL), and for human naive BCRs isolated by prior human B cell sorting studies (21, 43) (week 0, far left). Thick lines are medians; boxes show 25% and 75% quantiles; whiskers approximate 10% and 90% quantiles For median KD, “−” indicates median ≥ 100 μM. KD values were generally representative of multiple measurements. For the 778 KDs in this figure, 163 (21%) were measured once, 170 (21.9%) were measured twice, and 445 (57.2%) were measured three or more times.
VRC01-class iGLs had surprisingly high affinities, with a median KD of 119 nM, approximately 45-fold higher affinity than the median KD of 5.3 μM for eOD-GT8-specific human naive VRC01-class precursors (21, 43) (Fig. 7). The high VRC01-class iGL affinities were not likely due to the presence of affinity-enhancing mutations at CDR3 junctions, because iGL affinities were not higher for parental BCRs with higher SHM levels (fig. S30). Nor were the high VRC01-class iGL affinities due to bias for high affinity in the B cell sorting, as substantially lower affinity non-VRC01-class BCRs were recovered (Fig. 7). VRC01-class week -4 memory IgG BCRs, which in most cases were not likely to have served as precursors (discussed above), had moderate affinities, with a median KD of 5.9 μM, in a similar range as naive VRC01-class BCRs (Fig. 7). Evidently, the vaccine-induced VRC01-class IgG GC and memory B cells that survived GC competition originated predominantly from precursors with the very highest affinities (subset with median KD of 119 nM) among the naive VRC01-class precursors.
In contrast, only 43% of non-VRC01-class iGLs had detectable binding (KD < 100 μM), and the median KD was the limit of detection of our SPR assay (≥100 μM), approximately 840-fold lower in affinity than for VRC01-class iGLs (Fig. 7). Non-VRC01-class week -4 memory IgG BCRs also had low affinities, with a median KD of 16.0 μM, similar in magnitude to the median KD of 22.8 μM for non-VRC01-class human naive precursors (Fig. 7). The capacity for low affinity non-VRC01-class precursors to compete effectively for the CD4bs-specific response against higher affinity VRC01-class precursors was likely due to the higher non-VRC01-class BCR precursor frequency: among eOD-GT8 CD4bs-binding naive BCRs isolated using high-avidity probes to enhance recovery of low affinity clones, non-VRC01-class are more common than VRC01-class by a factor of approximately 170 (44).
After the first vaccination, VRC01-class affinities increased by an average factor of 4.8 over iGLs, to median KDs of 12, 31, and 31 nM in week 3 GC BCRs and week 4 and 8 memory BCRs, respectively (Fig. 7 and fig. S31). Non-VRC01-class median affinities increased by a much larger factor of >833 over iGLs, reaching a median KD of 120 nM in week 3 GC BCRs, but then declined 40-fold to median KDs of 3.7 and 6.0 μM in week 4 and 8 memory BCRs, respectively (Fig. 7 and fig. S31).
Following the second vaccination, affinities for VRC01-class memory BCRs increased by a factor of 12, from a median KD of 31 nM at week 8 to median KDs of 3 and 2 nM at weeks 10 and 16, respectively (Fig. 7). Similarly high affinities were found in PBs at 4–8 days after the boost (median KD, 1 nM). The step-like jump to higher affinities for VRC01-class BCRs in the periphery at weeks 9, 10, and 16 tracked with the step-like increase in mutation levels (Fig. 5A and 5C). VRC01-class GC BCRs at week 11 had lower median affinity (KD, 26 nM), but firm conclusions could not be drawn from that observation, because the data was obtained for only three participants (Fig. 7). Non-VRC01-class memory affinities increased by an average factor of 4.4 after the boost, from a median KD of 6.0 μM at week 8 to median KDs of 650 and 2100 nM at weeks 10 and 16, respectively (Fig. 7). Non-VRC01-class PBs (median KD, 7 nM) and week 11 GC BCRs (median KD, 0.7 nM) showed substantially higher affinities than memory BCRs at weeks 8, 10, or 16 (e.g. 3000-fold higher at week 11 than week 16), demonstrating that strong and rapid non-VRC01-class affinity maturation occurred in response to the week 8 boost, but the resulting high affinity BCRs either remained within GCs or populated the plasma compartment rather than the memory compartment. Thus, affinity maturation of the memory pool in response to the boost was more efficient for VRC01-class than non-VRC01-class B cells.
Overall, VRC01-class precursor BCRs started with a massive affinity advantage over CD4bs-specific, non-VRC01-class precursor BCRs (ratio of median KDs for iGLs, ≥840); this advantage declined but remained high after the first vaccination (ratio of median KDs at week 8, 194) and then increased again after the boost (ratio of median KDs at week 16, 1050). VRC01-class affinity gains were associated with both increased on-rates and decreased off-rates, but off-rate reduction was the larger effect (figs. S32 and S33). Thus, although B cell selection in the GC might occur under high avidity conditions (i.e. an array of BCRs on the surface of a B cell interacting with an array of eOD-GT8 60mer antigens on a follicular dendritic cell [FDC]), the process nevertheless selected BCRs with improvements in monovalent KD as well as on- and off-rate, perhaps by FDCs regulating antigen availability (57). VRC01-class affinities and on-rates both increased significantly with SHM, and off-rates decreased significantly with SHM, across all post-vaccination BCRs (p values for KD, kon, koff, respectively: <0.001, 0.0003, <0.0001) and in memory B cells at each timepoint (figs. S34–S36), indicating that SHM contributed to maintaining the VRC01-class affinity advantage over time.
Guiding SHM
A key requirement for germline-targeting priming immunogens is the induction of bnAb-precursor-derived GC and/or memory B cells capable of binding antigens that are more native-like than the priming immunogen within the target epitope (13, 39). This is necessary so that a more native-like immunogen can serve as a boost, to advance B cell maturation further toward bnAb development. eOD-GT8 was designed to have an “affinity gradient”, with stronger binding to bnAbs than to bnAb precursors, based on the hypothesis that if bnAb precursors could be primed, the affinity gradient would guide early SHM toward bnAb development and concomitantly induce bnAb-precursor-derived responses that bind to more native-like antigens (13, 21, 39). As a result, eOD-GT8 possesses a strong affinity gradient for VRC01-class BCRs, with approximately 230-fold higher affinity for bnAbs than for naive precursors (21, 43). To determine if eOD-GT8 60mer immunization selected for VRC01-class BCRs that bind more native-like immunogens, we first tested binding of ≥210 VRC01-class memory BCRs from the low dose group from weeks 4, 8, 10, and 16 to a native-like trimer (BG505 MD39, (22)) and a core-gp120 lacking the N276 glycan (core-e-2CC HxB2 N276D, (39)). We detected no trimer binding even at the highest trimer analyte concentrations tested (11 μM), even though the trimer analyte allowed for avidity in binding, but approximately 10% of the antibodies tested from weeks 10 and 16 showed weak binding to core-gp120 (fig. S37). We then tested binding of VRC01-class iGL and post-vaccination low dose group BCRs to eOD-GT6 (13) and four variants of eOD-GT6, each of which had a more native-like CD4bs compared to eOD-GT8 (fig. S38). Previous work demonstrated that eOD-GT6 had no detectable affinity for the vast majority of human naive VRC01-class precursors that bind eOD-GT8, which was explained by the fact that eOD-GT6 lacks several germline-targeting mutations present in eOD-GT8 (21). Consistent with that prior finding, we found here that eOD-GT6 and its four variants had very limited reactivity to VRC01-class iGL precursors. Whereas eOD-GT8 bound to 97% of iGLs with a median KD of 119 nM (Fig. 7), eOD-GT6 and its variants bound to only 2–39% of the iGLs and had median KDs of ≥100 μM (Fig. 8A). However, post-vaccination VRC01-class antibodies showed improved binding for eOD-GT6 and its variants, especially after the boost. For example, eOD-GT6 bound to 78% of BCRs tested at week 16, with an overall median KD of 6 μM (Fig. 8A), and the four more native-like variants of eOD-GT6 (GT6v2, GT6v3, GT6v4, and GT6-N276+ with an intact N276 glycosylation site) bound to 75%, 29%, 24%, and 30% of BCRs tested at week 16, respectively, in comparison to binding 39%, 17%, 2%, and 5% of iGLs, respectively (Fig. 8A). Thus, vaccination with eOD-GT8 60mer not only induced VRC01-class responses, but also selected for mutations in VRC01-class BCRs that conferred affinity for antigens with more native-like CD4bs epitopes. Post-vaccination non-VRC01-class antibodies showed substantially weaker binding to eOD-GT6 and variants (fig. S39) compared to VRC01-class antibodies (Fig. 8A), demonstrating that eOD-GT8 60mer vaccination also minimized the induction of competing responses capable of binding more native-like CD4bs epitopes. Finally, VRC01-class affinities for eOD-GT6 and all but one variant increased with affinities to eOD-GT8 (p-values for eOD-GT6, GT6v2, GT6v3, GT6v4, and GT6-N276+, respectively: <0.0001, <0.0001, 0.0002, 0.3809, 0.008; Fig. 8B). Therefore, higher affinity for eOD-GT8 translated into higher affinity for more native-like antigens, providing support for the hypothesis that engineering an affinity gradient into a germline-targeting priming immunogen can help guide SHM selected by that immunogen (13). Our data also support applicability of that hypothesis to boost immunogens, as previously proposed (26).
Fig. 8. SPR analysis of VRC01-class BCR affinities for eOD-GT6 and variants.
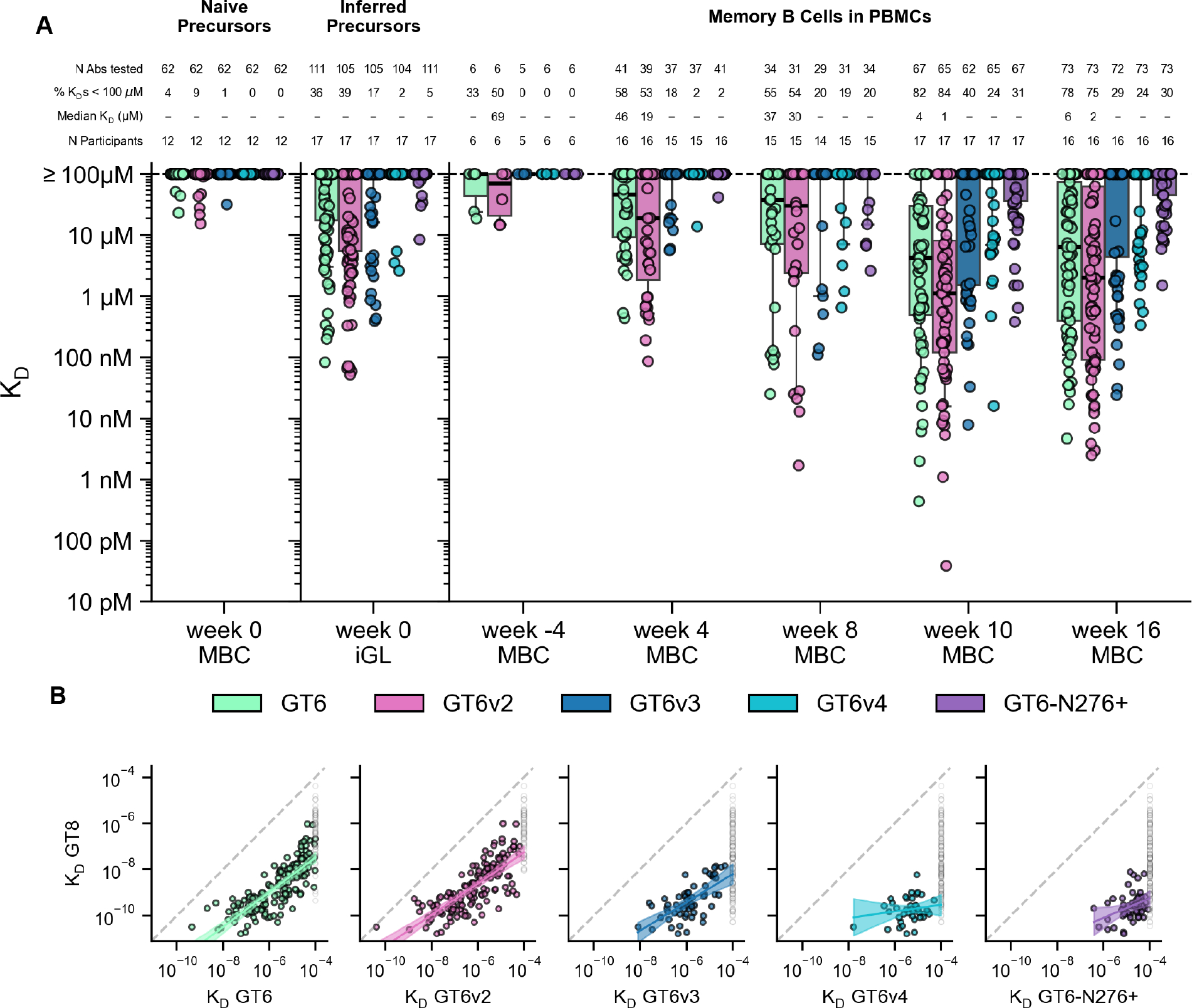
(A) Monovalent KD values for VRC01-class antibody ligands binding to monomeric eOD-GT6 and eOD-GT6 variant analytes (described in fig. S38), for memory BCRs from the low dose group post-vaccination (weeks 4–16) and pre-vaccination (week -4 and week 0 iGL), and for human naive BCRs isolated by prior human B cell sorting studies (21, 43) (week 0, far left). Thick lines are medians; boxes show 25% and 75% quantiles; whiskers approximate 10% and 90% quantiles. For median KD, “−” indicates median ≥ 100 μM. For the 1321 KDs in (A), 1171 (88.6%) were measured once, 48 (3.6%) were measured twice, and 102 (8.7%) were measured three times. (B) KD for eOD-GT8 versus KD for eOD-GT6 and variants, with estimated regression lines shown as solid lines and 95% confidence prediction intervals indicated by shading. Regressions were restricted to KDs <100 μM. KDs ≥100 μM for eOD-GT6 and variants are shown in grey. S, slope of regression line; P, p-value for slope; ns, not significant (p>0.05).
Discussion
Learning how to induce broadly neutralizing antibodies against pathogens with high antigenic diversity, such as HIV, influenza, hepatitis C virus, or the family of betacoronaviruses, represents a grand challenge for rational vaccine design. Germline-targeting vaccine design offers one potential strategy to meet this challenge. The strategy is predicated on the design of priming immunogens that consistently induce responses from rare bnAb-precursor B cells with pre-defined BCR features, select for at least a modicum of productive BCR maturation, and generate a pool of GC and/or memory B cells likely to be susceptible to boosting by more native-like immunogens. Here, in the first test of this strategy in humans, we found that the eOD-GT8 60mer/AS01B germline-targeting vaccine prime had an acceptable safety profile and induced the targeted VRC01-class IgG B cells with substantial frequencies in blood and lymph nodes consistently across vaccine recipients. We further demonstrated that the vaccine selected for bnAb-like BCR properties and favorable maturation, and consequently generated B cells with capacity to bind less engineered and more native-like forms of the CD4bs epitope. These findings establish clinical proof of concept for germline-targeting priming, support developing boost regimens to induce VRC01-class bnAbs, and encourage extending the strategy to other targets in HIV and other pathogens.
Developing germline-targeting priming immunogens for other classes of antibodies will be more challenging owing to the HCDR3 dominance of most antibodies that is not observed for VRC01-class bnAbs, but a generalized method for designing such immunogens has been described (25). Deploying the germline-targeting strategy effectively for other antibody classes will require knowledge of population frequencies of any required gene alleles, analogous to the VH1–2 *02 and *04 alleles here, as well as frequencies of recombination events that produce potential bnAb precursors, analogous to the production of 5-amino acid LCDR3s here.
Our BCR sequence hierarchical clustering analysis showed that VRC01-class responses derived from many independent recombination events in each participant. Thus, the ability to prime VRC01-class responses consistently across vaccine recipients was due in part to the immunogen having affinity for diverse VRC01-class precursors. Explicit engineering of priming immunogens with affinity for diverse precursors is one of the hallmarks of germline-targeting vaccine design that distinguishes this approach from others.
The robust serological and B cell immunogenicity observed here, especially the substantial frequencies of eOD-GT8- and CD4bs epitope-specific IgG memory B cells in PBMCs after only one or two vaccinations, was likely due largely to the combination of a high-valency, glycosylated nanoparticle immunogen with a strong adjuvant (13, 39, 47, 58). The strong immune responses and acceptable reactogenicity support the use of adjuvanted self-assembling nanoparticle vaccines in humans.
A major challenge for priming and maturation of responses from rare bnAb-precursor B cells is the competition from higher frequency non-bnAb B cells that can engage the same epitope (25–27, 39, 45–49). Here, VRC01-class responses remained in the minority of CD4bs-directed responses at all timepoints after prime and boost, but nevertheless maintained high positivity and exhibited favorable maturation. Our BCR sequence and affinity analyses provide insights into how that was achieved. The affinities required for priming of rare bnAb precursors were relatively high (81% were better than 3 μM and 50% better than 119 nM), orders of magnitude higher than for competitors, and the responses derived predominantly from naive B cells. Furthermore, the germline-targeting priming immunogen was able to: (i) select B cells encoding bnAb-like properties beyond the properties specifically targeted; (ii) stimulate previously matured GC and/or memory B cells by a booster immunization, leading to increased SHM and affinity of bnAb-precursors in the memory pool, with weaker increases for competitors; (iii) maintain a large affinity advantage for bnAb-precursors over competitors; and (iv) guide affinity maturation in bnAb-precursors toward bnAb development, likely by presenting an affinity gradient. Successful priming and boosting of bnAb-precursor B cells with such a multi-faceted set of desirable outcomes, even while the targeted cells remained in the minority of epitope-specific B cells, suggests that design of immunogens with appropriate affinities and affinity gradients can circumvent B cell competition as a barrier to steered maturation of bnAb-precursor responses. These findings provide support not only for the concept of germline-targeting priming but also for the broader concept of sequential vaccination to guide evolution of targeted responses.
Our finding that VRC01-class responses derived mostly from precursors with affinities better than 3 μM provides a potential benchmark for other germline-targeting efforts and accords with pre-clinical mouse model data on eOD-GT8 60mer and analogs (47–49), supporting the use of such models to predict human responses. Our data on VRC01-class and non-VRC01-class precursor affinities support the hypothesis that relatively high affinities are required for consistent priming of low frequency precursors, even for highly multivalent nanoparticle immunogens (13, 21, 25, 39), and also support the claim that precursor frequency and affinity are interdependent for determining B cell competitive fitness in GCs (47–49, 59). Given that interdependence, it will be important to determine if consistent priming by germline-targeting immunogens for lower frequency HCDR3-dominant bnAb-precursors (25) will require even higher affinities than observed here.
A germline-targeting vaccine prime should generate as large a pool of bnAb-like memory B cells as possible, to facilitate successful boosting by a more native-like immunogen. Prior human naive B cell sorting revealed that the frequency of VRC01-class B cells that bind eOD-GT8 with affinities better than 3 μM was approximately 1 in 900,000 among all human naive B cells (21, 43). Here, we estimated that, for the 81% of VRC01-class responses to eOD-GT8 60mer derived from precursors with affinity better than 3 μM, the expansion from naive B cells to IgG memory B cells was approximately 37-fold at week 8 (after one vaccination) and 250-fold at week 16 (after two vaccinations), averaging responses from the low and high dose groups. These expansion levels provide benchmarks for other germline-targeting priming vaccines. The expansion after one vaccination observed here was substantially stronger than the expansions into memory measured in five different mouse models with human naive VRC01-class precursors of affinity better than 3 μM and frequencies of either 1 in 1 million (two models, HuGL18 and HuGL17, with 2-fold expansion and >100-fold contraction, respectively (48)) or 1 in 10,000 (three models, CLK21, CLK19, and CLK09, with expansions of approximately 2.5-, 4.0-, and 5.5-fold, respectively (49)). Hence even precisely calibrated mouse models can underestimate the degree to which human germline-targeting vaccines can induce bnAb-precursor-derived memory B cell responses, which should be considered when evaluating other priming immunogens in pre-clinical experiments. A thorough and detailed comparison of the multidimensional human data from this trial (frequencies, SHM, bnAb properties, affinities) with the results of the many mouse models applied to eOD-GT8 60mer vaccination is warranted and may assist the selection and design of models that optimally predict human responses.
A key hypothesis underlying the germline-targeting vaccine design strategy is that sequential vaccination with increasingly more native-like boost immunogens will be capable of inducing bnAb development by driving bnAb-precursor B cells to undergo repeated rounds of affinity maturation. This could be achieved if each boost stimulated memory B cells to return to GCs and/or supplied new antigen to existing GCs. Raising questions about the practicality of generating sufficient mutation by sequential immunization, results from mouse studies with model antigens indicated that stimulation of previously matured IgG memory B cells generally induces differentiation to antibody-secreting cells rather than GC re-entry for further affinity maturation (60–63) and may suffer from clonality bottlenecks (60). Furthermore, repeated immunization with the same antigen was found to increase the number of memory B cells but not significantly increase SHM levels or affinities (61). On the other hand, sequential immunization with different immunogens can increase SHM of bnAb-precursor memory B cells in knockin mouse models (23, 29); and repeated boosting with HIV trimers in non-human primates or with SARS-CoV-2 mRNA vaccines in SARS-CoV-2-naive humans can increase SHM in antigen-specific memory B cell populations (64–66). Our human vaccination data showed that for VRC01-class responses, mutation levels and affinities in IgG memory BCRs increased following the boost, and polyclonality remained high after prime and boost. Thus, our data suggest that maturation of human B cells toward bnAb development by sequential vaccination remains plausible, especially considering that heterologous boosting should generate more mutation and diversification than the autologous boost studied here. In the future, it will be important to define the relative contributions of memory B cell re-entry to GCs versus antigen re-fueling of GCs, to sequential-vaccination-induced B cell maturation in humans. The degree to which one mechanism or the other dominates could have implications for optimizing design of boosting/shephering immunogens and regimens, and for developing strategies to monitor responses in clinical trials and pre-clinical models.
With efficient priming of VRC01-class responses established, major challenges remain ahead for sequential vaccination to shepherd these responses to bnAb development. The critical step will be to induce B cells that can bind fully native trimers with the N276 glycan intact. Succeeding at that step will likely require first taking one or more taking smaller steps to advance the maturation of VRC01-class bnAb-precursor-derived B cells to enable binding to increasingly more native-like forms of the CD4bs (26–29, 56), consistent with our SPR data showing that VRC01-class BCRs induced by eOD-GT8 60mer lack detectable affinity for a native trimer but do show at least low affinity for core-gp120 or eOD-GT6 variants. VRC01-class and non-VRC01-class BCRs induced by eOD-GT8 60mer should be helpful for identifying candidates for the first heterologous boost.
The consistent VRC01-class bnAb precursor priming demonstrated here represents an unprecedented level of vaccine control over the specificity of humoral responses, and as such, may herald a new era of precision vaccine design for HIV and other pathogens. By defining desired B cell responses at the molecular level, the germline-targeting vaccine design approach allows for a highly reductive and iterative design cycle to optimize vaccine discovery and development.
Materials and Methods
Study design
IAVI G001, with ClinicalTrials.gov registry number NCT03547245, was a phase 1, randomized, double-blind, placebo-controlled dose escalation study to evaluate the safety and immunogenicity of eOD-GT8 60mer vaccine adjuvanted with AS01B in HIV-uninfected, healthy adult volunteers. Two doses of 20 μg or 100 μg eOD-GT8 60mer with AS01B, or two doses of placebo were given by deltoid intramuscular injection eight weeks apart, with both immunizations given to the same arm. Placebo was the buffer used in the vaccine: DPBS containing 10% sucrose at pH 7.5. The consort diagram is shown in fig. S1.
The primary objectives of the study were to evaluate the vaccine for safety and tolerability and the capacity to induce Immunoglobulin G (IgG) B cell responses from rare precursors for VRC01-class bnAbs. The primary endpoints were the occurrence of adverse events, and the secondary endpoint was induction of eOD-GT8 60mer-specific, eOD-GT8 monomer-specific and CD4-binding-site (CD4bs)-specific serum binding antibody responses. The detection of VRC01-class responses in IgG memory B cells, IgG GC B cells, and plasmablasts were exploratory endpoints but nevertheless were the critical immunological readouts to judge vaccine efficacy. Additional exploratory immunological analyses included: (i) assessing the relative frequencies of the VRC01-class BCRs and competitors; (ii) measuring the changes in somatic hypermutation and eOD-GT8 binding affinities over time for both types of BCRs; (iii) evaluating a wide array of properties of the VRC01-class BCRs, to assess the potential for the BCRs to mature into bnAbs; and (iv) assessing the binding of VRC01-class and competitor BCRs to antigens with CD4bs epitopes closer to native HIV Env compared to the vaccine.
Participants and randomization:
Eligible participants were healthy male and female adults aged 18 through 50 years of age who were willing to undergo HIV testing, use an effective method of contraception, understood the study in the opinion of the investigator or designee, and provided written informed consent. Forty-eight participants who met all eligibility criteria were included in the study and were randomly assigned to receive vaccine or placebo within one of two groups. Group 1 included 18 low dose (20 μg) vaccine recipients and 6 placebo recipients, and Group 2 included 18 high dose (100 μg) vaccine recipients and 6 placebo recipients. Twenty-four participants were enrolled at each of two clinical sites: George Washington University (GWU) and Fred Hutchinson Cancer Research Center (FHCRC). There was no attempt to match the participants for any demographic category among the three study groups or between the two clinical sites. Participant demographics are given in table S1. Among enrolled participants, for sex at birth, approximately equal numbers were male (25/48; 52.1%) and female (23/48, 47.9%); the predominant race reported was White (33/48, 68.8%), with Asian and Multiracial (each at 5/48, 10.4%) being the next highest race categories reported. The ethnicity of “Not Hispanic and Not Latino” was reported for the majority of the participants (42/48, 87.5%). The median age and body mass index were 27 years and 25.6 kg/m2, respectively.
Oversight:
The trial was conducted under an Investigational New Drug (IND) application submitted to the US Food and Drug Administration, and was carried out in compliance with the protocol filed within the IND. The trial adhered to IAVI standard operating procedures in accordance with the guidelines formulated by the International Committee on Harmonization for Good Clinical Practice in clinical studies, and complied with applicable local standards and regulatory requirements including review and approval by the institutional review boards at FHCRC and GWU. The trial was overseen by a Protocol Safety Review Team and independent Safety Monitoring Committee.
Blinding:
Study site investigators, staff and volunteers were blinded in terms of vaccine versus placebo. An unblinded study pharmacist at each site was responsible for vaccine preparation and accountability. Staff carrying out immunological assays were blinded. Staff carrying out bioinformatic and statistical analyses were unblinded, which enabled analyses to be carried out during the trial and led to early planning and preparation for follow-on trials (IAVI G002, ClinicalTrials.gov Identifier: NCT05001373; and IAVI G003, NCT05414786).
Safety and tolerability monitoring:
Safety and tolerability were monitored during the trial by site Investigators, the IAVI Medical Monitor and the Protocol Safety Review Team. The safety and tolerability of the vaccine were evaluated by the Safety Monitoring Committee for, at minimum, the first 14 days after the first vaccination for all participants in the low dose group (Group 1) before escalating to the higher dose level (Group 2). Participants were followed up to 12 months after the final investigational product administration. Adverse events (AEs) were grouped by Medical Dictionary for Regulatory Activities Terminology (MedDRA) System Organ Class (SOC) and Preferred Term (PT). All AEs were graded for the entire duration of the study, using the National Institutes of Allergy and Infectious Diseases (NIAID) Division of AIDS (DAIDS) Table for Grading the Severity of Adult and Pediatric Adverse Events, Version 2.1, July 2017.
Immunological assays:
Serum antibody binding responses were assessed by Binding Antibody Multiplex Assay (BAMA), and serum antibody neutralization was assessed using TZM-bl neutralization assays. Frequencies of antigen-specific and CD4bs epitope-specific B cells were assessed by fluorescence-activated cell sorting (FACS). The primary immunological readout, the induction of VRC01-class IgG B cells, was assessed by CD4bs-specific single B cell sorting, B cell receptor (BCR) sequencing, and bioinformatic analysis. Polyclonality and genetic diversity of VRC01-class IgG BCR responses were assessed by bioinformatic analysis including hierarchical sequence clustering.
Definition of CD4bs-specific responses:
Assessment of serum or B cell binding to the eOD-GT8 CD4bs epitope was determined by differential binding to eOD-GT8 and eOD-GT8-KO11, a variant of eOD-GT8 with three mutations in the CD4bs (N280R, S365L, and F371R in HxB2 numbering) that essentially abrogates binding by VRC01-class precursor Abs and VRC01-class bnAbs (43, 46–49). In SPR experiments we measured no detectable binding to multiple VRC01-class human naive precursors and bnAbs at concentrations up to 30 μM of eOD-GT8-KO11 (not shown). eOD-GT8-KO11 was originally referred to as eOD-GT8-KO2 (43, 46, 47) but has subsequently been referred to as eOD-GT8-KO11 (48, 49). Additional details on how the eOD-GT8 and eOD-GT8-KO11 antigens were employed for BAMA and B cell sorting are provided below.
Power analysis and rationale for trial size
Group sizes for the study were selected to measure the primary hypothesis that the vaccine will induce VRC01-class IgG B cells with a response rate of at least 50% in at least one of the adjuvanted protein vaccine arms, as well as to satisfy the need to have enough endpoints for further characterization of that response. We powered the study to have high probability of observing at least 5 participants with a vaccine-induced VRC01-class IgG B cell response among participants receiving eOD-GT8 60mer in study Group 1 or 2 given that the true response rate for this class of B cells was at least 50% or greater among eOD-GT8 60mer recipients in either arm. We assumed a dropout rate of approximately 10%, which translated into an assumption that immunogenicity samples would be obtained from N=16 recipients of eOD-GT8 60mer in each of Groups 1 and 2. Under those assumptions, we determined that at least 5 positive responders for VRC01-class B cells would be required for the 95% confidence interval (CI) about the observed rate to be consistent with a true rate of 50%, because the 95% CI for an observed rate of 5/16=31.25% is 14.2% to 55.6%. Furthermore, power was 96.2% to detect 5 or more positive responders out of 16 when the true rate of response was 50% (fig. S40).
Vaccine and adjuvant
eOD-GT8 60mer was manufactured in accordance with current Good Manufacturing Practice (cGMP) regulations at Paragon BioServices, Inc (Baltimore, MD), as described in detail elsewhere (67). In summary, cGMP manufacture was accomplished by a combination of purification techniques following transient expression in suspension-adapted, cGMP-qualified VRC293 human embryonic kidney 293 (HEK293) cells generated at SAFC (Carlsbad, CA) from a Master Cell Bank generously provided by the National Institutes of Health (NIH) Vaccine Research Center (VRC) within the National Institute of Allergy and Infectious Diseases (NIAID). VRC293 cells were grown in serum-free Expi293 medium (Thermo Fischer), transfected with plasmid DNA encoding eOD-GT8 60mer (cGMP manufactured by Aldevron; Fargo, ND) and Polyethylenimide PEIpro®-HQ transfection reagent (cGMP manufactured by PolyPlus-Transfection SA; Illkirch, France), and expression was carried out in the presence of 14 μM Kifunensine (cGMP manufactured by GlycoSyn; Graceville, New Zealand). Benzonase endonuclease enzyme (high-purity grade, 250 U/μL, from Millipore; Burlington, MA, USA) was employed to remove residual host cell and plasmid DNA. The eOD-GT8 60mer cGMP clinical material was formulated at 1 mg/mL in 10% sucrose in phosphate-buffered saline (PBS) at pH 7.2, aliquoted at 0.4 mL volume in 2 mL Type 1 glass vials with stoppers (13 mm stopper, Rubber with Flurotec, Afton Scientific), sealed with sterile seals from Afton Scientific Corporation (Charlottesville, VA), and stored at −80°C. The material is currently on a stability testing program as per regulatory guidelines (stable for >36 months).
Quality control procedures were performed on the manufactured eOD-GT8 60mer nanoparticle to confirm its identity, determine protein concentration and purity, establish in-vitro potency, measure the nanoparticle size, and characterize N-linked glycans. Additionally, the clinical trial lot was tested to quantify host cell residual impurities (host cell proteins and host cell DNA), measure bioburden and bacterial endotoxin, determine subvisible particulate matter and confirm sterility. Quality control procedures included sandwich ELISA with GL-VRC01 for potency; high pressure liquid chromatography (HPLC) with tandem mass spectrometry (MS/MS) to confirm amino acid sequence; N-terminal Edman sequencing for further sequence confirmation; size exclusion chromatography (SEC) to assess purity; sedimentation velocity analytical ultracentrifugation (AUC-SV) for particle size distribution; dynamic light scattering (DLS) for average particle size; hydrophobic interaction liquid chromatography coupled to mass spectrometry (HILIC-FLD-MS/MS) for N-linked glycan profiling. eOD-GT8 60mer cGMP material had full glycan occupancy at only five of ten glycosylation sites, and had partial occupancy at three sites (67). Pre-clinical material that performed well in mouse models had full glycan occupancy at three sites and partial occupancy at four sites (43).
AS01B adjuvant is an adjuvant system composed of two immunoenhancers combined in a liposomal formulation consisting of dioleoyl phosphatidylcholine (DOPC) and cholesterol in phosphate-buffered saline solution. The immunoenhancers are: (1) 3-O-desacyl-4’-monophosphoryl lipid A (MPL), a derivative of lipopolysaccharide from the Gram-negative bacterium Salmonella minnesota, and (2) a saponin molecule (QS-21) purified from the bark of the tree Quillaja saponaria Molina. AS01B was manufactured and provided by GlaxoSmithKline Biologicals (Rixensart, Belgium). QS-21 was licensed by GSK from Antigenics LLC, a wholly owned subsidiary of Agenus Inc. (Delaware, USA). The administered dose of AS01B corresponded to 50 μg each of MPL and QS-21.
DPBS with 10% sucrose was manufactured by SAFC Biosciences.
Study procedures
Vaccine preparation:
Vaccine was diluted to the appropriate dose and mixed with adjuvant just prior to administration. For Group 1, the low dose (20 μg) group, eOD-GT8 60mer investigational product (IP) was first diluted 1:5 with DPBS Sucrose; then 0.15 mL of this diluted IP was transferred into a vial containing 0.65 mL AS01B; and finally, after gentle inversion to mix, 0.6 mL was withdrawn into a syringe and administered as an injection in the deltoid muscle of the non-dominant arm. For Group 2, the high dose (100 μg) group, 0.15 mL of eOD-GT8 60mer IP was transferred into a vial containing 0.65 mL AS01B, and after gentle inversion to mix, 0.6 mL was withdrawn into a syringe and administered as an injection in the deltoid muscle of the non-dominant arm. For all placebo assignments, 0.6 mL of DPBS Sucrose was withdrawn into a syringe and administered as an injection in the deltoid muscle of the non-dominant arm. Vaccine or placebo were administered at Day 0 and Week 8 (Day 56±7); for each participant, the first and second injections were administered in the same arm.
Schedule of procedures:
The full schedule of procedures is given in table S2.
Safety and tolerability:
Weekly follow-up visits were scheduled for study weeks 1–4 and 9–11, and additional follow-up visits were scheduled for weeks 16, 20, 32 and 56. Participants recorded local and systemic reactogenicity using a memory aid from Day 0 through Day 7 after vaccination. At each vaccination visit, vital signs were measured by study staff prior to vaccination and at least 30 minutes post-vaccination. Unsolicited adverse events (collected through open-ended questions) were collected from Day 0 through 28 days after the second (final) vaccination. Serious adverse events, medically attended adverse events and potential immune-mediated diseases (pIMDs) were collected during the entire study period through 12 months after the second dose administration. Potential immune-mediated diseases were a subset of adverse events that included autoimmune diseases and other inflammatory and/or neurologic disorders of interest which may not have had an autoimmune etiology.
Immunological sample collection and storage:
Leukapheresis was performed at two time points, once during screening at week -4 and once at study week 10 (approximately 14 days after the second vaccination). Peripheral blood mononuclear cells (PBMC) were collected at weeks -4, 4, 8, 10, and 16 after the 1st vaccination by leukapheresis or by venipuncture (whole blood with ACD anticoagulant) and were isolated by density gradient centrifugation and cryopreserved as aliquots of 20×106 or 50×106 cells. Separately, PBMC aliquots obtained by a single leukapheresis from an unvaccinated HIV-negative volunteer served as an internal negative control for flow cytometry panel and probe staining for every experiment. Ultrasound-guided fine needle aspirations (FNAs) of axillary lymph node(s) were performed at two time points, approximately 21 days after each vaccination. The procedure was performed by a board-certified radiologist using ultrasound guidance to avoid needle insertion into any adjacent structures. Plasmablast samples (10 mL PBMCs) were collected at week 9 (5–8 days after the second vaccination). Lymph node (LN) FNA and plasmablast samples were stored on wet ice but were not frozen before being subjected to cell sorting analyses within 24 hours of collection.
Fine needle aspiration from draining axillary lymph nodes
Aspirates were collected from the draining axillary lymph nodes by fine needle aspiration (FNA) at weeks 3 and 9. Ultrasound was used to visualize the axillary lymph nodes on the same side as the site of the most recent vaccination for lymph node sampling. Following site-specific standard FNA procedures, the overlying skin was swabbed generously with chlorhexidine, betadine, or similar skin disinfectant solution, and 1% lidocaine was injected subcutaneously as local anesthetic. A 22-gauge lumbar puncture needle attached to a 5 mL sterile syringe was then inserted into the largest and most accessible lymph node, and negative pressure was applied by withdrawing the syringe lunger approximately 2–3 mL while collecting sample over a 30 second period of ‘to and fro’ needle movement, or until first appearance of blood in syringe. Immediately after withdrawal of each needle from the study participant, syringe and needle contents were expelled into a 50mL conical tube containing R10 media, flushing out any remaining cells out of the syringe by detaching the lumbar needle from the syringe, withdrawing approximately 2 to 3 mL of fresh R10 media into the syringe barrel, reattaching it to the needle, and expelling the media into the 50mL conical tube; this flushing procedure was repeated a total of 4 times for each aspiration. The collection was repeated up to 4 times for each study participant, using a new sterile needle and syringe for each pass. After collection, the sample was transported to the laboratory on wet ice or cold packs as soon as possible. Lymph node FNA samples ranged in recovery from very few cells up to 4×107 cells (median recovery varied between sites from 2.4×106 to 4.7×106).
Preparation of lymphocytes from fine needle aspirates from axillary lymph nodes
The 50mL conical FNA sample was spun at 325 × g for 10 mins at 4°C (no brake). Supernatant was gently aspirated with a serological pipet down to about 500uL. If the cell pellet had any redness, cells were resuspended in 5mL of cold 1x red blood cell (RBC) lysis buffer (eBioscience) and incubated for 5 minutes at ambient temperature while agitating sample by gently pipetting up and down periodically with a P1000 pipette or using a tube rocker. Cells were washed with 40 mL cold 10% FBS/ 1xPBS. Samples were centrifuged at 325 × g for 10 minutes at 4°C. Supernatant was gently aspirated with serological pipette and the cell pellets were resuspended with 2 mL cold 10% FBS/ 1xPBS. Cell numbers and viability of the sample were determined using a hemocytometer, Muse cell analyzer, or Nexcelom Auto 2000.
Production of protein reagents for immunological assays
Reagents for B cell sorting:
Avi-tagged and biotinylated versions of eOD-GT8 monomer (21, 39) and eOD-GT8-KO11 monomer (different than the original eOD-GT8-KO) (43, 46), were produced by a cGLP lab under contract at Scripps Research. These proteins were produced as previously described (13) in FreeStyle 293F (Invitrogen) suspension cultures by transient transfection using 293Fectin (Invitrogen) of a pHLSec plasmid containing mammalian codon-optimized eOD with a C-terminal His6x affinity tag followed by an Avi tag. Protein was harvested from the supernatant after five days and purified by affinity chromatography with a HIS-TRAP column (GE Healthcare) followed by Superdex 75 size exclusion chromatography (GE Healthcare) using an AKTA chromatography system (GE Healthcare). Biotinylation was accomplished using BirA (Avidity), and the level of biotinylation was estimated by PAGE.
Reagents for BAMA:
Avi-tagged eOD-GT8 monomer was produced as described above. Non-avi-tagged eOD-GT8-KO11 monomer was produced following similar methods but was produced in the Schief lab at Scripps Research. eOD-GT8 60mer for BAMA assays was also produced in the Schief lab, in FreeStyle 293F (Invitrogen) suspension cultures by transient transfection using 293Fectin (Invitrogen) of a pHLSec plasmid containing mammalian codon-optimized eOD-GT8 60mer. Protein was harvested from the supernatant after five days and purified by lectin chromatography followed by size exclusion chromatography with a Superose 6 or Superose 6 Increase column (GE Healthcare). Lumazine synthase nanoparticle was produced by BlueSky Bioservices, in E.coli, using heat treatment (75°C for 30 min) of the supernatant from lysed cells followed by centrifugation to again obtain supernatant, and then size exclusion chromatography first with Superdex 75 and finally with Sephacryl 500.
Serum binding analysis by BAMA
Serum IgG responses were measured via a multiplex antigen panel (i.e. eOD-GT8, eOD-GT8-KO11, eOD-GT8 60mer, Lumazine synthase) to determine specificity and magnitude. A binding antibody multiplex assay (BAMA) (68–70) was modified by lengthening the primary antibody incubation period from 30 to 120 minutes for enhanced detection sensitivity of early bnAb precursors. The assay was validated for accuracy, specificity, precision, robustness and limit of detection/quantitation (LLOD/LLOQ). The LLOQ for detection of early bnAb precursors was 0.0041–0.0866 μg/ml. Antibodies were measured at day 0 (day of first vaccination), day 14 (two weeks post-first vaccination), day 28 (four weeks post-first vaccination, day 56 (day of second vaccination), day 70 (two weeks post-second vaccination), and day 112 (eight weeks post-second vaccination). Samples were serially diluted (1:50, 1:250, 1:1250, 1:6250, 1:31250, and 1:156250) and incubated for 120 minutes with a mixture of carboxlyated fluorescent MagPlex microsphere sets (Luminex) that were each covalently coupled to one of the antigens. Antigen-specific IgG was detected using a biotinylated detection antibody to human IgG Fc (Southern Biotech), followed by washing and incubation with Streptavidin PE (BD Pharmingen). Samples were acquired on a Bio-Plex instrument (Bio-Rad), and antibody levels were measured as median fluorescent intensity (MFI) from two wells and then averaged using the mean. The readout was background-subtracted median fluorescent intensity (MFI), where background refers to the antigen-specific plate-level control (i.e., a blank well containing antigen-conjugated beads run on each plate plus detection antibody). Additionally, a blank or reference bead was included to estimate non-specific antibody binding. Area under the titration curve (AUTC) was used as the magnitude measure of interest. AUTC was calculated using the trapezoid method with truncation in the case of negative background-adjusted MFI minus background-adjusted blank MFI (MFI*) values or MFI* values > 22000. Samples with blank MFI > 5000 were excluded from the analysis. The positive controls were GL-VRC01 (germline VRC01 mAb), VRC01 mAb, anti-6X HIS epitope tag (for His-tagged proteins), CH31 mAb, 2G12 mAb.
The positivity of a response was defined based on background-adjusted MFI values at the screening dilution level (1:50) except Lumazine synthase. Lumazine synthase was observed to have high baseline values across participants at the screening dilution, so filtering for high baseline MFI* was not applied and the response call for this antigen was made at the first dilution for which baseline MFI* was < 6500. Antigen-specific positivity thresholds were computed as the maximum of 100 and the 95th percentile of the baseline MFI* by antigen except Lumazine synthase which was set to 100. Samples from post-enrollment visits were declared to have a positive binding antibody response by BAMA if they met three criteria:
-
1
MFI* values were greater than the antigen specific positivity threshold.
-
2
MFI* values were greater than 3 times the baseline MFI* values.
-
3
Background-adjusted MFI values were greater than 3 times the baseline background-adjusted MFI values.
For differential binding to the CD4 binding site (CD4bs), defined as the difference in AUTC for binding to eOD-GT8 (AUTCRef) and eOD-GT8-KO11 (AUTCKO), the positivity of response was defined as a positive response to eOD-GT8 plus an additional criterion:
-
4
ΔAUTC > 0, where ΔAUTC was defined as AUTCRef − AUTCKO.
Pseudovirus production and neutralization assays
Neutralizing antibodies against HIV-1 pseudoviruses were measured as a function of reduction in Tat-regulated luciferase (Luc) reporter gene expression in TZM-bl cells (71, 72). Neutralization ID50 titers were measured from specimens obtained at days 0 (visit 3, baseline), 14 (visit 4, 2 weeks post-1st vaccination), and 70 (visit 8, 2 weeks post-2nd (final) vaccination). Neutralization titer was defined as the serum dilution at which relative luminescence units (RLUs) were reduced by 50% (ID50) relative to the RLUs in virus control wells (cell + virus only) after subtraction of background RLUs (wells with cells only).
A specialized panel of 426c Env-pseudotyped viruses was used to detect and characterize early intermediates of VRC01-class bnAbs. Some VRC01 early intermediates neutralize 426c in a manner that requires deletion of one or more N-glycans in the vicinity of the CD4bs. Neutralization may be enhanced by Man5-enrichment of remaining N-glycans that otherwise are processed into larger complex-type glycans. Man5-enrichment was achieved by producing Env-pseudotyped viruses in 293S GnTI− cells. 293T cells were used to produce Env-pseudotyped viruses with fully processed glycans. Mutation D279K was used to confirm CD4bs specificity. The viruses 426c.N276D/GnTI− and 426c.N276D.N460D.N463D/GnTI− can detect germline-reverted forms of VRC01, VRC07 and VRC20 but not germline-reverted forms of other CD4bs bnAbs (73).
A specialized panel of CH505 Env-pseudotyped viruses was used to detect and characterize early intermediates of CH235 and CH103 bnAb lineages. Neutralization by CH235 early intermediates is dependent on an N279K.G458Y double mutation and also requires Man5-enrichment of N-glycans that are otherwise processed into larger complex-type glycans. Man5-enrichment was achieved by producing Env-pseudotyped viruses in 293S GnTI− cells. 293T cells were used to produce Env-pseudotyped viruses with fully processed glycans. Mutation N280D was used to confirm CD4bs specificity. CH505.N279K.G458Y/GnTI− detects germline reverted and early intermediates of CH235 but not germline-reverted forms of other CD4bs bnAbs (74).
Neutralization by early intermediates of CH103 requires deletion of four N-glycans in the vicinity of the CD4bs, combined with Man5-enrichment of remaining N-glycans that otherwise are processed into larger complex-type glycans. Mutation S365P was used to confirm CD4bs specificity. CH505TF.gly4/GnTI− detects germline-reverted and early intermediates of CH103 (74).
Statistical analysis of neutralization assay data
Response to a viral isolate was considered to be positive if the neutralization titer was greater than or equal to 20. Response rates were calculated with 95% Wilson score intervals. Response magnitude analyses included vaccine-recipients only and included responders and non-responders (with truncated titer measurements). There were no detectable neutralizing antibody responses to the eOD-GT8 vaccine (low-dose or high-dose) that indicated elicitation of early intermediates of VRC01-class, CH235, or CH103 bnAbs. There was one low-level low-dose response (ID50 = 22.47) to CH0505TF.gly4.S365P.2/293S/GnTI− (CH103 precursor knock-out mutant) after the 2nd vaccination, but this was hard to interpret in the absence of a response to CH0505TF.gly4 (CH103 precursor). Negative results do not necessarily indicate a lack of bnAb precursors, since the virus panel only detects a subset of CD4bs bnAb precursors. There were no Tier 1A (MW965.26/293T/17 and MN.3/293T/17), placebo, or baseline responses. Due to overall lack of neutralization response, response rate and magnitude testing were not performed.
Overview of the VRC01-class B cell assay
VRC01-class B cell precursors were identified first based on their ability to bind to the CD4bs of the eOD-GT8 protein, determined using fluorescently-labeled eOD-GT8 proteins and a modified eOD-GT8 protein containing mutations specifically in the CD4bs epitope (eOD-GT8 KO11). Differential staining of B cells to the eOD-GT8 protein but not to the CD4bs mutant (eOD-GT8 KO11) indicated that B cells were specific to the CD4bs. Therefore, fluorescently-labeled eOD-GT8 and eOD-KO11 were combined with a flow cytometry antibody panel to identify and single-cell sort the specific B cell populations of interest: circulating memory B cells, plasmablasts and germinal center (GC) B cells. CD4bs-specific B cells bound two eOD-GT8 tetramers (GT8++) but not tetramers of eOD-GT8 KO11 (KO−). Since the B cell sorting and sequencing needed to be performed on fresh samples for plasmablasts and lymph node (LN) FNAs, the sorting and sequencing was established and performed at two laboratories that were in close proximity to the clinical sites. Procedures and reagents were harmonized (except where noted otherwise) and verified at the two laboratories (figs. S5–S10 and tables S10–S19).
PBMC samples from weeks -4, 4, 8, 10, and 16 after the first vaccination were sorted for CD4bs-specific IgG memory B cells; draining axillary lymph node cells, acquired by FNA at weeks 3 and 11, were sorted for CD4bs-specific IgG germinal center (GC) B cells; and PBMC samples at week 9 (5–8 days after the second vaccination) were sorted for CD4bs-specific IgD− plasmablasts (PBs) (Fig. 1A and tables S10–11). For each sample, RT-PCR and DNA sequencing were applied to as many CD4bs-specific IgG cells as possible, up to a maximum of two 96-well plates. DNA for HCs and LCs (kappa and lambda) was sequenced by the Sanger method, and selected samples from the low dose vaccine group were re-sequenced using next-generation sequencing. BCR sequences were subjected to quality filtering and bioinformatic analysis. Additional details of these procedures are described below.
Tetramer probe preparation
Tetramers were prepared at a 4:1 molar ratio of monomeric eOD-GT8 or KO11 proteins to streptavidin. The total volume of protein, 1x PBS, and protease inhibitor (catalog# 539131–10VL; MilliporeSigma; Burlington, MA, USA) was added to a microcentrifuge tube. Twenty percent of the total streptavidin volume was added and incubated with continuous rotation for 20 min at 4°C in the dark. The incremental addition of streptavidin was repeated 4 times until the total amount of streptavidin had been added to the protein. Tetramers were made fresh for each experiment (VRC) or stored at 4°C and kept to up to 1 month (FHCRC).
Monoclonal bead controls
Bead assays were performed on the day of sorting experiments to confirm the functionality of the tetramers by flow cytometry. Anti-mouse Ig kappa beads (BD Bioscience, La Jolla, CA, USA) were washed with 3 mL R10 in polystyrene FACS tubes and then centrifuged at 650 × g for 5 mins. Supernatants were discarded and beads were resuspended in 100μL R10. Each bead control was given 1μg of mouse anti-human IgG and incubated for 15 mins at room temperature. Beads were washed with 3mL R10 and resuspended with either 1μg of glVRC01 mAb (to test eOD-GT8 probes) or KG064 mAb (to test eOD-GT8-KO11 probes) in 100μL. Beads were incubated for 15 mins at room temperature, washed with 3mL R10 and then resuspended in 100μL R10. Both eOD-GT8 probes were added to the glVRC01 positive control beads as well as a negative control beads that did not receive glVRC01. The KO11 probe was added to the KG064 mAb-labeled beads as well as a bead-only control that did not receive KG064. Each tube was incubated for a minimum of 25 mins at 4°C. Beads were washed with 3mL R10, centrifuged, decanted and resuspended in up to 300μL R10 for collection.
Enrichment of B cells from cryopreserved PBMC samples
All five cryopreserved PBMC samples from a single participant were sorted in an experimental batch on a given day. Cryopreserved PBMCs were thawed, either by water bath or with use of Thawsome cryovial adapters, into warm benzonase (MilliporeSigma; Burlington, MA, USA) supplemented R10 [RPMI 1640 with 25mM HEPES buffer and L-glutamine (Thermo Fisher Scientific; Waltham, MA, USA) and 10% fetal bovine serum (FBS; Nucleus Biologics, San Diego, CA, USA)]. Samples were centrifuged at 300 × g for 12 minutes. The supernatant was decanted, and the cells were washed with R10. The cell recovery and viability were optionally determined using the Muse Cell Analyzer (MilliporeSigma; Burlington, MA, USA). B cells were enriched using the EasySep Human Pan-B Cell Enrichment Kit (StemCell Technologies; Vancouver, CA) and the Big Easy magnet (StemCell Technologies; Vancouver, CA) following the manufacturer’s instructions summarized here. Samples were resuspended at a concentration of 5×107 cells/mL with EasySep buffer. Fifty microliters of Enrichment Cocktail (per mL of sample) were added and mixed. The samples were incubated at room temperature for 10 min. Magnetic particles were vortexed for 30 seconds, and 75 μL magnetic particles (per mL of sample) were added and mixed with the samples and incubated at room temperature for 5 mins. EasySep Buffer was added to the sample up to 5 mL for samples under 2 mL (<108 cells) or 10 mL for samples greater than or equal to 2 mL (>108 cells). The sample was loaded into the EasySep magnet without the lid and incubated at room temperature for 5 mins. With the tube on the magnet, the cell suspension was pipetted off and into a new conical. The sample conical was removed from the magnet, and the beads were resuspended with the same amount of EasySep Buffer used previously and then mixed and re-incubated on the EasySep magnet (without lid) at room temperature for 5 mins. With the tube on the magnet, the cell suspension was pipetted off and combined with the previous matching sample. The cell number and viability of the sample was optionally determined.
Flow cytometry staining procedures
Cells, either fresh PBMCs from the plasmablast timepoint (week 9), fresh lymph node mononuclear cells (from FNAs at weeks 3 and 11), or enriched B cells from previously cryopreserved samples (at weeks -4, 4, 8, 10 and 16), were resuspended in 100uL 10%FBS/1x PBS and stained with the fluorescently labeled eOD-GT8 KO11 tetramer for 30 minutes at 4°C. Cells were washed with 10% FBS/1xPBS and then resuspended in a staining cocktail of antibodies and remaining eOD-GT8 tetramers diluted in Brilliant Buffer (BD Bioscience, La Jolla, CA, USA). The antibody staining panels were tissue dependent and included a panel for PBMC samples (memory B cells and plasmablasts) (table S10), and a separate panel for lymph node FNAs (table S11). After surface staining, the samples were resuspended at about 2×106 cells/mL in R10 containing the viability dye (7AAD) and maintained at 4°C until processed by flow cytometry.
Standardized templates for flow cytometry analysis
Standardized templates were designed using BD Diva software for analysis of bead assays, internal controls, and experimental samples for each sample type (fig. S7). These templates were replicated for each flow cytometry experiment to ensure consistent gating and labeling of samples. Templates were tested and compared between both test sites using aliquots of the internal control sample before trial samples were used. Since antigen-specific events were rare in the internal control sample which made setting gates difficult, we developed a standardized gating system that could be implemented at both sites referred to as the M.A.R.I.O gate.
Mathematically Articulated and Reasoned but Independently Optimized (M.A.R.I.O.) Gating
The analysis of fresh samples for the clinical trial required the use of equipment that was located near the clinical trial sites. The physical differences between flow cytometers, and the subjectivity of gating rare and hard to identify cell populations, can lead to high levels of variability in the sorting of populations like antigen-specific B cells. For this trial, we developed a method of standardizing the gating of antigen-specific B cells by flow cytometry. eOD-GT8 proteins were conjugated to two fluorophores, allowing us to identify fluorescently double-positive events and reduce the fluorophore-specific background that would be selected by using either fluorophore individually. Due to the variability between flow cytometers, developing a method to standardize the gating of antigen-specific events by flow cytometry was important for this trial. Here we utilized FMP (fluorescence minus probe) stained samples (fully stained samples without the addition of fluorescent probes) to determine the background for each channel being used to detect antigen-specific B cells. We then developed a gate based on the ratio of these fluorophores for each flow cytometer which could identify fluorescently well-balanced antigen-specific B cells while also reducing the amount of antigen-non-specific B cells captured by the gate.
FMP samples were used to calculate the coordinates for the 99th percentile of each probe-specific fluorophore using Flowjo software. This serves as the upper threshold of the probe negative gate (fig. S7A; C1). Similarly, twice the 99th percentile of each probe specific fluorophore was determined to serve as the lowest acceptable threshold for probe double positive-specific events (fig. S7A; C2). Axis points for each individual fluorophore were placed at coordinates representing ten times the 99th percentile for each individual fluorophore (fig. S7A; C4, C5). Additional axis points were added which are based on the proportion of the 99th percentiles for each fluorophore (fig. S7A; C6, C7). These bounds were used to define the area in which acceptable fluorescent probe double positive events were acceptable for sorting by each test facility and account for each facility’s individual cytometer characteristics (fig. S7B).
FACS Sorting
Individual target cells were index-sorted into empty skirted 96-well plates (Thermo Fisher Scientific; Waltham, MA, USA). Several wells were left empty for subsequent PCR controls. PBMC samples from weeks -4, 4, 8, 10, and 16 after the 1st vaccination were sorted until either four 96-well plates were filled with eOD-GT8 CD4bs-specific-specific IgG memory B cells (CD19+/CD20+/IgD−/IgG+/KO−/GT8++; fig. S8) or the sample was exhausted; two 96-well plates were subjected to RT-PCR and BCR sequencing, while the other two plates were reserved at −80°C. The number of PBMCs thawed and stained depended on the timepoint: 200×106 (FHCRC) or 250×106 (VRC) at week -4, 100×106 at week 4, 100×106 at week 8, 200×106 at week 10, and 100×106 at week 16.
In planning the trial procedures, we expected that GC B cells from LN FNAs and plasmablasts (PBs) would have lower levels of surface IgG, and hence we were uncertain if sorting of these samples would have sufficient detectable antigen-specific staining by flow cytometry. We therefore decided to sort two plates of phenotype-specific cells without regard to tetramer binding, followed by two plates of phenotype-specific cells that were also CD4bs-specific as determined by differential tetramer staining. Therefore, up to two plates of total GC B cells (CD19+ IgD− IgG+ CD20hi CD38hi) were sorted, as well as CD4bs-specific GC B cells (CD19+ IgD− IgG+ CD20hi CD38hi KO− GT8++) (fig. S9A). From fresh PBMCs at week 9, two plates of PBs (CD19+ CD27+ IgD− CD38hi) were sorted without regard to tetramer binding followed by the sorting of CD4bs-specific PBs (CD19+ CD27hi CD38hi IgD− KO− GT8++) (fig. S9B). Note that the PB gating strategy, which included CD38hi and did not gate on CD20, may have included CD38hiCD20hi B cells that have been described as activated B cells as opposed to PBs (10). A maximum of four 96-well plates of B cells were sorted for cDNA synthesis per time point. Plates were sealed, centrifuged at 800 × g for 1 min and immediately transferred to dry ice. Plates were stored at −80°C for at least overnight before proceeding. A total of 884 (384 at VRC; 500 at FHCRC) 96-well plates of sorted cells were produced and stored.
Standardized and direct analysis of sorting flow cytometry data
To allow for direct analysis of the flow cytometry data as it was analyzed during the sorts, the flow cytometry experiments were exported from Diva software as xml files along with the FCS files for the total recorded events and the index files for the sorted cells. These primary data were processed and concatenated at the Vaccine Immunology Statistical Center (VISC), using the Cytoverse suite of R packages. CytoML (75) is an R/Bioconductor package that enables cross-platform import, export, and sharing of gated cytometry data. Using CytoML we parsed the XML files created during the Diva experiments, which contained data transformations, compensation matrices, gates and their hierarchical relationships, sample meta-data and other information required to reproduce the gating analysis. Once the workspaces were imported into R, the analysis was faithfully reproduced for each sample, the gated cytometry data was visualized, and positive proportions for all populations were calculated. As a quality control measure, the calculated percent positive populations were compared to exported tables of populations from the Diva software using Spearman correlations. This data was processed using R (v4.1.1) and Cytoverse packages CytoML (v2.5.4), flowWorkspace (76) (v4.5.3), ggcyto (77) (v1.20.0), and cytolib (v2.5.3). Frequencies were measured either during the period of sorting (FHCRC) or until the samples were exhausted (VRC).
PCR and sequencing positive and negative controls
Early in the trial, the lysates from an immortalized VRC01-class naive B cell line, referred to as Immo-A1 (78), and sorted pooled donor CD19+ B cells were used as PCR controls. These were found to be insufficient as controls, so we developed synthetic positive controls to use for the remainder of the trial. Synthetic positive control DNA sequences compatible with our nested PCR protocol were generated for B cell receptor heavy and both light chains (kappa and lambda). Synthetic sequences were based on natural sequences identified from VRC01-class antibody sequences isolated from human PBMCs. The CDR3 amino acid junctions were altered to distinguish each of the synthetic controls from naturally occurring sequences and from each other by replacing them with amino acid “VRC” “VIP” and “DL” segments separated by chain-specific amino acids. Heavy chain CDR3 sequence tags coded for histidine (H) leading and separating the segments. Similarly, kappa chain CDR3 sequence tags coded for lysine (K) and lambda chain CDR3 sequences coded for leucine (L) leading and separating the segments. The sequence tags are shown below, and the full nucleotide sequences for the synthetic controls are given in table S19.
Sequence Tags:
5’ GVRCGVIPGDL 3’ 5’ ggcgtgcgctgcggcgtgattccgggcgatctg 3’
5’ KVRCKVIPKDL 3’ 5’ aaagtgcgctgcaaagtgattccgaaagatctg 3’
5’ LVRCLVIPLDL 3’ 5’ ctggtgcgctgcctggtgattccgctggatctg 3’
To further differentiate the synthetic controls by sequence as well as by physical size using gel electrophoresis for quality control purposes, novel sequences were inserted at both ends of the sequences. Inner and outer primer binding sites were attached to ensure these sequences would amplify with our nested PCR primer sets. Ultimately control sequences were ~50% mutated from natural sequences distributed throughout the sequence to ensure identification even with high mutation and poor sequence quality.
Synthetic controls were tested for primer binding capacity, in frame amino acid translation, Ig chain homology and gene usage and potential gBlock production issues. All controls were titered and rigorously tested prior to use in the trial.
Single-cell sequencing
In preparation for cDNA synthesis, plates were removed from −80°C storage and again centrifuged at 800 × g for 1 minute before being placed on wet ice. The cDNA synthesis reaction mix was constructed using the SuperScript III Reverse Transcriptase Kit (Invitrogen). The reaction mix consisted of 1x SuperScript III First Strand Buffer, 4.8 mM DTT and 200 U SuperScript III, as well as 20 U RNaseOUT (Invitrogen), 450 ng Random Hexamers (Gene Link) and 0.77 mM dNTP mix (GeneAmp). The mix was added to every well, including wells 12E through 12H to be used as negative controls. Thermal cycling parameters were 42°C for 10 min, 25°C for 10 min, 50°C for 60 min and 94°C for 5 min. Once complete, plates were frozen at −20°C. A total of 531 (267 at VRC, 264 at FHCRC) unique cDNA plates were generated for subsequent PCR amplification.
Amplification proceeded by three immunoglobulin chain-specific PCRs. The reaction mix generally consisted of 2 U HotStarTaq Plus DNA polymerase and 1x HotStar Plus PCR Buffer (Qiagen; Hilden, Germany), as well as 0.25 mM dNTP mix and 1.5 mM MgCl2. Each chain was amplified using a chain-specific pool of forward primers (IDT) and isotype-specific reverse primers (IDT) (tables S13, S15, S17). One reaction mix each was constructed containing 2.1 μM forward and 0.2 μM IgH reverse primers, 0.3 μM forward and 0.625 μM reverse Igκ primers, and 0.8 μM forward and 0.625 μM reverse Igλ primers. The template input was 4 μL of cDNA, including carrying forward 4 μL of each negative control from wells 12E through 12H. The cDNA plates were briefly vortexed and centrifuged before template addition. Thermal cycling parameters were 95°C for 5 min and 50 cycles of 95°C for 30 sec, either 52°C (IgH) or 58°C (Igκ and Igλ) for 30 sec and 72°C for 55 sec, followed by a final extension of 72°C for 10 min. Once complete, plates were frozen at −20°C.
An additional nested PCR was performed for all chains to increase specificity. The reaction mix consisted of 1 U Phusion High-Fidelity DNA Polymerase and 1x Phusion High-Fidelity Reaction Buffer (New England Biolabs; Ipswich, MA, USA), as well as 0.2 mM dNTP mix and 1x Q Solution (Qiagen; Hilden, Germany). Each chain was again amplified using a chain-specific pool of forward primers and isotype-specific reverse primers, with one reaction mix each containing 5.5 μM forward and 1.0 μM IgH reverse primers, 4.0 μM forward and 5.0 μM reverse Igκ primers, and 3.5 μM forward and 5.0 μM reverse Igλ primers (tables S14, S16, S18). The template input was 4 μL of chain-specific PCR product, again including carrying forward 4 μL of each negative control from wells 12E through 12H. In addition, chain-specific positive PCR controls (synthesized by IDT) were included in wells 12A through 12D. These synthetic constructs were added at 400 copies per well. The PCR plates, including the positive PCR controls, were briefly vortexed and centrifuged before template addition. Thermal cycling parameters were 98°C for 30 sec and 35 cycles of 98°C for 30 sec, either 58°C (IgH and Igλ) or 52°C (Igκ) for 30 sec and 72°C for 55 sec, followed by a final extension of 72°C for 10 min. Once complete, the plates were frozen at −20°C and shipped to Genewiz (South Plainfield, NJ) on dry ice. Genewiz conducted an enzymatic clean-up on the PCR products before conducting uni-directional Sanger sequencing using the reverse chain-specific constant region primers (3’Cg CH1, 3’CK 494, and 3’CL shown in tables S14, S16, S18). Sequencing data were obtained for a total of 3186 PCR plates (6x the total number of cDNA plates), not accounting for repeats. Quality control measures were taken for all sequence data to determine if sequencing of specific plates needed to be repeated. When PCR controls were not available, we required a minimum of 50% of the sequences on any plate to have identifiable and functional B cell receptor sequences. When PCR controls were available, we required >50% of the positive controls to have identifiable control sequences (2 to 4) and the negative controls to have <50% of the wells with identifiable sequences (1 or 0) (VRC) or we required the negative controls to have <50% of the wells with identifiable sequences (1 or 0) (FHCRC). Sequences quality was first checked with the Genewiz quality report to determine overall sequencing success for the plate. Sequences were then analyzed by IMGT/V-QUEST for productive B cell receptor sequences or sequences with identifiable B cell receptor gene usage. When synthetic PCR controls were used, all sequences were screened for contamination by the spiking of the controls using the CDR3 amino acid junction sequence tags mentioned previously. All data was transferred to the Vaccine Immunology Statistical Center (VISC) for data repository and analysis. However, sequencing plates that did not pass our criteria were investigated further and either sequenced again directly by Genewiz or PCR amplified from an earlier step with remaining cDNA or PCR material. The new plates were submitted to Genewiz for sequencing, and new sequence data was identified with an increased “Replicate” number.
Additional PCR and NGS BCR sequencing for low dose group
To control for potential sequencing errors in the uni-directional/single-read Sanger sequencing data, we resequenced selected samples from the low dose group using next generation sequencing (NGS). To perform NGS on selected cells, cDNA generated for Sanger sequencing was used as the starting point. Plates were removed from −80°C storage and centrifuged at 800 ×g for 1 minute before being placed on wet ice. Selected wells from different plates were collected into a new plate, and PCR was performed using the same nested method used for Sanger sequencing. The nested PCR product from the individual heavy and light chains were pooled in equal volumes of 10 μL from each plate into a single plate. The pooled plate was purified using AmpureXP beads (Beckman coulter Cat: A63881) in the ratio of 0.8X beads to PCR product for a total volume of 24 μL of beads to 30 μL of pooled PCR product. The purified product was eluted into 20 μL of RNase-free water. Each purified plate was quantified using the QuantIT dsDNA High sensitivity kit (Thermofisher Cat: Q33120) and normalized to 0.2ng/μL for optimal NGS library generation using the Illumina Nextera XT kit (Illumina Cat: FC1–31-1096). The normalized plates were processed for NGS library preparation using 1.25 μL of the product with 2.5 μL of Tagmentation buffer and 1.25 μL of the Amplicon Tagmentation mix provided in the Nextera kit. The plates were centrifuged at 800 ×g for 1 minute and placed in the thermocycler at 55°C for 10 minutes. Following the incubation, 1.25 μL of Neutralization buffer was added to the wells and incubated at room temperature for 5 minutes. The tagmented product was indexed using custom primers generated to multiplex 10 plates together in a single sequencing run. Each plate was indexed with 1.25 μL of a unique 10 base pair Mi7 index at a concentration of 10 μM and each well of the plate was indexed with 1.25 μL of a unique 10 base pair Mi5 index at a concentration of 10 μM, along with 3.75 μL of the NPM reaction mix provided in the Nextera XT kit. The PCR plates were centrifuged at 800 ×g for 1 minute and placed in the thermocycler for indexing. The thermocycler parameters used were 72°C for 3 minutes, 12 cycles of 95°C for 30 sec, 50°C for 30 sec, and 72°C for 1 min, followed by a final extension of 72°C for 5 min. The indexed libraries were further PCR purified using the AmpureXP beads at 0.8X concentration and eluted into 10 μL of RNase-free water. The purified product from 10 indexed plates was pooled together for loading onto the Illumina Miseq for sequencing. The final concentration of the pooled library was determined using the Kapa QPCR kit (Roche Cat: KK4835) and diluted to 4nM for sample denaturating according to the Illumina loading guidelines. The denatured sample was further diluted to 10 pM concentration for loading onto the Miseq V3 600 cycle kit (Illumina cat: MS-102–3003). Demultiplexed fastq files were retrieved from the Miseq and aligned to immunoglobulin genes using BALDR (79).
High quality NGS sequences with at least 100 Nextera reads were obtained for a total of 1,998 samples and were regarded as perfectly accurate. Comparison of high quality NGS sequences to corresponding Sanger sequences revealed perfect agreement for 82.9% of Sanger reads at the nucleotide level and 85.9% of Sanger reads at the amino acid level. There were differences of 1, 2, 3, 4, or 5 nucleotides for 7.8%, 2.3%, 1.3%, 0.65%, or 0.75% of Sanger reads, respectively, and differences of 1, 2, 3, 4, or 5 amino acids for 6.7%, 1.8%, 1.3%, 0.70%, or 0.50% of Sanger reads, respectively. Hence, the NGS data confirmed that >94.2% of the Sanger sequencing reads were accurate to within 3 nucleotides, and >95.6% of the Sanger sequencing reads were accurate to within 3 amino acids. We concluded that it was not necessary to carry out NGS resequencing on the remaining low dose samples or on any of the high dose samples. Nevertheless, a small fraction (<4.4%) of Sanger reads differed by more than 3 amino acids from the corresponding high quality NGS sequences. High quality NGS sequences were used in place of the corresponding Sanger sequences for downstream analysis in cases where Sanger and NGS differed by 1 to 19 nucleotides, as described in the NGS module of the BCR bioinformatic analysis pipeline below.
Sample identify confirmation for one case of accidental sample mislabeling
Comparison of serum antibody binding data from BAMA with antigen-specific B cell frequency data from cytometry suggested that samples from visits 8 and 10 (weeks 10 and 16) for placebo-recipient PubID_164 might have been accidentally swapped with samples from the same timepoints for vaccine-recipient PubID_153 during flow cytometry analysis. Short Tandem Repeat (STR) analysis along with confirmatory flow cytometry testing was used to confirm the identify of the samples. Duplicate samples were thawed and divided. Small aliquots were lysed and transported to two independent facilities for STR testing to confirm donor sample identity. Remaining samples were processed by trial flow cytometry protocols for comparison of cellular phenotypic data. Both STR testing facilities were in agreement with each other and the confirmatory flow cytometry data to confirm sample identities. This analysis confirmed that a sample labeling error of transposing the participant IDs had occurred during analysis by flow cytometry, effectively swapping the samples for the two participants at the timepoints listed above. The sample identity confirmation provided justification for the bioinformatic pipeline to correct for the effective “sample swap”, as described in the SWAP module below.
Bioinformatic BCR sequence processing
Database:
ABI files containing DNA sequence information from Sanger sequencing were organized in a central database at VISC in which each file was regarded as an entry in the database. The files were contained within folders such that each folder represented a 96-well plate. The folder and file naming structure was:
<Date Deposited>/<Plate Tracking Number>/<Participant ID>_<Visit Number>_<Plate Number>_<Round>_<Chain>_<Replicate>_<Well>.abi
Date Deposited was the date the ABI filed was deposited into the database. Plate Tracking Number was the Fedex tracking number associated with shipping the plate to Genewiz. Participant ID was the unique identifier assigned to each participant in the trial. Visit Number was the visit number as described in fig. S1 and table S2. Plate number was the unique number assigned to each 96-well plate sorted for each participant at each visit. Round was could be either 1 or 2, indicating the first or second of the nested PCR reactions; all samples submitted for sequencing were round 2. Chain was heavy, kappa, or lambda. Replicate was the unique index to indicate which one of several potential attempts at nested PCR and sequencing were made for this plate. Well was the unique number for each well in a 96-well plate. The database contained a total of 158,954 ABI files corresponding to 1,724 96-well plates, some of which were only partially filled. The database also contained a sample manifest that linked metadata to each well for which sequencing data was obtained. The data fields in the manifest included several important boolean variables, such as: (1) Has_sorted_cell, which was true if the well contained a sorted cell; (2) Is_negative_control, which was true if the well contained a negative control sample; (3) Is_positive_control, which was true if the well contained a positive control sample; and (4) Is_doublet, which was true if the well was known from FACS to contain a doublet (two sorted cells); and many other metadata about the sample (the sample manifest is available in the data repository; see Data and materials availability below). Sequences obtained by NGS for a small subset of samples from the low dose group were also deposited into the central database in a spreadsheet linking each NGS sequence to a unique sequence identifier that could be matched to the Sanger sequence identifiers. NGS sequences were incorporated into the data analysis as described below.
BCR analysis pipeline:
Preliminary analyses of BCR sequences were carried out during the trial, as the sequencing data was generated and deposited into the central database. This was necessary to obtain early readouts on the efficacy of the vaccine in order to help guide decision-making about additional pre-clinical and clinical experiments to build on the findings from this trial. The preliminary analyses also enabled us to select BCR sequences for production as soluble IgGs and subsequent characterization of binding affinities by SPR. Initially, and during most of the trial, the preliminary analysis pipeline employed Abstar (https://github.com/briney/abstar; (80)) for BCR gene assignment and annotation, coupled with custom scripts written in various programming languages for analysis. However, toward the end of the trial, and for the final analysis presented in this manuscript, we developed a more streamlined, adaptable, and transparent code base for analysis. The final, improved analysis pipeline used Sequencing Analysis and Data library for Immunoinformatics Exploration (SADIE, https://github.com/jwillis0720/sadie) for BCR gene assignment and annotation, coupled with a series of python modules, and reported BCR sequence characteristics in the standardized Adaptive Immune Receptor Repertoire (AIRR) format (81).
The final BCR analysis pipeline (fig. S10), a single installable python package, consisted of eleven independent modules that read as input the dataframe (a data structure that organizes tabular data) from the previous step and produced as output a new dataframe. The modules (FIND, MODEL, SPLIT, CORRECT, ANNOTATE, JOIN, NGS, TAG, SWAP, PAIR, PERSONALIZE, and MUTATION) are described below.
FIND:
The FIND module collected information from all ABI files in the central data repository. The filename, file path, nucleotide sequence, and Phred score (82, 83) were stored into a dataframe for subsequent analysis. A total of 158,954 ABI files were found.
MODEL:
The MODEL module tokenized the filename and path into expected metadata fields (e.g., verified that participant id was in our list of expected participant ids) and enforced consistent formatting for each field (e.g., rep0, or r0 became REP0 for the replicate field).
SPLIT:
The SPLIT module dropped entries that were duplicates or that had incomplete metadata fields assessed in the MODEL module. An entry was determined to be a duplicate if it contained the same metadata fields as well as the same nucleotide sequence and Phred score. Such duplicate entries resulted from duplicate manual uploads to the central data repository. After the SPLIT module, a total of 155,801 entries were available for subsequent analysis.
CORRECT:
The CORRECT module applied a curated list of updates to meta data fields and drops of certain sequences or plates that were requested by the FHCRC and VRC experimental labs. Updates were generally corrections to manual filenaming errors that violated the pre-defined naming format (common errors including entering “NA” instead of a valid field entry, or specifying the visit number with an invalid format). Drops were mostly either to eliminate wells for which no cell was known to be sorted, or to eliminate plates that had been named incorrectly and were resubmitted separately with correct naming. A total of 1,318 entries were updated, and 701 entries were dropped. A full list of updates and drops was stored in JSON format and can be reviewed in the GitHub repository. The CORRECT module then grouped all entries by participant ID, timepoint, plate, well, replicate, and chain, to ensure uniqueness. After the CORRECT module, a total of 155,100 entries were available for subsequent analysis.
ANNOTATE:
The ANNOTATE module used the Sequencing Analysis and Data library for Immunoinformatics Exploration (SADIE, https://github.com/jwillis0720/sadie) to annotate BCR sequences. The SADIE AIRR module ports IgBLAST (84) for analysis of nucleotide sequences and ensures that the data is represented in AIRR recombination schema that defines a data model, field names, data types, and encodings (81). In addition, SADIE AIRR provides a versioned and tested python package that includes the IgBLAST executable and allows automatic creation of custom IgBLAST databases, a process that is otherwise difficult. SADIE AIRR provides the user with scriptable, granular control over IgBLAST options via a python API or a command-line interface. SADIE AIRR will try to determine optimized V(D)J alignment penalties to find a productive recombination (adaptable penalty model) and will also correct insertions and deletions that are absent in the germline alignment using current IgBLAST (v 1.17.1). For analysis of the G001 light chain sequences, SADIE AIRR implementation of the adaptable penalty model was helpful for determining likely germline rearrangements for light chains with short LCDR3s with significant VJ gene overlap. In these cases, the use of adaptable penalties facilitated identification of likely boundaries for the V and J genes, which resulted in identification of a slightly increased number of productive recombinations. The adaptable penalty model started with the default value of −1 for both V and J gene penalties, and in that configuration identified 27,913 productive antibody light chain recombinations. Other (V, J) penalty combinations, of (−2, −1), (−1, −3), and (−3, −1), identified 683, 65, and 37 additional light chain recombinations, respectively, increasing the total number of light chain recombinations by 2.8%. The Annotate module joined all AIRR recombination data fields with the previous metadata fields for subsequent analysis. In addition, ANNOTATE calculated a “sliced” Phred score that was the mean Phred score computed over just the V(D)J portion of the sequence. This “sliced” Phred score was used in evaluation of sequence quality in subsequent modules. After the ANNOTATE module, a total of 155,100 entries were available for subsequent analysis.
JOIN:
As noted above, a manifest containing additional information about each well in each 96-well plate subjected to DNA sequencing was uploaded to the central data repository. The data fields in the manifest included the tokenized metadata from the FIND module as well as additional information. The JOIN module merged the manifest data with the annotated sequencing data from the ANNOTATE module. There were 22,707 entries with sequence information that could not be found in the manifest, of which only 633 were productive antibody sequences, and JOIN was unable to process those entries. After the JOIN module, 132,393 entries were available for subsequent analysis.
NGS:
The NGS module took the output from the resequencing pipeline, reannotated the sequences using SADIE AIRR, and updated the sequences and annotation fields (substituted the NGS sequences and their associated annotations in place of the corresponding Sanger sequences and annotations) if there were: (A) fewer than 20 nucleotide differences as measured by Levenshtein distance (85) between the Sanger and NGS; and (B) more than 100 V(D)J reads were produced by the NGS pipeline. When resequencing was carried out, the original Sanger sequence, the corrected sequence, and the Levenshtein distance were added to the dataframe for the subsequent analysis. Of 1,998 high quality NGS sequences, 1,656 (82.8%) sequences perfectly matched the corresponding Sanger sequence; 37 (1.9 %) differed from the corresponding Sanger sequence by more than 20 nucleotides and were not considered for correcting the Sanger sequence; and 304 (15.2%) differed from the corresponding Sanger sequence by 1 to 19 nucleotides and were used to correct the Sanger sequence.
TAG:
The tag module searched the productive sequences and performed a sequence alignment against the positive control sequences used in the PCR reaction, Synth or Immo (described in PCR and sequencing positive and negative controls). A positive match to a Synth or Immo heavy chain required that the HCDR3 nucleotide normalized identity to the true sequence was greater than 0.95 and the normalized VH gene distance to the true sequence was greater than 0.9. For kappa and lambda matches to Synth, the LCDR3 and VL gene distances to the true sequence were both required to be greater than 0.9. For kappa and lambda chain matches to Immo, the LCDR3 nucleotide normalized distance to the true sequence was required to be greater than 0.92; and the normalized distance to the true VK gene sequence was required to be greater than 0.9. In addition, for the kappa chain matches to Immo, an exact match was required to the n-addition nucleotide sequence (CG). These parameters were calculated by comparing control sequences from NGS with control sequences from Sanger, and adjusting the normalized distances until the controls were all identified by their Sanger sequences.
SWAP:
The SWAP module transposed all data in the current dataframe from participant PubID_153 at visits 8 and 10 with participant PubID_164. See Sample identify confirmation for one case of accidental sample mislabeling for additional details.
PAIR:
The pair module applied a series of filters to produce a final set of high quality BCR sequences including both heavy and light chains. The first filter eliminated non-final sequences, referred to as non-terminal replicates: for plates in which Sanger sequencing was attempted two or three times until the sequencing results passed quality controls (Single-cell sequencing), only the sequences from the final, or terminal, run were used. This filter eliminated 8,966 non-terminal replicate entries. The second filter eliminated positive control sequences that were either derived from a well labelled as a control in the sample manifest (n=6,898) or were identified from the TAG module (n=2,746). The third filter removed sequences that either had no antibody recombination detected by SADIE AIRR (n=2,536) or corresponded to negative control wells in the manifest (n=5,607). The fourth filter removed 1,677 heavy or light chain sequences identified in the manifest as doublets (cases of two cells sorted into a single well). The fifth filter removed 63,036 unproductive or incomplete V(D)J sequences that either contained a stop codon (unproductive; n=61,374) or contained a recombination that did not start at the first nucleotide of framework 1 and end at the last nucleotide of framework 4 (incomplete V(D)J; n=1,662). Of the incomplete V(D)J sequences, 505 were heavy chains less than 90 amino acids in length, and 836 were light chains less than 80 amino acids in length. The sixth filter removed 1,965 entries with mean Phred scores over the V(D)J sequence (“sliced” Phred scores) less than 30. The remaining 41,887 sequences, which included both heavy and light chains, were considered candidates for pairing. Sequences were then grouped by participant id, timepoint, plate and well. In the seventh and final filter, groups containing more than one heavy or light chain were eliminated (n=2,033), and unpaired heavy or light chains (n=12,102) were eliminated. The remaining groups were considered proper pairs, because they contained exactly one heavy chain and one light chain. This process yielded 13,876 total BCR sequences with paired heavy and light chains (9,862 heavy/kappa; 4,014 heavy/lambda). Of those, 11,372 were CD4bs-specific (sorted as KO−GT8++) and 2,504 were sorted as either GC B cell phenotype (from the first two plates in each FNA sort) or plasmablast phenotype (from the first two plates in each PB sort) without regard to eOD-GT8 tetramer binding (see FACS Sorting). Among the 2,504 BCRs sorted by phenotype only, 71 (2.8%) were VRC01-class, and 45 of those were produced by a single donor at a single timepoint (PubID 151 at week 3). We focused our subsequent analyses on the 11,372 BCRs sorted as CD4bs-specific.
PERSONALIZE:
The personalize module reannotated all BCR sequences with the personalized IGHV1–2 allele haplotype discovered in the genotype analysis (IGHV1–2 genotype analysis using IgDiscover). SADIE AIRR was run against a personalized reference set curated for each participant using the SADIE reference module. This allowed annotation only against the correct haplotype of IGHV1–2 alleles and thus a more accurate VH somatic mutation percentage assignment.
MUTATION:
The mutation module used SADIE ANARCI to number each VDJ amino acid sequence using the kabat numbering scheme. SADIE ANARCI is a port of the ANARCI program (86) that numbers amino acid sequences in a variety of numbering schemes. Both the germline V(D)J and mature V(D)J sequences were numbered in the Kabat scheme. The mutations were recorded and added to the final output dataframe.
Computing VRC01-class frequencies by combining frequencies from FACS and BCR sequencing
Memory B cell data.
The frequency of VRC01-class IgG memory B cells was estimated by multiplying two frequencies: (1) the frequency of CD4bs-specific (KO−GT8++) IgG memory B cells among all IgG memory B cells sorted by FACS; and (2) the frequency of VRC01-class BCRs among all CD4bs-specific IgG Memory B cells with sequenced BCRs. If the frequency of CD4bs-specific IgG memory B cells was zero, then the estimate for the VRC01-class frequency was also set to zero. In the case where the frequency of CD4bs-specific IgG memory B cells was positive but no CD4bs-specific IgG memory B cells were successfully sequenced, the estimate for the VRC01-class frequency was set to zero for post-vaccination samples or the CD4bs-specific frequency for baseline samples. This approach is conservative or detecting vaccine-induced responses.
Additionally, based on the data for IgG memory B cells from PBMC samples, we estimated the frequencies of VRC01-class B cells among three different populations of B cells in the periphery. First, as an estimate for the frequency of VRC01-class B cells among CD4bs-specific IgG memory B cells, we used the frequency of VRC01-class B cells among BCR-sequenced CD4bs-specific IgG memory B cells. Second, to obtain an estimate for the frequency of VRC01-class B cells among GT8-specific IgG memory B cells, we multiplied the frequency of VRC01-class B cells among CD4bs-specific IgG memory B cells by the ratio of CD4bs-specific IgG memory B cells to GT8-specific IgG memory B cells. Third, to obtain an estimate for the frequency of VRC01-class IgG memory B cells among all B cells, we multiplied the frequency of VRC01-class B cells among CD4bs-specific IgG memory B cells by the ratio of CD4bs-specific IgG memory B cells to all B cells.
GC B cell data.
The frequency of VRC01-class IgG GC B cells was estimated similarly as for memory B cells, by multiplying two frequencies: (1) the frequency of CD4bs-specific IgG GC B cells among all IgG GC B cells sorted by FACS; and (2) the frequency of VRC01-class BCRs among all CD4bs-specific IgG GC B cells with sequenced BCRs. If the frequency of CD4bs-specific IgG GC B cells was zero, then the estimate for the VRC01-class frequency was also set to zero. If the frequency of CD4bs-specific IgG GC B cells was positive but no CD4bs-specific IgG GC B cells were successfully sequenced, the estimate for the VRC01-class frequency was set to zero.
Analogous to procedures we followed with IgG memory B cells from PBMCs, based on the data for IgG GC B cells from FNA samples, we estimated the frequencies of VRC01-class B cells among three additional populations of B cells from FNA samples. First, as an estimate for the frequency of VRC01-class B cells among CD4bs-specific IgG GC B cells, we used the frequency of VRC01-class B cells among BCR-sequenced CD4bs-specific IgG GC B cells. Second, to obtain an estimate for the frequency of VRC01-class B cells among GT8-specific IgG GC B cells, we multiplied the frequency of VRC01-class B cells among CD4bs-specific IgG GC B cells by the ratio of CD4bs-specific IgG GC B cells to GT8-specific IgG GC B cells. Third, to obtain an estimate for the frequency of VRC01-class IgG GC B cells among all B cells, we multiplied the frequency of VRC01-class B cells among CD4bs-specific IgG GC B cells by the ratio of CD4bs-specific IgG GC B cells to all B cells.
Plasmablast data.
The frequency of VRC01-class IgD− plasmablasts (PBs) was also estimated similarly as for memory B cells, by multiplying two frequencies: (1) the frequency of CD4bs-specific IgD− PBs among all IgD− PBs sorted by FACS; and (2) the frequency of VRC01-class BCRs among all CD4bs-specific IgD− PBs with sequenced BCRs. If the frequency of CD4bs-specific IgD− PBs was zero, then the estimate for the VRC01-class frequency was also set to zero. If the frequency of CD4bs-specific IgD− PBs was positive but no CD4bs-specific IgD− PBs were successfully sequenced, the estimate for the VRC01-class frequency was set to zero.
Analogous to procedures we followed with memory B cells from PBMCs and GC B cells from FNAs, based on the data for IgD− PBs from week 9 PBMC samples, we estimated the frequencies of VRC01-class B cells among three additional populations of B cells in the periphery at week 9. First, as an estimate for the frequency of VRC01-class PBs among CD4bs-specific IgD− PBs, we used the frequency of VRC01-class PBs among BCR-sequenced CD4bs-specific IgD− PBs. Second, to obtain an estimate for the frequency of VRC01-class PBs among GT8-specific IgD− PBs, we multiplied the frequency of VRC01-class PBs among CD4bs-specific IgD− PBs by the ratio of CD4bs-specific IgD− PBs to GT8-specific IgD− PBs. Third, to obtain an estimate for the frequency of VRC01-class IgD− PBs among all B cells, we multiplied the frequency of VRC01-class cells among CD4bs-specific IgD− PBs by the ratio of CD4bs-specific IgD− PBs to all B cells.
Key B cell frequencies reported
B cell frequencies and B cell receptor (BCR) signatures were measured by flow cytometry (FACS) and BCR sequencing, respectively. In this section we list the key frequencies reported in the manuscript and/or in the supplementary data files. Additional frequencies not listed here are provided in the supplementary data files. Methods to estimate frequencies based on combined cytometry and BCR sequence analyses are given in the preceding section, Computing VRC01-class frequencies by combining frequencies from FACS and BCR sequencing.
The key frequencies reported for PBMC samples (weeks -4, 4, 8, 10, 16) were as follows.
From cytometry analysis:
Percent of IgG memory B cells that were GT8++ (regardless of KO binding status)
Percent of GT8++ IgG memory B cells that were KO−
Percent of IgG memory B cells that were CD4bs-specific
From combined cytometry and BCR sequence analysis:
Percent of CD4bs-specific IgG memory B cells that were VRC01-class
Percent of GT8-specific IgG memory B cells detected as VRC01-class
Percent of IgG memory B cells detected as VRC01-class
Percent of B cells detected as VRC01-class
The key frequencies reported for FNA samples (weeks 3 and 11) were as follows.
From cytometry analysis:
Percent of IgG GC B cells that were GT8++ (regardless of KO binding status)
Percent of GT8++ IgG GC B cells that were KO−
Percent of IgG GC B cells that were CD4bs-specific
From combined cytometry and BCR sequence analysis:
Percent of CD4bs-specific IgG GC B cells that were VRC01-class
Percent of GT8-specific IgG GC B cells detected as VRC01-class
Percent of IgG GC B cells detected as VRC01-class
Percent of B cells detected as VRC01-class
The key frequencies reported for PB samples (week 9) were as follows.
From cytometry analysis:
Percent of IgD− PBs that were GT8++ (regardless of KO binding status)
Percent of GT8++ IgD− PBs that were KO−
Percent of IgD− PBs that were CD4bs-specific
From combined cytometry and BCR sequence analysis:
Percent of CD4bs-specific IgD− PBs that were VRC01-class
Percent of GT8-specific IgD− PBs detected as VRC01-class
Percent of IgD− PBs detected as VRC01-class
Percent of B cells detected as VRC01-class
VRC01-class response calls
The criteria for determining if detection of one or more VRC01-class B cells in a sample represented a vaccine-induced VRC01-class response were as follows.
Memory B cell data.
For each post-baseline visit in which memory B cells from PBMCs were sorted (weeks 4, 8, 10, 16), detection of one or more VRC01-class B cells was labelled a vaccine-induced VRC01-class response if the estimated frequency of VRC01-class IgG memory B cells among all IgG memory B cells was greater than the corresponding baseline frequency. For participants with no sequences of CD4bs-specific memory B cells available at baseline, the baseline frequency was estimated as the frequency of CD4bs-specific (KO−GT8++) IgG memory B cells, which was a conservative approach.
GC B cell data.
For FNA samples (weeks 3 and 11), for which baseline data were not available, detection of one or more VRC01-class B cells was labelled a vaccine-induced VRC01-class response if the estimated frequency of VRC01-class IgG GC B cells among all IgG GC B cells was greater than 0.1% (1 in 1000). This was an arbitrary threshold, but we judged it to be reasonable considering that (a) none of the placebos produced any detectable VRC01-class B cells in FNA samples, and (b) 0.1% represents a substantial frequency for a single class of B cells (with specific BCR properties) within a polyclonal response.
Plasmablast data.
For PB samples (week 9), for which baseline data were also not available, detection of one or more VRC01-class plasmablasts was labelled a vaccine-induced VRC01-class response if the estimated frequency of VRC01-class IgD− PBs among all IgD− PB cells was greater than 0.1% (the same numerical threshold used for testing FNA samples). We judged this to be a reasonable threshold, because (a) none of the placebos produced any detectable VRC01-class B cells in PB samples, and (b) as noted above, 0.1% represents a substantial frequency for a single class of B cells (with specific BCR properties) within a polyclonal response.
Pre-trial evaluation of baseline VRC01-class IgG B cell frequency
During pre-trial development of the B cell sorting and BCR sequencing assay, we sought to determine an estimate for the baseline frequency of VRC01-class IgG B cells in humans. We applied the assay to leukapheresis samples from 35 healthy, HIV-unexposed donors, including approximately 12 billion PBMCs and approximately 29.2 million IgG+ memory B cells. From 142 BCRs with HC and LC sequenced out of 171 CD4bs-specific cells sorted, we detected no VRC01-class B cells, indicating a relatively low baseline frequency of VRC01-class B cells in the human IgG memory B cell repertoire. In contrast, we verified by sorting of human naive IgM+ B cells that the same assay isolated VRC01-class human naive precursors at frequencies similar to those reported previously (not shown).
IGHV1–2 genotype analysis using IgDiscover
Genotyping of the G001 participants was performed under an ethics approval from the National Ethical Review Agency of Sweden (decision #2021–01850). Total RNA was isolated from frozen PBMC samples prepared from each participant (n=48) using the Qiagen RNeasy kit and protocol. 200 ng of total RNA was used for cDNA synthesis with an IgM constant region primer that contained a Unique Molecular Identifier (UMI), according to the procedures described previously (87). Two independent 5’ multiplex libraries (L and U) were produced for each case using either a IGHV leader region specific primer set (L) or a primer set that targeted the 5’ UTR (U). The libraries were individually indexed and sequenced on the Illumina MiSeq platform using the V3 2 × 300 cycle kits (Illumina). The Read 1 and Read 2 files for each library were processed using the IgDiscover analysis pipeline as described previously (87, 88). The IgDiscover program (version 0.12.4.dev266+ge9e3119) enabled the analysis of IGHV1–2 allele content in each case, using the two independent libraries for high confidence allele identification.
BCR sequence hierarchical clustering
Average linkage agglomerative clustering (Fig. 3 and figs. S19–S20 and S24–S25) was performed using the SADIE cluster module. The module uses scikit-learn (89) to perform clustering specifically on BCR sequences with the AIRR annotation schema. Sequences were split into VRC01-class and non-VRC01-class and then grouped by HCDR3 and LCDR3 length. For each subgroup, the distance between two antibodies was the sum of Levenshtein distances between the corresponding HCDRs and LCDRs (HCDR1 distance + HCDR2 distance + HCDR3 distance + LCDR1 distance + LCDR2 distance + LCDR3 distance). To facilliate clustering of both mutated and un-mutated BCRs with a single distance cutoff, the “somatic pad” option was used to subtract all common somatic mutations from the total Levenshtein distance for any pair of BCRs (90). With this option, the minimal distance between two BCRs was zero. The sparse upper-triangular distance matrix was then saved into memory. Clustering was then performed using scikit-learn with the pre-computed distance matrix using average-linkage clustering and a distance cutoff of 3. The cluster labels were extracted and added to the output dataframe. The centroid of every cluster was computed as the lowest mean distance to every other member in the cluster. The all-vs-all, intra-cluster, and inter-centroid distances were then computed from the saved distance matrix.
Bioinformatic BCR sequence analysis other than VRC01-calls or clustering
BCR V gene assignments and mutation levels were determined using SADIE, which was also used in the ANNOTATION module described above. Mutation analysis for VH1–2 genes was made more accurate by accounting for the personalized IGHV1–2 genotypes as described in the PERSONALIZE module above. Statistical quantile analyses throughout the manuscript were carried out using R and Pandas (https://github.com/pandas-dev/pandas). Confidence intervals for the non-VRC01-class response rate (fig. S19C) were computed using the Wilson score method (91). Statistical comparisons for BCR mutation levels at different timepoints (Fig. 5, fig. S20, tables S46–S49), were carried out using a Wilcoxon signed-rank test for paired data using the VISCfunctions R package (https://github.com/FredHutch/VISCfunctions). Analyses of VRC01-class BCR features (Fig. 6 and figs. S16 and S24–S29) were carried out by custom python functions available as a package in the data repository for G001 (htttps://github.com/SchiefLab/G001). We detected few recurring VRC01-class clones at different timepoints, and therefore we did not carry out analyses of intra-clonal SHM over time as have been reported elsewhere (64). Inferred germline amino acid sequences were computed by reverting templated V, D, and J gene segments to their germline sequences for the alleles predicted on the basis of the antibody nucleotide sequence; in this process, VH1–2 allele predictions were made more accurate by restricting the allowed alleles for each participant to those experimentally determined by VH1–2 genotyping for that participant. When referring to control distributions from OAS (92, 93), we restricted to human, non-vaccinated, no disease state data from OAS. In data we report from OAS, each symbol refers to a separate individual.
Antibody selection and production for SPR
For IgG expression, we selected at least two VRC01-class and one non-VRC01-class BCR from each participant at each post-vaccination timepoint, attempting to represent the genetic diversity and mutation levels of the low dose BCRs. Although some mAbs failed to express, we carried out SPR analyses for 267 VRC01-class and 145 non-VRC01-class BCRs across all participants and post-vaccination timepoints (Fig. 7, fig. S29, and Data S2). To assess affinities of potential naive precursors to the post-vaccination BCRs, we evaluated eOD-GT8 binding to inferred-germline antibodies (iGLs) for 118 VRC01-class and 55 non-VRC01-class post-vaccination BCRs. For comparison we also measured eOD-GT8 binding to pre-vaccination IgG memory BCRs (6 of 7 VRC01-class BCRs from both dose groups and 53 of 55 non-VRC01-class BCRs from the low dose group) and human naive VRC01-class (n=62) and non-VRC01-class (n=72) precursors specific for the CD4bs of eOD-GT8 and isolated previously from 12 other HIV-unexposed individuals (21, 43) (Fig. 7 and Data S2).
Genes encoding the antibody Fv regions were synthesized by GenScript and cloned into antibody expression vectors pCW-CHIg-hG1, pCW-CLIg-hL2, and pCW-CLIg-hk. Monoclonal antibodies were produced by GenScript using transient transfection in the Expi293F system (ThermoFisher) and were purified using HiTrap MabSelect SuRe columns (Cytiva). Additional antibody production was conducted in house using transient transfection of HEK-293F cells (ThermoFisher) and purification using rProtein A Sepharose Fast Flow resin (Cytiva). We note that the antibodies were produced as IgG1, but our B cell sorting and BCR sequencing workflow did not distinguish between IgG subclasses.
Antigen production for SPR
His-tagged monomeric and trimeric antigens were produced by transient transfection of HEK-293F cells (ThermoFisher) and purified by immobilized metal ion affinity chromatography (IMAC) using HisTrap excel columns (Cytiva) followed be size-exclusion chromatography (SEC) using either Superdex 75 10/300 GL or Superdex 200 Increase 10/300 GL columns (Cytiva).
SPR
We measured the kinetics and affinities of antibody-antigen interactions on a Carterra LSA instrument using HC30M or CMDP sensor chips (Carterra) and 1x HBS-EP+ pH 7.4 running buffer (20x stock from Teknova, Cat. No H8022) supplemented with BSA at 1 mg/ml. We followed Carterra software instructions to prepare chip surfaces for ligand capture. In a typical experiment, approximately 2500 to 2700 RU of capture antibody (SouthernBiothech catalog #2047–01) in 10 mM Sodium Acetate pH 4.5 was amine coupled. The critical detail here was the concentration range of the amine coupling reagents and capture antibody. We used N-Hydroxysuccinimide (NHS) and 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimidehydrochloride (EDC) from Amine Coupling Kit (GE order code BR-1000–50). As per kit instruction 22–0510-62 AG, the NHS and EDC should be reconstituted in 10 ml of water each to give 11.5 mg/ml and 75 mg/ml respectively. However, the highest coupling levels of capture antibody were achieved by using 10 times diluted NHS and EDC during surface preparation runs. Thus, in our runs the concentrations of NHS and EDC were 1.15 mg/ml and 7.5 mg/ml. The concentrated stocks of NHS and EDC could be stored frozen in −20C for up to 2 month without noticeable loss of activity. The SouthernBiotech capture antibody was buffer exchanged into 10 mM Sodium Acetate pH 4.5 using Zeba spin desalting columns 7K MWCO 0.5ml (catalog # 89883 from Thermo) and was used at concentration 0.25 mg/ml with 20 minutes contact time. Phosphoric Acid 1.7% was our regeneration solution with 60 seconds contact time and injected three times per each cycle. Solution concentration of ligands was around 5 ug/ml and contact time was 3 min. Raw sensograms were analyzed using Kinetics software (Carterra), interspot and blank double referencing, Langmuir model. Analyte concentrations were quantified on NanoDrop 2000c Spectrophotometer using absorption signal at 280 nm. A typical SPR run tested 6 different analyte concentrations using a dilution factor of 4. Maximum analyte concentration for eOD-GT8 was generally 10 μM, except for weak binders which were generally re-run at higher maximum analyte concentrations of 50 or 118 μM. For eOD-GT6 and eOD-GT6 variant analytes, maximum analyte concentrations were 9 to 16 μM, but weak or expected weak binders were run at 37–63 μM. For MD39 trimer, maximum analyte concentration was 4 or 11 μM, but most interactions were tested at 11 μM. For core-gp120, maximum analyte concentration were 11 or 46 μM. For best results, analyte samples were buffer exchanged into the running buffer using desalting columns or dialysis. We typically covered a broad range of affinities in our runs, and the best referencing practices were different depending on how fast the off-rate was for each particular ligand. For fast off-rates (>9×10−3 1/s 1×10−2 1/s) we used automated batch referencing that included overlay y-align and higher analyte concentrations. For slow off-rates (≤ 9×10−3 1/s), we used manual process referencing that included serial y-align and lower analyte concentrations. After automated data analysis by the Carterra Kinetics software, we also performed additional filtering to remove datasets with highest response signals smaller than signals from negative controls. This additional filtering was performed automatically using an R-script. The script also identified ligands for which capture on the sensor chip was insufficient, and measurements involving those ligands were excluded from subsequent analyses. Many interactions were measured more than once. Whether or not multiple measurements were available, we used the following algorithm to select the representative measurement for any particular interation:
If no measurement resulted in a kinetic-fit KD from the Carterra Kinetics software analysis, the KD value was set to ≥100 μM.
If all available measurements had kinetic-fit KDs from the Carterra Kinetics software analysis that were ≥5 times the maximum analyte concentration used in the measurement, the KD value was set to ≥100 μM. The lowest max analyte concentration for which this was invoked was 37 μM, in which case the kinetic-fit KD of >185 μM was reported as ≥100 μM.
If at least one measurement had a kinetic-fit KD from the Carterra Kinetics software analysis, and if kon and koff were within range for the instrument (10 min−1M−1< kon< 1×109 min−1M−1; 1×10−5 min−1< koff < 5×10−1 min−1), the measurement with the lowest chi-squared fit value was chosen as the representative measure.
If all available measurements with a kinetic-fit KD had a koff that was out of range (this only occured with fast koff), then if the kinetic-fit KD was within a factor of 3 to the equilibrium-fit KD, the ratio of kinetic KD to equilibrium KD was calculated, the measurement with ratio closest to 1 was chosen as the representative, and equilibrium KD was reported for that measurement. If no kinetic-fit KD was within a factor of 3 to the equilibrium-fit KD, the KD was reported as ≥100 uM. The reporting of equilibrium-fit KDs was not common: in Fig. 7, n=2 of 267 post-vaccination VRC01-class KD values, and n=10 of 145 post-vaccination non-VRC01-class KD values, were reported as the equilibrium-fit KD.
We had no cases of kon out of range, thus the algorithm did not have to deal with that case.
SPR reproducibility:
The majority of the iGL and post-vaccination interactions reported in Fig. 7 were also measured using a different instrument (Biacore 8k) and different analyte preparations and often different ligand preparations. The KDs measured by the 8k were highly correlated with those measured by the Carterra LSA, and the conclusions from the two datasets were the same. Also, for the naive precursor KDs measured using the Carterra and reported in Fig. 7, we previously measured and published the same interactions using a different instrument (Proteon XPR) with different analyte and ligand preparations (21, 43); the results of the new and previous analyses were consistent.
Regression analyses involving SPR-measured quantities
Linear mixed effects models (LMEs) were used to analyze the linear dependence of one variable on another, for the following dependencies: (i) dependence of KD for eOD-GT6 or its variants on KD for eOD-GT8, for VRC01-class BCRs (Fig. 8B); (ii) dependence of iGL KD for eOD-GT8 on the number of mutations in the BCRs from which the iGL was inferred, for VRC01-class and non-VRC01-class BCRs (fig. S30B); (iii) dependence of eOD-GT8 KD on kon and koff, for VRC01-class and non-VRC01-class BCRs (fig. S32C); (iv) dependence of eOD-GT8 KD, kon, and koff, on the number of BCR V gene mutations (figs. S34–S36). LMEs were used to estimate the slope of each association [e.g. change in log(KD) per mutation]. Estimated associations only included antibodies for which the KD value was measureable (KD<100 μM). Each LME had fixed effects for the intercept and slope. Random effects for the intercept and slope were included for each participant, with the exception of estimation for a given week (figs. S34B, S35B, and S36B) or for non-VRC01-class iGL (fig. S30B), where a random effect for only the intercept was included. For non-VRC01-class BCRs at week 16 in figs. S34B, S35B, and S36B, only one antibody per participant was available, so estimates were based on a linear model (KD, kon, and koff). The estimated regression line and shaded 95% prediction interval were generated using a semi-parametric bootstrap method (using 1000 bootstrap datasets). LMEs were fit and p-values testing the null hypothesis that the fixed effect for slope is zero were evaluated using Satterthwaite’s degrees of freedom method using the lmerTest package in R (94). For a log10-transformed y-value depending linearly on , as in figs. S34–36 the effect of on is given by , where is the slope. This is equivalent to . Thus, in figs. S34–S36 where represents mutations and represents log(KD) or log(kon) or log(koff), an increase of mutations going from to will lead to a change in as: . For a log10-transformed y-value depending linearly on a log10-transformed x-value, as in Fig. 8B, the effect of on is given by , where is the slope. This is equivalent to . Thus, in Fig. 8B, where represents the KD for eOD-GT6 or a GT6 variant, and represents KD for eOD-GT8, an increase in the KD for eOD-GT6 or a GT6 variant by a factor of α will lead to a change in the KD for eOD-GT8 according to .
Statistical analysis
Placebos in both groups were pooled into a single control group for all statistical analyses. Confidence intervals for frequencies and response rates were based on the Wilson score method (91). Spearman correlations were used to evaluate the association between immune responses. To assess if response rates differed by group, Barnard’s exact test was used. To assess if response rates differed over time within a group, McNemar’s test was used to account for paired data. Response magnitude comparisons between groups were compared using the Wilcoxon rank-sum test. Response magnitude comparisons between time points were performed using the Wilcoxon signed-rank test to account for paired data. Comparisons of percent mutation levels over time (Fig. 5, fig. S20, and tables S46–S49) were performed using the Wilcoxon signed-rank test to account for paired data. Regression analyses were performed as described in Regression analyses involving SPR-measured quantities. Statistical quantile analyses were carried out using R and Pandas (https://github.com/pandas-dev/pandas). The R language and tidyverse R packages were used for graphical and statistical analysis (95, 96).
Figure generation
Most figures were generated with either Matplotlib (97) or a custom port of the Seaborn package that incorporates Wilson confidence intervals into the statistical analysis [ (98); https://github.com/jwillis0720/seaborn-fork], and with Adobe Illustrator for final composition. Figures S4 and S17 were produced using the ggplot package in R. Figure generation can be found in the accompanying data analysis repository (github.com/SchiefLab/G001/figures). Figures S5, S6, S8, and S9 were produced using Intaglio (https://intaglio.en.softonic.com/mac). Fig. S10 was produced using BioRender (BioRender.com), and the publishing license can be found in the data repository.
Supplementary Material
Acknowledgments:
We thank Pervin Anklesaria, Nina Russell, and Emilio Emini for discussions and trial planning; and Leo Stamatatos, Bruno Correia, Mihai Azoitei, Torben Schiffner, Jennifer Bohl, Steven Deeks, and Pat Fast for comments on the manuscript. At IAVI, we thank Harriet Park, Lisa Sunner, Iyore Ayanru, Hanlie Bester, Paige Neuenschwander, Dan Todd, Kathryn Rutkowski, Raleigh Edelstein, and Kathy Crisafi for clinical management; Jim Ackland for overseeing the toxicology study; Sunaina Hingorani, Andrew Elnatan, and Devika Zachariah for regulatory filings; and Vadim Tsvetnitsky, Eddy Sayeed, Vaneet Sharma, Jim Ackland, Kristen Syvertsen, Sam Pallerla, Sreenivas Avula, Polina Kishineskaya, Roslyn Platt, Natasha Williams, Reena Colacot, and Tom Hassell for manufacturing oversight and quality assurance; and Olayinka Fagbayi and Amelia Mosley for project management at the IAVI NAC at Scripps. At FHCRC, we thank David Berger, Gina Braun, and Kim Louis for clinical operations; Austin Varni, Todd Haight, Carol Marty, and Sarah Ameny for sample and assay management; Antje Heit for FNA method development; and Mingchao Shen and John Hural for laboratory operations. At GWU, we thank Elissa Malkin, Sarah Henn, Aimee Desrosiers, and Samantha Walker for clinical operations; and Larissa Scholte, Linda Schellhaas, and Lara Hoeweler for sample and assay management. Related to BAMA assays at Duke, we thank Tara McNair, Jessica Choi, Michael Archibald for technical expertise; Sheetal Sawant for data analysis; Mark Sampson and Angelina Sharak for data management; Judith Lucas for GCLP compliant laboratory environment; and Marcella Sarzotti-Kelsoe for quality assurance oversight. For neutralization assays at Duke, we thank Elizabeth Domin, Carley West, and Jiayu Chen. At VRC, we thank Jason M. Brenchley for flow cytometry support, and Madhu Prabhakaran and Britta Flach for technical advice. At IAVI and Scripps, we thank Devin Sok, Bryan Briney, Patrick Skog, David Nemazee, and Dennis Burton for a pre-clinical in vivo test of vaccine and adjuvant. At GSK, we thank Marguerite Koutsoukos, François Roman, Olivier van der Meeren, Clarisse Lorin, and Clémence Laugier for providing AS01B and for carrying out pre-clinical compatibility and toxicity studies. We also thank Colin Havenar-Daughton and Brianna Shakoor for sequences of CLK antibodies, Jeong Hyun Lee for providing human naive BCR sequences from (44), and Xuesong Wang for providing data from (49).
Funding:
This work was supported by the Bill and Melinda Gates Foundation Collaboration for AIDS Vaccine Discovery (CAVIMC INV-007368 to D.M. and G.D.T.; CCVIMC INV-007371 to R.A.K., A.B.M., and M.J.M.; VISC INV-008017 and INV-032929 to A.C.D.; VxPDC INV-008352 and INV-007375 to IAVI; and NAC INV-007522 and INV-008813 to W.R.S.), IAVI (including IAVI 167627819 to M.J.M., IAVI A08031 research collaboration agreement with Karolinska Intitutet for G.B.K.H., and other support to W.R.S.), the IAVI Neutralizing Antibody Center (NAC) to W.R.S., National Institute of Allergy and Infectious Diseases (NIAID) P01 AI094419 (HIVRAD Optimizing HIV immunogen-BCR interactions for vaccine development”) (to W.R.S.), UM1 Al100663 (Scripps Center for HIV/AIDS Vaccine Immunology and Immunogen Discovery) and UM1 AI144462 (Scripps Consortium for HIV/AIDS Vaccine Development) (to W.R.S. and M.J.M.); and UM1AI069481 (Seattle-Lausanne CTU), U19AI128914 (HIPC), and UM1AI068618 (HVTN LC) to M.J.M.; by the Swedish Research Council (grant 2017–00968 to G.B.K.H.), and by the Ragon Institute of MGH, MIT, and Harvard (to W.R.S.).
Footnotes
Competing interests: W.R.S. and S.M are inventors on patents filed by Scripps and IAVI on the eOD-GT8 monomer and 60mer immunogens.
Data and materials availability:
All BCR sequences and FACS analysis files produced in this study are available in the public data repository https://github.com/SchiefLab/G001, with the persistent identifier DOI: 10.5281/zenodo.7254574. The repository contains four separate modules: 1) FACS analysis; 2) BCR sequence analysis; 3) Combined B cell frequency and BCR sequencing analysis for VRC01 and non-VRC01 calls; and 4) Figure and table generation. Instructions for running each module of the repository are provided in the README. Sequences for antibody expression vectors were deposited in GenBank under accession numbers ON512569, ON5125670, and ON5125671. All other data are available in the main text or supplementary materials.
References and Notes
- 1.Fauci AS, An HIV Vaccine Is Essential for Ending the HIV/AIDS Pandemic. JAMA 318, 1535–1536 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Pegu A et al. , A Meta-analysis of Passive Immunization Studies Shows that Serum-Neutralizing Antibody Titer Associates with Protection against SHIV Challenge. Cell Host Microbe 26, 336–346 e333 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corey L et al. , Two Randomized Trials of Neutralizing Antibodies to Prevent HIV-1 Acquisition. N Engl J Med 384, 1003–1014 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gilbert PB et al. , Neutralization titer biomarker for antibody-mediated prevention of HIV-1 acquisition. Nature Medicine 28, 1924–1932 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiao X, Chen W, Feng Y, Dimitrov DS, Maturation Pathways of Cross-Reactive HIV-1 Neutralizing Antibodies. Viruses 1, 802–817 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiao X et al. , Germline-like predecessors of broadly neutralizing antibodies lack measurable binding to HIV-1 envelope glycoproteins: implications for evasion of immune responses and design of vaccine immunogens. Biochem Biophys Res Commun 390, 404–409 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dimitrov DS, Therapeutic antibodies, vaccines and antibodyomes. MAbs 2, 347–356 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pancera M et al. , Crystal structure of PG16 and chimeric dissection with somatically related PG9: structure-function analysis of two quaternary-specific antibodies that effectively neutralize HIV-1. Journal of virology 84, 8098–8110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou T et al. , Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science 329, 811–817 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonsignori M et al. , Analysis of a clonal lineage of HIV-1 envelope V2/V3 conformational epitope-specific broadly neutralizing antibodies and their inferred unmutated common ancestors. Journal of virology 85, 9998–10009 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scheid JF et al. , Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science 333, 1633–1637 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoot S et al. , Recombinant HIV envelope proteins fail to engage germline versions of anti-CD4bs bNAbs. PLoS pathogens 9, e1003106 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jardine J et al. , Rational HIV immunogen design to target specific germline B cell receptors. Science 340, 711–716 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McGuire AT et al. , Engineering HIV envelope protein to activate germline B cell receptors of broadly neutralizing anti-CD4 binding site antibodies. J Exp Med 210, 655–663 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haynes BF, Kelsoe G, Harrison SC, Kepler TB, B-cell-lineage immunogen design in vaccine development with HIV-1 as a case study. Nat Biotechnol 30, 423–433 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liao HX et al. , Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature 496, 469–476 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao F et al. , Cooperation of B cell lineages in induction of HIV-1-broadly neutralizing antibodies. Cell 158, 481–491 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bonsignori M et al. , Maturation Pathway from Germline to Broad HIV-1 Neutralizer of a CD4-Mimic Antibody. Cell 165, 449–463 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonsignori M et al. , Staged induction of HIV-1 glycan-dependent broadly neutralizing antibodies. Sci Transl Med 9, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saunders KO et al. , Targeted selection of HIV-specific antibody mutations by engineering B cell maturation. Science 366, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jardine JG et al. , HIV-1 broadly neutralizing antibody precursor B cells revealed by germline-targeting immunogen. Science 351, 1458–1463 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steichen JM et al. , HIV Vaccine Design to Target Germline Precursors of Glycan-Dependent Broadly Neutralizing Antibodies. Immunity 45, 483–496 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Escolano A et al. , Sequential Immunization Elicits Broadly Neutralizing Anti-HIV-1 Antibodies in Ig Knockin Mice. Cell 166, 1445–1458 e1412 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Medina-Ramirez M et al. , Design and crystal structure of a native-like HIV-1 envelope trimer that engages multiple broadly neutralizing antibody precursors in vivo. J Exp Med 214, 2573–2590 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steichen JM et al. , A generalized HIV vaccine design strategy for priming of broadly neutralizing antibody responses. Science 366, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Briney B et al. , Tailored Immunogens Direct Affinity Maturation toward HIV Neutralizing Antibodies. Cell 166, 1459–1470 e1411 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tian M et al. , Induction of HIV Neutralizing Antibody Lineages in Mice with Diverse Precursor Repertoires. Cell 166, 1471–1484 e1418 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parks KR et al. , Overcoming Steric Restrictions of VRC01 HIV-1 Neutralizing Antibodies through Immunization. Cell Rep 29, 3060–3072 e3067 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen X et al. , Vaccination induces maturation in a mouse model of diverse unmutated VRC01-class precursors to HIV-neutralizing antibodies with >50% breadth. Immunity 54, 324–339 e328 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verkoczy L et al. , Induction of HIV-1 broad neutralizing antibodies in 2F5 knock-in mice: selection against membrane proximal external region-associated autoreactivity limits T-dependent responses. Journal of immunology 191, 2538–2550 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bradley T et al. , HIV-1 Envelope Mimicry of Host Enzyme Kynureninase Does Not Disrupt Tryptophan Metabolism. Journal of immunology 197, 4663–4673 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang R et al. , Initiation of immune tolerance-controlled HIV gp41 neutralizing B cell lineages. Sci Transl Med 8, 336ra362 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alam SM et al. , Mimicry of an HIV broadly neutralizing antibody epitope with a synthetic glycopeptide. Sci Transl Med 9, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou T et al. , Quantification of the Impact of the HIV-1-Glycan Shield on Antibody Elicitation. Cell Rep 19, 719–732 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dubrovskaya V et al. , Targeted N-glycan deletion at the receptor-binding site retains HIV Env NFL trimer integrity and accelerates the elicited antibody response. PLoS pathogens 13, e1006614 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu K et al. , Epitope-based vaccine design yields fusion peptide-directed antibodies that neutralize diverse strains of HIV-1. Nat Med 24, 857–867 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fera D et al. , HIV envelope V3 region mimic embodies key features of a broadly neutralizing antibody lineage epitope. Nat Commun 9, 1111 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kong R et al. , Antibody Lineages with Vaccine-Induced Antigen-Binding Hotspots Develop Broad HIV Neutralization. Cell 178, 567–584 e519 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jardine JG et al. , HIV-1 VACCINES. Priming a broadly neutralizing antibody response to HIV-1 using a germline-targeting immunogen. Science 349, 156–161 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sharma VK et al. , Use of Transient Transfection for cGMP Manufacturing of eOD-GT8 60mer, a Self-Assembling Nanoparticle Germline-Targeting HIV-1 Vaccine Candidate. bioRxiv, (2022. (submitted)). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.West AP Jr., Diskin R, Nussenzweig MC, Bjorkman PJ, Structural basis for germline gene usage of a potent class of antibodies targeting the CD4-binding site of HIV-1 gp120. Proceedings of the National Academy of Sciences of the United States of America 109, E2083–2090 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou T et al. , Multidonor analysis reveals structural elements, genetic determinants, and maturation pathway for HIV-1 neutralization by VRC01-class antibodies. Immunity 39, 245–258 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Havenar-Daughton C et al. , The human naive B cell repertoire contains distinct subclasses for a germline-targeting HIV-1 vaccine immunogen. Sci Transl Med 10, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee JH et al. , Vaccine genetics of IGHV1–2 VRC01-class broadly neutralizing antibody precursor naive human B cells. NPJ Vaccines 6, 113 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dosenovic P et al. , Immunization for HIV-1 Broadly Neutralizing Antibodies in Human Ig Knockin Mice. Cell 161, 1505–1515 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sok D et al. , Priming HIV-1 broadly neutralizing antibody precursors in human Ig loci transgenic mice. Science 353, 1557–1560 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abbott RK et al. , Precursor Frequency and Affinity Determine B Cell Competitive Fitness in Germinal Centers, Tested with Germline-Targeting HIV Vaccine Immunogens. Immunity 48, 133–146 e136 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang D et al. , B cells expressing authentic naive human VRC01-class BCRs can be recruited to germinal centers and affinity mature in multiple independent mouse models. Proceedings of the National Academy of Sciences of the United States of America 117, 22920–22931 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang X et al. , Multiplexed CRISPR/CAS9-mediated engineering of pre-clinical mouse models bearing native human B cell receptors. EMBO J 40, e105926 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Havenar-Daughton C et al. , Rapid Germinal Center and Antibody Responses in Non-human Primates after a Single Nanoparticle Vaccine Immunization. Cell Rep 29, 1756–1766 e1758 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vigdorovich V et al. , Repertoire comparison of the B-cell receptor-encoding loci in humans and rhesus macaques by next-generation sequencing. Clin Transl Immunology 5, e93 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vazquez Bernat N et al. , Rhesus and cynomolgus macaque immunoglobulin heavy-chain genotyping yields comprehensive databases of germline VDJ alleles. Immunity 54, 355–366 e354 (2021). [DOI] [PubMed] [Google Scholar]
- 53.Herve C, Laupeze B, Del Giudice G, Didierlaurent AM, Tavares Da Silva F, The how’s and what’s of vaccine reactogenicity. NPJ Vaccines 4, 39 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duan H et al. , Glycan Masking Focuses Immune Responses to the HIV-1 CD4-Binding Site and Enhances Elicitation of VRC01-Class Precursor Antibodies. Immunity 49, 301–311 e305 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Umotoy J et al. , Rapid and Focused Maturation of a VRC01-Class HIV Broadly Neutralizing Antibody Lineage Involves Both Binding and Accommodation of the N276-Glycan. Immunity 51, 141–154 e146 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jardine JG et al. , Minimally Mutated HIV-1 Broadly Neutralizing Antibodies to Guide Reductionist Vaccine Design. PLoS pathogens 12, e1005815 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heesters BA, van der Poel CE, Das A, Carroll MC, Antigen Presentation to B Cells. Trends Immunol 37, 844–854 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Kato Y et al. , Multifaceted Effects of Antigen Valency on B Cell Response Composition and Differentiation In Vivo. Immunity 53, 548–563 e548 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dosenovic P et al. , Anti-HIV-1 B cell responses are dependent on B cell precursor frequency and antigen-binding affinity. Proceedings of the National Academy of Sciences of the United States of America 115, 4743–4748 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mesin L et al. , Restricted Clonality and Limited Germinal Center Reentry Characterize Memory B Cell Reactivation by Boosting. Cell 180, 92–106 e111 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Viant C et al. , Antibody Affinity Shapes the Choice between Memory and Germinal Center B Cell Fates. Cell 183, 1298–1311 e1211 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pape KA, Taylor JJ, Maul RW, Gearhart PJ, Jenkins MK, Different B cell populations mediate early and late memory during an endogenous immune response. Science 331, 1203–1207 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zuccarino-Catania GV et al. , CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat Immunol 15, 631–637 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Phad GE et al. , Extensive dissemination and intraclonal maturation of HIV Env vaccine-induced B cell responses. J Exp Med 217, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cho A et al. , Anti-SARS-CoV-2 receptor-binding domain antibody evolution after mRNA vaccination. Nature 600, 517–522 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Muecksch F et al. , Increased Memory B Cell Potency and Breadth After a SARS-CoV-2 mRNA Boost. Nature, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sharma VK et al. , Use of Transient Transfection for cGMP Manufacturing of eOD-GT8 60mer, a Self-Assembling Nanoparticle Germline-Targeting HIV-1 Vaccine Candidate. bioRxiv, 2022.2009.2030.510310 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tomaras GD et al. , Initial B-cell responses to transmitted human immunodeficiency virus type 1: virion-binding immunoglobulin M (IgM) and IgG antibodies followed by plasma anti-gp41 antibodies with ineffective control of initial viremia. Journal of virology 82, 12449–12463 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yates NL et al. , Vaccine-induced Env V1-V2 IgG3 correlates with lower HIV-1 infection risk and declines soon after vaccination. Sci Transl Med 6, 228ra239 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yates NL et al. , HIV-1 Envelope Glycoproteins from Diverse Clades Differentiate Antibody Responses and Durability among Vaccinees. Journal of virology 92, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Montefiori DC, Measuring HIV neutralization in a luciferase reporter gene assay. Methods in molecular biology 485, 395–405 (2009). [DOI] [PubMed] [Google Scholar]
- 72.Sarzotti-Kelsoe M et al. , Optimization and validation of the TZM-bl assay for standardized assessments of neutralizing antibodies against HIV-1. J Immunol Methods 409, 131–146 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.LaBranche CC et al. , HIV-1 envelope glycan modifications that permit neutralization by germline-reverted VRC01-class broadly neutralizing antibodies. PLoS pathogens 14, e1007431 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.LaBranche CC et al. , Neutralization-guided design of HIV-1 envelope trimers with high affinity for the unmutated common ancestor of CH235 lineage CD4bs broadly neutralizing antibodies. PLoS pathogens 15, e1008026 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Finak G, Jiang W, Gottardo R, CytoML for cross-platform cytometry data sharing. Cytometry A 93, 1189–1196 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Finak G, Jiang M, flowWorkspace: Infrastructure for representing and interacting with gated and ungated cytometry data sets. R package version 4.5.3. 2011. [Google Scholar]
- 77.Van P, Jiang W, Gottardo R, Finak G, ggCyto: next generation open-source visualization software for cytometry. Bioinformatics 34, 3951–3953 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Boswell KL et al. , Application of B cell immortalization for the isolation of antibodies and B cell clones from vaccine and infection settings. bioRxiv, 2022.2003.2029.485179 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Upadhyay AA et al. , BALDR: a computational pipeline for paired heavy and light chain immunoglobulin reconstruction in single-cell RNA-seq data. Genome Med 10, 20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Briney B, Burton DR, Massively scalable genetic analysis of antibody repertoires. bioRxiv https://www.biorxiv.org/content/early/2018/10/19/447813, (2018).
- 81.Breden F et al. , Reproducibility and Reuse of Adaptive Immune Receptor Repertoire Data. Front Immunol 8, 1418 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ewing B, Hillier L, Wendl MC, Green P, Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 8, 175–185 (1998). [DOI] [PubMed] [Google Scholar]
- 83.Ewing B, Green P, Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 8, 186–194 (1998). [PubMed] [Google Scholar]
- 84.Ye J, Ma N, Madden TL, Ostell JM, IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res 41, W34–40 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Navarro G, A guided tour to approximate string matching. ACM Comput. Surv. 33, 31–88 (2001). [Google Scholar]
- 86.Dunbar J, Deane CM, ANARCI: antigen receptor numbering and receptor classification. Bioinformatics 32, 298–300 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vazquez Bernat N et al. , High-Quality Library Preparation for NGS-Based Immunoglobulin Germline Gene Inference and Repertoire Expression Analysis. Front Immunol 10, 660 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Corcoran MM et al. , Production of individualized V gene databases reveals high levels of immunoglobulin genetic diversity. Nat Commun 7, 13642 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pedregosa F et al. , Scikit-learn: Machine Learning in Python. Journal of Machine Learning Research 12, 2825–2830 (2011). [Google Scholar]
- 90.Briney B, Le K, Zhu J, Burton DR, Clonify: unseeded antibody lineage assignment from next-generation sequencing data. Sci Rep 6, 23901 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Agresti A, Coull BA, Approximate is better than “exact” for interval estimation of binomial proportions. Am Stat 52, 119–126 (1998). [Google Scholar]
- 92.Kovaltsuk A et al. , Observed Antibody Space: A Resource for Data Mining Next-Generation Sequencing of Antibody Repertoires. Journal of immunology 201, 2502–2509 (2018). [DOI] [PubMed] [Google Scholar]
- 93.Olsen TH, Boyles F, Deane CM, Observed Antibody Space: A diverse database of cleaned, annotated, and translated unpaired and paired antibody sequences. Protein Sci, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kuznetsova A, Brockhoff PB, Christensen RHB, lmerTest Package: Tests in Linear Mixed Effects Models. Journal of Statistical Software 82, 1–26 (2017). [Google Scholar]
- 95.Wickham H et al. , Welcome to the Tidyverse. Journal of Open Source Software 4, (2019). [Google Scholar]
- 96.R Core Team, R: A Language and Environment for Statistical Computing. https://www.R-project.org/. 2019.
- 97.Hunter JD, Matplotlib: A 2D Graphics Environment. Computing in Science & Engineering 9, 90–95 (2007). [Google Scholar]
- 98.Waskom ML, seaborn: statistical data visualization. Journal of Open Source Software 6, 3021 (2021). [Google Scholar]
- 99.DeKosky BJ et al. , In-depth determination and analysis of the human paired heavy- and light-chain antibody repertoire. Nat Med 21, 86–91 (2015). [DOI] [PubMed] [Google Scholar]
- 100.Gristick HB et al. , Natively glycosylated HIV-1 Env structure reveals new mode for antibody recognition of the CD4-binding site. Nat Struct Mol Biol 23, 906–915 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schommers P et al. , Restriction of HIV-1 Escape by a Highly Broad and Potent Neutralizing Antibody. Cell 180, 471–489 e422 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cottrell CA et al. , Structural basis of glycan276-dependent recognition by HIV-1 broadly neutralizing antibodies. Cell Rep 37, 109922 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fornasari MS et al. , Sequence determinants of quaternary structure in lumazine synthase. Mol Biol Evol 21, 97–107 (2004). [DOI] [PubMed] [Google Scholar]
- 104.Andrews SF et al. , Activation Dynamics and Immunoglobulin Evolution of Pre-existing and Newly Generated Human Memory B cell Responses to Influenza Hemagglutinin. Immunity 51, 398–410 e395 (2019). [DOI] [PubMed] [Google Scholar]
- 105.Goel RR et al. , Distinct antibody and memory B cell responses in SARS-CoV-2 naive and recovered individuals following mRNA vaccination. Sci Immunol 6, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Stamatatos L et al. , mRNA vaccination boosts cross-variant neutralizing antibodies elicited by SARS-CoV-2 infection. Science, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pape KA et al. , High-affinity memory B cells induced by SARS-CoV-2 infection produce more plasmablasts and atypical memory B cells than those primed by mRNA vaccines. Cell Rep 37, 109823 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Seabright GE, Doores KJ, Burton DR, Crispin M, Protein and Glycan Mimicry in HIV Vaccine Design. J Mol Biol 431, 2223–2247 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Havenar-Daughton C, Abbott RK, Schief WR, Crotty S, When designing vaccines, consider the starting material: the human B cell repertoire. Curr Opin Immunol 53, 209–216 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Turner JS et al. , Human germinal centres engage memory and naive B cells after influenza vaccination. Nature 586, 127–132 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Turner JS et al. , SARS-CoV-2 mRNA vaccines induce persistent human germinal centre responses. Nature 596, 109–113 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Schiller JT, Markowitz LE, Hildesheim A, Lowy DR, in Plotkin’s Vaccines, Plotkin SA, Orenstein WA, Offit PA, Edwards KM, Eds. (Elsevier, 2018), pp. 430–455. [Google Scholar]
- 113.Van Damme P, Ward JW, Shouval D, Zanetti A, in Plotkin’s Vaccines, Plotkin SA, Orenstein WA, Offit PA, Edwards KM, Eds. (Elsevier, 2018), chap. 25, pp. 342–374. [Google Scholar]
- 114.Moon JE et al. , A Phase IIa Controlled Human Malaria Infection and Immunogenicity Study of RTS,S/AS01E and RTS,S/AS01B Delayed Fractional Dose Regimens in Malaria-Naive Adults. J Infect Dis 222, 1681–1691 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All BCR sequences and FACS analysis files produced in this study are available in the public data repository https://github.com/SchiefLab/G001, with the persistent identifier DOI: 10.5281/zenodo.7254574. The repository contains four separate modules: 1) FACS analysis; 2) BCR sequence analysis; 3) Combined B cell frequency and BCR sequencing analysis for VRC01 and non-VRC01 calls; and 4) Figure and table generation. Instructions for running each module of the repository are provided in the README. Sequences for antibody expression vectors were deposited in GenBank under accession numbers ON512569, ON5125670, and ON5125671. All other data are available in the main text or supplementary materials.