

Abstract
Purpose
Preclinical in vivo analyses of treatment responses are an important prerequisite to evaluate new therapeutics. Molecular in vivo imaging in the far red (FR)/near infra red (NIR) is a promising method, as it enables measurements at different time points in individual animals, thereby reducing the number of animals required, while increasing statistical significance. Here, we show the establishment of a method to monitor response to treatment using fluorescent cells, expressing the epidermal growth factor receptor (EGFR), a target already used in therapy.
Methods
We transfected A-431 tumour cells with the far red–emitting protein Katushka (Kat2), resulting in strong fluorescence allowing for the monitoring of tumour growth when implanted in BALB/c nu/nu mice with a CRi Maestro in vivo imager. We targeted A-431 cells with a previously reported immunotoxin (IT), consisting of the anti-EGFR antibody single-chain variable fragment (scFv) 425, fused to Pseudomonas aeruginosa Exotoxin A’ (ETA’). In addition, EGFR expression was verified using the 425(scFv) conjugated to a NIR dye BG-747 through a SNAP-tag linker.
Results
The results show the feasibility to evaluate response to treatment in vivo by FR imaging, while at the same location detecting EGFR expression. Treatment with 425(scFv)-ETA’ resulted in decelerated tumour growth, while not affecting the overall health of the animals. This is in contrast to treatment with Doxorubicin, which, although decreasing the tumour size, resulted in poor health.
Conclusions
We developed a novel method to non-invasively determine treatment responses by in vivo imaging of multiple parameters which showed the efficacy of 425(scFv)-ETA’.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-012-1219-3) contains supplementary material, which is available to authorized users.
Keywords: Fluorescence imaging, scFv immunotoxin, Katushka, EGF receptor, Immunotherapy, Treatment response
Introduction
Cancer accounts for 13% of all deaths worldwide (in 2004). Although common treatment like surgery and radiotherapy (WHO Cancer, Fact Sheet No. 297, 2009) yields increasing success, there is still room for improvement, especially in preventing the development of metastasis.
Development of novel therapeutics and therapeutic strategies for the treatment of cancer nowadays is an elaborate and expensive procedure. Therefore, in vivo immunodeficient mouse models using human tumour cell lines have been developed to mimic different human cancers. An effective and sensitive monitoring system to determine the in vivo effect in such animal models prior to start clinical trials is of great importance. Such system should be cost-effective, easy of use and have high predictive value. Classically, tumour growth is measured with a vernier calliper, and animals are killed at different time points, and ex vivo examination like pathology and specific staining is performed.
The major drawback of this approach is the necessity for separate groups per individual time point and consequently the intermittent nature. This inherently implies the use of large numbers of animals to achieve statistical relevance [1].
For continuous monitoring, in vivo imaging techniques are greatly gaining interest, in particular optical imaging, as it has the advantage that relatively simple equipment can be used. One groundbreaking development was the use of green fluorescent protein (GFP) [2], as it can easily be transfected into different cell types or used in fusion proteins. Due to its emission in the green, the use of GFP in fluorescence-based molecular imaging approaches, however, has several disadvantages like low penetration depth, high tissue absorption, as well as emission in the same spectral range as many endogenous autofluorescent molecules. These together lead to low signal and high background, limiting the use of GFP [3]. This can largely be avoided by using the far red (FR) to near infra red (NIR) window [1, 3–5]. The fluorescent protein Katushka (Kat2) has a very strong emission maximum at 635 nm. This in combination with high photostability compared to other red and FR proteins makes it a powerful tool for in vivo imaging [6].
In this study, we established the use of Kat2 for monitoring therapy in a human xenograft tumour model in hairless immunodeficient mice. To this end, we imaged the growth of subcutaneous tumours of epidermal growth factor receptor (EGFR)-expressing A431scM3 cells transfected with Kat2. A431scM3 are A-431-derived epidermoid cancer cells, which have been passaged in vivo to select for fast-growing cells. Because of their high EGFR expression, A-431 are suitable target cells to study the effect of therapeutic approaches directed to EGFR, which is overexpressed on a variety of cancer cells [7, 8]. Due to this, A-431 cells are often used in preclinical research [9, 10]. Already approved therapeutics targeting the EGFR are Gefitinib (Iressa) and Erlotinib (Tarceva) [11, 12] belonging to the class of tyrosine kinase inhibitors. Cetuximab [13] and Panitumumab [14] are monoclonal antibodies binding to the EGFR-blocking natural ligand binding sites. All these agents inhibit proliferation, differentiation and angiogenesis of EGFR-positive tumours improving the efficiency of conventional chemotherapy. However, development of resistance to anti-EGFR therapy is a huge problem for many patients [15]. Therefore, the need for other EGFR targeting therapeutics is high.
As EGFR-specific therapeutic, we used the 425(scFv)-Pseudomonas aeruginosa Exotoxin A’ (425(scFv)-ETA’) immunotoxin (IT), which has previously shown to have anti-tumour activity in a disseminated human pancreatic cancer mouse model [16, 17]. ITs are fusion proteins consisting of a cell-specific binding domain coupled to a toxic entity [18–22]. Due to their cell specificity, ITs show greatly reduced side effects in comparison with chemotherapeutics [23]. As toxic moieties, many molecules of different origins have been tested, but as yet, only two have been used in clinical studies: Diphtheria toxin and ETA [24–26]. Both toxins inhibit protein biosynthesis by ADP ribosylation of eukaryotic elongation factor 2, leading to cell death.
Cancer cells are known to adapt different cellular programmes coordinating regulatory circuits, allowing them to, for example, metastasise and invade tissues, sustain angiogenesis or proliferate infinitely [27, 28]. Additionally, receptor downregulation can be observed in some tumours as a mechanism to evade immunogenicity, which becomes a major problem when targeting those antigens. Recently, it was shown that Cetuximab treatment resulted in translocation of the EGFR to the nucleus [29]. Another study reports that treatment with Erlotinib leads to lower EGFR amounts, while not affecting gene expression levels [30]. Hence, it is important to check on antigen expression during therapy.
Preferably, one would like to determine multiple parameters like growth and marker expression in one go, using optical imaging.
Labelling of molecules with different fluorescent dyes can be used to track these molecules in vivo. With the SNAP-tag technology, small antibody fragments like scFvs or antibodies fused to O6-alkyl-DNA alkyltransferase (SNAP-tag) can be coupled to various fluorescent dyes or nanoparticles [31] and toxins. We recently published the coupling of 425(scFv)SNAP to the NIR dye BG-747 using this technology to efficiently detect EGFR-positive tumour cells in vivo. An additional advantage is the rapid systemic clearance of the fusion protein [32].
In this study, we established a system to test and quantify the therapeutic potential of a candidate drug, using optical imaging. To this end, we set up a model in immunocompromised mice using the Kat2-transfected, high EGFR-expressing epidermoid cancer cell line A431scM3 in combination with the EGFR-specific 425(scFv). This scFv was both used for detection, as imaging agent conjugated to BG-747 via SNAP-tag, as well as an IT linked to ETA’.
Materials and methods
Expression and purification of 425(scFv)-ETA’
After transformation of E. coli Rosetta 2 DE3 bacteria with pBM-425(scFv)-ETA’ DNA [16, 33], a 4L fermentation in synthetic medium was performed for protein expression [34]. Bacteria were harvested 18 h after isopropyl thiogalactoside induction yielding in a 480 g pellet. The pellet was resuspended in 960 ml cooled PBS containing 300 mM NaCl. Subsequently, bacterial lysis was performed using French Press (2 cycles). The lysed bacteria were spun down and the protein containing supernatant purified via immobilised metal-ion affinity chromatography (IMAC) using a “Streamline” Ni–NTA column, anion exchange chromatography using a 5 ml XK1620 column filled with Q-Sepharose FF matrix (GE Healthcare) and size exclusion chromatography using a 120 ml XK70 column filled with Superdex 75 matrix (GE Healthcare). Pure, full-length 70 kDa protein could be detected by SDS–PAGE and immunoblotting (Online Resource 1). The pure protein was aliquoted and lyophilised for long-term storage at −20°C. Before each experiment, one lyophilised aliquot was reconstituted with MilliQ and dialysed against PBS overnight.
Flow cytometric analyses
Binding of 1 μg 425(scFv)-ETA’ to A431scM3-Kat2 cells was confirmed via flow cytometry as described previously [16]. Bound protein was detected with anti-polyHistidin-antibody (Sigma, 1:250 in PBS) and FITC-labelled goat-anti-mouse as secondary antibody (Dianova, 1:250 in PBS). Measurements were taken with a FACSCalibur flow cytometer (BD Biosciences), and data were analysed with CellQuest Software (BD Biosciences).
Cell viability assays
In order to confirm 425(scFv)-ETA’ toxicity, an XTT-based cell viability assay was done. Purified 425(scFv)-ETA’ protein was pipetted in triplicates to complete RPMI-1640 medium (Invitrogen). Then, 1:5 dilutions were carried out before adding 5 × 103 A431scM3, A431scM3-Kat2 or L540cy cells per well. The assay was incubated for 72 h at 37°C and 5% CO2 in air. XTT + Phenazin (Serva) was added to each well and the assay incubated according to the manufacturer’s instructions for another 4 h. The conversion of the XTT substrate to the water-soluble formazan dye was measured at 450 and 630 nm (as reference) in an ELISA reader.
Cell culture
Human epidermoid cancer cell lines A431scM3 were derived from tumour specimen of subcutaneously implanted A-431 cells (DSMZ-No. ACC 91) in mice. A431scM3, A431scM3-Kat2 as well as the human embryonic kidney cell line HEK 293T (ATCC-No.: CRL-11268) and the Hodgkin lymphoma cell line L540cy [35] were cultivated in complex RPMI-1640 medium supplemented with 10% (v/v) foetal calf serum (FCS), 2 mM l-glutamine, 50 μg/ml Penicillin and 100 μg/ml Streptomycin. Culture was performed in a humidified atmosphere at 37°C and 5% CO2 in air. Cell lines obtained from DSMZ were all authenticated by cytogenetics, DNA typing, immunophenotyping and DNA analysis. Additionally, cells were regularly tested for mycoplasm and viral contaminations.
Generation of fluorescent tumour cells
Using the lipofection reagent Roti®-Fect (Roth), 1 × 105 A431scM3 cells were seeded in a 12-well plate and transfected with DNA encoding for the FR fluorescent protein Kat2 (pTag-Katushka2-N; Evrogen) according to the manufacturer’s instructions. Transfected cells were selected with G-418 (Nalgene) and sorted 2× with the FACSVantage Cell Sorter (Becton–Dickinson) before cultivation and use in experiments. Red fluorescent cells were detected by fluorescence microscopy (Leica, DM IL), as well as by flow cytometry analysis. The percentage of strongly fluorescent cells was enriched with every sorting cycle (Online Resource 2).
Pictures were taken with a Nikon Coolpix 4500 Camera.
Animals and injection of tumour cells
The experiments were officially approved by the local Animal Care and Use Review Committee. All animals received humane care in accordance with the requirements of the German Tierschutzgesetz, §8 Abs. 1 and in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institute of Health.
For all animal experiments, 6- to 8-week-old female BALB/c nu/nu mice (Charles River, Germany) were used. Fluorescent A431scM3-Kat2 cells were washed with PBS and harvested by treatment with 0.25% (w/v) Trypsin and 0.025% (w/v) EDTA. Trypsinisation was stopped with complex RPMI-1640 medium containing 10% (v/v) FCS, 2 mM l-glutamine, 50 μg/ml Penicillin and 100 μg/ml Streptomycin. Cells were spun down and resuspended in PBS; 5 × 106 A431scM3-Kat2 cells in ~20 μl were subcutaneously injected into the right hind limb of 18 mice. As in previous imaging experiments, mice were placed on a purified, chlorophyll-free diet (AIN93G, SSNIFF GmbH) 7 days before the imaging experiments were started [32].
Targeting of A431scM3-Kat2 cells in vivo
The 425(scFv)SNAP protein was purified from the supernatant of transfected HEK 293T cells [31, 36] by Ni–NTA affinity chromatography. Purified 425(scFv)SNAP protein was coupled to BG-747 dye, using a 1.5 molar excess of dye over protein. The coupling reaction was performed at RT for 1 h and at 4°C overnight. Excess of dye was removed by purification of the coupled protein via a PD10-column. The eluted protein was sterile filtered, and 0.75 nmol was retro-orbitally administered into 6 A431scM3-Kat2-bearing BALB/c nu/nu mice. Ten hours later, mice were imaged with the CRi Maestro system (CRi Inc., Woburn, MA, USA).
Treatment schedule
Treatment was started 11 days after cell injection, when tumours were palpable. Due to the success of the already described treatment protocol with 425(scFv)-ETA’ [17], we administered the IT in 4 single injections. Because of the high frequency of sedations during the imaging experiments, treatment was performed on 2 consecutive days and then on every other day (days 0, 1, 3 and 5). After an interval of 12 days, we performed a second treatment cycle (days 17, 18, 20 and 22). With every injection, the animals received 10 μg 425(scFv)-ETA’, which was determined as the half maximal tolerable dose [17], 100 μg Ribodoxo (generic name: Doxorubicin) as approximately half of the LD50 in nude mice [37] and PBS per single injection. Tumours were measured macroscopically with a vernier calliper as well as by fluorescence-based molecular imaging. Measurements were taken on days 0, 3, 5, 10, 13, 17, 20, 22, 24 and 26 (Fig. 1a).
Fig. 1.

Treatment with 425(scFv)-ETA’ and Doxorubicin. a Treatment was started at day 0 and performed in two cycles consisting each of 4 single injections. With each injection, mice received i.v. 10 μg 425(scFv)-ETA’, 100 μg Doxorubicin or PBS. Pictures were taken, and the tumour growth was calculated with the CRi Maestro System at all indicated time points. b All mice were imaged at different time points during, in between and after the treatment cycles. Based on the fluorescence signal, the tumour area could be determined, which is then calculated by the Maestro Software. The images show the tumour area of one representative mouse of each group at 3 different time points (days 0, 13 and 26). White light images of the mice and fluorescence images were overlaid. c The diagram shows the mean tumour size of 6 mice per group calculated by the Maestro Software throughout both treatment cycles. For comparison of the tumour growth between all groups, the mean tumour sizes on day 0 were set to 100%, while all other values were adjusted. The decelerated tumour growth of the group treated with 425(scFv)-ETA’ compared to the PBS control group was confirmed statistically significant (p < 0.05). d The diagram shows the mean tumour size of 6 mice per group measured with the vernier calliper and calculated according to the formula r1*r2*π throughout both treatment cycles. For comparison of the tumour growth between all groups, the mean tumour sizes on day 0 were set to 100%, while all other values were adjusted. The decelerated tumour growth of the group treated with 425(scFv)-ETA’ compared to the PBS control group was confirmed statistically significant (p < 0.05)
Imaging
Imaging was performed with the CRi Maestro system (CRi Inc., Woburn, MA, USA). Images were taken and analysed with the Maestro Spectral Imaging Software as described before [32]. For imaging of the Kat2 signal of A431scM3-Kat2 cells, we used the yellow filter set (630–850 nm). For the targeting experiments with A431scM3-Kat2 cells, we used the yellow filter set for detection of the Kat2 signal in combination with the deep red filter set (730–950 nm) for detection of the 425(scFv)SNAP-747 signal. With the Maestro software, it is possible to separate signals with different wavelengths based on the emission spectra of the respective dyes (Fig. 2).
Fig. 2.

Spectral libraries of Kat2 and 425(scFv)SNAP-747. Spectral libraries are used for a the measurement of the Kat2 signal and b for unmixing of the Kat2 and 425(scFv)SNAP-747 signals. The library is calculated from a cube acquired with multiple filter settings: yellow (630–850 nm) and deep red (730–950 nm). The red lines represent the Kat2 signal and the blue lines the BG-747 signal in both filter settings
Statistics
Treatment response with 425(scFv)-ETA’ (Maestro and vernier calliper measurements) was compared to the PBS control group using one-tailed t-test (GraphPad Prism, Version 4.0c). p < 0.05 was considered to be statistically significant.
Results
In vivo imaging of fluorescent cells
For the visualisation of A431scM3-Kat2 cells in vivo, 5 × 106 cells were subcutaneously injected into the right hind leg of BALB/c nu/nu mice. The imaging was performed immediately after tumour cell injection as well as after 11 days of tumour growth, when cells formed a palpable tumour.
Previous to the tumour inoculation, an image of the mice was acquired with the identical filter settings as the later tumour imaging (yellow filter set: 630–850 nm, 10-nm increments, exposure time: 100 ms). This was used to determine the background signal. After the tumours had developed, the animals were imaged with these settings, and the specific Kat2 signal from the known tumour location was selected. From these parameters, the spectral library was determined (Fig. 2a). This library was subsequently used to unmix all images at the different time points. The resulting images showed a very high signal-to-background ratio, which made it easy to determine the location of the tumours. In addition, the sharp delineation of the tumours allows for accurate measurement of their surface areas, even directly after cell inoculation (data not shown). Measurement after 11 days of tumour growth showed a clear signal at the site of tumour in all animals (Fig. 3).
Fig. 3.
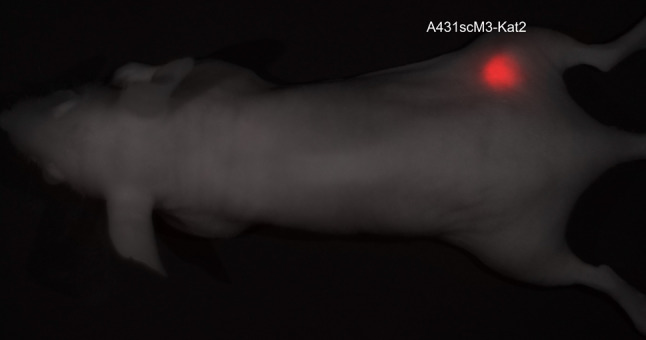
Visualisation of A431scM3-Kat2 cells in vivo. In vitro-transfected and sorted A431scM3-Kat2 cells were subcutaneously injected into the right hind leg of BALB/c nu/nu mice and visualised with the CRi Maestro system after 11 days of tumour growth. Measurement was taken with the yellow filter set (630–850 nm). Red fluorescent cells can be visualised without any background signal. Here, the picture of the fluorescence signal is merged with the white light picture of the mouse
Imaging of the treatment effect with 425(scFv)-ETA’ in vivo
After implantation of the fluorescent tumour cells into 18 mice, all mice showed palpable tumours varying in size at day 11. The mice were divided into 3 groups of 6 mice each with comparable tumour sizes as determined by macroscopic observation. Two treatment cycles, as depicted in Fig. 1a, were applied consisting of 4 injections each. The mice were treated with 10 μg 425(scFv)-ETA’ based on previous data [17], or 100 μg Doxorubicin (<50% of the LD50 in nude mice) [37] or PBS as control per intravenous (i.v.) injection. The animals were imaged according to the schedule (Fig. 1a), and tumour growth was monitored on the basis of the fluorescence signal at each imaging time point (Fig. 1a,b). The data show that 425(scFv)-ETA’ reduces tumour growth of small tumours at the beginning of treatment while decelerating tumour growth of larger tumours as can be observed in the course of treatment (Fig. 1c). This showed reproducibility and statistical significance (p < 0.05). Treatment with the chemotherapeutic Doxorubicin was more effective in terms of inhibition of tumour growth. However, despite using clinically relevant therapeutic concentrations (according to the instruction manual), Doxorubicin had severe side effects resulting in very poor health of the mice, including paralyses of the hind legs and up to 20% weight loss in the end leading to cachexia. Compared to PBS, all mice in the 425(scFv)-ETA’ group had a smaller mean tumour size and showed normal physical behaviour. The PBS-treated mice, however, showed limited mobility, mainly due to the large tumours.
Additionally, tumour growth was measured macroscopically with a vernier calliper (Fig. 1d).
The decelerated tumour growth of the group treated with 425(scFv)-ETA’ compared to the PBS control group was confirmed statistically significant (p < 0.05).
Determination of EGFR expression in vivo by targeting with 425(scFv)SNAP-747
Epidermal growth factor receptor expression of subcutaneously implanted A431scM3-Kat2 cells was confirmed by optical imaging using the same antibody fragment as in the IT, labelled with a NIR dye. According to previously published data, the BG-747 dye was conjugated to 425(scFv)SNAP. Purity of the protein as well as coupling efficiency was tested and confirmed equal as published recently [32]; 425(scFv)SNAP-747 was i.v. injected into 6 of the A431scM3-Kat2-bearing mice. For these imaging experiments, the imaging was done using the yellow and deep red filter sets in the multifilter acquisition mode and the predefined settings for these filters (yellow 630–850 nm plus deep red 730–950 nm; 10-nm increment). Exposure times were 25 ms for the yellow filter set (Kat2) and 3000 ms for deep red (BG-747). To distinguish the BG-747 signal, a pre-experimental image was acquired to determine the background signals. From these, the spectral library for both the Kat2 and the BG-747 signals was determined (Fig. 2b) and used for the unmixing of the images. Both signals could separately be identified, even when co-localised (Fig. 4). Both before and 10 h after injection, the mice were imaged. The resulting images showed that the 425(scFv)SNAP-747 was located at the site of the tumour as well as in the kidneys (Fig. 4).
Fig. 4.

Targeting of A431scM3-Kat2 with 425(scFv)SNAP-747. With BG-747 labelled 425(scFv)SNAP was i.v. applied into A431scM3-Kat2-bearing mice. The pictures shown here were taken 10 h after application. Measurement was taken with the yellow (630–850 nm) and deep red filter set (730–950 nm). a Composite image with all fluorescence signals (red A431scM3-Kat2, blue 425(scFv)SNAP-747, white background), b background signal, c pure A431scM3-Kat2 signal, d pure 425(scFv)SNAP-747 signal
Discussion
The use of in vivo molecular imaging, which allows for the real-time continuous monitoring of therapeutic effects over a prolonged period in a single animal, has considerably improved therapeutic evaluation. Due to its ease of use and versatile application to small animal in vivo experiments, especially optical imaging is increasingly used to address several questions in terms of molecule targeting or pharmacokinetics. Optical imaging techniques mainly comprise bioluminescence imaging and fluorescence imaging [1, 3]. For fluorescence optical imaging, a broad range of fluorophores has been developed today, which offers the possibility to track multiple probes simultaneously [4].
In this study, we report the feasibility of visualising treatment effects by Kat2 fluorescence-based molecular imaging. For a first proof-of-principle, we used cells with a high antigen expression as target, which were passaged through the mouse to select for cells that grew fast in vivo and have a high tumour take rate. By transfection of these A431scM3 tumour cells with DNA encoding for the highly stable and fluorescent protein Kat2, we established a method for easy in vivo monitoring based on the fluorescence signal, which is only dependent on the expression of Kat2 in the cells and not on external factors.
Transfected A431scM3 cells showed very strong fluorescence that even persisted for several weeks in culture. Continuous Kat2 expression does not alter the EGFR antigen expression nor apoptosis induction; both transfected and non-transfected cells showed similar sensitivity towards the 425(scFv)-ETA’ IT in vitro (Online Resource 3). Implantation of A431scM3-Kat2 cells induced tumour growth in 100% of BALB/c nu/nu mice. After determining the signal parameters, a spectral library was established that allowed for the unmixing of the images. Due to the strong fluorescence and the very low background signal from the emission of the Kat2 in the FR, this resulted in a clear delineation of the tumour area, which enables the exact measurement of the tumour and thereby increases statistical significance, compared to, for instance, measurements with a vernier calliper. This is also a clear difference to experiments, where GFP-transfected tumour cells are used, as in that case the entire body emits background fluorescence. This makes it more difficult to completely separate the specific signal from the background signal in, for example, feet or ears of the animals (results not shown).
Treatment efficacy was detectable by measuring the fluorescent area of the tumour. With this approach, no additional dye had to be injected, thus preventing the risk of potentially unbound fluorescent molecules circulating in the mice’s vasculature, which decreases the tumour-to-background ratio. This set-up was subsequently used to evaluate the effect of two different therapeutics against EGFR-expressing tumour cell lines in vivo.
Tumour-bearing mice were treated with 425(scFv)-ETA’ or Doxorubicin, a chemotherapeutic successfully used in the clinic for the treatment of cancer [38, 39], as positive control.
While treatment with 425(scFv)-ETA’ showed decelerated tumour growth compared to the PBS control group, treatment with Doxorubicin resulted in a shrinkage of the tumours.
The 425(scFv)-ETA’-treated mice, however, did not show any signs of the physical weakness or limitations observed in the Doxorubicin-treated animals. The arrest in tumour growth, combined with the lack of side effects, clearly demonstrates the high specificity of the 425(scFv)-ETA’ treatment. Changes in the treatment schedule would be required to optimise the therapeutic effect. Possibly, a combination therapy with a lower dose of Doxorubicin could combine both effects resulting in less side effects for the treatment of EGFR-positive tumours.
Parallel with the optical imaging, tumour size was also determined classically, using a vernier calliper, and we observed that this resulted in much wider variances and therefore the optical imaging data led to better statistics. This is in accordance with the data from Jensen et al. [40], who also found a systematic bias increasing with tumour size. Not only small tumours are difficult to grasp, but the measurement of large tumours can be difficult as they might grow into an irregular shape and extend deep into the tissue. Thus, with optical imaging, we were able to easily monitor treatment-induced changes in tumour growth, also of deeper localised parts of the tumour tissue. It is therefore preferred over vernier calliper measurements.
Next to the mere monitoring of tumour growth, assessment of additional parameters would be beneficial to specify treatment effects or detect changes in tumour physiology. Targeting a receptor on tumour cells may lead to downregulation of this receptor by the tumour cell as escape mechanism. We therefore looked at the expression of the EGFR using the same 425(scFv) as in the 425(scFv)-ETA’ fusion construct for treatment but fused to a SNAP-tag coupled to a fluorescent dye, considering a washout period of 11 days to prevent blocking of the binding by previously bound 425(scFv)-based IT. In particular, the 425(scFv)SNAP fusion protein has been shown to be useful by our laboratory in targeting EGFR-positive tumours in vivo [32].
In vivo optical imaging requires the use of fluorochromes that emit in the 650–900 nm range, and with normal optical imaging techniques using filters, it is difficult to discriminate between closely adjacent signals. Spectral analysis, as used by the Maestro, however, makes it possible to discriminate between two optical signals, not based on the colour, but on the exact shape of the spectrum [41]. The combination of the fluorochromes here used, Kat2 and BG-747, both emitting in the same range, could be used after determining the appropriate spectral library using the multifilter acquisition mode. This library could subsequently be used to detect a second, NIR fluorescent BG-747 dye next to the FR Kat2-labelled tumour, in the same mouse. The two signals could completely be separated even though they were in the same physical location, showing expression of the EGFR also after therapy. We could hereby affirm that the EGFR as a target on the A431scM3 cells is not downregulated during therapy and can be used as therapeutic target. Due to the renal clearance, the signal was also detected in the kidneys, which could be shown earlier [32].
Multicolour imaging is a method, which allows the simultaneous use of differentially labelled molecules. Due to the possibility to unmix different colours, there is a great flexibility in colour selection [42]; also, dyes with similar emission wavelengths can be distinguished. The use of differentially labelled molecules plays an important role, for example, in the diagnosis of different tumour species by their specific receptor expression pattern. After administration of a cocktail of different targeting moieties, subtraction of unspecific signals from the specific signal is possible and a huge advantage over radiolabelled imaging, where simultaneously acquired signals from different binders cannot be distinguished [43]. Activatable fluorescent probes have the advantage that they can be activated by different cancer-related factors, like endosome-specific enzymes or changes in pH upon internalisation. In this case, free dye can be distinguished from internalised probes. Thereby, these probes are helpful for localising specific tumour tissue, for example, metastases. Furthermore, when using activatable probes that are reversible, the discrimination between viable and dead tumour cells is possible. However, there are at present few activatable fluorescent probes suitable for in vivo imaging [44].
In our experiments, we employed solid tumours for a first, preclinical evaluation of new therapeutics enabling us to apply an easy method using “standard” commercially available dyes, which can rapidly be coupled to our targeting construct. This approach will be even more interesting for orthotopic and disseminated models, when the binding of fluorescently labelled molecules to tumours, which are not visible from outside, or metastases is examined.
In summary, we established an in vivo imaging method that allows for the monitoring of tumour growth and thereby therapeutic effects. We developed a robust and feasible method to monitor treatment for subcutaneous tumours by optical imaging using target cells, transfected with the strongly fluorescent, FR-emitting protein Kat2. Furthermore, we show that also additional parameters, such as target receptor expression, can be assessed even though the signal is in a similar spectral range and location, using the spectral analysis of the Maestro system.
Additionally, we could show that treatment with an EGFR-specific IT resulted in decelerated tumour growth. Improvement of 425(scFv)-ETA’ treatment schedule or a combination therapy with Doxorubicin might thus be more effective in tumour reduction as well as in overall health of the mice.
With molecular imaging, each animal represents its own statistical control throughout the whole experiment [1]. The results have less statistical variance, as the individual mice are monitored at different time points, as opposed to different animals for each time point. This also leads to a strong reduction in total animals required per experiment.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We thank Dirk Scheffler, Reinhard Rosinke and Peggy Jirak for excellent technical assistance. This publication is based upon work that was in part financed by the Bundesministerium für Bildung und Forschung (0315254A/B). The responsibility for the content of this publication lies with the author.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Theo Thepen and Stefan Barth contributed equally to the paper.
References
- 1.Contag PR. Whole-animal cellular and molecular imaging to accelerate drug development. Drug Discov Today. 2002;7:555–562. doi: 10.1016/S1359-6446(02)02268-7. [DOI] [PubMed] [Google Scholar]
- 2.Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 3.Alford R, Ogawa M, Choyke PL, Kobayashi H. Molecular probes for the in vivo imaging of cancer. Mol Biosyst. 2009;5:1279–1291. doi: 10.1039/b911307j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gong H, Kovar J, Little G, Chen H, Olive DM. In vivo imaging of xenograft tumors using an epidermal growth factor receptor-specific affibody molecule labeled with a near-infrared fluorophore. Neoplasia. 2010;12:139–149. doi: 10.1593/neo.91446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weissleder R. A clearer vision for in vivo imaging. Progress continues in the development of smaller, more penetrable probes for biological imaging. Nat Biotechnol. 2001;19:316–317. doi: 10.1038/86684. [DOI] [PubMed] [Google Scholar]
- 6.Shcherbo D, Merzlyak EM, Chepurnykh TV, Fradkov AF, Ermakova GV, Solovieva EA, et al. Bright far-red fluorescent protein for whole-body imaging. Nat Methods. 2007;4:741–746. doi: 10.1038/nmeth1083. [DOI] [PubMed] [Google Scholar]
- 7.Haigler H, Ash JF, Singer SJ, Cohen S. Visualization by fluorescence of the binding and internalization of epidermal growth factor in human carcinoma cells A-431. Proc Natl Acad Sci USA. 1978;75:3317–3321. doi: 10.1073/pnas.75.7.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tolmachev V, Rosik D, Wållberg H, Sjöberg A, Sandström M, Hansson M, et al. Imaging of EGFR expression in murine xenografts using site-specifically labelled anti-EGFR 111In-DOTA-Z EGFR:2377 Affibody molecule: aspect of the injected tracer amount. Eur J Nucl Med Mol Imag. 2010;37:613–622. doi: 10.1007/s00259-009-1283-x. [DOI] [PubMed] [Google Scholar]
- 9.Abdollahi A, Griggs DW, Zieher H, Roth A, Lipson KE, Saffrich R, et al. Inhibition of alpha(v)beta3 integrin survival signaling enhances antiangiogenic and antitumor effects of radiotherapy. Clin Cancer Res. 2005;11:6270–6279. doi: 10.1158/1078-0432.CCR-04-1223. [DOI] [PubMed] [Google Scholar]
- 10.Shao W, Zhao S, Liu Z, Zhang J, Ma S, Sato JD, et al. Inhibition of human tumor xenograft growth in nude mice by a conjugate of monoclonal antibody LA22 to epidermal growth factor receptor with anti-tumor antibiotics mitomycin C. Biochem Biophys Res Commun. 2006;349:816–824. doi: 10.1016/j.bbrc.2006.08.114. [DOI] [PubMed] [Google Scholar]
- 11.Cohen MH, Williams GA, Sridhara R, Chen G, Pazdur R. FDA drug approval summary: Gefitinib (ZD1839)(Iressa (R)) tablets. Oncologist. 2003;8:303–306. doi: 10.1634/theoncologist.8-4-303. [DOI] [PubMed] [Google Scholar]
- 12.Cohen MH, Johnson JR, Chen YF, Sridhara R, Pazdur R. FDA drug approval summary: erlotinib (Tarceva (R)) tablets. Oncologist. 2005;10:461–466. doi: 10.1634/theoncologist.10-7-461. [DOI] [PubMed] [Google Scholar]
- 13.Vincenzi B, Zoccoli A, Pantano F, Venditti O, Galluzzo S. Cetuximab: from bench to bedside. Curr Cancer Drug Targets. 2010;10:80–95. doi: 10.2174/156800910790980241. [DOI] [PubMed] [Google Scholar]
- 14.Giusti RM, Shastri KA, Cohen MH, Keegan P, Pazdur R. FDA drug approval summary: panitumumab (Vectibix) Oncologist. 2007;12:577–583. doi: 10.1634/theoncologist.12-5-577. [DOI] [PubMed] [Google Scholar]
- 15.Camp ER, Summy J, Bauer TW, Liu W, Gallick GE, Ellis LM. Molecular mechanisms of resistance to therapies targeting the epidermal growth factor receptor. Clin Cancer Res. 2005;11:397–405. [PubMed] [Google Scholar]
- 16.Bruell D, Stöcker M, Huhn M, Redding N, Küpper M, Schumacher P, et al. The recombinant anti-EGF receptor immunotoxin 425(scFv)-ETA’ suppresses growth of a highly metastatic pancreatic carcinoma cell line. Int J Oncol. 2003;23:1179–1186. doi: 10.3892/ijo.23.4.1179. [DOI] [PubMed] [Google Scholar]
- 17.Bruell D, Bruns CJ, Yezhelyev M, Huhn M, Müller J, Ischenko I, et al. Recombinant anti-EGFR immunotoxin 425 (scFv)-ETA’ demonstrates anti-tumor activity against disseminated human pancreatic cancer in nude mice. Int J Mol Med. 2005;15:305–313. [PubMed] [Google Scholar]
- 18.Barth S, Huhn M, Matthey B, Tawadros S, Schnell R, Schinköthe T, et al. Ki-4(scFv)-ETA’, a new recombinant anti-CD30 immunotoxin with highly specific cytotoxic activity against disseminated Hodgkin tumors in SCID mice. Blood. 2000;95:3909–3914. [PubMed] [Google Scholar]
- 19.Huhn M, Sasse S, Tur MK, Matthey B, Schinköthe T, Rybak SM, et al. Human angiogenin fused to human CD30 Ligand (Ang-CD30L) exhibits specific cytotoxicity against CD30-positive lymphoma. Cancer Res. 2001;61:8737–8742. [PubMed] [Google Scholar]
- 20.Tur M, Huhn M, Thepen T, Stöcker M, Krohn R, Vogel S, et al. Recombinant CD64-specific single chain immunotoxin exhibits specific cytotoxicity against acute myeloid leukemia cells. Cancer Res. 2003;63:8414–8419. [PubMed] [Google Scholar]
- 21.Stahnke B, Thepen T, Stöcker M, Rosinke R, Jost E, Fischer R, et al. Granzyme B-H22(scFv), a human immunotoxin targeting CD64 in acute myeloid leukemia of monocytic subtypes. Mol Cancer Ther. 2008;7:2924–2932. doi: 10.1158/1535-7163.MCT-08-0554. [DOI] [PubMed] [Google Scholar]
- 22.Tur MK, Neef I, Jost E, Galm O, Jäger G, Stöcker M, et al. Targeted restoration of down-regulated DAPK2 tumor suppressor activity induces apoptosis in Hodgkin lymphoma cells. J Immunother. 2009;32:431–441. doi: 10.1097/CJI.0b013e31819f1cb6. [DOI] [PubMed] [Google Scholar]
- 23.Pastan I, Hassan R, Fitzgerald DJ, Kreitman RJ. Immunotoxin therapy of cancer. Nat Rev Cancer. 2006;6:559–565. doi: 10.1038/nrc1891. [DOI] [PubMed] [Google Scholar]
- 24.Pastan I, Hassan R, FitzGerald DJ, Kreitman RJ. Immunotoxin treatment of cancer. Annu Rev Med. 2007;58:221–237. doi: 10.1146/annurev.med.58.070605.115320. [DOI] [PubMed] [Google Scholar]
- 25.Kreitman RJ, Wilson WH, Bergeron K, Raggio M, Stetler-Stevenson M, Fitzgerald DJ, et al. Efficacy of the anti-CD22 recombinant immunotoxin BL22 in chemotherapy-resistant hairy-cell leukemia. N Engl J Med. 2001;345:241–247. doi: 10.1056/NEJM200107263450402. [DOI] [PubMed] [Google Scholar]
- 26.Wayne AS, Kreitman RJ, Findley HW, Lew G, Delbrook C, Steinberg SM, et al. Anti-CD22 immunotoxin RFB4(dsFv)-PE38 (BL22) for CD22-positive hematologic malignancies of childhood: preclinical studies and phase I clinical trial. Clin Cancer Res. 2010;16:1894–1903. doi: 10.1158/1078-0432.CCR-09-2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 28.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li C, Iida M, Dunn EF, Ghia AJ, Wheeler DL. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene. 2009;28:3801–3813. doi: 10.1038/onc.2009.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arteaga CL. EGF receptor as a therapeutic target: patient selection and mechanisms of resistance to receptor-targeted drugs. J Clin Oncol. 2003;21:289s–291s. doi: 10.1200/JCO.2003.10.523. [DOI] [PubMed] [Google Scholar]
- 31.Kampmeier F, Ribbert M, Nachreiner T, Dembski S, Beaufils F, Brecht A, et al. Site-specific, covalent labeling of recombinant antibody fragments via fusion to an engineered version of 6-O-alkylguanine DNA alkyltransferase. Bioconjug Chem. 2009;20:1010–1015. doi: 10.1021/bc9000257. [DOI] [PubMed] [Google Scholar]
- 32.Kampmeier F, Niesen J, Koers A, Ribbert M, Brecht A, Fischer R, et al. Rapid optical imaging of EGF receptor expression with a single-chain antibody SNAP-tag fusion protein. Eur J Nucl Med Mol Imag. 2010;37:1926–1934. doi: 10.1007/s00259-010-1482-5. [DOI] [PubMed] [Google Scholar]
- 33.Matthey B, Engert A, Klimka A, Diehl V, Barth S. A new series of pET-derived vectors for high efficiency expression of Pseudomonas exotoxin-based fusion proteins. Gene. 1999;229:145–153. doi: 10.1016/S0378-1119(99)00038-4. [DOI] [PubMed] [Google Scholar]
- 34.Ribbert T, Thepen T, Tur MK, Fischer R, Huhn M, Barth S. Improved efficacy by increased valency, both in vitro, as well as in vivo in a chronic cutaneous inflammation model in hCD64 transgenic mice. Br J Dermatol. 2010;162:1–3. doi: 10.1111/j.1365-2133.2009.09612.x. [DOI] [PubMed] [Google Scholar]
- 35.Kapp U, Wolf J, Von Kalle C, Tawadros S, Röttgen A, Engert A, et al. Preliminary report: growth of Hodgkin’s lymphoma derived cells in immune compromised mice. Ann Oncol. 1992;3:S21–S23. doi: 10.1093/annonc/3.suppl_4.s21. [DOI] [PubMed] [Google Scholar]
- 36.Stocker M, Tur MK, Sasse S, Krüßmann A, Barth S, Engert A. Secretion of functional anti-CD30-angiogenin immunotoxins into the supernatant of transfected 293T-cells. Protein Expr Purif. 2003;28:211–219. doi: 10.1016/S1046-5928(02)00709-X. [DOI] [PubMed] [Google Scholar]
- 37.Johansen PB. Doxorubicin pharmacokinetics after intravenous and intraperitoneal administration in the nude mouse. Cancer Chemother Pharmacol. 1981;5:267–270. doi: 10.1007/BF00434396. [DOI] [PubMed] [Google Scholar]
- 38.Mizutani H, Tada-Oikawa S, Hiraku Y, Kojima M, Kawanishi S. Mechanism of apoptosis induced by doxorubicin through the generation of hydrogen peroxide. Life Sci. 2005;76:1439–1453. doi: 10.1016/j.lfs.2004.05.040. [DOI] [PubMed] [Google Scholar]
- 39.Ajaj KA, Graeser R, Fichtner I, Kratz F. In vitro and in vivo study of an albumin-binding prodrug of doxorubicin that is cleaved by cathepsin B. Cancer Chemother Pharmacol. 2009;64:413–418. doi: 10.1007/s00280-009-0942-8. [DOI] [PubMed] [Google Scholar]
- 40.Jensen MM, Jørgensen JT, Binderup T, Kjaer A. Tumor volume in subcutaneous mouse xenografts measured by microCT is more accurate and reproducible than determined by 18F-FDG-microPET or external caliper. BMC Med Imag. 2008;8:16. doi: 10.1186/1471-2342-8-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He X, Nie H, Wang K, Tan W, Wu X, Zhang P. In vivo study of biodistribution and urinary excretion of surface-modified silica nanoparticles. Anal Chem. 2008;80:9597–9603. doi: 10.1021/ac801882g. [DOI] [PubMed] [Google Scholar]
- 42.Kobayashi H, Longmire MR, Ogawa M, Choyke PL. Rational chemical design of the next generation of molecular imaging probes based on physics and biology: mixing modalities, colors and signals. Chem Soc Rev. 2011;40:4626–4648. doi: 10.1039/c1cs15077d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barrett T, Koyama Y, Hama Y, Ravizzini G, Shin IS, Jang BS, et al. In vivo diagnosis of epidermal growth factor receptor expression using molecular imaging with a cocktail of optically labeled monoclonal antibodies. Clin Cancer Res. 2007;13:6639–6648. doi: 10.1158/1078-0432.CCR-07-1119. [DOI] [PubMed] [Google Scholar]
- 44.Urano Y, Asanuma D, Hama Y, Koyama Y, Barrett T, Kamiya M, et al. Selective molecular imaging of viable cancer cells with pH-activatable fluorescence probes. Nat Med. 2009;15:104–109. doi: 10.1038/nm.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.