

Abstract
Background
Mutations to the co‐chaperone protein BAG3 (B‐cell lymphoma‐2–associated athanogene‐3) are a leading cause of dilated cardiomyopathy (DCM). These mutations often impact the C‐terminal BAG domain (residues 420–499), which regulates heat shock protein 70‐dependent protein turnover via autophagy. While mutations in other regions are less common, previous studies in patients with DCM found that co‐occurrence of 2 BAG3 variants (P63A, P380S) led to worse prognosis. However, the underlying mechanism for dysfunction is not fully understood.
Methods and Results
In this study, we used proteomics, Western blots, and myofilament functional assays on left ventricular tissue from patients with nonfailing, DCM, and DCM with BAG3 63/380 to determine how these mutations impact protein quality control and cardiomyocyte contractile function. We found dysregulated autophagy and increased protein ubiquitination in patients with BAG3 63/380 compared with nonfailing and DCM, suggesting impaired protein turnover. Expression and myofilament localization of BAG3‐binding proteins were also uniquely altered in the BAG3, 63/380 including abolished localization of the small heat shock protein CRYAB (alpha‐crystallin B chain) to the sarcomere. To determine whether these variants impacted sarcomere function, we used cardiomyocyte force‐calcium assays and found reduced maximal calcium‐activated force in DCM and BAG3 63/380. Interestingly, myofilament calcium sensitivity was increased in DCM but not with BAG3 63/380, which was not explained by differences in troponin I phosphorylation.
Conclusions
Together, our data support that the disease‐enhancing mechanism for BAG3 variants outside of the BAG domain is through disrupted protein turnover leading to compromised sarcomere function. These findings suggest a shared mechanism of disease among pathogenic BAG3 variants, regardless of location.
Keywords: autophagy, BAG3, dilated cardiomyopathy, sarcomere
Subject Categories: Contractile function, Myocardial Biology, Proteomics, Cardiomyopathy
Nonstandard Abbreviations and Acronyms
- BAG3
B‐cell lymphoma‐2–associated athanogene‐3
- DCM
dilated cardiomyopathy
- HSP
heat shock protein
- IPV
isoleucine‐proline‐valine
- MS
mass spectrometry
- Fmax
maximum calcium‐activated force
- SYNPO2
synaptopodin‐2
Research Perspective.
What Is New?
Impaired autophagic protein turnover and decreased cardiomyocyte contractile function with co‐occurrence of the BAG3 (B‐cell lymphoma‐2–associated athanogene‐3) P63A and P380S mutations suggests a shared mechanism of dysfunction between these and other previously characterized BAG3 variants, regardless of mutation location.
What Question Should Be Addressed Next?
To further support these findings, future steps should focus on obtaining a greater sample size of cardiomyocytes representing the BAG3 P63A and P380S mutations, as well as generation of patient‐derived and isogenic control human‐induced pluripotent stem cells for targeted in vitro analyses.
With increased power, we can address the remaining question of the extent to which these mutations specifically impair maximum cardiomyocyte contractile force in dilated cardiomyopathy and explore other underlying disease‐causing mechanisms within each specific BAG3 variant.
BAG3 (B‐cell lymphoma‐2‐associated athanogene‐3) is a widely expressed heat shock protein co‐chaperone with highest abundance in cardiac muscle, skeletal muscle, many types of cancer, and the central nervous system. 1 As an adaptor protein, numerous distinct functional roles have been identified for BAG3 in myocytes due to its unique combination of protein‐binding partners. These include maintenance of the sarcomeric cytoskeleton, 2 , 3 activation of macroautophagy, 4 , 5 inhibition of apoptosis, 6 and regulation of contractility through β‐adrenergic receptors and L‐type calcium channels. 7 , 8 Two recent comprehensive genetic studies independently concluded that BAG3 is 1 of 8 genes with a definitive causative link to genetic dilated cardiomyopathy (DCM). 9 , 10 Disease‐causing variants in BAG3 are also relatively abundant in the population and are estimated to account for at least 0.3% of all DCM cases. 10 Despite the definitive involvement of BAG3 mutations in causing cardiomyopathies, the molecular mechanisms that precipitate disease in patients with BAG3 variants remain poorly characterized.
Few studies have investigated the molecular mechanisms underlying disease with each of the dozens of known pathogenic and likely pathogenic BAG3 mutations. To date, only 3 different pathogenic variants have been studied in cell and animal models (P209L, E455K, R477H), and only 1 has been studied in human tissue (P209L). 11 , 12 , 13 , 14 These studies suggest a common mechanism of disease: compromised protein turnover leading to sarcomere structural and functional disorder. 2 , 11 , 12 , 13 Each of these 3 mutations lies in an important BAG3 functional domain: P209L occurs in the second isoleucine‐proline‐valine (IPV) motif of BAG3 that binds small heat shock proteins (HSPs), 15 and E455K/R477H occur in HSP70‐binding residues of the C‐terminal BAG domain (Figure 1A and 1B). 16 Notably, the majority of pathogenic BAG3 variants are truncating mutations where the BAG domain is absent at the protein level. 10 , 17 Thus, it is not surprising that compounds that disrupt the BAG3‐HSP70 complex formation also disrupt sarcomere integrity, prohibit autophagy, and are toxic for cardiomyocytes. 18 Disrupted HSP‐regulated protein quality control leading to compromised sarcomere integrity appears to be the shared mechanism of disease between these BAG3 variants. However, it is not clear how other disease‐associated mutations that do not impact the HSP‐binding domains contribute to disease.
Figure 1. Unbiased quantitative proteomics assessment suggests altered autophagy with BAG3 63/380 variants.
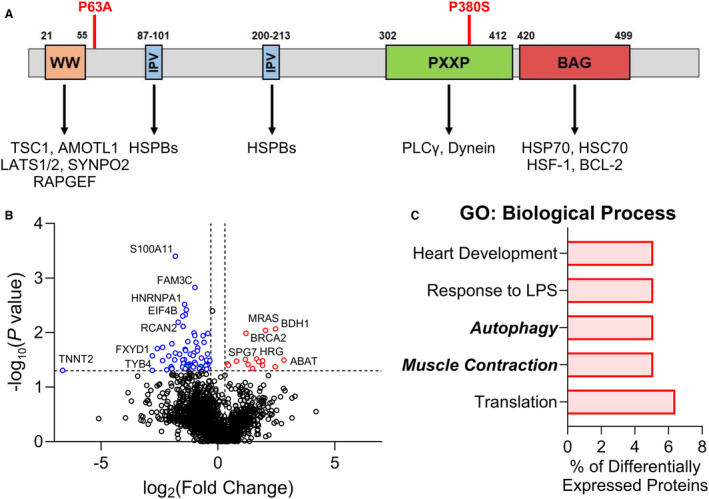
A, BAG3 protein sequence with domains and known BAG3‐binding partners; the BAG3 P63A mutation lies just outside of the WW domain, and the P380S mutation is located within the PXXP domain. B, Volcano plot of all proteins identified by bottom‐up mass spectrometry and analyzed by label‐free quantification; proteins with a +/− 0.3 log2 fold change in expression relative to nonfailing controls and a P value <0.05 were considered as having significantly different expression; upregulated proteins compared with nonfailing=red, downregulated proteins compared with nonfailing=blue; n=3 nonfailing, 2 BAG3 63/380; samples were not pooled for this analysis. C, DAVID GO analysis of the top 5 groups of differentially expressed proteins organized by biological process. AMOTL1 indicates angiomotin like 1; BCL‐2, B‐cell lymphoma 2; GO, gene ontology; HSC70, heat shock cognate protein 70; HSF‐1, heat shock transcription factor 1; HSPB/70, heat shock protein B/70; LATS1/2, large tumor suppressor kinase 1/2; LPS, lipopolysaccharide; PLCγ, phospholipase C γ; PXXP, proline‐rich; TSC1, tuberous sclerosis 1; and WW, tryptophan–tryptophan.
Two recently described BAG3 variants identified to co‐occur in patients with cardiomyopathy, while not causative of disease, were associated with worse clinical outcomes in DCM. 19 , 20 The variants, P63A and P380S, are located outside of the HSP‐binding domains of BAG3. Coexpression of these point mutations via adenovirus‐mediated gene transfer in AC16 cells resulted in increased apoptosis and reduced autophagy when cells were exposed to stress. 19 The finding of reduced autophagy suggests that, even with mutations outside of the IPV and BAG domains, a shared mechanism for dysfunction may exist. However, it is not known how autophagy is affected in patient cardiac tissue, if the BAG3 P63A/P380S variants impact the expression and stability of its HSP‐binding partners (as has been described for BAG domain mutations 13 ), or whether these mutations influence sarcomere function.
In the present study, we used left ventricle tissue from DCM patients with the BAG3 63/380 mutation (BAG3 63/380), patients with idiopathic DCM, and nonfailing rejected donor controls, to assess markers of autophagy and expression of key BAG3‐binding partners involved in mediating BAG3‐dependent autophagy. Additionally, we examined expression of these binding partners in the sarcomere‐enriched protein fraction and used biophysical assays to determine the impact of these mutations on function. We found that, as with mutations in the IPV and BAG domains, autophagy is dysregulated with the 63/380 variants compared with nonfailing and idiopathic DCM. Additionally, the expression and sarcomeric localization of BAG3‐binding partners were altered with these variants, and sarcomere function was reduced. Finally, local protein structure prediction suggested that the P63A mutation increases local beta sheet propensity, which may impact BAG3 N‐terminal protein‐binding partner affinity. Together, these data suggest a common molecular mechanism for pathogenic BAG3 variants through disrupted autophagic protein degradation, leading to compromised sarcomere contractile function.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethics Statement
All research involving human subjects was performed with strict adherence to the ethical guidelines established by the 1975 Declaration of Helsinki. These studies were performed after receiving written informed consent from patients or their legal representatives and with institutional review board approval of each of the participating centers: University of Pittsburgh, University of Colorado, and Loyola University Chicago.
Tissue Procurement
Human samples used were obtained from the Loyola Cardiovascular Research Institute, University of Pittsburgh, and University of Colorado biorepositories. The nonfailing control tissue was obtained from donor hearts rejected due to size, age, or other incompatibility. Idiopathic DCM and BAG3 63/380 mutation samples were obtained from hearts explanted during left ventricular assist device placement or transplant procedures.
Myofilament and Soluble Protein Enrichment
Frozen human left ventricular tissue was homogenized using a mechanical homogenizer in 1 mL of a standard rigor buffer (120 mmol/L CH3CO2K, 50 mmol/L HEPES, 4 mmol/L MgCl2, 5 mmol/L EGTA, pH 7.0) with 1% Triton X‐100 and protease and phosphatase inhibitors. The homogenate was left on ice for 20 minutes. After this incubation, the myofilament fraction was pelleted by centrifugation at 4 °C and 1800g for 2 minutes. The supernatant (containing the soluble fraction) was collected and stored at −80 °C. The pellet was washed with standard rigor buffer without Triton twice before being solubilized by resuspension in 8M urea/0.2% SDS. The resuspended pellet (containing the myofilament fraction) was sonicated and stored at −80 °C.
Western Blotting
Protein concentration was determined by bicinchoninic acid assay (Pierce). Samples were heated at 95 °C for 10 minutes in sample buffer (3:1 vol/vol SDS Tris‐Glycine Buffer [Life Technologies] and Bolt Reducing Agent [Fisher Scientific]) and loaded on 4% to 12% Tris‐Glycine gels (Invitrogen). After separation by electrophoresis, protein was transferred onto nitrocellulose membrane. Equal loading was assessed by Revert Total Protein Stain. After staining, the blot was blocked with Tris‐buffered saline and Intercept Blocking Buffer (LI‐COR Biosciences) (1:1 dilution) for 1 hour at room temperature. The blot was incubated overnight at 4 °C with agitation in blocking solution with primary antibody: BAG3 (Proteintech, 10 599‐1‐AP 1:4000), HSP70 (Proteintech, 10 995‐1‐AP, 1:4000), HSPB8 (Proteintech, 15 287‐1‐AP, 1:1000), Alpha B Crystallin (Proteintech, 15 808‐1‐AP, 1:2500), SYNPO2 (synaptopodin‐2; Proteintech, 25 453‐1‐AP 1:2500), Ubiquitin (Cytoskeleton Inc., AUB01, 1:500–1000), LC3 (CST, 2775S, 1:500–750), P62 (Proteintech, 18 420‐1‐AP, 1:2500), Troponin I (IPOC, MA‐1040, 1:2000), pS23/24 Troponin I (CST, 1:1000), α‐Actin (Sigma, A2172, 1:5000). The membrane was washed 3 times using Tris‐buffered saline with Tween before a final incubation of 1:1 blocking solution with tween and a near‐IR secondary antibody (LI‐COR Biosciences, 1:10 000) for 1 hour. The blot was washed 3 times using Tris‐buffered saline and imaged using an Azure c600. Densitometry analysis was performed using the LI‐COR Image Studio software with protein of interest signal normalized to total protein signal.
Myofilament Functional Assessment
Left ventricular tissue was mechanically homogenized in Isolation Buffer (8.91 mM KOH, 2 mmol/L EGTA, 7.11 mmol/L MgCl2, 10 mmol/L imidazole, 108.01 mmol/L KCl, 5.8 mmol/L adenosine triphosphate) containing 0.5% Triton X‐100 with 3 one‐second pulses at 7000 RPM and left on ice for 20 minutes to demembranate the myocytes. Myocytes were passed through a 70‐μm filter and pelleted by centrifugation at 120g and resuspended in Isolation Buffer without triton. Myocytes were attached by UV‐curing glue (NOA61, Thor Labs) to 2 pins, 1 attached to a calibrated force transducer (Kronex, AE801) and the other to a Piezo length controller. Calcium‐activated force measurements were performed at 6 different calcium concentrations (0.79, 0.97, 1.28, 2.41, 3.84, and 46.8 μmol/L), and the force produced was normalized to the myocyte cross‐sectional area, as previously described. 21 Data were fitted to a Hill Curve to determine the myocyte maximum force and EC50 (calcium concentration required to elicit half‐maximal force). All experiments were performed at room temperature and a sarcomere length of 2.1 μm as measured by fast Fourier transform.
Mass Spectrometry
The differentially expressed proteins from samples of nonfailing and failing human hearts were identified by using a modified in stage tip method and mass spectrometry analysis as described in detail previously. 22 , 23 Briefly, the hearts were homogenized in M‐PER buffer (Mammalian Protein Extraction Reagent, Thermo Fisher Scientific) containing protease and phosphatase inhibitors. Guanidinium hydrochloride buffer (6M) was then added, and the homogenate was heated for 5 minutes at 95 °C. Lys‐C was then added and the lysate was diluted 5‐fold with 25 mmol/L Tris, 10% acetonitrile buffer. The proteins were digested for 4 hours at 37 °C, and a second extensive digestion was achieved by overnight incubation at 37 °C with trypsin. The digestion was stopped by acidification with 3% trifluoroacetic acid followed by centrifugation at 2000 RCF for 5 minutes. The supernatant was collected and loaded onto an activated in‐house‐made cation stage tip, and the peptides were eluted into 6 fractions. The desalted tryptic peptide samples were then loaded onto an Acclaim PepMap 100 precolumn (C18, 75 μm×2 cm, Thermo Scientific) and separated by Easy‐Spray PepMap RSLC C18 column with an emitter (2 μm, 100Å, 50 μm×15 cm, Thermo Scientific) by an Easy nLC system with Easy Spray Source (Thermo Scientific). To elute the peptides, a mobile‐phase gradient was run using increasing concentration of acetonitrile. Peptides were loaded in buffer A (0.1% formic acid) and eluted with a nonlinear 145‐minute gradient as follows: 0% to 25% buffer B (0.1% formic acid, 85% acetonitrile) for 80 minutes, 25% to 40% B for 20 minutes, 40% to 60% B for 20 minutes and 60% to 100% B for 10 minutes. All percentages were volume/volume. The column was then washed with 100% buffer B for 5 minutes, 50% buffer B for 5 minutes, and reequilibrated with buffer A for 4 minutes. The flow rate was maintained at 300 nL/min.
Electron spray ionization was delivered at a spray voltage of −2000 V. Tandem mass spectrometry fragmentation was performed on the 5 most abundant ions in each spectrum using collision‐induced dissociation with dynamic exclusion (excluded for 10.0 s after 1 spectrum), with automatic switching between mass spectrometry (MS) and tandem MS modes. The complete system was entirely controlled by Xcalibur software (Thermo Fisher Scientific). Mass spectra processing was performed using Proteome Discoverer version 2.4 (Thermo Fisher Scientific). The generated deisotoped peak list was submitted to 3 separate databases: Mascot, MS Amanda 2.0, and Sequest HT. All 3 databases’ search parameters were set as follows: species, homo sapiens; enzyme, trypsin with maximal 2 missed cleavages; minimum peptide length, 4; maximum peptide length, 144; static modification, carbamidomethyl/+57.021 Da(C); 10 ppm precursor mass tolerance and 0.6 Da fragment mass tolerance for tandem MS fragment ions. Gene ontology analyses of the differentially expressed proteins were performed using the DAVID Bioinformatics platform. 24 , 25
Statistical Analysis
Data analysis and graphical representation were performed with Prism 9 (GraphPad software), unless otherwise noted. Comparisons of 2 groups were performed by 2‐tailed Student's t‐test. Comparisons of 3 groups were performed by 1‐way ANOVA. When a significant interaction was identified, multiple comparisons were performed using Tukey's post hoc test. For all Western blot data, due to small sample size, analysis was performed by 1‐way permutational AVOVA using RStudio (R Foundation for Statistical Computing). Experimental group blinding was not done.
Results
Dysregulated Autophagy With BAG3 63 /380 Mutations
Earlier work with the BAG3 P63A and P380S variants found that they were associated with a worse prognosis in DCM compared with DCM from nongenetic causes. 19 The mechanism of dysfunction remains poorly defined, but neither variant is in an HSP‐binding domain of BAG3 (Figure 1A). The mechanisms of dysfunction with the 63/380 variants are critical to understand, as there are multiple other disease‐associated BAG3 variants that are also located outside of these domains. Thus, with institutional review board approval, we procured left ventricular tissue from explanted hearts or pretransplant endomyocardial biopsies collected from patients with nonfailing (n=4; rejected donor hearts), idiopathic DCM (n=5; explanted hearts), and BAG3 63/380 (n=2; left ventricular assist device cores) from patients at the University of Pittsburgh, University of Colorado, and Loyola University Chicago (Table) and examined the molecular impact of these variants to determine broadly how BAG3 variants outside of the HSP‐binding domains may be pathogenic. To begin, we performed an unbiased quantitative proteomics screen of the left ventricular proteome in BAG3 63/380 compared with nonfailing. Label‐free quantification MS showed 78 proteins with significantly altered abundance in the BAG363/380 hearts compared with nonfailing (Figure 1B). Gene ontology biological process analysis revealed that these proteins were primarily involved in protein translation, muscle contraction, and autophagy (Figure 1C).
Table 1.
Patient Clinical Characteristics
| Characteristic | Nonfailing controls | Dilated cardiomyopathy | BAG3 (P63A/P380S) |
|---|---|---|---|
| Number | 4 | 5 | 2 |
| Female, % | 75 | 40 | 0.0 |
| Age, y, mean±SD | 59.3±8.5 | 49.0±18.0 | 35.0±1.4 |
| Ethnicity, % | |||
| Non‐Hispanic | 100 | 100 | 100 |
| Hispanic | 0 | 0 | 0 |
| LVEF, mean±SD | 66.3±5.3 | 22.4±22.5 | NA |
LVEF indicates left ventricular ejection fraction; and NA, not applicable.
Because BAG3 is an established regulator of protein degradation by autophagy, 26 we next examined whether autophagy was altered in the tissue of patients with BAG3 63/380. During maturation of autophagosomes, the early phagophore‐associated LC3‐I protein is converted to LC3‐II; therefore, relative expression of these 2 forms of LC3 can provide insight into autophagic activity. 27 By Western blot, we found that while LC3‐I protein expression was consistent among the 3 groups, LC3‐II levels were increased in the BAG3 63/380 samples, suggesting altered autophagy (Figure 2A through 2C). We next assessed the expression of another autophagy marker, P62, which is necessary for connecting ubiquitinated protein substrates to the autophagosome to facilitate their removal. We found that P62 levels increased by 37% and 23% in the patients with BAG3 63/380 compared with nonfailing DCM and DCM, respectively, although statistical significance was not reached (Figure 2D). Protein ubiquitination levels, however, were found to be significantly increased with BAG3 63/380 compared with both the nonfailing and DCM groups (Figure 2E).
Figure 2. Dysregulated autophagy in left ventricle of patients with BAG3 63/380.
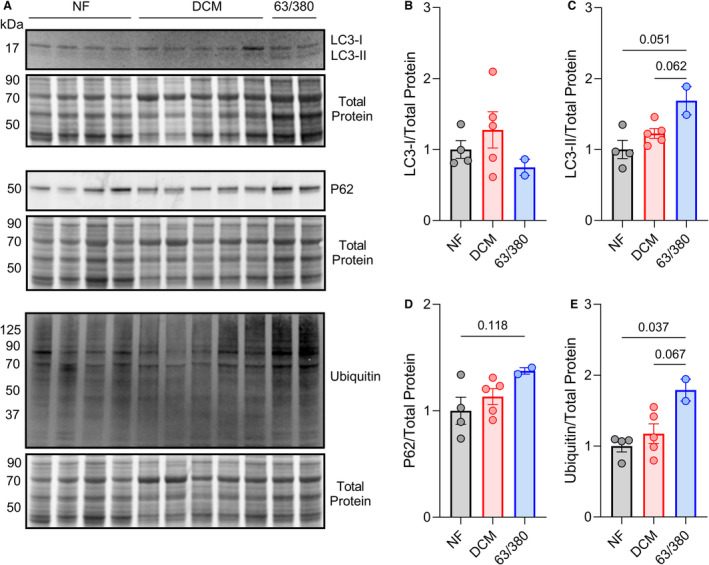
A, Western blots for LC3, P62, and ubiquitin in the triton‐soluble protein fraction of left ventricles from patients with NF, DCM, and BAG3 63/380 mutation, with corresponding Revert total protein stain. B through E, Quantitative densitometry analysis of the LC3‐I (B), LC3‐II (C), P62 (D), and ubiquitin (E) signals normalized to total protein; n=4 NF, 5 DCM, 2 BAG3 63/380; data are presented as the mean±standard error and were analyzed by 1‐way permutational ANOVA. DCM indicates dilated cardiomyopathy; NF, nonfailing; and LC3, microtubule‐associated proteins 1A/1B light chain 3B.
Altered Expression of BAG3‐Binding Proteins in the Left Ventricle of Patients With BAG3 63 /380
The involvement of BAG3 in autophagy stems from mediating the formation and regulation of a multichaperone protein assembly termed the chaperone‐assisted selective autophagy complex. Through its 2 IPV motifs, BAG3 associates with the adenosine triphosphate–independent small HSPs, which bind to denatured proteins and prevent their aggregation. The interaction of HSPBs with BAG3 allows for the connection of their cargo with HSP70—bound to BAG3's C‐terminal BAG domain—and subsequent ubiquitination and trafficking to an autophagosome. SYNPO2 assists with autophagosome maturation and engulfment of the chaperone‐assisted selective autophagy complex with its associated substrates (Figure 1A). 4 , 28 Due to our observation of disrupted autophagy in the BAG3 63/380 tissue, we hypothesized that this could result from a mutation‐dependent effect on the expression/localization of chaperone‐assisted selective autophagy complex proteins.
We used Western blot to assess expression of BAG3, HSP70, SYNPO2, HSPB8, and HSPB5 (also known as CRYAB) in the triton‐soluble/cytosolic protein fraction of the nonfailing, DCM, and BAG3 63/380 samples (Figure 3A and 3B). BAG3 protein expression was reduced in DCM as reported previously 19 , 29 , 30 ; however, no change was observed for the patients with BAG3 63/380. HSP70 and CRYAB protein expression were similar between nonfailing and BAG3 63/380. However, HSPB8 and SYNPO2 levels were each reduced by ≈50% in the BAG3 63/380 tissue (Figure 3C through 3G). A similar decrease was found in the DCM group for HSPB8, suggesting a mutation‐independent change in expression in DCM, which we have previously shown is due to the reduction in BAG3 levels. 2 , 18 However, for SYNPO2, the DCM group displayed a ≈60% increase, suggesting the observed decrease in SYNPO2 in patients with BAG3 63/380 was a mutation‐specific effect.
Figure 3. Assessment of BAG3‐binding partners in the soluble protein fraction.
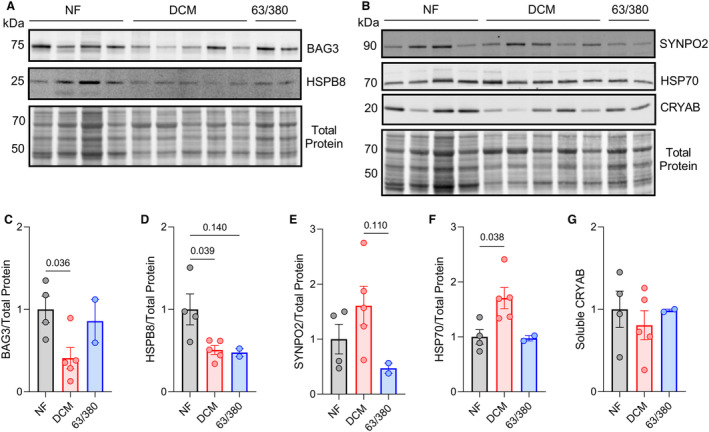
A, Western blot for BAG3 and HSPB8 in the triton‐soluble protein fraction with corresponding Revert total protein stain. B, Western blot for SYNPO2, HSP70, and CRYAB in the triton‐soluble protein fraction with corresponding Revert total protein stain. C through G, Quantitative densitometry analysis of BAG3 (C), HSPB8 (D), SYNPO2 (E), HSP70 (F), and CRYAB (G) signals normalized to total protein signal. For all, n=4 NF, 5 DCM, 2 BAG3 63/380; data are presented as the mean±standard error and were analyzed by 1‐way permutational ANOVA. CRYAB indicates crystallin alpha B; DCM, dilated cardiomyopathy; HSP70/B8, heat shock protein 70/β‐8; NF, nonfailing; and SYNPO2, synaptopodin 2.
Our previous work showed that BAG3 and its binding partners localized to the sarcomere under proteotoxic stress conditions and that this localization was partially BAG3 dependent. 2 Therefore, we next investigated expression of these proteins in the myofilament protein fraction to determine if BAG3 63/380 disrupts their sarcomeric localization. Using Western blot on myofilament‐enriched protein lysates, we found similar expression in the nonfailing, DCM, and BAG3 63/380 samples for BAG3, HSPB8, SYNPO2, and HSP70 (Figure 4A through 4F). However, while CRYAB levels were similar between nonfailing and DCM, myofilament CRYAB was completely lost in patients with BAG3 63/380 (Figure 4G).
Figure 4. Assessment of BAG3‐binding partners in the myofilament protein fraction.
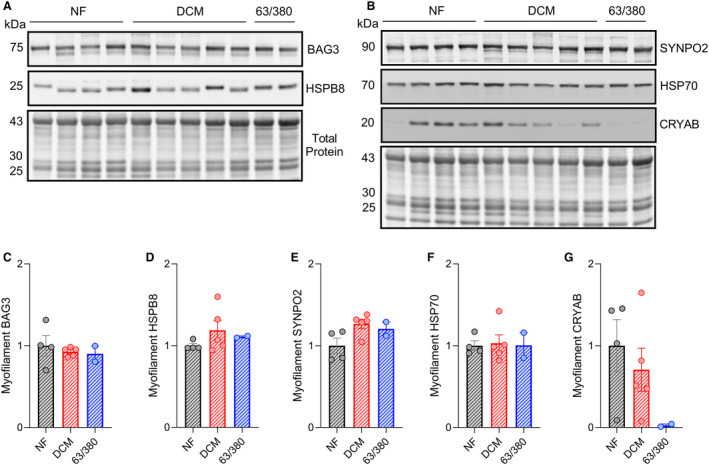
A, Western blot for BAG3 and HSPB8 in the myofilament protein fraction with corresponding Revert total protein stain. B, Western blot for SYNPO2, HSP70, and CRYAB in the myofilament protein fraction with corresponding Revert total protein stain. C through G, Quantitative densitometry analysis of BAG3 (C), HSPB8 (D), SYNPO2 (E), HSP70 (F), and CRYAB (G) signals normalized to total protein signal. For all, n=4 NF, 5 DCM, 2 BAG3 63/380; data are presented as the mean±standard error. CRYAB indicates crystallin alpha B; DCM, dilated cardiomyopathy; HSP70/B8, heat shock protein 70/β‐8; NF, nonfailing; and SYNPO2, synaptopodin 2.
BAG3 63 /380 Cardiomyocytes Display Sarcomere Contractile Dysfunction and Increased Myofilament Protein Ubiquitination
Because several previous studies identified BAG3 as an important factor for maintaining sarcomere structure and function, 2 , 3 , 12 , 18 , 31 , 32 we next assessed myofilament function in cardiomyocytes from the nonfailing, DCM, and BAG3 63/380 samples using the skinned myocyte force‐calcium assay. Maximum calcium‐activated force (Fmax) was significantly reduced in the DCM group compared with nonfailing (15.27±0.71 versus 24.1±1.62). Notably, Fmax was also considerably reduced in the BAG3 63/380 cardiomyocytes (11.41±1.44) and trended toward statistical significance when compared with the DCM group (P=0.0897) (Figure 5A and 5B). Cardiomyocyte calcium sensitivity was also increased in DCM compared with nonfailing, as indicated by reduced EC50. However, the EC50 in the BAG3 63/380 cardiomyocytes was not significantly different from nonfailing (Figure 5C).
Figure 5. Cardiomyocytes from BAG3 63/380 patients exhibit reduced sarcomere function and increased myofilament protein ubiquitination.
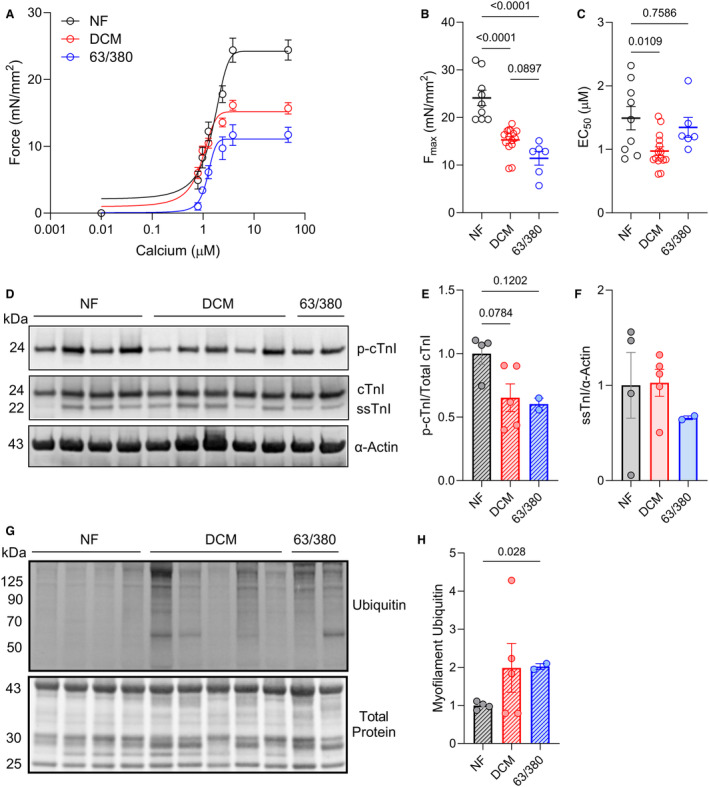
A. Skinned cardiomyocyte force‐calcium curves for the NF, DCM, and BAG3 63/380 samples. B and C, Summary data for cardiomyocyte maximal calcium‐activated force (Fmax) (B) and EC50 (calcium concentration required to elicit half‐maximal force) (C) from the 3 groups; n=9 NF cardiomyocytes from 3 samples (1 sample not used due to lack of remaining tissue), 15 DCM from 5 samples, 6 BAG3 63/380 from 2 samples. D, Western blot for phosphorylated (S23/S24) cardiac troponin I (cTnI), total TnI, and sarcomeric α‐Actin. E, Phosphorylated cTnI signal normalized to total cTnI signal. F, Slow skeletal TnI (ssTnI) signal normalized to α‐Actin. G. Western blot for ubiquitin in the myofilament protein fraction with corresponding total protein stain. H, Ubiquitin signal normalized to total protein. All data are presented as the mean±standard error and were analyzed by 1‐way ANOVA. (p‐)cTnI indicates (phosphorylated) cardiac troponin I; DCM, dilated cardiomyopathy; NF, nonfailing; and ssTnI, slow skeletal troponin I.
Altered myofilament calcium sensitivity in heart failure is often reflective of changes in the phosphorylation status of troponin I. Troponin I phosphorylation at S23/S24 by protein kinase A regulates myofilament calcium sensitivity. 33 In heart failure, troponin I phosphorylation decreases, thus increasing the calcium sensitivity. 34 , 35 To determine if the differences in calcium sensitivity observed in our study could be explained by troponin I phosphorylation, we used Western blot for total troponin I and S23/24 phosphorylated troponin I in myofilament protein lysates. Similar qualitative decreases in troponin I phosphorylation were observed in the DCM and BAG3 63/380 groups compared with nonfailing (Figure 5D and 5E). However, protein expression of the lowly expressed slow skeletal muscle troponin I isoform were qualitatively lower in the BAG3 63/380 samples (Figure 5F).
BAG3 had previously been shown to mediate removal of misfolded sarcomeric proteins by autophagy, and decreased expression/mutation to BAG3 was associated with increased myofilament protein ubiquitination. 2 We therefore next assessed total ubiquitin levels in the myofilament protein fraction by Western blot. BAG3 63/380 samples displayed a significant increase in myofilament ubiquitin compared with nonfailing (Figure 5G and 5H). This increase was consistent with that observed in the DCM group.
BAG3 P63A Variant Is Predicted to Alter Local Protein Secondary Structure
The P63A and P380S BAG3 variants are caused by missense mutations in exons 2 and 4 of BAG3's 4 coding exons (Figure 1A). Within the protein structure, P63 is slightly outside of a tryptophan–tryptophan domain (AA 21–55), which facilitates interactions with proline‐rich domains of many BAG3‐binding partners including SYNPO2 (Figure 1A). The P380S variant is located in BAG3's own proline‐rich domain, which interacts with the motor protein dynein to transport its misfolded protein substrates to degradation pathways (Figure 1A). 36 , 37
One previous study of a pathogenic BAG3 variant found that replacing proline with leucine at amino acid 209 was predicted to alter local protein secondary structure by increasing beta‐sheet propensity. 38 This resulted in increased exposure of hydrophobic residues on BAG3, making the protein more prone to aggregation. Because the mutations of interest in our 2 patients were also at proline residues, we hypothesized that a similar effect on secondary structure might be observed. We used PepFold 39 to predict whether replacing the prolines at 63 and 380 to alanine and serine, respectively, altered the predicted secondary structure. The P63A mutation greatly increased the predicted β‐sheet propensity and decreased disorder (Figure 6A). No overt differences were predicted by replacing the proline at residue 380 with serine (Figure 6B).
Figure 6. Secondary structure prediction suggests increased β‐sheet propensity and reduced disorder with P63A variants.
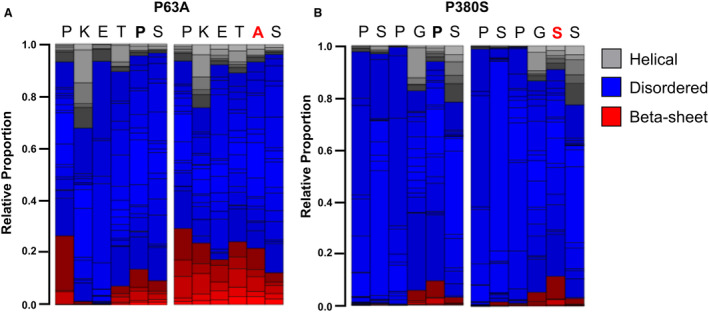
A, PepFold predicted secondary peptide structure with the P63A variant compared with the wild‐type BAG3 peptide sequence. B, Pepfold predicted secondary peptide structure with the P380S variant compared with the wild‐type BAG3 peptide sequence.
Discussion
Mutations in BAG3 are associated with the development of DCM. 9 , 10 The most common pathogenic mutations are truncating variants where the HSP70‐binding C‐terminal BAG domain is absent. 17 Several point mutations in the IPV and BAG domains are also linked to cardiomyopathies through altered HSP70 binding and small HSP (HSPB) stability. 13 , 38 The disease‐causing mechanism for these mutations is therefore suggested to be through disrupting BAG3‐dependent turnover of HSP70 clients through autophagy, which leads to sarcomere dysfunction. 2 , 12 , 13 Interestingly, 2 BAG3 variants outside of the IPV and BAG domains (P63A and P380S) were recently found to co‐occur in a subset of patients and contribute to worse prognosis in DCM through impairing BAG3's stress responsiveness. 19 , 20 We hypothesized that these mutations would also disrupt BAG3‐dependent protein turnover and sarcomere function, thus highlighting a common mechanism for pathogenesis. To test our hypothesis, we compared left ventricular tissue from 2 patients with DCM with the BAG3 63/380 variants to tissue from nonfailing donors and patients with idiopathic DCM.
Previous examination of these variants using adenoviral expression in AC16 cells identified reduced autophagy and increased cell death. 20 Herein, we found that autophagy was also dysregulated in the myocardium of patients harboring these mutations. While studying autophagy at a fixed time point in tissue does not allow for the assessment of autophagic flux, the collective increases in LC3‐II, P62, and ubiquitin protein expression suggests a buildup of late‐stage autophagosomes. This observation is further supported by our gene ontology analysis of the proteomics data, which identified autophagy as the third most altered protein group by biological process. Similar observations were made with human left ventricular tissue from a patient with the P209L BAG3 mutation. 11 A failure of autophagosome–lysosome fusion and subsequent impaired degradation of autophagosome contents in the absence of functional BAG3 is supported by previous work. 40
The association of BAG3 with HSP70 is required for maintaining stability of the small heat shock proteins HSPB6, HSPB8, and CRYAB. 13 , 18 , 31 In the present study, we found reduced expression of HSPB8 in patients with the 63/380 variants; however, the reduction matched that observed in the DCM group, suggesting a disease‐dependent decrease in expression. CRYAB protein expression was also not impacted in the BAG3 63/380 tissue, but myofilament localization was completely abrogated. We do not expect these variants to impact the association of BAG3 with CRYAB because they do not lie in the IPV motifs, and even mutations in a single IPV motif have previously been shown not to impact binding. 38 From this, it is unclear how CRYAB localization is so drastically impacted in these patients. One hypothesis is that aggregation of BAG3 in the cytosol might facilitate CRYAB coaggregation, a common feature of heart failure, 41 , 42 thus precluding its cytoskeletal localization. Such an effect would be detrimental to cardiomyocyte function, as CRYAB is a known cochaperone for both desmin and titin. 43 Our data also support that these variants may disrupt stability of the BAG3 adaptor protein SYNPO2, which we have previously shown to require BAG3. 18 We hypothesize that this is due to impaired binding resulting from the P63A mutation, which is adjacent to the SYNPO2 binding tryptophan–tryptophan domain.
PepFold analysis of peptide secondary structure aberrations with the BAG3 variants predicted that switching an alanine for proline at residue 63 would increase local β‐sheet propensity, which may impair the ability to bind SYNPO2. Earlier work with another BAG3 proline mutation (P209L) found the same prediction and went on to show experimentally that the increased β‐sheet content was associated with increased propensity for aggregation. 38 If the prediction in our study is true, an aggregation‐prone BAG3 would likely lead to a buildup of misfolded proteins. This hypothesis is supported by our data indicating that total protein ubiquitination, a readout of proteins marked for degradation but that fail to be removed, increases in the patients with BAG3 63/380 compared with both nonfailing and DCM. In addition, the fact that BAG3 protein levels with the variants do not decrease, as they do with DCM alone, which may be due to increased aggregation.
Previous work from our group and others has shown that BAG3‐dependent autophagy is fundamental for sarcomere structural and functional maintenance. 2 , 3 , 13 We previously proposed that retention of old/misfolded proteins in the sarcomere with reduced BAG3 or BAG3 mutations was an underlying mechanism of reduced myofilament Fmax in heart failure. 2 In the present study, we found reduced Fmax in both the DCM and BAG3 63/380 groups, with BAG3 63/380 displaying the lowest force. However, due to lack of power, it is not clear whether these variants have an additive detrimental effect on Fmax in DCM. One feature that appears different between the patients with DCM and patients with BAG3 63/380 is the myofilament calcium sensitivity. Calcium sensitivity is typically increased in heart failure due to reduced PKA‐mediated phosphorylation of troponin I.33 While this correlation was present in the DCM group, in the BAG3 63/380 group, sensitivity matched nonfailing controls despite a reduction in phospho–troponin I. One potential explanation might be reduced expression of the slow skeletal troponin I isoform, which has previously been associated with altered calcium sensitivity. 3 However, future studies will be required to validate this.
Study Limitations
The study is limited by the low sample number in the BAG3 63/380 group. However, we were unable to find additional samples, as genetic cardiomyopathy tissue from the same mutations is rare. The observations made with this sample size should inform future studies in cultured cardiomyocytes. Future work should focus on generating induced pluripotent stem cell–derived cardiomyocytes and isogenic controls from patients with the P63A and P380S variants. We were unable to directly measure autophagy flux in human tissue, as this would require pharmacological inhibition of autophagy before tissue collection. However, our previous in vitro studies of these variants in AC16 cells also identified disrupted autophagy. 19 Previous work indicated that both reduced BAG3 protein levels and BAG3 mutations alter sarcomere structure, which likely underlies the frequently observed reduced contractile function. 11 , 14 , 33 Unfortunately, due to limited tissue amount, ultrastructural assessment was not possible in this study. In addition, the quantitative proteomic assessment of protein expression does not include a DCM group, which makes it difficult to conclude how DCM with the BAG3 variants differs from idiopathic DCM at the global proteome level. Finally, the patients with DCM were not subjected to genetic testing to rule out other possible contributing variants, as there were no indications of familial disease.
Conclusions
Patients with DCM carrying the BAG3 P63A and P380S variants display worse clinical outcomes. 19 Using tissue from 2 patients with these variants, we identified dysregulated autophagy and reduced sarcomere function in the myocardium. Our data suggest that the disease‐enhancing mechanism for BAG3 variants outside of the BAG domain (as with previously characterized IPV and BAG domain variants) may also occur through disrupted autophagic protein turnover leading to compromised sarcomere function. These findings suggest a shared mechanism of disease among pathogenic BAG3 variants, regardless of location. Thus, there may be therapeutic potential for broad targeting of autophagy in patients with heart failure stemming from all disease‐associated BAG3 variants, not only those impacting the HSP‐binding domains.
Sources of Funding
This study was supported by funding from the National Institutes of Health (HL136737 to J.A.K., and HL91799; HL12309 to A.M.F.) and the American Heart Association (predoctoral fellowship 20PRE35170045 to T.G.M.).
Disclosures
A.M. Feldman is the founder of Renovacor, Inc, and holds equity in the company. He is the chief scientific advisor, is a member of the scientific advisory board, and serves as a consultant to the company for which he receives a stipend. No funds from Renovacor were used in this study. The remaining authors have no disclosures to report.
Acknowledgments
Drs Martin, McTiernan, Bristow, Feldman, and Kirk conceived the project idea. Drs Martin, Gerhard, Merali, and Dubey and H. Pak, C.Merali, and B. Lemster performed the experiments and analyzed the data. Drs Martin and Kirk designed the figures and wrote the manuscript with input from all authors.
This manuscript was sent to Rebecca D. Levit, MD, Associate Editor, for review by expert referees, editorial decision, and final disposition.
For Sources of Funding and Disclosures, see page 12.
REFERENCES
- 1. Kirk JA, Cheung JY, Feldman AM. Therapeutic targeting of BAG3: considering its complexity in cancer and heart disease. J Clin Invest. 2021;131:e149415. doi: 10.1172/JCI149415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Martin TG, Myers VD, Dubey P, Dubey S, Perez E, Moravec CS, Willis MS, Feldman AM, Kirk JA. Cardiomyocyte contractile impairment in heart failure results from reduced BAG3‐mediated sarcomeric protein turnover. Nat Commun. 2021;12:2942. doi: 10.1038/s41467-021-23272-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hishiya A, Kitazawa T, Takayama S. BAG3 And Hsc70 interact with Actin capping protein CapZ to maintain myofibrillar integrity under mechanical stress. Circ Res. 2010;107:1220–1231. doi: 10.1161/CIRCRESAHA.110.225649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ulbricht A, Gehlert S, Leciejewski B, Schiffer T, Bloch W, Höhfeld J. Induction and adaptation of chaperone‐assisted selective autophagy CASA in response to resistance exercise in human skeletal muscle. Autophagy. 2015;11:538–546. doi: 10.1080/15548627.2015.1017186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Arndt V, Dick N, Tawo R, Dreiseidler M, Wenzel D, Hesse M, Fürst DO, Saftig P, Saint R, Fleischmann BK, et al. Chaperone‐assisted selective autophagy is essential for muscle maintenance. Curr Biol. 2010;20:143–148. doi: 10.1016/j.cub.2009.11.022 [DOI] [PubMed] [Google Scholar]
- 6. Zhang J, He Z, Xiao W, Na Q, Wu T, Su K, Cui X. Overexpression of BAG3 attenuates hypoxia‐induced cardiomyocyte apoptosis by inducing autophagy. Cell Physiol Biochem. 2016;39:491–500. doi: 10.1159/000445641 [DOI] [PubMed] [Google Scholar]
- 7. Myers VD, Tomar D, Madesh M, Wang JF, Song J, Zhang XQ, Gupta MK, Tahrir FG, Gordon J, McClung JM, et al. Haplo‐insufficiency of Bcl2‐associated athanogene 3 in mice results in progressive left ventricular dysfunction, β‐adrenergic insensitivity, and increased apoptosis. J Cell Physiol. 2018;233:6319–6326. doi: 10.1002/jcp.26482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feldman AM, Gordon J, Wang JF, Song J, Zhang XQ, Myers VD, Tilley DG, Gao E, Hoffman NE, Tomar D, et al. BAG3 regulates contractility and Ca2+ homeostasis in adult mouse ventricular myocytes. J Mol Cell Cardiol. 2016;92:10–20. doi: 10.1016/j.yjmcc.2016.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jordan E, Peterson L, Ai T, Asatryan B, Bronicki L, Brown E, Celeghin R, Edwards M, Fan J, Ingles J, et al. Evidence‐based assessment of genes in dilated cardiomyopathy. Circulation. 2021;144:7–19. doi: 10.1161/CIRCULATIONAHA.120.053033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mazzarotto F, Tayal U, Buchan RJ, Midwinter W, Wilk A, Whiffin N, Govind R, Mazaika E, De Marvao A, Dawes TJW, et al. Reevaluating the genetic contribution of monogenic dilated cardiomyopathy. Circulation. 2020;141:387–398. doi: 10.1161/CIRCULATIONAHA.119.037661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schänzer A, Rupp S, Gräf S, Zengeler D, Jux C, Akintürk H, Gulatz L, Mazhari N, Acker T, Van Coster R, et al. Dysregulated autophagy in restrictive cardiomyopathy due to Pro209Leu mutation in BAG3. Mol Genet Metab. 2018;123:388–399. doi: 10.1016/j.ymgme.2018.01.001 [DOI] [PubMed] [Google Scholar]
- 12. McDermott‐Roe C, Lv W, Maximova T, Wada S, Bukowy J, Marquez M, Lai S, Shehu A, Benjamin I, Geurts A, et al. Investigation of a dilated cardiomyopathy–associated variant in BAG3 using genome‐edited iPSC‐derived cardiomyocytes. JCI Insight. 2019;4:e128799. doi: 10.1172/jci.insight.128799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fang X, Bogomolovas J, Wu T, Zhang W, Liu C, Veevers J, Stroud MJ, Zhang Z, Ma X, Mu Y, et al. Loss‐of‐function mutations in co‐chaperone BAG3 destabilize small HSPs and cause cardiomyopathy. J Clin Invest. 2017;127:3189–3200. doi: 10.1172/JCI94310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kimura K, Ooms A, Graf‐Riesen K, Kuppusamy M, Unger A, Schuld J, Daerr J, Lother A, Geisen C, Hein L, et al. Overexpression of human BAG3P209L in mice causes restrictive cardiomyopathy. Nat Commun. 2021;12:3575. doi: 10.1038/s41467-021-23858-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fuchs M, Poirier DJ, Seguin SJ, Lambert H, Carra S, Charette SJ, Landry J. Identification of the key structural motifs involved in HspB8/HspB6‐Bag3 interaction. Biochem J. 2010;425:245–255. doi: 10.1042/BJ20090907 [DOI] [PubMed] [Google Scholar]
- 16. Sondermann H, Scheufler C, Schneider C, Höhfeld J, Hartl FU, Moarefi I. Structure of a bag/Hsc70 complex: convergent functional evolution of Hsp70 nucleotide exchange factors. Science. 2001;291:1553–1557. doi: 10.1126/science.1057268 [DOI] [PubMed] [Google Scholar]
- 17. Domínguez F, Cuenca S, Bilińska Z, Toro R, Villard E, Barriales‐Villa R, Ochoa JP, Asselbergs F, Sammani A, Franaszczyk M, et al. Dilated cardiomyopathy due to BLC2‐associated Athanogene 3 (BAG3) mutations. J Am Coll Cardiol. 2018;72:2471–2481. doi: 10.1016/j.jacc.2018.08.2181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Martin TG, Delligatti CE, Muntu NA, Stachowski‐Doll MJ, Kirk JA. Pharmacological inhibition of BAG3‐HSP70 with the proposed cancer therapeutic JG‐98 is toxic for cardiomyocytes. J Cell Biochem. 2021;123:128–141. doi: 10.1002/jcb.30140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Myers VD, Gerhard GS, McNamara DM, Tomar D, Madesh M, Kaniper S, Ramsey FV, Fisher SG, Ingersoll RG, Kasch‐Semenza L, et al. Association of Variants in BAG3 with cardiomyopathy outcomes in African American individuals. JAMA Cardiol. 2018;3:929–938. doi: 10.1001/jamacardio.2018.2541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Feldman AM, Gordon J, Wang J, Song J, Zhang XQ, Myers VD, Tomar D, Gerhard GS, Khalili K, Cheung JY. Novel BAG3 variants in African American patients with cardiomyopathy: reduced β‐adrenergic responsiveness in excitation–contraction. J Card Fail. 2020;26:1075–1085. doi: 10.1016/j.cardfail.2020.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Papadaki M, Holewinski RJ, Previs SB, Martin TG, Stachowski MJ, Li A, Blair CA, Moravec CS, Van Eyk JE, Campbell KS, et al. Diabetes with heart failure increases methylglyoxal modifications in the sarcomere, which inhibit function. JCI Insight. 2018;3:e12164. doi: 10.1172/jci.insight.121264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barrero CA, Datta PK, Sen S, Deshmane S, Amini S, Khalili K, Merali S. HIV‐1 Vpr modulates macrophage metabolic pathways: a SILAC‐based quantitative analysis. PLoS One. 2013;8:e68376. doi: 10.1371/journal.pone.0068376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boden G, Homko C, Barrero CA, Stein TP, Chen X, Cheung P, Fecchio C, Koller S, Merali S. Excessive caloric intake acutely causes oxidative stress, GLUT4 carbonylation, and insulin resistance in healthy men. Sci Transl Med. 2015;7:304re7. doi: 10.1126/scitranslmed.aac4765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 25. Sherman BT, Hao M, Qiu J, Jiao X, Baseler MW, Lane HC, Imamichi T, Chang W. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022;50:W216–W221. doi: 10.1093/nar/gkac194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stürner E, Behl C. The role of the multifunctional bag3 protein in cellular protein quality control and in disease. Front Mol Neurosci. 2017;10:177. doi: 10.3389/fnmol.2017.00177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Loos B, Du Toit A, Hofmeyr JHS. Defining and measuring autophagosome flux–concept and reality. Autophagy. 2014;10:2087–2096. doi: 10.4161/15548627.2014.973338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Klimek C, Kathage B, Wördehoff J, Höhfeld J. BAG3‐mediated proteostasis at a glance. J Cell Sci. 2017;130:2781–2788. doi: 10.1242/jcs.203679 [DOI] [PubMed] [Google Scholar]
- 29. Feldman AM, Begay RL, Knezevic T, Myers VD, Slavov DB, Zhu W, Gowan K, Graw SL, Jones KL, Tilley DG, et al. Decreased levels of BAG3 in a family with a rare variant and in idiopathic dilated cardiomyopathy. J Cell Physiol. 2014;229:1697–1702. doi: 10.1002/jcp.24615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Martin TG, Tawfik S, Moravec CS, Pak TR, Kirk JA. BAG3 expression and sarcomere localization in the human heart are linked to HSF‐1 and are differentially affected by sex and disease. Am J Physiol Heart Circ Physiol. 2021;320:H2339–H2350. doi: 10.1152/ajpheart.00419.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Judge LM, Perez‐Bermejo JA, Truong A, Ribeiro AJ, Yoo JC, Jensen CL, Mandegar MA, Huebsch N, Kaake RM, So PL, et al. A BAG3 chaperone complex maintains cardiomyocyte function during proteotoxic stress. JCI Insight. 2017;2:e94623. doi: 10.1172/jci.insight.94623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ding Y, Dvornikov AV, Ma X, Zhang H, Wang Y, Lowerison M, Packard RR, Wang L, Chen J, Zhang Y, et al. Haploinsufficiency of mechanistic target of rapamycin ameliorates bag3 cardiomyopathy in adult zebrafish. Dis Model Mech. 2019;12:dmm040154. doi: 10.1242/dmm.040154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kentish JC, McCloskey DT, Layland J, Palmer S, Leiden JM, Martin AF, Solaro RJ. Phosphorylation of troponin I by protein kinase a accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle. Circ Res. 2001;88:1059–1065. doi: 10.1161/hh1001.091640 [DOI] [PubMed] [Google Scholar]
- 34. Bodor GS, Oakeley AE, Allen PD, Crimmins DL, Ladenson JH, Anderson PAW. Troponin I phosphorylation in the normal and failing adult human heart. Circulation. 1997;96:1495–1500. doi: 10.1161/01.CIR.96.5.1495 [DOI] [PubMed] [Google Scholar]
- 35. Wijnker PJM, Murphy AM, Stienen GJM, van der Velden J. Troponin I phosphorylation in human myocardium in health and disease. Neth Heart J. 2014;22:463–469. doi: 10.1007/s12471-014-0590-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gamerdinger M, Kaya AM, Wolfrum U, Clement AM, Behl C. BAG3 mediates chaperone‐based aggresome‐targeting and selective autophagy of misfolded proteins. EMBO Rep. 2011;12:149–156. doi: 10.1038/embor.2010.203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu Z, Graham K, Foote M, Liang F, Rizkallah R, Hurt M, Wang Y, Wu Y, Zhou Y. 14‐3‐3 protein targets misfolded chaperone‐associated proteins to aggresomes. J Cell Sci. 2013;126:4173–4186. doi: 10.1242/jcs.126102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Meister‐Broekema M, Freilich R, Jagadeesan C, Rauch JN, Bengoechea R, Motley WW, Kuiper EFE, Minoia M, Furtado GV, van Waarde MAWH, et al. Myopathy associated BAG3 mutations lead to protein aggregation by stalling Hsp70 networks. Nat Commun. 2018;9:5342. doi: 10.1038/s41467-018-07718-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maupetit J, Derreumaux P, Tuffery P. PEP‐FOLD: an online resource for de novo peptide structure prediction. Nucleic Acids Res. 2009;37:W498–W503. doi: 10.1093/nar/gkp323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Su F, Myers VD, Knezevic T, Wang JF, Gao E, Madesh M, Tahrir FG, Gupta MK, Gordon J, Rabinowitz J, et al. Bcl‐2‐associated athanogene 3 protects the heart from ischemia/reperfusion injury. J Clin Invest. 2016;1:e90931. doi: 10.1172/jci.insight.90931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang X, Klevitsky R, Huang W, Glasford J, Li F, Robbins J. αB‐crystallin modulates protein aggregation of abnormal Desmin. Circ Res. 2003;93:998–1005. doi: 10.1161/01.RES.0000102401.77712.ED [DOI] [PubMed] [Google Scholar]
- 42. Zhang H, Rajasekaran NS, Orosz A, Xiao X, Rechsteiner M, Benjamin IJ. Selective degradation of aggregate‐prone CryAB mutants by HSPB1 is mediated by ubiquitin‐proteasome pathways. J Mol Cell Cardiol. 2010;49:918–930. doi: 10.1016/j.yjmcc.2010.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Martin TG, Kirk JA. Under construction: the dynamic assembly, maintenance, and degradation of the cardiac sarcomere. J Mol Cell Cardiol. 2020;148:89–102. doi: 10.1016/j.yjmcc.2020.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]