

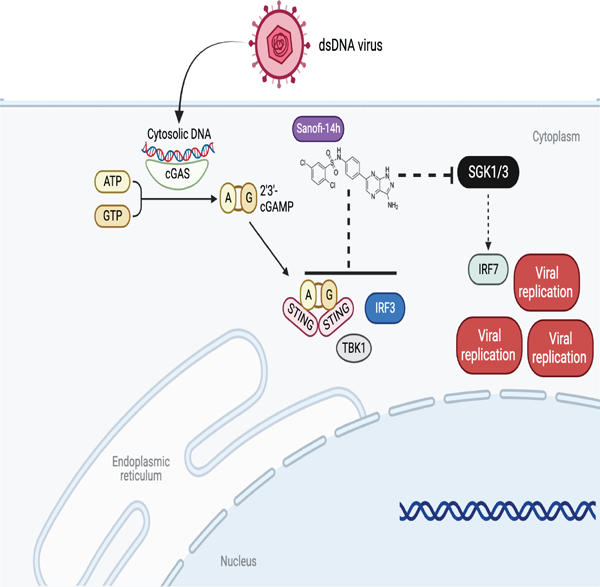
SUMMARY
Type 1 IFN expression is critical in the innate immune response, but aberrant expression is associated with autoimmunity and cancer. Here we identify N-[4-(1H46 pyrazolo[3,4-b] pyrazin-6-yl)-phenyl]-sulfonamide (Sanofi-14h), a compound with preference for inhibition of the AGC family kinase SGK3, as an inhibitor of Ifnb1 gene expression in response to STING stimulation of macrophages. Sanofi-14h abrogated SGK activity and also impaired activation of the critical TBK1/IRF3 pathway downstream of STING activation, blocking interaction of STING with TBK1. Deletion of SGK1/3 in a macrophage cell line did not block TBK1/IRF3 activation but decreased expression of transcription factors, such as IRF7 and STAT1, required for the innate immune response. Other AGC kinase inhibitors blocked TBK1 and IRF3 activation suggesting common action on a critical regulatory node in the STING pathway. These studies reveal both SGK-dependent and SGK-independent mechanisms in the innate immune response and indicate an approach to block aberrant Ifnb1 expression.
Graphical Abstract
eTOC
Cells respond to pathogens via protective signaling pathways. Castillo Cabrera et al demonstrate that SGK1 and SGK3 (serum-and glucocorticoid-regulated kinases 1 and 3) regulate the STING-driven IFNβ response. SGK inhibitors block STING activation, while SGK1/3 deletion leads to loss of important transcription factors IRF7 and STAT1.
INTRODUCTION
Type I interferons (IFNs) are cytokines that play critical roles in the innate immune response by inhibiting viral replication, promoting apoptosis of infected cells, recruiting and activating antigen presenting cells, and eliciting the adaptive immune response through activation of T and B cells1. However, dysregulated expression of interferons promotes autoimmune diseases2, inflammation3, and immunosuppressive stages of infection4. Underscoring the importance of interferons in the immune response, many pathogens express proteins to block interferon production leading to enhanced pathogenicity5. Understanding the regulation of IFN expression is critical for elucidating driving events and sequelae in infectious and chronic diseases.
Expression of type I IFNs (α/β) is induced by stimulation of pathogen receptors including Toll-like receptors (TLR) and nucleic acid receptors such as cGAS1,6. In the case of cGAS, binding to cytoplasmic DNA leads to the enzymatic generation of the cyclic dinucleotide 2’,3’ cGAMP7. Binding of bacteria- or cGAS-derived cyclic dinucleotides/cGAMP by STING leads to activation of the kinase TANK-binding kinase 1 (TBK1) and to the phosphorylation of transcription factors interferon regulatory factor (IRF) 3 and 7, which directly promote transcription of type I IFN and other immune response genes8–11. Secreted type I IFN signals through its receptor (IFNAR) to repress pathogen replication in infected and bystander cells and to activate transcription of a second wave of interferon-stimulated genes (ISGs). ISG expression downstream of IFN requires IFNAR-mediated activation of the JAK-STAT pathway with activating phosphorylation of the STAT1/STAT2/IRF9 (ISGF3) complex as well as STAT312,13.
Serum- and glucocorticoid-regulated kinases (SGK1-3) are members of the AGC family of serine/threonine kinases with homology to the AKT subfamily14. SGK3 is widely expressed, whereas SGK2 expression is restricted to certain organs, and SGK1 is expressed in most tissues but with dynamic regulation15. SGK3 uniquely contains a phosphoinositide-binding PX domain at the N-terminus which interacts with phosphatidylinositol-3-phosphate to control SGK3 localization at endosomes16. The activation of all three kinases is similar to that for AKT and is controlled by phosphorylation by PI3K/PDK1 and mTORC217. Substrates for SGK family substrates are members of the N-myc downstream regulated (NDRG) family in addition to several Na+/K+ ion channels 18–22. All three SGKs are linked to cancer by promoting drug resistance and by controlling pathways important for cancer progression15. Bago et al demonstrated that PI3K/Akt inhibition in breast cancer cells leads to the upregulation of SGK3 to drive mTORC1 activity and tumor growth23, and Ranzuglia et al proposed SGK2 as regulator of autophagy that modulates drug resistance in cancer cells24. SGK1 is the most characterized of the three in cancer with roles in driving tumorigenesis, proliferation, metastasis and drug resistance25.
SGK inhibition has been proposed as a therapeutic approach in cancer and SGK inhibitors have been developed and tested with this objective26. GSK650394, SI113 and EMD638683 are the most widely used inhibitors of SGK in cancer studies and have been shown to block cell proliferation and to decrease tumor growth alone or in combination with other inhibitors in animal models 27–29. A family of N-[4-(1H-pyrazolo[3,4-b] pyrazin-6-yl)-phenyl]-sulfonamides have been described to have more potent and selective inhibitory effects on SGK than previous inhibitors30. Furthermore, one derivative of these compounds, named Sanofi-14h, has been used in breast cancer models in vivo and in vitro and in combination with AKT inhibitors 23. Another derivative (Sanofi-17a) has been studied in a mouse explant model of osteoarthritis31. Notably, Sanofi-14h shows higher activity towards SGK3 as compared with SGK1, but also has significant activity against kinases such as S6K1 (P70S6K1) and MLK323. Sanofi-17a preferentially inhibits SGK1 in vitro and has decreased activity against other AGC kinases including SGK3 and S6K31.
Related to innate immunity, SGK1 negatively regulates the TLR4/NF-κB response in mice32, promotes macrophage polarization to an M1 phenotype33, and controls macrophage migration in vascular inflammation34. SGK3 was shown to regulate Ca2+-mediated dendritic migration in response to lipopolysaccharides (LPS) in mice35. SGK1 has been also reported to phosphorylate IKKα in Neuro2A cells and to phosphorylate IKKβ in breast cancer cells to increase NF-κB activity36,37.
We show here that Sanofi-14h blocks the phosphorylation of TBK1 and IRF3, along with the dimerization and nuclear translocation of IRF3 following treatment with a STING agonist, thus blocking IFNβ gene induction. Deletion of SGK1 and SGK3 in macrophages phenocopies the transcriptional defect of Sanofi-14h-treated cells. However, the SGK1/3-deleted cells retain TBK1 and IRF3 phosphorylation downstream of STING, and Sanofi-14h is able to block TBK1/IRF phosphorylation in SGK1/3-null cells, indicating both SGK-dependent and -independent effects in the response of macrophages to STING activation. The effect of loss of SGK1 and SGK3 on IFNβ gene expression is through loss of basal expression of several key IRF and STAT family members. Interestingly, Sanofi-14h blocks the recruitment of TBK1 and IRF3 into the STING complex. Analysis of other AGC kinase inhibitors across different drug classes revealed that many suppress IRF3 activation albeit with different efficacy. These studies reveal unexpected effects of AGC kinase inhibitors on downstream STING-controlled signaling and that SGK1/3 control expression of critical transcription factors involved in the innate immune response.
RESULTS
Sanofi-14h blocks IFNβ gene induction induced by STING ligands and HSV-1
N-[4-(1H-pyrazolo[3,4-b] pyrazin-6-yl)-phenyl]-sulfonamide (Sanofi-14h) suppressed IFNβ mRNA induction in murine RAW264.7 macrophages treated with the STING agonist DMXAA38 and in THP-1 and human monocyte-derived macrophages (hMDMs) treated with the STING agonist DiABZI39. Sanofi-14h blocked IFNβ mRNA induction in a dose-dependent manner with 50% inhibition measured between 500nM and 1μM in RAW264.7 cells while 10μM had a similar effect on human THP-1 cells and hMDMs [Figure 1A]. Additionally, we analyzed the effect of Sanofi-14h on replication of the dsDNA herpes simplex virus (HSV-1). We found that Sanofi-14h increased replication of HSV-1 in RAW264.7 macrophages as measured through plaque-forming assays [Figure 1B]. Consistently, Sanofi-14h treatment blocked induction of IFNβ mRNA in HSV-1 infected cells [Figure 1B].
Figure 1: SGK inhibitor Sanofi-14h impairs IFN expression and STING/TBK1/IRF3 signaling in macrophages.

A) RAW264.7 cells were pre-treated with Sanofi-14h at the indicated concentrations, whereas THP-1 and hMDM were pre-treated with 10 μM, for 2 hours and then treated with STING ligands DMXAA (10μg/mL, RAW cells) or diABZI (1μM) for an additional 2 hours. RT-qPCR of IFNβ mRNA expression was normalized to GAPDH for RAW264.7 and hMDM or 18S for THP1. B) RAW264.7 cells were pre-treated with Sanofi-14h (5 μM) for 24 h and then infected with HSV-1 KOS (MOI 0.1). Images shows representative plaque assays and graphs depict the HSV-1 PFU (left) and IFNβ mRNA (right). C) RAW264.7 macrophages (left panel) were pretreated with Sanofi-14h at the indicated doses for 2h prior to stimulation with DMXAA (10μg/mL) for 1h and 2h and lysates were immunoblotted. BMDM (right panel) were pretreated with Sanofi-14h (10 μM) for 2h and prior to DMXAA (10μg/mL) for 30 min, 1h and 2h. Graph shows IFNβ mRNA expression normalized to GAPDH in BMDM. pNDRG1(T346) was a used a control to measure Sanofi-14h activity in both cell lines. D) Dimerization (left panel) and nuclear localization (right panel) of IRF3 after pretreatment with Sanofi-14h at different doses for 2h and DMXAA for 1h. Lamin A/C and β-tubulin indicate nuclear and cytoplasmic fractions respectively. E) Effects of Sanofi-14h are upstream of IFNβ signaling. RAW cells were pretreated with Sanofi-14h (10 μM) or vehicle for 2h prior to treatment with DMXAA for 2h. A subset was treated with exogenous IFNβ (10 ng/mL) for the last 15 minutes. Immunoblots depict total and phosphorylated STAT3 and TBK1. BMDM: Bone Marrow Derived Macrophages, PFU: Plaque-Forming Unite, HSV: Herpes Simplex Virus. Data are representative of three different experiments (* p<0.05).
Sanofi-14h blocks the STING-induced TBK1-IRF3 signaling cascade
STING activation in response to cytoplasmic DNA/viral infection leads to the phosphorylation and activation of the kinase TBK1 which phosphorylates the transcription factor IRF3, leading to its subsequent dimerization and nuclear translocation to promote transcription of the IFNβ gene 13,40. To determine whether Sanofi-14h affects this signaling pathway, we treated RAW264.7 cells with different doses of the inhibitor prior to stimulation with DMXAA. Sanofi-14h pre-treatment led to a dose-dependent inhibition of activating phosphorylation of TBK1, IRF3, and STING [Figure 1C]. STING phosphorylation is characterized by the reduction in its electrophoretic mobility. Consistent with the ability of Sanofi-14h to block induction of IFNβ, treatment with this inhibitor led to a marked decrease in STAT3 phosphorylation [Figure 1C], which occurs downstream of IFN receptor signaling. We observed that submicromolar doses of Sanofi-14h efficiently blocked phosphorylation of the canonical SGK target NDRG1, while higher doses of Sanofi-14h (>5 μM) potently impaired TBK1 and IRF3 phosphorylation [Figure 1C]. Both SGK1 protein [Figure 1C] and mRNA levels [Suppl. Figure 1A] are induced by DMXAA treatment and this increase is blocked in a dose-dependent manner by Sanofi-14h. These results indicate that the SGK1 gene is activated in the STING-regulated pathway.
Additionally, we assayed STING-induced signaling and IFNβ expression in primary mouse bone marrow-derived macrophages (BMDMs) treated with Sanofi-14h. Sanofi-14h efficiently blocked phosphorylation of NDRG1 in BMDMs [Figure 1C]. In addition, Sanofi-14h blocked induction of IFNβ mRNA and blocked activating phosphorylation of STAT3, TBK1, and IRF3 in primary macrophages [Figure 1C].
After phosphorylation, IRF3 dimerizes and translocates to the nucleus to engage target genes for activation40. To further address downstream effects of Sanofi-14h on STING-induced signaling, we measured IRF3 dimerization and nuclear translocation in the presence of the drug following DMXAA stimulation of RAW264.7 cells. We observed that Sanofi-14h inhibited both dimerization and nuclear accumulation of IRF3 in a dose-dependent manner [Figure 1D]. Because STAT3 Y705 phosphorylation was blocked by Sanofi-14h, we asked whether this effect was dependent on type 1 IFN expression or via an intrinsic interaction between STAT3 and either SGK1/3 or TBK1. In this regard, TBK1 has been reported to phosphorylate STAT3 at S754 and to modulate its function in the STING pathway in macrophages41. We co-treated RAW264.7 cells with exogenous IFNβ, DMXAA and Sanofi-14h. We observed that treatment with exogenous IFNβ rescues the STAT3 phosphorylation inhibition in the presence of Sanofi-14h but does not rescue TBK1 phosphorylation inhibition promoted by Sanofi-14h treatment [Figure 1E]. This result indicates that the loss of p-STAT3 induced by Sanofi-14h is dependent on loss of IFNβ production.
The effects of Sanofi-14h on STING/TBK1/IRF3 activation are largely independent of SGK1 and SGK3
In RAW264.7 cells, SGK3 is expressed at relatively high levels and SGK1 is expressed at lower levels but is inducible upon STING stimulation [see Figure 2D, and suppl. Fig 1A]. SGK2 is not detected in these cells and is not upregulated with SGK1 or SGK3 deletion [Suppl. Figure 1B]. To determine whether SGK1 and/or 3 are involved in control of IFNβ production and in STING-induced signaling, we generated single and double knockouts (KOs) of SGK1 and SGK3 in RAW264.7 cells [Figure 2D]. We asked whether SGK1/3 KO cells phenocopy cells treated with Sanofi-14h prior to STING agonist treatment. First, we showed that loss of either SGK1 or SGK3 reduced IFNβ mRNA induction following DMXAA treatment [Figure 2A]. Next, we analyzed the effect of HSV1 infection on wild-type and SGK1/3 KO cells. RAW264.7 macrophages with single or double deletion of SGK1 or SGK3 showed an increased cytopathic effect after HSV-1 infection [Figure 2B]. SGK1/3 KO macrophages showed an increase in viral production and decreased IFNβ transcription. The effect of SGK3 deletion on IFNβ mRNA was more pronounced than SGK1 deletion [Figure 2B]. We generated primary bone marrow-derived macrophages (BMDMs) from SGK3 null mice 42 and infected them with HSV in vitro. SGK3 null BMDMs showed increased viral replication as compared with wildtype cells [Figure 2C]. Thus, deletion of SGK1/3 in macrophages impairs in vitro antiviral defense similar to treatment with Sanofi-14h.
Figure 2: Deletion of SGK1 and SGK3 impairs IFNβ production and STAT3 phosphorylation without affecting STING/TBK1/IRF3 phosphorylation.

A) IFNβ mRNA expression in single and double (dKO) RAW264.7 SGK1/3 KO cells with (top segment) and without (bottom segment) DMXAA treatment for 2h. B) RAW264.7 SGK KO cells infected with HSV-1 KOS. Images show plaque assay for single and dKO cells. Graphs indicated HSV-1 PFU and IFNβ mRNA production. C) HSV-1 PFU in SGK3 KO BMDMs. D) Cells were treated with DMXAA for 1h and 2h and lysates were immunoblotted for indicated proteins. SGK1 and SGK3 protein levels confirm knockout and pNDRG1 indicates SGK activity. E) Dimerization (left panel) and nuclear localization (right panel) of IRF3 after DMXAA treatment for 1h in SGK1 KO, SGK3 KO, and SGK1/3 dKO cells. Lamin A/C and β-tubulin indicate nuclear and cytoplasmic fractions, respectively. F) Immunoblot representing the treatment of SGK1/3 dKO cells with DMXAA (2 h total) with and without exogenous IFNβ (15 min). G) WT and SGK1/3 dKO cells were treated with Sanofi-14h for 2h prior to DMXAA stimulation for 2h. Lysates were immunoblotted for indicated proteins (left panel) and IFNβ mRNA was assayed by RT-PCR (right graph). Data are representative of three different experiments (* p<0.05).
We next examined the signaling consequences of SGK deletion in response to DMXAA. We found that SGK3 KO cells demonstrate loss of basal NDRG1 phosphorylation, indicating that in RAW264.7 macrophages SGK3 is the primary NDRG1 kinase [Figure 2D]. SGK1 KO, SGK3 KO, and dual SGK1/3 KO cells exhibit decreased STAT3 Y705 phosphorylation in response to DMXAA treatment, in agreement with reduced IFNβ expression. Surprisingly, SGK1 KO, SGK3 KO, and double KO (dKO) cells have largely intact DMXAA-induced phosphorylation of TBK1 and STING [Figure 2D]. Consistent with this finding, SGK1/3 KO cells have largely unimpaired IRF3 phosphorylation after STING stimulation [Figure 2D]. DMXAA-stimulated dimerization of IRF3 is likewise unaffected by single or dual deletion of SGK1 or SGK3 [Figure 2E]. Fractionation of the nuclear and cytoplasmic compartments showed reduced nuclear accumulation of phosphorylated STAT3 but not IRF3 in SGK3 KO and SGK1/3 dKO cells [Figure 2E]. To test if SGK1/3 KO affects STAT3 through IFN expression or another mechanism, we treated WT and SGK1/3 dKO cells with recombinant IFNβ and found that IFNβ rescues the loss of STAT3 phosphorylation in the dKO cells, indicating that SGK1/3 activate STAT3 via control of IFNβ expression [Figure 2F].
As indicated in Figure 2D, loss of SGK1/3 did not block the phosphorylation of TBK1 or IRF3 downstream of DMXAA stimulation, yet Sanofi-14h treatment blocked activation of TBK1 as well as IRF3 in dKO cells [Figure 2G]. While knockout of SGK1/3 reduced IFNβ mRNA induction downstream of STING treatment, Sanofi-14h further reduced that response in these cells [Figure 2G]. Collectively, these data indicate that Sanofi-14h inhibition of TBK1/IRF3 phosphorylation during STING activation is largely independent of SGK1/3 inhibition and that SGK1/3 function through a distinct mechanism to promote STING-induced IFNβ mRNA production.
SGK1 and SGK3 control baseline and inducible expression of innate immune regulators
To address mechanisms whereby SGK1 and SGK3 regulate IFNβ expression we performed RNA sequencing (RNAseq) on WT and SGK1/3 dKO RAW264.7 cells with and without DMXAA stimulation. Unsupervised clustering of unstimulated samples showed clear delineation of WT and SGK-null samples with a large number of genes dysregulated in SGK1/3 KO cells. Gene ontology analysis revealed basal downregulation of genes associated with pattern recognition receptor (PRR) signaling, innate immune response, interferon production, and responses to viral infection in cells lacking SGK1 and SGK3 [Figure 3A]. These SGK-dependent genes include IFN regulating transcription factors Irf7 and Irf9 [Figure 3A, C]. Volcano plots show SGK-dependent baseline expression of other innate immune genes and Interferon-Stimulated Genes (ISGs) including Ifi44, Il1rn, Usp14, Siglec1, and Oas3. Gene set enrichment analysis (GSEA) comparing dysregulated genes in WT and SGK1/3 dKO cells at baseline indicated loss of gene sets related to host defense with no other strongly significant gene sets identified.
Figure 3: SGK1 and SGK3 KO blocks expression of Interferon Stimulated Genes (ISGs) and innate immunity regulators.
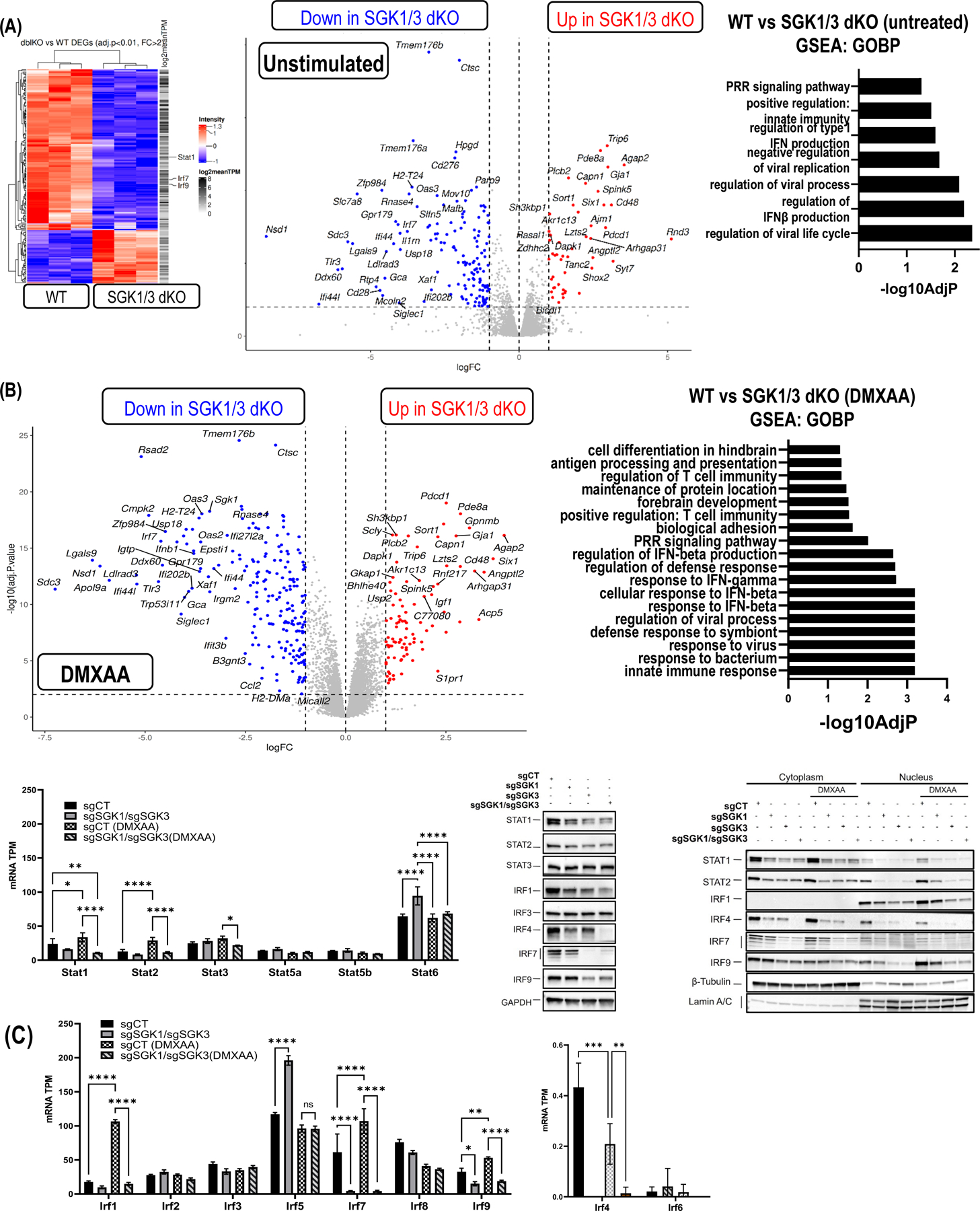
A) Heatmap (left panel) depicts differentially expressed genes (DEGs) in unstimulated WT and SGK1/3 dKO RAW264.7 cells. Blue indicates downregulated and red indicates upregulated DEGs. Volcano plot (middle graph) depicts the expression and statistical significance of DEGs in unstimulated cells with highlighting of immune-related or Interferon Stimulated Genes (ISGs). Right graph shows Gene Set Enrichment Analysis (GSEA) representing significantly altered pathways in unstimulated SGK1/3 dKO cells relative to WT. B) Volcano plot (left graph) of gene expression in SGK1/3 dKO cells compared to WT after 2h DMXAA treatment. Significance was determined by an adjusted p value of <0.05 and a fold change >2 in either direction. GSEA (right graph) of the most affected pathways after DMXAA treatment in SGK1/3 dKO cells. C) IRF and STAT mRNA expression in Transcripts Per Million (TPM) (*p<0.05) in SGK1/3 dKO cells with and without DMXAA (2h). Immunoblot of IRFs and STATs protein expression at basal conditions (left panel) and their nuclear localization (right panel) after DMXAA treatment for 1h.
We next examined gene expression by RNAseq in STING-activated cells and found striking loss of IFN and ISG pathway genes in SGK1/3 dKO cells stimulated with DMXAA relative to WT cells [Figure 3B]. The extent and significance of downregulation is enhanced after DMXAA stimulation relative to the basal state. GSEA confirmed the loss of interferon and host defense pathway transcription in SGK1/3 dKO cells. Other gene sets impacted by SGK1/3 deletion include those involved in antigen processing, T cell stimulation, cell adhesion, and cell differentiation. Examination of individual transcription factors showed that in addition to the baseline defect in expression of Irf7 and Irf9 SGK1/3 dKO macrophages do not upregulate Irf1, Irf7, Irf9, Stat1 or Stat2 RNA after DMXAA treatment [Figure 3B and 3C]. We extended these findings by immunoblotting which demonstrated a baseline decrease of STAT1, STAT2, IRF1, IRF7, and IRF9 protein levels in dKO cells [Figure 3C]. SGK3 appears to control IRF7, IRF9 and STAT2 preferentially as compared with SGK1. Despite relatively low SGK1 protein expression in RAW264.7 cells, SGK1 KO cells show enhanced loss of IRF4 as compared with SGK3 KO cells. Consistent with the expression defects, we observed that SGK1/3 deletion diminished nuclear translocation of IRF4, IRF9, STAT1 and STAT2 and IRF1 in SGK1/3 KO cells [Figure 3C]. IRF9 and STAT1 are part of Interferon Stimulated Gene Factor 3 (ISGF3)43, which is responsible for inducing expression of Interferon Stimulated Genes (ISGs), and IRF1 is also involved in the regulation of ISGs in an IFN-dependent and -independent manner44. STAT2 is also part of the ISGF3 complex43 which partially explains (along with a block in IRF1 expression) why the vast majority of affected genes are ISGs. Together, these results indicate that SGK1 and SGK3 regulate the expression of IFNβ and ISGs by controlling expression of innate immune regulators IRF1, IRF4, IRF7, IRF9, STAT1 and STAT2 at the RNA level and prior to STING-induced signaling.
To determine the individual pathway contributions under basal conditions and in the DMXAA response regulated by each kinase we performed RNA sequencing on single SGK1 and SGK3 KO cells. Under unstimulated conditions SGK1 KO cells had impaired expression of 308 genes and SGK3 KO cells had decreased expression of 122 genes (as compared with WT cells). SGK1/SGK3 double KO decreased the expression of 135 genes as compared with WT cells. The overlap of downregulated genes between SGK1 and SGK3 KO cells was 48, indicating common pathways of regulation but also indicating unique functions for the two kinases. The overlap between the SGK1/3 KO cells and the SGK1 KO cells was 59 genes and overlap between the double KO cells with the SGK3 KO cells was 82 [Figure 4A]. 64 genes were upregulated with loss of SGK1 while 114 genes were upregulated with loss of SGK3. 45 genes were upregulated with loss of SGK1 and SGK3 [Figure 4A]. Pathway analysis revealed that genes regulated by SGK1 are involved in processes related to translation, homeostasis regulation, cellular response to IFNβ and response to viral infection, whereas SGK3 dependent genes have a more significant role in innate immune response and viral life cycle control in addition to functions in cellular response to IFNβ and viral infection [Figure 4A].
Figure 4: SGK1 and SGK3 individually control different subsets of genes under basal and STING stimulation:
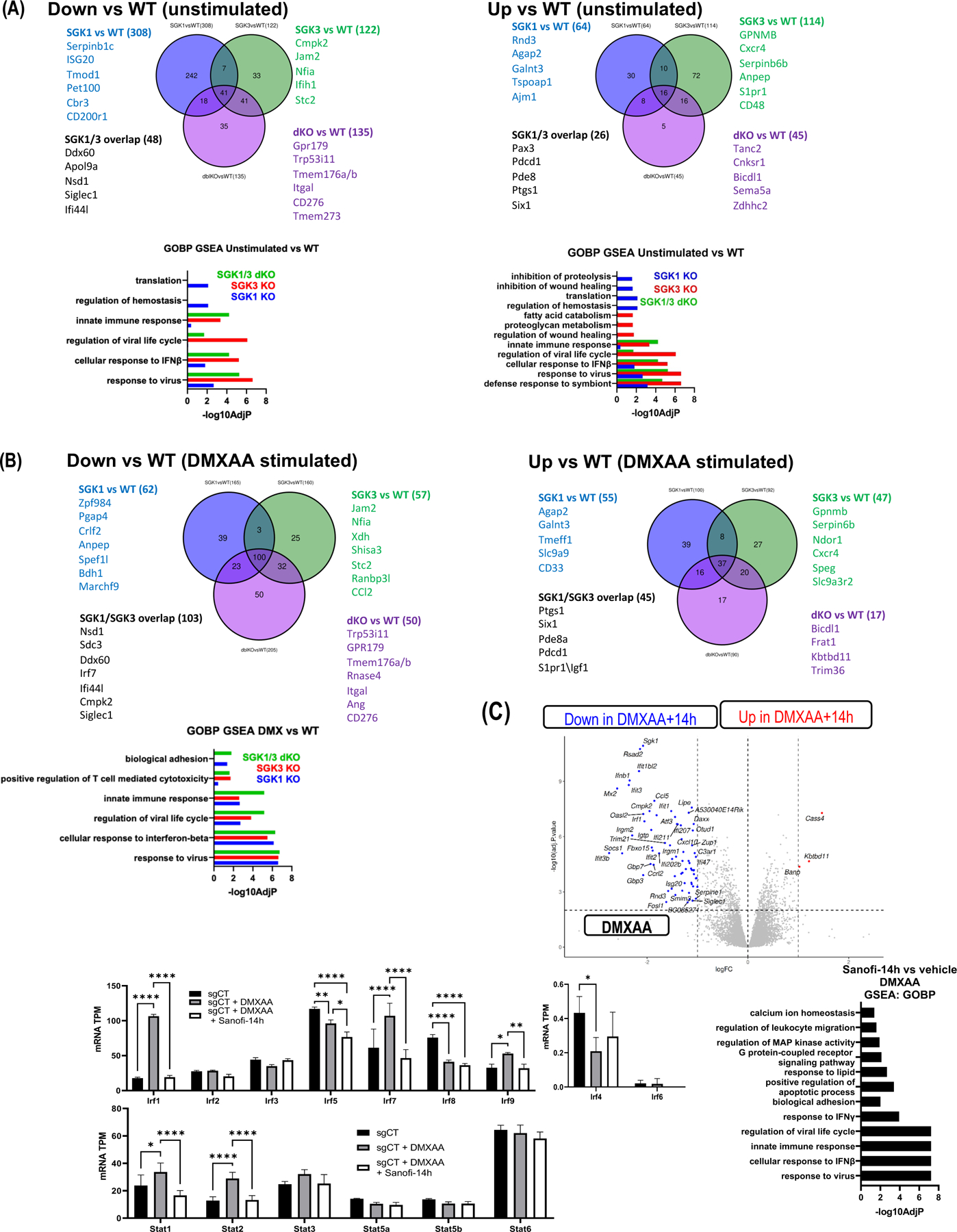
A) Venn diagrams depicting the overlap of down- and up-regulated genes among unstimulated SGK1 KO, SGK3 KO, and SGK1/3 dKO cells relative to WT. GSEA showing affected pathways in individual and double knockouts relative to WT (lower panel). E) As in (D), comparison of gene expression changes in single and double knockouts after DMXAA stimulation. F) Volcano plot (left graph) indicating DEGs between vehicle- or Sanofi-14h-treated cells after DMXAA stimulation. GSEA (right graph) indicating pathway effects of Sanofi-14h on DMXAA-stimulated gene expression. (Bottom panel) Expression of individual IRF and STAT mRNAs with and without Sanofi-14h after DMXAA treatment. All experiments were repeated at least three times (* p<0.05).
Under DMXAA stimulation 165 genes were downregulated SGK1 knockout cells and 160 were downregulated in SGK3 cells as compared with WT, with an overlap of 103 genes. In SGK1/3 knockout cells 205 genes were downregulated following DMXAA stimulation, with the majority of these genes (approximately 75%) overlapping with SGK1 and SGK3 knockout cells [Figure 4B]. These results indicate both unique and overlapping functions for SGK1 and SGK3 in promoting gene expression in macrophage cells. Genes controlled by both SGK1 and SGK3 are significant for cellular response to IFNβ and viral infection, whereas SGK3 remains the main kinase with respect to the innate immune response pathways and regulation of viral cycle as well as biological adhesion and positive regulation of T cell mediated cytotoxicity in the DMXAA response [Figure 4B]. Together, these data indicate that both SGK1 and SGK3 have individual and overlapping roles in macrophage physiology in both basal and stimulated conditions. Under basal conditions SGK3 primes the cells to respond to viral infection, while SGK1 is upregulated under DMXAA stimulation potentially to assist in this response by contributing to SGK3 functions and exercising its own individual roles.
Sanofi-14h blocks DMXAA-stimulated Interferon and ISG gene expression in macrophages
We next asked whether effects of Sanofi-14h treatment overlap the observed transcriptomic effect of SGK1/3 deletion in RAW264.7 cells. Because high dose (5–10μM) Sanofi-14h efficiently blocks TBK1 activation [Figure 1C], we selected a lower dose (500nM) of Sanofi-14h that inhibits SGK1 and SGK3 (as assayed by NDRG1 phosphorylation) but largely leaves TBK1 activation intact. Similar to SGK1/3 deletion, 500nM Sanofi-14h inhibited the inducible expression of Ifnβ1 and Stat1 as well as several ISGs (including Mx1, Gbp5, ISG15, Ifit1/2/3, Rsad2) and chemotaxis-related genes (Ccl5, Cccrl2) [Figure 4C]. GSEA analysis confirmed the similar transcriptomic effect of SGK inhibition and deletion, showing that the most affected genes are those related to pathogen sensing. Moreover, RNAseq confirmed that Sanofi-14h at a dose of 500nM has a similar effect as deletion of SGK1 and SGK3 in terms of IFN and ISG expression. However, only Irf1, Stat1 and Stat2 RNA are impaired by Sanofi-14h to a similar level as in SGK dKO cells. Irf7 and Irf9 RNA levels were not significantly reduced by Sanofi-14h during the short (2 h pretreatment, 2 h stimulation) exposure to drug, suggesting they are not wholly responsible for Ifnβ1 regulation by Sanofi-14h in this time frame [Figure 4C]. The less broad effect of Sanofi-14h is likely explained by acute responses of the cells versus the stable loss of SGK1/3. These data confirm that Sanofi-14h and SGK1/3 regulate STING-driven gene expression responses in macrophage cells.
Sanofi-14h disrupts inducible TBK1-STING interaction
We asked whether doses of Sanofi-14h that inhibit TBK1 activation do so by blocking induced STING-TBK1 interaction40. To address this, we performed immunofluorescence to measure TBK1 presence on the ER/GOLGI. We observed that TBK1 formed aggregates or puncta after DMXAA stimulation across the cytoplasm and a portion of these puncta localize to the ER/Golgi space. When treated with Sanofi-14h, the total number of puncta formed by TBK1 significantly decreased and the puncta localized to the ER/GOLGI appear less organized and more diffuse than the ones localized to the same space under DMXAA stimulation alone [Figure 5A]. To extend these results, we co-immunoprecipitated STING and TBK1 in the presence or absence of Sanofi-14h under DMXAA stimulation and found that DMXAA induced robust interaction between STING and TBK1 as expected, and Sanofi-14h impaired DMXAA-induced TBK1-STING interaction [Figure 5B]. Additionally, Sanofi-14h blocked DMXAA-induced phosphorylation of STING as indicated by a reduction in the slower migrating STING form [Figure 5B]42. We next examined whether Sanofi-14h treatment alters subcellular trafficking of TBK1 and IRF3 following STING activation. We isolated cytoplasmic and membranous (containing the ER, mitochondria and Golgi) fractions after DMXAA treatment. We observed recruitment and phosphorylation of TBK1 and IRF3 in the membrane fraction as expected [Figure 5C]. Pretreatment with Sanofi-14h prior to DMXAA prevented TBK1 and IRF3 translocation/retention and subsequent STING activation in the ER [Figure 5C]. These results support the hypothesis that Sanofi-14h inhibits STING pathway signaling by preventing TBK1-STING interaction in the ER, thus blocking subsequent recruitment and phosphorylation of IRF3, thus inhibiting this key step in the activation pathway.
Figure 5: Sanofi-14h disrupts TBK1/STING interaction.
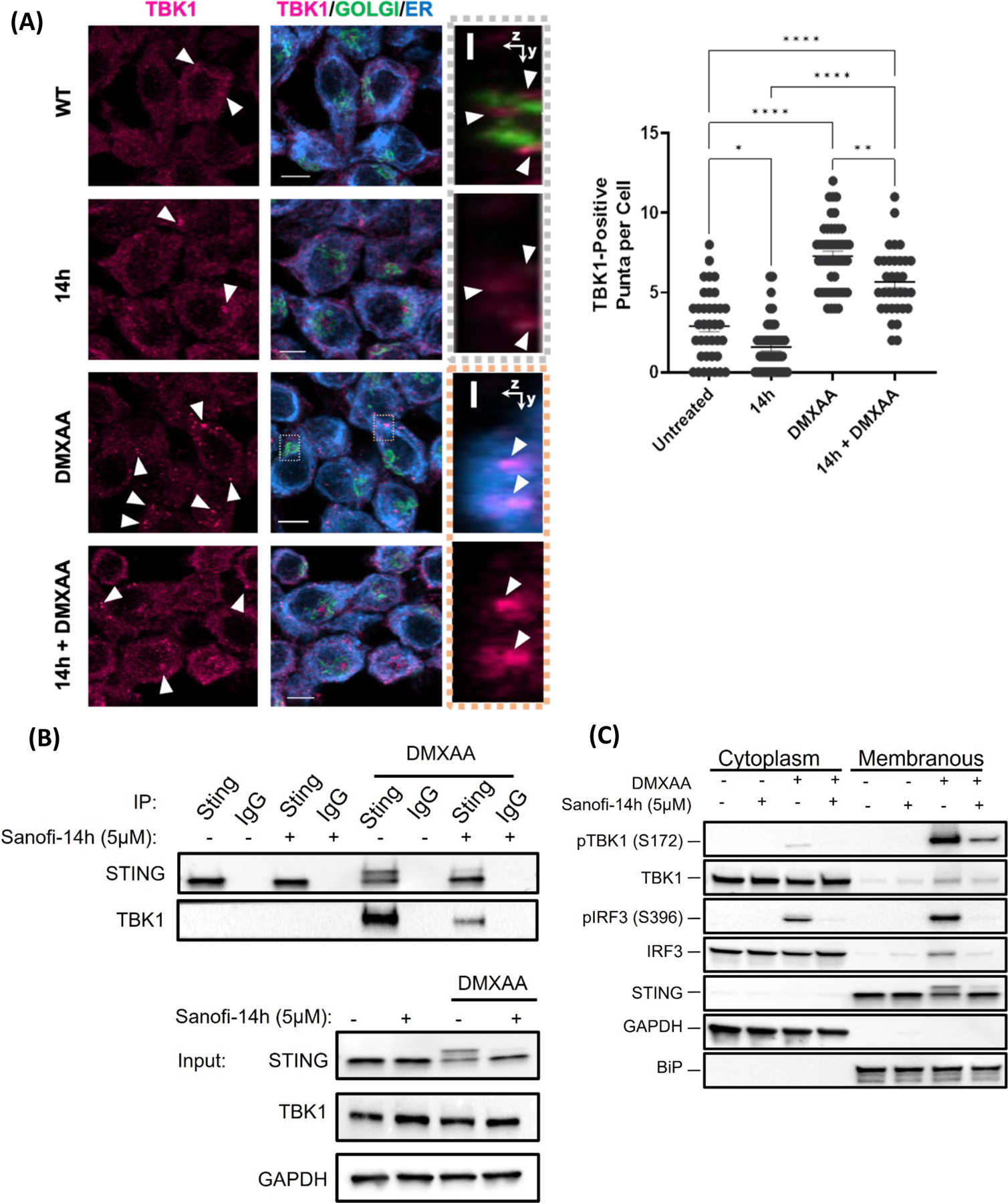
A) Immunofluorescence (left) of RAW264.7 and TBK1 puncta quantification (right) after co-treatment with 10 mM of Sanofi-14h (2h) and 10μg/mL DMXAA (1h). Arrows indicate TBK1 puncta. B) Effects of Sanofi-14h on STING and TBK1 interaction after DMXAA treatment for 1h. STING was immunoprecipitated and the interaction was detected by TBK1 immunoblot. Immunoprecipitation with non-specific IgG serves as a negative control and inputs show the levels of both proteins in total lysate. C) RAW264.7 cells were fractionated and immunoblotted for TBK1 and IRF3 translocation into the ER-Golgi after Sanofi-14h (2h) and DMXAA treatment for 1h. Data is representative of 3 separate experiments (* p<0.05).
Class effects of SGK inhibition on STING/TBK1/IFN signaling
Using an IRF-dependent luciferase reporter system in RAW264.7 macrophages (Invivogen), we tested DMXAA-stimulated cells pre-treated with 4 different SGK inhibitors across a 10,000-fold concentration range. For this study we synthesized and tested Sanofi-17a, a structural derivative of Sanofi-14h [Figure 6A], that was recently described31 and which preferentially targets SGK1 over SGK3. We included three other small molecules, GSK650394, SI113, and EMD638683, with established SGK inhibitory activity27–29. We found that each of these SGK inhibitors, with the exception of EMD638683, blocked DMXAA-stimulated IRF3-promoter activity with an IC50 of 3.334 μM and 5.612 μM, respectively [Figure 6B]. Sanofi-17a was less effective than the other SGK inhibitors in the reporter assay with an IC50 of 9.006 μM whereas Sanofi-14h was the most effective with an IC50 of 1.728 μM. Since Sanofi-17a is a derivative of Sanofi-14h, we examined the effects of Sanofi-17a on STING-activated signaling. Similar to Sanofi-14h, Sanofi-17a at low micromolar concentrations effectively blocked NDRG1 phosphorylation [Figure 6B]. Sanofi-17a inhibited TBK1, STAT3, and IRF3 phosphorylation induced by DMXAA with 50% inhibition of TBK1 phosphorylation seen at ~10μM, similar to but less effective as compared with Sanofi-14h [Figure 6B and 1C].
Figure 6: Other SGK inhibitors have variable effects on IRF DMXAA driven activation.

A) Structures of Sanofi-14h and Sanofi-17a. B) Effect of SGK inhibitors on DMXAA-stimulated IRF luciferase activity. Raw-Dual Luciferase cells were seeded in 96 well plates overnight and pretreated with indicated inhibitors for 2h prior to DMXAA stimulation for 24h. Luciferase signal indicates type I IFN production. X axis depicts log10 inhibitor concentration. Immunoblot (right panel) shows phosphorylation of indicated proteins after 2h DMXAA treatment. C) Immunoblot of RAW264.7 cells pre-treated with 10 μM Sanofi-14h followed by treatment with LPS at the indicated times. Data is representative of 3 separate experiments. D) Immunoblot (right panel) depicting effects of p70S6K (LY258702) and Akt (MK2206) inhibition on markers of the DMXAA response compared to Sanofi-14h. IRF activation luciferase assay (left graph) after co-treatment with DMXAA and indicated doses of LY258702 and MK2206. E) Co-immunoprecipitation (left panel) showing the effects of LY258702 (2 h pre-treatment) on TBK1/STING interaction after DMXAA treatment for 1 h. Data is representative of 3 separate experiments.
It was reported that interaction between the TBK1/STING complex with IRF3 is dependent on the structural activity of P70S6K145 without affecting TBK1/STING interaction. In this regard, we examined phosphorylation of ribosomal S6 protein, a substrate of the S6 kinase family, based on reports that P70S6K regulates the STING/TBK1/IRF3 complex45. DMXAA stimulated S6 phosphorylation in RAW264.7 cells and this was blocked by Sanofi-17a in the low micromolar range [Figure 6B]. Sanofi-17a also impaired DMXAA-stimulated phosphorylation of SGK3 T320 at 500nM [Figure 6B]. SGK3 T320 is reported to be a direct phosphorylation substrate of 3-phosphoinositide-dependent protein kinase 1 (PDK1)46. The sensitivity of SGK3 T320 phosphorylation to inhibition by Sanofi-17a may represent either loss of feedback from SGK3 to PDK1 or an undescribed component of SGK3 autophosphorylation at that site. Thus, Sanofi-17a blocks phosphorylation of S6 around 1μM but is less effective at blocking phosphorylation of TBK1 and IRF3.
Predicted non-SGK targets of Sanofi-14h do not mediate TBK1 phosphorylation
We then tested whether Sanofi-14h directly targets TBK1 by co-treating RAW264.7 cells with LPS, a known TBK1 activator in macrophages. We observed that phosphorylation of two canonical TBK1 targets, IRF3 and p6247,48, were not affected and that phosphorylation of TBK1 was only partly decreased [Figure 6C]. Additionally, we performed an in vitro kinase assay using TBK1 and IRF3 and various doses of Sanofi-14h and found that this inhibitor does not prevent phosphorylation of IRF3 by TBK1 in a cell-free setting [Suppl. Figure 2A]. These results suggest that Sanofi-14h doesn’t directly target TBK1.
Because SGK inhibitors are reported to have activity against other AGC kinases like P70S6K1 or Akt23, we next asked whether the signaling effects of Sanofi-14h treatment overlap with those of Akt or P70S6K inhibitors. In RAW264.7 cells stimulated with DMXAA, Akt inhibition by MK2206 or P70S6K inhibition by LY2584702 had no effect on STAT3 Y705 phosphorylation while Sanofi-14h at the same dose completely blocked phospho-STAT3 [Figure 6D], consistent with loss of IFNβ production by this inhibitor. Akt inhibition had no measurable effect on DMXAA-stimulated phosphorylation of TBK1 or IRF3. P70S6K inhibition partly abrogated IRF3 phosphorylation while leaving TBK1 phosphorylation largely intact, consistent with reports that S6K1 interacts with STING45. When assayed using the IRF3-luciferase reporter, Akt and P70S6K inhibitors had weak effects on reporter activity with IC50 greater than 50μM, while Sanofi-14h was significantly more potent [Figure 6D]. To further test whether P70S6K inhibition has the same effect as Sanofi-14h we assayed the interaction between TBK1 and STING by co-immunoprecipitation. LY2584702 did not alter the STING-TBK1 interaction or STING phosphorylation after DMXAA treatment [Figure 6E], suggesting that Sanofi-14h is unlikely to work by the same mechanism as S6K inhibition. The lack of effect on TBK1 phosphorylation, IFN expression, or STING-TBK1 interaction by Akt or P70S6K inhibitors makes it unlikely that Sanofi-14h affects the TBK1/IFN pathway via these kinases (see Discussion).
Finally, we co-treated RAW264.7 cells with DMXAA and inhibitors for Tie2, MLK1/3 and MAP4K3/5 which, in addition to Akt and p70S6K, were predicted to be targeted by Sanofi-14h in vitro23. We found that none of those inhibitors could prevent TBK1 phosphorylation in response to STING activation as well as Sanofi-14h. Interestingly, the MLK1/3 inhibitor prevented IRF3 activation and STAT3, IRF3, and NDRG1 phosphorylation without influencing TBK1 phosphorylation [Suppl. Fig 2B and 2C], which suggests that multiple kinase inhibitor classes can block NDRG1 phosphorylation and IRF3 activation without blocking TBK1 itself and that Sanofi-14h is not inhibiting these kinases to block TBK1 activation in the STING pathway.
DISCUSSION
Inhibitors of the SGK family have been developed and tested for cancer as well as other disease indications, such as osteoarthritis 23,31. These studies showed that other kinases are targeted by these inhibitors, as Sanofi-14h was shown to have significant activity against S6K1 and MLK3 23,31. Our studies revealed an unexpected effect of Sanofi-14h on inhibition of IFNβ mRNA and protein induction following treatment of macrophages cells with the STING agonist DMXAA (Fig. 1A and Suppl. Figure 2D) and this result was extended to effects on viral replication and virus-induced IFNβ (Fig. 1B).
We analyzed the effects of Sanofi-14h on DMXAA-induced STING signaling and found that it blocked phosphorylation of TBK1 and IRF3 as well an IFN-driven STAT3 phosphorylation (Fig. 1C). The effect of Sanofi-14h was shown to be at the level of blocking the induced interaction between STING and TBK1 (Fig. 5A – C), thus at an early stage in the STING-induced signaling pathway (and see below). Consistent with the effect of SGK inhibition by Sanofi-14h, loss of SGK1/3 blocked STING-induced IFNβ mRNA induction (Fig. 2A). However, loss of SGK1/3 in the macrophage cells did not block the induction of phosphorylation of TBK1 and IRF3, indicating an SGK-independent mechanism for regulatory event (Fig. 2D).
We performed RNA sequencing on the SGK1/SGK3 dKO cells at baseline and under DMXAA-stimulated conditions and compared the results to WT cells (Fig. 3A, B). In SGK1/3 KO cells we found markedly reduced expression of direct and indirect regulators of IFNβ production IRF1, IRF4, IRF7, IRF9, STAT1 and STAT2 at basal and stimulated conditions at the protein and RNA levels (Fig. 3C). IRF7 is particularly critical for IFNβ expression in response to STING activation because it directly engages the IFNβ promoter along with IRF347 whereas IRF1 predominantly drives expression of ISGs such as OAS2, BST2 and RNASEL. IRF1 also promotes expression of IRF944 which, along with STAT1 and STAT3, forms the ISGF3 complex, further promoting IRF7 expression48. Multiple IRF and STAT genes were highly dependent on SGK3, while IRF4 appears uniquely dependent on SGK1. Thus, SGK1-dependent IFNβ expression and downstream STAT3 phosphorylation (Fig. 2A, 2D) may reflect contribution of IRF4 either to IFNβ transcription or macrophage polarization49.
To better understand the individual contributions of SGK1 and SGK3 basal and DMXAA-stimulated gene expression, we also performed RNA sequencing of SGK1 KO and SGK3 KO cells (Fig. 4A, B). Gene set enrichment analysis revealed that SGK3 is vital for cellular priming against viral infection and the regulation of the subsequent IFN response at basal levels, whereas SGK1 has a role in other processes such as adhesion despite its low expression levels in these cells. Under DMXAA stimulation, however, they both have a similar effect on controlling the antiviral response (Fig. 4A, B).
To address whether SGK1/3 play a role in immediate STING-induced signaling, we treated cells with 500nM Sanofi-14h, a dose that is preferential for targeting SGK1/3 over blocking TBK1 activation, and performed RNAseq studies (Fig. 4A). The results demonstrated that Sanofi-14h blocked expression of the same genes group affected by SGK1 and SGK3 knockout namely Irf1, Irf7, Irf9 STAT1 and STAT2, which were inhibited prior to STING activation in dKO cells. However, in the specific case of Irf7 and Irf9 this short pre-treatment with Sanofi-14h only partially impaired their expression, though these transcripts are under-expressed in SGK KO cells (Fig. 3C). This may relate to the short time frame of inhibition compared to CRISPR deletion and to the half-life of Irf7 and Irf9 mRNAs. Likewise, the effects of Sanofi-14h at 500nM were phenotypically similar to those of the dKO cells in terms of the gene sets that were affected after DMXAA treatment. IFNβ production, response to viral cycle, and response to bacterium were among such gene sets. These data suggest that SGK controls expression of IFNβ mRNA by positively regulating IRF7 expression and this may occur via a mechanism involving IRF1, IRF9, STAT1 and STAT2 but future experiments will be necessary to define this response.
The ability of Sanofi-14h treatment to inhibit activation of TBK1 led us to examine a panel of SGK and AGC family kinase inhibitors for their effects on STING-induced signaling. In this regard, we synthesized Sanofi 17a31, a derivative of Sanofi-14h, which preferentially targets SGK1 (Fig. 6A)31. We found that Sanofi-14h, Sanofi-17a, GSK650394, and SI113 all inhibited STING-driven IRF activation in a reporter assay with similar potency. The SGK inhibitor EMD638683, reported to exhibit preference for SGK1 over SGK3 50, was ineffective at blocking IRF activation (Fig. 6B). Sanofi-17a blocked phosphorylation of TBK1 and IRF3, although less efficiently than Sanofi-14h, with inhibitory effects around 10 μM (Fig. 6B) whereas Sanofi-14h blocks TBK1 and IRF3 in the ~1 μM range (Fig. 1C). Consistently, Sanofi-17a blocked the IRF-dependent reporter less effectively compared to Sanofi-14h (Fig. 6B). Overall, our studies show that structurally dissimilar SGK inhibitors such as Sanofi-14h, SI113, and GSK650394 all inhibit the STING-induced innate immune response.
It was reported that the AGC family kinase S6K1 associates with STING to promote recruitment of IRF3, but not TBK1, into the STING complex, in the viral-regulated innate immune response45. Interestingly, this mechanism was reported to not require the catalytic activity of S6K1. In this regard, we analyzed effects of different AGC kinase inhibitors on phosphorylation of S6 and on the DMXAA-induced downstream signaling response. Sanofi-14h has been shown (in cell-free assays) to block S6K1 in a range similar to that of SGK3 and SGK123, while Sanofi 17a was reported to have little to no effect on S6K1 activity 31. Interestingly, we observed that phosphorylation of S6 is induced by DMXAA (Fig. 6B) suggesting the involvement of an S6 kinase in the STING-induced response. Both Sanofi-17a and Sanofi-14h inhibited the phosphorylation of S6 induced by DMXAA (Fig. 6B, D). The Akt inhibitor MK2206 prevented phosphorylation of S6 (though not as effectively as Sanofi-14h) but did not block the induced phosphorylation of TBK1 or IRF3 in the STING pathway. The P70S6K inhibitor LY2584702 effectively impeded the induced phosphorylation of S6, but less effectively blocked phosphorylation of IRF3. Consistently neither the Akt inhibitor nor the P70S6K inhibitor affected phosphorylation of STAT3 or activation of the IRF3 reporter (Fig. 6D). While Sanofi-14h impaired recruitment of TBK1 to the STING complex in response to STING stimulation, LY2584702 did not inhibit recruitment of TBK1 and did not hinder phosphorylation of STING which is controlled by TBK1 recruitment (Fig. 6E). These studies show that S6K and Akt inhibition do not phenocopy SGK inhibition and suggests that Akt and S6K do not mediate the effect of Sanofi-14h and related compounds on TBK1 and IRF3 activation.
To determine if the effects of Sanofi-14h were exclusive to the STING pathway we co-treated cells with LPS, which potently induces TBK1 phosphorylation, in combination with Sanofi-14h and found that this inhibitor only partially affected TBK1 phosphorylation while phosphorylation of IRF3 remained unchanged (Fig. 6C) These data indicate the possibility that the effect of Sanofi-14h is mostly relevant for the STING pathway and are not directly targeting TBK1, although future studies must be performed to prove this hypothesis. Finally, we treated RAW264.7 cells with inhibitors of 5 different kinases predicted to be affected by Sanofi-14h and discovered that none of these inhibitors affected TBK1 phosphorylation (Suppl. Figure 2C), but that MLK1/3 inhibitor URCM-099 markedly impaired phosphorylation of STAT3, IRF3 and NDRG1, suggesting that MLK1/3 may phosphorylate proteins downstream of TBK1 but that they are not the target of Sanofi-14h. Future studies will test the role of MLK1/3 in innate immune signaling.
Our study highlights SGK1 and SGK3 as important, targetable regulators of the STING/IFN and IRF/IFN pathways. Future studies are directed at the mechanism of action of Sanofi-14h and other SGK/AGC inhibitors relative to the STING/TBK1/IRF3 axis. We propose a model related to the ability of Sanofi14h to block TBK1 activation in the STING pathway and whereby SGK1 and SGK3 control the expression of key regulators pathway in the innate immune response pathway (Figure 7). This work implicates currently existing SGK inhibitors as useful modulators of IFN signaling and points to the SGK kinase family as a target in IFN-related diseases.
Figure 7: Model of SGK regulation of STING/TBK1 signaling.

Sanofi-14h inhibits phosphorylation of TBK1 through an unknown mechanism preventing STING and IRF3 phosphorylation, dimerization and nuclear translocation in the presence of 2’3’-cGAMP and DMXAA. SGK1/3 promote IRF7 expression and stabilization under basal and DMXAA stimulated conditions in macrophages. Adapted from “cGAS detects Cytosolic dsDNA”, BioRender.com (2022), https://app.biorender.com/biorender-templates
LIMITATIONS OF THE STUDY
While this study is thorough, it has notable limitations. There is no identification of Sanofi-14h targets which could reveal more about TBK1 regulation. A comparison between chronic Sanofi-14h exposure and CRISPR KO cells is also missing and the shown effects are only under STING stimulation which omits other related pathways like RIG-I. Furthermore, despite detailing molecular mechanism the study lacks essential in vivo validation.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
For any further communication regarding protocols or material availability please contact Albert S. Baldwin at albert_baldwin@med.unc.edu
Materials availability
The gRNA plasmids and the RAW264.7 SGK1/3 KO cells generated in this study are available upon request.
This study did not generate any novel reagents
Sanofi-14h can be bought at the MCR pure reagent center of the University of Dundee in the United Kingdom by emailing them at MRCPPUreagents@dundee.ac.uk.
Alternatively, it can be bought in MedChem Express under the name SGK1-IN-2 9(14h) with catalog number HY-135893
Data and code availability
RNAseq data has been uploaded to NCBI GEO with accession number GSE243816 and it is publicly accessible.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available upon request from the lead contact.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell lines and culture
RAW 264.7 (mouse, male), THP-1 cells (human, male), Vero (African green monkey, female) and HEK293T/17(human, female) cells were purchased from the Tissue Culture Facility (TCF) at UNC. RAW264.7, Vero and HEK293T/17 were cultured in DMEM High glucose medium (Gibco, 11995-065) supplemented with 10% fetal bovine Serum (FBS) (Avantor, 97068-085) and 1% penicillin/streptomycin (Gibco, 15140122) in an atmosphere of 5% CO2 and 95% humidity at 37°C. THP-1 cells were cultured in RPMI (Gibco, 11875093) with 10% FBS, 1% pen/strep, and 0.05 mM beta-mercaptoethanol (Gibco, 31350010) and differentiated into macrophages with 50nM PMA (Sigma, P1585-1MG) for 48 hours and rest for 24h after differentiation. Human (male) monocytes were derived from healthy donor PBMCs (UNC IRB 22-2450) and differentiated into macrophages for 7 days in 50ng/mL human M-CSF (Peprotech, 300–25). HEK293T/17 cells were maintained in 10 cm plates and passaged after reaching 90% confluency with 2 mL of Trypsin-EDTA (0.25%) (Gibco, 25200056) at 37°C for 2 minutes until passage 50. RAW-Dual Lucia (Mouse, male, Invivogen, rawd-ismip) cells were maintained in DMEM complemented with 100 μg/mL of Normocin (Invivogen, ant-nr-1), 10% heat inactivated FBS, 200μg/mL of Zeocin (Invivogen, ant-zn-05) and 1% penicillin/streptomycin(P/S). All cell lines were regularly tested for mycoplasma.
Lentivirus production and RAW264.7 cell infection
Lentiviral particles were produced as described with some modification51. HEK293T/17 cells (0.75x106 per well) were plated in 6 well plates and after 20 hr cells were co-transfected with the 2μg of TLVC2-gRNA, 1μg of psPAX2 (Addgene #12260) and 0.5μg pMD2.G (Addgene #12259) in a ratio of 4:2:1 using FuGENE HD (Promega, E2311) and OptiMEM according to the manufacturer’s instructions. Media was changed the next day and after 48h the viral particles were collected by filtration (0.45 μM filter). RAW 264.7 cells were plated at a density of 0.75x106 cells per well in a 6 well plates and were infected using 1 mL of lentivirus, 1 mL of fresh regular DMEM, 8μg/mL Polybrene (Santa Cruz Biotechnology, sc-134220) and spin infection (1372 RCF for 1hr at RT). Cells were re-infected the next day and selected the following day with Puromycin (2.5μg/mL) (Gibco, A1113803).
SGK1, SGK3 and double SGK1/3 CRISPR/CAS9 cell line generation
After the non-infected control died from selection a portion of the cells was frozen with BAMBANKER FREEZE freezing media (Fisher Scientific, NC9582225) and the rest was plated in 10 cm without puromycin and supplemented with 1μg/mL of doxycycline (Sigma, D5207-1G) which was refreshed every day for 7 days. Cells were sorted based their GFP “high” signal and plated in 96 well plates at a ratio of 1 cell/well. Clones were screened for SGK1 or SGK3 KO through immunoblot (Figure 2D). The double SGK1 and SGK3 KO RAW 264.7 cells were generated by Synthego Corporation (Redwood City, CA, USA) as previously described52 and single clones were isolated in the same way as the one for the single KOs.
Bone marrow derived macrophages preparation
The SGK3 knockout mice (adult mice, both sexes, 6–12 weeks old) were a kind gift from Dr. David Pearce at the University of California San Francisco and have been described previously53. All procedures involving mice handling were in accordance to approved protocols by the University of North Carolina Chapel Hill (IACUC protocol 20–167). Bone marrow from WT and SGK3 KO mice was extracted as previously described 54. Bone marrow was harvested from mice as previously described55, and cultured in L929-cell condition media (LCCM) to generate macrophages 56. Cells were seeded in non-tissue culture treated 10 cm plates (Fisherbrand, FB08755712) in media (complete media) containing RPMI1640 (Corning, 10-040-CV), 20% LCCM, 10% FBS, 1%(P/S), 2 mM of L-Glutamine, and 0.05 mM of beta-mercaptoethanol. After 24 hours cells were washed twice with PBS to remove non-adherent cells and fresh complete media was added. Media was changed on days 3 and 5 prior to scraping, counting, and replating. On day 7 media was changed to regular media without LCCM for 24 hours prior to experiments.
METHOD DETAILS
Cell treatments
RAW264.7 DMXAA (Cayman Chemical, 14617) treatments were carried out at a 10 μg/mL dose for all experiments at a density of 3.5x106 cells in 60 mm plates (immunoblots, RT-qPCR and RNA seq) and 35x106 cells in 10 cm plates (co-IP and cell fractionations). Recombinant IFNβ treatment was performed at 10 ng/mL for 15 min. LPS treatment (Invivogen, tlrl-eblps) was carried out at the indicated times at a dose of 100 ng/mL. Co-treatments with Sanofi-14h (MRC Pure Reagents) and MK2206 (MCE, HY-108232) and LY2584702 (MCE, HY-12493) were performed at the indicated doses. THP-1 and hMDM were treated with 1μM of diABZI (SelleckChem, S8796) at the indicated time. RAW-Dual Lucia cells were plated in 96 well plates at a density of 0.2x106 cells per well and let to rest until the next morning. Cells were treated with inhibitors for AKT (MK2206), P70S6K, (LY2584702), Tie2 (Tie2 kinase inhibitor, MCE, HY-100556), MLK1/3 (URMC-099, MCE, HY-12599), MAP4K3/5 (DMX-5804, MCE, HY-111754), and SGK inhibitors GSK650394 (MCE, HY-15192 ), EMD638683 (MCE, HY-15193) and SI113 (MCE, HY-117357) for 2 hours and then were stimulated with DMXAA for an additional 24h. The luciferase assay was performed following the manufacturer’s instructions.
Cloning
Guide RNAs targeting mSGK1 exon 5(GATTTCAGGTTCTTCTGGCT) and mSGK3 exon 3 (TTCAAGACATTAAATGCAG) were designed using the online tool CHOPCHOP (http://chopchop.cbu.uib.no/) and cloned into TLCV2 (Addgene plasmid # 87360,) as described previously57, 58. Plasmids were transformed into One Shot™ Stbl3™ Chemically Competent E. coli (Invitrogen, C737303) and plated in ampicillin(100ug/mL) plates overnight at 37°C. Colonies were picked and sequenced using the LNX primer (AGCTCGTTTAGTGAACCGTCAGATC).
Immunofluorescence
RAW264.7 cells (1.2x106/well) were plated on 18mm glass coverslips (EMS; 72222-01) in 12 well plates and incubated overnight. After treatment with Sanofi-14h and DMXAA, cells were immediately fixed with 4% Paraformaldehyde for 10 min at RT and rinsed 3 times with PBS for 5 min each. Unless otherwise stated, all washes were done for 5 minutes with PBS using gentle rocking. Cells were then permeabilized with 0.2% Triton-X for 5 min at RT and quicky rinsed with PBS before blocking in a 5%BSA/%NGS solution for 30 min at RT. Samples were then incubated at 4 °C overnight with the TBK1(1:200) antibody. Samples were washed 3 times after primary antibody incubation and then incubated with Rhodamine Red-X (goat anti-rabbit) (ThermoFisher; R-6394) secondary antibody for 1.5h at RT. Samples were again rinsed 3 times with PBS before incubating with AF647-conjugated Calreticulin (Abcam, ab196159, 1:200) for 1h at RT. All antibodies were diluted in 1% BSA/PBS. Cells were rinsed 3 times with PBS and then quickly rinsed with ddH2O. Coverslips were mounted on slides using Fluorogel mounting media (Life Technologies, 17985-10) and dried overnight dried overnight at RT. Images were acquired using the 63x objective on a Zeiss LSM800 confocal microscope and processed using Imaris. Cells were analyzed for TBK1-positive puncta by manually counting using multi-point tool in ImageJ and statistical analysis were done using GraphPad Prism 9.0.
Immunoblot
Immunoblots were performed utilizing lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1mM EDTA, 1mM EGTA, 1 mM Beta-Glycerol-Phosphate, 2.5 mM Sodium-Pyrophosphate, 1% Triton-100X, 0.1% SDS) complemented with fresh protease (Sigma, 11697498001) and phosphatase inhibitors (Sigma, P0044-5ML). Proteins were transferred using the Trans-Blot Turbo RTA Mini 0.2 µm Nitrocellulose Transfer Kit (Bio-Rad, 1704270) in the Trans-Blot Turbo Transfer System (Bio-Rad 1704150). Membranes were then blocked for 1 hat RT in 5% dry milk and blotted with primaries overnight at 4 °C, after three washes with TBST the membranes were blotted with secondary for 1 hat RT in blocking buffer. The three washes were repeated and the membranes were imaged using Clarity Western ECL (Bio-Rad, 1705060) in the ChemiDoc Imaging System. Images were processed with the ImageJ software (https://imagej.nih.gov/ij/).
Co-immunoprecipitation
STING immunoprecipitation was performed as previously described 45. In brief, after treatment samples were lysed in IP buffer (20 mM Tris-HCl pH 7.4, 137 mM NaCl, 1 mM EDTA, 1% NP-40, 10% glycerol) complemented with fresh protease and phosphatase inhibitors. 700 μg of protein were mixed and rotated with 0.5 μg of STING antibody for 3h at 4 °C after which 25 μL of Dynabeads Protein G (Invitrogen, 10004D) were added to each sample and rotated for an additional hour. Samples were washed three times for 5 min with IP buffer at 4 °C. After the last wash, the supernatant was discarded, and samples were boiled in 4x loading buffer with 10% of beta mercaptoethanol for 5 min prior to SDS-PAGE and immunoblotting. Clean-Blot™ IP Detection Reagent (Thermo Fisher, 21230) was used for detection to avoid Ig heavy chain.
Nuclear fractionation
Cytoplasmic and nuclear fractions were obtained as previously described59 with modifications. Cells were washed with PBS and centrifuged at 2,300 RCF for 30 seconds and the supernatant discarded. Cells were resuspended in 250 μL of 0.1% NP-40 diluted in cold PBS (with protease and phosphatase inhibitors) and centrifuged at 4 °C at 2000 RCF for 2 min. The supernatant (cytoplasmic fraction) was removed without disturbing the nuclear pellet. The nuclear pellet was washed by gently resuspending with 1 mL of the 0.1% NP40 buffer and samples were centrifuged for 2 min at 2,000 RCF at 4 °C. The supernatant was discarded and the pellets were resuspended in RIPA buffer and sonicated for 10 min (at high with 30 sec on and 30 Sec off) in a water bath sonicator. After sonication pellets were centrifuged 4 °C at 16, 000 RCF for 10 minutes and protein from both fractions were quantified and immunoblotted. β-Tubulin was used as a cytoplasmic control while Lamin A/C was used as nuclear control.
ER fractionation
Cytoplasmic and membranous fractionation was performed as previously described60. Cells were washed with PBS after treatment, collected and centrifuged at 2, 300 RCF for 30 seconds. The supernatant was removed, and cells were resuspended in buffer 1 (200μg/mL Digitonin, 150 mM NaCl, 50 mM HEPES pH7.4 (Boston Bioproducts, NC0703855), ddH2O, fresh protease and phosphatase inhibitor) and put on ice for 10 min. Afterwards, they were centrifuged for 2 min at 2000 RCF at 4 °C and the supernatant (cytoplasmic fraction) was removed, and the pellets were washed with 1 mL of cold PBS. Pellets were resuspended in buffer 2 (1% NP-40, 150 mM NaCl, 50 mM HEPES pH7.4, ddH2O, fresh protease and phosphatase inhibitor) and left on ice for 30 min. After centrifugation, the supernatant was kept (membranous organelles) and the pellets were discarded. Samples were then quantified and immunoblotted.
Dimerization
The IRF3 dimerization procedure was adapted from the literature61 as follows: after treatment cells collected and lysed in dimerization Buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1mM EDTA, 1mM EGTA, 1 mM Beta-Glycerol-Phosphate, 2.5 mM Sodium-Pyrophosphate, 1% Triton-100X) for 10 min on ice. Samples were centrifuged for 10 min at maximum speed at 4 °C and protein was quantified. Proteins were mixed with 2x native sample buffer (Bio-Rad, 1610738) and resolved for 35 min at 200 V in precast gels with native running buffer (25 mM Tris and 192 mM glycine) complemented with 0.2% sodium deoxycholate in the cathode chamber prior to transfer and immunoblotting.
Reverse Transcriptase Quantitative PCR (RT-qPCR)
Total RNA was extracted using the Quick-RNA miniprep kit (Zymo Research, R1055). cDNA was prepared with the iScript™ cDNA Synthesis Kit (Bio-Rad, 1708891) and RT-qPCR was performed utilizing the iTaq™ Universal Probes Supermix (Bio-Rad, 1725131) according to the manufacture’s instruction. Mouse Taqman probes were ordered from ThermoFisher: IFNβ (Mm00439552_s1), SGK1 (Mm00441380_m1), SGK3 (Mm01227735_m1) and GAPDH (Mm99999915_g1). Human Taqman probes were also ordered from ThermoFisher: IFNβ (Hs01077958_s1) and GAPDH (Hs02786624_g1).
Herpes Simplex Virus (HSV)
HSV-1 KOS 62 was a kind gift from Dr. Steve Bachenheimer (University Of North Carolina at Chapel Hill) and was expanded as described63. Briefly, Vero cells were grown to 100% confluency and infected with HSV (0.01 MOI) in 10 cm plates. 4 days after infection the supernatant was harvested and centrifuged at 700 RCF for 10 min, then 25–30 mL of supernatant was ultracentrifuged in 5 mL of 20% sucrose in sterile ddH2O (overlayed on the supernatant) at 64, 512 RCF for 3h at 4 °C. The supernatant was discarded and the reminder viral solution was resuspended in PBS to a 1:100 ratio of the original supernatant from the Vero cells infection. The virus was then aliquoted and stored at −80 °C. Viral stocks were titled on Vero cells.
HSV infection of RAW 264.7 cells and BMDM
WT and KO RAW 264.7 cells were seeded (0.45x106 cells/well) in 12 well plates, then infected the next day with 1 mL of DMEM (without serum or P/S) containing HSV KOS (less pathogenetic, yet still infective HSV strain)64 (MOI 0.1) and left in the incubator for 1h. After this time, the media was replaced with fresh complete DMEM and cells were returned to the incubator. The cell supernatant was collected 24 and 48h after infection, centrifuged for 15 min at 1, 000 RCF and was used to perform a plaque assay in Vero cells as mentioned above. The resulting WT and KO pellets were used for RT-qPCR to determine the expression of IFNβ mRNA. WT RAW 264.7 (0.25x106 cells/well) were seeded in 12 well plates and treated the day after with 5 μM of Sanofi-14h for 24h. After pre-treatment cells were infected and the plaque assay was performed as described. BMDM were infected in 24 well plates (0.35x106 cells/well) with HSV (MOI 5) in 300 μL of DMEM for 1h. The media then was replaced with regular RPMI without LCCM, cells were returned to the incubator and after 24h the supernatant was harvested, centrifuged at 1, 972 RCF for 15 min and used to performed the plaque assay.
Sanofi-17a synthesis
Compound Sanofi-17a was synthesized according to the reported procedure using trans-1,4-cyclohexanediol instead of mixtures31. The final deprotection of THP was performed using HCl in diethyl ether and dichloromethane to minimize the by-product formation using alcoholic solvents. 1 hNMR spectra of Sanofi-17a matched the reported values.
RNA sequencing
WT and KO RAW 264.7 cells were plated and treated with DMXAA and Sanofi-14h (0.5 μM) for the indicated times. The RNA was extracted as described and sent to Novogene for sequencing. The details of the strategy used can be found in their website (https://www.novogene.com/us-en/services/research-services/transcriptome-sequencing/mrna-sequencing/).
In vitro kinase assay
The invitro kinase assay was performed as previously described65 with some modifications. Reactions were carried out at 30°C, 500 rpm for 30 minutes with “cold” ATP (Sigma, A6559-25UMO) and recombinant GST-TBK1(SignalChem, T02-10G) and Flag-IRF3 (Active-Motif, 31544) with and without various doses of Sanofi-14h.
QUANTIFICATION AND STATISTICAL ANALYSIS
Experimental analysis
GraphPad Prism version 9.0.0 for MAC was used to perform One-way ANOVA followed by Dunnett’s multiple comparisons test and the resulting statistical significance was defined by a P <0.05(*) based on standard error of the mean (SEM) calculations in figures 1B, 1C, 2A, 2C, 2G, 3B, 3C, 3F, 4A, 5B and supplemental figures 1A and 2B. This software was also use to calculate the inhibitors’ IC50 values based on the Nonlinear fit function. For all experiments a minimum sample size (N) of 3 was maintained. The shown figures are representative of these biological replicates. RT-qPCR results in figures 1C, 2A and supplemental figure 1A had at least 3 technical replicates.
RNA Sequencing analysis
Paired-end RNA-seq data in FASTQ format were mapped to the mouse reference genome, GRCm39 and Gencode mouse vM27 transcript annotation, using the spliced transcripts alignment to a reference (STAR) software66. Gene expression estimates as count of mapped sequence reads, and transcripts per million mapped reads (TPM) were estimated using the transcriptome assembler, StringTie267. Differential gene expression analysis was performed using linear models and the Bioconductor R package, limma, and voom normalization, starting from gene expression count68,69. Gene set enrichment analysis were performed on log2 transformed fold-changes (logFC) ranks, with the Bioconductor R package, fgsea70. Volcano plots and heatmaps were generated using the R package, ggplot2. ggplot271 and ComplexHeatmap72. Batch-corrected TPMs by ComBat method were obtained using the Bioconductor R package, sva73, and were used as starting input for the heatmaps. Unless indicated otherwise, the default differentially expressed genes (DEGs) high-lighted in volcano plots and heatmaps were filtered at Benjamini-Hochberg FDR adjusted p-value<0.01, and absolute logFC>1, and average log2 TPM>0.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| phospho-STAT3 Thr705 | Cell Signaling Technologies | Cat# 9145S |
| STAT3 | Cell Signaling Technologies | Cat# 12640S |
| phospho-TBK1 Ser172 | Cell Signaling Technologies | Cat# 5483S |
| TBK1 | Cell Signaling Technologies | Cat# 38066S |
| phospho-IRF3 Ser396 | Cell Signaling Technologies | Cat# 29047S |
| IRF3 | Cell Signaling Technologies | Cat# 4302S |
| STING | Cell Signaling Technologies | Cat# 50494S |
| phosphor-NDRG1 Thr346 | Cell Signaling Technologies | Cat# 5482 |
| NDRG1 | Cell Signaling Technologies | Cat# 9408 |
| GAPDH | Cell Signaling Technologies | Cat# 5174S |
| b-Tubulin | Cell Signaling Technologies | Cat# 2146S |
| Lamin A/C | Cell Signaling Technologies | Cat# 4777 |
| SGK1 | Cell Signaling Technologies | Cat# 12103 |
| SGK3 | Cell Signaling Technologies | Cat# 8154 |
| BiP | Cell Signaling Technologies | Cat# 3177T |
| STAT1 | Cell Signaling Technologies | Cat# 14994S |
| STAT2 | Cell Signaling Technologies | Cat# 4597S |
| IRF1 | Cell Signaling Technologies | Cat# 8478S |
| IRF4 | Cell Signaling Technologies | Cat# 62834T |
| IRF7 | Cell Signaling Technologies | Cat# 72073 |
| IRF9 | Cell Signaling Technologies | Cat# 28845S |
| IFNβ | Cell Signaling Technologies | Cat# 97450 |
| Anti-Rabbit | Promega | Cat# W4018 |
| Anti-Mouse | Promega | Cat# W4028 |
| Anti-Rabbit (IF) | ThermoFisher | Cat# R-6394 |
| Calreticulin | Abcam | Cat# ab196159 |
| Bacterial and virus strains | ||
| One Shot™ Stbl3™ Chemically Competent E. coli | Invitrogen | Cat# C737303 |
| Lentiviral particles | In house | |
| HSV-1 KOS | The University Of North Carolina at Chapel Hill | Dr. Steve Bachenheimer, laboratory |
| Biological samples | ||
| Human Monocytes | Healthy donor | UNC IRB 22-2450 |
| Bone marrow derived macrophages | Dr. David Pearce laboratory at the University of California San Francisco | SGK3 KO mice |
| Chemicals, peptides, and recombinant proteins | ||
| Human M-CSF | Peprotech | Cat# 300-25 |
| DMXAA | Cayman Chemical | Cat# 14617 |
| diABZI | SelleckChem | Cat# S8796 |
| LPS | Invivogen | Cat# tlrl-eblps |
| Sanofi-14h | MRC Pure Reagents | https://mrcppureagents.dundee.ac.uk/ |
| Sanofi-14h (SGK1-IN-2) | MedChemExpress | Cat# HY-135893 |
| GSK650394 | MedChemExpress | Cat# HY-15192 |
| EMD638683 | MedChemExpress | Cat# HY-15193 |
| SI113 | MedChemExpress | Cat# HY-117357 |
| GST-TBK1 | SignalChem | Cat# T02-10G |
| Flag-IRF3 | Active-Motif | Cat# 31544 |
| URMC-099 | MedChemExpress | Cat# HY-100556 |
| Tie2 Kinase inhibitor | MedChemExpress | Cat# HY-12599 |
| DMX-5084 | MedChemExpress | Cat# HY-111754 |
| MK2206 | MedChemExpress | Cat# HY-108232 |
| LY2584702 | MedChemExpress | Cat# HY-12493 |
| Polybrene | Santa Cruz Biotechnology | Cat# sc-134220 |
| Doxycycline hyclate | Sigma | Cat# D5207-1G |
| PMA | Sigma | Cat# P1585-1MG |
| Recombinant IFNβ | R&D systems | Cat# 8234-MB-010/CF |
| Sanofi-17a | In house | No catalog number |
| FuGENE HD | Promega | Cat# E2311 |
| Critical commercial assays | ||
| Mouse Ifnb1 taqman probe | ThermoFisher | Mm00439552_s1 |
| Mouse SGK1 taqman probe | ThermoFisher | Mm00441380_m1 |
| Mouse SGK3 taqman probe | ThermoFisher | Mm01227735_m1 |
| Mouse GAPDH taqman probe | ThermoFisher | Mm99999915_g1 |
| Human Ifnb1 taqman probe | ThermoFisher | Hs01077958_s1 |
| Human GAPDH taqman probe | ThermoFisher | Hs02786624_g1 |
| Deposited data | ||
| RNAseq of mouse RAW264.7 macrophages with CRISPR deletion of SGK1, SGK3, or SGK1/3 or treated with SGKi Sanofi-14h with or without DMXAA stimulation. | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE243816 | GSE243816 |
| Experimental models: Cell lines | ||
| L929 | ATCC | Cat# CCL-1 |
| RAW264.7 | UNC Tissue culture facility | No catalog number |
| THP-1 | UNC Tissue culture facility | No catalog number |
| Vero | UNC Tissue culture facility | No catalog number |
| HEK293T/17 | UNC Tissue culture facility | No catalog number |
| RAW-Dual Lucia | Invivogen | Cat# rawd-ismip |
| Experimental models: Organisms/strains | ||
| SGK3 knockout mouse | Dr. David Pearce, UCSF | MGI: Sgk3tm1Dpea |
| Oligonucleotides | ||
| gRNA targeting mSGK1 exon 5 | Integrated DNA technologies | GATTTCAGGTTCTT CTGGCT |
| gRNA targeting mSGK3 exon 3 | Integrated DNA technologies | TTCAAGACATTAAA TGCAG |
| LNX primer | Integrated DNA Technologies | AGCTCGTTTAGTG AACCGTCAGATC |
| Software and algorithms | ||
| STAR | Reference 66 | doi:10.1093/bioinformatics/bts635 |
| Prism | Dotmatics | https://www.graphpad.com/features |
| Image J | NIH | https://imagej.nih.gov/ij/download.html |
| StringTie2 | Reference 67 | doi:10.1186/s13059-019-1910-1 |
| Bioconductor R package, Lima and Voon normalization | References 68 and 69 | doi:10.1093/nar/gkv007 and doi:10.1186/gb-2014-15-2-r29 |
| R package fsea | References 70 | doi:10.1101/060012 |
| R package ggplot2. ggplot2 | References 71 | doi: 10.1007/978-0-387-98141-3_9 |
| R Package ComplexHeatmap | References 72 | doi:10.1093/bioinformatics/btw313 |
| R package sva | References 73 | doi:10.1093/bioinformatics/bts034 |
| Biorrender | Biorender | https://www.biorender.com/ |
SIGNIFICANCE.
Here we identify a role for the Serum- and Glucocorticoid-Regulated Kinases SGK1 and SGK3 in regulating innate immunity downstream of STING signaling. We show that SGK inhibitors such as Sanofi-14h or CRISPR deletion of SGK1 or SGK3 impairs the ability of macrophages to produce type 1 interferons and their associated transcriptional signature. SGK loss or inhibition markedly impairs macrophage defense against the DNA virus HSV-1. Sanofi-14h impairs STING binding of STING to the kinase TBK1, translocation of STING to membrane structures, and phosphorylation of STING, TBK1, and IRF3. Intriguingly, deletion of SGKs leaves these signaling events intact but impairs baseline expression of key transcription factors such as IRF7 and STAT1. While SGK inhibitors phenocopy the transcriptional effects of SGK deletion, SGKi can block IFN expression even in SGK1/3 double knockout macrophages, pointing to another target that participates in SGKi-mediated IFN suppression. We examine other members of the AGC kinase family and kinases predicted to be secondary targets of Sanofi-14h and find that none efficiently inhibit STING-activated IFN expression.
Highlights.
Sanofi-14h inhibits IFNβ production under STING stimulation.
This SGK inhibitor hinders STING-mediated TBK1 activation, not via SGK1/3
SGK1/3 are required for macrophage expression of IRF7 and STAT1.
Other AGC kinase inhibitors affect TBK1/IRF but do not impair IFNβ expression.
ACKNOWLEDGMENTS
We thank the members of the Baldwin and Hagan Labs for their technical and intellectual support, and Drs. Matthew McPeek, Alexia Perryman and Karel Alcedo for insightful discussion. We thank Dr. David Pearce at UCSF for providing the SGK3 KO mice. This research was supported by NIH grants R35-CA197684 to ASB and K08HL143271-01A1 and 1R03HL155249-01 to RSH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTEREST
The authors declare no competing financial interests or conflicting external relationships.
INCLUSION AND DIVERSITY
One of more of the authors of this paper self-identifies as an underrepresented ethnic minority. One of more of the authors of this paper self-identifies as member of the LGBTQ+ community.
References
- 1.McNab F, Mayer-Barber K, Sher A, Wack A, and O’Garra A (2015). Type I interferons in infectious disease. Nat Rev Immunol 15, 87–103. 10.1038/nri3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choubey D, and Moudgil KD (2011). Interferons in autoimmune and inflammatory diseases: Regulation and roles. Journal of Interferon and Cytokine Research 31, 857–865. 10.1089/jir.2011.0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davidson S, Crotta S, McCabe TM, and Wack A (2014). Pathogenic potential of interferon αβ in acute influenza infection. Nat Commun 5. 10.1038/ncomms4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biron CA (2001). Interferons α and β as immune regulators - A new look. Immunity 14, 661–664. 10.1016/S1074-7613(01)00154-6. [DOI] [PubMed] [Google Scholar]
- 5.Kim YM, and Shin EC (2021). Type I and III interferon responses in SARS-CoV-2 infection. Exp Mol Med 53, 750–760. 10.1038/s12276-021-00592-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wan D, Jiang W, and Hao J (2020). Research Advances in How the cGAS-STING Pathway Controls the Cellular Inflammatory Response. Front Immunol 11, 1–25. 10.3389/fimmu.2020.00615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ablasser A, and Chen ZJ (2019). CGAS in action: Expanding roles in immunity and inflammation. Science (1979) 363. 10.1126/science.aat8657. [DOI] [PubMed] [Google Scholar]
- 8.Sun L, Wu J, Du F, Chen X, and Chen ZJ (2013). Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science (1979) 339, 786–791. 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang X, Bai X chen, and Chen, Z.J. (2020). Structures and Mechanisms in the cGAS-STING Innate Immunity Pathway. Immunity 53, 43–53. 10.1016/j.immuni.2020.05.013. [DOI] [PubMed] [Google Scholar]
- 10.Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV, et al. (2015). Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science (1979) 347. 10.1126/science.aaa2630. [DOI] [PubMed] [Google Scholar]
- 11.Yamashiro LH, Wilson SC, Morrison HM, Karalis V, Chung JYJ, Chen KJ, Bateup HS, Szpara ML, Lee AY, Cox JS, et al. (2020). Interferon-independent STING signaling promotes resistance to HSV-1 in vivo. Nat Commun 11. 10.1038/s41467-020-17156-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mogensen TH (2019). IRF and STAT transcription factors - From basic biology to roles in infection, protective immunity, and primary immunodeficiencies. Front Immunol 10, 1–13. 10.3389/fimmu.2018.03047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barber GN (2014). STING-dependent cytosolic DNA sensing pathways. Trends Immunol 35, 88–93. 10.1016/j.it.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 14.Castel P, Ellis H, Bago R, Toska E, Razavi P, Carmona FJ, Kannan S, Verma CS, Dickler M, Chandarlapaty S, et al. (2016). PDK1-SGK1 Signaling Sustains AKT-Independent mTORC1 Activation and Confers Resistance to PI3Kα Inhibition. Cancer Cell 30, 229–242. 10.1016/j.ccell.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Cristofano A (2017). SGK1: The Dark Side of PI3K Signaling. Curr Top Dev Biol 123, 49–71. 10.1016/bs.ctdb.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guerriero I, Monaco G, Coppola V, and Orlacchio A (2020). Serum and glucocorticoid-inducible kinase 1 (Sgk1) in nsclc therapy. Pharmaceuticals 13, 1–25. 10.3390/ph13110413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pearce LR, Komander D, and Alessi DR (2010). The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol 11, 9–22. 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- 18.Villoslada P, Vila G, Colafrancesco V, Moreno B, Fernandez-Diez B, Vazquez R, Pertsovskaya I, Zubizarreta I, Pulido-Valdeolivas I, Messeguer J, et al. (2019). Axonal and Myelin Neuroprotection by the Peptoid BN201 in Brain Inflammation. Neurotherapeutics 16, 808–827. 10.1007/s13311-019-00717-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gasser JA, Inuzuka H, Lau AW, Wei W, Beroukhim R, and Toker A (2014). SGK3 Mediates INPP4B-Dependent PI3K Signaling in Breast Cancer. Mol Cell 56, 595–607. 10.1016/j.molcel.2014.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murray JT, Campbell DG, Morrice N, Auld GC, Shpiro N, Marquez R, Peggie M, Bain J, Bloomberg GB, Grahammer F, et al. (2004). Exploitation of KESTREL to identify NDRG family members as physiological substrates for SGK1 and GSK3. Biochemical Journal 384, 477–488. 10.1042/BJ20041057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gamper N, Fillon S, Feng Y, Friedrich B, Lang PA, Henke G, Huber SM, Lang F, Kobayashi T, and Cohen P (2002). K+ channel activation by all three isoforms of serum- and glucocorticoid-dependent protein kinase SGK. Pflugers Arch 445, 60–66. 10.1007/s00424-002-0873-2. [DOI] [PubMed] [Google Scholar]
- 22.Ahmed M, Fezai M, Uzcategui NL, Hosseinzadeh Z, and Lang F (2016). SGK3 Sensitivity of Voltage Gated K + Channel K v1.5 (KCNA5). Cellular Physiology and Biochemistry 38, 359–367. 10.1159/000438636. [DOI] [PubMed] [Google Scholar]
- 23.Bago R, Sommer E, Castel P, Crafter C, Bailey FP, Shpiro N, Baselga J, Cross D, Eyers PA, and Alessi DR (2016). The hVps34‐ SGK 3 pathway alleviates sustained PI3K/Akt inhibition by stimulating mTORC 1 and tumour growth. EMBO J 35, 1902–1922. 10.15252/embj.201693929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ranzuglia V, Lorenzon I, Pellarin I, Sonego M, Dall’Acqua A, D’Andrea S, Lovisa S, Segatto I, Coan M, Polesel J, et al. (2020). Serum- and glucocorticoid-inducible kinase 2, SGK2, is a novel autophagy regulator and modulates platinum drugs response in cancer cells. Oncogene 39, 6370–6386. 10.1038/s41388-020-01433-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sang Y, Kong P, Zhang S, Zhang L, Cao Y, Duan X, Sun T, Tao Z, and Liu W (2021). SGK1 in Human Cancer: Emerging Roles and Mechanisms. Front Oncol 10, 1–17. 10.3389/fonc.2020.608722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Basnet R, Gong GQ, Li C, and Wang MW (2018). Serum and glucocorticoid inducible protein kinases (SGKs): a potential target for cancer intervention. Acta Pharm Sin B 8, 767–771. 10.1016/j.apsb.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang X, Lan C, Zhou J, Fu W, Long X, An Y, Jiao G, Wang K, Li Y, Xu J, et al. (2017). Development of a new analog of SGK1 inhibitor and its evaluation as a therapeutic molecule of colorectal cancer. J Cancer 8, 2256–2262. 10.7150/jca.19566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Towhid ST, Liu GL, Ackermann TF, Beier N, Scholz W, Fuchß T, Toulany M, Rodemann HP, and Lang F (2013). Inhibition of colonic tumor growth by the selective SGK inhibitor EMD638683. Cellular Physiology and Biochemistry 32, 838–848. 10.1159/000354486. [DOI] [PubMed] [Google Scholar]
- 29.D’Antona L, Amato R, Talarico C, Ortuso F, Menniti M, Dattilo V, Iuliano R, Gigliotti F, Artese A, Costa G, et al. (2015). SI113, a specific inhibitor of the Sgk1 kinase activity that counteracts cancer cell proliferation. Cellular Physiology and Biochemistry 35, 2006–2018. 10.1159/000374008. [DOI] [PubMed] [Google Scholar]
- 30.Halland N, Schmidt F, Weiss T, Saas J, Li Z, Czech J, Dreyer M, Hofmeister A, Mertsch K, Dietz U, et al. (2015). Discovery of N-[4-(1H-pyrazolo[3,4-b]pyrazin-6-yl)-phenyl]-sulfonamides as highly active and selective SGK1 inhibitors. ACS Med Chem Lett 6, 73–78. 10.1021/ml5003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Halland N, Schmidt F, Weiss T, Li Z, Czech J, Saas J, Ding-Pfennigdorff D, Dreyer MK, Strübing C, and Nazare M (2021). Rational Design of Highly Potent, Selective, and Bioavailable SGK1 Protein Kinase Inhibitors for the Treatment of Osteoarthritis. J Med Chem 10.1021/acs.jmedchem.1c01601. [DOI] [PubMed] [Google Scholar]
- 32.Zhou H, Gao S, Duan X, Liang S, Scott DA, Lamont RJ, and Wang H (2015). Inhibition of serum-and glucocorticoid-inducible kinase 1 enhances TLR-mediated inflammation and promotes endotoxin-driven organ failure. FASEB Journal 29, 3737–3749. 10.1096/fj.15-270462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ren J, Han X, Lohner H, Liang R, Liang S, and Wang H (2021). Serum- and Glucocorticoid-Inducible Kinase 1 Promotes Alternative Macrophage Polarization and Restrains Inflammation through FoxO1 and STAT3 Signaling. The Journal of Immunology 207, 268–280. 10.4049/jimmunol.2001455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Borst O, Schaub M, Walker B, Schmid E, Münzer P, Voelkl J, Alesutan I, Rodríguez JM, Vogel S, Schoenberger T, et al. (2015). Pivotal role of serum- and glucocorticoid-inducible kinase 1 in vascular inflammation and atherogenesis. Arterioscler Thromb Vasc Biol 35, 547–557. 10.1161/ATVBAHA.114.304454. [DOI] [PubMed] [Google Scholar]
- 35.Schmid E, Bhandaru M, Nurbaeva MK, Yang W, Szteyn K, Russo A, Leibrock C, Tyan L, Pearce D, Shumilina E, et al. (2012). SGK3 Regulates Ca 2+ Entry and Migration of Dendritic Cells. Cellular Physiology and Biochemistry 30, 1423–1435. 10.1159/000343330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tai DJC, Su CC, Ma YL, and Lee EHY (2009). SGK1 phosphorylation of IκB kinase α and p300 up-regulates NF-κB activity and increases N-methyl-D-aspartate receptorNR2A and NR2B expression. Journal of Biological Chemistry 284, 4073–4089. 10.1074/jbc.M805055200. [DOI] [PubMed] [Google Scholar]
- 37.Zhang L, Cui R, Cheng X, and Du J (2005). Antiapoptotic effect of serum and glucocorticoid-inducible protein kinase is mediated by novel mechanism activating IκB kinase. Cancer Res 65, 457–464. [PubMed] [Google Scholar]
- 38.Prantner D, Perkins DJ, Lai W, Williams MS, Sharma S, Fitzgerald KA, and Vogel SN (2012). 5,6-Dimethylxanthenone-4-acetic acid (DMXAA) activates stimulator of interferon gene (STING)-dependent innate immune pathways and is regulated by mitochondrial membrane potential. Journal of Biological Chemistry 287, 39776–39788. 10.1074/jbc.M112.382986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu Q, Hu H, Liu H, Shen H, Yan Z, and Gao L (2020). A synthetic STING agonist inhibits the replication of human parainfluenza virus 3 and rhinovirus 16 through distinct mechanisms. Antiviral Res 183, 104933. 10.1016/j.antiviral.2020.104933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hopfner KP, and Hornung V (2020). Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat Rev Mol Cell Biol 21, 501–521. 10.1038/s41580-020-0244-x. [DOI] [PubMed] [Google Scholar]
- 41.Hsia HC, Hutti JE, and Baldwin AS (2017). Cytosolic DNA promotes signal transducer and activator of transcription 3 (STAT3) phosphorylation by TANK-binding kinase 1 (TBK1) to restrain STAT3 activity. Journal of Biological Chemistry 292, 5405–5417. 10.1074/jbc.M116.771964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grahammer F, Artunc F, Sandulache D, Rexhepaj R, Friedrich B, Risler T, McCormick JA, Dawson K, Wang J, Pearce D, et al. (2006). Renal function of gene-targeted mice lacking both SGK1 and SGK3. Am J Physiol Regul Integr Comp Physiol 290, 945–950. 10.1152/ajpregu.00484.2005. [DOI] [PubMed] [Google Scholar]
- 43.Tsai MH, Pai LM, and Lee CK (2019). Fine-tuning of type I interferon response by STAT3. Front Immunol 10, 1–10. 10.3389/fimmu.2019.01448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Panda D, Gjinaj E, Bachu M, Squire E, Novatt H, Ozato K, and Rabin RL (2019). IRF1 maintains optimal constitutive expression of antiviral genes and regulates the early antiviral response. Front Immunol 10, 1–16. 10.3389/fimmu.2019.01019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang F, Alain T, Szretter KJ, Stephenson K, Pol JG, Atherton MJ, Hoang HD, Fonseca BD, Zakaria C, Chen L, et al. (2016). S6K-STING interaction regulates cytosolic DNA-mediated activation of the transcription factor IRF3. Nat Immunol 17, 514–522. 10.1038/ni.3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bruhn MA, Pearson RB, Hannan RD, and Sheppard KE (2013). AKT-independent PI3-K signaling in cancer - Emerging role for SGK3. Cancer Manag Res 5, 281–292. 10.2147/CMAR.S35178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ning S, Pagano JS, and Barber GN (2011). IRF7: Activation, regulation, modification and function. Genes Immun 12, 399–414. 10.1038/gene.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Platanitis E, Demiroz D, Schneller A, Fischer K, Capelle C, Hartl M, Gossenreiter T, Müller M, Novatchkova M, and Decker T (2019). A molecular switch from STAT2-IRF9 to ISGF3 underlies interferon-induced gene transcription. Nat Commun 10, 1–17. 10.1038/s41467-019-10970-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sasaki K, Terker AS, Pan Y, Li Z, Cao S, Wang Y, Niu A, Wang S, Fan X, Zhang MZ, et al. (2021). Deletion of myeloid interferon regulatory factor 4 (Irf4) in mouse model protects against kidney fibrosis after ischemic injury by decreased macrophage recruitment and activation 10.1681/ASN.2020071010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ackermann TF, Boini KM, Beier N, Scholz W, Fuchß T, and Lang F (2011). EMD638683, a Novel SGK Inhibitor with Antihypertensive Potency. Cellular Physiology and Biochemistry 28, 137–146. 10.1159/000331722. [DOI] [PubMed] [Google Scholar]
- 51.Kalidasan V, Ng WH, Ishola OA, Ravichantar N, Tan JJ, and Das KT (2021). A guide in lentiviral vector production for hard-to-transfect cells, using cardiac-derived c-kit expressing cells as a model system. Sci Rep 11, 1–11. 10.1038/s41598-021-98657-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li XL, Pongor L, Tang W, Das S, Muys BR, Jones MF, Lazar SB, Dangelmaier EA, Hartford CCR, Grammatikakis I, et al. (2020). A small protein encoded by a putative lncrna regulates apoptosis and tumorigenicity in human colorectal cancer cells. Elife 9, 1–44. 10.7554/eLife.53734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCormick JA, Feng Y, Dawson K, Behne MJ, Yu B, Wang J, Wyatt AW, Henke G, Grahammer F, Mauro TM, et al. (2004). Targeted Disruption of the Protein Kinase SGK3/CISK Impairs Postnatal Hair Follicle Development. Mol Biol Cell 15, 4278–4288. 10.1091/mbc.e04-01-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marim FM, Silveira TN, Lima DS, and Zamboni DS (2010). A Method for Generation of Bone Marrow-Derived Macrophages from Cryopreserved Mouse Bone Marrow Cells. PLoS One 5, 1–8. 10.1371/journal.pone.0015263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zamboni DS, and Rabinovitch M (2003). Nitric oxide partially controls Coxiella burnetii phase II infection in mouse primary macrophages. Infect Immun 71, 1225–1233. 10.1128/IAI.71.3.1225-1233.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Englen MD, Valdez YE, Lehnert NM, and Lehnert BE (1995). Granulocyte/macrophage colony-stimulating factor is expressed and secreted in cultures of murine L929 cells. J Immunol Methods 184, 281–283. 10.1016/0022-1759(95)00136-X. [DOI] [PubMed] [Google Scholar]
- 57.Sanjana #1, N.E., Shalem #1, O., and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening HHS Public Access Supplementary Material. Nat Methods 11, 783–784. 10.1038/nmeth.3047.Improved. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boukhaled GM, Harding S, and Brooks DG (2021). Opposing Roles of Type I Interferons in Cancer Immunity. Annual Review of Pathology: Mechanisms of Disease 16, 167–198. 10.1146/annurev-pathol-031920-093932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Suzuki K, Bose P, Leong-Quong RY, Fujita DJ, and Riabowol K (2010). REAP: A two minute cell fractionation method. BMC Res Notes 3, 294. 10.1186/1756-0500-3-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Holden P, and Horton W (2009). Crude subcellular fractionation of cultured mammalian cell lines. BMC Res Notes 2. 10.1186/1756-0500-2-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Iwamura T, Yoneyama M, Yamaguchi K, Suhara W, Mori W, Shiota K, Okabe Y, Namiki H, and Fujita T (2001). Induction of IRF-3/-7 kinase and NF-κB in response to double-stranded RNA and virus infection: Common and unique pathways. Genes to Cells 6, 375–388. 10.1046/j.1365-2443.2001.00426.x. [DOI] [PubMed] [Google Scholar]
- 62.Macdonald SJ, Mostafa HH, Morrison LA, and Davido DJ (2012). Genome Sequence of Herpes Simplex Virus 1 Strain KOS. J Virol 86, 6371–6372. 10.1128/jvi.00646-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang L, Mo J, Swanson KV, Wen H, Petrucelli A, Gregory SM, Zhang Z, Schneider M, Jiang Y, Fitzgerald KA, et al. (2014). NLRC3, a Member of the NLR Family of Proteins, Is a Negative Regulator of Innate Immune Signaling Induced by the DNA Sensor STING. Immunity 40, 329–341. 10.1016/j.immuni.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Macdonald SJ, Mostafa HH, Morrison LA, and Davido DJ (2012). Genome Sequence of Herpes Simplex Virus 1 Strain KOS. J Virol 86, 6371–6372. 10.1128/jvi.00646-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hu L, Xie H, Liu X, Potjewyd F, James LI, Wilkerson EM, Herring LE, Xie L, Chen X, Cabrera JC, et al. (2020). TBK1 Is a Synthetic Lethal Target in Cancer with VHL Loss. Cancer Discov 10, 460–475. 10.1158/2159-8290.CD-19-0837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kovaka S, Zimin AV, Pertea GM, Razaghi R, Salzberg SL, and Pertea M (2019). Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol 20, 1–13. 10.1186/s13059-019-1910-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Law CW, Chen Y, Shi W, and Smyth GK (2014). voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15, R29. 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Korotkevich G, Sukhov V, and Sergushichev A (2019). Fast gene set enrichment analysis. Preprint of BioRxiv. bioRxiv [Google Scholar]
- 71.Wickham H (2009). Scales, axes and legends. In ggplot2 (Springer New York; ), pp. 91–113. [Google Scholar]
- 72.Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 73.Leek JT, Johnson WE, Parker HS, Jaffe AE, and Storey JD (2012). The SVA package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883. 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNAseq data has been uploaded to NCBI GEO with accession number GSE243816 and it is publicly accessible.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available upon request from the lead contact.