

Abstract
Retrotransposons are highly enriched in the animal genome1–3. Their activation can rewrite host DNA information and fundamentally impact host biology1–3. While developmental activation of retrotransposons can bring hosts benefits, such as against virus infection, uncontrolled activation promotes diseases or potentially drives aging1–5. Upon activation, retrotransposons use their mRNA as templates to synthesize double-stranded DNA for making new insertions in the host genome1–3,6. While the reverse transcriptase encoded by them can synthesize the 1st-strand DNA1–3,6, how the 2nd-strand DNA is generated remains largely unknown. Here we report retrotransposons hijack the alternative end-joining (alt-EJ) DNA repair process from the hosts for a circularization step to synthesize their 2nd-strand DNA. We applied Nanopore sequencing to examine the fates of replicated retrotransposon DNA, and found that 10% of them achieve new insertions, while 90% exist as extrachromosomal circular DNA (eccDNA). Using eccDNA production as a readout, further genetic screens identified factors from alt-EJ as essential for retrotransposon replication. alt-EJ drives the 2nd-strand synthesis of the LTR-retrotransposon DNA via a circularization process, thus is necessitated for eccDNA production and new insertions. Together, our study reveals that alt-EJ is essential in driving the propagation of parasitic genomic retroelements. Our work uncovers a novel conserved function of this understudied DNA repair process, and provides a new perspective to understand, and potentially control, retrotransposon life cycle.
Retrotransposons abundantly occupy the genomes of nearly all animals, comprising almost 38% of human DNA1–3. During evolution, given their ability to mobilize and (re)wire gene regulation, retrotransposons bring one source of genome dynamics to (re)writing DNA sequences. Within one generation, the nucleic acids generated during their replication cycles can be highly immunogenic and their mobilization step produces DNA breaks and generates mutations1–3. In the past, retrotransposon activation was largely considered as deleterious to the hosts, from causing animal infertility, contributing to diseases, such as cancer, hemophilia, or neurodegenerative disorders, to potentially driving aging1–3. Notably, recent studies showed that programmed retrotransposon activation during animal development can bring hosts benefits, such as fending off invading viruses4,5. Despite the fundamental impacts brought from these parasitic genomic elements, how retrotransposons fulfill their life cycle to modulate host physiology and pathology remains unclear.
Our previous efforts established Drosophila oogenesis as a platform to precisely characterize retrotransposon activity at the mobilization level within an animal7. We found that retrotransposons rarely mobilize in germline stem cells7, which upon differentiation produce developing oocytes and supporting nurse cells8. Instead, retrotransposons use nurse cells as factories to massively manufacture themselves like viruses 7. Then they transport the virus-like particles into the oocyte and mobilize into the genome that will be transmitted to the next generation7. Leveraging this unique biological system that allows us to spatiotemporally follow their activation process, we sought to characterize how retrotransposons generate new copies of integrated DNA and (re)write the host germline genome.
RESULTS
Engineered reporter mainly produces eccDNA
The Drosophila genome is enriched with both DNA transposons and retrotransposons, which comprise LTR and non-LTR retrotransposons9,10. For these transposon families, very few can achieve mobilization into oocytes7. Among them, the LTR-retrotransposon HMS-Beagle displays the highest mobilization rate in oocytes7,11. To thoroughly investigate its mobilization, we generated a fly strain carrying one copy of eGFP-tagged HMS-Beagle (Extended Data Fig. 1a). Landing it into a specific site of the fly genome, this eGFP-tagged HMS-Beagle serves as the sole precursor for any newly integrated copies that harbor eGFP sequences (Fig. 1a,b). To potentially capture the bona fide mobilization events from this tagged HMS-Beagle within oocytes, we sequenced their genome using Nanopore technology, which can directly read DNA up to mega bases without PCR amplification.
Fig. 1 |. HMS-Beagle predominantly produces eccDNA upon activation.
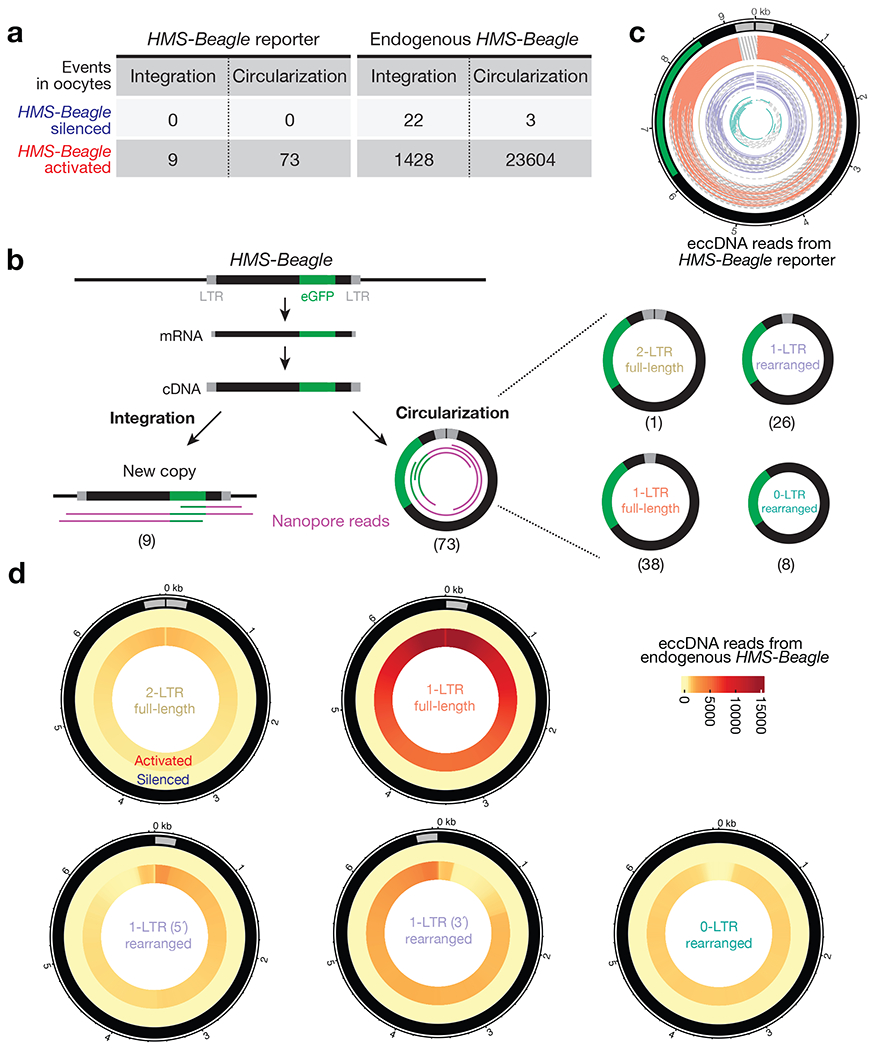
a, Table to summarize the outcomes of replicated HMS-Beagle DNA detected in Drosophila oocytes. HMS-Beagle activation is achieved by suppressing Aub and Ago3 during oogenesis. b, Workflow to characterize the integration and circularization (eccDNA) events from an engineered HMS-Beagle reporter. The Nanopore sequencing reads were classified as integration by having flanking sequences mapped to the genome or as eccDNA by containing end-to-end junction sites. The eccDNA was further classified into four categories based on their structures. The numbers within parentheses denote the number of reads identified for each type of event. c, eccDNA reads from engineered HMS-Beagle reporter. Each circle represents a read: the solid part represents the sequenced region, and the dashed line represents the gap filled computationally. Salmon: reads supporting 1-LTR full-length circles; gold: one read supporting 2-LTR full-length circle; purple: reads supporting 1-LTR rearranged circles, likely resulting from autointegration; dark green: eccDNA reads do not contain intact LTR. d, Circos plot showing eccDNA reads from endogenous HMS-Beagle. Color scores indicate the mapping coverage throughout the full-length HMS-Beagle consensus. Outer layer: eccDNA reads from HMS-Beagle silenced oocytes. Inner layer: eccDNA reads from HMS-Beagle activated oocytes. Data for this figure were generated from flies carrying sh-aub and sh-ago3 to trigger transposon activation.
As expected, in the oocytes with transposons silenced (Extended Data Fig. 1b), the eGFP-tagged HMS-Beagle was only detected at its original landing site (Extended Data Fig. 1c). We next triggered transposon activation by depleting Aub and Ago3 (Extended Data Fig. 1b), which are two key factors from a small RNA (piRNA) based silencing system that suppresses transposons during Drosophila oogenesis12–14. Under this condition, we detected 9 new insertions from the tagged HMS-Beagle with 28X genome coverage (Fig. 1a and Extended Data Fig. 1d), consistent with our previous finding that HMS-Beagle preferentially targets the oocyte genome for integration7.
Remarkably, manually analyzing the eGFP-derived reads that did not support integration indicated the formation of circular DNA. Since we prepared genomic libraries by the Tn5 tagmentation method, this linearizes any circular DNA molecules. Sequencing such molecules allows us to quantify these DNA circles by searching for reads that cover the end-to-end junctions. By examining these events, we found that no circles formed when HMS-Beagle is silenced (Fig. 1a). However, upon triggering its activation (Extended Data Fig. 1b), we observed 73 reads that support the production of circular DNA from eGFP-tagged HMS-Beagle with 28-fold genome coverage (Fig. 1a,b), which is 8.1-fold more abundant than observed integration events. Given that HMS-Beagle has one LTR at each end, a head-to-tail circle would generate a junction read possessing two LTRs. However, among these 73 circle-supporting reads, only one has a junction with two LTRs (Fig. 1b,c), suggesting that the formation of 2-LTR full-length circles is a rare event. In contrast, 38 reads have junctions covering the start and end positions of the engineered HMS-Beagle reporter by containing one LTR (Fig. 1b,c), indicating that 1-LTR full-length circles are the dominant circular form. Among the remaining 34 reads that encompass partial HMS-Beagle sequences (termed as “rearranged circles”), 26 still have one intact LTR (termed as “1-LTR rearranged”). The remaining 8 reads do not contain intact LTRs (termed as “0-LTR rearranged”). Given their circular nature, we designated these HMS-Beagle-derived circles as extrachromosomal circular DNA (eccDNA). Collectively, our data suggest that upon activation, our engineered HMS-Beagle abundantly produced eccDNA as 1-LTR full-length circles, but achieved far fewer integration events.
Endogenous copies preferentially form eccDNA
Our findings from the engineered HMS-Beagle prompted us to explore whether its endogenous copies also form circular DNA upon activation. We accordingly mined our Nanopore sequencing data to characterize the reads that support either integrated DNA or eccDNA events from endogenous HMS-Beagle. For control oocytes, in which transposons are silenced, we detected 0.8 potential integrations and 0.1 potential eccDNA from endogenous HMS-Beagle per genome coverage (Fig. 1a). These integration events most likely reflect polymorphisms between the genome of the fly strain used in this study and the Drosophila reference genome, hence defining the false-positive rate of our methodology on probing transposition events.
Upon transposon activation, we detected 1428 integrations and 23604 eccDNAs from endogenous HMS-Beagle loci with 28X genome coverage (Fig. 1a,d), highlighting that 94.3% of the replication products from HMS-Beagle form circles. Similar to the eGFP tagged reporter, endogenous HMS-Beagle appears to also primarily form 1-LTR full-length circles: 54.7% of eccDNA reads support 1-LTR full-length circles, 6.4% of eccDNA reads indicate 2-LTR full-length circles, and 38.9% of eccDNA reads are derived from rearranged HMS-Beagle circles (Fig. 1d). We concluded that, consistent with the observations from our reporter, endogenous HMS-Beagle also dominantly forms 1-LTR eccDNA.
To validate the formation of eccDNA, we designed a set of divergent PCR primers, which would only give a PCR product upon the circularization of HMS-Beagle DNA (Fig. 2a and Extended Data Fig. 2). Given that HMS-Beagle replication occurs within the ovary during oogenesis7, we reasoned that their eccDNA is readily generated within ovaries before egg laying. By using ovary DNA as a template, performing PCR with divergent primers generated two products with distinct sizes (Extended Data Fig. 2). Sanger sequencing revealed that the upper faint band is derived from eccDNA with two LTRs (Extended Data Fig. 2). In contrast, the lower sharp band contains PCR product from 1-LTR eccDNA (Extended Data Fig. 2). Thus, our data consistently indicate that upon activation, endogenous HMS-Beagle preferentially generates eccDNA, especially in the form of 1-LTR circles.
Fig. 2 |. Factors from the alt-EJ process drive 1-LTR full-length eccDNA formation.
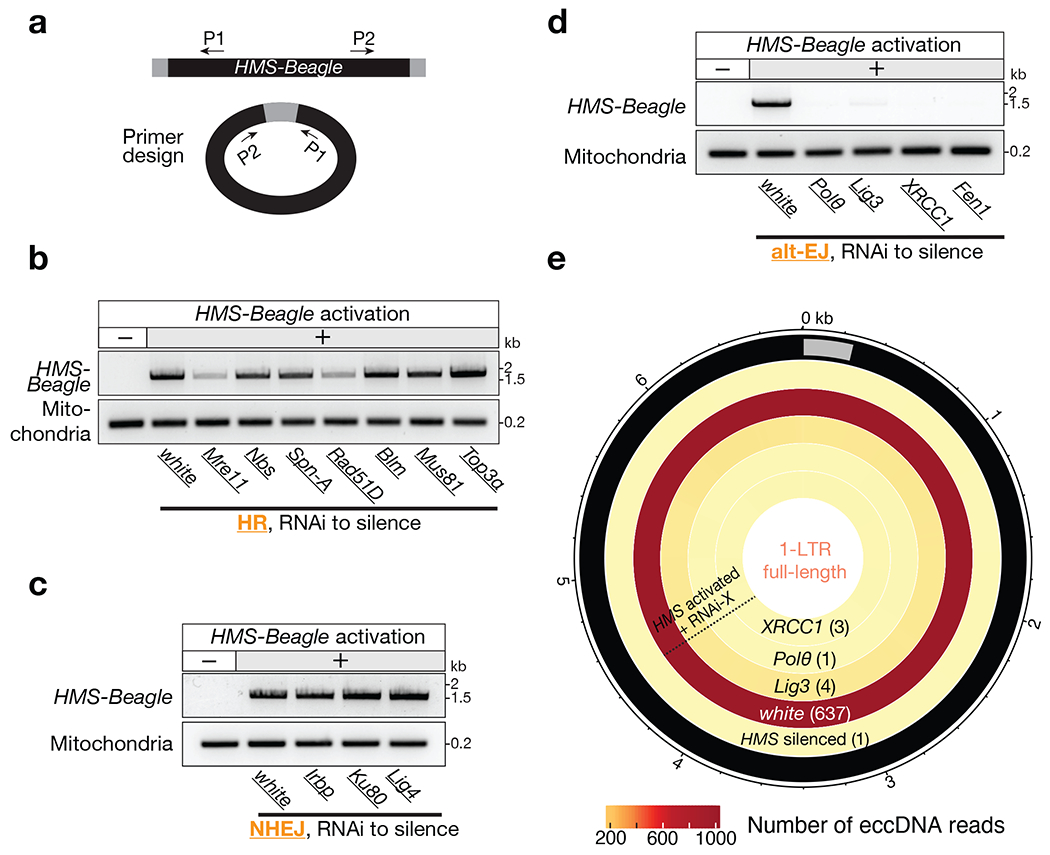
a, Schematic of the design of divergent primers to identify HMS-Beagle eccDNA. b-d, The representative gel image to show whether components from the HR (b), NHEJ (c), or alt-EJ (d) process are required for the formation of 1-LTR full-length eccDNA. e, Circos plot showing 1-LTR full-length eccDNA reads from endogenous HMS-Beagle. Color scores indicate the mapping coverage throughout the full-length HMS-Beagle consensus. The numbers within parentheses denote the number of reads identified from each genotype. Data for this figure were generated from flies carrying sh-aub to trigger transposon activation.
Sequencing genomic DNA by tagmentation can indicate the formation of eccDNA by capturing the end-to-end junctions. However, this method lacks the power to validate their circularity or to reconstruct the complete circular sequences of eccDNA. To obtain strong and direct evidence of circle formation, we established a Nanopore-based eccDNA sequencing method (eccDNA-Seq, Extended Data Fig. 3a). Our method appears to outperform recently published protocols for capturing large eccDNA: While the published protocols produced reads with N50 < 5,000 bp15,16, our method consistently generated reads with N50 at around 15,000 bp (Supplementary Table 1). Applying eccDNA-Seq to normal fly ovaries generated reads that mainly mapped to the mitochondrial genome (83.8% of total reads, Extended Data Fig. 3b), which is circular. Given that mitochondria only take ~0.6% of the Nanopore sequencing space when the total fly ovarian DNA is sequenced (Extended Data Fig. 3b), we conclude that our eccDNA-Seq can enrich circular DNA by 139-fold. This highlights the high efficiency of our method on enriching circular DNA for sequencing. Spiking in a plasmid as an internal control for circular DNA quantification revealed that the amount of mitochondrial DNA remains unchanged upon transposon activation (Extended Data Fig. 3c). Hence, the eccDNA-Seq reads from transposons were normalized to mitochondrial DNA reads across samples. In these control samples, HMS-Beagle generated very few, if any, eccDNA (Extended Data Fig. 3d). By contrast, upon its activation, we detected 13362 HMS-Beagle eccDNA (Extended Data Fig. 3d). Consistent with our findings from genomic sequencing and PCR-based method, 61.32% of HMS-Beagle circles detected by eccDNA-Seq were 1-LTR full-length circles (Extended Data Fig. 3d). In summary, our eccDNA-Seq data provide strong and direct evidence of the formation of eccDNA from HMS-Beagle in vivo.
alt-EJ is required for eccDNA production
We next sought to understand the mechanism of eccDNA biogenesis, thus potentially providing insights into retrotransposon DNA replication cycle. Retrotransposons use their RNA transcripts as a template for reverse transcription to generate DNA for subsequent integration17. This replication intermediate could be the source for eccDNA biogenesis. This hypothesis predicts that depleting transposon RNA during oogenesis would abrogate eccDNA production. Indeed, once we depleted HMS-Beagle RNA by RNAi (Extended Data Fig. 4a), their eccDNA production was abolished (Extended Data Fig. 4b). Hence, our data indicate that retrotransposon-derived eccDNA is produced from their DNA replication intermediates, leaving their original genomic loci intact. This is different from the previously reported mechanisms on driving eccDNA formation, which involve either genomic DNA fragmentation or recombination within the genome15,18–20.
For exogenous retroviruses and retroviral elements embedded in the host genome, it has been proposed that the homologous recombination (HR) pathway can mediate the recombination of the two LTRs from the replicated linear DNA for the formation of 1-LTR circles6,21. To understand how HMS-Beagle forms 1-LTR eccDNA, we first tested the function of HR machinery proteins during this process. However, after individually depleting 7 key factors linked to this pathway during oogenesis––Nbs, Spn-A (Drosophila homolog of mammalian Rad51), Rad51D, Blm, Mre11, Mus81, and Top3a––HMS-Beagle still formed 1-LTR circles (Fig. 2b). These data argue against a previously proposed function from the HR pathway in eccDNA biogenesis. Additionally, silencing of key factors from the non-homologous end joining (NHEJ) pathway (Irbp, Ku80, or DNA ligase 4) also had no impact on the formation of HMS-Beagle-derived eccDNA (Fig. 2c).
To systematically characterize the factors that are essential for HMS-Beagle eccDNA formation, we performed a candidate-based RNAi screen to individually deplete 123 factors (from 135 alleles) that are known to function in DNA repair or DNA damage response (Supplementary Table 2). After depleting each factor during oogenesis, we examined the production of HMS-Beagle 1-LTR circles by the PCR method we established (Extended Data Fig. 2). Among these factors, 23 lead to lethality, impeding any further investigation (Supplementary Table 2). From the rest of the candidates, there were four factors screened as essential for HMS-Beagle 1-LTR eccDNA production: DNA polymerase θ (Polθ, encoded by the polQ gene), XRCC1, DNA ligase 3 (Lig3), and Fen1 (Fig. 2d, Extended Data Fig. 5, and Supplementary Table 2). Interestingly, all four of these factors have been proposed to work coordinately for the alternative end-joining (alt-EJ) DNA repair process (also known as the microhomology-mediated end-joining, MMEJ) 22–24.
To further validate the function of alt-EJ factors on driving eccDNA production, we performed eccDNA-Seq upon individually depleting three of the identified factors: Polθ, XRCC1, and Lig3 (depleting Fen1 leads to semi-lethality, impeding obtaining enough DNA for sequencing). eccDNA-Seq generated consistent data with the PCR results: silencing each of these alt-EJ factors completely abolished the biogenesis of 1-LTR eccDNA from HMS-Beagle (Fig. 2e).
Circularization for 2nd-strand synthesis
How does the alt-EJ process license eccDNA production from HMS-Beagle? It is possible that alt-EJ is required for transposon activation, and accordingly is necessitated for eccDNA production. To test this possibility, we performed Nanopore RNA-Seq and found the expression of HMS-Beagle transcripts is unaltered upon depletion of alt-EJ factors (Extended Data Fig. 6). This suggests that the alt-EJ factors are not required for retrotransposon transcription. Instead, here we provide evidence that alt-EJ is essential for the synthesis of HMS-Beagle 2nd-strand DNA via a circularization process, thus is necessitated for eccDNA biogenesis (Fig. 3a).
Fig. 3 |. Blocking alt-EJ process abrogates DNA synthesis and all eccDNA production from HMS-Beagle.

a, A model to depict how alt-EJ-mediated circularization drives DNA synthesis, thus is essential for eccDNA production and mobilization. Step 1: tRNA fragment pairs with the primer binding site (PBS) to initiate the 1st-strand DNA synthesis via reverse transcription to form RNA:DNA hybrid. Step 2: RNase H activity to remove RNA from the RNA:DNA hybrid, but leaving a polypurine tract (PPT). Step 3: PPT initiates the 2nd-strand DNA synthesis for the 3′-LTR. Step 4: alt-EJ-mediated circularization drives the synthesis of the remaining of the 2nd-strand DNA. b, qPCR to quantify the relative abundance of single-stranded HMS-Beagle DNA. The bars report mean ± standard deviation from three biological replicates (n=3). p values were calculated with a two-tailed, two-sample unequal variance t test. c, Circos plot showing 2-LTR full-length eccDNA reads from endogenous HMS-Beagle. Color scores indicate the mapping coverage throughout the full-length HMS-Beagle consensus. The numbers within parentheses denote the number of reads identified from each genotype. These circles are likely generated by joining the 2 LTRs together via NHEJ. d, Circos plot showing rearranged eccDNA reads from endogenous HMS-Beagle. Color scores indicate the mapping coverage throughout the full-length HMS-Beagle consensus. The numbers within parentheses denote the number of reads identified for each genotype. 1-LTR rearranged circles are likely generated by autointegration events: LTR to attack its own interstitial sequences in cis. 0-LTR rearranged circles are possibly the by-products of autointegration. Data for this figure were generated from flies carrying sh-aub to trigger transposon activation.
Previous research on yeast LTR-retrotransposons and retroviruses has laid groundwork on the replication cycle of these elements6,17,25,26. They first transcribe genomic RNA6. Using the 3′-end of a tRNA to pair with its primer binding site (PBS) sequence, the RNA transcripts are reverse transcribed into the 1st-strand DNA (Fig. 3a, step 1)6. After finishing the 1st-strand DNA synthesis, the reverse transcriptase uses its RNase H activity to digest most of the retroviral RNA from the DNA-RNA hybrid except a short RNA sequence at the 3′ end––the polypurine tract (PPT, Fig. 3a, step 2)6,27,28. PPT serves as a primer to synthesize the 3′-LTR and the PBS sequence (by using the tRNA as template) for the 2nd-strand DNA (Fig. 3a, step 3)6,28. To replicate the rest of the 2nd-strand sequence that is upstream of the PPT site, it has been proposed that the PBS sequences between the 1st and 2nd-strand retroviral DNA can anneal with each other6,29. Known as the “2nd-strand transfer” (Fig. 3a, step 4), this step converts the 3’ end of the 2nd strand DNA as the priming site for the synthesis of rest strand6,29. Despite the essentiality of this step during the life cycle of retrotransposons and retroviruses, what mediates this process remains unknown.
Different from other DNA repair pathways, alt-EJ primes DNA synthesis by annealing a short homology (3–25 bp)22,24. Notably, the PBSs for retroviral elements are in general ≤ 18 nt6,30. Its apparent function in mediating microhomology formation led us hypothesize that alt-EJ circularizes the two DNA strands by annealing their PBS homology (Fig. 3a). This circularization step initiates the subsequent 2nd-strand DNA synthesis. This would produce a non-covalent circle with two fates: either fill the nick to dominantly generate covalent 1-LTR eccDNA or convert it into linear DNA with 2 LTRs (Fig. 3a). The linear DNA can serve as precursors for three subsequent outcomes: forming 2-LTR circles; using its LTR to attack its own interstitial sequences in cis to generate rearranged circles (known as autointegration); inserting into host genomic DNA in trans for integration (Fig. 3a). Our model predicts that impeding the alt-EJ process would halt 2nd-strand replication. This would lead to a higher single-stranded DNA ratio and the abolished biogenesis of the linear full-length double-stranded DNA, which serves as precursors for all downstream outcomes. To rigorously test our model, we correspondingly quantified the production of single-stranded DNA (Fig. 3b and Extended Data Fig. 7), 2-LTR eccDNA (Fig. 3c), rearranged eccDNA (Fig. 3d), linear full-length double-stranded DNA (Fig. 4a), and integration events upon the depletion of alt-EJ factors (Fig. 4b).
Fig. 4 |. Blocking alt-EJ process abrogates HMS-Beagle mobilization.
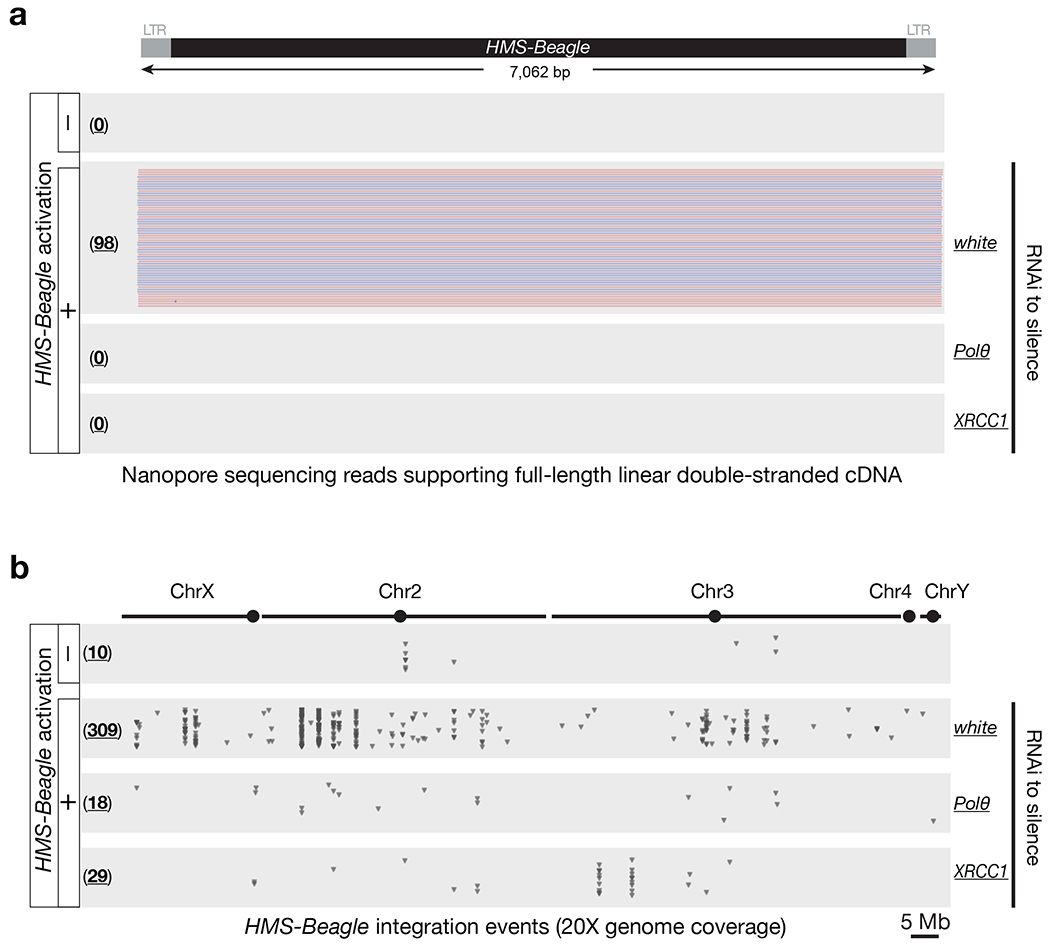
a, Nanopore sequencing to directly examine the biogenesis of full-length double-stranded linear DNA from replicated HMS-Beagle. Red- and blue-colored reads reflect the sequenced plus and minus strands respectively. b, Dot plots to display the new integrations from HMS-Beagle. Each triangle represents an integration event detected by Nanopore genome-seq. The numbers in parentheses present the total amounts of integration events detected. The numbers of integration detected under transposon-silenced condition likely represent the false-positive rates from our methodology. Data for this figure were generated from flies carrying sh-aub to trigger transposon activation.
First, we examined whether HMS-Beagle DNA extracted from the alt-EJ-perturbated ovaries is more sensitive to the treatment of Nuclease P1, an endonuclease that digests single-stranded DNA. Indeed, upon Nuclease P1 treatment, while HMS-Beagle DNA from control ovaries remained unchanged, silencing Polθ or Lig3 or XRCC1 led to > 2-fold reduction of HMS-Beagle DNA (Fig. 3b). To further measure the amount of single-stranded DNA, we used Mab3034 antibodies that preferentially binds single-stranded DNA to perform immunoprecipitation31. Upon individually depleting alt-EJ factors, the amount of HMS-Beagle single-stranded DNA increased significantly. Altogether, these data suggest that the alt-EJ process is essential for the completion of the 2nd-strand synthesis to produce double-stranded DNA.
Next, we used Nanopore ligation-based sequencing method to directly examine the biogenesis of linear full-length double-stranded DNA. While triggering transposon activation resulted in the production of double-stranded DNA with 2 intact LTRs flanking each end (Fig. 4a), individually silencing Polθ or XRCC1 completely abolished the formation of this essential precursor for all downstream events (Fig. 4a), such as the biogenesis of 2-LTR and rearranged eccDNA or integration events. Our data further suggest that alt-EJ drives the conversion process from single-stranded to double-stranded DNA.
Furthermore, we detailedly quantified the production of 2-LTR and rearranged eccDNA upon silencing of alt-EJ factors (Fig. 3c,d). Here we report the eccDNA-Seq reads by normalizing them per mitochondrial genome coverage. For 2-LTR full-length circles, we detected 11 of them from HMS-Beagle after triggering its activation (Fig. 3c). However, individually silencing Polθ or Lig3 or XRCC1 completely abolished their biogenesis (Fig. 3c). Similarly, the number of rearranged circles also dropped to the background level upon suppressing the alt-EJ process (Fig. 3d). Triggering HMS-Beagle activation generated 286 rearranged eccDNA that contain one intact LTR (1-LTR rearranged circles), indicative of autointegration events. Meanwhile, there were 100 rearranged HMS-Beagle circles that do not contain intact LTRs (0-LTR rearranged circles, Fig. 3d), which likely are generated as the by-products of autointegration events. However, for both categories of rearranged circles, we detected ≤ 6 circles with Polθ or Lig3 or XRCC1 silenced (Fig. 3d). Our data thus further support a function of the alt-EJ pathway in the DNA replication process of LTR-retrotransposons.
Lastly, we tested whether impeding the alt-EJ process would abrogate not only eccDNA production, but also HMS-Beagle mobilization. To test this, we individually depleted Polθ or XRCC1 (flies with Lig3 silenced lay very few mature oocytes), and then examined transposon mobilization rates in oocytes. For each genotype, DNA from somatic carcasses was sequenced to construct individual genomes, which serve as the reference to precisely define new transposon integrations in oocytes. With the same genome coverage (20X), we detected 309 new insertion events from HMS-Beagle when the alt-EJ process is undisturbed (Fig. 4b). However, once this process is blocked to abolish eccDNA biogenesis, the new insertion events also drastically decreased: 18 insertions upon Polθ depletion, and 29 insertions upon XRCC1 depletion (Fig. 4b). Collectively, our data indicate that alt-EJ is essential for the generation of double-stranded HMS-Beagle DNA that serves as a precursor for both circularization and integration.
Conserved function of alt-EJ
Besides using piRNA pathway perturbation to study the function of alt-EJ on retrotransposon DNA replication during oogenesis, we sought to further investigate its role under normal developmental conditions. We recently found that the retrotransposon mdg4, also known as Gypsy, naturally mobilizes in somatic tissues5. Particularly, mdg4 appears to only mobilize at the pupal stage5, when flies are regenerating new somatic tissues for adulthood. Accordingly, as an indication of the completion of the mdg4 DNA replication, we monitored mdg4 eccDNA production at different developmental stages. We found that mdg4 specifically generated eccDNA at the pupal stage, but not other developmental stages (Fig. 5a,b), consistent with the time window when mobilization events are detected. Notably, silencing the alt-EJ factors suppressed mdg4 eccDNA production (Fig. 5c). These results suggest that the alt-EJ pathway is also essential for retrotransposon replication in somatic tissues.
Fig. 5 |. mdg4 and mammalian IAP form eccDNA via the alt-EJ factors.
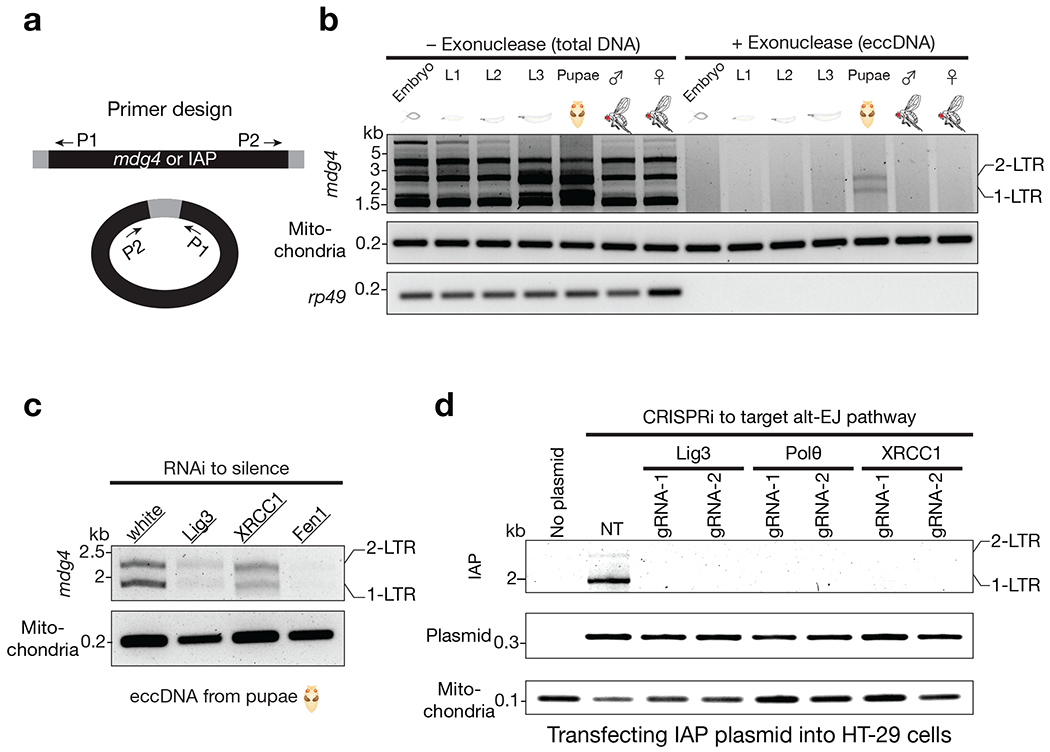
a, Schematic design of the divergent primers to detect mdg4 or IAP eccDNA. b, mdg4 retrotransposon produces both 1-LTR and 2-LTR eccDNA at the pupal stage, the time window when mobilization occurs. Performing PCR using total DNA as template produced non-specific bands. Using exonuclease to enrich eccDNA generated two PCR products corresponding to 1-LTR and 2-LTR eccDNA respectively, as confirmed by Sanger sequencing. c, eccDNA production from mdg4 depends on alt-EJ factors. d, Suppressing the factors from alt-EJ repair process blocks IAP eccDNA biogenesis. The PCR products corresponding to 1-LTR and 2-LTR eccDNA were confirmed by Sanger sequencing. NT (non-targeting) is a random gRNA without a targeting site in the human genome.
Do mobile elements from different species also employ the alt-EJ process for their DNA replication? By using eccDNA production as a readout, we investigated the function of alt-EJ in the replication cycle of Intracisternal A-Particle (IAP), a mouse LTR-retrotransposon. IAP presents ~2,800 full-length copies in the mouse genome and its activation contributes to ~6% of all pathogenic mutations32. To unambiguously examine IAP activity, previous work established a procedure to introduce IAP into cultured human cells33,34, which do not contain IAP in their own genome. Following this procedure, we monitored IAP eccDNA formation in human cells and found that IAP indeed generated circular DNA, including both 1-LTR and 2-LTR circles (Fig. 5d). Notably, disrupting the reverse transcriptase activity, but not the integrase function, leads to abolished biogenesis of both 1-LTR and 2-LTR eccDNA (Extended Data Fig. 8a–c). Using eccDNA production as a readout, we next asked whether alt-EJ is essential for IAP DNA replication. Notably, upon individually depleting the human orthologs of the factors identified in Drosophila that function in the retrotransposon life cycle (Polθ, XRCC1, and Lig3; Extended Data Fig. 8d–f), IAP failed to manufacture both 1-LTR and 2-LTR eccDNA (Fig. 5d). Meanwhile, suppressing NHEJ only blocks 2-LTR eccDNA production (Extended Data Fig. 9), suggesting that the 2-LTR eccDNA is formed by joining the two ends of the replicated linear double-stranded precursors. Altogether, these findings suggest a conserved function of alt-EJ in driving the retrotransposon replication cycle in metazoan.
DISCUSSION
The current view posits that retroviral elements synthesize their DNA largely for integration, and the remaining unintegrated ones can form 1-LTR eccDNA via homologous recombination between the 2 LTRs2,3,6,21,35. Although these models were proposed more than four decades ago and since then have been extensively cited16,17,28, the direct evidence to support them is still lacking. Our data do not support these models. Instead, by combining the power of Nanopore long-read sequencing with our robust genetic system, here we report that retrotransposons hijack alt-EJ-mediated circularization to dominantly produce 1-LTR eccDNA, but achieve far fewer integrations (Extended Data Fig. 10). Instead of merely being produced as the dead-end byproducts, these circles can potentially serve some biological purposes, such as transcribing mRNA to initiate a new round of replication cycle or breaking internally and then inserting into the genome to rewrite the genetic information. Moreover, given that circular DNA is highly potent for inducing innate immunity15, it is possible that retrotransposon-derived eccDNA can serve as immune regulators. Notably, we recently found that during the developmental time window when the mdg4 eccDNA is manufactured, mdg4 activation licenses the host’s immune system for future antiviral responses5.
alt-EJ was initially viewed as a merely back-up pathway for canonical DNA repair22,24. By performing genetic screens in vivo, here we uncovered its conserved function in the replication cycle of parasitic genetic mobile elements. Expression of both alt-EJ factors and retrotransposons is tightly controlled. Notably, both of them appear to maintain a high activity during embryogenesis, or under aging or pathological progression, such as cancer2,22,24,36–40. Under these conditions, alt-EJ likely drives the replication of retrotransposon DNA, enabling eccDNA production and mobilization from retrotransposons, thereby ultimately contributing to disease progression or driving evolution.
METHODS
Fly strains, housing, and husbandry conditions
All flies were grown on standard agar-corn medium. Female flies aged 3-7 days were selected for experiments unless otherwise noted. Fly alleles used for the genetic screens are listed in Supplementary Table 2 and the rest of the alleles used in this study are listed in Supplementary Table 3. Flies carrying vas-Gal4 were used in this study to achieve germline-specific gene silencing. For Fig. 1 and Extended Data Fig. 1–4, sh-aub and sh-ago3 double RNAi flies were used for genome-seq and eccDNA-Seq. These flies were maintained at 25°C. For Fig. 2–4, Extended Data Fig. 5,6, only sh-aub was used to block the piRNA pathway. Meanwhile, to facilitate the genetic screen, tub-Gal80ts was introduced into the genetic background to achieve conditional RNAi silencing. For the targeted screen, crosses were set at 18°C for 3 days and then shifted to 29°C to activate the RNAi constructs. For the Oxford Nanopore cDNA-seq and eccDNA-seq experiments, crosses were set and kept at 18°C for 9 days then shifted to 29°C for 7 days. For the genome-seq from oocytes, F1 virgins with desired genotypes were collected and crossed with w1118 males. F2 embryos laid within 6 hours were collected for DNA extraction. To detect mdg4 eccDNA from different developmental stages, the mixed genders of ac5c-Gal4 > sh-white flies from embryo to pupa stages and 2-5 days old adult male and female flies were collected respectively (Fig. 5b). For Fig. 5c, flies carrying ac5c-Gal4 were used to silence indicated factors.
Transgenic flies: eGFP reporter and sh-HMS-Beagle
The construct of HMS-Beagle transposition reporter was generated by using Counter-Selection BAC Modification Kit (GENE BRIDGES, Cat# K002). The BAC clone p[acman]-CH322-33A08 was used in this study to serve as template. The eGFP reporter was landed into the 6,242th position of HMS-Beagle.
To construct the plasmid for HMS-Beagle silencing, DNA fragments of sh-RNA were synthesized (sequences are listed in Supplementary Table 4) and cloned into the NheI and EcoRI sites of VALIUM20. All of the constructs were verified by colony PCR and Sanger sequencing. All plasmids were site-specifically landed into the fly genome at the attP2 site.
RNA-FISH
Stellaris RNA FISH probe set for HMS-Beagle was from a previous study and RNA-FISH experiments were performed as described7. Briefly, 3 pairs of ovaries were dissected in cold PBS and fixed for 20 minutes in 4% formaldehyde. Ovaries were washed once with PBST and twice with PBS, and then immersed in 70% (v/v) ethanol for 8 hours at 4°C. Then, ovaries were washed once with Wash Buffer A (LGC Biosearch Tech, Cat# SMF-WA1-60) at room temperature for 5 minutes, then incubated with 50 ml Hybridization Buffer (LGC Biosearch Tech, Cat# SMF-HB1-10) containing probe set (125 nM) for hybridization overnight at 37°C. Next, ovaries were washed twice with Wash Buffer A for 30 minutes at 37°C and once with Wash Buffer B (LGC Biosearch Tech, Cat# SMF-WB1-20) for 5 minutes at room temperature. Samples were mounted with 20 μl Vectashield Mounting Medium (VECTOR LABORATORIES INC MS, Cat # 101098-042). Images were acquired on Leica SP5 inverted microscope. All images were assembled in Adobe Photoshop and Illustrator.
eccDNA-Seq library preparation and Oxford Nanopore sequencing
For eccDNA sequencing, total DNA from ovaries was extracted by using Quick-gDNA MicroPrep Kit (Zymo Research, Cat # D3021). After removing linear DNA, rolling circle amplification, and debranching, the library was prepared with the Ligation Sequencing Kit (Oxford Nanopore, Cat #SQK-LSK109). Detailedly, 2 μg of total DNA was mixed with 2 μl Plasmid-safe DNase (Lucigen, Cat # E3110K), 5 μl 10xPlasmid-safe buffer, 2 μl 100 mM ATP (Thermo Scientific, Cat # R0441), and ultrapure water (Thermo Scientific, Cat #10977023) to 50 μl. On a thermocycler machine, the mixture was incubated at 37 °C for 3 hours. Then 2 μl Plasmid-safe DNase and 1 μl ATP were added to the mixture. The mixture was further incubated at 37°C for 16 hours and 70°C for 30 minutes on a thermocycler machine. Then 50 μl AMPure XP beads (Beckman Coulter, Cat # A63881) was used to purify DNA. The concentration of the purified circular DNA was measured the by Qubit dsDNA HS Assay kit (Thermo Scientific, Cat # Q33231). The RCA reaction was set as following: 2 ng circular DNA, 5 μl 10x Phi29 DNA Polymerase buffer, 1 μl Phi29 DNA Polymerase (New England Biolabs, Cat # M0269L), 2.5 μl 10 mM dNTP (Qiagen, Cat # 201901), 2.5 μl Exo Resistant Random Primer (Thermo Scientific, Cat # SO181), and ultrapure water to 50 μl. The mixture was incubated at 30 °C for 16 hours and 65 °C for 10 minutes on a thermocycler machine. The RCA product was purified by iso-propanol precipitation and debranched by T7 Endonuclease I (New England Biolabs, Cat # 0302L). The short fragments were eliminated by Short-Read Eliminator XL kit (Circulomics, Cat # SS-100-111-01). Then the library was constructed following the Oxford Nanopore SQK-LSK109 protocol. All libraries were sequenced in R9.4 flow cells on a GridION instrument according to the manufacturer’s instructions.
AFM imaging
To prepare the sample for AFM imaging, 2-5 ng DNA was diluted in low salt buffer (25 mM HEPES pH 7.5, 10 mM MgCl2, and 50 mM NaCl) to a total volume of 10 μl. The entire mixture was deposited on a freshly cleaved mica surface (Ted Pella Inc., Cat # 50) and incubated for 1 minute before being rinsed with 30 μl ultrapure water three times. The mica surface was then dried using compressed air. Imaging was performed using an Asylum Cypher Atomic Force Microscope equipped with AC240TS-R3 probe (Oxford Instruments, Cat # 803.OLY.AC240TS-R3) in ACMoleculeAir mode, and images were processed using Gwyddion 2.52.
Nanopore RNA-Seq library preparation
For RNA sequencing, fly ovaries were dissected on ice-cold PBS. The polyA+ RNA was extracted by Magnetic mRNA Isolation Kit (New England Biolabs, Cat # S1550S) following the manufacturer’s instructions with a minor modification: The LBB was incubated with 100 μl 1x Turbo DNase buffer and 3 μl Turbo DNase (Thermo Scientific, Cat # AM2239) at 37°C for 30 minutes and RNA was eluted by 100 μl Elution Buffer. The RNA was purified again by RNA Clean & Concentrator-5 (Zymo Research, Cat # R1016), and RNA concentration was measured by Qubit RNA HS Assay kit (Thermo Scientific, Cat # Q32852). Next, 700 ng of RNA was used to prepare cDNA library following the Oxford Nanopore SQK-DCS109 protocol. All libraries were sequenced in R9.4 flow cells on a GridION instrument according to the manufacturer’s instructions.
Nanopore Genome-Seq library preparation
For genome-seq, DNA from F2 embryos was extracted by using Quick-gDNA MicroPrep Kit (Zymo Research, Cat # D3021). Then 400-700 ng DNA was used to prepare library with either Ligation Sequencing Kit (Fig. 4; Oxford Nanopore, Cat # SQK-LSK109) or Rapid Sequencing Kit (Fig. 1; Oxford Nanopore, Cat #SQK-RAD004). All the libraries were sequenced in R9.4 flow cells on a GridION instrument according to the manufacturer’s instructions.
Divergent PCR
One hundred ng total DNA (in 10 μl volume) was mixed with 1 μl 10xPlasmid-safe DNase buffer, 0.5 μl Plasmid-safe DNase, and 0.5 μl 100 mM ATP. The mixture was incubated at 37 °C for 16 hours on thermocycler and followed by 70 °C for 30 minutes. One μl of the digested DNA was used for divergent PCR using CloneAMP HiFi PCR Premix (Takara Bio, Cat # 639398), Gotaq Green Master Mix (Promega, Cat # M7123), or 2x Phanta Max Master Mix (Vazyme, Cat # P515). The primer sequences are listed in Supplementary Table 4.
Cell culture and IAP plasmid transfection
HT-29 cells were cultured in RPMI 1640 (ThermoFisher, Cat# 11875093), 10% FBS (Cytvia SH30396.03), and 1% Penicillan/Streptomycin (ThermoFisher, Cat# 15140122) at 37 °C and 5% CO2. HT-29 cells were seeded at 300,000 cells per well in 6-well plates with complete media and allowed to grow overnight. Two ml of culture media was replaced the next day prior to transfection. IAP 440N1 WT plasmid was a generous gift from Dr. Marie Dewannieux. Five μg of plasmids were delivered to cells via Lipofectamine™ 3000 Transfection Reagent (ThermoFisher, Cat# L3000001) according to manufacturer’s recommended protocol. Cells were incubated with transfection mixture for 24 hours and then incubated with fresh media. Total DNA was collected 48 hours post-transfection, and eccDNA was isolated according to procedure detailed above. To remove remaining transfected plasmid, one hundred ng total DNA (in 10 μl volume) was mixed with 0.5 μl DpnI and 1 μl of CutSmart™ Buffer (NEB, Cat# R0176S). Mixture was incubated at 37 °C for 30 minutes and 70 °C for 20 minutes on thermocycler. For transfection control amplification, DpnI digestion was excluded. One μl 10x Plasmid-safe DNase buffer, 0.5 μl Plasmid-safe DNase, and 0.5 μl 100 mM ATP was then added to the mixture. The mixture was incubated at 37 °C for 2 hours. An additional 0.3 μl 10x Plasmid-safe DNase buffer, 0.5 μl Plasmid-safe DNase, and 0.5 μl 100 mM ATP were added and incubated for 16 hours on thermocycler and followed by 70 °C for 30 minutes. Divergent PCR products were analyzed with 0.8% agarose gel and imaged with Bio-Rad ChemiDoc XRS System (Bio-Rad, Cat# 1708265).
CRISPRi to deplete proteins in HT-29 cells
To construct the hUbC-dCas9-ZIM3-KRAB-hU6-sgRNA-PuroR (ZIM3-One) plasmid, the dCas9-ZIM3-KRAB was cloned from pLX303-ZIM3-KRAB-dCas9 (Addgene, Cat# 154472) into a hU6-sgRNA-PuroR vector (a gift from Dr. Kris Wood). sgRNAs targeting the promoter region were designed by using an online tool (http://chopchop.cbu.uib.no/) and are listed in Supplementary Table 4. An additional guanine was appended to the sgRNAs that do not start with a guanine. Each sgRNA was cloned into ZIM3-One plasmid and lentiviruses were produced. HT-29 cells were transduced with lentiviruses for 24 hours and selected with 2 μg/ml puromycin for 10-14 days. The bulk cells were collected to evaluate the depletion efficiency by either Western blot or RT-qPCR.
Gene mutation by CRISPR-Cas9 in HEK293T cells
The sgRNAs were derived from the MinLib CRISPR guide RNA library and are listed in Supplementary Table 4. To ensure efficiency, an additional guanine was appended to the sgRNAs that do not start with a guanine. Each sgRNA was cloned into pU6-(BbsI)-CBh-Cas9-T2A-BFP plasmid (Addgene, Cat # 64323) and verified by Sanger sequencing.
HEK293T cells were cultured in DMEM supplemented with GlutaMAX (ThermoFisher, Cat # 10569-010), 10% FBS (Cytvia SH30396.03), and 1% Penicillan/Streptomycin (ThermoFisher, Cat # 15140122) at 37°C with 5% CO2. Cells were seeded at 300,000 cells per well in 6-well plates with complete media and allowed to grow overnight. The next day, two ml of culture media was replaced prior to transfection. Transfection was carried out by delivering three μg of plasmid to cells using Lipofectamine™ 3000 Transfection Reagent (ThermoFisher, Cat # L3000001) according to manufacturer’s recommended protocol. Cells were incubated with transfection mixture for 24 hours and then incubated with fresh media for an additional 48 hours. Then cells were sorted for BFP signal using a Beckman Coulter Astrios EQ High-Speed Cell Sorter. The collected cells were plated into 6-cm dishes and allowed to recover for 48 hours post-sorting. Next, the cells were dissociated and diluted to 30 cells/ml. One hundred μl of the cell suspension was distributed into 96-well plates per well. Single clones were allowed to grow into stable colonies over a period of approximately 10-14 days. Finally, the colonies were validated for mutation by Sanger sequencing the site of sgRNA directed mutation.
Western blot
Cells were lysed in RIPA buffer (Thermo Scientific, Cat # 89900) with 1x complete protease inhibitor cocktail (Roche, Cat # 4693159001). The lysate was resolved by SDS-PAGE gels and analyzed by immunoblotting with indicated primary antibodies. The following primary antibodies were used: anti-DNA ligase 3 (Proteintech, Cat # 26583-1-AP; 1:1000), anti-XRCC1 (Proteintech, Cat # 21468-1-AP; 1:1000), and anti-β-Actin (Proteintech, Cat # 66009-1-Ig; 1:10000). Secondary antibodies include: anti-mouse and anti-rabbit IgG-HRP (Thermo Scientific, Cat # G-21040 and # G-21234; 1:5000). The membrane was developed by SuperSignal West Pico PLUS Chemiluminescent Substrate Kit (Thermo Scientific, Cat # 34577) according to the manufacturer’s instructions.
RNA purification and RT-qPCR
The total RNA of fly embryos or cells was extracted by using mirVana miRNA Isolation Kit (Thermo Scientific, Cat # AM1560). Ten μg of total RNA was treated with 2 μl Turbo DNase (Thermo Scientific, Cat # AM2238) at 37 °C for 30 minutes. After DNase treatment, the RNA was purified by RNA Clean & Concentrator-5 (Zymo Research, Cat # R1016). The complementary DNA (cDNA) was synthesized by using iScript cDNA Synthesis Kit (Bio-Rad, Cat # 1708890). RT-qPCR was performed with two technical replicates by using SsoFast EvaGreen (Bio-Rad, Cat # 1725204) on a CFX96 Real-time System (Bio-Rad). Fold changes for mRNA were calculated using the ΔΔCt method. Rp49 was used as the internal control for quantifying fly gene expression. RR18S was used as the internal control for quantifying human gene expression. The primer sequences for qPCR of each gene were listed in Supplementary Table 4. p values were calculated from at least three independent biological replicates using a two-tailed, two-sample unequal variance t test (Excel, Microsoft).
ssDNA immunoprecipitation and qPCR
Two-thousand ng of total DNA in a 50 μl volume was mixed with 5 μl 10x CutSmart buffer and 1 μl Xho1 (NEB, Cat # R0146S). The mixture was incubated at 37 °C for 4 hours and heat-inactivated at 65 °C for 20 minutes. Five μl of the mixture was set aside as input DNA and the remaining mixture was split into 1 ml PBST (10 mM NaH2PO4, 175 mM NaCl, pH 7.4, 0.1% Triton X-100) and incubated overnight at 4 °C with 2 μg anti-ssDNA antibody (Sigma, Cat #MAB3034, Clone 16-19) or 2 μg normal mouse IgG (Sigma, Cat # 12–371). The DNA-antibody complex was captured using 20 μl Dynabeads Protein G (Thermo Fisher, Cat # 10004D) for 2 hours at 4 °C. The beads were sequentially washed three times with PBST. The DNA was eluted by 80 μl elution buffer (10 mM Tris-HCl, 300 mM NaCl, 5mM EDTA, 0.5% SDS, pH 8.0) containing 5 μg Proteinase K (Zymo Research, Cat # D3001–2-20) at 55 °C for 1 hour with vigorous vortexing. The eluted DNA was then purified by Phenol/Chloroform (Thermo Fisher, Cat #15593–049) and qPCR was performed by using SsoFast EvaGreen (Bio-Rad, Cat # 1725204) on a CFX96 Real-time System. Fold changes were calculated using the ΔΔCt method, with input DNA used for normalization and set the sh-white & sh-aub control as 1.
Nuclease P1 treatment and qPCR
Forty ng total DNA (in 10 μl volume) was mixed with 1 μl 10x NEBuffer r1.1, 0.5 μl Xho1 (NEB, Cat # R0146S), and with or without 0.1 μl nuclease P1 (NEB, Cat # M0660S). The mixture was incubated at 37 °C for 4 hours on thermocycler and followed by 75 °C for 20 minutes. One μl of the digested DNA was used for qPCR, which was performed with two technical replicates by using SsoFast EvaGreen (Bio-Rad, Cat # 1725204) on a CFX96 Real-time System (Bio-Rad). Fold changes for mRNA were calculated using the ΔΔCt method. Rp49 was used as the internal control for quantifying DNA. The primer sequences for qPCR of each gene were listed in Supplementary Table 4. p values were calculated from at least three independent biological replicates using a two-tailed, two-sample unequal variance t test (Excel, Microsoft).
Spike-in and qPCR
The total DNA from ovary was extracted by using Quick-gDNA MicroPrep Kit (Zymo Research, Cat # D3021). One hundred ng total DNA (in 50 μl volume) was mixed with 5 ng plasmid (contains CopGFP), 2 μl Plasmid-safe DNase (Lucigen, Cat #E3110K), 5 μl 10x Plasmid-safe buffer, 2 μl 100 mM ATP (Thermo Scientific, Cat #R0441), and ultrapure water (Thermo Scientific, Cat #10977023). The mixture was incubated at 37°C for 16 hours and 70°C for 30 minutes on thermocycler. Then 45 μl AMPure XP beads (Beckman Coulter, Cat #A63881) was used to purify DNA and eluted with 10 μl water. One μl purified DNA was used to perform qPCR by using SsoFast EvaGreen (Bio-Rad, Cat #1725204) on a CFX96 Real-time System (Bio-Rad). CopGFP was used as the internal control for quantifying mitochondria DNA. The primer sequences for qPCR of each gene were listed in Supplementary Table 4. p values were calculated from at least three independent biological replicates using a two-tailed, two-sample unequal variance t test (Excel, Microsoft).
Nanopore sequencing reads pre-processing and mapping
All sequencing data and analytical pipelines will be released upon paper acceptance. The fast5 files generated by the Nanopore GridION machine were used as input in MinKNOW version 21.05.25 (MinKNOW core 4.3.12). Guppy 5.0.16 is integrated into the MinKNOW. The basic data preprocessing parameters are the following: Basecall model = High-accuracy base-calling; Read filtering = 9; The passed fastq files produced by MinKNOW were used for further quality control. Adapter sequences were detected and trimmed by porechop (0.2.4) with parameters: --extra_end_trim 0 --discard_middle. This setting only removes the adapter sequencing detected at the beginning and the end of the reads, if the adapter sequence is detected in the middle of the reads, the reads were filtered out. Output files of the porechop were used for further analysis. Reads were mapped to the reference genome of Drosophila melanogaster version dm6 (GCA_000001215.4) and the transposon consensus sequences respectively. The transposon consensus reference used in this study can be found at https://github.com/ZhaoZhangZZlab/eccDNA_formation_2021/tree/main/Reference/. This transposon dataset contains 121 transposons, classified at the sub-family level. Reads mapping was performed using the minimap2 (2.17-r941)41 software with parameter settings -ax map-ont -Y -t 16 to keep the soft clipping sequences for all supplementary alignments in the SAM output. Mapped results were converted to the bam format, sorted by reference coordinates, and indexed by samtools (1.12)42. Data visualization was achieved by R (4.1.2) and Python (3.9.12). The software IGV (2.12.0)43 was used to visualize mapping results.
Reads identification from the engineered and the endogenous HMS-Beagle
Sequencing reads were first mapped to HMS-Beagle consensus sequences. Mapped reads were further mapped to the GFP sequences, which can be found at https://github.com/ZhaoZhangZZlab/eccDNA_formation_2021/tree/main/Reference/. Reads mapped to both HMS-Beagle consensus and the GFP sequences were considered from engineered HMS-Beagle products. Reads only mapped to HMS-Beagle consensus but not to the GFP sequence were considered from the endogenous HMS-Beagle.
eccDNA reads selection from the genomic libraries (Tn5 method prepared)
To detect transposon-derived eccDNA, we first mapped the reads to the HMS-Beagle consensus sequence and the dm6 genome. Since HMS-Beagle from the linear DNA is typically encompassed by genomic sequences, reads carrying HMS-Beagle sequence flanked by genomic sequences were filtered out. Only reads entirely mapped to HMS-Beagle were considered as candidates for eccDNA construction and classification.
eccDNA identification and classification
For both the reads selected from the genomic-seq or eccDNA-Seq, chimeric alignments are suggestive of structural variation in genomic DNA sequencing. Therefore, reads with supplementary alignments were used to identify the junction-junction site of the circular DNA. In general, the two alignments from the same read were compared as a pair. The reads were identified as transposon-derived circular DNA reads if the pair of alignments met the following conditions: 1) they mapped to the same strand; 2) the spliced sites of the two alignments were adjacent to each other, overlapping with each other, or closer than 100 bp; 3) the two alignments were in convergent configuration. To further classify the transposon-derived eccDNA based on their structures, the following strategies were applied: (1) the reads were classified as 1-LTR full-length eccDNA if both mapping sites on the transposons were at the start and end of the transposon sequences, and the supplementary alignments were overlapping with each other by the length of LTR; The reads were classified as 2-LTR full-length eccDNA if both mapping sites on the transposon were at the start and end of the transposon sequence, and the supplementary alignments do not overlap with each other. The reads were classified as 1-LTR rearranged eccDNA if only one mapping site is at either the start or the end of the transposon, and the other mapping site is in the middle of the transposon. The reads were classified as non-LTR rearranged eccDNA if both mapping sites on the transposon were in the middle of the transposon. Of note, by following this criterion, reads that only had partial of LTR sequences were classified as non-LTR fragments. Considering the higher sequencing error rate of the Nanopore technology, we allowed 100 bp flexibility when setting the coordinates cutoffs. The abundance of each eccDNA type is represented by the number of reads that are classified into each type. In the figures, the circos (circlize 0.4.14)44 plot density indicated the mapping coverage of each eccDNA type. The coverage was generated by bedtools (v2.29.2)45 suite, using parameters bedtools genomecov -bga.
Integration events detection
To identify the integration events, reads were first mapped to the transposon consensus. Candidate reads supporting integrations were selected if they met the following criteria: 1) reads mapped to transposon by at least 500 bp; 2) reads mapped to either the start or the end of the transposons. Next, the selected reads were mapped to the dm6 reference genome to determine the junction sites. Reads carrying transposon-genome chimeric structures that are not present in the reference genome are considered as potential integration events. Reads aligned to a single transposon but multiple genomic regions were likely due to the repetitive nature of the landing sites. For these reads, the genomic landing locations were assigned based on the best mapping results. Reads with only one transposon-genome chimeric configuration were likely from the insertions with the sequences only spanning partial of the transposons. Reads with multiple transposon-genome chimeric configurations were likely from insertions with the sequences spanning the entire transposon and the flanking sequences from both sides. These reads were minorities in the population because it requires the reads long enough to cover the full-length transposons. These reads were further examined by their flanking sequence features. If the flanking genomic sequences were from the same chromosome, and the breakpoints were adjacent to each other, the reads were selected as insertions. Otherwise, the reads are excluded. In addition, reads were filtered out if they carry structures that two transposons join from the ends because these reads are unlikely generated from the insertion events. To characterize the transposon insertion loci, the insertion events were clustered based on the genome coordinates. The events that had breakpoints closer than 100 bp were grouped into one cluster. Any insertions shared between the oocytes and their corresponding somatic carcasses or shared by two or more datasets were removed because these were likely resulted from the polymorphisms between the fly genomes used in this study and the reference dm6 genome. Final integration events were represented in a bed file with the information including transposon name, insertion location, and the number of reads that support the insertion cluster.
Normalization
Genomic sequencing data were normalized by the genome coverage. The size factors were calculated by the total number of bases in the library divided by the effective genome size of dm6 (142,573,017 bp). eccDNA sequencing data were normalized by the coverage of the mitochondrial genome. The size factors were calculated by the total number of reads mapped to the mitochondrial genome divided by the size of the dm6 chrM (19,524 bp). The number of eccDNA was normalized by the equation:
Where N is the number of replicates, cn and sn are the raw eccDNA and the size factor in each replicate, respectively. And is the average sequencing depth of all data sets. Silencing DNA Ligase 3 likely affects the mitochondria numbers within the ovary. Hence, the eccDNA data from sh-Lig3 flies were further normalized by the number of reads from the inactive transposons.
Statistics and reproducibility
Statistical tests were calculated by using GraphPad Prism (v8) and Microsoft Excel (v16.67). The significance test of different groups was determined by a two-tailed, two sample unequal variance t test. The experiments in Fig. 2b–d as well as Extended Data Figs. 2c, and 8c were repeated at least three times independently with similar results. Similarly, the experiments in Fig. 5b–d, along with Extended Data Figs. 8d–f and 9a,b were independently repeated twice, yielding consistent results. Biological replicates were used for all the independent experiments. The micrographs shown in Extended Data Figs. 2b, 4a are representative of two independently conducted experiments, with similar results obtained.
Extended Data
Extended Data Fig. 1 |. The engineered HMS-Beagle reporter dominantly forms eccDNA.
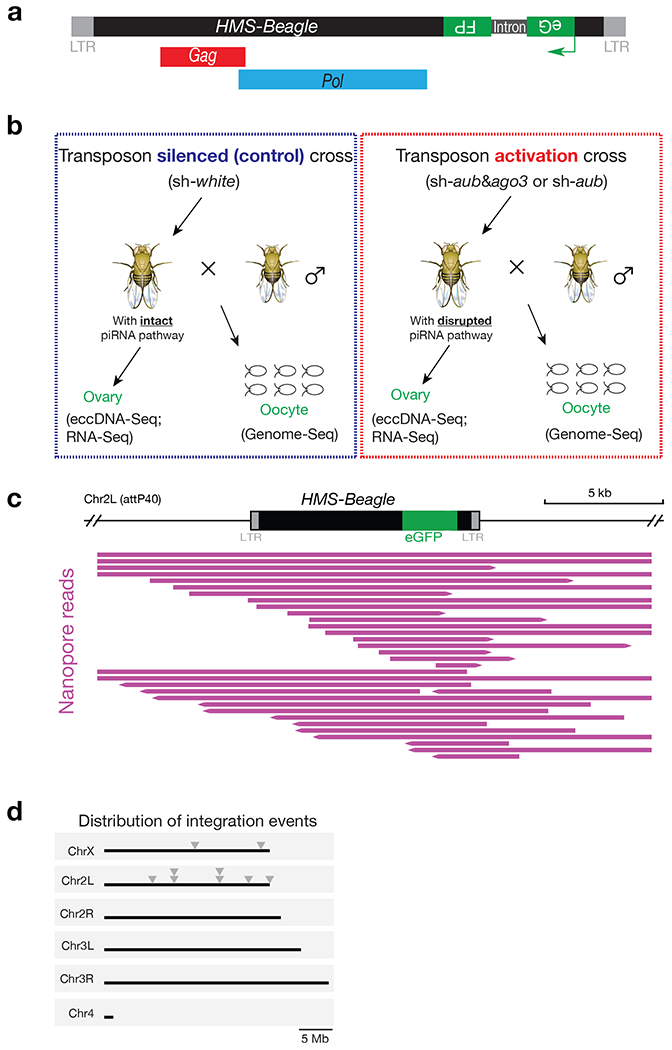
a, Schematic design of the HMS-Beagle reporter. An eGFP reporter is inserted into the 3’ UTR of HMS-Beagle sequence in an antisense direction. b, Fly cross scheme to collect samples for measuring the potential integration and eccDNA events from transposon-silenced and transposon-activated flies. c, Integrative Genomics Viewer (IGV) alignments showing reads mapped to HMS-Beagle reporter locus in the genome from embryos laid by transposon-silenced females. Individual purple horizonal bar represents a unique Nanopore read containing eGFP sequence. All of the reads contained at least one of the LTRs and extended to the adjacent region, indicating they were aligned to the original genomic locus of the reporter. d, The distribution of new integrations from engineered HMS-Beagle reporter on Drosophila genome. Each triangle represents a new integration event.
Extended Data Fig. 2 |. PCR based assay to measure HMS-Beagle eccDNA.

a, schematic of the design of divergent primers to identify retrotransposon eccDNA. b, AFM imaging to visualize the shapes of DNA. Exonuclease digestion significantly enrich eccDNA for detection in panel c. Scale bar, 500 nm. c, the representative gel image showing retrotransposons predominantly form 1-LTR circles. Performing PCR using total DNA as template produced non-specific bands, likely resulting from the nested transposon fragments resided within the linear genome. Using exonuclease to enrich eccDNA generated two PCR products corresponding to 1-LTR and 2-LTR eccDNA respectively. d and e, Sanger sequencing to validate the formation of HMS-Beagle eccDNA. The PCR products for the very right lane of panel c were cloned into plasmid vector and 11 corresponding colonies were sequenced. Ten of the 11 colonies are from 1-LTR eccDNA (d). One colony is from 2-LTR eccDNA (e). Notably, this 2-LTR eccDNA has 34 bp deletion at the end-end junction site, indicating it is formed by the error-prone NHEJ pathway. This conclusion is further supported by Extended Data Fig. 9.
Extended Data Fig. 3 |. eccDNA-Seq to provide direct evidence of circle formation.

a, Schematic of the eccDNA-Seq workflow. After extracting total DNA, linear DNA was removed by Plasmid-safe DNase digestion. eccDNA was amplified by Phi29 DNA polymerase through rolling circle amplification. And the sequencing libraries were prepared and sequenced on a Nanopore instrument. b, The proportion of eccDNA-Seq and Genome-Seq reads mapped to mitochondria (black regions), transposons (pink regions), and the rest of the genome (gray regions). All samples were from fly ovaries. The Genome-Seq libraries were made by the tagmentation method and sequenced by the Nanopore platform to capture circular DNA, such as the mitochondrial genome. c, Bar graph showing qPCR results of mitochondrial DNA copies detected by two sets of primers respectively. The relative abundances are normalized to the spike-in plasmid. The mitochondrial DNA copies are essentially unchanged upon transposon activation in Drosophila ovary. The bars report mean ± standard deviation from three biological replicates (n=3). p values were calculated with a two-tailed, two-sample unequal variance t test. d, Circos plots showing the number of the eccDNA-Seq reads for the four classes of HMS-Beagle circles. From the outer layer to inward: 1-LTR full-length circles, 2-LTR full-length circles, 1-LTR-rearranged circle, and non-LTR rearranged circles.
Extended Data Fig. 4 |. eccDNA production from HMS-Beagle requires its mRNA intermediates.
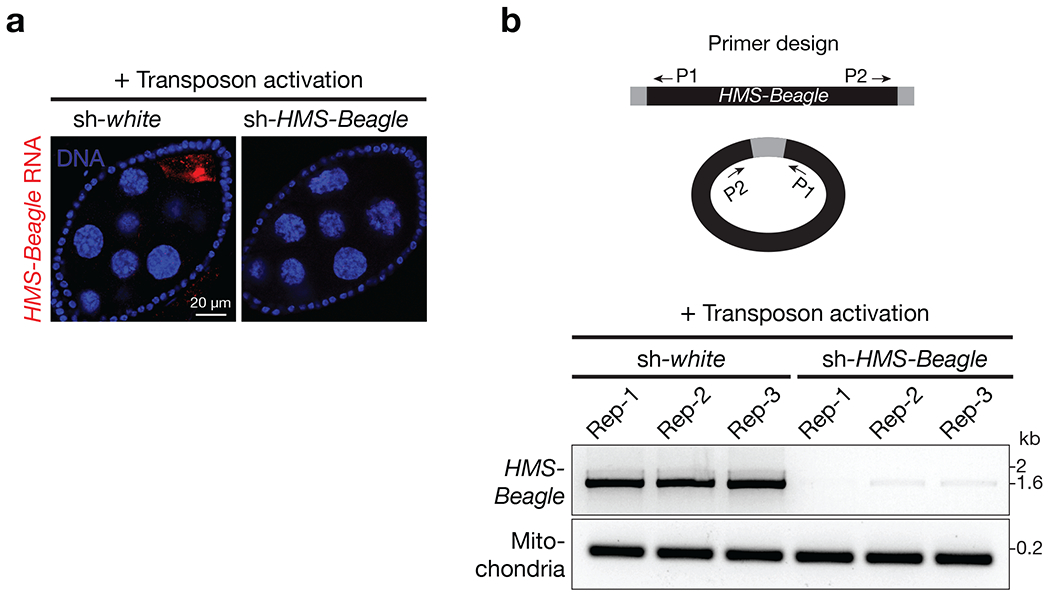
a, RNA-FISH to detect HMS-Beagle mRNA. All flies carrying sh-aub to activate transposons in germline cells. Further introducing sh-white (serving as a control) into the animals does not change transposon activity: HMS-Beagle remains activated. Upon introducing sh-HMS-Beagle construct to silence it, its RNA was undetectable by RNA-FISH. Scale bar, 20 μm, b, Top: primer design to detect HMS-Beagle eccDNA (Extended Data Fig. 2). Bottom: The representative gel image of PCR products showing that HMS-Beagle eccDNA production was abolished when its mRNA production was suppressed by RNAi. Each genotype has three biological replicates.
Extended Data Fig. 5 |. Confirmation of the RNAi silencing efficiency in oocytes.
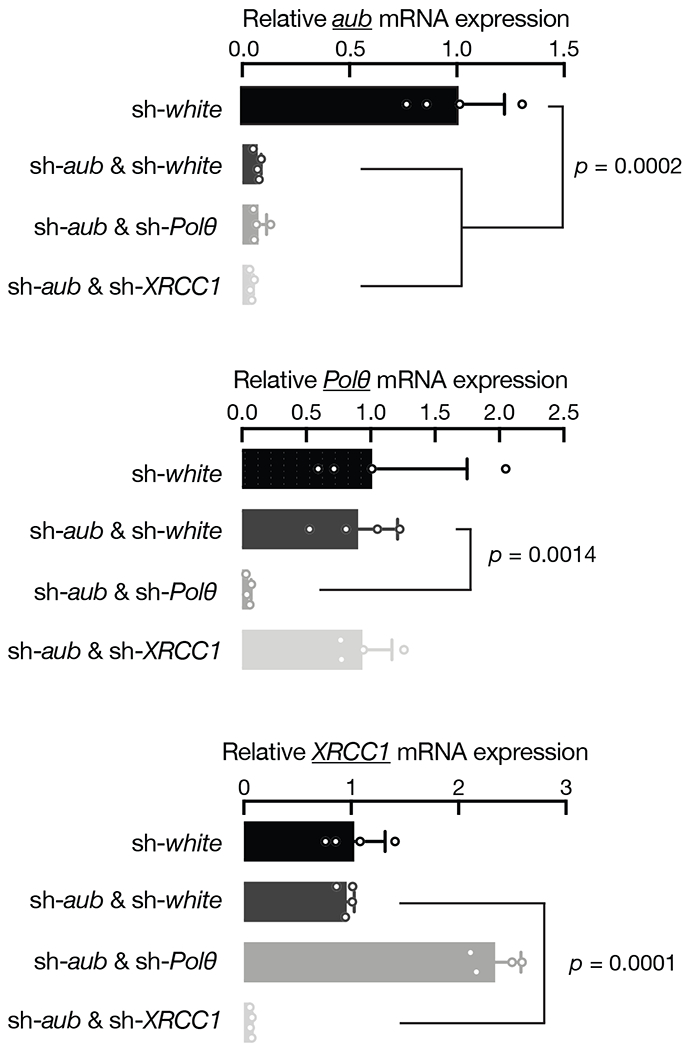
RT-qPCR showing the depletion efficiency of indicated genes by germline-specific RNAi. Relative mRNA levels were normalized to rp49 gene. The bars report mean ± standard deviation from four biological replicates (n=4). p values were calculated with a two-tailed, two-sample unequal variance t test. Silencing Lig3 or Fen1 made flies barely lay eggs/oocytes, impeding a validation of the RNAi silencing efficiency for them.
Extended Data Fig. 6 |. HMS-Beagle mRNA remains unchanged upon depletion of the components from alt-EJ process.
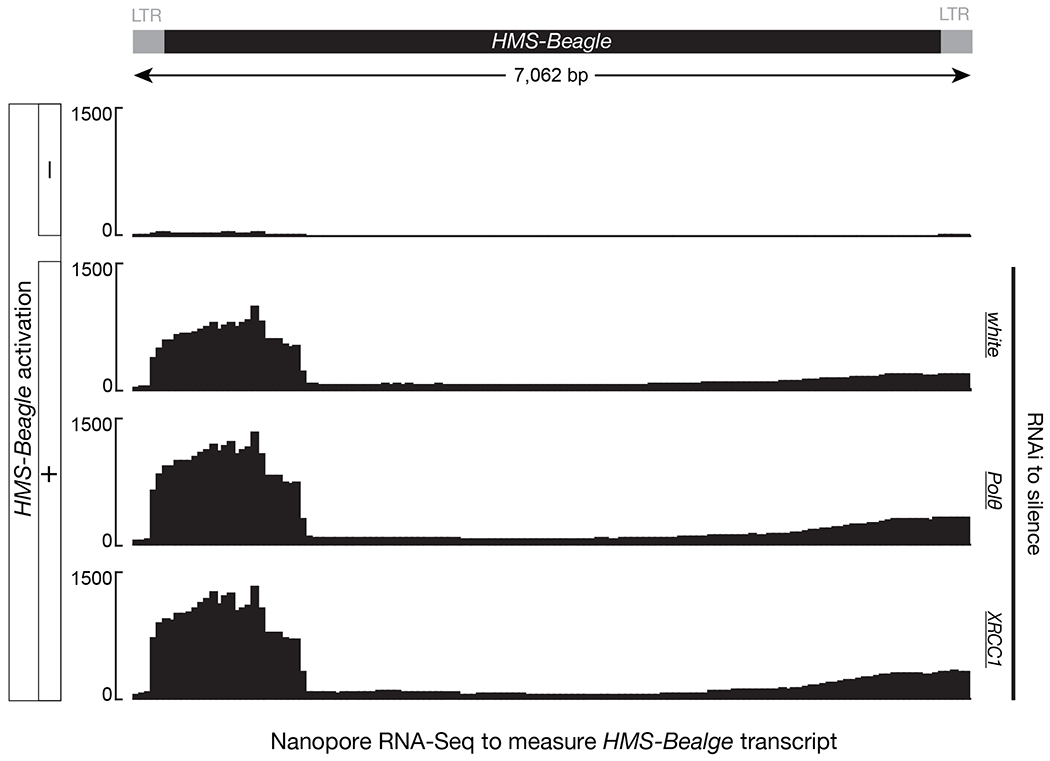
Transposon activation was achieved by silencing Aub in germline cells. The Y-axis is normalized reads count.
Extended Data Fig. 7 |. Immunoprecipitation assay to measure the accumulation of HMS-Beagle single-stranded DNA upon alt-EJ suppression.

The bars report mean ± standard deviation from four biological replicates (n=4). P values were calculated with a two-tailed, two-sample unequal variance t test. Although Mab3034 antibodies used in this experiment have 10-fold higher affinity for single-stranded DNA than double-stranded DNA31, they still can bind HMS-Beagle genomic double-stranded DNA across all samples. This would mask the difference of single-stranded DNA across samples and lead to underestimation of the amount of accumulated single-stranded DNA upon alt-EJ inhibition.
Extended Data Fig. 8 |. IAP needs its reverse transcriptase, but not integrase, activity for eccDNA biogenesis.
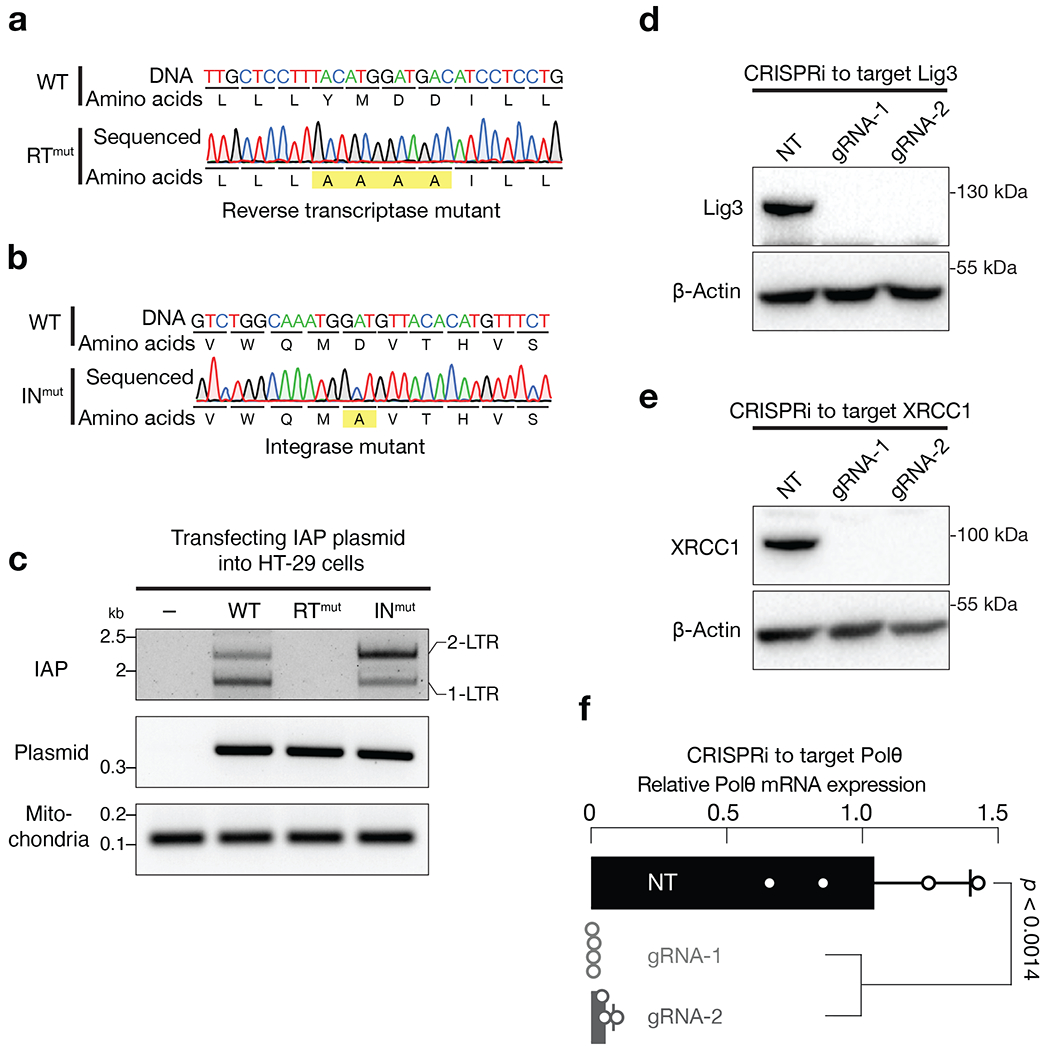
a, Sanger sequencing to validate the IAP reverse transcriptase mutant. b, Sanger sequencing to validate the IAP integrase mutant. c, PCR based assay to measure the production of IAP eccDNA. The very left lane was the condition without introducing IAP plasmid. d-e, Either immunoblotting (d and e) or RT-qPCR (f) to test the silencing efficiency of CRISPRi on depleting the alt-EJ factors. For each gene, two gRNAs were designed. NT (non-targeting) is a random gRNA without a targeting site. For RT-qPCR, relative mRNA levels were normalized to the RR18S gene. The bars report mean ± standard deviation from four biological replicates (n=4). Statistical significance were calculated with a two-tailed, two-sample unequal variance t test. The p value for gRNA-1 is 0.0011, while for gRNA-2 is 0.0014.
Extended Data Fig. 9 |. NHEJ pathway is essential for 2-LTR eccDNA biogenesis.
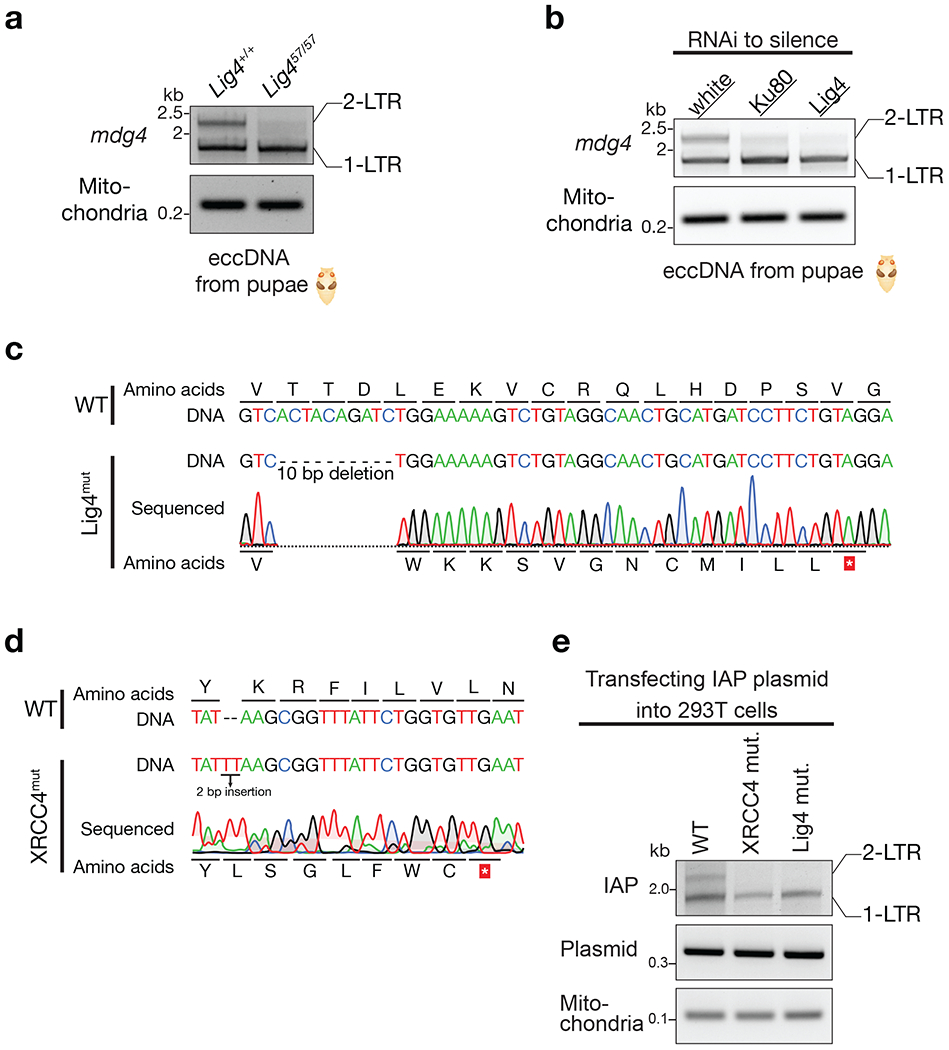
a, Mutating Lig4 abolishes 2-LTR eccDNA production for mdg4 retrotransposon. b, Silencing Ku80 or Lig4 by RNAi reduces mdg4 2-LTR eccDNA formation. c, Sanger sequencing to validate lig4 mutation of the 293T cells. d, Sanger sequencing to validate XRCC4 mutation of the 293T cells. e, mutating either Lig4 or XRCC4 abolishes 2-LTR eccDNA production for IAP retrotransposon.
Extended Data Fig. 10 |. Detailed model of the replication cycle of LTR-retrotransposons supported by our study.
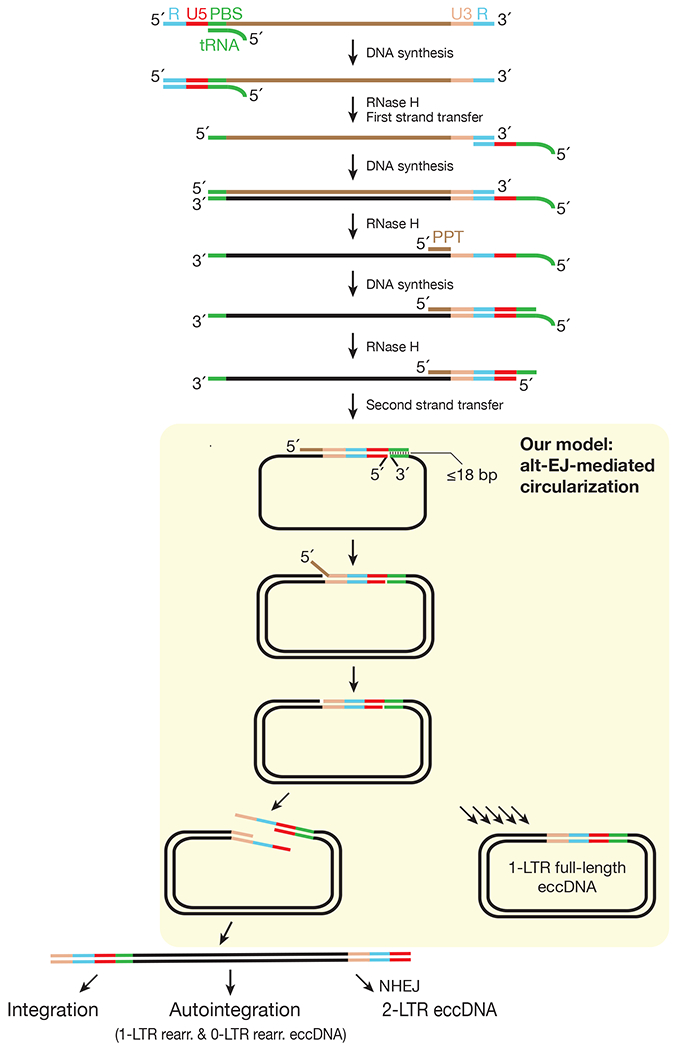
Our data support alt-EJ factors mediate a circularization step for retrotransposon 2-nd strand DNA synthesis. While this step can generate full-length linear double-stranded DNA for integration, it appears to dominantly produce 1-LTR eccDNA.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Marie Dewannieux and Kris Wood for providing plasmids, Julius Brennecke, Xin Chen, Jeff Sekelsky, and BDSC for providing fly stocks. We thank members from ZZ lab and David MacAlpine for critical suggestions, and Don Fox, Xiao-Fan Wang, Bryan Cullen, Lin Lin, and David MacAlpine for reading the manuscript. We thank Dorothy Erie and Piotr Marszalek for critical suggestions on AFM sample preparation. This work was supported by the grants to Z.Z. from the Pew Biomedical Scholars Program and the National Institutes of Health (DP5 OD021355 and R01 GM141018), and to D.A.R from the National Cancer Institute (P01CA247773).
Footnotes
COMPETING INTERESTS
Z.Z, F.Y., and W.S. are co-inventors on a US provisional patent application filed by Duke University related to this work.
DATA AND CODE AVAILABILITY
The sequencing data were deposited to the National Center for Biotechnology Information (NCBI) with accession number PRJNA794176. The sequence of the eGFP-tagged HMS-Beagle can be found at: https://github.com/ZhaoZhangZZlab/eccDNA_formation_2021/tree/main/Reference. All related code is available at https://github.com/ZhaoZhangZZlab/eccDNA_formation_2021
REFERENCES
- 1.Wells JN & Feschotte C A Field Guide to Eukaryotic Transposable Elements. Annual review of genetics (2020). 10.1146/annurev-genet-040620-022145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kazazian HH Jr. & Moran JV Mobile DNA in Health and Disease. The New England journal of medicine 377, 361–370 (2017). 10.1056/NEJMra1510092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fueyo R, Judd J, Feschotte C & Wysocka J Roles of transposable elements in the regulation of mammalian transcription. Nature reviews. Molecular cell biology (2022). 10.1038/s41580-022-00457-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frank JA et al. Evolution and antiviral activity of a human protein of retroviral origin. Science 378, 422–428 (2022). 10.1126/science.abq7871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang L et al. Retrotransposon activation during Drosophila metamorphosis conditions adult antiviral responses. Nature genetics 54, 1933–1945 (2022). 10.1038/s41588-022-01214-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Telesnitsky A & Goff SP in Retroviruses (eds Coffin JM, Hughes SH, & Varmus HE) (1997). [Google Scholar]
- 7.Wang L, Dou K, Moon S, Tan FJ & Zhang ZZ Hijacking Oogenesis Enables Massive Propagation of LINE and Retroviral Transposons. Cell (2018). 10.1016/j.cell.2018.06.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie T & Spradling AC A Niche Maintaining Germ Line Stem Cells in the Drosophila Ovary. Science 290, 328–330 (2000). 10.1126/science.290.5490.328 [DOI] [PubMed] [Google Scholar]
- 9.Kaminker JS et al. The transposable elements of the Drosophila melanogaster euchromatin: a genomics perspective. Genome biology 3, RESEARCH0084 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wicker T et al. A unified classification system for eukaryotic transposable elements. Nature reviews. Genetics 8, 973–982 (2007). 10.1038/nrg2165 [DOI] [PubMed] [Google Scholar]
- 11.Lammel U & Klambt C Specific expression of the Drosophila midline-jumper retro-transposon in embryonic CNS midline cells. Mechanisms of development 100, 339–342 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Siomi MC, Sato K, Pezic D & Aravin AA PIWI-interacting small RNAs: the vanguard of genome defence. Nature reviews. Molecular cell biology 12, 246–258 (2011). 10.1038/nrm3089 [DOI] [PubMed] [Google Scholar]
- 13.Li C et al. Collapse of germline piRNAs in the absence of Argonaute3 reveals somatic piRNAs in flies. Cell 137, 509–521 (2009). 10.1016/j.cell.2009.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vagin VV et al. A distinct small RNA pathway silences selfish genetic elements in the germline. Science 313, 320–324 (2006). 10.1126/science.1129333 [DOI] [PubMed] [Google Scholar]
- 15.Wang Y et al. eccDNAs are apoptotic products with high innate immunostimulatory activity. Nature (2021). 10.1038/s41586-021-04009-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henriksen RA et al. Circular DNA in the human germline and its association with recombination. Molecular cell 82, 209–217 e207 (2022). 10.1016/j.molcel.2021.11.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boeke JD, Garfinkel DJ, Styles CA & Fink GR Ty elements transpose through an RNA intermediate. Cell 40, 491–500 (1985). [DOI] [PubMed] [Google Scholar]
- 18.Shoshani O et al. Chromothripsis drives the evolution of gene amplification in cancer. Nature 591, 137–141 (2021). 10.1038/s41586-020-03064-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Libuda DE & Winston F Amplification of histone genes by circular chromosome formation in Saccharomyces cerevisiae. Nature 443, 1003–1007 (2006). 10.1038/nature05205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moller HD et al. Formation of Extrachromosomal Circular DNA from Long Terminal Repeats of Retrotransposons in Saccharomyces cerevisiae. G3 6, 453–462 (2015). 10.1534/g3.115.025858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown PO in Retroviruses (eds Coffin JM, Hughes SH, & Varmus HE) (1997). [Google Scholar]
- 22.Brambati A, Barry RM & Sfeir A DNA polymerase theta (Poltheta) - an error-prone polymerase necessary for genome stability. Current opinion in genetics & development 60, 119–126 (2020). 10.1016/j.gde.2020.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mateos-Gomez PA et al. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 518, 254–257 (2015). 10.1038/nature14157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramsden DA, Carvajal-Garcia J & Gupta GP Mechanism, cellular functions and cancer roles of polymerase-theta-mediated DNA end joining. Nature reviews. Molecular cell biology 23, 125–140 (2022). 10.1038/s41580-021-00405-2 [DOI] [PubMed] [Google Scholar]
- 25.Lauermann V & Boeke JD Plus-strand strong-stop DNA transfer in yeast Ty retrotransposons. The EMBO journal 16, 6603–6612 (1997). 10.1093/emboj/16.21.6603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heyman T, Agoutin B, Friant S, Wilhelm FX & Wilhelm ML Plus-strand DNA synthesis of the yeast retrotransposon Ty1 is initiated at two sites, PPT1 next to the 3’ LTR and PPT2 within the pol gene. PPT1 is sufficient for Ty1 transposition. Journal of molecular biology 253, 291–303 (1995). 10.1006/jmbi.1995.0553 [DOI] [PubMed] [Google Scholar]
- 27.Tanese N, Telesnitsky A & Goff SP Abortive reverse transcription by mutants of Moloney murine leukemia virus deficient in the reverse transcriptase-associated RNase H function. J Virol 65, 4387–4397 (1991). 10.1128/JVI.65.8.4387-4397.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Finston WI & Champoux JJ RNA-primed initiation of Moloney murine leukemia virus plus strands by reverse transcriptase in vitro. J Virol 51, 26–33 (1984). 10.1128/JVI.51.1.26-33.1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rhim H, Park J & Morrow CD Deletions in the tRNA(Lys) primer-binding site of human immunodeficiency virus type 1 identify essential regions for reverse transcription. J Virol 65, 4555–4564 (1991). 10.1128/JVI.65.9.4555-4564.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Le Grice SF “In the beginning”: initiation of minus strand DNA synthesis in retroviruses and LTR-containing retrotransposons. Biochemistry 42, 14349–14355 (2003). 10.1021/bi030201q [DOI] [PubMed] [Google Scholar]
- 31.Hu Z, Leppla SH, Li B & Elkins CA Antibodies specific for nucleic acids and applications in genomic detection and clinical diagnostics. Expert Rev Mol Diagn 14, 895–916 (2014). 10.1586/14737159.2014.931810 [DOI] [PubMed] [Google Scholar]
- 32.Gagnier L, Belancio VP & Mager DL Mouse germ line mutations due to retrotransposon insertions. Mobile DNA 10, 15 (2019). 10.1186/s13100-019-0157-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dewannieux M, Dupressoir A, Harper F, Pierron G & Heidmann T Identification of autonomous IAP LTR retrotransposons mobile in mammalian cells. Nature genetics 36, 534–539 (2004). 10.1038/ng1353 [DOI] [PubMed] [Google Scholar]
- 34.Schorn AJ, Gutbrod MJ, LeBlanc C & Martienssen R LTR-Retrotransposon Control by tRNA-Derived Small RNAs. Cell 170, 61–71 e11 (2017). 10.1016/j.cell.2017.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shank PR et al. Mapping unintegrated avian sarcoma virus DNA: termini of linear DNA bear 300 nucleotides present once or twice in two species of circular DNA. Cell 15, 1383–1395 (1978). 10.1016/0092-8674(78)90063-6 [DOI] [PubMed] [Google Scholar]
- 36.Grow EJ et al. Intrinsic retroviral reactivation in human preimplantation embryos and pluripotent cells. Nature 522, 221–225 (2015). 10.1038/nature14308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang J et al. Primate-specific endogenous retrovirus-driven transcription defines naive-like stem cells. Nature 516, 405–409 (2014). 10.1038/nature13804 [DOI] [PubMed] [Google Scholar]
- 38.Macfarlan TS et al. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 487, 57–63 (2012). 10.1038/nature11244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pang M, McConnell M & Fisher PA The Drosophila mus 308 gene product, implicated in tolerance of DNA interstrand crosslinks, is a nuclear protein found in both ovaries and embryos. DNA Repair (Amst) 4, 971–982 (2005). 10.1016/j.dnarep.2005.04.020 [DOI] [PubMed] [Google Scholar]
- 40.Vaidya A et al. Knock-in reporter mice demonstrate that DNA repair by non-homologous end joining declines with age. PLoS genetics 10, e1004511 (2014). 10.1371/journal.pgen.1004511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Danecek P et al. Twelve years of SAMtools and BCFtools. Gigascience 10 (2021). 10.1093/gigascience/giab008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gu Z, Gu L, Eils R, Schlesner M & Brors B circlize Implements and enhances circular visualization in R. Bioinformatics 30, 2811–2812 (2014). 10.1093/bioinformatics/btu393 [DOI] [PubMed] [Google Scholar]
- 43.Li H Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018). 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparinggenomic features. Bioinformatics 26, 841–842 (2010). 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robinson JT et al. Integrative genomics viewer. Nat Biotechnol 29, 24–26 (2011). 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data were deposited to the National Center for Biotechnology Information (NCBI) with accession number PRJNA794176. The sequence of the eGFP-tagged HMS-Beagle can be found at: https://github.com/ZhaoZhangZZlab/eccDNA_formation_2021/tree/main/Reference. All related code is available at https://github.com/ZhaoZhangZZlab/eccDNA_formation_2021