

Abstract
Long interspersed nuclear elements (LINEs) play essential roles in shaping chromatin states, while the factors that cooperate with LINEs and their roles in higher‐order chromatin organization remain poorly understood. Here, we show that MATR3, a nuclear matrix protein, interplays with antisense LINE1 (AS L1) RNAs to form a meshwork via phase separation, providing a dynamic platform for chromatin spatial organization. MATR3 and AS L1 RNAs affect the nuclear localization of each other. After MATR3 depletion, the chromatin, particularly H3K27me3‐modified chromatin, redistributes in the cell nuclei. Topologically associating domains (TADs) that highly transcribe MATR3‐associated AS L1 RNAs show decreased intra‐TAD interactions in both AML12 and ES cells. MATR3 depletion increases the accessibility of H3K27me3 domains adjacent to MATR3‐associated AS L1, without affecting H3K27me3 modifications. Furthermore, amyotrophic lateral sclerosis (ALS)‐associated MATR3 mutants alter biophysical features of the MATR3‐AS L1 RNA meshwork and cause an abnormal H3K27me3 staining. Collectively, we reveal a role of the meshwork formed by MATR3 and AS L1 RNAs in gathering chromatin in the nucleus.
Keywords: 3D genome, amyotrophic lateral sclerosis, antisense LINE1, MATR3, phase separation
Subject Categories: Chromatin, Transcription & Genomics; RNA Biology
The regulatory role of retrotransposons in genome organization is poorly explored. This study shows that nuclear matrix protein MATR3 and antisense L1 RNAs phase‐separate into a meshwork that scaffolds higher‐order chromatin architecture, particularly H3K27me3‐modified chromatin.
Introduction
Noncoding RNAs (ncRNAs) can act as structural molecules participating in genome organization, mostly through interacting with the chromatin near their transcription loci (Quinodoz et al, 2021). RNA‐binding proteins (RBPs) contribute to the in cis interaction, and ncRNAs may promote liquid–liquid phase separation (LLPS) of RBPs, further facilitating the chromatin compaction (A & Weber, 2019; Pandya‐Jones et al, 2020; Elguindy & Mendell, 2021; Quinodoz et al, 2021). Besides conventional ncRNAs, repeat‐element‐derived RNAs play important roles in the organization of higher‐order chromatin architecture. For example, telomeric repeat‐containing RNA TERRA and major satellites (MajSAT) RNAs are essential for the higher‐order organization of telomeres (Cusanelli et al, 2013) and the pericentric heterochromatin (Huo et al, 2020), respectively. Furthermore, C0T‐1 repeat RNAs (including LINEs and SINEs) associate with euchromatin and the nascent repeat‐rich RNAs function as scaffolds countering DNA compaction (Hall et al, 2014; Creamer et al, 2021).
LINE1 (L1) repeat elements comprise 17 and 19% genomic regions in human and mouse, respectively (Mouse Genome Sequencing Consortium et al, 2002; Taylor et al, 2013). Based on the observation that the X chromosome has higher L1 composition than autosomes, it has been suggested the role of L1 elements as “boosters” during X‐chromosome inactivation (XI) (Boyle et al, 1990; Lyon, 1998; Bailey et al, 2000). Further investigations showed that silent LINEs are involved in heterochromatin compartmentalization at early XI, and the actively transcribed LINEs (young LINEs) help to spread the inactive state to escape‐prone regions, promoting facultative heterochromatin compaction at late stage of XI (Chaumeil et al, 2006; Chow et al, 2010). L1 repeat RNAs were shown to interact with the chromatin domains from where they were transcribed (Hall et al, 2014; Lu et al, 2020). Functionally, sense‐transcribed L1 RNAs can act as the nuclear scaffold, recruiting Nucleolin/KAP1 proteins to inactivate 2C‐related Dux‐loci (Percharde et al, 2018). Further studies showed that N6‐methyladenosine (m6A) marks on these chromatin‐associated L1 RNAs help regulating chromatin accessibility by recruiting m6A reader YTHDC1 or m6A eraser FTO, further affecting histone modification on nearby chromatin (Liu et al, 2020; Chen et al, 2021; Wei et al, 2022). As for L1 RNAs in higher‐order chromatin organization, a recent study demonstrated that L1 elements and their transcripts may instruct the segregation of inactive B compartments, and depletion of sense‐transcribed L1 RNAs in ESCs disrupted the homotypic contacts between L1‐rich chromosome regions (Lu et al, 2021); CBX5/HP1α and L1 RNAs may phase‐separate into larger liquid droplets and promote heterochromatin compaction (Lu et al, 2021). Of note, L1 elements and their transcripts are widespread distributed in nucleus, while HP1α proteins mainly distribute as large foci in the nucleus, suggesting other factors may also work with L1 RNAs in chromatin structure organization.
In this study, we revealed functional roles of antisense‐transcribed L1 (AS L1) RNAs and a nuclear matrix (NM) protein Matrin‐3 (MATR3) in chromatin organization. NM is a network‐like nuclear structure proposed as the platform for various functions in the nucleus (Berezney & Coffey, 1974, 1977). Although it has long been hypothesized that NM scaffolds the chromatin organization (Paulson & Laemmli, 1977; Jackson et al, 1990), the roles of NM components in 3D genome organization just began to be revealed. Recently, we demonstrated that NM proteins SAF‐A/HNRNPU and SAFB can regulate the higher‐order organization of euchromatin and heterochromatin at the genome‐wide scale, respectively (Fan et al, 2018; Huo et al, 2020). Interestingly, the functional roles of SAF‐A/HNRNPU and SAFB in chromatin organization were dependent on chromatin‐associated RNAs (Nozawa et al, 2017; Huo et al, 2020). MATR3 is one of the first identified NM components that presents unique physicochemical properties (Nakayasu & Berezney, 1991). Functionally, MATR3 is implicated in pre‐mRNA splicing (Coelho et al, 2015; Uemura et al, 2017; Attig et al, 2018) and affects biological processes including pluripotency maintenance (Niimori‐Kita et al, 2018; Pollini et al, 2021) and X‐chromosome inactivation (XI) (Chu et al, 2015; Pandya‐Jones et al, 2020). A recent 3D genome study in erythroid cells showed that MATR3 stabilizes the chromatin occupancy of CTCF and cohesin at a subset of sites; loss of MATR3 affects weak‐insulated topologically associating domains (TADs) and accelerates cell fate transition (Cha et al, 2021). Here, we show that MATR3 mediates chromatin interaction by interacting with AS L1 RNAs, which may enhance our understandings of repeat RNAs in chromatin organization.
Results
MATR3 regulates the spatial organization of nuclear chromatin
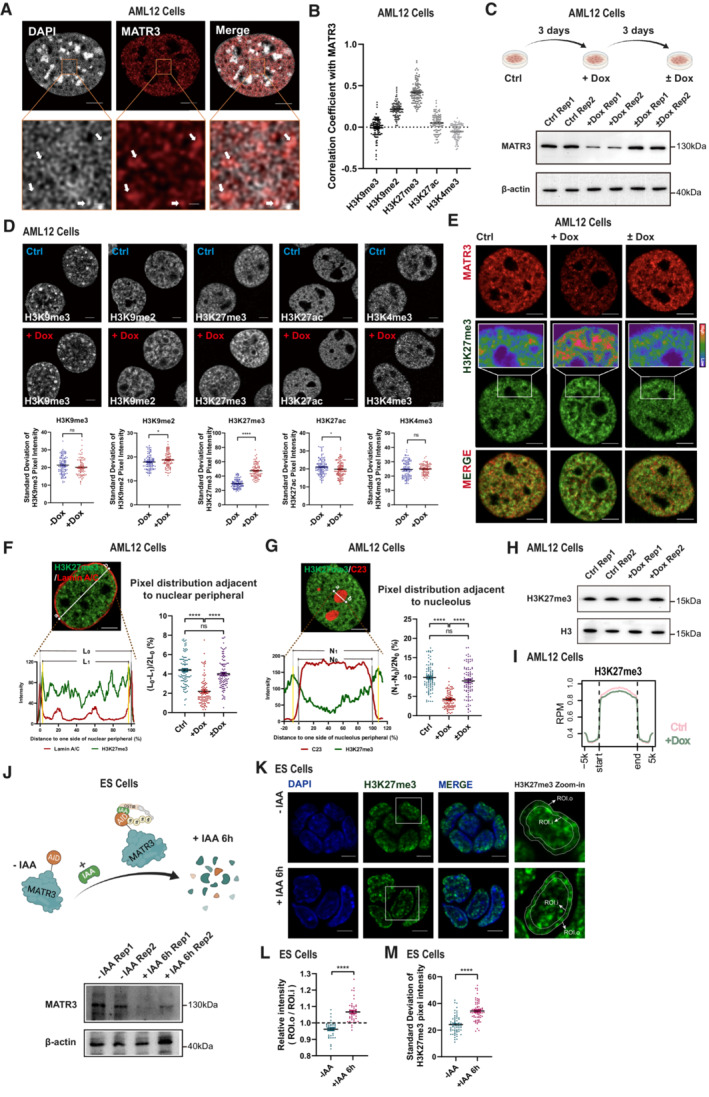
To investigate the fine‐scale localization pattern of MATR3 in the nucleus, we performed super‐resolution fluorescence microscopy and immuno‐electron microscopy in mouse hepatocytes (AML12 cells), which had been applied in studying nuclear architecture previously (Fan et al, 2018; Huo et al, 2020). The imaging data showed that MATR3 proteins organize into network‐like structures with some concentrated puncta, which were adjacent to chromatin fibers in the nucleus (Figs 1A and EV1A). To determine the chromatin types associated with MATR3, we measured the correlation coefficient (r) between MATR3 and histone marks on randomly selected regions of interest (ROI) in the nuclei. The nuclear distribution of MATR3 showed the highest correlation with H3K27me3, comparing to other histone marks, including H3K9me3, H3K9me2, H3K27ac, or H3K4me3 (Figs 1B and EV1B).
Figure 1. MATR3 regulates the spatial organization of chromatin.
-
ASuper‐resolution fluorescence microscopy images showing relative distribution between MATR3 and DAPI. Scale bars, 5 μm (Upper) or 0.5 μm (Lower). Arrows point to the representative MATR3/DAPI co‐staining regions.
-
BCoefficient of correlation between MATR3 and histone modification H3K9me3 (n = 105), H3K9me2 (n = 98), H3K27me3 (n = 107), H3K27ac (n = 97) and H3K4me3 (n = 99) in AML12 cells. Quantifications were performed on randomly selected ROIs in cell nuclei. Also see Fig EV1B. Each point represents one cell.
-
C(Upper) Schematic diagram of dox‐inducible shRNA system for MATR3 knockdown and MATR3 rescue in AML12 cells. (Lower) Western blotting detected the expression level of MATR3 after 3 days of Dox treatment (+Dox) and followed by 3 days of Dox removal (±Dox) in AML12 cells. Rep, replicate.
-
D(Upper) Representative cross‐section images showing the distribution of histone modifications upon Ctrl and MATR3 knockdown (+Dox). (Lower) Quantify the distribution pattern of histone modifications by Standard Deviation of Pixel Intensity in cell nuclei. For H3K9me3, n = 102 (Ctrl) or 84 (+Dox); for H3K9me2, n = 100 (Ctrl) or 101 (+Dox); for H3K27me3, n = 98 (Ctrl) or 98 (+Dox); for H3K27ac, n = 117 (Ctrl) or 124 (+Dox); for H3K27me3, n = 98 (Ctrl) or 98 (+Dox); for H3K4me3, n = 107 (Ctrl) or 97 (+Dox). Each point represents one cell. The P‐values were calculated using unpaired two‐tailed Student's t‐test; ns, not significant, *P < 0.05, ****P < 0.0001. Error bars indicate mean ± s.e.m.
-
ERepresentative cross‐section images showing nuclear localization of MATR3 and H3K27me3 upon Ctrl, MATR3 knockdown (+Dox) and MATR3 rescue (±Dox). Scale bars, 5 μm.
-
FRelative distribution of H3K27me3 and Lamin A/C. L0, region between nuclear membrane (position of nuclear membrane was determined by the X‐axis of the Lamin A/C pixel peaks on both sides). L1, region between two H3K27me3 pixel peaks that closest to the nuclear membrane. Quantify changes in H3K27me3 distribution adjacent to nuclear peripheral in Ctrl (n = 95), MATR3 knockdown (+Dox) (n = 92) and MATR3 rescue (±Dox) (n = 98) cells by formula of (L0‐L1)/2L0 (%). Scale bars, 5 μm. Each point represents one cell. The P‐values were calculated using unpaired two‐tailed Student's t‐test; ns, not significant, ****P < 0.0001. Error bars indicate mean ± s.e.m.
-
GRelative distribution of H3K27me3 and C23. N0, region between nucleolus membrane (position of nucleolus membrane was determined by the X‐axis of the half‐peaks on both sides). N1, region between two H3K27me3 pixel peaks that are closest to the nucleolus membrane. Quantify changes in H3K27me3 distribution adjacent to nucleolus in Ctrl (n = 91), MATR3 knockdown (n = 91) and MATR3 rescue (n = 92) cells by formula of (N1‐N0)/2N0 (%). Scale bars, 5 μm. Each point represents one cell. The P‐values were calculated using unpaired two‐tailed Student's t‐test; ns, not significant, ****P < 0.0001. Error bars indicate mean ± s.e.m.
-
HH3K27me3 modification level upon Ctrl and MATR3 knockdown (+Dox) as detected by western blotting. Rep, replication.
-
IAverage enrichment of H3K27me3 ChIP‐seq signal at peaks regions with 5 kb upstream and downstream flanking regions in Ctrl and shMatr3 from AML12 cells.
-
J(Upper) Schematic diagram of IAA‐inducible rapid protein degradation system for MATR3 in ES cells. (Lower) Western blotting detection of the efficiency of MATR3 knockdown in ES cells after 6 h addiction of 500 μM IAA or equal volume of alcohol (−IAA). Rep, replication.
-
KRepresentative cross‐section images showing nuclear localization of H3K27me3 in ES cells after 6 h addiction of 500 μM IAA (+IAA 6 h) or equal volume of alcohol (−IAA). Middle: The zoom‐in view of H3K27me3 in one ES cell. The outer dotted line colocalizes with nuclear membrane; the inner dotted line has the 80% diameter of the outer dotted line. The average pixel intensity of regions within outer dotted line (ROI.o) and regions within inner dotted line (ROI.i) were measured separately for each cell. Scale bars, 5 μm.
-
LStatistics for relative intensity (ROI.o/ ROI.i) after 6 h addiction of 500 μM IAA (+IAA 6 h) (n = 46) or equal volume of alcohol (−IAA) (n = 38). Each point represents one cell. The P‐values were calculated using unpaired two‐tailed Student's t‐test; ****P < 0.0001. Error bars indicate mean ± s.e.m.
-
MStandard deviation of H3K27me3 pixel intensity after 6 h addiction of 500 μM IAA (+IAA 6 h) (n = 68) or equal volume of alcohol (−IAA) (n = 60). Each point represents one cell. The P‐values were calculated using unpaired two‐tailed Student's t‐test; ****P < 0.0001. Error bars indicate mean ± s.e.m.
Source data are available online for this figure.
Figure EV1. MATR3 modulates redistribution of chromatin in nuclei.

-
AImmuno‐electron microscopy analysis of MATR3 protein distribution labeled by DAB in the AML12 cell. The solid arrows point to the individual DAB signal and the hallow arrows point to the clustered DAB signals, both of which represent MATR3 proteins. NM, nuclear membrane; Nu, nucleolus. Scale bars, (i): 2 μm; (ii): 0.5 μm; (iii), (iv): 0.2 μm.
-
B(Upper) Super‐resolution fluorescence microscopy images showing relative distribution between MATR3 and histone modifications (H3K9me3, H3K9me2, H3K27me3, H3K27ac and H3K4me3) in AML12 cells. (Lower) Line charts showing pixel intensity of each channel on the regions of interest (ROI). r, coefficient of correlation. Scale bars, 5 μm.
-
C(Left) Representative cross‐section images showing nuclear localization of MATR3 and H3K27me3 upon Ctrl, MATR3 knockdown (+Dox) and MATR3 rescue (±Dox). (Right) Line charts showing pixel intensity of each channel on the ROIs. Scale bars, 10 μm.
-
DPixel intensity of ROIs showing relative distribution of H3K27me3 and Lamin A/C upon Ctrl, MATR3 knockdown (+Dox) and MATR3 rescue (±Dox). L0, region between nuclear membrane. L1, region between two H3K27me3 pixel peaks that closest to the nuclear membrane. Scale bars, 5 μm.
-
EPixel intensity of ROIs showing relative distribution of H3K27me3 and C23. N0, region between nucleolus membrane (position of nucleolus membrane on X‐axis determined by C23 pixel half‐peaks on both sides). N1, region between two H3K27me3 pixel peaks that closest to the nucleolus membrane. Scale bars, 5 μm.
-
FStandard deviation of H3K27me3 pixel intensity of Ctrl (n = 98), MATR3 knockdown (+Dox) (n = 100) and MATR3 rescue (±Dox) (n = 94). The P‐values were calculated using unpaired two‐tailed Student's t‐test; *P < 0.05, ****P < 0.0001. Error bars indicate mean ± s.e.m.
-
GGenome browser of the H3K27me3 enriched region in Ctrl and MATR3 knockdown (+Dox) samples.
-
HImmunofluorescent detection of the efficiency of MATR3 knockdown in ES cells after 6 h addiction of 500 μM IAA or equal volume of alcohol (−IAA). Scale bars, 10 μm.
To reveal the biological function of MATR3 proteins in chromatin regulation, we established a doxycycline (Dox)‐inducible, short hairpin RNA (shRNA)‐based RNAi system (Wiederschain et al, 2009) in AML12 cells. Using this system, 3 days of Dox treatment can generate a knockdown of MATR3, and 3 days after Dox removal can restore the expression level consistent to the control group (Fig 1C). We first investigated the chromatin changes within regions of different chromatin types by immunofluorescence staining. To quantify the dispersion degree of fluorescence signal, we measured the standard deviation (SD) values of pixel intensity in the whole nuclear region. After MATR3 depletion, SD values of H3K9me2 and H3K27me3 increased, and that of H3K27ac decreased. Among these histone marks, H3K27me3 showed the most significant redistribution (P < 0.0001; Fig 1D). For the following investigation, therefore, we took H3K27me3 as the representative mark to probe chromatin organization changes. H3K27me3 staining in the control group showed a relatively diffused distribution pattern with some irregular foci in the nucleus. After depletion of MATR3, H3K27me3 staining presented larger and brighter foci in the inner nucleus as well as near the nuclear periphery and the nucleolus (Figs 1E and EV1C). To quantify the redistribution of H3K27me3‐modified chromatin toward nuclear periphery or nucleolus, we analyzed the pixel signal distribution on randomly selected ROIs in Lamin A/C or C23 (a nucleolus marker) co‐stained cells. The data indicated that H3K27me3‐modified chromatin became significantly closer to the nuclear periphery and the nucleolus after MATR3 depletion (Figs 1F and G, and EV1D and E). Importantly, Dox removal for 3 days restored the H3K27me3 pixel intensity SD values and spatial distribution relative to nuclear periphery and nucleolus (Figs 1E–G and EV1D–F), suggesting a direct regulatory role of MATR3 on chromatin organization. We confirmed that the changes in H3K27me3 staining were not a result of the Dox treatment or the cellular response to the shRNA, as no H3K27me3 alteration was observed in Dox‐treated control cells that transfected with the scrambled shRNA (Appendix Fig S1A). Next, we asked whether the changes in H3K27me3 staining were due to the alteration of H3K27me3 modification levels. However, the total H3K27me3 level and the genome‐wide profiles were largely unchanged upon MATR3 depletion, as indicated by western blotting (Fig 1H) and ChIP‐seq (Figs 1I and EV1G) in AML12 cells. Therefore, the changes in H3K27me3 staining reflected the spatial redistribution of chromatin rather than the alteration of histone modification levels.
To rule out the potential off‐target of RNAi methods and other secondary effects after knockdown, we established an auxin (IAA)‐inducible rapid protein degradation system (Natsume et al, 2016) in mouse embryonic stem cells (ESCs), which can generate MATR3 degradation within 6 h (Figs 1J and EV1H). As AML12 cells cannot be cultured in clones, the acute MATR3 degradation is technically difficult in this cell line. In ES cells, we first evaluated colocalization between MATR3 and different types of chromatins. H3K27me3‐modified chromatin showed positive correlation with MATR3, although the correlation in ES cells was weaker than that in AML12 cells (Appendix Fig S1B and C). After rapid degradation of MATR3, H3K27me3 in ES Cells showed larger and brighter staining near the nuclear periphery (Fig 1K and L), and the SD of H3K27me3 pixel intensity significantly increased accordingly (Fig 1M). Collectively, the acute degradation of MATR3 in ES Cells and RNAi‐based MATR3 knockdown in AML12 cells resulted in similar spatial changes in chromatin as probed by H3K27me3 staining.
MATR3 interacts with nuclear RNAs including antisense LINE1 RNAs
Nuclear RNAs were shown to participate in chromatin spatial organization (Rinn & Guttman, 2014; Nozawa & Gilbert, 2019). As MATR3 acts as an RNA‐binding protein (Salton et al, 2011; Uemura et al, 2017; Attig et al, 2018), we wondered whether MATR3's functional role in chromatin organization is RNA‐dependent. According to cell fractionation assay (Bhatt et al, 2012) followed by western blotting detection, most MATR3 proteins present in chromatin‐associated fractions in control cells, but greatly delocalized from chromatin after RNA pol II inhibitor dichloro‐ribofuranosyl benzimidazole (DRB) treatment (75 μM, 12 h) or RNase A treatment (10 μg/ml, 1 h; Fig 2A). Interestingly, immunofluorescent images showed that treating cells with RNA transcription inhibitor (75 μM DRB or 50 μg/ml α‐amanitin for 24 h) or RNase A (10 μg/ml for 1 h) disrupted network of MATR3 proteins and led them forming into spheroidal puncta (Fig 2B; Appendix Fig S2A and B). Pixel signal of MATR3 and H3K27me3 is positively correlated (r = 0.66) in the control cell, but the correlation turned negative in the DRB (r = −0.36) or RNase A (r = −0.13) treated cells (Fig 2B; Appendix Fig S2B). We observed that 29% of cells showed the spheroidal MATR3 puncta after 1 h treating with 75 μM DRB, and the percentage of cells containing MATR3 puncta increased with treating time (Fig EV2A and B). The overall RNA decay and the secondary consequences caused by protein synthesis inhibition (Tamm, 1984) could contribute to this phenomenon. Therefore, we looked into the nuclear distribution of MATR3 truncations, which lack the RNA‐binding ability. Of note, RNA recognition motifs (RRM1/2) deletion made MATR3 distribute as spheroidal foci, and the impact of RRM2 deletion was more prominent (Appendix Fig S2C). These data suggested that the interaction between MATR3 and RNAs is necessary for MATR3 meshwork formation and MATR3‐chromatin association.
Figure 2. MATR3 associates with nuclear RNAs including repeat elements‐derived transcripts.
-
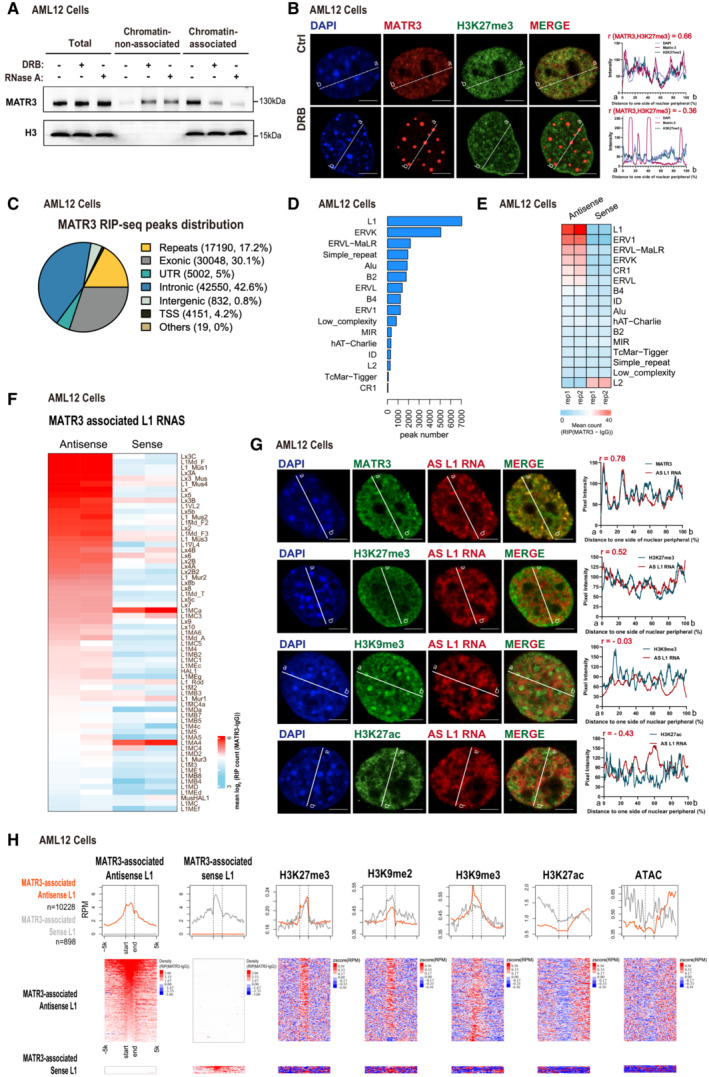
AWestern blotting showing the distribution of MATR3 proteins in chromatin‐non‐associated and chromatin‐associated extracts before and after DRB (75 μM for 12 h) or RNase A (pretreat with 0.05% Triton x‐100 for 30s, followed by 10 μg/ml RNase A for 1 h) treatment in AML12 cells. Representative of two independent replicates with similar results.
-
B(Left) The representative cross‐section image showing nuclear distribution of DAPI, MATR3 and H3K27me3 before and after 24 h treating of 75 μM DRB in AML12 cells. (Right) Line charts showing pixel intensity of each channel on the ROIs. r, coefficient of correlation. Scale bars, 5 μm.
-
CPie chart showing the MATR3 RIP‐seq peaks in relation to the genomic features. For each MATR3 RIP‐seq peak, we found its overlap with the genomic features and annotated. The priority of features is TSS, UTR, exonic, intronic, repeats, intergenic and others.
-
DThe number of MATR3 RIP‐seq peaks in repetitive elements (REs) from AML12 cells.
-
EHeatmap of MATR3 RIP‐seq sense and antisense signal of repetitive elements from AML12 cells. All RE copies with the RIP (MATR3 ‐IgG) count number ≥ 10 are kept. For each RE family, copies from antisense and sense of two replicates are merged. Then, compute the RIP (MATR3 ‐IgG) count number for RE copies and compute the mean count of RIP (MATR3 ‐IgG) of each sample. The color indicates the mean reads count of RIP (MATR3 ‐IgG) for each RE family.
-
FHeatmap of MATR3 RIP‐seq antisense and sense signal for MATR3 associated L1 subfamilies. L1 subfamilies are considered as MATR3 associated if the subfamily contains more than 50 copies. The copies with RIP (MATR3 ‐IgG) count number ≥ 10 are kept. L1 RNAs are ranked by antisense mean reads count. The color indicates the mean reads count of RIP (MATR3 ‐IgG) for each L1 subfamily.
-
G(Left) Representative cross‐section images showing relative distribution between AS L1 RNA with MATR3 and with histone modification marks (H3K27me3, H3K9me3 and H3K27ac) in AML12 cells. Probes for RNA FISH were designed toward the consensus sequence of antisense L1_Mus1 RNAs. (Right) Line charts showing pixel intensity of each channel on the ROIs. Scale bars, 5 μm. r, coefficient of correlation.
-
HNormalized average density of the marks (top) and heatmaps(bottom) for the two groups of L1 loci. The two groups are antisense L1 RNAs or sense L1 RNAs that interacted with MATR3. L1 loci with RIP (MATR3 ‐IgG) count number ≥ 10 of antisense RNA were identified as MATR3‐associated antisense L1, and the same cutoff for MATR3‐associated sense L1.
Source data are available online for this figure.
Figure EV2. MATR3 associates with antisense L1 RNAs.

-
ARepresentative cross‐section images showing distribution pattern for MATR3 in cell nuclei after 0, 1, 2, 6, 12, 24 h treating with 75 μM DRB. Scale bars, 10 μm.
-
BStatistics for MATR3 distribution pattern in cell nuclei after 0 (n = 500), 1 (n = 578), 2 (n = 560), 6 (n = 495), 12 (n = 489), 24 (n = 490) hours treating with 75 μM DRB.
-
CHeatmap of MATR3 RIP‐seq sense and antisense mean reads count in repetitive elements in ES cells. All RE copies with the RIP (MATR3 ‐IgG) count number ≥ 10 are kept. For each RE family, RE copies from antisense and sense of two replicates are merged. Then, compute the RIP (MATR3 ‐IgG) count number for RE copies and compute the mean count of RIP (MATR3 ‐IgG) of each sample.
-
DGenomic distribution of MATR3‐AS L1 RNAs loci in AML12 cells.
-
E(Left) Genome browser of the MATR3 RIP, RepeatMasker and Refseq genes. Targeting loci of the primers were noted on the screenshot. (Right) DNA gel images of RT–PCR of MATR3 RIP products (and input RNAs).
-
FScatter plot of RIP‐seq antisense for MATR3‐associated L1 copies (AS L1) and host genes' expression. ASL1 copies that with RIP (MATR3 ‐IgG) count number ≥ 10 and located within gene body regions are kept. X‐axis: RIP (MATR3 ‐IgG) count of ASL1 copies. Y‐axis: the FPKM of ASL1's host genes as detected by RNA‐seq. Pearson correlation coefficient (PCC) between RIP (MATR3 ‐IgG) count number of ASL1 copies and FPKM of host genes.
-
GRNA decay curve for AS L1 RNAs, Neat1 RNA and Actb RNA after blocking transcription with 5 μg/ml actinomycin D before and after MATR3 depletion. RNA remaining was measured relative to 18 s RNA at 0 h by qPCR. The RNA decay assay was performed in three independent biological replicates (N = 3). Fit modeled by one‐phase decay using nonlinear least squares regression. Error bars indicate mean ± s.d.
-
H(Left) Representative cross‐section images showing relative distribution between AS L1 RNA with MATR3 in ES cells. (Right) Line charts showing pixel intensity of each channel on the ROIs. Scale bars, 5 μm.
To further identify MATR3‐associated RNAs in chromatin organization, we performed strand‐specific RNA immunoprecipitation sequencing (RIP‐seq) in AML12 and ES cells with an anti‐MATR3 antibody. In total, 42.6% of RIP‐seq peaks were associated with intronic regions of annotated genes (Fig 2C), which is in accordance with MATR3 PAR‐CLIP data from human neuronal cells (Uemura et al, 2017) and eCLIP data from HepG2 cells (Van Nostrand et al, 2020). Notably, 17.2% of MATR3 RIP‐seq peaks distributed at repeat sequences in AML12 cells (Fig 2C). Among a total of 17,190 repeat sequences associated MATR3 RIP‐seq peaks, 40.99% are localized in L1 loci (Fig 2D). The degree of MATR3 RIP‐seq signals was strongly enriched at antisense (AS) L1 loci (Fig 2E; Appendix Fig S2D). Furthermore, for most of L1 subfamilies, the interactions between MATR3 and AS L1 RNAs were stronger than those between MATR3 and sense L1 RNAs (Fig 2F). Our data in ES cells (Fig EV2C; Appendix Fig S2E) and previous MATR3 eCLIP conducted in HepG2 cells (Van Nostrand et al, 2020) indicated the same results.
Next, we examined the genomic distribution of the L1 elements that transcribed MATR3‐associated AS L1 RNAs. Of note, 88% of them located in introns of annotated genes (Fig EV2D). It raised the question whether MATR3‐associated AS L1 RNAs are independent RNA fragments or parts of host gene transcripts. We thus conducted RT–PCR with MATR3 RIP samples at two intronic L1 loci transcribed AS L1 RNAs; for each locus, we designed two pairs of primers: one targeted the borders of RIP peaks (primer 1 or 3), and the other targeted ~ 200 bp flanking regions (primer 2 or 4). The former can detect RNA fragments about 300 nt with both input and MATR3 RIP products, while the latter cannot detect RNA with MATR3 RIP products (Fig EV2E). Besides, the enrichment degree of MATR3‐associated AS L1 in RIP‐seq is not correlative to the expression level of their host genes (Fig EV2F). Additionally, we compared the half‐life of AS L1 RNAs with those of Actb mRNA and Neat1, the architectural lncRNA for paraspeckles (Fig EV2G). In AML12 cells, the half‐life of the AS L1 RNAs is 2–3 h (Fig EV2G), similar to that of Neat1 (2.54 h), although much shorter than that of Actb mRNA (35.22 h). Therefore, MATR3‐associated AS L1 RNAs could be mainly independent RNA fragments; however, we cannot exclude the possibility that MATR3 interacts with some AS L1‐containing unspliced pre‐mRNAs or other long transcripts.
In order to visualize nuclear distribution of AS L1 RNAs, we performed fluorescent in situ hybridization (FISH) in AML12 cells. Probes for RNA FISH were designed according to the consensus sequence of antisense transcripts from L1_Mus 1 subfamily, which ranked high on MATR3‐associated AS L1 subfamilies (Fig 2F). Remarkably, nuclear localization of AS L1 RNAs was highly correlated with that of MATR3 in both AML12 (r = 0.78; Fig 2G) and ES (r = 0.62) cells (Fig EV2H), displaying a meshwork‐like structure in the nucleus. Imaging data in AML12 cells showed that AS L1 RNAs were positively correlated with H3K27me3‐modified chromatin (r = 0.52), but negatively correlated with H3K27ac‐modified chromatin (r = −0.43; Fig 2G). Integrated analyses of RIP‐seq and ChIP‐seq data showed that MATR3‐associated ASL1 elements are enriched for H3K27me3 and H3K9me3, but depleted of H3K27ac (Fig 2H), which suggested the cis‐acting role of AS L1 RNAs. Of note, H3K27me3 and H3K9me3 are not fully overlapped but enriched at the end and start boundaries of ASL1 elements, respectively (Fig 2H, top panel).
Together, our genomic and imaging data revealed an extensive interaction between MATR3 and AS L1 RNAs.
MATR3‐AS L1 meshwork shapes the nuclear chromatin architecture
Next, we investigated the functional roles of the MATR3‐AS L1 meshwork in higher‐order chromatin organization. We began to test whether AS L1 RNAs affect MATR3's function in chromatin organization by treating AML12 cells with antisense oligonucleotides (ASOs) to knockdown AS L1 RNAs. We designed five ASOs, targeting the scrambled sequence, the sense or antisense transcripts of a MATR3‐associated L1 subfamily (L1_Mus1) and a MATR3‐non‐associated L1 subfamily (L1_MA7), respectively (Figs 2F and EV3A). Sense and antisense L1_Mus1 ASOs treatment for 24 h caused MATR3 forming into spheroidal foci in 10 and 69% of the cells (Fig EV3A and B), in accordance with the results that MATR3 is more associated with the antisense than the sense L1_Mus1 RNAs (Fig 2F). As additional negative controls, both sense and antisense L1_MA7 ASO treatment had no effects on MATR3 localization (Fig EV3A and B). Thus, we took the ASO targeting the antisense L1_Mus1 RNA (hereafter referred to as ASL1 ASO) for the following investigation. After 6 h of AS L1 ASO treatment, 22% of cells showed MATR3 forming into spheroidal foci in the nucleus; after 12 and 24 h, this kind of cells increased to 53 and 65%, respectively. Moreover, a small portion of cells appeared spheroidal foci in both nucleus and cytoplasm (Fig EV3C and D).
Figure EV3. AS L1 RNAs depletion affects nuclear distribution of MATR3.

-
ARepresentative RNA FISH (with probes targeting antisense L1_Mus1 RNAs) and immunofluorescence images showing nuclear distribution of AS L1 RNAs and MATR3 after 24 h treating with different ASOs in AML12 cells. Scales bars, 10 μm.
-
BStatistical data for intracellular distribution of MATR3 proteins after 24 h treating with Scramble (n = 200), antisense L1_Mus1 (n = 198), sense L1_Mus1 (n = 195), antisense L1_MA7 (n = 200) and sense L1_MA7 (n = 200) in AML12 cells.
-
CThe representative intracellular distribution of MATR3 proteins before and after treating with AS L1 ASOs in AML12 cells. Scales bars, 5 μm.
-
DStatistical data for intracellular distribution of MATR3 proteins after 0 h (n = 1,000), 6 h (n = 1,280), 12 h (n = 1,504) and 24 h (n = 1,430) treating with AS L1 ASOs in AML12 cells.
-
E, FThe representative cross‐section image showing nuclear distribution of MATR3 and HNRNPU (E) and SAFB (F) before and after 24 h treating with antisense L1 ASOs in AML12 cells. Scales bars, 5 μm.
-
G(Left) The representative cross‐section image showing nuclear distribution of MATR3 and FUS before and after 24 h treating with antisense L1 ASOs in AML12 cells. (Right) The zoom‐in MERGE image showing relative distribution of MATR3 and FUS. Line charts showing pixel intensity of each channel on the ROIs. Scales bars, 5 μm.
-
HScatter plot of antisense RNA expression changes in L1_Mus1 subfamily with genes expression of host between AS L1 ASO and scramble ASO treatment samples. Mus1 copies located within gene body regions are kept. X‐axis: antisense RNA expression fold changes log2(AS L1 ASO/scramble ASO) of Mus1 copies. Y‐axis: the FPKM fold changes in L1_Mus1's host genes. Pearson correlation coefficient (PCC) between antisense RNA expression changes in Mus1 copies and expression changes in host genes.
To determine the specificity of MATR3 redistribution upon AS L1 RNA knockdown, we tested SAF‐A/HNRNPU and SAFB, which are also nuclear matrix proteins. Unlike MATR3, they did not form the spherical foci after ASL1 depletion (Fig EV3E and F). We also examined the RNA‐binding protein FUS, which was reported to condensate into droplets after RNase A treatment (Maharana et al, 2018). After treating with AS L1 ASO for 24 h, FUS proteins kept the dispersed distribution in most nuclear areas while some of them assembled around the MATR3 foci (Fig EV3G). The interaction between FUS and MATR3 (Kamelgarn et al, 2016) may contribute to this phenomenon.
As most AS L1 elements are located on introns, we conducted rRNA‐off and strand‐specific RNA‐seq to examine whether the host genes that containing AS L1 in introns were knocked down after AS L1 ASO treatment. The expression changes in these genes were subtle and not correlative to the changes in the intronic AS L1 RNAs (PCC = 0.04; Fig EV3H). Therefore, the consequence of AS L1 ASO treatment was not the secondary effects by the degradation of host genes.
Subsequently, we looked into the effects on chromatin structure after AS L1 ASO treatment. Immunofluorescent staining revealed that after AS L1 knockdown, MATR3 foci disassociated from H3K27me3‐modified chromatin (r = 0.66 in Ctrl ASO; r = −0.11 in AS L1 ASO), resulted in a redistribution of H3K27me3 which shown as larger foci in the nucleus (Fig 3A). Furthermore, SD of H3K27me3 pixel intensity significantly increased in AS L1 ASO‐treated cells (Fig 3B). The spatial alteration of H3K27me3 upon AS L1 depletion is similar as the phenomenon caused by MATR3 knockdown.
Figure 3. MATR3 cooperates with AS L1 RNA in chromatin organization.

-
A(Left) The representative cross‐section image showing nuclear distribution of MATR3 and H3K27me3 before and after 12 h treating with antisense L1 ASOs in AML12 cells. (Right) Line charts showing pixel intensity of each channel on the ROIs. r, coefficient of correlation. Scale bars, 5 μm.
-
BStandard deviation of H3K27me3 pixel intensity before (n = 98) and after (n = 97) 12 h treating with antisense L1 ASOs in AML12 cells. The P‐values were calculated using unpaired two‐tailed Student's t‐test; ****P < 0.0001. Error bars indicate mean ± s.e.m.
-
C(Left) The representative cross‐section image showing nuclear distribution of H3K27me3 and AS L1 RNA before and after MATR3 knockdown (Dox treatment for 3d) in AML12 cells. (Right) The normal distribution curve for the AS L1 pixel intensity. Scale bars, 5 μm.
-
DSkewness of antisense L1 RNA pixel intensity in Ctrl (n = 63) and shMatr3 cells (n = 66). The P‐values were calculated using unpaired two‐tailed Student's t‐test; ns, ****P < 0.0001. Error bars indicate mean ± s.e.m.
-
ERepresentative images showing nuclear colocalization of AS L1 RNAs with wild‐type and truncated GFP‐MATR3 proteins in AML12 cells. Scale bars, 5 μm.
-
FCoefficient of correlation between AS L1 RNA with wild‐type and truncated GFP‐MATR3 proteins. WT (n = 98), ▵ZF1 (n = 83), ▵ZF2 (n = 89), ▵RRM1 (n = 91), ▵RRM2 (n = 94). The P‐values were calculated using unpaired two‐tailed Student's t‐test; *P < 0.05, ****P < 0.0001. Error bars indicate mean ± s.e.m.
-
GRepresentative cross‐section images showing nuclear localization of H3K27me3 upon Ctrl, MATR3 knockdown and exogenous GFP‐MATR3 (WT/▵ZF1/▵ZF2/▵RRM1/▵RRM2) overexpression in AML12 cells. Scale bars, 5 μm.
-
HStandard deviation of H3K27me3 pixel intensity upon Ctrl (n = 50), MATR3 knockdown (n = 50), GFP‐MATR3 (WT) rescue (n = 48), GFP‐MATR3 (▵ZF1) rescue (n = 47), GFP‐MATR3 (▵ZF2) rescue (n = 46), GFP‐MATR3 (▵RRM1) (n = 45) and GFP‐MATR3 (▵RRM2) rescue (n = 49). The P‐values were calculated using unpaired two‐tailed Student's t‐test; ns, not significant, **P < 0.01, ****P < 0.0001. Error bars indicate mean ± s.e.m.
Source data are available online for this figure.
We further investigated whether loss of MATR3 could affect AS L1 RNAs. Compared with the meshwork‐like organization in control cells, the distribution of AS L1 RNAs appeared to be more dispersed in MATR3‐depleted cells (Fig 3C). To quantify these changes, we analyzed the Gaussian fit distribution curve of the fluorescence signal. Distribution of AS L1 RNAs in control cells is right‐skewed with a mean value greater than mode value and a skewness greater than zero, which suggested the presence of concentrated distribution in partial areas. After MATR3 depletion; however, the fluorescence signals of AS L1 RNAs nearly followed a standard Gaussian distribution with a mean value nearly equal to mode value and a skewness close to zero, which suggested a random distribution (Fig 3C and D).
MATR3 protein has two zinc finger (ZF) domains and two RNA recognition motifs (RRMs; Hibino et al, 2006). We wondered which domains are necessary for MATR3 to interact with AS L1 RNAs. We expressed GFP‐tagged MATR3 truncations (▵ZF1, ▵ZF2, ▵RRM1, and ▵RRM2) in endogenous MATR3‐depleted AML12 cells and examined their colocalization with AS L1 RNAs. The deletion of ZF1 or ZF2 did not affect the MATR3 distribution and its association with AS L1 RNAs. The deletion of RRM1 and RRM2 both affect MATR3 distribution, but only RRM2 deletion abolished MATR3's association with AS L1 RNAs (Fig 3E and F). Therefore, RRM2 domain is essential for MATR3 to interact with AS L1 RNAs. To examine the effects of these truncations on chromatin structure, we depleted the endogenous MATR3 with shRNA and re‐expressed WT or truncated MATR3 to rescue the phenotype. The WT MATR3 completely rescued changes in H3K27me3 that caused by MATR3 knockdown, while ▵ZF1, ▵ZF2, and ▵RRM1 truncations partially rescued the phenotype. However, H3K27me3 in ▵RRM2‐expressed cells showed no significant difference compared with the MATR3‐depleted cells (Fig 3G and H), indicating that RRM2 is essential for the chromatin organization function of MATR3.
Collectively, these results indicated that AS L1 RNAs and MATR3 affect cellular localization of each other and loss of either of them could lead to the redistribution of chromatin. MATR3 interplay with AS L1 RNAs by the RRM2 domain, and they act together to form a meshwork‐like structure that helps to shape the chromatin architecture in cell nuclei.
Phase separation facilitates the formation of MATR3‐AS L1 meshwork
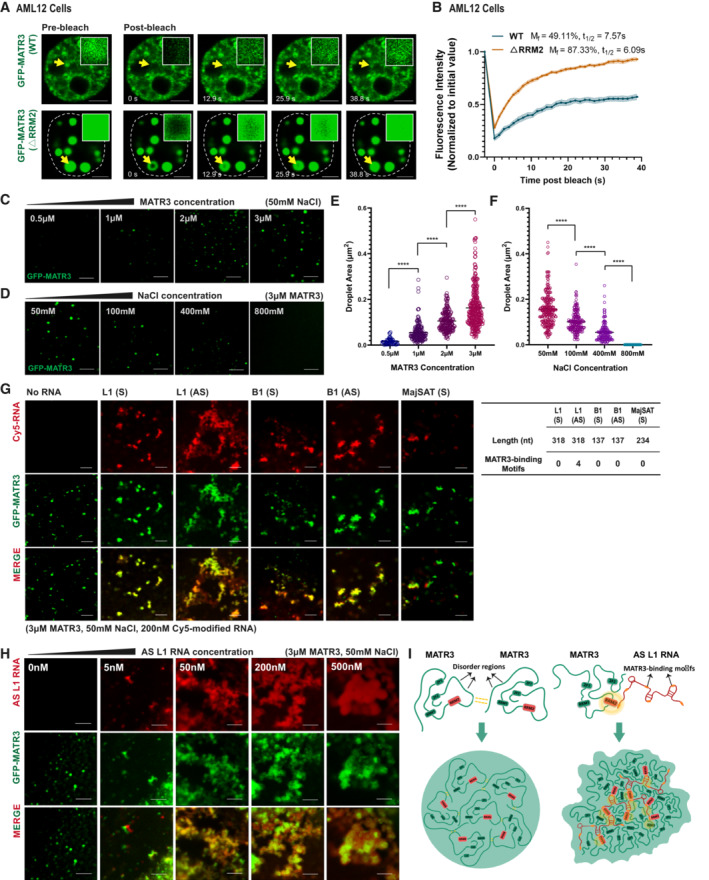
To investigate the physical characters of the meshwork formed by MATR3 and AS L1 RNAs, we first tested the dynamic feature in GFP‐MATR3‐expressed AML12 cells with fluorescence recovery after photobleaching (FRAP) assay. Both the wild‐type MATR3 and the RRM2‐depleted MATR3 showed partial recovery of fluorescence intensity, while the mobile fraction of RRM2‐depleted MATR3 (87.33%) was much higher than that of the wild‐type protein (49.11%) (Fig 4A and B).
Figure 4. AS L1 RNAs facilitate meshwork‐like organization of MATR3 proteins.
-
ARepresentative images of the GFP‐MATR3 (WT/▵RRM2) FRAP experiments in AML2 cells. Solid arrows indicate the bleached points. Scale bars, 5 μm.
-
BThe fluorescence recovery curve of the GFP‐MATR3 (WT/▵RRM2) corrected for fluorescence decay. Data are expressed as the mean ± s.e.m. (WT, n = 11; ▵RRM2, n = 11).
-
CRepresentative images of droplet formation assays with different concentrations of GFP‐MATR3 proteins. NaCl concentration, 50 mM. Scale bars, 5 μm.
-
DRepresentative images of droplet formation assays with different NaCl concentrations. GFP‐MATR3 protein concentration, 3 μM. Scale bars, 5 μm.
-
EAreas of MATR3 protein droplets formed in different protein concentration (3 μM: n = 315; 2 μM: n = 196; 1 μM: n = 165; 0.5 μM: n = 50). The P‐values were calculated using unpaired two‐tailed Student's t‐test; ****P < 0.0001.
-
FAreas of MATR3 protein droplets formed in different NaCl concentration (50 mM: n = 248; 100 mM: n = 196; 400 mM: n = 127; 800 mM: none). The P‐values were calculated using unpaired two‐tailed Student's t‐test; ****P < 0.0001.
-
G(Left) Representative images of droplet formation assays by GFP‐MATR3 with different in vitro‐transcribed RNAs (Sense L1, Antisense L1, Sense B1, Antisense B1, MajSAT). RNA concentration, 200 nM. GFP‐MATR3 protein concentration, 3 μM. NaCl concentration, 50 mM. (Right) The table showing the nucleotide number and the number of MATR3‐binding motifs on the in vitro‐transcribed RNAs. Scale bars, 5 μm.
-
HRepresentative images of droplet formation assays by GFP‐MATR3 with different concentration (0, 5, 50, 200, 500 nM) of AS L1 RNAs. GFP‐MATR3 protein concentration, 3 μM. NaCl concentration, 50 mM. Scale bars, 5 μm.
-
ISchematic representation for MATR3‐MATR3 droplets formation and MATR3‐AS L1 RNA meshwork formation.
Source data are available online for this figure.
Recent studies have revealed dynamic nuclear compartments driven by the liquid–liquid phase separation (LLPS) of proteins and/or RNAs (Stanek & Fox, 2017; Lyon et al, 2021). To test the phase separation potential of MATR3 proteins, we expressed and purified GFP‐tagged MATR3 proteins and performed the in vitro droplet formation assay. As expected, GFP‐MATR3 proteins formed spherical assemblies at room temperature; area of these assemblies increased with higher protein concentration and decreased with higher NaCl concentration (Fig 4C–F). When incubating 3 μM of GFP‐MATR3 proteins with total RNAs extracted from AML12 cells, 5–10 ng/μl of total RNAs facilitated GFP‐MATR3 proteins to form larger irregular particles, while more than 20 ng/μl of total RNAs buffered these particles (Appendix Fig S3A). These results are in agreement with reported features of phase‐separated proteins (Maharana et al, 2018).
To examine the roles of repeat RNAs in shaping MATR3 condensates in vitro, we incubated GFP‐MATR3 proteins with various in vitro transcribed, Cy5‐modified repeat RNAs and observed their droplet formation behaviors. The B1 and major satellites (MajSAT) RNAs were transcribed from full‐length B1 or MajSAT elements, respectively. As the full‐length L1 element is too long (~ 6 kb), the L1 RNAs were transcribed from a 318 bp consensus sequence of L1 Md_F2 elements (Appendix Fig S3B). Intriguingly, AS L1 RNAs facilitated MATR3 proteins forming into mesh‐like assemblies in vitro. While under the same molecule concentration, other repeat RNAs only made MATR3 droplets to undergo a slight deformation (Fig 4G).
To further interpret the action mechanisms of each repeat RNAs, we analyzed the numbers of MATR3‐binding motifs (Uemura et al, 2017) on them. As expected, only the AS L1 sequence contains MATR3‐binding motifs. Furthermore, motif density on the 318 nt‐antisense L1 Md_F2 RNA fragment (4/318) is comparable to that on full‐length antisense L1 Md_F2 RNA (89/5948) and is much higher than that on full‐length sense L1 Md_F2 RNA (4/5948) (Appendix Fig S3B). Hence, we suggest that AS L1 RNA has a higher affinity to bind MATR3 proteins. In cell nuclei, MATR3 could stochastically interact with different RNAs due to electrostatic forces. Those nonspecifically interacting RNAs would lower MATR3′s saturation concentration and buffer the MATR3 liquids at high RNA concentration (Maharana et al, 2018; Wang et al, 2018a). While the specifically interacting RNAs like AS L1 RNAs could form multivalent interaction with MATR3 proteins, that may enhance the overall avidity (Bhat et al, 2021) and promote the meshwork‐like assembly formation.
Furthermore, when incubating 3 μM of MATR3 proteins with different concentrations of AS L1 RNAs, 5–200 nM AS L1 RNAs could help the formation of the meshwork‐like structure (Fig 4H). According to a quantitative proteomics analysis, there are on average 3,240,673 MATR3 protein molecules in each mammalian cell (Hein et al, 2015). the estimated nuclear concentration of MATR3 protein is about 5.6 μM (close to 3 μM MATR3 concentration within in vitro system). In addition, based on RNA‐seq data combined with experimental measurements, the estimated molar concentration of the 318 nt‐AS L1 RNA is about 469 nM (calculation details described in Materials and Methods). We suggest the in vitro system containing 200 nM 318 nt‐AS L1 RNA fragment (Fig 4H) could nearly mimic the average stoichiometric ratio between MATR3 and AS L1 RNAs in vivo.
In summary, we suggest that MATR3 proteins have the potential to undergo liquid–liquid phase separation and the weak interaction between proteins' disorder regions contributes to this behavior. When MATR3 proteins interact with AS L1 RNAs via the RRM2 domain, the affinity between molecules increased and the mobility decreased, thereby facilitating the higher‐order MATR3‐AS L1 meshwork formation (Fig 4I).
3D genome organization changes upon MATR3 depletion
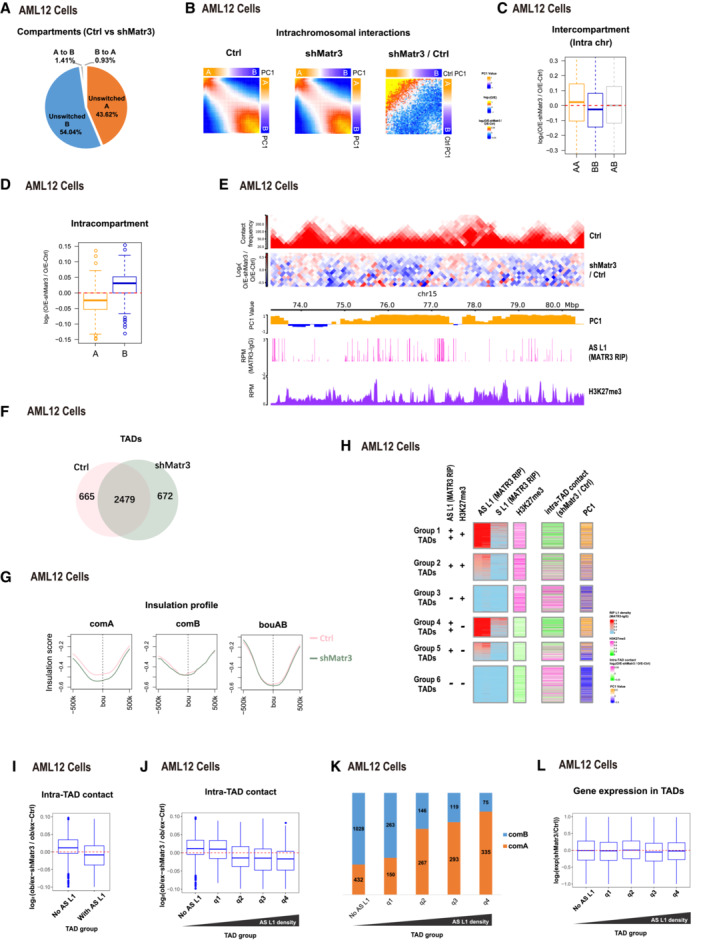
To study the role of MATR3 on 3D genome organization, we performed Hi‐C in control and Matr3‐depleted AML12 cells. We used DpnII and obtained over 140 million uniquely aligned read pairs per replicate (Appendix Fig S4A). Biological replicates from the same condition were highly correlated (Appendix Fig S4B), and then, we merged replicates for further analysis. We next examined intrachromosomal interactions at different resolutions and found that the Hi‐C contact maps in these two samples were similar (Appendix Fig S4C). Most of the compartments (97.7%) were unswitched after MATR3 knockdown (Fig 5A). However, the degree of genome compartmentalization, as visualized by heatmaps of average contacts, showed compartment‐specific alteration: increased in AA but decreased in BB compartments (Fig 5B). Consistently, the interactions between compartment A regions increased but decreased between compartment B regions (Fig 5C), and the interactions within compartment regions showed opposite trends (Fig 5D).
Figure 5. 3D genome organization changes on compartments and TADs upon MATR3 depletion.
-
APercentages of compartment status at unswitched A, unswitched B, B switched to A and A switched to B between Ctrl and shMatr3.
-
BAverage contact enrichment between pairs of 100 kb loci in Ctrl, shMatr3 and the comparison between them. All the 100 kb loci are arranged by Ctrl PC1 values in decreasing order and divided into 50 quantiles. Average enrichment of PC1 values is calculated for each quantile.
-
CChanges in contacts between compartment regions from the same (AA or BB) and different (AB) type in Ctrl and shMatr3. Data are represented as boxplots based on log2(O/E‐shMatr3/O/E‐Ctrl) values per pair. The number of compartment A and B are 1,456 and 1,573. The box of the boxplot covers the 50% of the values between 1st quartile and 3rd quartile of each group; The horizontal line of box represents the median value of each group; Red dashed lines indicate the zero.
-
DChanges in contacts within A or B compartment regions between Ctrl and shMatr3. Data are represented as boxplots based on log2(O/E‐shMatr3/O/E‐Ctrl) values per compartment region. The box of the boxplot covers the 50% of the values between 1st quartile and 3rd quartile of each group; Boxplot whiskers represent the minimal and maximal values of each group; The horizontal line of box represents the median value of each group; Red dashed lines indicate the zero.
-
ESnapshot of an example region, showing Hi‐C, H3K27me3 ChIP‐seq and AS L1 in MATR3 RIP‐seq in Ctrl and/or shMatr3 samples, using HiCExplorer. The values on the y‐axis for Hi‐C contact and O/E heatmap are iced normalized read counts at 100 kb resolution. The values on the y‐axis for ChIP‐seq and RIP‐seq are average reads per million of mapped reads (RPM).
-
FVenn diagram shows the common and sample‐specific TADs between Ctrl and shMatr3 samples. The TADs that overlapped length/TAD length > 0.8 both in Ctrl and shMatr3 samples were identified as common TADs.
-
GInsulation strength at TAD boundaries within compartment A, B regions or AB boundaries.
-
HHeatmap showing six groups of TADs based on average enrichment of AS L1 MATR3 RIP‐seq signal and H3K27me3 ChIP‐seq signal from ctrl cells. Enrichment of AS L1 in MATR3 RIP‐seq classified into strong enriched (++), enriched (+) and none (−) levels. Enrichment of H3K27me3 classified into enriched (+) and none (−) levels. Average enrichment of intra‐TAD contact and PC1 is calculated for each TAD.
-
IIntra‐TAD contacts changes in TADs associated with MATR3‐AS L1 RNAs TADs and nonassociated TADs according to density of anti‐MATR3 RIP‐seq signal of AS L1 RNAs. The y‐axis showing log2 contacts changes between shMatr3 and ctrl cells. The contacts are the density of the observed contacts normalized by the density of the expected contacts. The number of TADs associated with MATR3‐AS L1 RNAs and nonassociated are 1,658 and 1,486. The box of the boxplot covers the 50% of the values between 1st quartile and 3rd quartile of each group; Boxplot whiskers represent the minimal and maximal values of each group; The horizontal line of box represents the median value of each group; Red dashed lines indicate the zero.
-
JIntra‐TAD contacts changes in TADs nonassociated with MATR3‐AS L1 RNAs and four quantile groups associated with MATR3‐AS L1 RNAs ranking by increasing in MATR3‐AS L1 RNAs density. The number of TADs nonassociated with MATR3‐AS L1 RNAs and associated four quantile groups is 1,486, 415, 415, 415, and 413. The box of the boxplot covers the 50% of the values between 1st quartile and 3rd quartile of each group; Boxplot whiskers represent the minimal and maximal values of each group; The horizontal line of box represents the median value of each group; Red dashed lines indicate the zero.
-
KPercentage of TADs located in compartment A or B regions from five TAD groups (same groups in [J]).
-
LBoxplot shows gene expression changes for TAD groups from (J). The number of genes in each group are 2,050, 2,094, 2,356, 2,071 and 1,605. The box of the boxplot covers the 50% of the values between 1st quartile and 3rd quartile of each group; Boxplot whiskers represent the minimal and maximal values of each group; The horizontal line of box represents the median value of each group; Red dashed lines indicate the zero.
Source data are available online for this figure.
We also observed the chromatin interaction changes within TADs changed upon MATR3 knockdown (Fig 5E). We then investigated the Hi‐C data at TAD level. After MATR3 depletion, 78.8% TAD boundaries were overlapped between Ctrl and shMATR3 samples (Fig 5F). By calculating the insulation score, TAD boundaries located in compartment A regions increased in boundary strength, while TAD boundaries located in compartment B regions and AB boundaries hardly changed (Fig 5G). Furthermore, we categorized TADs into six groups according to the intensity of MATR3‐associated AS L1 and H3K27me3 (Fig 5H). All the six groups of TADs altered the local contacts, and the degree of change showed no clear correlation with the extent of H3K27me3 enrichment. However, the intra‐TAD interaction decreased in AS L1‐high TADs (groups 1 and 4), but increased in AS L1‐low TADs (groups 3 and 6); in the intermediate groups (2 and 5), intra‐TAD interaction increased with the decrease in MATR3‐associated AS L1 intensity (Fig 5H).
To further examine the roles of MATR3‐AS L1 meshwork in chromatin interactions at the TAD level, we compared TADs with or without MATR3‐associated AS L1 RNAs, as indicated by MATR3 RIP‐seq data. After MATR3 knockdown, the intra‐TAD contacts within no‐AS L1 TADs increased while decreased within with‐AS L1 TADs (Fig 5I); TADs with the higher AS L1 density exhibited a greater degree of reduction in terms of intra‐TAD contacts (Fig 5J). Furthermore, TADs with the higher AS L1 density were more enriched in compartment A regions, while TADs with no‐AS L1 density were highly enriched in compartment B regions (Fig 5K). The overall gene expression changes exhibited no trend along with AS L1 density in TADs (Fig 5L).
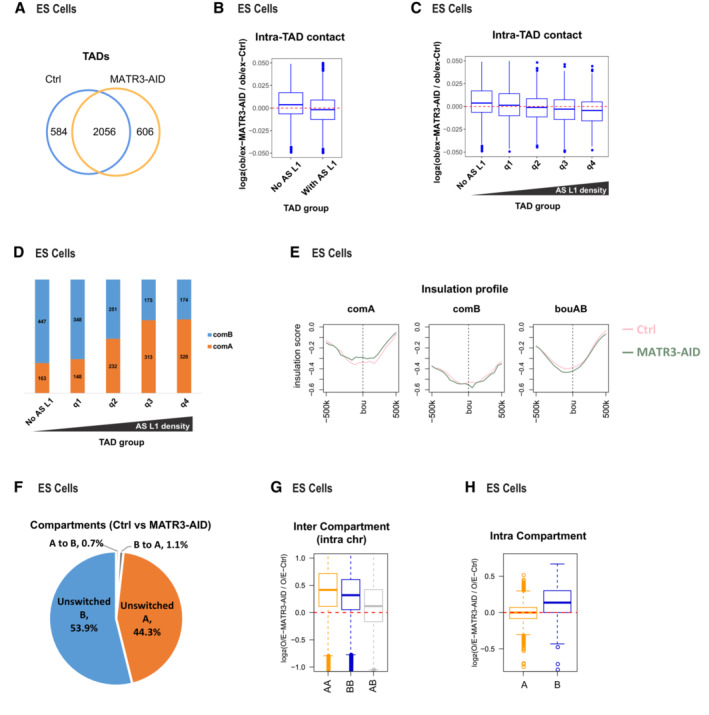
Additionally, we examined the 3D genome changes upon acute MATR3 degradation in mouse ES cells. We conducted Hi‐C with IAA‐treated (6 h) and control cells (Appendix Fig S5). The fast degradation allowed us to detect the direct effects on the 3D genome caused by MATR3 loss in ES cells. Most of (77.9%) TAD boundaries were overlapped between control and MATR3‐AID samples (Fig EV4A). Of note, after degradation of MATR3 in ES cells, TADs with higher MATR3‐associated AS L1 density presented a more loss of local contacts (Fig EV4B and C) and a greater enrichment in A compartments (Fig EV4D); these results are highly consistent with the those observed in AML12 cells. However, TAD boundaries strength decreased in compartment A (Fig EV4E). Furthermore, 98.2% compartments were unswitched (Fig EV4F), while the intercompartmental interactions increased in both A and B compartments (Fig EV4G), and the intracompartmental interactions increased in B compartments (Fig EV4H). There are some differences between ES and AML12 cells on TAD boundary strength and intercompartmental interactions, which could be attributed to the secondary effects by long‐time shRNA treatment in AML12 cells, or the cell type‐specific interactions within chromosomes (Jin et al, 2013; Rao et al, 2014). We have reported previously that AML12 and ES cells showed different patterns on chromatin compartmentalization (Yan et al, 2020).
Figure EV4. 3D genome organization as revealed by Hi‐C with ES cells.
-
AVenn diagram shows the common and sample‐specific TADs between Ctrl and MATR3‐AID samples from ES cells. The TADs that overlapped length/TAD length > 0.8 both in Ctrl and MATR3‐AID samples were identified as common TADs.
-
BIntra‐TAD contacts changes in MATR3‐AS L1 RNAs associated and nonassociated TADs from ESC cells. For each TAD, we calculate the density of the anti‐MATR3 RIP‐seq signal of AS L1 RNAs. TADs are grouped into “No AS L1” and “with AS L1” by density greater than 0 or not. The number of TADs associated with MATR3‐AS L1 RNAs and nonassociated are 2001 and 639. The box of the boxplot covers the 50% of the values between 1st quartile and 3rd quartile of each group; Boxplot whiskers represent the minimal and maximal values of each group; The horizontal line of box represents the median value of each group; Red dashed lines indicate the zero.
-
CIntra‐TAD contacts changes in TADs nonassociated with MATR3‐AS L1 RNAs and four quantile groups associated with MATR3‐AS L1 RNAs ranking by increasing in MATR3‐AS L1 RNAs density from ES cells. The number of TADs nonassociated with MATR3‐AS L1 RNAs and associated four quantile groups is 639, 500, 500, 500 and 501. The box of the boxplot covers the 50% of the values between 1st quartile and 3rd quartile of each group; Boxplot whiskers represent the minimal and maximal values of each group; The horizontal line of box represents the median value of each group; Red dashed lines indicate the zero.
-
DPercentage of TADs located in compartments A or B regions from five TAD groups (same groups in C) from ES cells.
-
EInsulation strength at TAD boundaries within compartment A, B regions or at AB boundaries of Ctrl and MATR3‐AID from ES cells.
-
FPercentages of compartment status at unswitched A, unswitched B, B switched to A and A switched to B between Ctrl and MATR3‐AID from ES cells.
-
GChanges in contacts between compartment regions from the same (AA or BB) and different (AB) type in Ctrl and MATR3‐AID from ES cells. The y‐axis showing log2 contacts changes between MATR3‐AID and ctrl cells. The contacts are the density of the observed contacts normalized by the density of the expected contacts. The box of the boxplot covers the 50% of the values between 1st quartile and 3rd quartile of each group; The horizontal line of box represents the median value of each group; Red dashed lines indicate the zero.
-
HChanges in contacts within A or B compartment regions between Ctrl and MATR3‐AID from ES cells. Data are represented as boxplots based on log2(O/E‐MATR3‐AID / O/E‐Ctrl) values per compartment region. The number of compartment A and B are 938 and 991. The box of the boxplot covers the 50% of the values between 1st quartile and 3rd quartile of each group; Boxplot whiskers represent the minimal and maximal values of each group; The horizontal line of box represents the median value of each group; Red dashed lines indicate the zero.
Collectively, our Hi‐C data in both AML12 and ES cells suggested that MATR3‐AS L1 meshwork facilitates clustering of chromatin within TADs highly transcribed MATR3‐associated AS L1 RNAs.
MATR3‐AS L1 meshwork maintains the expression stability of essential genes
We further asked whether the interplay between MATR3 and AS L1 RNAs contributes to gene regulation. As most of the MATR3‐associated AS L1 loci locate in annotated genes (Fig EV2D), we focused on the genes which contain MATR3‐associated AS L1 on gene body. After MATR3 knockdown, the genes containing MATR3‐associated AS L1 were more susceptible to change their expression level (Appendix Fig S6A).
Subsequently, to investigate the functional relevance of MATR3‐AS L1 meshwork, we examined genes that contain MATR3‐associated AS L1 in intronic regions. MATR3 RIP‐seq data in AML12 cells and ES cells were used for these analyses. Genes with AS L1 signals were highly overlapped between the two types of cells (Appendix Fig S6B). Gene Oncology (GO) and KEGG enrichment analyses indicated that the common genes were significantly enriched in survival‐related pathways including cell cycle, cellular response to DNA damage stimulus and chromatin organization (Appendix Fig S6C). This partially explained the observation that cell growth was greatly impeded after MATR3 knockdown (Appendix Fig S6D). Furthermore, the AS L1‐associated differentially expressed genes (DEGs) were significantly enriched in terms related to CNS development (neuron projection morphogenesis) and liver function (Cholesterol metabolism with Bloch and Kandutsch−Russell pathways; Appendix Fig S6E).
MATR3 depletion alters the structure of H3K27me3‐modified chromatin
To obtain a higher resolution, genome‐wide view about the dynamics of H3K27me3‐modified chromatin, we conducted SAMMY‐seq (Sequential Analysis of Macro‐Molecules accessibilitY), which was reported to be more sensitive than Hi‐C in detecting structure and accessibility changes in heterochromatin (Sebestyen et al, 2020). DNA of control and MATR3‐depleted AML12 cells was extracted from four fractions: the CSK buffer‐soluble fraction (S1), DNase I‐sensitive fraction (S2), DNase I‐resistant fraction (S3), and the high‐salt resistant fraction (S4) (Fig 6A). We examined the S4 vs S2 domains (refer to as SAMMY domain thereafter), which are robust to detect the dynamics of heterochromatin, including H3K9me3 and H3K27me3‐marked chromatin; the higher the SAMMY‐seq signal, the lower the accessibility (Sebestyen et al, 2020). The SAMMY‐seq signals of control and MATR3 knockdown samples were consistent at the larger scale, while showing noticeable changes at some smaller regions (Fig 6B). Overall, 672 SAMMY domains were common between the two conditions and accounted for 43.08% of the genome; 856 domains (7.01% of the genome) were lost, while 397 domains (4.21% of the genome) were gained in MATR3‐depleted AML12 cells (Fig 6C). As indicated by the average profiles, after MATR3 knockdown, lost or gained domains showed the strong reduction or increase in SAMMY‐seq signals, and the common domains also displayed a mild decrease in the domain borders (Fig 6D). We then examined average profiles of H3K27me3 on these SAMMY domains. The H3K27me3 level was the highest in the lost SAMMY domains, and for both common and gained domains, H3K27me3 level was lower in the center but higher on the borders (Fig 6E). Of note, for all the three types of domains, H3K27me3 level was unchanged after MATR3 knockdown (Fig 6E).
Figure 6. Higher‐order structure changes in H3K27me3‐modified chromatin as detected by SAMMY‐Seq.

-
ARepresentative Western blot on chromatin fractionation experiments of Ctrl and shMatr3 cells. Each fraction was loaded with the same number of cells. GAPDH, histone H3, Lamin A/C were used as positive controls for S1, S2 and S3, S4, respectively.
-
BGenome browser view of the SAMMY‐Seq and H3K27me3 ChIP‐seq in Ctrl and shMatr3 (replicate 1 and 2 merged) cells at a representative region. Gray boxes under each SAMMY‐seq track showing the SAMMY‐seq domains. The shadows showing the shMatr3 loss SAMMY‐seq domains.
-
CThe genome coverage of SAMMY domains (S4. vs. S2) from AML12 cells. SAMMY domains are grouped into common, shMatr3‐lost and shMatr3‐gained domains by their genomic location between ctrl and shMatr3.
-
D, EAverage SAMMY‐seq signal (S4 vs. S2) (D) and average H3K27me3 ChIP‐seq signal (E) around SAMMY domains (groups as in C) with 200 kb upstream and downstream flanking regions of ctrl and shMatr3 from AML12 cells.
-
F, GAverage H3K27me3 ChIP‐seq signal (S4. vs. S2) (F) and average SAMMY‐seq signal (G) around H3K27me3 domains with 200 kb upstream and downstream flanking regions of ctrl and shMatr3 from AML12 cells. We calculate the overlapped region length between the H3K27me3 domains and three groups of SAMMY domains (same groups in [C]). For each H3K27me3 domain, the ratio of overlap regions (overlap length/H3K27me3 peak length) ≥ 0.6 was assigned to its overlapped SAMMY domains.
-
HAverage MATR3 RIP‐seq sense and antisense signal at L1 loci around H3K27me3 domains (groups as in [F]) with 200 kb upstream and downstream flanking regions from AML12 cells.
-
IDEGs and the proportion of DEGs in relative to all genes in common, shMatr3‐lost and shMatr3‐gained SAMMY domains, respectively.
-
JHistogram plots showing the DEGs number in 100 kb bins around SAMMY domains boundaries. Y‐axis is the DEGs number in each bin.
Source data are available online for this figure.
Next, we took a closer look at H3K27me3 regions overlapped with SAMMY domains. H3K27me3 level was consistent between control and MATR3 knockdown (Fig 6F). However, the SAMMY‐seq signals were reduced in H3K27me3 regions overlapped with lost and common domains (Fig 6G), indicating an increase in accessibility at these regions. Furthermore, MATR3‐associated AS L1 elements were highly enriched near the boundaries of these H3K27me3 regions (Fig 6H). Taken together, our SAMMY‐seq data indicated that H3K27me3‐modified chromatin domains adjacent to AS L1 elements increase their accessibility upon MATR3 knockdown, without changing the H3K27me3 modifications. It should be noted that SAMMY‐seq is more sensitive to detect heterochromatin structure, and those H3K27me3 regions located at A compartments may not be detected by this assay.
To investigate the biological consequences by the structure changes in SAMMY domains, we counted numbers of DEGs in these regions. The number of DEGs associated with lost domains was significantly higher than that of common domains (Fig 6I; P < 0.001, Chi‐Square test). Furthermore, DEGs tended to be enriched at the borders of lost and gained, but not the common SAMMY domains (Fig 6J). GO analysis of these structure‐associated genes indicated that some terms were related to hepatocytes functions, such as lipid transport, cholesterol metabolism, and P450 pathway (Appendix Fig S7A–C).
As H3K27me3 is deposited and recognized by polycomb repressive complex 1 and 2 (PRC1 and PRC2; Margueron & Reinberg, 2011; Schuettengruber et al, 2017; Blackledge & Klose, 2021), we studied the regulatory relationship between MATR3 and PRC1 component BMI‐1 and PRC2 component EZH2. The co‐staining and the co‐immunoprecipitation results indicated that MATR3 interacted with BMI‐1 and EZH2 in cell nuclei (Fig EV5A and B). After depletion of MATR3, both BMI‐1 and EZH2 exhibited larger and brighter foci (Fig EV5C and D), and the significant SD increase in pixel intensity can be detected (Fig EV5E). However, MATR3 depletion did not change the protein level of BMI‐1 and EZH2 or their association with chromatin (Fig EV5F). Consistently, our imaging data showed that the redistributed BMI‐1 and EZH2 maintained their colocalization with H3K27me3 (Fig EV5G and H). As previous studies reported that the chromatin occupancy of PRC2 could be dependent on RNA‐PRC2 interaction (Long et al, 2020), we conducted the RIP–qPCR to examine whether MATR3 affect the RNA‐binding ability of EZH2. Besides the well‐known EZH2‐binding lncRNAs Malat1 and Neat1, we also examined the liver‐specific EZH2‐binding RNA Hnf1aos1 (Chen et al, 2018; Wang et al, 2018b). The RNAs were enriched in anti‐EZH2 RIP samples compared with IgG, while MATR3 depletion did not affect the enrichment (Fig EV5I). On the contrary, knockdown of BMI‐1 and EZH2 did not affect the protein level of MATR3 (Fig EV5J) and did not abolish the MATR3‐AS L1 RNA association (Fig EV5K and L). These data suggested that MATR3 regulated the localization of PcG proteins, but the latter did not modulate MATR3.
Figure EV5. MATR3 regulates the localization of PcG proteins.

-
A(Left) Representative cross‐section images showing relative distribution between MATR3 and PcG proteins BMI‐1 and EZH2. (Right) Line charts showing pixel intensity of each channel on the ROIs. r represents coefficient of correlation. Scale bar, 5 μm.
-
B(Upper) Western blotting of anti‐MATR3 immunoprecipitation (anti‐IgG IP as negative control) products hybridized with MATR3, EZH2 and BMI‐1 antibodies. (Lower) Western blot of anti‐EZH2 immunoprecipitation (anti‐IgG IP as negative control) products hybridized with EZH2 and MATR3 antibodies.
-
C, DThe representative cross‐section image showing nuclear distribution of BMI‐1 (C) and EZH2 (D) in Ctrl and shMatr3 (+Dox 3d) cells. Scale bar, 10 μm.
-
E(Upper) Standard deviation of BMI‐1 pixel intensity upon Ctrl (n = 86) and shMatr3 (+Dox 3d) (n = 89) cells. ****P < 0.0001, Student's t‐test. Error bars represent SD. (Lower) Standard deviation of EZH2 pixel intensity upon Ctrl (n = 86) and shMatr3 (+Dox 3d) (n = 97) cells. The P‐values were calculated using unpaired two‐tailed Student's t‐test; ****P < 0.0001. Error bars indicate mean ± s.e.m.
-
FWestern blotting showing the distribution of BMI‐1 and EZH2 proteins in chromatin‐non‐associated and chromatin‐associated extracts before and after MATR3 knockdown (+Dox 3d) in AML12 cells. Representative of two independent replicates with similar results.
-
G, H(Left) The representative cross‐section image showing nuclear distribution of BMI‐1 (G) and EZH2 (H), and colocalization with H3K27me3 upon Ctrl and MATR3 knockdown (+Dox 3d). (Right) Line charts showing pixel intensity of each channel on the ROIs. r, coefficient of correlation Scale bar, 5 μm.
-
IThe RNA level of Gapdh, Malat1, Neat1 and Hnf1aos1 in anti‐EZH2 RIP products relative to that in anti‐IgG RIP products as detected by RT–qPCR. For each sample, the relative RNA level was normalized to Actb. The RIP–qPCR assay was performed in six independent biological replicates (n = 6). The P‐values were calculated using unpaired two‐tailed Student's t‐test; ns, not significant. Error bars indicate mean ± s.d.
-
J(Upper) Western blotting showing the protein level of MATR3 and BMI‐1 in shCtrl and shBmi‐1 cells; (Lower) Western blotting showing the protein level of MATR3 and EZH2 in shCtrl and shEzh2 cells.
-
KThe representative images showing nuclear colocalization of MATR3 with AS L1 RNA in shCtrl, shBMI‐1 and shEZH2 AML12 cells. Scales bar, 5 μm.
-
LCoefficient of correlation between MATR3 and AS L1 RNA in shCtrl (n = 42), shBMI‐1 (n = 48) and shEZH2 (n = 46) AML12 cells. The P‐values were calculated using unpaired two‐tailed Student's t‐test; ns, not significant. Error bars indicate mean ± s.e.m.
Amyotrophic lateral sclerosis‐associated MATR3 mutations lead to chromatin redistribution
Multiple mutations of MATR3 were reported to be associated with neurodegenerative diseases including amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), vocal cord and pharyngeal weakness with distal myopathy (VCPDM), and early onset neurodegeneration (EON; Johnson et al, 2014; Marangi et al, 2017; Kao et al, 2020; Fig 7A). Nevertheless, how MATR3 dysfunction contributed to the pathology remained unclear. As the S85C and F115C mutations were reported to have more supportive pedigree data (Johnson et al, 2014) and had been studied the pathogenicity in knock‐in mice models (Kao et al, 2020; Van Bruggen et al, 2021), we focused on the action mechanism of these two MATR3 mutants for subsequent investigation. Aberrant LLPS behavior of neurodegenerative‐disease‐associated proteins was found to be pathogenic (Zbinden et al, 2020). Therefore, we tested whether mutated MATR3 proteins lead to aberrant phase separation. Predictor of natural disordered regions (PONDR) algorithm suggested that most ALS‐associated mutations could change the PONOR score of MATR3 (Appendix Fig S8A). Particularly, the S85C mutation leads a disorder region shifting to an ordered one (Fig 7B).
Figure 7. ALS‐associated mutants lead to reorganization of nuclear chromatin in N2A cells.

-
ASchematic diagram of degenerative‐disease‐associated mutations on MATR3 protein.
-
BThe order/disorder regions of MATR3 (WT/S85C/F115C) protein predicted by the PONDR algorithm.
-
CMATR3 knockdown and GFP‐tagged WT/S85C/F115C MATR3 protein replacement in N2A cells. The efficiency of endogenous MATR3 knockdown and exogenous GFP‐MATR3 (WT/S85C/F115C) overexpression as detected by western blotting. Representative of two independent replicates with similar results.
-
DRepresentative images of the GFP‐MATR3 (WT/S85C/F115C) FRAP experiments in N2A cells. Solid arrows point to the bleached points. Scale bars, 5 μm.
-
EThe fluorescence recovery curve of the GFP‐MATR3 (WT/S85C/F115C) corrected for fluorescence decay in N2A cells. Data are expressed as the mean ± s.e.m. (n = 9).
-
FRepresentative images of droplet formation assays with different concentrations of GFP‐hMATR3 (WT/S85C) proteins. NaCl concentration, 50 mM. Scale bars, 3 μm.
-
GRepresentative cross‐section images showing nuclear localization of H3K27me3 upon Ctrl, MATR3 knockdown and exogenous MATR3 (WT/S85C/F115C) overexpression in N2A cells. Scale bars, 5 μm.
-
HStandard deviation of H3K27me3 pixel intensity upon Ctrl (n = 93), MATR3 knockdown (n = 100), GFP‐MATR3 (WT) rescue (n = 104), GFP‐MATR3 (S85C) rescue (n = 82) and GFP‐MATR3 (F115C) rescue (n = 120). The P‐values were calculated using unpaired two‐tailed Student's t‐test; ns, not significant, *P < 0.05, ****P < 0.0001. Error bars indicate mean ± s.e.m.
Source data are available online for this figure.
We established a Dox‐inducible MATR3 knockdown system in mouse neuroblastoma N2A cells, a common cell model for neurodegenerative disease studies, and then transfected GFP‐tagged wild‐type or the mutants of MATR3 (S85C and F115C) to replace the endogenous MATR3 protein (Fig 7C). FRAP assay was used to investigate their dynamic features. We showed that 20 s after photobleaching, the fluorescence intensity of GFP‐WT, GFP‐F115C, and GFP‐S85C proteins recovered for 57, 43, and 20%, respectively, suggesting that these mutations make MATR3 proteins less dynamic in the nucleus (Fig 7D and E). Furthermore, we purified GFP‐tagged human MATR3 WT and S85C proteins and performed in vitro droplet formation assay. The WT MATR3 formed into highly dynamic droplets, while the S85C MATR3 proteins were much less dynamic and finally assembled into the fiber‐like structure (Fig 7F; Movies EV1 and EV2). To determine whether the mutation on MATR3 would alter its capacity to interact with AS L1 RNAs, we co‐stained MATR3 proteins (WT/S85C/F115C) with AS L1 RNAs in N2A cells. Both wild‐type and mutated MATR3 are well‐colocalized with AS L1 RNAs in N2A cells (Appendix Fig S8B and C).
Finally, we asked whether the MATR3 mutants could change nuclear chromatin distribution. As H3K27me3‐modified chromatin showed the highest correlation with MATR3 in N2A cells (Appendix Fig S8D and E), we also examined changes in H3K27me3 in this cell line. After MATR3 depletion, H3K27me3 greatly redistributed in the nucleus with brighter foci appeared; GFP‐WT overexpression completely rescued this phenotype. However, neither GFP‐S85C nor GFP‐F115C rescued the redistribution of H3K27me3 (Fig 7G and H). These data suggested that ALS‐associated MATR3 mutants have changes in their physical state, which could further lead to an abnormal chromatin organization.
Discussion
Based on the data presented, we proposed a hypothetical model that MATR3 proteins and AS L1 RNAs phase‐separate into a dynamic meshwork that facilitates clustering of chromatin (Fig 8). Mechanistically, MATR3 interacts with AS L1 RNAs via the RRM2 domain, thereby gathering chromatin regions that transcribed AS L1 RNAs.
Figure 8. Hypothesized model on how MATR3‐AS L1 RNA meshwork organizes the 3D structure of chromatin.

This possible model proposes that MATR3 proteins and AS L1 RNAs phase‐separate into a dynamic meshwork, thereby facilitating the 3D organization of chromatin that transcribed AS L1 RNAs.
Notably, our imaging and genomics data (particularly SAMMY‐seq) indicated the stronger structure changes in H3K27me3‐modified chromatin upon MATR3 knockdown (Figs 1E and 6G). This could be due to the enrichment of AS L1 elements near H3K27me3 regions (Figs 2H and 6H). The interaction between MATR3 and PcG proteins may further enhance the association of H3K27me3‐modified chromatin with MATR3/AS L1 meshwork. Furthermore, the secondary effects caused by overall transcription changes after MATR3 or AS L1 depletion could also contribute to the chromatin structure alteration.
In this paper, we demonstrated the functional roles of AS L1 RNAs in chromatin organization. However, many questions regarding AS L1 RNAs remain to be answered. First of all, it remains unclear how AS L1 RNAs retain on the chromatin. The AS L1 RNAs are possibly to form the R‐loop or the RNA:DNA triplex with L1 DNA loci (Li & Fu, 2019; Stamidis & Zylicz, 2023), in cis or in trans, which needs further investigation. Besides, the biogenesis of AS L1 RNAs is still unclear. What is the regulation mechanism underlying AS L1 RNA transcription? Are there other partners of AS L1 RNAs except for MATR3? What is the function of AS L1 RNAs in development and diseases? Previous studies regarding LINEs‐derived RNAs seldomly distinguish their transcription orientation (Jachowicz et al, 2017; Liu et al, 2018; Marasca et al, 2022) or just focused on sense LINEs (Chow et al, 2010; Percharde et al, 2018; Chen et al, 2021; Wei et al, 2022). The functions and mechanisms of AS L1 RNAs in biological processes have not been well discussed. Based on ENCODE eCLIP datasets, AS L1 RNAs are associated with nine RBPs (including MATR3, HNRNPM, and SUGP2), and MATR3 eCLIP dataset in HepG2 cells showed the highest AS L1 RNA enrichment (Van Nostrand et al, 2020). In this study, we revealed that AS L1 RNAs take part in chromatin organization by interplaying with MATR3. It may provide new insights for understanding biological functions of antisense repeat RNAs. Moreover, there are a great number of natural antisense transcripts (NATs) in human cells which is almost equal to sense transcripts, most (> 91%) of them are ncRNAs (Reis et al, 2005). NATs were suggested to interfere with the expression of sense mRNA through gene silencing, nuclear retention, epigenetic silencing, or other mechanisms (Faghihi & Wahlestedt, 2009; Reis & Poirier, 2021). Further investigations are needed to reveal the independent role of NATs, especially in the process of 3D genome organization.
NM was observed to be a network in high‐salt extracted nucleus since a half‐century ago (Berezney & Coffey, 1974). However, there have been few clear microscopic imaging data supporting for the existence of NM in living cells, especially the inner NM (Razin et al, 2014). Our super‐resolution fluorescence microscopy (Fig 1A) and immuno‐electron microscopy (Fig EV1A) data showed that MATR3, the representative NM protein, organizes as a meshwork and locate on chromatin fibers in the intact nucleus, similar to previous observations in the extracted nucleus (Lagarkova et al, 1998). NM is an RNA–protein skeleton, and most protein components are RNA‐binding proteins (Davie, 1997). Previous work used to take NM as an entirety and suggested a consistent mode of action for NM proteins. Our data in this paper and our previous work on SAF‐A/HNRNPU and SAFB, performed in the same cell line, indicate that different NM proteins act to regulate different chromatin regions (Fan et al, 2018; Huo et al, 2020). Their preferential interaction RNAs may account for the discrepancy.
Previous work reported that N‐terminal of MATR3 could form liquid‐like droplets in the cell nuclei of C2C12 mouse myoblasts, suggesting a phase separation potential for MATR3 (Gallego‐Iradi et al, 2019). This is further proved by the in vitro droplet formation assays in our study. In addition, we demonstrate that MATR3 proteins and AS L1 RNAs comprise a nuclear scaffold, and they function together to maintain a partially dynamic environment for chromatin regulation. There have been a number of instances showing noncoding RNAs and RBPs undergo LLPS, which promote cellular subcompartment formation (Rippe, 2022). In the nucleolus, higher concentration of rRNA may strengthen heterotypic interactions between nucleolus marker proteins (e.g., FBL and NPM1) and increase the nucleolus size (Lafontaine et al, 2021). The cytoplasmic lncRNA NORAD nucleate droplet formation of Pumilio RBPs (PUM1, PUN2); depletion of NORAD leads to dispersal of PUM proteins (Elguindy & Mendell, 2021). These studies revealed that under physiological conditions, higher RNA/protein ratio contributes to larger size of liquid‐like phases. However, MATR3 proteins self‐organize into liquid‐like droplets in cell nuclei when depletion of RNAs; the existence of AS L1 RNAs may abolish the weak interaction between MATR3 proteins and resulted in a gel‐like meshwork formation. This indicates that an increase in the AS L1 RNA/MATR3 ratio may facilitate a liquid‐to‐gel phase transition. We suggest that the gel‐like meshwork structure reconciles the dynamics and stability, which is suitable for widespread chromatin regions to switch states in response to spatiotemporal cues.
A study on the engineered ALS/FTD model mice indicated an abnormal heterochromatin structure and increased staining of H3K27me3 in cortices (Zhang et al, 2019). It suggested that the pathogenesis of ALS may be partially explained by dysregulation of chromatin organization. Recently, knock‐in mice models were developed to mimic the two ALS‐associated MATR3 mutants (S85C and F115C), and only S85C mice recapitulated pathological features of ALS (Kao et al, 2020; Van Bruggen et al, 2021). In this study, we revealed that S85C shows greater effects on the chromatin mislocalization and protein dynamics than F115C does, which indicated a relevance between physical property variation and the pathogenicity. Besides S85C and F115C, other mutations also caused great changes in PONDR score, suggesting that the MATR3 mutants are probably to follow the similar mechanism of action. However, genetic mutations could not be the sole reason accounting for MATR3's pathogenicity in ALS. According to previous reports, more than 60% of ALS brain samples exhibited an abnormal cytoplasm localization of MATR3 (Tada et al, 2018). That percentage is much higher than the mutation frequency of MATR3 gene (Johnson et al, 2014; Lin et al, 2015; Leblond et al, 2016). Hence, there are likely to be other mechanisms relating to this abnormal phenotype. For example, aberrant expression of MATR3‐associated RNAs may disrupt the physiological stoichiometry of MATR3:RNA in the cell nuclei, which could further affect the MATR3 localization and its association with chromatin. Due to the physical change, the MATR3 mutants are more likely to form insoluble aggregates after losing interaction with RNAs. Notably, a previous work in Drosophila model of ALS has reported an altered L1 RNA expression (Krug et al, 2017). Nevertheless, we could not illustrate the change of MATR3 expression and AS L1 RNA constitution during ALS development, due to the lack of the strand‐specific transcriptome datasets for ALS patients (and controls). Further investigation focusing on AS L1 RNAs may provide new insights into ALS pathogenesis.
Materials and Methods
shRNA system and stable express system in AML12 cells
The mouse hepatocyte cell line alpha mouse liver 12 (AML12; CRL‐2254, ATCC, Manassas, VA) were cultured in DMEM/F12 (11320033, Thermo Fisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (16140071, Gibco, Grand Island, NY), ITS Liquid Media Supplement (100×, 41400045, Gibco), and 40 ng/ml dexamethasone (D4902, Sigma, Darmstadt, Germany) at 37°C and 5% CO2. For Dox‐inducible MATR3 knockdown system (and scramble), plasmids were constructed by cloning the target sequences (the Matr3 shRNA target: GAGACCGATCTTGCTAATTTA; the Scramble shRNA target: ACTCGACACTATAGTATCTCA) to the pLKO‐Tet‐On vector (Wiederschain et al, 2009) and then transfect AML12 cells by lentivirus‐based system. To generate stable cell lines, AML12 cells were selected in the presence of 1 μg/ml puromycin for 1 day. One μg/ml dox was used to induce MATR3 depletion. For BMI‐1/EZH2 knockdown system, the 21‐mer sequences (the Ctrl shRNA target: ACTCGACACTATAGTATCTCA; the BMI‐1 shRNA target: CCAGATTGACGTCATGTATGA; the EZH2 shRNA target: GTGTGATAGCACAAAATTTTT) were cloned into pLKO.1‐TRC vector. For stable express cell lines, the GFP‐tagged MATR3 cDNA was cloned into Lv‐ef1a‐blasticidin‐tre‐MCS plasmid as described (Biscotti et al, 2015). GFP‐tagged truncations (▵ZF1, ▵ZF2, ▵RRM1 and ▵RRM2) were further constructed based on this plasmid using TOYOBO KOD‐401 Kit, using primers as described (Malik et al, 2018). Plasmid transfection in AML12 cells was accomplished by lentivirus‐based system. To generate stable cell lines, AML12 cells were selected in the presence of 1 μg/ml puromycin and 4 μg/ml blasticidin for 2 days. One μg/ml dox was used to induce GFP‐MATR3 (or its truncations) expression. For exogenous truncations rescue assay, the shMatr3‐resistant mutation was first introduced to Lv‐ef1a‐blasticidin‐tre‐GFP‐MATR3 plasmid. And then GFP‐tagged truncations (▵ZF1, ▵ZF2, ▵RRM1, and ▵RRM2) were further constructed based on this plasmid. Then, plasmids were transfected with Lipofectamine™ 3000 Transfection Reagent (L3000150, Thermo Fisher Scientific, Waltham, MA) into shMatr3 AML12 cells.
Rapid protein degradation system in mouse ES cells
The mouse ES cell line (E14TG2a) was cultured in 2i/LIF conditions as described (Li et al, 2008). The auxin (IAA)‐inducible MATR3 degradation system was developed in mouse ESCs according to the rapid protein depletion methods established by Natsume et al (2016). First, the parental cells were generated by introducing the vector encoding constitutive cytomegalovirus‐controlled auxin responsive F‐box protein (CMV‐OsTIR1) at the safe harbor ROSA26 using CRISPR/Cas (Nakao et al, 2016). Cells were further selected in the presence of 1 μg/ml puromycin. After 7 days, colonies were picked for further selection in a 96‐well plate and the genotype was checked by genomic PCR. Based on this parental cell line, an in‐frame mAID cassette was introduced after the last codon of MATR3 gene by CRISPR/Cas (sgRNA target on Matr3 gene: ATAAATTGGCAGAAGAACGG). Plasmids were transfected with Lipofectamine™ 3000 Transfection Reagent (L3000150, Thermo Fisher Scientific, Waltham, MA). To ensure AID‐tagging on both alleles, two short homology donor vectors containing neomycin and hygromycin resistance markers were transfected simultaneously into cells. Cells were further selected in the presence of 2 mg/ml neomycin and 200 μg/ml hygromycin. After 7 days, colonies were picked for further selection in a 96‐well plate and the genotype was checked by genomic PCR. 0.5 mM IAA (dissolved in alcohol) was used to induce MATR3 degradation.
Dox‐inducible shRNA system and mutant transfection in N2A cells
Mouse Neuro 2a cells (N2A; CCL‐131, ATCC, Manassas, VA) were cultured in DMEM (Dulbecco's Modified Eagle's medium, Hyclone) supplemented with 10% fetal bovine serum, nonessential amino acids (11140050, Thermo Fisher Scientific) at 37°C and 5% CO2. Dox‐inducible MATR3 knockdown system in N2A cells was established in the same way as in AML12 cells. To generate the shMATR3‐resistant MATR3 cDNA, we introduced a synonymous mutation of shMATR3 targeting sites into the Lv‐ef1a‐GFP‐MATR3 plasmid. And based on this plasmid, we further introduced MATR3 mutants (S85C/F115C) using TOYOBO KOD‐401 Kit. Lv‐ef1a‐GFP‐MATR3 WT/S85C/F115C (resistant) plasmids were separately transfected into the dox‐inducible shMATR3 N2A cells, using standard Polyethylenimine (PEI)‐based transfection approach.
Western blotting and immunofluorescence
For western blotting, the cell lysates were blotted against primary antibodies and the blots were visualized with peroxidase‐coupled secondary antibodies using ProteinSimple FluorChem M gel imaging system. For immunofluorescence, cells were cultured on the glassy coverslip, fixed by 4% formaldehyde for 10 min, treated by 0.5% Triton X‐100 for 10 min, blocked by 4% BSA for 30 min at room temperature, cultured by diluted primary antibody overnight at 4°C, followed by adding second‐fluorescence antibody for 1 h at room temperature, and stained with DAPI. Most experiments involving MATR3 antibody were performed by Bethyl (A300‐591A), which recognizes carboxy‐terminal of MATR3 protein and has higher antibody titers. There was an exception: for detecting the MATR3 degradation efficiency in mouse ES cells. As mAID‐tag blocked the antigen‐binding sites of the carboxy‐terminal MATR3 protein, which could not be recognized by Bethyl (A300‐591A), we used the amino‐terminal MATR3 antibody (Abcam, ab51081) as an alternative. Other antibody used in this paper as follows: anti‐H3K9me3 (ABclonal, A2360), anti‐H3K9me2 (ABclonal, A2359), anti‐H3K27me3 (ABclonal, A16199), anti‐H3K27ac (Active Motif, 39,133), anti‐H3K4me3 (ABclonal, A2357), anti‐β‐actin (Proteintech, 66,009‐1‐Ig), anti‐H3 (Abcam, ab1791), anti‐BMI‐1 (Santa Cruz, SC‐390443), and anti‐EZH2 (CST, 5246).
RNA fluorescence in situ hybridization (RNA FISH)
RNA fluorescence in situ hybridization was performed using RNAscope® Multiplex Fluorescent Reagent Kit v2 (ACD, 323100). Probes for AS L1 RNA were designed by ACD company (Advanced Cell Diagnostics, Hayward, CA, USA) based on the consensus sequence on antisense L1_Mus1 RNAs. The product code is 1084381‐C1 (Advanced Cell Diagnostics, Hayward, CA, USA). The RNAscope experiment was performed according to the standard RNAscope protocol without protease treating, and then the slides were refixed with 4% PFA followed by the standard protocol of immunofluorescence.
Immuno‐electron microscopy
Cells were fixed with 4% formaldehyde and incubated with the primary antibody. The HRP secondary antibody was used to detect the primary antibody, and then using DAB (P0203, Beyotime) for the HRP staining, the product is EM‐visible. After that, the samples were prepared for conventional transmission electron microscope. In brief, cells were fixed with 2.5% glutaraldehyde, followed by 1% osmium tetroxide treating, gradient dehydration with alcohol, dehydrated with acetone, and embedded with resin. The sections were imaged at the EM facility of ION (Institute of Neuroscience, Shanghai, China).
Chromatin immunoprecipitation sequencing (ChIP‐Seq) assay
ChIP experiments were performed as previously described (Wen et al, 2008), antibody against H3K9me3 (Abcam, ab8898), H3K9me2 (Abcam, ab1220), H3K27me3 (ABclonal, A16199), and H3K27ac (Active Motif, 39,133) were used. Cells were fixed with 1% formaldehyde for 10 min at room temperature. The libraries were prepared using the VAHTS Universal DNA Library Prep Kit for Illumina V3 (Vazyme, ND607‐01) followed by next‐generation sequencing (NGS) using the Illumina HiSeq X Ten system.
Assay for transposase‐accessible chromatin with high‐throughput sequencing (ATAC‐seq)
ATAC‐seq experiments were performed as described (Buenrostro et al, 2015). For each sample, 4 × 104 AML12 cells were used. The transposition reaction was incubated at 37°C for 40 min. The libraries were prepared using the TruePrep DNA Library Prep Kit V2 for Illumina (Vazyme, TD501‐01) followed by next‐generation sequencing (NGS) using the Illumina HiSeq X Ten system.
Analysis of subcellular protein fractions
Cells were washed once with 1× PBS and were processed into chromatin‐non‐associated and chromatin‐associated fractions as described (Bhatt et al, 2012). For RNase A treatment, final concentration of 10 μg/ml RNase A (EN0531, Thermo Fisher Scientific) was added to the lysis buffer. The extracts were diluted with equal volume 1× SDS loading buffer, and proteins in each fraction were detected by western blotting.
Co‐immunoprecipitation assay and RNA immunoprecipitation sequencing (RIP‐Seq)
RIP experiments were performed as previously described (Rinn et al, 2007). Briefly, AML12 cells were cross‐linked with UV (4,000 J) and the cell nuclei were extracted and sonicated. Then, incubated the supernatant with anti‐IgG (CST, 2729), anti‐MATR3 (Bethyl, A300‐591A) or anti‐EZH2 (CST, 5246) antibodies overnight at 4°C. After wash for 3 times in RIP buffer and elution in 65°C, the RNA samples were extracted with Trizol. Genomic DNA was digested with DNase I (EN0523, Thermo Fisher Scientific) at 37°C for 1 h. The libraries were prepared using the VAHTS Total RNA‐seq (H/M/R) Library Prep Kit for Illumina (Vazyme, NRM603‐01) followed by next‐generation sequencing (NGS) using the Illumina HiSeq X Ten system. For anti‐MATR3 RIP‐qPCR test, the following primers were used: primer 1‐f (CCCAGCCCCTTGTTAAATACC); primer 1‐r (TGGGGCACACATGAACCTT); primer 2‐f (CACTCTCCTGTCTTGCCTCT); primer 2‐r (CACACACACACAAAGCAATCAAA); primer 3‐f (ACTGTTGGTGGGATTGCAAG); primer 3‐r (TGGGTTCTTTCCAGCTTCTG); primer 4‐f (CCCACTTCACTCCAGTCAGA); primer 4‐r (AGTTTCTGGGCTAATATCCACTT). For anti‐EZH2 RIP‐qPCR test, the following primers were used: Actb‐f (GGTCATCACTATTGGCAACG), Actb‐r (ACGGATGTCAACGTCACACT), Gapdh‐f (AACTTTGGCATTGTGGAAGG), Gapdh‐r (GGATGCAGGGATGATGTTCT), Malat1‐f (CTTTGCGGGTGTTGTAGGTT), Malat1‐r (ACAAAACTGGGAGGTTGTGC), Neat1‐f (TGAGTGCTTTGCCACTGAAT), Neat1‐r (TAACAGCTTCCCCTCTGCTC), Hnf1aos1‐f (ACCTGGCAATCATCCTTTCA), Hnf1aos1‐r (GCTCATTCCTGGGTAGCAGA).
RNA decay assay
RNA transcription was blocked with 5 μg/ml actinomycin D. Cells were collected after 0, 2, 4, 6, 8, and 12 h and RNAs were extracted with Trizol. For RT–qPCR, the following primers were used: AS L1 RNA‐1‐f (CTGTTGAGGGACATCTGGGT); AS L1 RNA‐1‐r (CCAATACCTCTCCTGGGCAT); AS L1 RNA‐2‐f (CCAGCTTGCAATCCCATCAG); AS L1 RNA‐2‐r (CCACCTCACACCAGTCAGAA); Neat1‐f (TGAGTGCTTTGCCACTGAAT); Neat1‐r (TAACAGCTTCCCCTCTGCTC).
Antisense oligonucleotides (ASOs) treatment
Cells were adherently cultured to about 60% confluency, and then, the antisense oligonucleotides were added to the culture medium using Lipofectamine™ RNAiMAX transfection regent (13778030, Thermo Fisher Scientific). The final concentration of ASOs is 50 mM. ASO target for AS L1 RNA was chosen on the consensus sequence on antisense L1_Mus1 RNAs. The following sequences were used: Scramble ASO (CCUUCCCTGAAGGTTCCUCC; Huo et al, 2020); Antisense L1_Mus1 (AS L1)‐ASO (GAAGGTATAGGGAACUUUCG); Sense L1_Mus1‐ASO (CGAAAGTTCCCTATACCUUC); Antisense L1_MA7‐ASO (CUUAAAAAAGAAGGAAAUCC); Sense L1_MA7‐ASO (GGAUUTCCTTCTTTTUUAAG).
Fluorescent recovery after photobleaching (FRAP)
The FRAP was performed as described before (Huo et al, 2020). Briefly, captured one image at prebleach, and then an approximately 1 mm2 region was selected to bleach once with maximum light intensity; following images were captured every 1.3 s on a confocal laser scanning microscopy (Leica TCS SP5, Wetzlar, Germany). Image data analyses were performed using LAS AF Lite software (Leica Microsystems, Wetzlar, Germany). The mobile fraction and the half‐life of recovery was calculated as described before (Day et al, 2012).
In vitro droplet formation assay
Protein purification. The GFP‐mouse MATR3 (or human MATR3 WT/S85C) cDNA was cloned into the pCAG‐flag‐6 × His plasmid and then transfected into HEK293T cells. The proteins were first purified with Ni Agarose 6 FF (AOGMA) with the AKTA system (GE Healthcare Life Sciences) and further purified with anti‐Flag affinity beads. After concentrated in an Amicon Ultracel‐50K spin concentrator to exchange the storage buffer (50 mM Tris–HCl [pH 7.5], 200 mM NaCl, 1 mM DTT, 10% glycerol), the proteins were stored in −80°C after flash freezing in liquid nitrogen.
In vitro RNA transcription. cDNA of L1, B1 and MajSAT containing T7 promoter sequence and restriction sites hanging on both sides (in opposite orientation) were cloned to the PUC57 vector. And then, linearize the vector with endonuclease for sense‐ or antisense‐ oriented transcription, separately. RNAs were transcribed in vitro using TranscriptAid T7 High Yield Transcription Kit (K0441, Thermo Fisher Scientific). UTP in this kit was replaced by Cy5‐modified UTP for RNA labeling. The sequences of B1 and MajSAT were obtained from previous reports (Lehnertz et al, 2003; Vassetzky et al, 2003), and the representative L1 sequence was a 318 bp consensus sequence of L1 Md_F2 elements.
In vitro droplet formation. Proteins or the mix of protein and RNAs were diluted in PCR tubes at different concentrations in reaction buffer (50 mM Tris–HCl (pH 7.5), 5 mM DTT, 0.1% Triton x‐100 and 50–800 mM NaCl). Then, 3 μl mixed liquid was dropped on the living cell chamber and the images were captured with confocal laser scanning microscopy (Leica TCS SP5, Wetzlar, Germany). Image data analyses were performed using LAS AF Lite software (Leica Microsystems, Wetzlar, Germany).
In vivo protein and RNA concentration estimation
The radius of cell nuclei was measured on the confocal images using LAS AF Lite software (Leica Microsystems, Wetzlar, Germany). The average radius of AML12 cell nuclei is 6.1 μm. So, the average volume of nuclei was calculated to be 9.5 × 10−7 μl. Assuming the MATR3 are equally distributed in cell nuclei, the 3,240,673 molecules in one cell nuclei can be converted to the concentration of 5.7 μM. For AS L1 RNA concentration, we first measured the total RNA amount of 500 k AML12 cells to be 12,294 ng. Excluding 80% proportion of the rRNA, the total RNA is 0.005 ng per cell. Then, we analyzed proportion of total AS L1 RNAs by RNA‐seq data (library prepared using Ribo‐off depletion kit). On average, 1.52% of mapped reads are AS L1 RNAs. Therefore, the nuclear concentration of total AS L1 RNAs can be calculated to be 74.7 ng/μl. The molecular weight of the 318 nt‐AS L1 RNA fragment is 159,128.6 g/mol. So, the concentration of AS L1 RNAs like 318 nt‐AS L1 RNA fragment in cell nuclei could be 469.4 nM.
In situ Hi‐C experiment
The in situ Hi‐C libraries were prepared as previously described (Rao et al, 2014). In brief, cells (2.5 millions/sample) were cross‐linked by 1% formaldehyde at 25°C for 10 min. Then, the cell chromatin was digested with 100 U MboI at 37°C overnight. Subsequently, filled in the restriction fragment overhangs with biotin at 37°C for 1.5 h and performed proximity ligation at room temperature for 4 h. After protein degradation and cross‐link reversal, DNA was purified by ethanol precipitation and was sheared to a size of 300–500 bp. After size selection, the biotinylated DNA was pulled down by streptavidin beads and then prepared for Illumine sequencing. The libraries were sequenced via the Illumina HiSeq X Ten system at Annoroad Gene Technology. Two biological replicates were performed for both control and MATR3‐depleted AML12 cells.
SAMMY‐seq experiment
The SAMMY‐seq experiment was conducted as previously described (Marasca et al, 2016; Sebestyen et al, 2020). In brief, 4 million AML12 cells were used for one sample. After 5 min treatment of 600 μl CSK buffer at 4°C, the supernatant was collected as S1. The pellets were washed and treated with 10 U Turbo DNase in 200 μl CSK buffer for 1 h at 37°C, the supernatant was collected as S2. Then, the pellets were washed and extracted with 2 M NaCl in CSK buffer for 10 min at 4°C. After centrifugation, the supernatant was collected as S3. The pellets were washed and solubilized in 8 M urea, and this fraction was collected as S4. For DNA sequencing, 50% volume of each fraction was extracted by phenol/chloroform and sonicated into 300–700 bp fragments. The DNA libraries were prepared using the VAHTS Universal DNA Library Prep Kit for Illumina V3 (Vazyme, ND607‐01) followed by next‐generation sequencing (NGS) using the Illumina HiSeq X Ten system. The remaining 50% of each fraction was lysed with SDS loading buffer and prepared for western blotting test.
Image processing and quantification
The images of immunofluorescence or RNA FISH were obtained with the confocal laser scanning microscopy (Leica TCS SP5, Wetzlar, Germany). To achieve the comparable image data between groups, the microscope parameters were kept unchanged in each set of experiments. To obtain the super‐resolution images, we conducted confocal laser scanning microscopy using Leica TCS SP8 STED system, followed by processing the images with a deconvolution software Huygens Ver. Image data analysis was performed using LAS AF Lite software (Leica Microsystems, Wetzlar, Germany) and ImageJ software (ImageJ Software, National Institutes of Health, Bethesda, MD, USA).
ChIP‐seq data and ATAC‐seq data processing
CutAdapt v.1.16 (Martin, 2011) was used to remove adaptor sequences from raw reads of ChIP‐seq and ATAC‐seq. Reads were mapped to mouse genome (mm10) using Bowtie (v1.2.3) (Langmead et al, 2009). Duplicate reads were excluded and kept only one read for each genomic site. BedGraph files were normalized for total mapped read counts using genomecov from bedtools v2.29.2 (Quinlan & Hall, 2010). The normalized read density bigwig tracks were used for visualization with Integrative Genomics Viewer (IGV; Robinson et al, 2011; Thorvaldsdottir et al, 2013). H3K27me3 peaks were called using SICER (Zang et al, 2009). To generate the ChIP‐Seq signal distribution for interested regions, we calculated the average ChIP‐Seq signal across these regions.
RIP‐Seq data processing
RIP‐Seq reads were mapped to mouse reference genome (mm10) using HISAT2 (v2.1.0; Kim et al, 2019), with parameters: ‐‐rna‐strandness RF ‐‐dta. Peaks were called using MACS (v1.4.2) with parameters: ‐‐nomodel ‐‐nolambda (Zhang et al, 2008). Read counts were generated using HTSeq‐count (v0.6.1; Anders et al, 2015). BedGraph files were generated using genomecov from bedtools (v2.29.2; Salton et al, 2011). Integrative Genomics Viewer (IGV) was used for data visualization (Robinson et al, 2011, Thorvaldsdottir et al, 2013). To identify the Matr3‐associated L1 RNAs from RIP‐seq, L1 loci with RIP (MATR3 ‐IgG) count number ≥ 10 are kept (Fig 2F).
Hi‐C data analysis
Mapping and matrix generation. For Hi‐C data, paired‐end reads were mapped, processed, and iterative correction (ICE) using HiC‐Pro software (version 2.11.4) (https://github.com/nservant/HiC-Pro; Servant et al, 2015). Read pairs were mapped to the mouse mm10 reference genome (https://hgdownload.soe.ucsc.edu/downloads.html#mouse) with end‐to‐end algorithm and “‐very‐sensitive” option. Singleton, multimapped, dumped, dangling, self‐circle paired‐end reads, and PCR duplicates were removed after mapping. To eliminate the possible effects on data analyses of variable sequencing depths, we randomly sampled equal numbers read pairs from each sample for downstream analyses. Valid read pairs were used to generate raw contact matrices at 100‐kb, 250‐kb, and 1‐Mb resolutions and applied iterative correction (ICE) on them. We converted .all ValidPairs files to .hic files by the script hicpro2juicebox.sh from HiC‐Pro utilities. HiCExplorer (version 3.5.3) (https://hicexplorer.readthedocs.io/en/3.5.3/; Ramirez et al, 2018) was used to compute correlations between replicates and plot contact maps for ICE normalized matrices.
Identification of A and B Compartments. A and B compartments were identified as described previously (Lieberman‐Aiden et al, 2009). For normalized contact matrix at 100‐kb resolution, expected matrix was calculated as the sum of contacts per genomic distance divided by the maximal possible contacts and then converted to a Pearson correlation matrix. Then, principal component analysis was performed on the correlation matrix. The first principal eigenvector (PC1) for each bin was used to calculate the overlap ratio with H3K27ac ChIP‐seq peaks to assign each bin to A or B compartment.
TAD calling. TADs were called by the insulation score method using cworld software (https://github.com/dekkerlab/cworld-dekker) as previously described (Crane et al, 2015). Insulation scores were calculated by the cworld script “matrix2insulation.pl.” at 100‐kb resolution matrix with the parameters “‐‐im iqrMean ‐‐is 500000 ‐‐ids 250000 ‐‐nt 0.3.” The topologically associated domains were identified by the cworld script “insulation2tads.pl,” and the 0.3 min boundary strength was set as the threshold.
Aggregate contact frequency to compartments. In order to examine the relationship between PC1 values and contact frequency, we sorted PC1 values for all 100 kb bins in decreasing order and divided into 50 equal quantiles (Fig 5B). We then built the 50 × 50 grid of 2D interval that x‐axis and y‐axis are 50 equal quantiles described above. Then in a 50 × 50 2D interval, we assigned the intrachromosomal contacts to pairs of 100 kb loci in each grid and calculated the average contact frequency. We then plotted heatmap to show average contact frequency enrichment in Ctrl and shMatr3 samples and the fold change of contact frequency between them.
Average contact frequency for compartment regions. To quantify the average enrichment of contacts at compartment level, we first connected the continuous bins that have the same state of PC1 values in Ctrl defined as compartment regions. We then calculated average log ratio of the observed and the expected contacts for A/B intracompartment regions and intercompartment region pairs between the same type (AA and BB) and different types (AB) in Ctrl and shMatr3. Finally, changes in compartment regions between Ctrl and shMatr3 were calculated and represented as boxplots (Fig 5C and D).
Insulation score was computed by cworld software (https://github.com/dekkerlab/cworld-dekker; Crane et al, 2015) at 50 kb resolution using function matrix2insulation.pl with parameters ‐‐is 1,000,000 ‐‐ids 200,000 ‐‐nt 0.1.
TADs classified into MATR3‐AS L1 RNAs associated TADs and nonassociated TADs according to density of anti‐MATR3 RIP‐seq signal of AS L1 RNAs within TADs (Fig 5H). MATR3‐AS L1 RNAs associated TADs classified into four quantile groups by increasing in MATR3‐AS L1 RNAs density (Fig 5I). GO/KEGG analysis was performed using online software metascape (https://metascape.org/gp/index.html#/main/; Appendix Fig S5D and F).
Strand‐specific RNA‐Seq
The libraries were prepared using the VAHTS Stranded mRNA‐seq Library Prep Kit for Illumina (Vazyme, NR602‐01) followed by next‐generation sequencing (NGS) using the Illumina HiSeq X Ten system. Reads were mapped to mouse reference genome (mm10) using HISAT2 v2.1.0 (Kim et al, 2019), with parameters: ‐‐rna‐strandness RF. FPKM values were calculated using StringTie v1.3.5 (Pertea, Pertea et al, 2015) based on Refgene annotation from the UCSC genome browser, with parameters: ‐‐rf. Genes with FPKM ≥ 1 were considered to be expressed. Read counts were generated using HTSeq‐count v0.6.1 (Anders et al, 2015). Differentially expressed genes (DEGs) were calculated using the R package DESeq2 v1.26.0 (Love et al, 2014), Expression genes were considered differentially expressed if DESeq2 P‐value < 0.1 and | log2(fold changes) | ≥ 1. BedGraph format files were calculated using genomecov from bedtools v2.29.2 (Quinlan & Hall, 2010). Integrative Genomics Viewer (IGV) was used for data visualization in genome (Robinson et al, 2011; Thorvaldsdottir et al, 2013).
SAMMY‐seq data analysis
SAMMY‐seq reads were mapped to mouse reference genome (mm10) using Bowtie (v1.2.3; Langmead et al, 2009). Duplicate reads were excluded and kept only one read for each genomic site. We generated the bigwig and bedgraph files of log2(S2/S4) at 10 kb bin using bedtools (v2.29.2; Quinlan & Hall, 2010). Integrative Genomics Viewer (IGV) was used for data visualization (Robinson et al, 2011; Thorvaldsdottir et al, 2013). SAMMY domains were determined using package DNAcopy (http://bioconductor.org/packages/DNAcopy/). The log2(S2/S4) profiles were smoothed using the default smooth.CNA process. Smoothed samples were then segmented using the segment command with parameter: alpha = 0.01, min.width = 2, undo.splits = “none.”
Statistical analyses
Image data analyses were performed using LAS AF Lite software (Leica Microsystems, Wetzlar, Germany) and ImageJ software (ImageJ Software, National Institutes of Health, Bethesda, MD, USA). Statistical analyses were performed using GraphPad Prism (8.0) and Microsoft Excel. Representative data with ≥ 3 independent experiments were expressed as the mean ± s.d. or mean ± s.e.m. Significance testing was accomplished using unpaired two‐tailed Student's t‐test. No statistical method was used to predetermine the sample size. No data were excluded from the analyses. The western blots were performed in two or three independent biological replicates with similar results, and a representative blot was shown. The ChIP‐seq, RIP‐seq, ATAC‐seq, and Hi‐C were all performed in two independent biological replicates. The details for each experiment are also provided in the figure legends.
Author contributions
Yuwen Zhang: Conceptualization; data curation; formal analysis; investigation; visualization; methodology; writing – original draft; writing – review and editing. Xuan Cao: Data curation; software; formal analysis; investigation; visualization; methodology; writing – review and editing. Zehua Gao: Data curation; investigation; writing – review and editing. Xuying Ma: Data curation; software; formal analysis; investigation; visualization. Qianfeng Wang: Data curation; methodology. Xiaoxuan Xu: Data curation. Xiumei Cai: Project administration. Yan Zhang: Project administration. Zhao Zhang: Data curation; methodology. Gang Wei: Data curation; supervision; methodology; writing – review and editing. Bo Wen: Conceptualization; resources; data curation; formal analysis; supervision; funding acquisition; investigation; visualization; methodology; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix S1
Expanded View Figures PDF
Movie EV1
Movie EV2
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank members of the Wen laboratory for their support and suggestions. We thank Yalin Huang and Jin Li for assistance with fluorescence microscopy. This work was supported by the National Natural Science Foundation of China (32130019 to B.W.) and the National Key Research and Development Program of China (2021YFA1100203 to B.W.).
EMBO reports (2023) 24: e57550
Contributor Information
Gang Wei, Email: weigang@picb.ac.cn.
Bo Wen, Email: bowen75@fudan.edu.cn.
Data availability
High‐throughput sequencing data have been deposited to GEO with the following accession number. AML12 H3K27me3 ChIP‐seq (GSE200241, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE200241); AML12 Hi‐C (GSE200243, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE200243); AML12 RIP‐seq (GSE200244, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE200244); AML12 mRNA‐seq (GSE200245, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE200245); AML12 rRNA‐off RNA‐seq (GSE228646, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE228646); AML12 SAMMY‐Seq (GSE228141, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE228141); ESC Hi‐C (GSE225500, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE225500); ESC RIP‐seq (GSE225501, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE225501).
References
- A P, Weber SC (2019) Evidence for and against liquid‐liquid phase separation in the nucleus. Noncoding RNA 5: 50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W (2015) HTSeq—a python framework to work with high‐throughput sequencing data. Bioinformatics 31: 166–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attig J, Agostini F, Gooding C, Chakrabarti AM, Singh A, Haberman N, Zagalak JA, Emmett W, Smith CWJ, Luscombe NM et al (2018) Heteromeric RNP assembly at LINEs controls lineage‐specific RNA processing. Cell 174: 1067–1081.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Carrel L, Chakravarti A, Eichler EE (2000) Molecular evidence for a relationship between LINE‐1 elements and X chromosome inactivation: the Lyon repeat hypothesis. Proc Natl Acad Sci USA 97: 6634–6639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezney R, Coffey DS (1974) Identification of a nuclear protein matrix. Biochem Biophys Res Commun 60: 1410–1417 [DOI] [PubMed] [Google Scholar]
- Berezney R, Coffey DS (1977) Nuclear matrix. Isolation and characterization of a framework structure from rat liver nuclei. J Cell Biol 73: 616–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat P, Honson D, Guttman M (2021) Nuclear compartmentalization as a mechanism of quantitative control of gene expression. Nat Rev Mol Cell Biol 22: 653–670 [DOI] [PubMed] [Google Scholar]
- Bhatt DM, Pandya‐Jones A, Tong AJ, Barozzi I, Lissner MM, Natoli G, Black DL, Smale ST (2012) Transcript dynamics of proinflammatory genes revealed by sequence analysis of subcellular RNA fractions. Cell 150: 279–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biscotti MA, Canapa A, Forconi M, Olmo E, Barucca M (2015) Transcription of tandemly repetitive DNA: functional roles. Chromosome Res 23: 463–477 [DOI] [PubMed] [Google Scholar]
- Blackledge NP, Klose RJ (2021) The molecular principles of gene regulation by Polycomb repressive complexes. Nat Rev Mol Cell Biol 22: 815–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle AL, Ballard SG, Ward DC (1990) Differential distribution of long and short interspersed element sequences in the mouse genome: chromosome karyotyping by fluorescence in situ hybridization. Proc Natl Acad Sci USA 87: 7757–7761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, Greenleaf WJ (2015) ATAC‐seq: a method for assaying chromatin accessibility genome‐wide. Curr Protoc Mol Biol 109: 21.29.1–21.29.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha HJ, Uyan O, Kai Y, Liu T, Zhu Q, Tothova Z, Botten GA, Xu J, Yuan GC, Dekker J et al (2021) Inner nuclear protein Matrin‐3 coordinates cell differentiation by stabilizing chromatin architecture. Nat Commun 12: 6241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaumeil J, Le Baccon P, Wutz A, Heard E (2006) A novel role for Xist RNA in the formation of a repressive nuclear compartment into which genes are recruited when silenced. Genes Dev 20: 2223–2237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Cai J, Wang Q, Wang Y, Liu M, Yang J, Zhou J, Kang C, Li M, Jiang C (2018) Long noncoding RNA NEAT1, regulated by the EGFR pathway, contributes to glioblastoma progression through the WNT/beta‐catenin pathway by scaffolding EZH2. Clin Cancer Res 24: 684–695 [DOI] [PubMed] [Google Scholar]
- Chen C, Liu W, Guo J, Liu Y, Liu X, Liu J, Dou X, Le R, Huang Y, Li C et al (2021) Nuclear m(6)a reader YTHDC1 regulates the scaffold function of LINE1 RNA in mouse ESCs and early embryos. Protein Cell 12: 455–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow JC, Ciaudo C, Fazzari MJ, Mise N, Servant N, Glass JL, Attreed M, Avner P, Wutz A, Barillot E et al (2010) LINE‐1 activity in facultative heterochromatin formation during X chromosome inactivation. Cell 141: 956–969 [DOI] [PubMed] [Google Scholar]
- Chu C, Zhang QC, da Rocha ST, Flynn RA, Bharadwaj M, Calabrese JM, Magnuson T, Heard E, Chang HY (2015) Systematic discovery of Xist RNA binding proteins. Cell 161: 404–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho MB, Attig J, Bellora N, Konig J, Hallegger M, Kayikci M, Eyras E, Ule J, Smith CW (2015) Nuclear matrix protein Matrin3 regulates alternative splicing and forms overlapping regulatory networks with PTB. EMBO J 34: 653–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane E, Bian Q, McCord RP, Lajoie BR, Wheeler BS, Ralston EJ, Uzawa S, Dekker J, Meyer BJ (2015) Condensin‐driven remodelling of X chromosome topology during dosage compensation. Nature 523: 240–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creamer KM, Kolpa HJ, Lawrence JB (2021) Nascent RNA scaffolds contribute to chromosome territory architecture and counter chromatin compaction. Mol Cell 81: 3509–3525.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusanelli E, Romero CA, Chartrand P (2013) Telomeric noncoding RNA TERRA is induced by telomere shortening to nucleate telomerase molecules at short telomeres. Mol Cell 51: 780–791 [DOI] [PubMed] [Google Scholar]
- Davie JR (1997) Nuclear matrix, dynamic histone acetylation and transcriptionally active chromatin. Mol Biol Rep 24: 197–207 [DOI] [PubMed] [Google Scholar]
- Day CA, Kraft LJ, Kang M, Kenworthy AK (2012) Analysis of protein and lipid dynamics using confocal fluorescence recovery after photobleaching (FRAP). Curr Protoc Cytom Chapter 2: Unit2.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elguindy MM, Mendell JT (2021) NORAD‐induced Pumilio phase separation is required for genome stability. Nature 595: 303–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faghihi MA, Wahlestedt C (2009) Regulatory roles of natural antisense transcripts. Nat Rev Mol Cell Biol 10: 637–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H, Lv P, Huo X, Wu J, Wang Q, Cheng L, Liu Y, Tang QQ, Zhang L, Zhang F et al (2018) The nuclear matrix protein HNRNPU maintains 3D genome architecture globally in mouse hepatocytes. Genome Res 28: 192–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego‐Iradi MC, Strunk H, Crown AM, Davila R, Brown H, Rodriguez‐Lebron E, Borchelt DR (2019) N‐terminal sequences in matrin 3 mediate phase separation into droplet‐like structures that recruit TDP43 variants lacking RNA binding elements. Lab Invest 99: 1030–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall LL, Carone DM, Gomez AV, Kolpa HJ, Byron M, Mehta N, Fackelmayer FO, Lawrence JB (2014) Stable C0T‐1 repeat RNA is abundant and is associated with euchromatic interphase chromosomes. Cell 156: 907–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak IA, Weisswange I, Mansfeld J, Buchholz F et al (2015) A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163: 712–723 [DOI] [PubMed] [Google Scholar]
- Hibino Y, Usui T, Morita Y, Hirose N, Okazaki M, Sugano N, Hiraga K (2006) Molecular properties and intracellular localization of rat liver nuclear scaffold protein P130. Biochim Biophys Acta 1759: 195–207 [DOI] [PubMed] [Google Scholar]
- Huo X, Ji L, Zhang Y, Lv P, Cao X, Wang Q, Yan Z, Dong S, Du D, Zhang F et al (2020) The nuclear matrix protein SAFB cooperates with major satellite RNAs to stabilize heterochromatin architecture partially through phase separation. Mol Cell 77: 368–383.e7 [DOI] [PubMed] [Google Scholar]
- Jachowicz JW, Bing X, Pontabry J, Boskovic A, Rando OJ, Torres‐Padilla ME (2017) LINE‐1 activation after fertilization regulates global chromatin accessibility in the early mouse embryo. Nat Genet 49: 1502–1510 [DOI] [PubMed] [Google Scholar]
- Jackson DA, Dickinson P, Cook PR (1990) The size of chromatin loops in HeLa cells. EMBO J 9: 567–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin F, Li Y, Dixon JR, Selvaraj S, Ye Z, Lee AY, Yen CA, Schmitt AD, Espinoza CA, Ren B (2013) A high‐resolution map of the three‐dimensional chromatin interactome in human cells. Nature 503: 290–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JO, Pioro EP, Boehringer A, Chia R, Feit H, Renton AE, Pliner HA, Abramzon Y, Marangi G, Winborn BJ et al (2014) Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci 17: 664–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamelgarn M, Chen J, Kuang L, Arenas A, Zhai J, Zhu H, Gal J (2016) Proteomic analysis of FUS interacting proteins provides insights into FUS function and its role in ALS. Biochim Biophys Acta 1862: 2004–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao CS, van Bruggen R, Kim JR, Chen XXL, Chan C, Lee J, Cho WI, Zhao M, Arndt C, Maksimovic K et al (2020) Selective neuronal degeneration in MATR3 S85C knock‐in mouse model of early‐stage ALS. Nat Commun 11: 5304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL (2019) Graph‐based genome alignment and genotyping with HISAT2 and HISAT‐genotype. Nat Biotechnol 37: 907–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug L, Chatterjee N, Borges‐Monroy R, Hearn S, Liao WW, Morrill K, Prazak L, Rozhkov N, Theodorou D, Hammell M et al (2017) Retrotransposon activation contributes to neurodegeneration in a drosophila TDP‐43 model of ALS. PLoS Genet 13: e1006635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafontaine DLJ, Riback JA, Bascetin R, Brangwynne CP (2021) The nucleolus as a multiphase liquid condensate. Nat Rev Mol Cell Biol 22: 165–182 [DOI] [PubMed] [Google Scholar]
- Lagarkova MA, Svetlova E, Giacca M, Falaschi A, Razin SV (1998) DNA loop anchorage region colocalizes with the replication origin located downstream to the human gene encoding Lamin B2. J Cell Biochem 69: 13–18 [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblond CS, Gan‐Or Z, Spiegelman D, Laurent SB, Szuto A, Hodgkinson A, Dionne‐Laporte A, Provencher P, de Carvalho M, Orru S et al (2016) Replication study of MATR3 in familial and sporadic amyotrophic lateral sclerosis. Neurobiol Aging 37: 209.e17–209.e21 [DOI] [PubMed] [Google Scholar]
- Lehnertz B, Ueda Y, Derijck AA, Braunschweig U, Perez‐Burgos L, Kubicek S, Chen T, Li E, Jenuwein T, Peters AH (2003) Suv39h‐mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr Biol 13: 1192–1200 [DOI] [PubMed] [Google Scholar]
- Li X, Fu XD (2019) Chromatin‐associated RNAs as facilitators of functional genomic interactions. Nat Rev Genet 20: 503–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Tong C, Mehrian‐Shai R, Jia L, Wu N, Yan Y, Maxson RE, Schulze EN, Song H, Hsieh CL et al (2008) Germline competent embryonic stem cells derived from rat blastocysts. Cell 135: 1299–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman‐Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO et al (2009) Comprehensive mapping of long‐range interactions reveals folding principles of the human genome. Science 326: 289–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KP, Tsai PC, Liao YC, Chen WT, Tsai CP, Soong BW, Lee YC (2015) Mutational analysis of MATR3 in Taiwanese patients with amyotrophic lateral sclerosis. Neurobiol Aging 36: 2005.e1–2005.e4 [DOI] [PubMed] [Google Scholar]
- Liu N, Lee CH, Swigut T, Grow E, Gu B, Bassik MC, Wysocka J (2018) Selective silencing of euchromatic L1s revealed by genome‐wide screens for L1 regulators. Nature 553: 228–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Dou X, Chen C, Chen C, Liu C, Xu MM, Zhao S, Shen B, Gao Y, Han D et al (2020) N (6)‐methyladenosine of chromosome‐associated regulatory RNA regulates chromatin state and transcription. Science 367: 580–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Y, Hwang T, Gooding AR, Goodrich KJ, Rinn JL, Cech TR (2020) RNA is essential for PRC2 chromatin occupancy and function in human pluripotent stem cells. Nat Genet 52: 931–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JY, Shao W, Chang L, Yin Y, Li T, Zhang H, Hong Y, Percharde M, Guo L, Wu Z et al (2020) Genomic repeats categorize genes with distinct functions for orchestrated regulation. Cell Rep 30: 3296–3311.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JY, Chang L, Li T, Wang T, Yin Y, Zhan G, Han X, Zhang K, Tao Y, Percharde M et al (2021) Homotypic clustering of L1 and B1/Alu repeats compartmentalizes the 3D genome. Cell Res 31: 613–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon MF (1998) X‐chromosome inactivation: a repeat hypothesis. Cytogenet Cell Genet 80: 133–137 [DOI] [PubMed] [Google Scholar]
- Lyon AS, Peeples WB, Rosen MK (2021) A framework for understanding the functions of biomolecular condensates across scales. Nat Rev Mol Cell Biol 22: 215–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharana S, Wang J, Papadopoulos DK, Richter D, Pozniakovsky A, Poser I, Bickle M, Rizk S, Guillen‐Boixet J, Franzmann TM et al (2018) RNA buffers the phase separation behavior of prion‐like RNA binding proteins. Science 360: 918–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik AM, Miguez RA, Li X, Ho YS, Feldman EL, Barmada SJ (2018) Matrin 3‐dependent neurotoxicity is modified by nucleic acid binding and nucleocytoplasmic localization. Elife 7: e35977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marangi G, Lattante S, Doronzio PN, Conte A, Tasca G, Monforte M, Patanella AK, Bisogni G, Meleo E, La Spada S et al (2017) Matrin 3 variants are frequent in Italian ALS patients. Neurobiol Aging 49: 218.e1–218.e7 [DOI] [PubMed] [Google Scholar]
- Marasca F, Marullo F, Lanzuolo C (2016) Determination of polycomb group of protein compartmentalization through chromatin fractionation procedure. Methods Mol Biol 1480: 167–180 [DOI] [PubMed] [Google Scholar]
- Marasca F, Sinha S, Vadala R, Polimeni B, Ranzani V, Paraboschi EM, Burattin FV, Ghilotti M, Crosti M, Negri ML et al (2022) LINE1 are spliced in non‐canonical transcript variants to regulate T cell quiescence and exhaustion. Nat Genet 54: 180–193 [DOI] [PubMed] [Google Scholar]
- Margueron R, Reinberg D (2011) The polycomb complex PRC2 and its mark in life. Nature 469: 343–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet J 17: 10 [Google Scholar]
- Mouse Genome Sequencing Consortium , Waterston RH, Lindblad‐Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M et al (2002) Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562 [DOI] [PubMed] [Google Scholar]
- Nakao H, Harada T, Nakao K, Kiyonari H, Inoue K, Furuta Y, Aiba A (2016) A possible aid in targeted insertion of large DNA elements by CRISPR/Cas in mouse zygotes. Genesis 54: 65–77 [DOI] [PubMed] [Google Scholar]
- Nakayasu H, Berezney R (1991) Nuclear matrins: identification of the major nuclear matrix proteins. Proc Natl Acad Sci USA 88: 10312–10316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsume T, Kiyomitsu T, Saga Y, Kanemaki MT (2016) Rapid protein depletion in human cells by auxin‐inducible degron tagging with short homology donors. Cell Rep 15: 210–218 [DOI] [PubMed] [Google Scholar]
- Niimori‐Kita K, Tamamaki N, Koizumi D, Niimori D (2018) Matrin‐3 is essential for fibroblast growth factor 2‐dependent maintenance of neural stem cells. Sci Rep 8: 13412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa RS, Gilbert N (2019) RNA: nuclear glue for folding the genome. Trends Cell Biol 29: 201–211 [DOI] [PubMed] [Google Scholar]
- Nozawa RS, Boteva L, Soares DC, Naughton C, Dun AR, Buckle A, Ramsahoye B, Bruton PC, Saleeb RS, Arnedo M et al (2017) SAF‐A regulates interphase chromosome structure through oligomerization with chromatin‐associated RNAs. Cell 169: 1214–1227.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya‐Jones A, Markaki Y, Serizay J, Chitiashvili T, Mancia Leon WR, Damianov A, Chronis C, Papp B, Chen CK, McKee R et al (2020) A protein assembly mediates Xist localization and gene silencing. Nature 587: 145–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson JR, Laemmli UK (1977) The structure of histone‐depleted metaphase chromosomes. Cell 12: 817–828 [DOI] [PubMed] [Google Scholar]
- Percharde M, Lin CJ, Yin Y, Guan J, Peixoto GA, Bulut‐Karslioglu A, Biechele S, Huang B, Shen X, Ramalho‐Santos M (2018) A LINE1‐nucleolin partnership regulates early development and ESC identity. Cell 174: 391–405.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL (2015) StringTie enables improved reconstruction of a transcriptome from RNA‐seq reads. Nat Biotechnol 33: 290–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollini D, Loffredo R, Maniscalco F, Cardano M, Micaelli M, Bonomo I, Licata NV, Peroni D, Tomaszewska W, Rossi A et al (2021) Multilayer and MATR3‐dependent regulation of mRNAs maintains pluripotency in human induced pluripotent stem cells. iScience 24: 102197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinodoz SA, Jachowicz JW, Bhat P, Ollikainen N, Banerjee AK, Goronzy IN, Blanco MR, Chovanec P, Chow A, Markaki Y et al (2021) RNA promotes the formation of spatial compartments in the nucleus. Cell 184: 5775–5790.e30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Bhardwaj V, Arrigoni L, Lam KC, Gruning BA, Villaveces J, Habermann B, Akhtar A, Manke T (2018) High‐resolution TADs reveal DNA sequences underlying genome organization in flies. Nat Commun 9: 189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES et al (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159: 1665–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razin SV, Iarovaia OV, Vassetzky YS (2014) A requiem to the nuclear matrix: from a controversial concept to 3D organization of the nucleus. Chromosoma 123: 217–224 [DOI] [PubMed] [Google Scholar]
- Reis RS, Poirier Y (2021) Making sense of the natural antisense transcript puzzle. Trends Plant Sci 26: 1104–1115 [DOI] [PubMed] [Google Scholar]
- Reis EM, Louro R, Nakaya HI, Verjovski‐Almeida S (2005) As antisense RNA gets intronic. OMICS 9: 2–12 [DOI] [PubMed] [Google Scholar]
- Rinn J, Guttman M (2014) RNA function. RNA and dynamic nuclear organization. Science 345: 1240–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E et al (2007) Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129: 1311–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rippe K (2022) Liquid‐liquid phase separation in chromatin. Cold spring Harb Perspect biol 14: a040683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP (2011) Integrative genomics viewer. Nat Biotechnol 29: 24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salton M, Elkon R, Borodina T, Davydov A, Yaspo ML, Halperin E, Shiloh Y (2011) Matrin 3 binds and stabilizes mRNA. PLoS One 6: e23882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettengruber B, Bourbon HM, Di Croce L, Cavalli G (2017) Genome regulation by Polycomb and Trithorax: 70 years and counting. Cell 171: 34–57 [DOI] [PubMed] [Google Scholar]
- Sebestyen E, Marullo F, Lucini F, Petrini C, Bianchi A, Valsoni S, Olivieri I, Antonelli L, Gregoretti F, Oliva G et al (2020) SAMMY‐seq reveals early alteration of heterochromatin and deregulation of bivalent genes in Hutchinson‐Gilford progeria syndrome. Nat Commun 11: 6274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant N, Varoquaux N, Lajoie BR, Viara E, Chen CJ, Vert JP, Heard E, Dekker J, Barillot E (2015) HiC‐pro: an optimized and flexible pipeline for hi‐C data processing. Genome Biol 16: 259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamidis N, Zylicz JJ (2023) RNA‐mediated heterochromatin formation at repetitive elements in mammals. EMBO J 42: e111717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanek D, Fox AH (2017) Nuclear bodies: news insights into structure and function. Curr Opin Cell Biol 46: 94–101 [DOI] [PubMed] [Google Scholar]
- Tada M, Doi H, Koyano S, Kubota S, Fukai R, Hashiguchi S, Hayashi N, Kawamoto Y, Kunii M, Tanaka K et al (2018) Matrin 3 is a component of neuronal cytoplasmic inclusions of motor neurons in sporadic amyotrophic lateral sclerosis. Am J Pathol 188: 507–514 [DOI] [PubMed] [Google Scholar]
- Tamm I (1984) HeLa cell RNA and protein syntheses. Effects of long‐term treatment with 5,6‐dichloro‐1‐beta‐D‐ribofuranosylbenzimidazole (DRB). Biochem Pharmacol 33: 551–557 [DOI] [PubMed] [Google Scholar]
- Taylor MS, LaCava J, Mita P, Molloy KR, Huang CR, Li D, Adney EM, Jiang H, Burns KH, Chait BT et al (2013) Affinity proteomics reveals human host factors implicated in discrete stages of LINE‐1 retrotransposition. Cell 155: 1034–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP (2013) Integrative genomics viewer (IGV): high‐performance genomics data visualization and exploration. Brief Bioinform 14: 178–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura Y, Oshima T, Yamamoto M, Reyes CJ, Costa Cruz PH, Shibuya T, Kawahara Y (2017) Matrin3 binds directly to intronic pyrimidine‐rich sequences and controls alternative splicing. Genes Cells 22: 785–798 [DOI] [PubMed] [Google Scholar]
- Van Bruggen R, Maksimovic K, You J, Tran DD, Lee HJ, Khan M, Kao CS, Kim JR, Cho W, Chen XXL et al (2021) MATR3 F115C knock‐in mice do not exhibit motor defects or neuropathological features of ALS. Biochem Biophys Res Commun 568: 48–54 [DOI] [PubMed] [Google Scholar]
- Van Nostrand EL, Pratt GA, Yee BA, Wheeler EC, Blue SM, Mueller J, Park SS, Garcia KE, Gelboin‐Burkhart C, Nguyen TB et al (2020) Principles of RNA processing from analysis of enhanced CLIP maps for 150 RNA binding proteins. Genome Biol 21: 90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassetzky NS, Ten OA, Kramerov DA (2003) B1 and related SINEs in mammalian genomes. Gene 319: 149–160 [DOI] [PubMed] [Google Scholar]
- Wang J, Choi JM, Holehouse AS, Lee HO, Zhang X, Jahnel M, Maharana S, Lemaitre R, Pozniakovsky A, Drechsel D et al (2018a) A molecular grammar governing the driving forces for phase separation of prion‐like RNA binding proteins. Cell 174: 688–699.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Xie Y, Li L, He Y, Zheng D, Yu P, Yu L, Tang L, Wang Y, Wang Z (2018b) EZH2 RIP‐seq identifies tissue‐specific Long non‐coding RNAs. Curr Gene Ther 18: 275–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Yu X, Yang L, Liu X, Gao B, Huang B, Dou X, Liu J, Zou Z, Cui XL et al (2022) FTO mediates LINE1 m(6)a demethylation and chromatin regulation in mESCs and mouse development. Science 376: 968–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen B, Wu H, Bjornsson H, Green RD, Irizarry R, Feinberg AP (2008) Overlapping euchromatin/heterochromatin‐ associated marks are enriched in imprinted gene regions and predict allele‐specific modification. Genome Res 18: 1806–1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederschain D, Wee S, Chen L, Loo A, Yang G, Huang A, Chen Y, Caponigro G, Yao YM, Lengauer C et al (2009) Single‐vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle 8: 498–504 [DOI] [PubMed] [Google Scholar]
- Yan Z, Ji L, Huo X, Wang Q, Zhang Y, Wen B (2020) G9a/GLP‐sensitivity of H3K9me2 demarcates two types of genomic compartments. Genomics Proteomics Bioinformatics 18: 359–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang C, Schones DE, Zeng C, Cui K, Zhao K, Peng W (2009) A clustering approach for identification of enriched domains from histone modification ChIP‐Seq data. Bioinformatics 25: 1952–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zbinden A, Perez‐Berlanga M, De Rossi P, Polymenidou M (2020) Phase separation and neurodegenerative diseases: a disturbance in the force. Dev Cell 55: 45–68 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W et al (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Guo L, Gonzales PK, Gendron TF, Wu Y, Jansen‐West K, O'Raw AD, Pickles SR, Prudencio M, Carlomagno Y et al (2019) Heterochromatin anomalies and double‐stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science 363: eaav2606 [DOI] [PMC free article] [PubMed] [Google Scholar]