

Abstract
For several decades, few substantial therapeutic advances have been made for patients with acute myeloid leukaemia. However, since 2017 unprecedented growth has been seen in the number of drugs available for the treatment of acute myeloid leukaemia, with several new drugs receiving regulatory approval. In addition to advancing our therapeutic armamentarium, an increased understanding of the biology and genomic architecture of acute myeloid leukaemia has led to refined risk assessment of this disease, with consensus risk stratification guidelines now incorporating a growing number of recurrent molecular aberrations that aid in the selection of risk-adapted management strategies. Despite this promising recent progress, the outcomes of patients with acute myeloid leukaemia remain unsatisfactory, with more than half of patients ultimately dying from their disease. Enrolment of patients into clinical trials that evaluate novel drugs and rational combination therapies is imperative to continuing this progress and further improving the outcomes of patients with acute myeloid leukaemia.
Introduction
Acute myeloid leukaemia is a malignant disorder of haemopoietic stem cells characterised by clonal expansion of abnormally differentiated blasts of myeloid lineage. Consequences of this proliferation of immature myeloid cells include accumulation of immature progenitors (blasts) with impairment of normal haemopoiesis, leading to severe infections, anaemia, and haemorrhage. Some patients might also present with extramedullary disease, including involvement of the CNS.1 Prompt diagnosis and initiation of acute myeloid leukaemia directed therapy is imperative, especially when rapid proliferation of malignant blasts is accompanied by tumour lysis syndrome or disseminated intravascular coagulation, both of which can be rapidly fatal without aggressive supportive management and treatment of the underlying acute myeloid leukaemia.2
This Seminar provides an overview of the most recent advances in genomics, prognostication, and therapeutics for acute myeloid leukaemia. We aim to provide an understanding of the complex interactions of disease-related and patient-related factors that both affect outcomes of patients with acute myeloid leukaemia and help to guide therapeutic decisions. In light of recent regulatory approval of several promising new drugs for acute myeloid leukaemia, we focus on how they have altered the therapeutic algorithm for patients with acute myeloid leukaemia and on their therapeutic strategies.
Epidemiology
Acute myeloid leukaemia is the most common acute type of leukaemia in adults, accounting for 1·3% of new cancer cases in the USA and affecting an estimated 0·5% of the population at some point in their lifetime.3 Although acute myeloid leukaemia can occur in any age group, acute myeloid leukaemia is predominantly a disease in older adults, with a median age at diagnosis of 68 years. The incidence of acute myeloid leukaemia is rising, partly due to an increasing prevalence of therapy-related acute myeloid leukaemia as more patients with cancer treated with cytotoxic chemotherapy are cured of their primary malignancy.4 Several genetic and environmental risk factors have been identified that predispose individuals to the development of acute myeloid leukaemia (appendix). Germline predisposition to acute myeloid leukaemia might be more common than previously thought, although, despite a robust history and genomic testing, most patients still do not have a clear predisposing factor for acute myeloid leukaemia.5 History of antecedent haematological disorders, including the myelodysplastic syndromes or myeloproliferative neoplasms, is also associated with a substantially increased likelihood of progression of acute myeloid leukaemia.6,7
Pathogenesis
Advances in stem cell biology and large, comprehensive genomic analyses have greatly improved our understanding of the mechanisms by which acute myeloid leukaemia develops. Although leukaemogenesis is still incompletely understood, acute myeloid leukaemia is believed to originate from the oncogenic transformation of a haemopoietic stem cell or of progenitors that have reacquired stem cell-like properties of self-renewal.8 The resultant self-renewing leukaemic stem cell is capable of maintaining the malignant clone. These leukaemic stem cells are both rare and quiescent, making them particularly resistant to cytotoxic chemotherapy and contributing to relapse.9 Progenitors from leukaemic stem cells undergo further genetic events, leading to karyotypic and molecular heterogeneity of the bulk leukaemic population, with multiple coexisting, competing clones present at the time of diagnosis.10–12 However, of note, the pathogenesis of acute myeloid leukaemia is probably quite different between subtypes of acute myeloid leukaemia;13 therefore, no singular model of pathogenesis is likely to account for all cases.
Specific mutations occur early in leukaemogenesis and might provide a selective advantage for clonal expansion of haemopoietic stem cells and eventual progression to acute myeloid leukaemia. In particular, epigenetic mutations of DNMT3A, TET2, and ASXL1 have been identified in preleukaemic haemopoietic stem cells decades before the development of acute myeloid leukaemia, suggesting that these are early founder events that precede leukaemogenic transformation.14,15 Expanded clones containing these somatic mutations can be identified in the peripheral blood or bone marrow of patients without evidence of overt haematological malignancy. This is a newly defined entity called clonal haemopoiesis of indeterminate potential (CHIP).16 CHIP has been identified in 10% of patients older than 65 years of age, with an incidence that increases with age, and predisposes to acute myeloid leukaemia and other haematological malignancies, including myelodysplastic syndromes.17,18 Notably, the rate of transformation of CHIP into overt haematological disease is about 0·5–1% per year.16 Through incompletely understood mechanisms, CHIP is also associated with an increased risk of atherosclerotic cardiovascular disease.18,19
In roughly 10% of patients with acute myeloid leukaemia, the development of the disease is preceded by exposure to cytotoxic chemotherapy (particularly alkylating agents or topoisomerase inhibitors) or ionising radiation, usually as treatment for a primary malignancy.4 In some cases, direct genotoxic effects from chemotherapy or irradiation can serve as the leukaemogenic event, directly leading to the development of acute myeloid leukaemia. However, emerging data suggest that some patients harbour CHIP before treatment for their primary malignancy and that these patients are at increased risk for the development of therapy-related myeloid neoplasms such as acute myeloid leukaemia.20,21 Some somatic mutations associated with these clones (eg, TP53 mutations, which are present in up to 37% of therapy-related myeloid neoplasms22) are relatively resistant to chemotherapy and, therefore, have a competitive advantage over healthy haemopoietic stem cells when exposed to cytotoxic drugs. These mutations have been hypothesised to be enriched in the post chemotherapy bone marrow and, in the case of TP53 mutations, contribute to genomic instability and the acquisition of new leukaemogenic mutations during regenerative haemopoiesis.23
Genomics
Acute myeloid leukaemia is characterised by several recurrent mutations that affect disease biology and phenotype, response to therapy, and risk of subsequent relapse (table 1).24 Great strides have been made in understanding the genomic diversity of acute myeloid leukaemia and how these various aberrations interact to affect disease phenotype and prognosis.25,26 Although the number of mutations per acute myeloid leukaemia genome or exome is lower than for most other cancers,27 with an average of only five recurrent mutations per acute myeloid leukaemia genome,11 at least one driver mutation can be identified in 96% of patients with de-novo acute myeloid leukaemia, and 86% of patients have two or more driver mutations.26 Tremendous diversity exists in the overlap of these mutations and the subclonal genomic architecture of the disease. In addition to informing prognosis, some of these mutations serve as potential targets for acute myeloid leukaemia directed therapies.28
Table 1:
Recurrent genomic mutations in newly diagnosed acute myeloid leukaemia in adults
| Frequency | Impact on prognosis | Comments | |
|---|---|---|---|
| FLT3 | 20–25% (ITD) and 5–10% (D835 TKD) | Inferior survival for ITD mutations and prognostic significance of D835 TKD mutations unclear | More common in NK acute myeloid leukaemia (<35% for ITD mutations), FLT3-ITD mutation with high allelic burden (ie, ≥0·5) associated with worse prognosis29,30 than lower allelic burden, prognosis affected by concomitant NPM1 mutation status, and prognostic significance not fully established with widespread use of FLT3 inhibitors |
| NPM1 | About 30% | Superior survival in the absence of high allelic burden FLT3-ITD mutation | More common in NK acute myeloid leukaemia (<60%) than in acute myeloid leukaemia with cytogenetic abnormalities, increased incidence in younger patients, coexisting chromosomal abnormalities do not affect prognosis31,32, substantial association with concomitant FLT3, IDH1/2, and DNMT3A mutations,26 and can be used to monitor for minimal residual disease33 |
| CEBPA | About 10% | Superior survival (only if biallelic) | More common in NK acute myeloid leukaemia (<20%) than in acute myeloid leukaemia with cytogenetic abnormalities, increased incidence in younger patients, coexisting chromosomal abnormalities do not affect prognosis,34 and germline mutations with familiar predisposition to acute myeloid leukaemia have been described35 |
| KIT | About 10% | Inferior survival in CBF acute myeloid leukaemia | More common in CBF acute myeloid leukaemia (present in 25–35%) than in non-CBF, poor prognosis more notable in acute myeloid leukaemia with t(8;21) than with inv(16), KIT inhibitors (eg, dasatinib) are being evaluated in clinical trials of CBF acute myeloid leukaemia |
| DNMT3A | About 20% | Conflicting reports on impact on survival | More common in NK acute myeloid leukaemia (<35%) than in acute myeloid leukaemia with cytogenetic abnormalities, increased incidence in older adults, CHIP mutation18, inferior prognosis particularly when present with other mutations (eg, IDH2R140)26, prognosis affected by type of DNMT3A mutation (ie, R882 vs non-R882) and patient age |
| IDH1 and IDH2 | 5–15% (IDH1) and 10–20% (IDH2) | Conflicting reports on impact on survival | More common in NK acute myeloid leukaemia (<30%) than in acute myeloid leukaemia with cytogenetic abnormalities, IDH1 and IDH2R140 are associated with concomitant NPM1 mutations, IDH2R172 can represent distinct acute myeloid leukaemia disease subtype,26 enasidenib (IDH2 inhibitor) has been approved for use in relapsed or refractory IDH2-mutated acute myeloid leukaemia, and IDH1 inhibitors are in clinical development |
| NRAS | About 15% | Conflicting reports on impact on survival | Associated with NPM1 and biallelic CEPBA mutations, and with inv(16) or t(16;16) and inv(3) or t(3;3), superior outcomes with NRASG12/G13 mutation in presence of NPM1 and DNMT3A mutations,26 and RAS pathway inhibitors are in clinical development |
| TET2 | 5–20% | Conflicting reports on impact on survival | More common in NK acute myeloid leukaemia (<25%) than in acute myeloid leukaemia with cytogenetic abnormalities, increased incidence in older adults, CHIP mutation,18 mutually exclusive with IDH1 and IDH2 mutations |
| ASXL1 | 5–15% | Inferior survival | Increased incidence in older adults, CHIP mutation,18 associated with secondary acute myeloid leukaemia that has progressed from antecedent haematologic malignancy36 |
| RUNX1 | 5–20% | Inferior survival | Increased incidence in older adults, associated with secondary acute myeloid leukaemia that has progressed from antecedent haematologic malignancy,37 and germline mutations with familiar predisposition to acute myeloid leukaemia have been described38 |
| TP53 | 5–20% | Inferior survival | Increased incidence in older adults, and associated with complex karyotype, monosomal karyotype, and secondary acute myeloid leukaemia (from antecedent haematological malignancy or therapy related) |
ITD=internal tandem duplication. TKD=tyrosine kinase domain. NK=normal karyotype. CBF=core-binding factor. CHIP=clonal haemopoiesis of indeterminate potential.
As conventional cytogenetics have an established prognostic impact for acute myeloid leukaemia, consideration of both karyotype and mutations is necessary, to classify acute myeloid leukaemia subtypes in a clinically meaningful way. To further refine prognostication of acute myeloid leukaemia, a large, comprehensive analysis of acute myeloid leukaemia genomics was done using targeted sequencing of 1540 patients with acute myeloid leukaemia.26 By incorporating cytogenetic analysis into genomic profiling, 11 mutually exclusive subtypes of acute myeloid leukaemia were identified, unambiguously classifying 80% of patients into distinct disease subgroups. In addition to eight well established acute myeloid leukaemia subsets, defined by gene fusions or the presence of an NPM1 mutation or biallelic CEBPA mutations, three new heterogeneous subtypes of acute myeloid leukaemia were defined. These groups included acute myeloid leukaemia with mutations of genes regulating RNA splicing (eg, SRSF2 and SF3B1), chromatin (eg, ASXL1), or transcription (eg, RUNX1), acute myeloid leukaemia with mutation of TP53 or cytogenetically visible copy number alterations, and, provisionally, acute myeloid leukaemia with IDH2R172 mutation. Further studies are needed to confirm the prognostic significance of these novel acute myeloid leukaemia subgroups and their justification as distinct disease entities.
Diagnosis
A diagnosis of acute myeloid leukaemia requires identification of 20% or more myeloid blasts (eg, myeloblasts, monoblasts, or megakaryoblasts) with morphological assessment of the peripheral blood or bone marrow. Exceptions to this blast cutoff, in which acute myeloid leukaemia can still be diagnosed, include isolated extramedullary acute myeloid leukaemia (ie, myeloid sarcoma) or the presence of recurrent karyotypic or molecular aberrations that are pathognomonic for acute myeloid leukaemia. These acute myeloid leukaemia-defining genomic changes consist of t(8;21) forming the RUNX1-RUNX1T1 fusion and inv(16) or t(16;16) forming CBFB-MYH11, which define core-binding factor acute myeloid leukaemia, and the t(15;17) fusion gene PML-RARA, which defines acute promyelocytic leukaemia.
In addition to morphological assessment of peripheral blood and bone marrow, immunophenotyping by flow cytometry39 is used at the time of diagnosis to confirm the myeloid origin of malignant blast populations and to aid in further categorisation of acute myeloid leukaemia subtype. Cytogenetic analysis and screening for commonly occurring gene mutations and rearrangements should also be done. Such screening is necessary, because, although some myeloid neoplasms are characterised primarily by morphological and immunophenotypic assessment, the 2016 WHO update recognises 11 acute myeloid leukaemia subgroups defined by the presence of specific recurrent genetic abnormalities, including balanced translocations, gene fusions, or single molecular mutations.40 Further to testing for NPM1, CEBPA, and RUNX1, which each define specific acute myeloid leukaemia subtypes, additional genomic testing for FLT3-internal tandem duplication (ITD), TP53, and ASXL1 should also be done, as mutations in these genes have prognostic importance and have been incorporated into consensus risk stratification guidelines.41
Risk stratification
The outcome of acute myeloid leukaemia is heterogeneous, with both patient-related and disease-related factors contributing to an individual patient’s likelihood of achieving response to therapy and long-term survival. Accurate prognostication of acute myeloid leukaemia is imperative, as postremission therapies (eg, consolidation chemotherapy vs haemopoietic stem cell transplantation [HSCT] for patients in first remission) are assigned largely according to a patient’s anticipated risk of relapse in the absence of HSCT and their anticipated risk of non-relapse mortality with HSCT.42 Of note, the risk categories established by the consensus guidelines are not fixed and might evolve with emerging therapies, requiring continued reassessment of their prognostic importance. In addition to pretreatment characteristics, such as cytogenetics or molecular mutations, depth of response (ie, presence or absence of minimal residual disease) achieved with initial therapy is emerging as a powerful factor in assessing relapse risk.43
Pretreatment factors
Pretreatment factors that affect prognosis for patients with acute myeloid leukaemia can be divided into those related to the patient’s ability to tolerate therapy and those related to the inherent chemosensitivity or chemoresistance of the disease itself, although some overlap between these two features exists. Patient-related variables that affect a patient’s ability to receive adequate antileukaemic therapy include advanced age, poor performance status, and the presence of clinically significant medical comorbidities. Patients with these risk factors have higher treatment-related mortality when treated with intensive chemotherapy.44 Due, in part, to their poor tolerance of intensive treatment, the outcomes of many older patients (particularly those patients 60 years of age and older) are significantly worse than their younger counterparts (<60 years).45 However, less intensive therapies might not be adequate to result in long-term remission.46 Although higher treatment-related mortality could account for some of the differential outcomes observed between older and younger patients with acute myeloid leukaemia, older patients have more adverse-risk cytogenetic and molecular abnormalities, which also contribute to their poorer outcomes.36,47 Similarly, the presence of an antecedent haematological disorder (eg, myelodysplastic syndromes or myeloproliferative neoplasms) or previous treatment with cytotoxic chemotherapy or irradiation are historical factors that are associated with poorer response to acute myeloid leukaemia directed therapy and shorter survival.48
Genetic analyses, including both karyotyping and screening for recurrent gene fusions and molecular mutations, provide important information about disease biology and strongly inform prognostic assessment, which in turn is used to guide decisions about postremission therapy (table 2).41 Based on karyotypic analysis, the favorable risk group includes patients with core-binding factor acute myeloid leukaemia—eg, t(8;21) or inv(16)—whereas the adverse risk group includes patients with complex karyotype (defined as three or more cytogenetic abnormalities) or specific chromosomal aneuploidies (eg, −5/−5q, −7, and −17/17p).49 However, cytogenetics cannot effectively risk stratify many patients with acute myeloid leukaemia, as up to 50% of adult patients present with cytogenetically normal acute myeloid leukaemia.50 Historically, the majority of patients with a normal karyotype, in whom outcomes are particularly heterogeneous, were classified into an intermediate-risk group.50 In this large subset of patients with cytogenetically normal acute myeloid leukaemia and in patients without a well established prognostic karyotypic abnormality, identification of recurrent gene mutations is especially important for risk stratification. The presence of biallelic mutations of CEBPA defines an acute myeloid leukaemia subset with relatively favorable prognosis,51,52 whereas mutations of RUNX1, ASXL1, or TP53 are associated with a high risk of relapse and are classified as adverse risk.24,37,53
Table 2:
Genomic risk stratification of acute myeloid leukaemia
| Favourable | Intermediate | Adverse | |
|---|---|---|---|
| Cytogenetic | t(8;21)(q22;q22.1) for RUNX1-RUNX1T1, and inv(16)(p14.1q22) or t(16;16) (p13.1;q22) for CBFB-MYH11 | t(9;11)(p21.3;q23.3) for MLL3-KMT2A*, and cytogenetic abnormalities not classified as favourable or adverse | t(6;9)(p23;q34.1) for DEK-NUP214; t(v;11q23.3) for KMT2A rearranged; t(9;22)(p34.1;q11.2) for BCR-ABL1; inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2) for GATA2 and MECOM (EVI1); −5 or del(5q), −7, and-17/abn(17p); complex karyotype†; and monosomal karyotyped‡ |
| Molecular | Mutated NPM1 without FLT3-ITD or with FLT3-ITDlow§, and biallelic mutated CEBPA | Mutated NPM1 and FLT3-ITDhigh§, and wild-type NPM1 without FLT3-ITD or with FLT3-ITDlow§ (without adverse-risk genetic lesions) | Wild-type NPM1 and FLT3-ITDhigh§, mutated RUNX1¶, mutated ASXL1¶, and mutated TP53‖ |
Table is adapted from Dohner and colleagues,41 the European LeukemiaNet 2017 recommendations. Prognostic impact of a marker is treatment-dependent and might change with new therapies. ITD=internal tandem duplication.
The presence of t(9;11)(p21.3;q23.3) takes precedence over rare, concurrent adverse-risk gene mutations.
Three or more unrelated chromosomal abnormalities in the absence of one of the WHO-designated recurring translocations or inversions—ie, t(8;21), inv(16) or t(16;16), t(9;11), t(v;11)(v;q23.3), t(6;9), inv(3) or t(3;3) for acute myeloid leukaemia with BCR-ABL1.
Presence of a single monosomy (excluding loss of X or Y) in association with at least one additional monosomy or structural chromosomal abnormalities (excluding core-binding factor acute myeloid leukaemia).
Low and high allelic ratios are defined as less than 0·5 and 0·5 and higher, respectively; semiquantitative assessment of FLT3-ITD allelic ratio (with DNA fragment analysis) is determined as the ratio of the area under the FLT3-ITD curve divided by area under the FLT3-wild type curve. Studies29,30 indicate that acute myeloid leukaemia with NPM1 mutation and FLT3-ITD low allelic ratio might also have a more favourable prognosis and patients should not be routinely assigned to allogeneic haemapoietic stem cell transplant.
Should not be used as an adverse prognostic marker if they co-occur with favourable-risk acute myeloid leukaemia.
TP53 mutations are significantly associated with acute myeloid leukaemia with a complex and monosomal karyotype
Mutations of different genes frequently interact in complex ways to affect prognosis, associations that are just beginning to be elucidated on the basis of analyses with large annotated genomic databases.26 Thus, in many cases, the prognostic impact of a specific gene mutation can only be understood in the context of the other genomic aberrations. For example, NPM1 and FLT3-ITD mutation status interact to affect prognosis, and knowledge of the mutational status of both of these genes, as well as the FLT3-ITD allelic ratio, are required to fully assess relapse risk in an individual patient.41 Data have suggested that DNMT3A mutation status could in part mediate the effect of this prognostic association between NPM1 and FLT3-ITD.26 Prognostic gene–gene interactions have also been described with respect to mutation status of NRAS, DNMT3A, and NPM1, and DNMT3A and IDH2R140, further adding to the complexity of devising a comprehensive genomics-based risk stratification algorithm for acute myeloid leukaemia.26
Post-treatment factors
A patient’s response to acute myeloid leukaemia directed therapy is a strong determinant of future outcomes. Achievement of a complete remission requires bone marrow assessment showing less than 5% blasts with recovery of peripheral blood elements (ie, neutrophil count >1000 per μL and platelet counts >100 000 per μL) and no evidence of extramedullary disease. Less stringent criteria for response, in which blasts decrease to less than 5% but with incomplete peripheral blood recovery, are associated with less favourable outcomes than in patients achieving complete remission, but better than non-responders.54
More sensitive tests for minimal (also called measurable) residual disease, allow for better discrimination of relapse than does morphological assessment alone.55 The two methods in routine clinical practice for minimal residual disease detection are multiparameter flow cytometry (MFC) and quantitative real-time PCR, which both have their own advantages and disadvantages.56 MFC-based minimal residual disease assessment relies on comparison of leukaemia-associated immunophenotypes (ie, aberrant patterns of antigen expression found on leukaemic blasts) between diagnostic and remission samples. In the absence of a baseline sample, flow cytometric analysis of the remission sample evaluating different than normal immunophenotype can also be used to detect minimal residual disease.57 By contrast, real-time PCR requires the presence of a defined target (eg, fusion transcripts, such as PML-RARA, or gene mutations, such as NPM1) at diagnosis that can be monitored with high sensitivity in follow-up remission peripheral blood or bone marrow samples. Both MFC-based and real-time PCR-based minimal residual disease measurements are highly prognostic for long-term survival in acute myeloid leukaemia.33,58–61 In one study of patients with acute myeloid leukaemia undergoing HSCT, those in complete remission but with detectable minimal residual disease by MFC had similar post-transplant relapse and overall survival to those individuals transplanted when not in morphological remission.62 These studies have supported the development of a new acute myeloid leukaemia response criterion: complete remission without minimal residual disease.41
Treatment
General approach
With the approval of several new drugs in 2017, the frontline management of acute myeloid leukaemia is rapidly changing, and rapid, targeted genomic analysis is becoming increasingly necessary to identify genomic and molecular changes that inform the selection of appropriate upfront therapy (table 3).63 Another important consideration in designing a treatment plan for a patient with newly diagnosed acute myeloid leukaemia is to determine whether they are suitable candidates to receive intensive chemotherapy (figure 1). Such assessment is based largely on the anticipated treatment-related mortality of this approach,64 which defines the patient’s fitness for a given anti-leukemic therapy. Thus, fitness is primarily influenced by patient-related factors such as advanced age, performance status, and pretreatment co-morbidities. Although the predicted mortality can be affected by patient age, it is important to consider all patient-related and disease-related factors when designing an appropriate treatment plan. Owing to better supportive care measures and better selection of patients for intensive versus less intensive acute myeloid leukaemia directed therapies, treatment-related mortality with intensive therapy in large clinical trials has declined over the past two decades from 15–20% to less than 5% in many studies.65
Table 3:
Drugs approved for acute myeloid leukaemia by the US Food and Drug Administration in 2017
| Drug class | Approved indication | Clinical outcomes | |
|---|---|---|---|
| Midostaurin | Multi-targeted kinase inhibitor (FLT3, VEGFR2, PDGFR, and KIT) | Newly diagnosed FLT3-mutated acute myeloid leukaemia in adults (given in combination with chemotherapy) | Chemotherapy and midostaurin vs chemotherapy alone: median overall survival 74·7 months vs 25·6 months (HR 0·78; p=0·009)72 |
| Enasidenib | IDH2 inhibitor | Relapsed or refractory IDH2-mutated acute myeloid leukaemia | Overall response rate 40·3%, complete remission 19·3%, and median overall survival 9·3 months73 |
| CPX-351 | Cytotoxic chemotherapy (liposomal 5:1 molar ratio of cytarabine and daunorubicin) | Newly diagnosed therapy-related acute myeloid leukaemia or acute myeloid leukaemia with myelodysplasia-related changes in adults | CPX-351 vs 7 + 3 regimen: median overall survival 9·6 months vs 6·0 months (HR 0·69; p=0·005)74 |
| Gemtuzumab ozogamicin | Anti-CD33 antibody-drug conjugate | Newly diagnosed CD33-positive acute myeloid leukaemia in adults and relapsed or refractory CD33-positive acute myeloid leukaemia in adults or paediatric patients aged 2 years and older | For newly diagnosed, chemotherapy plus gemtuzumab ozogamicin vs chemotherapy alone: median event free survival 15·6 months vs 9·7 months (HR 0·58; p=0·0003),75 and gemtuzumab ozogamicin vs best supportive care: median overall survival 4·9 months vs 3·6 months (HR 0·69; p=0·005).76 For relapsed or refractory, overall response rate 33·3%, complete remission 26·3%, and median overall survival 8·4 months77 |
HR=hazard ratio.
Figure 1: Management of acute myeloid leukaemia in adults.
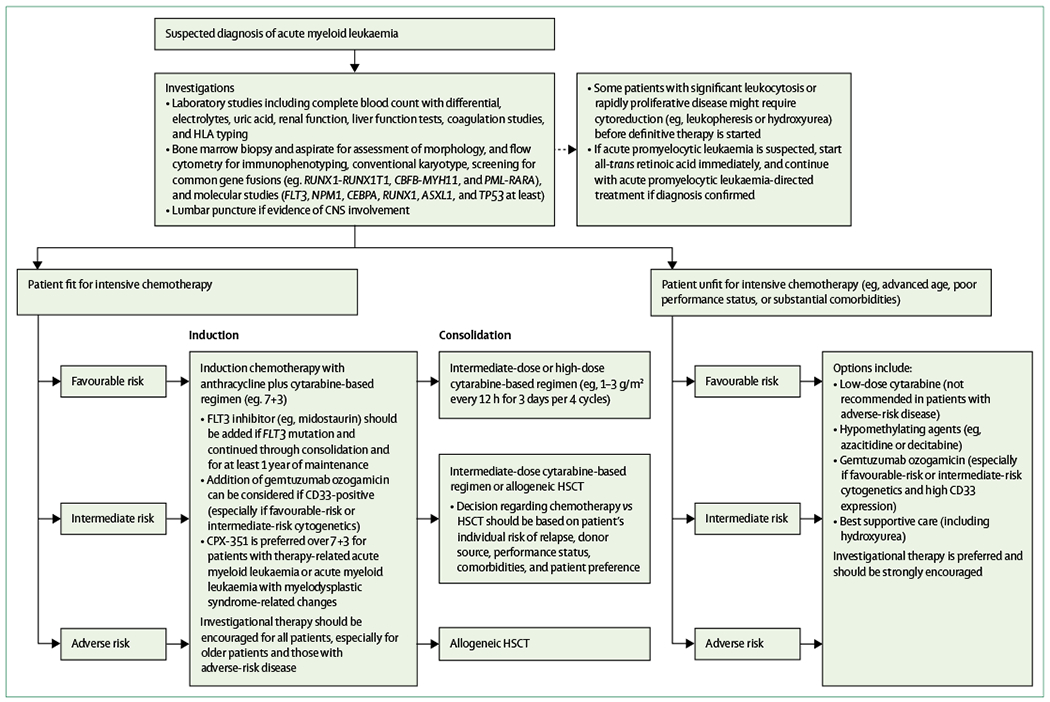
HLA=human leukocyte antigen. HSCT=haemopoietic stem cell transplant.
For patients who achieve remission with induction therapy, appropriate selection of postremission therapy is essential.66 Postremission therapies are generally selected by balancing the treatment-related mortality and morbidity associated with HSCT in first remission with the patient’s risk of relapse. Patients with an increased risk of relapse in the absence of HSCT (eg, >35%) are usually considered for HSCT in first remission. Various models exist to help determine a patient’s candidacy for HSCT, accounting for both non-relapse mortality associated with transplantation and the relapse risk with and without HSCT.67 Patients with favourable disease-related features generally receive postremission consolidation chemotherapy, whereas those individuals with adverse-risk disease are usually offered HSCT in first remission.42 For those patients with intermediate-risk disease, no consensus exists regarding the optimal postremission therapy, and treatment should be individualised on the basis of full assessment of relapse risk, patient fitness, adequacy of a suitable donor, and patient preference.
Induction therapy
For over four decades, a combination of cytarabine and an anthracycline has been the standard induction regimen for patients deemed suitable for intensive acute myeloid leukaemia therapy.68,69 With the 7+3 regimen of 7 days of infused cytarabine (100–200 mg/m2 daily) plus 3 days of an anthracycline (eg, daunorubicin or idarubicin), complete remission of 60–85% was achieved in patients younger than 60 years and 40–60% was achieved in patients aged 60 years or older, respectively.70 In an attempt to increase these responses, various studies have evaluated the optimal dosing of the anthracycline and cytarabine for acute myeloid leukaemia induction, as well as whether the addition of a third drug to this backbone can further improve long-term outcomes (appendix).71
Gemtuzumab ozogamicin is an anti-CD33 antibody-drug conjugate that carries calicheamicin, a potent DNA-damaging toxin. In a meta-analysis of five randomised clinical trials enroling patients 15 years of age and older with newly diagnosed acute myeloid leukaemia, the addition of gemtuzumab ozogamicin to induction chemotherapy was shown to improve overall survival at 6 years compared with standard induction chemotherapy (6-year overall survival 34·3% vs 30·6%; odds ratio 0·90, 95% CI 0·82–0·98; p=0·01).78 This beneficial effect is most pronounced among patients with favourable-risk or intermediate-risk cytogenetics. Notably, gemtuzumab ozogamicin initially gained regulatory approval in the USA in 2000, but was subsequently withdrawn from the market in June, 2010, after one randomised trial of intensive chemotherapy with or without gemtuzumab ozogamicin in patients 18–60 years of age with acute myeloid leukaemia found an excess of early mortality, which reduced the clinical benefit in the gemtuzumab ozogamicin containing group. However, owing to promising results from several subsequent randomised trials, gemtuzumab ozogamicin was again approved by the US Food and Drug Administration in September, 2017, for treatment of adults with either newly diagnosed or relapsed or refractory CD33-positive acute myeloid leukaemia. The approval includes single-drug use, as well as combined (in a fractionated dosing schedule) with standard chemotherapy, and, importantly, includes paediatric patients.
Few effective treatments are available for patients with acute myeloid leukaemia who harbour adverse risk features. CPX-351 is a liposomally encapsulated formulation of cytarabine and daunorubicin that preserves a 5:1 molar ratio and has several theoretical advantages over the standard 7 + 3 regimen, including delivery of the drugs at a more sustained synergistic ratio, bypassing drug efflux pumps, and prolonged drug exposure in the bone marrow.79 Initial randomised phase 2 studies in both the frontline and relapsed or refractory settings showed improved proportions of patients who achieved an objective response compared with conventional therapies, especially in patients with poor-risk features, including secondary acute myeloid leukaemia (ie, arising from a preceding haematological disorder or therapy related).80,81 A subsequent randomised phase 3 trial was done in older patients (aged 60–75 years) with secondary acute myeloid leukaemia or de-novo acute myeloid leukaemia with myelodysplastic syndromes related cytogenetic abnormalities.74 Compared with standard 7 + 3 induction, CPX-351 resulted in higher proportions of patients with an objective response and longer survival (median overall survival 9·6 months vs 6·0 months; hazard ratio [HR] 0·69; p=0·005), without an increase in early mortality or toxicity, although data on absolute survival benefit are still awaited. CPX-351 therefore represents a reasonable treatment option for patients with secondary acute myeloid leukaemia who are considered suitable for intensive acute myeloid leukaemia therapy.
Mutations of the FLT3 gene are present in about 30% of patients with newly diagnosed acute myeloid leukaemia.24,82 Many FLT3 inhibitors have been developed. When used by themselves in patients with FLT3 mutations, they induce a rapid decrease in peripheral blood and bone marrow blasts in a substantial percentage of patients.83,84 However, most responses are partial or without full haemopoietic recovery, and typically of short duration. Thus, FLT3 inhibitors have been combined with chemotherapy in patients harbouring FLT3 mutations.85,86 Midostaurin is an oral multitargeted kinase inhibitor that is active against FLT3. In a large, multicenter phase 3 study, patients younger than 60 years of age with newly diagnosed FLT3-positive acute myeloid leukaemia (either ITD or a point mutation in the tyrosine kinase domain) received standard induction followed by consolidation chemotherapy (or HSCT, if indicated) and were randomly assigned to receive either midostaurin or placebo.72 Midostaurin was given with induction and consolidation, followed by up to an additional year of maintenance midostaurin. Compared with the placebo group, the midostaurin group had a significant improvement in overall survival, both in patients with FLT3-ITD and those with FLT3 kinase domain mutations (4-year overall survival 51·4% vs 44·3%; median overall survival 74·7 months vs 25·6 months; HR 0·78; 95% CI 0·63–0·96; p=0·009]). Based on these results, midostaurin was approved in the USA and is now standard therapy for patients with a FLT3 mutation in combination with conventional therapy.
Postremission therapy
Consolidation chemotherapy with a cytarabine-based regimen is standard of care for patients who achieve remission after induction chemotherapy and in whom HSCT is not recommended, owing to favourable disease-related factors, high expected transplant-related mortality, or lack of suitable donor availability.66 In patients who were given cytarabine and were younger than 60 years of age, the decreased relapse and prolonged survival appeared to be dose dependent.87 Therefore, in younger patients with acute myeloid leukaemia, single-drug high-dose cytarabine, typically at a dose of 3 g/m2 every 12 h over 3 days of each cycle, is the most commonly used consolidation strategy, although multiple variations of the dose and schedule have been reported, and such high doses of cytarabine are unlikely to be required for optimal antileukaemic activity.88 Four consolidation cycles are generally administered, although the optimal number of consolidation cycles has not been firmly established. With this high-dose cytarabine consolidative regimen, long-term survival of about 50% has been achieved in patients younger than 60 years of age who achieve complete remission with induction chemotherapy.87 The benefit of high-dose cytarabine consolidation is largely limited to those with favourable-risk or cytogenetically normal acute myeloid leukaemia, which represent subtypes of acute myeloid leukaemia that are generally more chemosensitive.89 Older patients (≥60 years) are less likely to benefit from or tolerate intensive cytarabine, except perhaps the small proportion of those with favourable-risk cytogenetics.
The addition of other drugs to the cytarabine backbone for consolidation might also be beneficial in some acute myeloid leukaemia subgroups. For example, midostaurin should be given with consolidation for patients with FLT3-mutated acute myeloid leukaemia.72 In one study of younger patients (18–59 years) with intermediate-risk or adverse-risk cytogenetics, the addition of the purine nucleoside analogue clofarabine to intermediate-dose cytarabine significantly prolonged relapse-free survival, although no overall survival benefit was observed.90 After consolidation chemotherapy, no role for further maintenance in the management of acute myeloid leukaemia has been established. However, several maintenance strategies, such as targeted drugs (eg, FLT3 inhibitors) and immune approaches (eg, checkpoint inhibitors and monoclonal antibodies), are being investigated.
Several variables are part of the considerations for HSCT, in addition to proper patient selection, including the optimal preparative regimen, donor, and stem cell source (appendix). The mechanistic role of allogeneic HSCT in acute myeloid leukaemia is in two parts: the high-dose preparative conditioning regimen provides antileukaemic cytoreduction, and, perhaps more importantly, engrafted donor T cells exert an immunological graft-versus-leukaemia effect to further eliminate residual leukaemic cells.91 Allogeneic HSCT improves outcomes of patients with both poor-risk and intermediate-risk acute myeloid leukaemia, although the magnitude of benefit in patients with intermediate-risk disease is lower, probably due to the substantial heterogeneity of this group and because chemotherapy is generally more effective than in patients with poor-risk disease.92–94 For these intermediate-risk patients, minimal residual disease status might have a particularly informative role in defining the risk-benefit ratio of HSCT for a given patient, although the optimal use of minimal residual disease to inform such decisions remains controversial.95 Although HSCT for high-risk patients with acute myeloid leukaemia consistently decreases the frequency of relapse compared with chemotherapy alone, many patients still relapse. In an effort to improve outcomes for these high-risk patients, several studies have evaluated the use of post-HSCT maintenance with mixed results.96 Studies evaluating post-HSCT hypomethylating agents, lenalidomide, and FLT3 inhibitors are in progress.
Patients unfit for intensive therapy
As almost half of patients with acute myeloid leukaemia are older than 70 years of age at the time of diagnosis, frequently with comorbidities and poor performance status, many are considered unfit for intensive chemotherapy.64 Of note, this assessment is often subjective and some older patients can still benefit from intensive chemotherapy.97 Several less intensive therapies are commonly used for these unfit patients, including low-dose cytarabine and hypomethylating agents.45 Although these regimens are associated with lower treatment-related mortality than intensive chemotherapy, long-term outcomes for older patients (≥60 years) with acute myeloid leukaemia remain dismal, with a median survival of about 6–9 months. Therefore, enrolment in a clinical trial should always be considered for these patients.
In patients deemed unfit for intensive chemotherapy, low-dose cytarabine (eg, 20 mg subcutaneously twice daily for 10 days administered every 4 weeks) is associated with a complete remission of 15–25% and improves overall survival compared with hydroxyurea plus best supportive care.98 However, response to low-dose cytarabine is minimal in patients with adverse karyotype. Given the potential for haematological toxicity with low-dose cytarabine and the high proportion of older adults with poor-risk cytogenetics, low-dose cytarabine is a suboptimal strategy for the majority of older patients with acute myeloid leukaemia.
The hypomethylating agents azacitidine and decitabine are epigenetic therapies that inhibit DNA methylation and are believed to lead to re-expression of silenced tumour suppressor genes, which can, in part, explain their efficacy in both myelodysplastic syndromes and acute myeloid leukaemia.99 In a randomised study comparing azacitidine (75 mg/m2 subcutaneously daily for 7 days administered every 4 weeks) with conventional care regimens (eg, standard induction chemotherapy, low-dose cytarabine, or supportive care only) in patients with acute myeloid leukaemia aged 65 years or older, azacitidine significantly improved survival (median overall survival 12·1 months vs 6·9 months, HR 0·76; 95% CI, 0·60–0·96; p=0·019), an effect that was seen across subgroups, including in patients with poor-risk cytogenetics.100 A randomised trial comparing decitabine (20 mg/m2 intravenously daily for 5 days administered every 4 weeks) with low-dose cytarabine or supportive care in a similar population of older patients (≥65 years) with acute myeloid leukaemia showed a modest overall survival benefit for patients treated with decitabine.101 The proportion of patients who achieve an objective response with hypomethylating agents is 20–30%, which is lower than that observed with intensive chemotherapy.70 However, overall survival is similar or possibly superior compared with what can be expected with intensive chemotherapy in these older patients with acute myeloid leukaemia, probably driven by lower treatment-related mortality with these less intensive strategies.102,103 Of note, epigenetic therapy could be particularly beneficial in patients harbouring TP53 mutations, which confer resistance to traditional cytotoxic chemotherapy.104
In several studies, gemtuzumab ozogamicin alone has been shown to be effective in older adults (>60 years), including those who are deemed unfit for intensive chemotherapy. In a randomised study of gemtuzumab ozogamicin treatment versus best supportive care, low-dose gemtuzumab ozogamicin (6 mg/m2 on day 1 and 3 mg/m2 on day 8 of induction, followed by monthly doses of 2 mg/m2 as consolidation) resulted in a response of 27% and improved overall survival compared with the control group, without an increase in adverse events.76
The benefit of gemtuzumab ozogamicin was seen across most subgroups, especially patients with high CD33 expression status and those with favourable or intermediate-risk cytogenetics. With the reapproval of gemtuzumab ozogamicin, future studies evaluating its optimal use in older patients with acute myeloid leukaemia, including combination strategies, are needed.
Relapsed or refractory acute myeloid leukaemia
With standard chemotherapy, long-term survival for patients with acute myeloid leukaemia is achieved in only 35–45% of those younger than 60 years of age and 10–15% of those aged 60 years and older.41 Relapsed disease and the associated leukemia-associated complications are the most common causes of death. Acute myeloid leukaemia relapse is associated with a substantial increase in molecular complexity, with multiple new subclones and mutations identified at the time of relapse, contributing to increased resistance to cytotoxic chemotherapy.15,105–107 For patients in first relapse, the median survival is roughly 6 months with only about 10% of patients achieving long-term survival.108,109 Predictors for poorer outcomes in patients with first acute myeloid leukaemia relapse include a duration of first remission of 6 months or less, unfavourable karyotype, previous HSCT, and advanced age.108 Patients with primary induction failure or multiply relapsed disease have especially poor outcomes.
For patients with primary induction failure or who develop relapsed disease, the goal of further antileukaemic therapy is to achieve remission and proceed to allogeneic HSCT, which offers the best chance of cure.110 For older (>70 years) or unfit patients with relapsed or refractory disease and in whom allogeneic HSCT is not feasible, further treatment is largely palliative. For patients with a first remission duration longer than 1 year, retreatment with an intermediate-dose or high-dose cytarabine-containing regimen should be considered, as this subset of patients often relapse with somewhat chemosensitive disease.108 This regimen should still be followed by HSCT whenever feasible. However, for patients with a shorter first remission duration or with primary induction failure, there is no consensus reinduction regimen. In a large, multicenter trial of 381 patients with relapsed or refractory acute myeloid leukaemia, most of whom had multiply relapsed disease, patients were randomly assigned to the experimental therapy (elacytarabine) or investigator’s choice of one of seven commonly used acute myeloid leukaemia salvage regimens, including high-dose cytarabine, multidrug chemotherapy, hypomethylating agents, hydroxyurea, or supportive care.111 Overall survival did not differ among any of the therapies administered. The proportion of patients who achieved an objective response to salvage therapy was 20–25% and the median survival was 3–4 months regardless of the regimen received, highlighting the need for more effective therapies for patients with relapsed or refractory acute myeloid leukaemia and the necessity of enrolling these patients in clinical trials.
As knowledge of the genomic landscape of acute myeloid leukaemia continues to grow, therapies targeting specific pathogenic mutations are likely to have an increasing role in the management of acute myeloid leukaemia and improve outcome. One such example is enasidenib, an oral, selective inhibitor of mutant IDH2. When mutated, IDH2 has pro-leukaemic properties mediated by epigenetic phenomena; the use of enasidenib reverts these effects and induces differentiation of malignancy myeloblasts.112 IDH2 mutations are identified in 10–20% of patients with acute myeloid leukaemia.113,114 In a phase 1/2 trial of patients with IDH2-mutant relapsed or refractory acute myeloid leukaemia, enasidenib was associated with overall response of about 40%, complete remission of about 20%, and median survival of 9·3 months.73 In August, 2017, enasidenib was approved in the USA for use in patients with relapsed or refractory acute myeloid leukaemia harbouring an IDH2 mutation. As retrospective studies suggest that intensive chemotherapy is also effective in patients with IDH2 mutations,115 future combinations of IDH2 inhibitors with chemotherapy might improve these outcomes. Clinical trials of other targeted therapies for relapsed or refractory acute myeloid leukaemia are continuing, including drugs targeting mutant FLT3, IDH1, and RAS.63 Incorporation of several of these drugs into frontline acute myeloid leukaemia management is also being investigated.
Acute myeloid leukaemia in the paediatric patient
Acute myeloid leukaemia in childhood accounts for 20% of paediatric leukaemias, with 5·1% of patients with acute myeloid leukaemia being diagnosed at younger than 20 years of age.3 Several genetic syndromes have been associated with the development of acute myeloid leukaemia in childhood (appendix). As paediatric acute myeloid leukaemia is a relatively rare entity, much of the approach regarding prognostic factors and treatment are derived from data and studies in adult patients. Risk stratification for paediatric acute myeloid leukaemia is, therefore, largely based on the same genetic categories that have been established in adult acute myeloid leukaemia.41 However, some notable differences exist in the prevalence of different karyotypic and molecular abnormalities between adult and paediatric populations. For example, acute myeloid leukaemia that presents in childhood is associated with higher rates of t(8;21), inv(16), and rearrangements of 11q23 (KMT2A).116 By contrast, mutations of NPM1 and FLT3-ITD appear to be less frequent in children than in their adult counterparts.117 As in adults, flow cytometric minimal residual disease response to acute myeloid leukaemia directed therapy is a powerful prognostic factor in the paediatric population.118–120
Treatment of paediatric acute myeloid leukaemia uses a multidrug cytarabine and anthracycline-based induction, followed by either post-remission consolidative chemotherapy or HSCT.121 Similar to the approach in adults, HSCT in first remission is generally reserved for patients with high-risk disease features, including poor response to induction chemotherapy or poor-risk cytogenetics, although proper patient selection remains controversial.122 With contemporary treatment, the proportion of patients cured in some subgroups of paediatric acute myeloid leukaemia can approach 70%.123 The anti-CD33 antibody-drug conjugate gemtuzumab ozogamicin has shown particular promise in further improving these outcomes. A randomised phase 3 study in children and young adults (≤29 years) with newly diagnosed acute myeloid leukaemia showed that the addition of gemtuzumab ozogamicin to standard chemotherapy was associated with a significant improvement in 3-year event-free survival (53·1% vs 46·9%; HR 0·83; 95% CI 0·70–0·99; p=0·04), although no overall survival benefit was observed.124 In a subsequent analysis, this benefit was primarily restricted to the around 50% of patients who have the CC genotype of a CD33 splicing polymorphism.125 As with adult acute myeloid leukaemia, supportive care has an essential role in improving outcomes in paediatric acute myeloid leukaemia.126
Future directions
Despite progress in recent years, with several new drugs gaining regulatory approval for the treatment of adults with acute myeloid leukaemia since 2017, many important questions remain. Ongoing efforts to understand the genomic background of acute myeloid leukaemia, including the mechanisms by which each mutation drives the disease phenotype and how these mutations interact with one another to affect risk of relapse, will be crucial, not only in risk stratification of acute myeloid leukaemia, but also in developing novel targeted therapies and rational combinations. The development of novel minimal residual disease assays might also further refine selection of patients with HSCT in first remission.127 For example, mutation clearance using next-generation sequencing of recurrent myeloid gene mutations done on remission bone marrow has been associated with improved survival, but is still a research tool.128,129 Ultrasensitive methods for minimal residual disease detection with digital droplet PCR to target low amounts of residual gene mutations are also being developed.130 Ultimately, comprehensive prognostic models are needed that incorporate both pretreatment prognostic factors (eg, karyotype and molecular mutations) with minimal residual disease status to improve assessment of relapse risk. Furthermore, whether a reliable measure of minimal residual disease could serve as a surrogate endpoint for regulatory drug approval is also an area of active investigation and controversy.43,131
The development of future therapies for acute myeloid leukaemia must be informed by an increased understanding of the biology of acute myeloid leukaemia, including its heterogeneity and subclonal nature, as well as an appreciation of the presence of leukaemic stem cells that are generally chemoresistant and serve as an important reservoir of disease that can lead to relapse after initial response to therapy (figure 2). Many compounds with novel and diverse mechanisms of action are actively being tested in clinical trials, including drugs such as monoclonal antibody constructs against CD33 or CD123, bromodomain and extra terminal protein inhibitors, and B-cell lymphoma 2 (BCL-2) inhibitors, all of which are capable of targeting and potentially eliminating leukaemic stem cells.9 Given the substantial advances made with immunotherapy in the field of solid oncology, efforts are also ongoing to investigate immune-based therapies in acute myeloid leukaemia, including the use of checkpoint inhibitors (eg, anti-CTLA-4 and anti-PD1 monoclonal antibodies) and novel chimeric antigen receptor T-cell therapies that target epitopes highly expressed on acute myeloid leukaemia blasts.132 Several novel epigenetic therapies are in clinical trials, including guadecitabine (SGI-110), an oral formulation of azacitidine (CC-486), and IDH1 inhibitors (eg, AG-120; table 4). Because the role of the microenvironment is increasingly recognised as being protective, allowing leukaemic cells to survive the effect of therapy, treatment strategies directed at reversing this protective effect are actively being pursued.
Figure 2: Examples of selected novel therapeutic strategies in acute myeloid leukaemia.
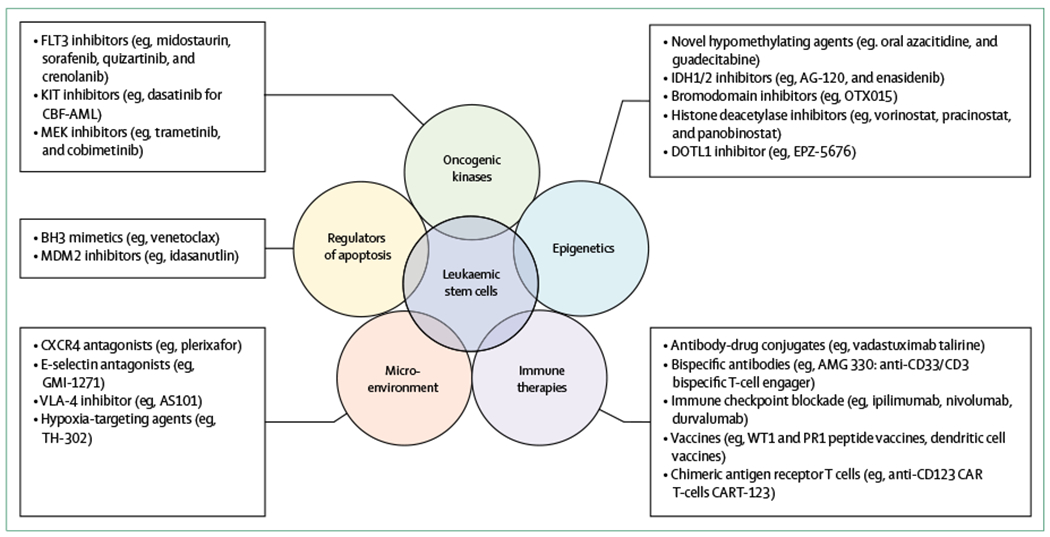
Five therapeutic targets being developed for the management of acute myeloid leukaemia are highlighted, as well as representative compounds that are in clinical trials. These examples are not intended to be all inclusive. Strategies that eliminate leukaemia stem cells, which are generally chemoresistant and serve as a reservoir for relapse, are needed to achieve cure.
Table 4:
Selected ongoing randomised, phase III frontline trials for adults with acute myeloid leukaemia
| Specific inclusion criteria | Trial identifier | |
|---|---|---|
| Patients eligible for intensive chemotherapy | ||
|
| ||
| Dasatinib (multitargeted kinase inhibitor) plus chemotherapy vs chemotherapy alone | Core-binding factor acute myeloid leukaemia | NCT02013648 |
| Quizartinib (FLT3 inhibitor) plus chemotherapy vs chemotherapy alone | Acute myeloid leukaemia with FLT3-ITD mutation | NCT02668653 |
|
| ||
| Patients not eligible for intensive chemotherapy | ||
|
| ||
| Venetoclax (BCL-2 inhibitor) plus azacitidine vs azacitidine alone | NA | NCT02993523 |
| Venetoclax (BCL-2 inhibitor) plus low-dose cytarabine vs low-dose cytarabine alone | NA | NCT03069352 |
| Pracinostat (histone deacetylase inhibitor) plus azacitidine vs azacitidine alone | NA | NCT03151408 |
| Ivosidenib (IDH1 inhibitor) plus azacitidine vs azacitidine alone | Acute myeloid leukaemia with IDH1 mutation | NCT03173248 |
| Guadecitabine (novel hypomethylating agent) vs treatment choice | NA | NCT02348489 |
|
| ||
| Patients in remission after induction and consolidation | ||
|
| ||
| Gilteritinib (FLT3 inhibitor) vs placebo | Acute myeloid leukaemia with FLT3-ITD mutation in first remission | NCT02927262 |
| Oral azacitidine vs placebo | NA | NCT01757535 |
ITD=internal tandem duplication.
We have outlined the ongoing phase 3 trials enrolling patients with newly diagnosed acute myeloid leukaemia or those who have achieved remission with standard therapy (ie, maintenance trials; table 4). Most of these studies combine investigational drugs with chemotherapy or hypomethylating agents, an approach that is most likely to maximise the benefit of these novel drugs.133 Multi-arm, biomarker-driven trials (such as the BEAT AML study134) offer a novel way to rationally select therapies for patients on the basis of specific genomic alterations present at diagnosis and for testing multiple compounds within a single umbrella trial. However, given the genomic complexity of acute myeloid leukaemia, whether single drugs targeting a specific genomic alteration will be sufficient to eradicate the disease is unclear. Therefore, strategies targeting more universal pathways (eg, immune-based therapies) might be a more successful therapeutic strategy. Beyond mutation-driven or antigen-driven drug selection, the use of in-vitro drug screening or functionality testing (eg, BH3 profiling to predict sensitivity to BCL-2 inhibitors) or the individualised study of induced pluripotential stem cells derived from primary acute myeloid leukaemia cells could further personalise acute myeloid leukaemia directed therapy.135,136 To evaluate the growing list of promising drugs for acute myeloid leukaemia, increased enrolment of patients with acute myeloid leukaemia into clinical trials will be needed to rapidly investigate the safety and efficacy of these promising therapies and ultimately bring them to the general population so that all patients can benefit. However, as data accumulate from rigorously monitored trials, in which patients are carefully selected for enrolment, we must be judicious in extrapolating these results to general practice.137 As we now have improved tools to approach these complex tasks, the work is ready to be done.
Supplementary Material
Search strategy and selection criteria.
We searched the Cochrane Library and PubMed for relevant randomised trials and other high-quality studies (eg, systematic reviews and meta-analyses) published in English between Jan 1, 2007 and Oct 1, 2017. We used the search terms “acute myeloid leukaemia” or “AML” in combination with the terms “genomics”, “outcomes”, “prognosis”, and “treatment”. We largely selected publications from the past 5 years, but did not exclude commonly referenced and highly regarded older publications. We also searched the reference lists of articles identified by this search strategy and selected those we deemed relevant. Our reference list was modified based on comments from peer reviewers.
Footnotes
See Online for appendix
Declaration of interests
We declare no competing interests.
References
- 1.Ganzel C, Manola J, Douer D, et al. Extramedullary disease in adult acute myeloid leukemia is common but lacks independent significance: analysis of patients in ECOG-ACRIN Cancer Research Group trials, 1980–2008. J Clin Oncol 2016; 34: 3544–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zuckerman T, Ganzel C, Tallman MS, Rowe JM. How I treat hematologic emergencies in adults with acute leukemia. Blood 2012; 120: 1993–2002. [DOI] [PubMed] [Google Scholar]
- 3.National Cancer Institute. Cancer stat facts: leukemia—acute myeloid leukemia (AML). 2017. https://seer.cancer.gov/statfacts/html/amyl.html (accessed Sept 21, 2017).
- 4.McNerney ME, Godley LA, Le Beau MM. Therapy-related myeloid neoplasms: when genetics and environment collide. Nat Rev Cancer 2017; 17: 513–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leonard JP, Martin P, Roboz GJ. Practical implications of the 2016 revision of the World Health Organization classification of lymphoid and myeloid neoplasms and acute leukemia. J Clin Oncol 2017; 35: 2708–15. [DOI] [PubMed] [Google Scholar]
- 6.Adès L, Itzykson R, Fenaux P. Myelodysplastic syndromes. Lancet 2014; 383: 2239–2252. [DOI] [PubMed] [Google Scholar]
- 7.Tefferi A, Pardanani A. Myeloproliferative neoplasms: a contemporary review. JAMA Oncol 2015; 1: 97–105. [DOI] [PubMed] [Google Scholar]
- 8.Lane SW, Gilliland DG. Leukemia stem cells. Semin Cancer Biol 2010; 20: 71–76. [DOI] [PubMed] [Google Scholar]
- 9.Pollyea DA, Jordan CT. Therapeutic targeting of acute myeloid leukemia stem cells. Blood 2017; 129: 1627–35. [DOI] [PubMed] [Google Scholar]
- 10.Shlush LI, Mitchell A, Heisler L, et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 2017; 547: 104–08. [DOI] [PubMed] [Google Scholar]
- 11.The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013; 368: 2059–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012; 150: 264–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steffen B, Müller-Tidow C, Schwäble J, Berdel WE, Serve H. The molecular pathogenesis of acute myeloid leukemia. Crit Rev Oncol Hematol 2005; 56: 195–221. [DOI] [PubMed] [Google Scholar]
- 14.Shlush LI, Zandi S, Mitchell A, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014; 506: 328–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012; 481: 506–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015; 126: 91–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014; 371: 2477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014; 371: 2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jaiswal S, Natarajan P, Silver AJ, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med 2017; 377: 111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takahashi K, Wang F, Kantarjian H, et al. Preleukaemic clonal haemopoiesis and risk of therapy-related myeloid neoplasms:a case-control study. Lancet Oncol 2017; 18: 100–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gibson CJ, Lindsley RC, Tchekmedyian V, et al. Clonal hematopoiesis associated with adverse outcomes after autologous stem-cell transplantation for lymphoma. J Clin Oncol 2017; 35: 1598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ok CY, Patel KP, Garcia-Manero G, et al. TP53 mutation characteristics in therapy-related myelodysplastic syndromes and acute myeloid leukemia is similar to de novo diseases. J Hematol Oncol 2015; 8: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong TN, Ramsingh G, Young AL, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 2015; 518: 552–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012; 366: 1079–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bullinger L, Dohner K, Dohner H. Genomics of acute myeloid leukemia diagnosis and pathways. J Clin Oncol 2017; 35: 934–46. [DOI] [PubMed] [Google Scholar]
- 26.Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 2016; 374: 2209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawrence MS, Stojanov P, Mermel CH, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014; 505: 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shafer D, Grant S. Update on rational targeted therapy in AML. Blood Rev 2016; 30: 275–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pratcorona M, Brunet S, Nomdedeu J, et al. Favorable outcome of patients with acute myeloid leukemia harboring a low-allelic burden FLT3-ITD mutation and concomitant NPM1 mutation: relevance to post-remission therapy. Blood 2013; 121: 2734–38. [DOI] [PubMed] [Google Scholar]
- 30.Schlenk RF, Kayser S, Bullinger L, et al. Differential impact of allelic ratio and insertion site in FLT3-ITD-positive AML with respect to allogeneic transplantation. Blood 2014; 124: 3441–49. [DOI] [PubMed] [Google Scholar]
- 31.Haferlach C, Mecucci C, Schnittger S, et al. AML with mutated NPM1 carrying a normal or aberrant karyotype show overlapping biologic, pathologic, immunophenotypic, and prognostic features. Blood 2009; 114: 3024–32. [DOI] [PubMed] [Google Scholar]
- 32.Falini B, Macijewski K, Weiss T, et al. Multilineage dysplasia has no impact on biologic, clinicopathologic, and prognostic features of AML with mutated nucleophosmin (NPM1). Blood 2010; 115: 3776–86. [DOI] [PubMed] [Google Scholar]
- 33.Ivey A, Hills RK, Simpson MA, et al. Assessment of minimal residual disease in standard-risk AML. N Engl J Med 2016; 374: 422–33. [DOI] [PubMed] [Google Scholar]
- 34.Schlenk RF, Taskesen E, van Norden Y, et al. The value of allogeneic and autologous hematopoietic stem cell transplantation in prognostically favorable acute myeloid leukemia with double mutant CEBPA. Blood 2013; 122: 1576–82. [DOI] [PubMed] [Google Scholar]
- 35.Tawana K, Rio-Machin A, Preudhomme C, Fitzgibbon J. Familial CEBPA-mutated acute myeloid leukemia. Semin Hematol 2017; 54: 87–93. [DOI] [PubMed] [Google Scholar]
- 36.Lindsley RC, Mar BG, Mazzola E, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015; 125:1367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gaidzik VI, Teleanu V, Papaemmanuil E, et al. RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features. Leukemia 2016; 30: 2160–68. [DOI] [PubMed] [Google Scholar]
- 38.Preudhomme C, Renneville A, Bourdon V, et al. High frequency of RUNX1 biallelic alteration in acute myeloid leukemia secondary to familial platelet disorder. Blood 2009; 1l3: 5583–87. [DOI] [PubMed] [Google Scholar]
- 39.Peters JM, Ansari MQ. Multiparameter flow cytometry in the diagnosis and management of acute leukemia. Arch Pathol Lab Med 2011; 135: 44–45. [DOI] [PubMed] [Google Scholar]
- 40.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016; 127: 2391–405. [DOI] [PubMed] [Google Scholar]
- 41.Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017; 129: 424–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cornelissen JJ, Blaise D. Hematopoietic stem cell transplantation for patients with AML in first complete remission. Blood 2016; 127: 62–70. [DOI] [PubMed] [Google Scholar]
- 43.Hourigan CS, Gale RP, Gormley NJ, Ossenkoppele GJ, Walter RB. Measurable residual disease testing in acute myeloid leukaemia. Leukemia 2017; 31: 1482–90. [DOI] [PubMed] [Google Scholar]
- 44.Kantarjian H, O’Brien S, Cortes J, et al. Results of intensive chemotherapy in 998 patients age 65 years or older with acute myeloid leukemia or high-risk myelodysplastic syndrome: predictive prognostic models for outcome. Cancer 2006; 106: 1090–98. [DOI] [PubMed] [Google Scholar]
- 45.Klepin HD, Rao AV, Pardee TS. Acute myeloid leukemia and myelodysplastic syndromes in older adults. J Clin Oncol 2014; 32: 2541–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ossenkoppele G, Lowenberg B. How I treat the older patient with acute myeloid leukemia. Blood 2015; 125: 767–74. [DOI] [PubMed] [Google Scholar]
- 47.Creutzig U, Zimmermann M, Reinhardt D, et al. Changes in cytogenetics and molecular genetics in acute myeloid leukemia from childhood to adult age groups. Cancer 2016; 122: 3821–30. [DOI] [PubMed] [Google Scholar]
- 48.Granfeldt Ostgard LS, Medeiros BC, Sengelov H, et al. Epidemiology and clinical significance of secondary and therapy-related acute myeloid leukemia: a national population-based cohort study. J Clin Oncol 2015; 33: 3641–49. [DOI] [PubMed] [Google Scholar]
- 49.Grimwade D, Hills RK, Moorman AV, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010; 116: 354–65. [DOI] [PubMed] [Google Scholar]
- 50.Martelli MP, Sportoletti P, Tiacci E, Martelli MF, Falini B. Mutational landscape of AML with normal cytogenetics: biological and clinical implications. Blood Rev 2013; 27: 13–22. [DOI] [PubMed] [Google Scholar]
- 51.Wouters BJ, Lowenberg B, Erpelinck-Verschueren CA, van Putten WL, Valk PJ, Delwel R. Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood 2009; 113: 3088–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taskesen E, Bullinger L, Corbacioglu A, et al. Prognostic impact, concurrent genetic mutations, and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML patients: further evidence for CEBPA double mutant AML as a distinctive disease entity. Blood 2011; 117: 2469–75. [DOI] [PubMed] [Google Scholar]
- 53.Kadia TM, Jain P, Ravandi F, et al. TP53 mutations in newly diagnosed acute myeloid leukemia: clinicomolecular characteristics, response to therapy, and outcomes. Cancer 2016; published online July 26. DOI: 10.1002/cncr.30203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen X, Xie H, Wood BL, et al. Relation of clinical response and minimal residual disease and their prognostic impact on outcome in acute myeloid leukemia. J Clin Oncol 2015; 33: 1258–64. [DOI] [PubMed] [Google Scholar]
- 55.Grimwade D, Freeman SD. Defining minimal residual disease in acute myeloid leukemia: which platforms are ready for “prime time”? Blood 2014; 124: 3345–55. [DOI] [PubMed] [Google Scholar]
- 56.Ravandi F, Jorgensen JL. Monitoring minimal residual disease in acute myeloid leukemia: ready for prime time? J Natl Compr Cancer Netw 2012; 10: 1029–36. [DOI] [PubMed] [Google Scholar]
- 57.Walter RB, Gooley TA, Wood BL, et al. Impact of pretransplantation minimal residual disease, as detected by multiparametric flow cytometry, on outcome of myeloablative hematopoietic cell transplantation for acute myeloid leukemia. J Clin Oncol 2011; 29: 1190–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Freeman SD, Virgo P, Couzens S, et al. Prognostic relevance of treatment response measured by flow cytometric residual disease detection in older patients with acute myeloid leukemia. J Clin Oncol 2013; 31: 4123–31. [DOI] [PubMed] [Google Scholar]
- 59.Terwijn M, van Putten WL, Kelder A, et al. High prognostic impact of flow cytometric minimal residual disease detection in acute myeloid leukemia: data from the HOVON/SAKK AML 42A study. J Clin Oncol 2013; 31: 3889–97. [DOI] [PubMed] [Google Scholar]
- 60.Balsat M, Renneville A, Thomas X, et al. Postinduction minimal residual disease predicts outcome and benefit from allogeneic stem cell transplantation in acute myeloid leukemia with NPM1 mutation:a study by the Acute Leukemia French Association group. J Clin Oncol 2017; 35: 185–93. [DOI] [PubMed] [Google Scholar]
- 61.Ravandi F, Jorgensen J, Borthakur G, et al. Persistence of minimal residual disease assessed by multiparameter flow cytometry is highly prognostic in younger patients with acute myeloid leukemia. Cancer 2017; 123: 426–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Araki D, Wood BL, Othus M, et al. Allogeneic hematopoietic cell transplantation for acute myeloid leukemia: time to move toward a minimal residual disease-based definition of complete remission? J Clin Oncol 2016; 34: 329–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kadia TM, Ravandi F, Cortes J, Kantarjian H. Toward individualized therapy in acute myeloid leukemia: a contemporary review. JAMA Oncol 2015; 1: 820–28. [DOI] [PubMed] [Google Scholar]
- 64.Podoltsev NA, Stahl M, Zeidan AM, Gore SD. Selecting initial treatment of acute myeloid leukaemia in older adults. Blood Rev 2017; 31: 43–62. [DOI] [PubMed] [Google Scholar]
- 65.Othus M, Kantarjian H, Petersdorf S, et al. Declining rates of treatment-related mortality in patients with newly diagnosed AML given ‘intense’ induction regimens: a report from SWOG and MD Anderson. Leukemia 2014; 28: 289–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schlenk RF. Post-remission therapy for acute myeloid leukemia. Haematologica 2014; 99: 1663–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Elsawy M, Sorror ML. Up-to-date tools for risk assessment before allogeneic hematopoietic cell transplantation. Bone Marrow Transplant 2016; 51: 1283–300. [DOI] [PubMed] [Google Scholar]
- 68.Yates JW, Wallace HJ Jr, Ellison RR, Holland JF. Cytosine arabinoside (NSC-63878) and daunorubicin (NSC-83142) therapy in acute nonlymphocytic leukemia. Cancer Chemother Rep 1973; 57: 485–88. [PubMed] [Google Scholar]
- 69.Yates J, Glidewell O, Wiernik P, et al. Cytosine arabinoside with daunorubicin or adriamycin for therapy of acute myelocytic leukemia: a CALGB study. Blood 1982; 60: 45–62. [PubMed] [Google Scholar]
- 70.Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med 2015; 373: 1136–52. [DOI] [PubMed] [Google Scholar]
- 71.Short NJ, Ravandi F. Acute myeloid leukemia: past, present, and prospects for the future. Clin Lymphoma Myeloma Leuk 2016; 16 (suppl): S25–29. [DOI] [PubMed] [Google Scholar]
- 72.Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med 2017; 377: 454–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017; 130: 722–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lancet JE, Uy GL, Cortes JE, et al. Final results of a phase III randomized trial of CPX-351 versus 7+3 in older patients with newly diagnosed high risk (secondary) AML. Proc Am Soc Clin Oncol 2016;34 (suppl): 7000 (abstr). [Google Scholar]
- 75.Castaigne S, Pautas C, Terre C, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet 2012; 379: 1508–16. [DOI] [PubMed] [Google Scholar]
- 76.Amadori S, Suciu S, Selleslag D, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol 2016; 34: 972–79. [DOI] [PubMed] [Google Scholar]
- 77.Taksin AL, Legrand O, Raffoux E, et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia 2007; 21: 66–71. [DOI] [PubMed] [Google Scholar]
- 78.Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol 2014; 15: 986–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stein EM, Tallman MS. Emerging therapeutic drugs for AML. Blood 2016; 127: 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lancet JE, Cortes JE, Hogge DE, et al. Phase 2 trial of CPX-351, a fixed 5:1 molar ratio of cytarabine/daunorubicin, vs cytarabine/daunorubicin in older adults with untreated AML. Blood 2014; 123: 3239–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cortes JE, Goldberg SL, Feldman EJ, et al. Phase II, multicenter, randomized trial of CPX-351 (cytarabine:daunorubicin) liposome injection versus intensive salvage therapy in adults with first relapse AML. Cancer 2015; 121: 234–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kihara R, Nagata Y, Kiyoi H, et al. Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia 2014; 28: 1586–95. [DOI] [PubMed] [Google Scholar]
- 83.Perl AE, Altman JK, Cortes J, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia:a multicentre, first-in-human, open-label, phase 1–2 study. Lancet Oncol 2017; 18: 1061–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cortes J, Perl AE, Döhner H, et al. Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukaemia: an open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol 2018; 19: 889–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ravandi F, Cortes JE, Jones D, et al. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J Clin Oncol 2010; 28: 1856–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stone RM, Fischer T, Paquette R, et al. Phase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia. Leukemia 2012; 26: 2061–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mayer RJ, Davis RB, Schiffer CA, et al. Intensive postremission chemotherapy in adults with acute myeloid leukemia. N Engl J Med 1994; 331: 896–903. [DOI] [PubMed] [Google Scholar]
- 88.Lowenberg B. Sense and nonsense of high-dose cytarabine for acute myeloid leukemia. Blood 2013; 121: 26–28. [DOI] [PubMed] [Google Scholar]
- 89.Bloomfield CD, Lawrence D, Byrd JC, et al. Frequency of prolonged remission duration after high-dose cytarabine intensification in acute myeloid leukemia varies by cytogenetic subtype. Cancer Res 1998; 58: 4173–79. [PubMed] [Google Scholar]
- 90.Thomas X, de Botton S, Chevret S, et al. Randomized phase II study of clofarabine-based consolidation for younger adults with acute myeloid leukemia in first remission. J Clin Oncol 2017; 35: 1223–30. [DOI] [PubMed] [Google Scholar]
- 91.Gupta V, Tallman MS, Weisdorf DJ. Allogeneic hematopoietic cell transplantation for adults with acute myeloid leukemia: myths, controversies, and unknowns. Blood 2011; 117: 2307–18. [DOI] [PubMed] [Google Scholar]
- 92.Koreth J, Schlenk R, Kopecky KJ, et al. Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: systematic review and meta-analysis of prospective clinical trials. JAMA 2009; 301: 2349–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dohner K, Paschka P. Intermediate-risk acute myeloid leukemia therapy: current and future. Hematology Am Soc Hematol Educ Program 2014; 2014: 34–43. [DOI] [PubMed] [Google Scholar]
- 94.Stelljes M, Krug U, Beelen DW, et al. Allogeneic transplantation versus chemotherapy as postremission therapy for acute myeloid leukemia: a prospective matched pairs analysis. J Clin Oncol 2014; 32: 288–96. [DOI] [PubMed] [Google Scholar]
- 95.Luskin MR, Stone RM. Can minimal residual disease determination in acute myeloid leukemia be used in clinical practice? J Oncol Pract 2017; 13: 471–80. [DOI] [PubMed] [Google Scholar]
- 96.Rashidi A, Walter RB, Tallman MS, Appelbaum FR, DiPersio JF. Maintenance therapy in acute myeloid leukemia: an evidence-based review of randomized trials. Blood 2016; 128: 763–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sorror ML, Storer BE, Elsawy M, et al. Intensive versus non-intensive induction therapy for patients (pts) with newly diagnosed acute myeloid leukemia (AML) using two different novel prognostic models. Blood 2016; 128: 216. [Google Scholar]
- 98.Burnett AK, Milligan D, Prentice AG, et al. A comparison of low-dose cytarabine and hydroxyurea with or without all-trans retinoic acid for acute myeloid leukemia and high-risk myelodysplastic syndrome in patients not considered fit for intensive treatment. Cancer 2007; 109: 1114–24. [DOI] [PubMed] [Google Scholar]
- 99.Itzykson R, Fenaux P. Epigenetics of myelodysplastic syndromes. Leukemia 2014; 28: 497–506. [DOI] [PubMed] [Google Scholar]
- 100.Dombret H, Seymour JF, Butrym A, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015; 126: 291–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kantarjian HM, Thomas XG, Dmoszynska A, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol 2012; 30: 2670–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Quintas-Cardama A, Ravandi F, Liu-Dumlao T, et al. Epigenetic therapy is associated with similar survival compared with intensive chemotherapy in older patients with newly diagnosed acute myeloid leukemia. Blood 2012; 120: 4840–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dhulipala VC, Extermann M, Al Ali N, et al. Survival comparison amongst commonly used frontline regimens in patients age 70 years and older with acute myeloid leukemia (AML): a single-institution study of over 600 patients. Blood 2015; 126: 2505. [Google Scholar]
- 104.Welch JS, Petti AA, Miller CA, et al. TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N Engl J Med 2016; 375: 2023–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kronke J, Bullinger L, Teleanu V, et al. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood 2013; 122: 100–08. [DOI] [PubMed] [Google Scholar]
- 106.Nakano Y, Kiyoi H, Miyawaki S, et al. Molecular evolution of acute myeloid leukaemia in relapse: unstable N-ras and FLT3 genes compared with p53 gene. Br J Haematol 1999; 104: 659–64. [DOI] [PubMed] [Google Scholar]
- 107.Estey E, Keating MJ, Pierce S, Stass S. Change in karyotype between diagnosis and first relapse in acute myelogenous leukemia. Leukemia 1995; 9: 972–76. [PubMed] [Google Scholar]
- 108.Breems DA, Van Putten WL, Huijgens PC, et al. Prognostic index for adult patients with acute myeloid leukemia in first relapse. J Clin Oncol 2005; 23: 1969–78. [DOI] [PubMed] [Google Scholar]
- 109.Pemmaraju N, Kantarjian H, Garcia-Manero G, et al. Improving outcomes for patients with acute myeloid leukemia in first relapse: a single center experience. Am J Hematol 2015; 90: 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kurosawa S, Yamaguchi T, Miyawaki S, et al. Prognostic factors and outcomes of adult patients with acute myeloid leukemia after first relapse. Haematologica 2010; 95: 1857–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Roboz GJ, Rosenblat T, Arellano M, et al. International randomized phase III study of elacytarabine versus investigator choice in patients with relapsed/refractory acute myeloid leukemia. J Clin Oncol 2014; 32: 1919–26. [DOI] [PubMed] [Google Scholar]
- 112.Amatangelo MD, Quek L, Shih A, et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood 2017; 130: 732–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010; 17: 225–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.DiNardo CD, Ravandi F, Agresta S, et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am J Hematol 2015; 90: 732–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Paschka P, Schlenk R, Weber D, et al. Outcome of patients with refractory or relapsed AML with IDH1 and IDH2 mutations after conventional salvage therapy: a study of the German-Austrian AML study group (AMLSG). European Hematology Association; Copenhagen, Denmark; June 12, 2016. S809. [Google Scholar]
- 116.Grimwade D, Walker H, Oliver F, et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. Blood 1998; 92: 2322–33. [PubMed] [Google Scholar]
- 117.Meshinchi S, Arceci RJ. Prognostic factors and risk-based therapy in pediatric acute myeloid leukemia. Oncologist 2007; 12: 341–55. [DOI] [PubMed] [Google Scholar]
- 118.Inaba H, Coustan-Smith E, Cao X, et al. Comparative analysis of different approaches to measure treatment response in acute myeloid leukemia. J Clin Oncol 2012; 30: 3625–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Loken MR, Alonzo TA, Pardo L, et al. Residual disease detected by multidimensional flow cytometry signifies high relapse risk in patients with de novo acute myeloid leukemia: a report from Children’s Oncology Group. Blood 2012; 120: 1581–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.van der Velden VH, van der Sluijs-Geling A, Gibson BE, et al. Clinical significance of flowcytometric minimal residual disease detection in pediatric acute myeloid leukemia patients treated according to the DCOG ANLL97/MRC AML12 protocol. Leukemia 2010; 24: 1599–606. [DOI] [PubMed] [Google Scholar]
- 121.Hann IM, Stevens RF, Goldstone AH, et al. Randomized comparison of DAT versus ADE as induction chemotherapy in children and younger adults with acute myeloid leukemia. Results of the Medical Research Council’s 10th AML trial (MRC AML10). Blood 1997; 89: 2311–18. [PubMed] [Google Scholar]
- 122.Taga T, Tomizawa D, Takahashi H, Adachi S. Acute myeloid leukemia in children: current status and future directions. Pediatrics Int 2016; 58: 71–80. [DOI] [PubMed] [Google Scholar]
- 123.Kolb EA, Meshinchi S. Acute myeloid leukemia in children and adolescents: identification of new molecular targets brings promise of new therapies. Hematology Am Soc Hematol Educ Program 2015; 2015: 507–13. [DOI] [PubMed] [Google Scholar]
- 124.Gamis AS, Alonzo TA, Meshinchi S, et al. Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: results from the randomized phase III Children’s Oncology Group trial AAML0531. J Clin Oncol 2014; 32: 3021–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lamba JK, Chauhan L, Shin M, et al. CD33 splicing polymorphism determines gemtuzumab ozogamicin response in de novo acute myeloid leukemia: report from randomized phase III Children’s Oncology Group trial AAML0531. J Clin Oncol 2017; 35: 267–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sung L, Aplenc R, Alonzo TA, Gerbing RB, Lehrnbecher T, Gamis AS. Effectiveness of supportive care measures to reduce infections in pediatric AML: a report from the Children’s Oncology Group. Blood 2013; 121: 3573–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hokland P, Ommen HB, Mule MP, Hourigan CS. Advancing the minimal residual disease concept in acute myeloid leukemia. Semin Hematol 2015; 52: 184–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Klco JM, Miller CA, Griffith M, et al. Association between mutation clearance after induction therapy and outcomes in acute myeloid leukemia. JAMA 2015; 314: 811–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jongen-Lavrencic M, Grob T, Hanekamp D, et al. Molecular minimal residual disease in acute myeloid leukemia. N Engl J Med 2018; 378: 1189–99. [DOI] [PubMed] [Google Scholar]
- 130.Parkin B, Londono-Joshi A, Kang Q, Tewari M, Rhim AD, Malek SN. Ultrasensitive mutation detection identifies rare residual cells causing acute myelogenous leukemia relapse. J Clin Invest 2017; 127: 3484–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017; 129: 424–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Grosso DA, Hess RC, Weiss MA. Immunotherapy in acute myeloid leukemia. Cancer 2015; 121: 2689–704. [DOI] [PubMed] [Google Scholar]
- 133.Estey E, Levine RL, Lowenberg B. Current challenges in clinical development of “targeted therapies”: the case of acute myeloid leukemia. Blood 2015; 125: 2461–66. [DOI] [PubMed] [Google Scholar]
- 134.No authors listed. Study seeks new AML therapies. Cancer Discov 2016; 6: 1297–98. [DOI] [PubMed] [Google Scholar]
- 135.Lam SS, He AB, Leung AY. Treatment of acute myeloid leukemia in the next decade—towards real-time functional testing and personalized medicine. Blood Rev 2017; 31: 418–25. [DOI] [PubMed] [Google Scholar]
- 136.Chao MP, Gentles AJ, Chatterjee S, et al. Human AML-iPSCs reacquire leukemic properties after differentiation and model clonal variation of disease. Cell Stem Cell 2017; 20: 329–44.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lichtman SM, Harvey RD, Damiette Smit MA, et al. Modernizing clinical trial eligibility criteria: recommendations of the American Society of Clinical Oncology-Friends of Cancer Research Organ Dysfunction, Prior or Concurrent Malignancy, and Comorbidities Working Group. J Clin Oncol 2017; 35: 3753–59. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.