

Late-onset ataxias are clinically and etiologically diverse. Patients rarely have defining clinical features, and many remain classified as idiopathic, despite extensive clinical, metabolic, and genetic investigations. Here we show that mutations in a gene known to cause hereditary spastic paraplegia (SPG7) are a major cause of unexplained ataxia presenting in mid-adult life.
Methods.
Exome sequencing in 2 undiagnosed ataxia patients identified compound heterozygous SPG7 mutations, not previously considered likely in the absence of pyramidal signs. This prompted us to prospectively study SPG7 in 70 other probands with undiagnosed ataxia and pyramidal signs attending routine follow-up over a 12-month period. Other sporadic and inherited causes of ataxia were excluded, including inflammatory, metabolic, neoplastic, and sporadic degenerative ataxia; spinocerebellar ataxia 1, 2, 3, 6, 7, 10, 12, and 17; dentatorubral-pallidoluysian atrophy; and Friedreich ataxia. Mutations in SPG7 were detected by Sanger sequencing of all 17 coding exons and multiplex ligation-dependent probe amplification analysis (MRC-Holland kit P213-B1, Amsterdam, the Netherlands). All patients provided written informed consent.
Results.
Exome sequencing identified 2 SPG7 mutations in patient 1 (c.1529C>T/p.Ala510Val and c.1715C>T/p.Ala572Val) and 2 in patient 3 (c.1529C>T/p.Ala510Val and c.1192C>T/p.Arg398*). These were confirmed by Sanger sequencing, and present in their affected siblings (patients 2 and P4, respectively). The variants were heterozygous in the unaffected parents, and were previously reported as pathogenic.1 No other recessive mutations in relevant disease genes were identified (tables e-1 to e-4 on the Neurology® Web site at Neurology.org).
Of the 70 patients subsequently studied, 13 had likely recessive mutations (4 homozygous and 9 compound heterozygous). Two patients had novel mutations (c.1225_1229del/p.Glu409Arg_fs49* and c.2228T>C/p.Ile743Thr). All patients had the c.1529C>T/p.Ala510Val mutation on at least one allele. All patients were of British descent. No rearrangements were detected. The clinical features are summarized in table 1.
Table 1.
Clinical features and molecular findings in 17 patients with SPG7 mutations
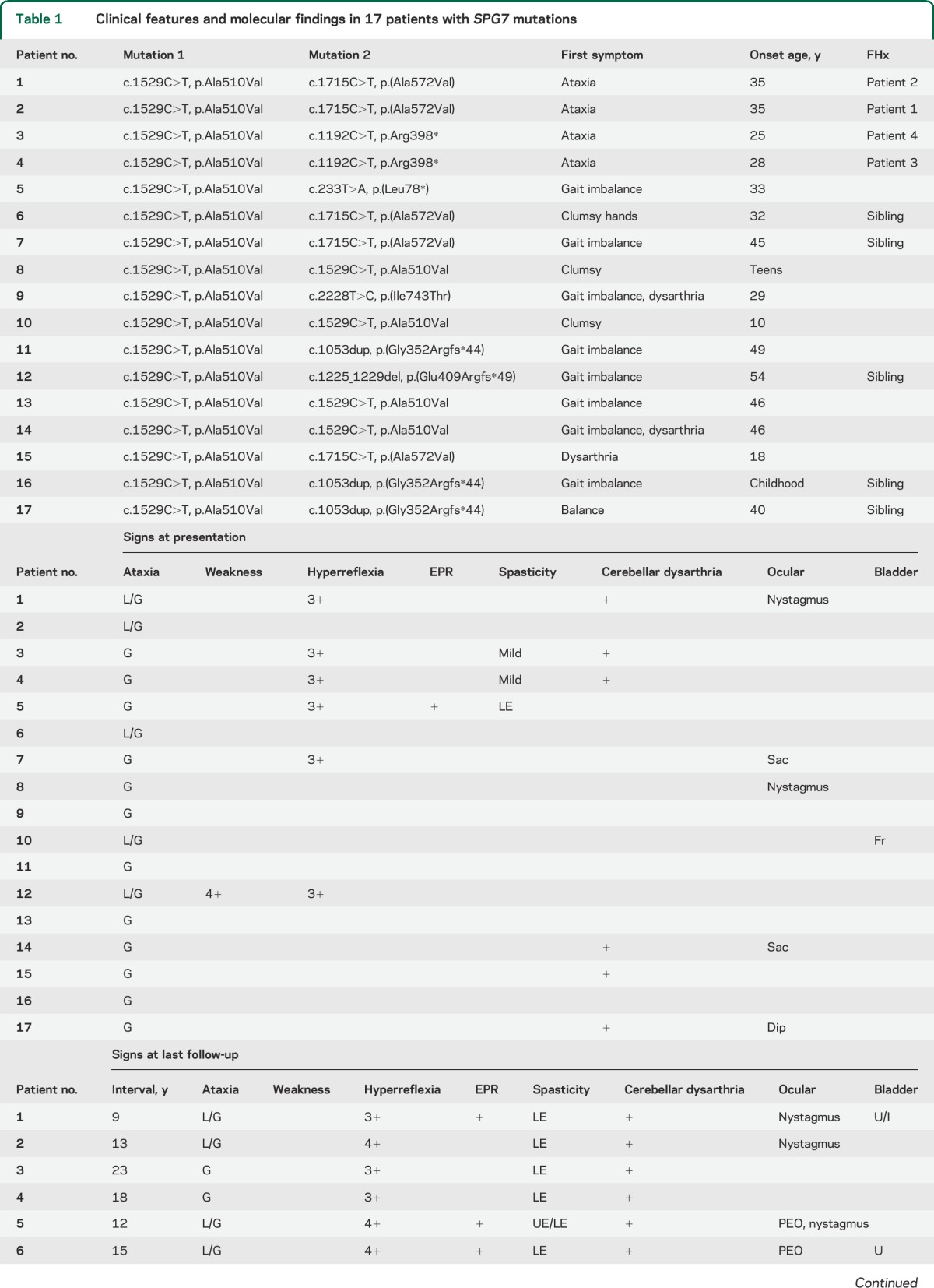
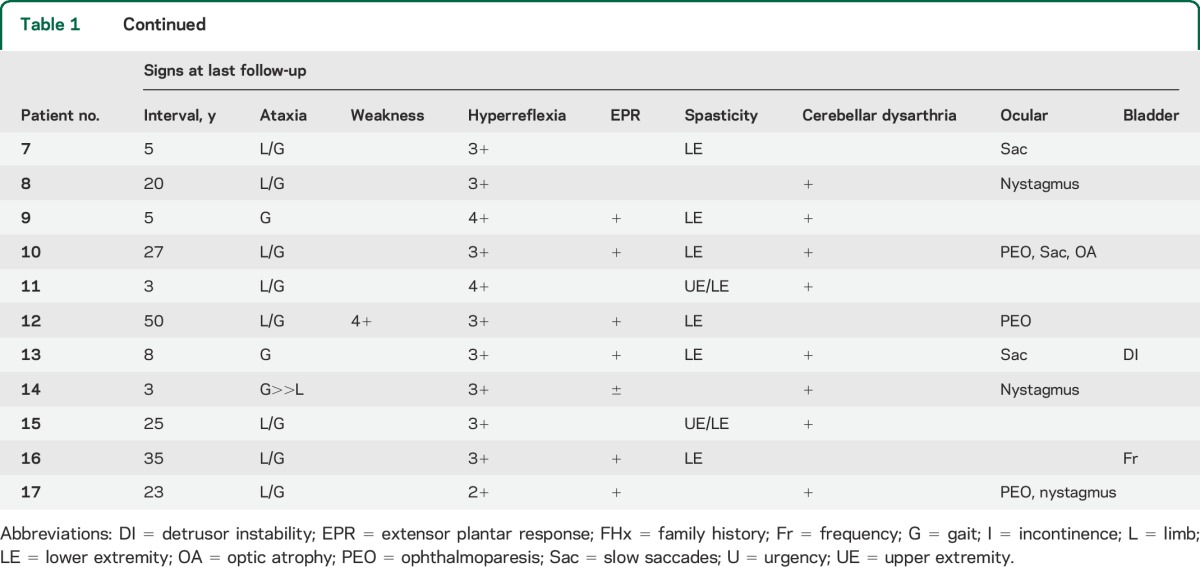
At initial presentation, all the patients presented with symptoms of ataxia or gait disturbance (mean age 36.3 years, SD 12.5). Midline ataxia was present in all patients at onset, with gait ataxia present in all patients and ocular signs in 5. Eleven patients (65%) had no pyramidal signs (normal reflexes, and no spasticity). Ocular findings were present in 5 (29%) patients.
On follow-up examination, most patients (76%) developed appendicular ataxia. All developed clear pyramidal signs: 12 had overt spasticity, and the remainder developed brisk tendon reflexes or extensor plantar responses. Ocular findings were present in 11 (65%), with nystagmus being the most common finding and partial ophthalmoparesis or slow saccades in 7 (41%). Twelve patients had brain MRI, with 11 (93%) showing cerebellar atrophy. Cervical spine MRI (n = 5) was normal in 4, with 1 patient having a likely incidental thoracic syrinx. Muscle biopsy identified cytochrome c oxidase–negative fibers in 2 patients, multiple mtDNA deletions in 1 patient, and coenzyme Q10 deficiency in a single patient.
Discussion.
We were surprised to find likely pathogenic SPG7 mutations in 18.6% of patients with unexplained ataxia. Although these patients did not have pure ataxia on follow-up, it was the predominating feature, and the patients had been clinically diagnosed and investigated for ataxic disorders. In our SPG7 patients, even after an average follow-up of 16.8 years, the pyramidal signs were subtle in many, endorsing our conclusion that a gene identified in patients with autosomal recessive hereditary spastic paraplegia should be considered in adults with unexplained ataxia. Eight (57%) of the probands had no relevant family history, so SPG7 should even be considered in sporadic cases. Combining these findings with another novel clinical presentation of SPG7,2 we provide the first minimum prevalence of SPG7-related disease at 0.72/100,000, making this a common cause of inherited ataxia, comparable with both autosomal dominant spinocerebellar ataxia (1.59/100,000)3 and Friedrich ataxia (1.8/100,000).4 This is probably an underestimate given the late presentation of some cases. All of our patients had p.Ala510Val, reaffirming the pathogenicity of this allele, which has been considered a low frequency polymorphism (0.3463% in EVS, dbSNP rs61755320). This study illustrates the advantage of exome sequencing in neurogenetic disorders, where genes initially shown to cause one classical phenotype (such as hereditary spastic paraplegia) can also cause other phenotypes in a subgroup of patients (such as ataxia).
SPG7 encodes paraplegin, which is a component of the mitochondrial AAA protease, and the binding partner of AFG3L2.5 Both paraplegin and AFG3L2 are highly expressed in Purkinje neurons,6 and mutations in AFG3L2 cause spinocerebellar ataxia type 28.7 This explains why the phenotypic spectrum of SPG7 includes a predominantly ataxic presentation. It will be interesting to see whether specific mutations predispose to an ataxic or spastic presentation when cohorts increase in size. However, given the diverse nature of the clinical presentations, a more inclusive disease name such as parapleginopathy may avoid the misleading expectation that spasticity always predominates in this condition. Future study should address the relative likelihoods of these various presentations along the phenotypic spectrum of paraplegin-related diseases.
Supplementary Material
Footnotes
Editorial, page 1070
Supplemental data at Neurology.org
Author contributions: Gerald Pfeffer: study design, data analysis, manuscript authorship. Angela Pyle: data analysis. Helen Griffin: data analysis. Jack Miller: data analysis. Valerie Wilson: data analysis. Lisa Turnbull: data analysis. Katherine Fawcett: data analysis. David Sims: data analysis. Gail Eglon: data analysis. Marios Hadjivassiliou: data analysis. Rita Horvath: data analysis. Andrea Németh: data analysis. Patrick F. Chinnery: study design, data analysis, manuscript authorship, study supervision.
Study funding: G.P. is the recipient of a Bisby Fellowship from the Canadian Institutes of Health Research. P.F.C. is an Honorary Consultant Neurologist at Newcastle-upon-Tyne Foundation Hospitals NHS Trust, is a Wellcome Trust Senior Fellow in Clinical Science (101876/Z/13/Z), and is a UK NIHR Senior Investigator. P.F.C. receives additional support from the Wellcome Trust Centre for Mitochondrial Research (096919Z/11/Z), the Medical Research Council (UK) Centre for Translational Muscle Disease Research (G0601943), and EU FP7 TIRCON, and the National Institute for Health Research (NIHR) Newcastle Biomedical Research Centre based at Newcastle-upon-Tyne Hospitals NHS Foundation Trust and Newcastle University. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health.
Disclosure: The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures. The Article Processing Charge was paid by Wellcome Trust.
References
- 1.Klebe S, Depienne C, Gerber S, et al. Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy. Brain 2012;135:2980–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pfeffer G, Gorman GS, Griffin H, et al. Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain 2014;137:1323–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Craig K, Keers SM, Archibald K, Curtis A, Chinnery PF. Molecular epidemiology of spinocerebellar ataxia type 6. Ann Neurol 2004;55:752–755. [DOI] [PubMed] [Google Scholar]
- 4.Schulz JB, Boesch S, Burk K, et al. Diagnosis and treatment of Friedreich ataxia: a European perspective. Nat Reviews Neurol 2009;5:222–234. [DOI] [PubMed] [Google Scholar]
- 5.Koppen M, Metodiev MD, Casari G, Rugarli EI, Langer T. Variable and tissue-specific subunit composition of mitochondrial m-AAA protease complexes linked to hereditary spastic paraplegia. Mol Cell Biol 2007;27:758–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sacco T, Boda E, Hoxha E, et al. Mouse brain expression patterns of Spg7, Afg3l1, and Afg3l2 transcripts, encoding for the mitochondrial m-AAA protease. BMC Neurosci 2010;11:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Bella D, Lazzaro F, Brusco A, et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat Genet 2010;42:313–321. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.