

Abstract
Alzheimer’s disease (AD) is an age-associated neurodegenerative disorder with multifactorial etiology, intersecting genetic and environmental risk factors, and a lack of disease-modifying therapeutics. While the abnormal accumulation of lipids was described in the very first report of AD neuropathology, it was not until recent decades that lipid dyshomeostasis became a focus of AD research. Clinically, lipidomic and metabolomic studies have consistently shown alterations in the levels of various lipid classes emerging in early stages of AD brains. Mechanistically, decades of discovery research have revealed multifaceted interactions between lipid metabolism and key AD pathogenic mechanisms including amyloidogenesis, bioenergetic deficit, oxidative stress, neuroinflammation, and myelin degeneration. In the present review, converging evidence defining lipid dyshomeostasis in AD is summarized, followed by discussions on mechanisms by which lipid metabolism contributes to pathogenesis and modifies disease risk. Furthermore, lipid-targeting therapeutic strategies, and the modification of their efficacy by disease stage, ApoE status, and metabolic and vascular profiles, are reviewed.
Keywords: alzheimer’s disease, fatty acid, lipid metabolism, therapeutics
Introduction
Alzheimer’s disease (AD) is the most prevalent form of dementia. Worldwide, approximately 50 million people suffer from AD, and this number is projected to reach 82 million in 2030 and 152 million in 2050 [1]. AD is clinically characterized by progressive memory deficits, cognitive impairment, and other behavioral changes accompanied by structural abnormalities in the brain [2,3]. Neuropathologically, AD features extracellular aggregation of β-amyloid (Aβ) forming senile plaques, intraneuronal aggregation of hyperphosphorylated tau forming neurofibrillary tangles (NFT), and loss of neurons and synapses in the hippocampus and neocortex among other brain regions [2,3]. Biomarkers of these three key features constitute the core of a biological framework—the amyloid, tau, neurodegeneration (AT(N)) system—to define disease onset and progression [4].
While diagnosed in different stages, AD develops along a continuum with progressive accumulation of biomarkers, neural damage, and cognitive decline [4]. Before disease onset, preclinical AD is characterized by subtle cognitive decline and accumulated Aβ deposits, and the subsequent transition to mild cognitive impairment (MCI) is associated with a high probability of progressing to AD dementia [5]. There are two types of AD—early-onset and late-onset. The late-onset form (LOAD), which accounts for more than 95% of total cases [6,7], has a multifactorial nature and poorly understood molecular mechanisms. The early-onset, and autosomal dominant, form of AD (familial AD; fAD), caused by rare mutations in amyloid processing genes including APP (amyloid precursor protein), PSEN1 (presenilin-1), and PSEN2, accounts for ~ 1% of total cases [8].
In the past few decades, therapeutics targeting the Aβ cascade have predominantly failed at various stages of clinical trials [9–12], calling for refined and personized intervention strategies and/or alternative therapeutic targets [13,14]. Despite the recent U.S. FDA approval of AD drug aducanumab, which raised debates on its ambiguous efficacy and safety profile [15], there is still a lack of disease-modifying or preventative strategy beyond symptom alleviation.
Beyond pathological hallmarks involving Aβ and tau, AD brains are characterized by microglia-mediated inflammation, as well as metabolic abnormalities encompassing glucose hypometabolism, mitochondrial dysfunction, oxidative stress, and lipid dyshomeostasis [16–20]. Hypometabolism of glucose is the best-known form of metabolic dysfunction in AD. It not only occurs as an early event in AD-vulnerable regions in the brain [21,22] but its presence prior to amyloid or tau detection results in the fastest rate of disease progression [23]. In addition to glucose metabolism, abundant clinical investigations and epidemiological studies have strongly connected disrupted lipid metabolism with altered disease risk and the pathogenesis and progression of AD. In fact, lipid-related abnormalities are among the initial neuropathological findings identified by Alois Alzheimer in 1907 [24].
Lipids are a major class of nutrients that are vital for all organisms. They represent a diverse group of biomolecules that are structurally and functionally involved in a wide variety of cellular processes and tissue functions. As the body’s second most lipidated organ—only after adipose tissue–10% to 12% of the fresh weight, and more than 50% of the dry weight, of the brain is composed of lipids [25,26]. Major lipid species in the brain can be generally categorized to phospholipids, sphingolipids, glycerolipids, fatty acids, and sterols, with phospholipids accounting for ~ 50% of total lipid content [27]. Except for cholesterol, all lipids with various backbones carry at least one fatty acid (acyl) chain. For cholesterol, it can also form a cholesteryl ester (CE) by attaching to a fatty acid chain (esterification). Functionally, these lipids are key components of cellular membranes including synapses and myelin sheath; they can serve to transduce signaling to regulate a range of biological processes; and in some circumstances, lipids can be used as bioenergetic fuels [28].
Given the significance of various lipids in brain physiology, it is not surprising that accumulating research has discovered complex and diverse mechanisms that connect lipid metabolism with AD-related pathophysiologies. Further, development of lipid-targeting therapeutics has exhibited various degrees of efficacy against AD pathologies. In this review, I will summarize clinical evidence that demonstrates altered lipid metabolism in AD, known mechanisms that connect lipid metabolism and AD etiology, and the potential of AD therapeutics that target lipid metabolism.
Correlating lipids with AD: clinical evidence
Changes in lipid levels in AD
Fatty acids
Fatty acids are building blocks for all lipid classes except cholesterol. They are hydrocarbon chains with varying lengths terminated with carboxylic acid groups (Fig. 1). Elevated levels of free fatty acids, and their metabolic intermediates acyl-carnitines and acyl-CoA, are neurotoxic and can induce mitochondrial uncoupling and bioenergetic dysfunction [29,30]. Perturbations to brain fatty acid metabolism in AD are suggested by altered levels of free fatty acid levels. While the total free fatty acid levels are higher in the cerebrospinal fluid (CSF) of AD brains [31], each subclass of fatty acid show diverse shifts.
Fig. 1.
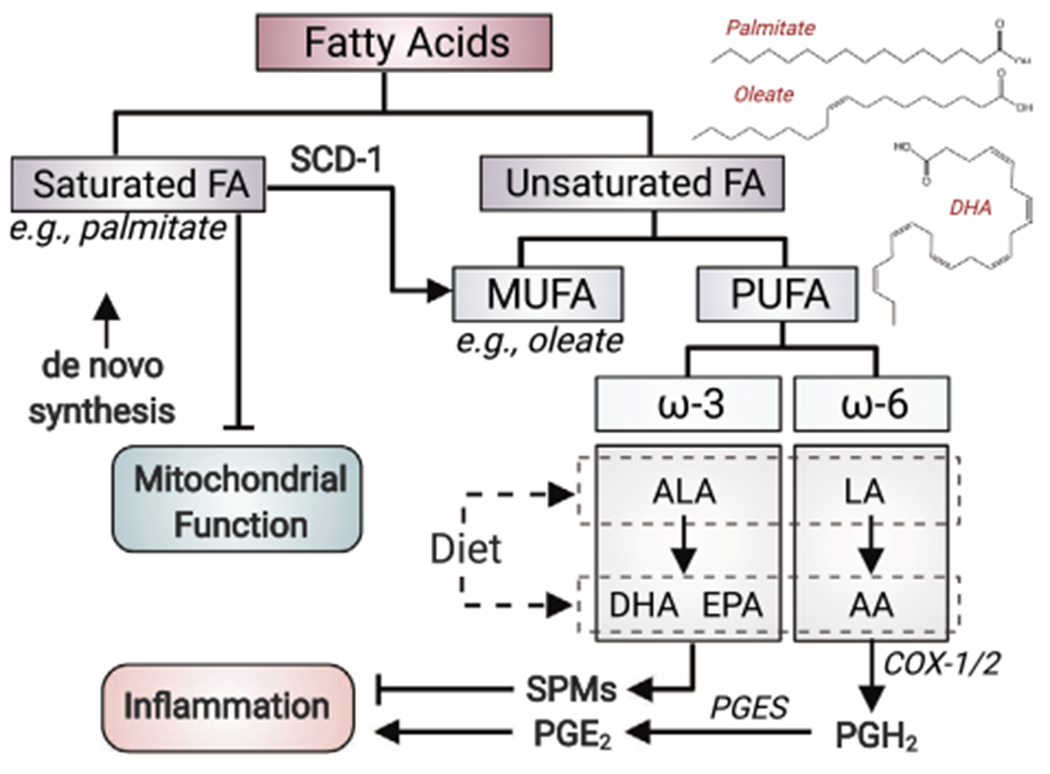
Classification of fatty acids. Fatty acids can be classified to saturated- and unsaturated fatty acids, and the unsaturated ones can be further grouped to MUFA and PUFA. Chemical structures of palmitate (saturated fatty acid), oleate (MUFA) and DHA (PUFA) are shown as examples of each class. Saturated fatty acids are negative regulators of mitochondrial function. MUFA can be generated from saturated fatty acid by SCD-1. ω-3 PUFAs, such as DHA and EPA generated from α-linolenic acid (ALA) or from diet, are precursors of SPMs, which are involved in inflammation resolution. AA, a ω-6 PUFA generated from LA or obtained from diet, elicits pro-inflammatory effect via the production of prostaglandin E2 (PGE2) by cyclooxygenase 1/2 (COX1/2) and prostaglandin E synthase (PGES). ALA and LA are essential fatty acids that can only be obtained from diet for human. PGH2, Prostaglandin H2.
Fatty acid can be classified by their saturation status to saturated- and unsaturated fatty acid, and the latter can be further grouped to monounsaturated- (MUFA) and polyunsaturated fatty acid (PUFA) (Fig. 1). Changes in the abundance of unsaturated fatty acid are found associated with AD in multiple studies. Across these studies, both brain (cortex, middle frontal gyrus, and inferior temporal gyrus)- and plasma levels of unsaturated fatty acids, including ω-3 PUFAs and a MUFA (oleic acid), are found lower in AD individuals than cognitively normal controls, and these changes lead to an overall reduced unsaturation index [32,33]. As the most abundant PUFA in the brain, docosahexaenoic acid (DHA) levels are also lower in AD brains, particularly in regions affected by AD such as the hippocampus [34,35]. Importantly, DHA levels are positively correlated with cognitive performance. Corresponding to the declines in unsaturated fatty acids and the unsaturation index, even-chain saturated fatty acids are increased in the CSF of AD patients [31].
While ω-3 fatty acids are believed to be anti-inflammatory, arachidonic acid (AA) of the ω-6 fatty acid family is largely pro-inflammatory, and the levels of free AA increase in AD brains. Such an increase is associated with the decrease in its precursor linoleic acid (LA), and a pro-inflammatory- and pro-oxidative state in the CSF [35]. Corresponding to the increase in free AA, the levels of AA in phospholipids are reduced in the hippocampus of AD subjects [36]. AA can be produced from phospholipids by the hydrolysis at the sn-2 acyl bond by phospholipase A2 (PLA2) [37]. Consistent with the shift in AA pool from being phospholipid-bound towards the free form, total PLA2 activity, as well as the expression of the Ca2+-dependent cytosolic PLA2 (cPLA2) are higher in the cortex and CSF of AD brains [38,39].
Lipid rafts are cholesterol- and sphingolipid-enriched membrane structure, where most of the proteins involved in synaptic transmission and the amyloidogenic secretases are located [40,41]. Consistent with changes at tissue level, the fatty acid profiles of lipid rafts in AD brains are characterized by lower ω-3 PUFA and MUFA (primarily oleic acid), and a reduced unsaturation index for phospholipid acyl chains in the cortex [42,43]. Of note, these changes also manifest in the earliest stages of AD in entorhinal cortex and frontal cortex [44]. These data suggest that lipid rafts are potentially in the center of altered lipid homeostasis in early stages of Alzheimer’s brains.
Glycerophospholipids and sphingolipids
The two most abundant lipid classes in the brain are glycerophospholipids and sphingolipids. They are both amphiphilic lipids with two hydrophobic fatty acyl chains and a hydrophilic head attached to an alcohol-based backbone. Glycerophospholipids represent the major form of phospholipids in cell membranes. For each glycerophospholipid molecule, two fatty acyl tails and one phosphate polar head are attached to a glycerol backbone. Across lipidomic studies with postmortem brains of MCI and AD subjects, decreased levels of glycerophospholipid contents including phosphatidylcholine (PC) [45,46], phosphatidylinositol (PI) [47,48], and phosphatidylethanolamines (PE) [45,46] are detected in multiple brain regions, primarily those vulnerable to AD pathology such as the hippocampus and the cortex.
Sphingolipids, including ceramide, sphingomyelin, and glycosphingolipid, are a lipid class containing a long-chain sphingoid base backbone (primarily sphingosine that is de novo synthesized from serine and palmitoyl-CoA). Ceramide is the key molecule in the synthesis, recycling, and degradation of other sphingolipids. Sphingomyelin consist of a phosphatidylcholine group attached to a ceramide and can also be classified as phospholipid (sphingophospholipid). An elevation in ceramide levels occurs early in AD brains, including the frontal and temporal cortices [49–51], which is coupled with a reduction in sphingomyelin levels [52,53]. It should be noted that sphingomyelin changes in AD are disease stage- and brain region-specific [54,55]. Gene and protein expression studies further suggest that changes in sphingomyelin and ceramide are associated with upregulated levels of genes involved in ceramide de novo synthesis and sphingomyelin degradation and downregulated expression of genes for sphingomyelin synthesis [52,56]. As increased ceramide levels promote lipid peroxidation, oxidative stress, mitochondrial dysfunction, and neuronal death [57,58], these findings collectively suggest a critical role of dysregulated sphingolipid metabolism in early stages of AD.
Sulfatides represent another class of ceramide-derived lipids that are enriched in myelin. Similar to sphingomyelins, sulfatide levels in both the gray matter and the white matter decline at prodromal and early stages of AD [51,59]. Galactosylceramide, another class of lipids found in myelin, is also depleted in the hippocampus of early-stage AD brains proceeding NFT pathology [60]. The early loss of sulfatide and galactosylceramide in AD can be metabolically traced to the loss of their common biosynthetic precursor, very long chain ceramide, and the reduced activity of the enzyme catalyzing its synthesis, ceramide synthase 2 [60]. Functionally, reductions in these myelin-specific lipids are associated with myelin degeneration and loss of white matter integrity in the AD brains (discussed in section “Myelin degeneration and white matter integrity” below).
Glycerolipids
Glycerolipids are mono-, di-, or tri-esterified glycerol, corresponding to mono-, di-, and tri-acylglycerol (MAG, DAG, and TAG), respectively. They share the same glycerol backbone with glycerophospholipids but have variable numbers of acyl groups and do not carry a phosphate group. While TAG levels are not changed in AD brains, both MAGs and DAGs are elevated in the frontal cortex of MCI and AD brains [61–63], suggesting disrupted glycerolipid metabolism as an early event in AD. Consistently, lipid droplets (LDs), the organelles that serve as a storage of neutral lipids [64], accumulate in AD [65]. The expression of perilipin-2 (Plin2, a specific surface marker of LD), but not other perilipin family members, is selectively upregulated in the frontal cortex [66]. Increased neutral lipids and LDs in post-mortem AD brains, AD patient-derived fibroblasts [67], and peripheral blood mononuclear cells [68] collectively signify disrupted neutral lipid metabolism in AD (see reviews [69,70]).
Cholesterol and cholesteryl esters
With its 2% of total body weight, the brain contains 25% of the total cholesterol [71]. Because of the blood-brain barrier (BBB), the majority of brain cholesterol is de novo synthesized in the central nervous system (CNS) [72]. Most studies suggest that the levels of both circulating and brain cholesterol are elevated in AD patients than in control cases [50,73–75], although unchanged brain cholesterol levels are observed in some other reports [76,77], which could be related to different brain regions and disease stages across these studies. Notably, increased cholesterol is also seen in the cores of senile plaques in human brain [78], and brain cholesterol levels are positively correlated with the severity of AD [50]. Under physiological conditions, excess cellular cholesterol is esterified to CEs, a form of neutral lipids stored in LDs together with TAG. Cholesterol esterification is catalyzed by acyl CoA:cholesterol acyltransferase 1 (ACAT1, also known as sterol O-acyltransferase 1) [79]. In the entorhinal cortex of human AD brains, total CE levels are elevated by ~ 1.8 fold, which is resembled in AD mouse models [62,80].
Collectively, across various lipid classes discussed above, it can bse summarized that alterations in lipid raft fatty acid compositions, ceramide, sphingomyelin, sulfatide, and glycerolipid (MAG and DAG) temporally emerge in early stages of AD and/or spatially locate to brain regions that are affected by pathology at first. These lines of evidence support an early, and potentially initiating, role of lipid metabolism in AD.
Lipid-related risk factors for AD
The critical, and potentially initiating, role of lipid metabolism in AD is further supported by the intimate relationships between key AD risk factors and various aspects of lipid metabolism (Fig. 2). Such factors include both genetic risk factors identified from genome-wide association studies (GWAS) and environmental or lifestyle risks discovered from epidemiological studies [3].
Fig. 2.
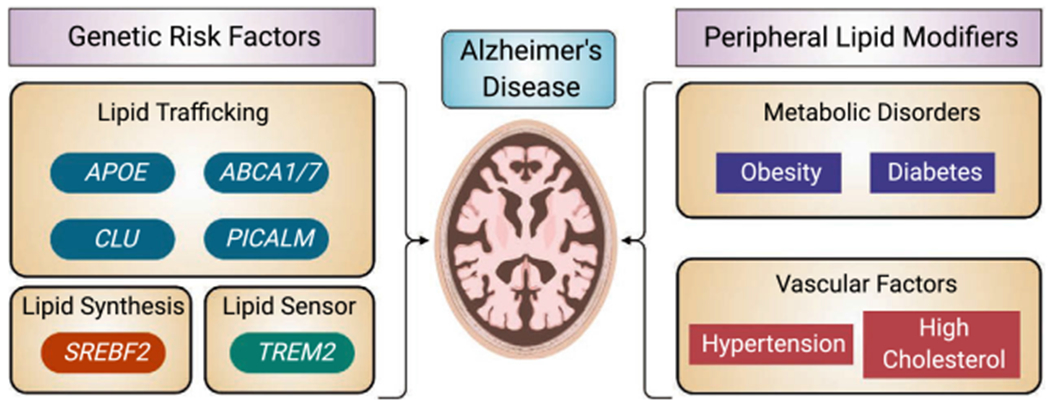
Lipid-related risk factors in AD. A variety of AD genetic risk factors are involved in different aspects of lipid metabolism including lipid trafficking, lipid synthesis and lipid signaling. Peripheral lipid modifiers including metabolic and vascular risk factors also alter AD risks.
Genetic risk factors
Among the top risk genes for the sporadic LOAD, APOE, TREM2, APOJ, PICALM, ABCA1, and ABCA7 are all directly involved in lipid trafficking or metabolism [81] (Fig. 2). A key regulator of cholesterol metabolism, SREBP-2 is also genetically associated with altered AD risk [82]. While this section focuses on risk factors for the predominant sporadic AD (LOAD), causal genes for fAD (APP, PSEN1 and PSEN2) also modulate lipid metabolism, which will be discussed in detail in section “Amyloid metabolism” below.
Apolipoprotein E
The human APOE gene encodes the 34KD ApoE protein, which is best known as a lipid-binding protein responsible for transporting lipids across organs in the periphery, and between cells in the brain [83]. Compared with the most common ε3 isoform, the ε4 isoform of ApoE (ApoE4) is the strongest genetic risk factor for LOAD [84,85] whereas the ε2 isoform (ApoE2) significantly reduces the risk [86,87]. Each ApoE4 allele increases AD risk by 3 ~ 4-fold, while lowers the age at onset by ~ 8 years [88–90]. Beyond its role in regulating Aβ production and clearance, the pathological effect of ApoE4 is also driven by lipid-centric mechanisms [91]. Consistent with its loss-of-function as an intercellular lipid carrier relative to ApoE3, ApoE4 carriers have higher plasma levels of total cholesterol and TAG but reduced high-density lipoprotein (HDL)-cholesterol; in contrast, plasma levels of total cholesterol and HDL-cholesterol in ApoE2 carriers are lower and higher, respectively [92,93]. Moreover, the link between ApoE4 and disrupted lipid metabolism is supported by studies showing that ApoE4 enhances the activity of the AA-producing cPLA2 [94], whereas ApoE4-related pathologies can be alleviated by DHA-rich diet but exacerbated by high-cholesterol diet [95].
TREM2
Triggering receptor expressed on myeloid cells 2 (TREM2), primarily expressed on microglial surfaces, mediates phagocytosis and inflammatory response by these resident myeloid cells in the brain [96,97]. TREM2 loss-of-function mutations cause an early-onset dementia called Nasu-Hakola disease, which is characterized by severe myelin loss and neurodegeneration [98,99]. A rare variant of TREM2 (R47H) is associated with a significant increase in AD risk [100,101]. Beyond its interaction with Aβ, TREM2 has been identified as a sensor of phospholipids, lipoproteins and apolipoproteins [102–104].
Clusterin
Clusterin (ApoJ, gene symbol CLU) is identified as an AD risk gene based on the association between multiple single nucleotide polymorphisms (SNPs) at the CLU locus and altered disease risk [105,106]. Beyond its role in the folding of secreted proteins as a molecular chaperone and its effect on Aβ aggregation, clusterin is also involved in lipid transport and metabolism in both the brain and the periphery [107].
PICALM
As another major AD risk gene [105], phosphatidylinositol binding clathrin assembly protein (PICALM) is involved in clathrin-mediated endocytosis, which is essential in the internalization and transport of not only proteins but also lipids in lipoprotein particles [108]. Interestingly, the Aβ clearance role of PICALM is mediated by its binding to the low-density lipoprotein receptor related protein 1 (LRP1) [109].
ABCA1 and ABCA7
Both ABCA1 and ABCA7 (ATP-binding cassette subfamily A member 1 and 7) are members of the ABC transporter family, sharing 54% sequence identity [110]. ABCA1 initiates the efflux of lipids such as cholesterol and phospholipids by loading them to lipid-free lipoproteins, with ApoE being the primary substrate for such lipidation in the brain [111,112]. A loss-of-function mutation in ABCA1 is associated with low plasma levels of ApoE and higher AD risk [111]. ABCA7 is also involved in the transport of cholesterol and phospholipids [113]. Human-based genetic and epigenetic studies have identified multiple SNPs, variants, alternative splicings, and methylations of ABCA7 gene that contribute to its loss-of-function, altered lipid- and Aβ metabolism and increased AD risks [110,114].
SREBP-2
Sterol regulatory-element binding proteins (SREBPs) are a family of transcriptional factors that regulate the biosynthesis of lipids [115]. As the master regulator of cholesterol synthesis, SREBP-2 is found connected with AD risk and progression. SREBF2 (gene encoding SREBP-2) polymorphism (rs2269657) is associated with LOAD biomarkers and gene expression, and brain SREBF2 mRNA levels are negatively correlated with the age at death [82]. SREBP-2 could thus be a potential risk factor for AD (also see section “Fatty acid and cholesterol synthesis” below).
Metabolic and vascular factors
The essential role of lipid homeostasis in the etiology of AD is also supported by the modification of AD risks by multiple lipid-related factors—either positively or negatively.
It has been established epidemiologically that metabolic disorders such as obesity and type 2 diabetes (T2D) are associated with increased risk for AD (Fig. 2), and diets enriched in saturated fatty acids are linked to learning and memory deficits [116–121]. Midlife, but not late-life overweight or obesity is associated with increased AD risk and an earlier onset [122,123]. Multiple studies show that higher systemic insulin resistance in AD patients is positively correlated with Aβ deposition in brain [124,125]. Interestingly, in a study conducted in non-dementia subjects, midlife, but not late-life systemic insulin resistance is associated with greater brain Aβ burdens 15 years later [126]. Notably, recent neuropathological studies suggest that the association between T2D and AD pathology (Aβ deposits and NFTs) is not significant [127,128]. Despite the lack of evidence supporting a direct effect of T2D on AD pathogenesis, it has been suggested that metabolic mechanisms that are commonly affected in both disorders, such as dyslipidemia, oxidative stress, and cerebrovascular function, mediate their epidemiological connection [127,128].
Acylcarnitines are intermediates of fatty acid metabolism during fatty acid transport into the mitochondria by the carnitine-carrier system. Two pilot studies suggest that serum levels of acetyl-l-carnitine and other acyl-carnitines decrease along the continuum from cognitively normal to MCI, and to AD [129,130]. These studies suggest a decreased systemic fatty acid catabolism in AD that may indicate altered fatty acid degradation in the brain. Interestingly, the decline in circulating acyl-carnitines is also associated with lower levels of ketone bodies [130], which are the major alternative substrate that the brain can utilize upon glucose insufficiency or hypometabolism. These findings connecting midlife alterations in systemic lipid metabolism with late life AD risk collectively underline an early role of abnormalities in lipid metabolism in modifying AD susceptibility.
While the cholesterol pool of the CNS is separate from its systemic counterpart, high circulating levels of cholesterol, familial hypercholesterolemia, and low circulating HDL-cholesterol are known risk factors for AD [131–133] (Fig. 2). High midlife cholesterol levels increase the risk of AD by 2.8-fold [134]. Persistent midlife hypertension is another significant risk factor for late life incidence of both all-cause dementia and AD [135,136] independent of cardiovascular diseases [137]. In contrast, people with a normal cardiovascular profile have a lower 10-year risk of all-cause dementia and of AD [138]. In a separate cohort, a comprehensive cardiovascular health score considering both metabolic and vascular factors in midlife (diet, body mass index, physical activity, fasting glucose, blood pressure, and blood cholesterol) is associated with a lower risk of dementia later in life [139], although this study did not separate AD from other dementia etiologies (such as vascular dementia).
These findings, along with the alterations in lipid species occurring early in the brain, consistently suggest that both central- and peripheral lipid dysregulation play an important role in the initial stages of AD.
Deciphering lipid metabolism in AD: mechanistic links
Lipid homeostasis in AD - the dynamics of lipid synthesis, transport and degradation
Because of their essential roles, the levels of lipid species are finely regulated for their homeostasis and functionality in the brain. In this section, key players and mechanisms involved in the regulation of lipid homeostasis will be discussed in the context of AD. Given that fatty acids are the essential building blocks for nearly all lipid species and excess fatty acids are associated with lipotoxicity to both neuronal and non-neuronal cells via multiple mechanisms [140–144] (reviewed in [145]), the discussion will be centered on the anabolic and catabolic metabolisms and the transport of fatty acids, with connections to the overall lipid profile in the brain (Fig. 3).
Fig. 3.
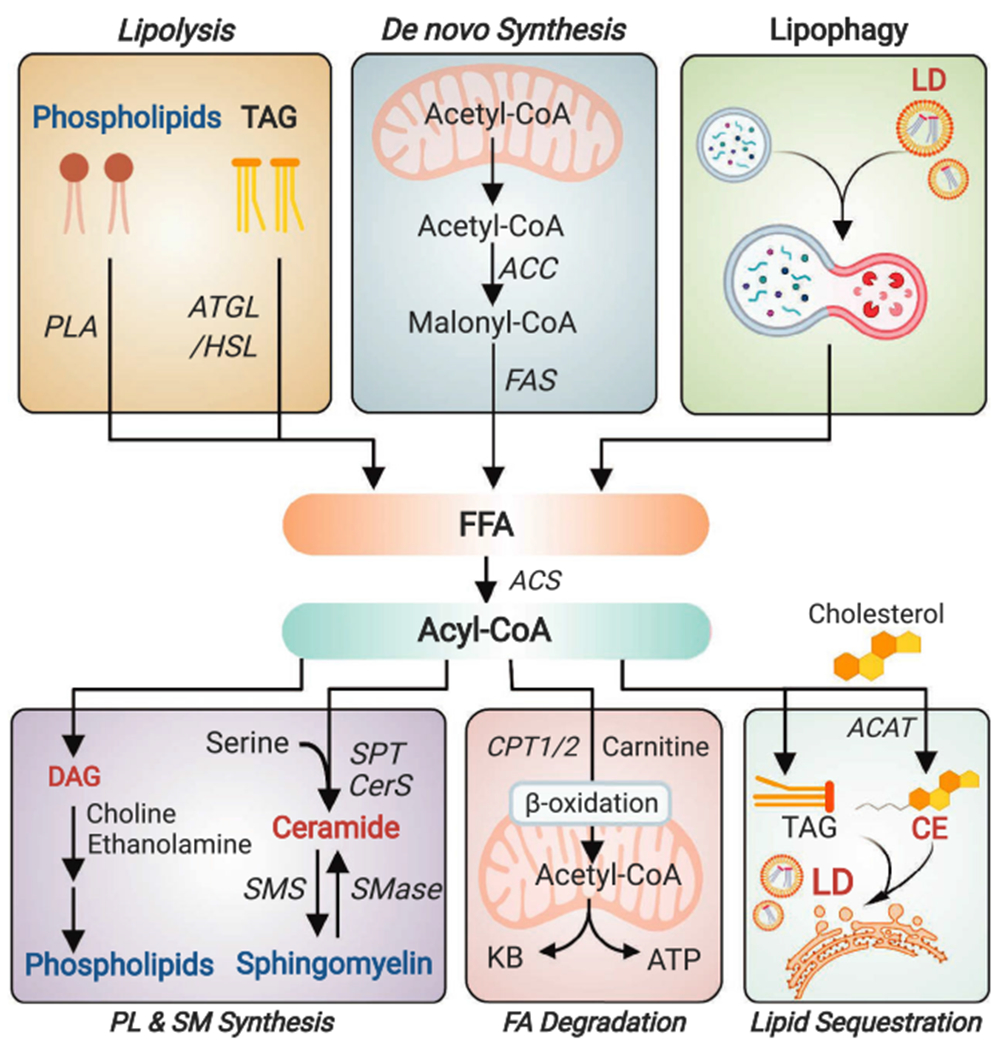
Fatty acids in the center stage of the dynamic regulation of brain lipid homeostasis. Free fatty acids can be generated from: 1) lipolysis of phospholipids by PLA and TAG by adipose triglyceride lipase (ATGL) or hormone-sensitive lipase (HSL); 2) de novo synthesis from acetyl-CoA involving key enzymes ACC and fatty acid synthase; or 3) autophagic degradation of lipophagy. After being converted to acyl-CoA by various forms of ACS, fatty acids can be: 1) used to synthesize phospholipid, ceramide and sphingomyelin (SM); 2) transported into the mitochondria by carnitine palmitoyltransferase 1/2 (CPT1/2) for β-oxidation, which further generates acetyl-CoA for ATP production or ketogenesis (primarily occurs in astrocytes); or 3) sequestered in LDs in the forms of TAG or cholesterol ester (CE; acyl chain attached to cholesterol by ACAT). DAG, diacylglycerol; SMS, sphingomyelin synthase; SMase, sphingomyelinase; KB, ketone body; ACAT, acyl CoA:cholesterol acyltransferase.
Fatty acid and cholesterol biosynthesis
While short- and medium-chain fatty acids are transported from the periphery, long- and very-long-chain fatty acids are primarily de novo synthesized in the brain from acetyl-CoA [146]. Additionally, brain fatty acids can be generated by phospholipase A2 (PLA2)-catalyzed phospholipid hydrolysis [147], and potentially by lipophagy [148] (Fig. 3). In the brain of a fAD mouse model overexpressing mutant APP and PSEN1 (APP/PS1), increased palmitic acid (C16) levels are accompanied by upregulated protein levels of fatty acid synthase (FAS) [149]. In human AD cortex, FAS protein levels are also elevated, especially at areas around the plaques [149,150]. Consistently, another key enzyme controlling fatty acid synthesis, acetyl-CoA carboxylase (ACC), is activated in fAD mouse brain as suggested by a decrease in its inhibitory phosphorylation [151].
Uncontrolled cholesterol synthesis in brain is also associated with worsened AD-related phenotypes. Increased cholesterol synthesis by overexpressing SREBP-2 in APP/PS1 mice accelerates oxidative damage, amyloid accumulation, neuroinflammation, and cognitive decline, and remarkably, it induces tau hyperphosphorylation and NFT formation in the absence of tau transgene or mutation [152]. Conversely, astrocyte-specific (astrocytes are the major cholesterol-synthesizing cells in the brain) SREBF2 knockout dramatically reduces both Aβ- and tau pathologies [153]. A recent study suggests that tau, in the opposite direction, alters the distribution and signaling of SREBPs in AD [154]. Overall, these findings validate the detrimental effect of SREBP-2 on AD mortality in humans [82].
Fatty acid oxidative degradation
The oxidative degradation of free fatty acids via β-oxidation is primarily in the domain of mitochondria, and secondarily, peroxisomes. While the role of fatty acids, as fuel substrates, in meeting the energetic demand of the brain remains unclear [145,155], a timely degradation of free fatty acids, especially those peroxidized ones, is critical for brain health because of their negative effect on cellular function [29]. Compared with the oxidative degradation of glucose, fatty acid β-oxidation by mitochondria is associated with higher production of reactive oxygen species (ROS). Correspondingly, neurons, which have high metabolic rate but limited ROS-detoxification capacity [156], possess low β-oxidation activity.
Astrocytes are a highly heterogeneous population of neural cells and represent the primary brain cell type degrading fatty acids [141,157–159] (Fig. 3). Relative to neurons, the metabolic role of astrocytes in AD and neurodegeneration has just started to be revealed. In the context of AD, a recent study by our group suggests that compromised astrocytic degradation of fatty acids could constitute another potential mechanism underlying lipid dysregulation in brain [160]. In astrocytes, human ApoE4 knockin induces a metabolic shift towards enhanced glucose metabolism and reduced fatty acid β-oxidation, which subsequently elicits lipid accumulation in astrocytes. Consistently, this metabolic shift in ApoE4 astrocytes is accompanied by an increase in fragmented mitochondria [160], which are less capable of distributing and oxidizing fatty acid than fused ones [161]. ApoE4-induced astrocytic lipid accumulation is also seen in immortalized cells [162] and human iPSC-derived cells [163]. Further, our group developed an ex vivo brain fatty acid β-oxidation assay with acute brain slices that enables metabolic assessment of brain lipid catabolism [164]. Using this method, a reduced fatty acid degradation and increased TAG accumulation in the hippocampus of young ApoE4 knockin mice is observed, confirming the unique role of astrocytes in brain fatty acid degradation. These findings are consistent with a previous report in Drosophila that loss of a mitochondrial fatty acid importer (carnitine palmitoyltransferase 2; CPT2) leads to LD accumulation in the brain [165]. In line with reduced fatty acid oxidation capacity associated with ApoE4 and potentially AD, the mRNA levels of peroxisome proliferator-activated receptor α (PPARα), the nuclear receptor promoting fatty acid degradation, is significantly reduced in AD brains [166].
Beside astrocytes, microglia also express enzymes for β-oxidation [167], and a growing body of literature suggest fatty acid metabolism as a determinant of microglial phenotype. Evidence in macrophages suggest that their inflammatory response and polarization are modulated by fatty acid oxidation [168]. Inhibition of macrophage β-oxidation exacerbates palmitate-induced inflammatory activation, whereas enhancement of fatty acid oxidation reduces TAG accumulation and lipid-induced inflammation [169,170]. In microglia, mRNA levels of fatty acid oxidation genes are repressed by pro-inflammatory activation by lipopolysaccharides (LPS) or interferon-γ (IFN-γ) [171]. Consistently, interventions that activate mitochondrial function and facilitate microglial metabolic reprogramming alleviate neuroinflammation and promote Aβ clearance [172–175].
The peroxisome is another organelle that performs β-oxidation, specifically for very-long-chain fatty acids (VLCFAs; >= 22 carbons) [176]. Evidence for the impairment of peroxisomal function in advanced AD stages include the accumulation of VLCFA in AD brains with Braak stages V-VI, the increased peroxisomal volume in the soma of neurons, and a loss of peroxisomes in neuronal processes with hyperphosphorylated tau [177]. Intriguingly, this study suggests that peroxisomal dysfunction has a stronger association with NFT than with neurite plaques. Studies also suggest the role of glial peroxisomes in AD-relevant brain phenotypes such as demyelination and neuroinflammation. In oligodendrocytes, a loss of functional peroxisomes causes demyelination, axonal degeneration, and neuroinflammation, which is coupled with an accumulation of VLCFAs, and these neurotoxic VLCFAs further lead to neurodegenerative phenotypes relevant to AD [176]. Likewise, peroxisomal dysfunction of β-oxidation in microglia caused by the loss of multi-functional protein-2 (MFP2) elicits a profound pro-inflammatory response [178].
Fatty acid and cholesterol sequestration in lipid droplets (LDs)
LDs are one of the first layers of protection against free fatty acid - or cholesterol-related lipotoxicity by sequestering them as the neutral TAGs and CEs [64] (Fig. 3). LDs are detected in multiple cell types in brain including astrocyte, microglia, ependymal cell and neuron across aging-, AD-, and other neurodegenerative brains (reviewed in [69]). While the exact role of LD in AD pathogenesis remains to be determined, elevated LD accumulation in AD is consistent with the function of these organelles as sequesters of peroxidized fatty acids as well as unmodified PUFAs that are prone to peroxidation [69]. In a fAD mouse model overexpressing mutant APP, PSEN1, and MAPT (3xTG), LD accumulation in ependymal cells of the subventricular zone proceeds amyloid and tau pathology and suppresses neural stem cell proliferation [179]. Further, LD-accumulating microglia have pro-inflammatory and defective phagocytosis phenotype and possess transcriptional signatures overlapping with that of AD microglia [180]. In cultured neurons, ApoE4 induces suppressed fatty acid sequestering in LDs, which leads to elevated free fatty acid levels, disrupted bioenergetics, and synaptic dysfunction [160]. Moreover, CEs are upstream regulators of both Aβ and tau pathologies during early AD development [181].
It should be noted that while LDs are increased in aging and neurodegenerative diseases, they serve as a protective and adaptive mechanism to control lipid-mediated neurotoxicity by buffering excess free fatty acid and cholesterol, particularly during the initial stages of these lipid-implicated abnormalities. This notion is supported by the finding that blockage of glial LD accumulation causes more severe neurodegeneration [182,183].
Intercellular lipid transport
Fatty acid transport
Because of their low fatty acid degradation capacity, neurons rely on glia, primarily astrocytes, to clear and degrade excessive free fatty acids and neutral lipids in LDs [141,160,182,183]. Upon oxidative stress or neuronal stimulation, peroxidized fatty acids are transported from neurons to astrocyte, via an ApoE-dependent mechanism [141,182] (Fig. 4). Our recent study suggests that neuron-to-astrocyte transport of lipids is impaired by ApoE4 [160]. Lipids within neuronal LDs are eliminated upon astrocyte exposure and transferred to astrocytes, with ApoE4 in either neurons or astrocytes diminishing the transport efficiency. Further, we demonstrate that the clearance of neuronal LDs is integral to the metabolic and neurotrophic support provided by astrocytes: when the export of neuronal LD is inhibited, the metabolic enhancement of neurons by astrocyte is abrogated [160]. Notably, this study also suggests a differential role of supplemental ApoE3 versus ApoE4 in regulating the clearance of neuronal lipids. Supplemental recombinant ApoE3, but not recombinant ApoE4, promotes astrocyte-induced clearance of neuronal LDs, which suggests ApoE3 as an enhancer, while ApoE4 as a negative regulator, of neuronal lipid clearance [160].
Fig. 4.
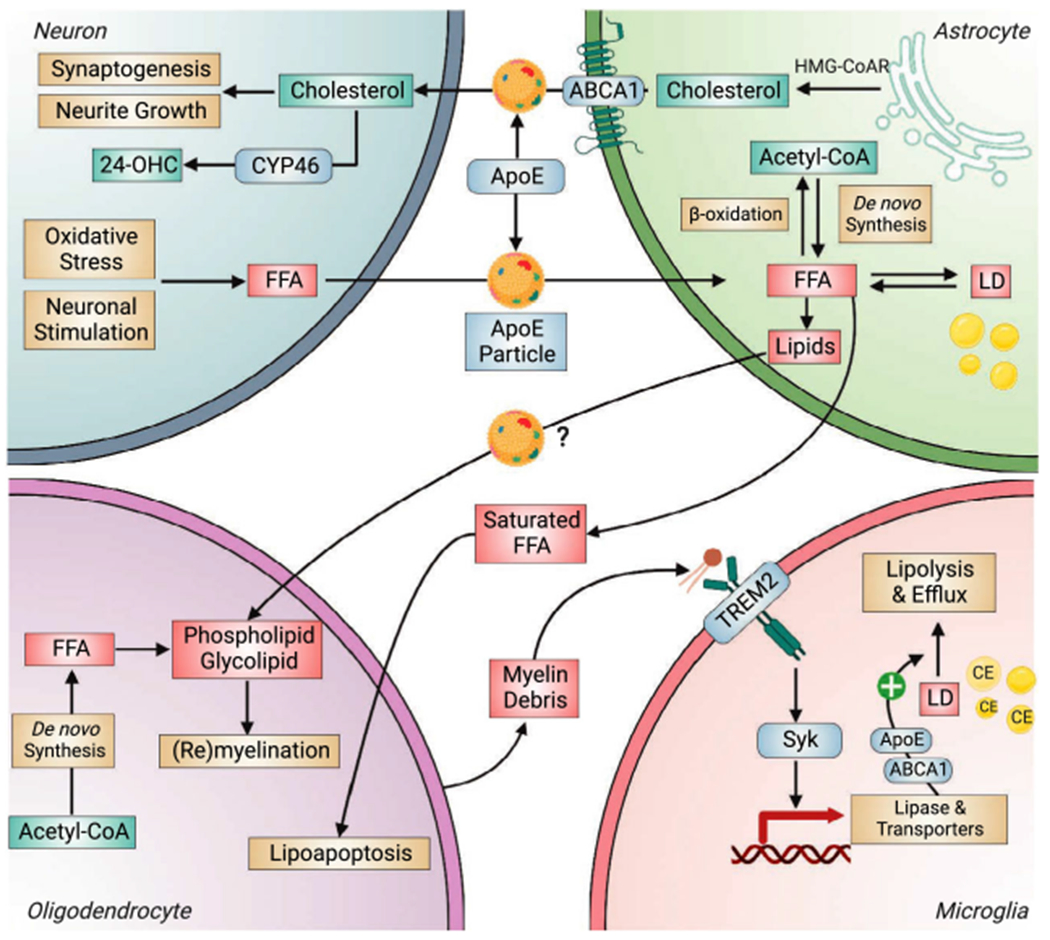
Intercellular lipid transport in the brain. Cholesterol synthesized in astrocyte by HMG-CoA reductase (HMG-CoAR) is packaged and exported in ApoE-containing lipoproteins (ApoE-particles) by ATP-binding cassette transporter A1 (ABCA1) and subsequently transported to neuron for neurite growth and synaptogenesis or for conversion to 24-hydroxycholesterol (24-OHC) by CYP46 (cholesterol 24-hydroxylase). Neurons, upon stimulation or oxidative stress, release fatty acids in ApoE-particles to astrocytes for degradation by mitochondrial β-oxidation, or for storage in LDs. fatty acids and other lipids required for oligodendrocyte-mediated myelination or remyelination are synthesized by both oligodendrocyte and astrocyte, and ApoE particles are potentially involved in astrocyte-to-oligodendrocyte lipid transport. Excessive astrocyte-derived saturated fatty acids induce oligodendrocyte death via lipoapoptosis pathway. Lipids from myelin debris can bind to and activate TREM2 signaling to induce the expression of lipid metabolism genes to facilitate the lipolysis of CE-rich LDs and efflux of lipids. Syk, spleen tyrosine kinase.
In postmortem human brains, increased LDs in the choroid plexus are significantly correlated with worsened cognitive function and AD neuropathology including neuroinflammation [184]. Further, such a correlation resembles the phenotypes seen in ApoE knockout mice or hyperlipidemic ApoE4 mice (on high-fat diet). This study further supports the connection between ApoE regulation of brain LD metabolism and AD pathogenesis. In addition to ApoE, other AD risk factor genes such as ABCA1, ABCA7, and PICALM are also involved in the formation of glial LDs [185]. Collectively, these studies suggest that impairment in lipid clearance and transport could underlie lipid dyshomeostasis and reduced neurotrophic function observed in aging and AD brains.
Cholesterol transport
Cholesterol in brain is essential for neuronal function due to their roles in membrane fluidity, vesicle formation, and synaptic transmission [186]. Different from fatty acid transport, intercellular transport of cholesterol is primarily from glia to neurons (Fig. 4). This mechanism is important for synaptic function and sterol homeostasis because: (a) the cholesterol required for synaptogenesis in neurons is transported from astrocytes because of the suppression of cholesterol biosynthetic pathway in neuron [187] and (b) cholesterol needs to be hydroxylated to 24-hydroxycholesterol in neurons before excretion from the brain [188,189]. When cholesterol synthesis in astrocytes is suppressed by conditional SREBP-2 knockout, synaptic, behavioral and motor functions are impaired [190]
ApoE is the primary cholesterol transporter in the brain by facilitating its efflux from astrocytes and its uptake by neurons [191,192]. Upon ApoE knockout, cholesterol biosynthesis and hence its levels are reduced in the brain [193], and these mice develop age-associated cognitive deficits [194]. Further, the depletion of LRP1, a major neuronal receptor for ApoE-containing lipoproteins, induces lipid dysregulation, neuroinflammation and synapse loss [195]. In addition to cholesterol, astrocyte-to-neuron ApoE particles are found to carry miRNAs to inhibit neuronal cholesterol biosynthesis, which subsequently elevates neuronal levels of acetyl-CoA (substrate for cholesterol synthesis) and epigenetically regulates the expression of neuronal genes for memory consolidation [196]. With regard to the effect of ApoE genotype on these processes, ApoE4 is consistently shown to be associated with reduced capacity in binding, secreting, and transporting cholesterol and other lipids [197,198]. Together, these studies suggest that disrupted intercellular exchanges of fatty acids and cholesterol contribute to lipid dyshomeostasis in AD, and furthermore, they unanimously highlight the detrimental effect of ApoE4 in these mechanisms.
Lipid interaction with AD pathogenic mechanisms
Amyloid metabolism
Aβ peptides are generated from the sequential cleavage of APP by β- and γ-secretases [199]. Such a process, also known as the amyloidogenic pathway, is believed to occur within membrane microdomains (lipid rafts) [200,201], which are enriched in cholesterol and sphingolipids [202]. It is thus not surprising that cholesterol and sphingolipids affect amyloidogenesis (Fig. 5). Further, multiple surface receptors of the low-density lipoprotein receptor family, such as LRP1, LRP1B, SORL1, and ApoER2 have been connected with APP metabolism, mostly involving their role in endocytic trafficking (reviewed in [203]).
Fig. 5.
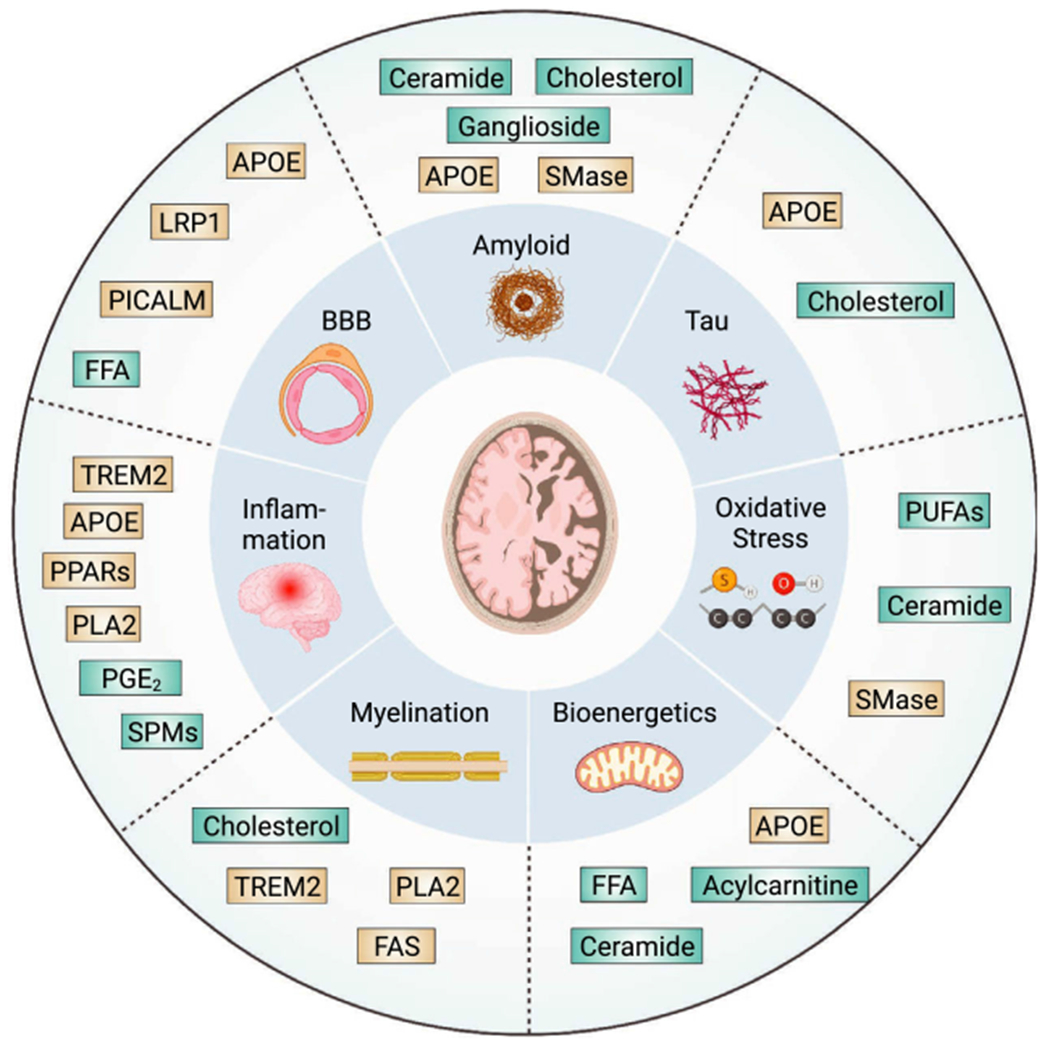
Interactions between lipid metabolism and AD pathogenic mechanisms. Lipid-related factors could contribute to AD pathogenesis via interactions with key mechanisms involved in AD etiology. Inner circle, key AD mechanisms; outer circle, lipid factors affecting each mechanism. Boxes in cyan are lipid species, and boxes in gold represent lipid regulators.
Cholesterol
A large volume of studies suggests that amyloidogenic pathway is regulated by the levels of cholesterol and CE [17,204]. The activity of both β- and γ-secretases is enhanced by high cholesterol and inhibited by low cholesterol [203]. The mechanism of cholesterol on Aβ production is recently furthered by superresolution imaging [153]. It is shown that suppressed cholesterol synthesis in astrocytes substantially reduces amyloid and tau pathology in a mouse model of AD. This effect is mediated by decreased cholesterol in neurons, which facilitate APP to traffic out of lipid rafts, where β- and γ-secretases are localized. Outside of the lipid rafts, APP is instead converted to the neuroprotective [205] and nonamyloidogenic soluble APP-α (sAPP-α) by α-secretase [153,206].
Sphingolipid
In the case of sphingolipid, ceramide is proposed as a key contributor to AD pathophysiology by affecting the production of Aβ and hyperphosphorylation of tau. When sphingomyelinase, the enzyme hydrolyzing sphingomyelin to ceramide, is inhibited, γ-secretase activity and Aβ secretion is reduced [207]. This finding is likely related to the effect of ceramide promoting amyloid biogenesis in lipid rafts by stabilizing β-secretase [208].
The intimate connection between Aβ generation and cholesterol- and sphingolipid metabolism is further manifested by the regulation in the reverse direction, where Aβ42 and Aβ40 reduces sphingomyelin and cholesterol levels by activating neutral sphingomyelinases and by inhibiting hydroxymethylglutaryl-CoA reductase activity, respectively [207]. Likewise, fibrillar Aβ induces sphingomyelinase activity and the subsequent ceramide production via ROS-dependent mechanisms [207,209,210] (also see the Oxidative Stress section below). These mechanisms are consistent with the reduction in sphingomyelin levels in human AD brains [52]. Thus, the bidirectional effect between Aβ production and ceramide levels could form a vicious cycle that exacerbate initial ceramide accumulation in AD brains [211]. Upstream of Aβ peptides, APP and γ-secretase are also important regulators of lipid metabolism via modulating the synthesis and turnover of cholesterol, the accumulation of acylglycerols, and the homeostasis of lipoproteins [212–215].
Mitochondria-associated membranes
Mitochondria are the powerhouses of the cell and play fundamental roles in processes such as redox homeostasis, apoptosis, steroid biosynthesis, calcium homeostasis, and cell fate regulation [216,217]. Mitochondria perform these functions by forming a dynamic network with close interactions with other components in the cell including the endoplasmic reticulum (ER) [218–220]. Intraneuronally, lipid metabolism and Aβ metabolism converge at the mitochondria-associated membranes (MAMs), a unique intracellular interface between the mitochondria and the ER [221]. MAMs are enriched in cholesterol, sphingomyelin, and proteins involved in phospholipid exchange, calcium signaling, and redox regulation. Structurally, MAMs are similar to lipid rafts [221]. Accumulating evidence has established the essential role of MAMs in lipid synthesis and trafficking [41,222]. MAM regulation of Aβ production is suggested by the enriched β- and γ-secretases and elevated β-secretase cleaved product of APP (C99) localized to MAMs [223,224]. Intriguingly, this mechanism is connected to reduced mitochondrial function, via a cascade initiated from increased C99 to enhanced sphingomyelin hydrolysis by sphingomyelinases and to increased ceramides [223,225]. Besides C99, Aβ peptides are also known to directly modulate the sphingomyelinase-ceramide pathway [210,226].
Conversely, fatty acids could directly affect axonal Aβ generation by regulating palmitoylation and enhancing MAM activity [227]. Palmitoylated APP is enriched in lipid rafts including MAMs, where it serves as a preferred substrate towards β-secretase-mediated cleavage and amyloidogenesis; upon inhibition of ACAT, an enzyme that transfers fatty acyl groups, the levels of both palmitoylated APP and Aβ are substantially reduced [228]. These findings are consistent with the upregulated MAM activity, disrupted phospholipid synthesis, and accumulated neutral lipids in AD- and ApoE40-positive human cells [229,230].
Gangliosides
Gangliosides are a group of complex glycosphingolipids with one or more sialic acids connected to the sugar chain and are abundant in the rafts of plasma and organelle membranes [231]. GM1, a ganglioside that contains one sialic acid residue is known to act as a seed for Aβ binding (by forming GM1-bound Aβ) and aggregation, leading to the formation of amyloid fibrils [232]. In this process, GM1 clusters are formed because of disrupted endosomal function or lipid metabolism, which interact with Aβ and lead to the sequential formation of preamyloid β-sheet-rich oligomers and tape-like amyloid fibrils [233].
ApoE
ApoE is involved in almost all aspects of Aβ pathology, from its production, aggregation, and deposition, to its degradation and clearance (reviewed in [234]). Functionally, ApoE isoforms differentially affect the size, composition, and extent of lipidation of ApoE particles. Compared with ApoE3 group, ApoE in ApoE4 carriers is less lipidated while ApoE in ApoE2 carriers is more lipidated [235,236]. Such isoform-dependent lipidation status also pertains to ApoE particles in the CSF: those in E2/E3 subjects have larger sizes, whereas those in E3/E4 subjects have smaller sizes, relative to E3/E3 controls [237]. Further, the smaller size and lower lipidation rate of ApoE4 particles are associated with reduced receptor binding ability and resultantly, compromised lipid carrying and trafficking capacity [160,198,236].
Compared with cognitively normal individuals, those with MCI have higher CSF levels of lipid-depleted (non-lipidated) Aβ, which is less likely to be cleared by enzymatic degradation, phagocytosis, or BBB transport out of the brain [234,238]. Moreover, the lipidation status of CSF Aβ is modified by ApoE genotype, with ApoE4 carriers exhibiting reduced lipidation of both Aβ and ApoE itself, which could be relevant to the differential stability of ApoE/Aβ complexes [239]. Further, the lipidation status of ApoE and Aβ in CSF is reduced after a 4-week high saturated fat and high glycemic dietary intervention, whereas the same parameters are increased by diet low in saturated fat and glycemic index [238]. This study corroborates the close relationships among AD pathology, ApoE status and lipid profile. Consistent with findings in human CSF, a higher deposition of Aβ in ApoE4 mouse brains is associated with a longer half-life of Aβ [240], highlighting the central role of ApoE in Aβ clearance.
Two competitive bindings among ApoE, lipids, and Aβ have been proposed [234]: (a) Aβ may compete with lipids for ApoE binding, and (b) ApoE may compete with Aβ for their common cell surface receptors. Therefore, it can be speculated that disruptions to lipid metabolism could affect Aβ metabolism, and vice versa. In the case of ApoE4, its reduced lipidation and transport efficiency could affect both Aβ clearance and intercellular lipid exchange and subsequent degradation. Further, the potential competition between accumulated Aβ and uncleared lipids for lipid carriers could further exacerbate the lagged clearance of both species.
Tau
Relative to the intimate and bidirectional relationship between Aβ and lipids, the interaction between tau and lipids is less understood, and existing studies suggest their connections are be primarily indirect – in many instances through Aβ [17]. This is likely related to the fact that tau is a cytosolic microtubule-associated protein, and their phosphorylation and aggregation do not occur at lipid-rich membrane structures. Moreover, as tau pathology is primarily intraneuronal, glia-centered mechanisms of lipid synthesis and catabolism are unlikely to be directly affected by tau. Nevertheless, several studies provide initial evidence that brain cholesterol accumulation and high dietary cholesterol are associated with increased tau phosphorylation and aggregation [241,242] (Fig. 5). Intraneuronally, the lipid-rich ER-mitochondria interaction site (MAMs) is also regulated by tau [243]. Additionally, a significant association between T2D and CSF tau levels is detected in ApoE4 carriers but not in noncarriers [244].
Neuroinflammation
Neuroinflammation is a key element in the pathogenesis and progression of AD, signatured by increased pro-inflammatory cytokines, decreased anti-inflammatory responses, and amplified reactivity of glial cells including microglia and astrocytes [245]. The immunomodulatory properties of lipids, in particular fatty acids and their derivatives, were discovered decades ago. Depending on the class of the lipids and the cell type in which they are located, both pro- and anti-inflammatory responses can be induced [246].
Specialized pro-resolution mediators
Lipid metabolism can be involved in modulating neuroinflammation via a class of lipid-derived signaling molecules called specialized pro-resolution mediators (SPMs). SPMs, including resolvins, lipoxins, protectins, and maresins, are produced from PUFA (primarily ω-3 PUFAs such as DHA and eicosapentaenoic acid (EPA)) and orchestrate the resolution of acute responses by activating cellular events that repress inflammatory processes and restore tissue homeostasis [247] (Fig. 1). This mechanism is used by both the periphery and the CNS to avoid persistent, chronic inflammation. In AD, mounting evidence reveals an impairment of the resolution of inflammation, which contributes to chronic neuroinflammation and exacerbated AD pathology [248]. In human AD brains, multiple SPMs are decreased in AD-affected brain regions compared with controls, whereas the protein expression of SPM receptors is upregulated [249,250]. Importantly, a positive correlation exists between CSF levels of SPMs (LXA4 and RvD1) and cognitive function across control- and AD cases [251]. In AD mouse models, SPM administration not only restores the decline in hippocampal SPM levels but also reduces pro-inflammatory cytokines and glial reactivity and alleviates Aβ and tau pathology [252,253]. Lipid metabolism is also linked with neuroinflammation via the expression of anti-inflammatory genes induced by lipid-sensing nuclear receptors such as liver X receptors (LXRs) and PPARs [254,255].
Prostaglandins
In contrast to the pro-resolution SPMs, PUFAs (primarily AA generated from PLA2-catalyzed phospholipid hydrolysis) are also substrates of pro-inflammatory prostaglandins, particularly prostaglandin E2 (PGE2), through sequential reactions catalyzed by cyclooxygenase-1 (COX-1) and COX-2, and prostaglandin E synthases [256] (Fig. 1). CSF levels of PGE2 is elevated in early-, but not late-, stage of AD [257,258]. In fAD mouse models, deletion of PGE2 receptors alleviates neuroinflammation, oxidative stress, Aβ deposition, and memory deficit via microglia-centered mechanisms [259–261]. PGE2, via its EP2 receptor, promotes microglial glucose sequestration and mitochondrial bioenergetic deficits. By blocking microglial EP2 signaling, cellular energy metabolism, brain inflammatory state, and synaptic function are restored [262]. These findings are consistent with retrospective studies supporting the effect of long-term use of non-steroidal anti-inflammatory agents (NSAIDs, inhibitors of cyclooxygenases) on reducing AD risks [263–266], although such a beneficial effect is not observed in randomized trials [267,268]. Notably, pro-inflammatory fatty acids generated from phospholipid hydrolysis are also involved in Aβ-mediated neurotoxicity. cPLA2 activity and the production of AA can be dose-dependently activated by Aβ in neuronal cultures, whereas cPLA2 inhibition diminishes Aβ-induced neurotoxicity and protects fAD mice from memory and learning deficits [269]. Spatially, lipid rafts may play a critical role in initiating inflammatory response in glial cells involving cholesterol- and sphingolipid metabolism, where high cholesterol levels are associated with clustering of inflammatory proteins and elevated inflammation [270].
Collectively, lipid metabolism, via the production of both pro-inflammatory and pro-resolving lipid-derived messengers, contributes to the chronic and unresolved neuroinflammation in AD (Fig. 5).
Oxidative stress
Oxidative stress refers to oxidative damage to cell constituents caused by accumulation of ROS and reactive nitrogen species (RNS) that overwhelm cellular antioxidative capacity [217,271]. Increased generation of ROS and oxidative stress is a hallmark of AD and contributes to the neurotoxic environment with disease progression [16]. Compared with glial cells, neurons are particularly vulnerable to oxidized redox environment becausse of their limited antioxidative capacity.
Oxidative stress in brain is largely manifested by lipid peroxidation, owning to its high oxidation-prone lipid content. In AD brains, amyloid plaques and their immediate surroundings are characterized by the presence of oxidized lipids, whereas in cognitively normal subjects, the levels of oxidized lipids are lower even in the presence of plaques [272]. The interactions between lipids and oxidative stress in AD are three-fold. First, some lipid species are known as ROS inducers. In addition to the direct effect on Aβ, ceramides promote ROS generation in AD [273]. Second, selective lipids, primarily PUFAs are prone to lipid peroxidation to generate highly reactive electrophilic aldehydes including 4-hydroxy-2-nonenal (4-HNE), acrolein, and malondialdehyde [274]. Accordingly, markers for lipid peroxidation are increased early in AD brains along with reduced PUFA levels [275–277], and ApoE4 is associated with enhanced lipid peroxidation [278]. Last, ROS such as H2O2 can directly [279] or indirectly (via cPLA2 and AA) [280] activate sphingomyelinases, which catalyze the hydrolysis of sphingomyelin to generate ceramide. Consistently, protein levels of acid sphingomyelinase are increased in the cortex of AD patients and fAD mice [52,281,282]. In fAD mouse models, Aβ deposition and memory impairment are improved after genetic- or pharmacological reduction of acid- or neutral sphingomyelinases [282,283]. These three mechanisms constitute the interplays between oxidative stress and lipid dyshomeostasis implicated in AD pathogenesis (Fig. 5).
Myelin degeneration and white matter integrity
Around 40% of the human brain contains white matter that consists of densely packed fibers, and myelin is the major component of these structures [284]. In the CNS, myelin sheath is a plasma membrane extension of oligodendrocyte that wraps around neuronal axons to facilitate rapid nerve conduction and efficient propagation of action potential [285]. About 80% of the dry weight of myelin is lipid [286]. Accumulation of fatty acids and glycosphingolipids are coupled with myelin dysfunction in multiple inherited lysosomal and peroxisomal diseases [287]. In addition to their role as key inducer of multiple sclerosis, white matter loss and demyelination is also observed in AD brains [288]. White matter hyperintensities are correlated with CSF levels of Aβ and tau in individuals with MCI or preclinical AD [289,290].
Glycolipids and phospholipids together comprise the majority of myelin membrane lipids, and both classes use fatty acids as structural blocks for their synthesis. Altered fatty acid metabolism could thus contribute to AD-related structural and functional abnormalities of white matter. It is recently reported that fatty acid synthesis in oligodendrocytes is essential for myelination with correct lipid composition [291] (Fig. 5). Further, lipid biosynthesis by astrocytes is also required for myelination via astrocyte-to-oligodendrocyte transport [292], suggesting complementary roles of these two types of glia in supplying lipid building blocks for myelination (Fig. 4). While astrocyte-derived fatty acids are important for myelination, excess saturated fatty acids released from reactive astrocytes also mediate the toxic effect that induces oligodendrocyte death [140].
Besides the interaction with Aβ [293], TREM2 can also be bound by lipid species and lipoproteins [102–104]. Remarkably, the AD-related R47H mutation impairs its lipid-detecting function [102]. More recently, there has been convincing evidence on the direct role of TREM2 in orchestrating myelin debris clearance and remyelination by regulating cholesterol esterification and LD metabolism in microglia (Fig. 4), where TREM2 deficiency elicits dysregulatied lipid metabolism and transport [294,295]. These findings suggest cholesterol metabolism and lipid signaling as key players in TREM2 polymorphism-associated myelin degeneration and increased AD risk.
Blood-brain barrier (BBB) integrity
The BBB plays an essential role in both separating the brain from, and connecting the brain to, the peripheral circulation by forming an interface for transcytotic exchange of substances including lipids [296]. BBB dysfunction is manifested in early stages of AD irrespective of changes in Aβ or tau biomarkers [297,298]. Aβ endothelial transcytosis and clearance across the BBB is mediated by lipid related genes PICALM and LRP1 [109] (Fig. 5). While it remains unclear how lipid metabolism contributes to disrupted BBB integrity in AD, recent studies suggest that lipid profile and metabolic status in both the brain and the periphery modulate BBB function. Mfsd2a, a phospholipid flippase and transporter expressed by BBB endothelial cells, suppresses transcytosis and ensures BBB integrity [299]. It is discovered that, as a major DHA transporter across BBB, Mfsd2a increases DHA-containing phospholipids in CNS endothelial cells, which inhibit the caveolae-mediated transcytosis and maintains BBB integrity [300]. This is consistent with (a) the increased BBB permeability in AD, (b) lowered DHA levels in AD brains, and (c) DHA-free diet accelerates cognitive decline in AD mice [301]. In another study focusing on the effect of peripheral lipid profile, obesity-associated high circulating saturated fatty acids lead to elevated CSF levels of palmitate and increased BBB permeability, and high brain levels of palmitate further elicit neuroinflammation and impair synaptic and cognitive functions [302–304].
Targeting lipid metabolism against AD: therapeutic potential
Activating lipid-sensitive nuclear receptors
Retinoid X receptors (RXRs), LXRs and PPARs belong to the nuclear receptor superfamily, and they are the master regulators of both peripheral and central lipid homeostasis by transactivating genes involved in lipid metabolism [305]. Multiple therapeutic strategies for AD have been investigated by targeting these receptors.
LXR/RXR
LXRs, including LXRα and LXRβ, form heterodimers with RXRs, and their primary ligands are oxysterols, products of cholesterol hydroxylation [305]. Natural RXR ligands include retinoic acid, linoleic acid, linolenic acid, and DHA. Besides LXR, RXR can form dimers with itself, or with PPARs and retinoic acid receptors (RARs) [306]. LXR functions as a sensor of intracellular cholesterol levels and promotes the transcription of genes involved in cholesterol efflux, including ApoE [307]. LXRβ is the major LXR form expressed in brain, 3-to 5-fold higher than LXRα [308]. LXRβ knockout mice exhibit neuronal loss and impaired motor coordination, accompanied by lipid accumulation, astrogliosis, and axonal atrophy [309].
The LXR/RXR dimer can be activated by either LXR or RXR ligands. LXR agonists (e.g., GW3965 and T0901317) promote Aβ clearance, attenuate neuroinflammation, and mitigate cognitive impairments in fAD mice, and these effects are primarily mediated by ABCA1 and ApoE [310,311]. Bexarotene, an RXR agonist and a drug for cutaneous T-cell lymphoma, also stimulates the clearance of Aβ peptides and improves cognitive deficit [312] by upregulating ABCA1 activity and reversing the hypolipidation of ApoE4 [313] in AD mouse models. However, clinical studies of Bexarotene suggest no effect on Aβ metabolism or cognitive function [314,315]. Interestingly, animal studies reported a sexual dimorphism of Bexarotene in modulating synaptic function in a fAD mouse model carrying five mutations to APP and PSEN1 (5xFAD), with male mice more responsive than female [316].
PPARγ
PPARs are initially discovered for their role in promoting peroxisomal proliferation before they are regarded as major regulators of lipid- and carbohydrate metabolism [317]. PPARγ is the most investigated and targeted PPAR in AD research. In peripheral tissues, PPARγ regulates lipogenesis and insulin sensitization [318]. Earlier studies have connected the effect of PPARγ agonists against AD-like pathologies with anti-inflammatory mechanisms including the suppression of pro-inflammatory transcription factors such as NFκB and STATs [246,319], further underlining the intimate connection between lipid metabolism and inflammatory response.
In animal models, PPARγ agonists attenuate Aβ pathology and improve cognitive function by modulating both the production and clearance of Aβ [320–326]. In human studies, despite initial favorable outcomes from trials with PPARγ agonist pioglitazone [327,328], recent report on a large-scale phase III trial in people at risk of AD shows that pioglitazone fails to delay the onset of MCI [329]. A similar scenario applies to the clinical evaluation of another PPARγ agonist of the thiazolidinedione (TZD) class, rosiglitazone, for which predominantly negative results are obtained from larger trials, whether rosiglitazone is used alone [330–332] or as adjunctive therapy [333]. Notably, a phase II trial in patients with mild to moderate AD finds that rosiglitazone, at a higher dose, can selectively improve cognition in ApoE4 noncarriers but not ApoE4 carriers [334]. This interesting interaction between ApoE4 and a lipid modulator further highlight the critical role of lipid metabolism in AD pathogenesis and susceptibility.
Since existing PPARγ agonists have a low to moderate ability to cross the BBB, the question arises as to whether next generation PPARγ agonists with improved brain penetration can elicit improved efficacy. It is also plausible that many of the beneficial effects seen in animal models and early phase trials are relevant to the insulin sensitizing and mitochondria promoting properties of TZD signaling [335], whereas the activation of lipogenic pathway downstream of PPARγ is not an effective therapeutic target. This speculation is consistent with the increased expression of PPARγ mRNA in the frontal cortex of human AD brains [166]. Given the accumulation of many toxic lipid species and reduced lipid catabolism in AD brains, the activation of PPARα, which promotes lipid degradation, may represent a better strategy to intervene lipid dysregulation in AD (see section “Reducing lipid accumulations” below).
Reducing lipid accumulations
PPARα controls the expression of genes encoding key enzymes of fatty acid oxidation, such as carnitine palmitoyl transferases (CPTs) and acyl-CoA oxidase [336]. PPARα is highly expressed in tissues with higher fatty acid oxidation capacity such as liver, heart, kidney, and skeletal muscle. In the brain, PPARα is predominantly expressed in astrocytes [167,337], which is consistent with the unique capability of astrocytes in performing fatty acid oxidation [29]. Beyond their role in astrocytes, PPARα in neurons maintains spatial learning and memory by regulating the CREB pathway [338].
In the context of AD pathology, PPARα modulates amyloid metabolism by activating the non-amyloidogenic α-secretase while inhibiting the amyloidogenic β-secretase [339,340]. While the pathophysiological role of PPARα in neurodegeneration remains elusive, initial interventional studies in rodent models suggest that endogenous ligands or exogenous agonists of PPARα promote synaptic function of hippocampal neurons and protect them from oxidative damage and Aβ toxicity [341,342]. In vivo, PPARα agonist Wy-14643 alleviates AD-like pathologies and memory deficits in the APP/PS1 mice [343,344]. Further, PPARα agonists elicit anti-inflammatory effects and promote survival in a mouse model of amyotrophic lateral sclerosis (ALS) [345], suggesting a common mechanistic role of PPARα across neurodegenerative diseases.
Acetyl-L-carnitine (ALCAR) is an ester of the trimethylated amino acid L-carnitine, and it can be metabolized to acetyl-CoA and carnitine, which can serve as tricarboxylic acid (TCA) cycle fuels, and facilitators of fatty acid transport into mitochondria, respectively [346]. Serum levels of ALCAR and CSF levels of l-carnitine are decreased in AD [129,347]. In primary neurons, ALCAR ameliorates Aβ-oligomer-evoked changes in mitochondrial respiration, fragmentation, and movement [348]. Animal studies show that ALCAR improves age-associated mitochondrial dysfunction and cognitive deficit [349,350]. However, meta-analyses of AD clinical trials with ALCAR yield conflicting and inconclusive results regarding the effect of ALCAR on cognitive measures [351,352].
Besides promoting lipid degradation using PPARα agonists, another strategy to prevent the accumulation of toxic fatty acids, amyloidogenic ceramides, and pro-inflammatory prostaglandins is to inhibit the de novo synthesis of fatty acid in the brain. It is found recently that an inhibition of FAS in fAD mice alleviates lipid peroxidation, neuroinflammation, and cognitive loss [149]. In line with this report, another compound that reduces brain inflammation and cognitive deficit in fAD mice, CAD-31, elicits its protective effect by simultaneously promoting fatty acid oxidation and inhibiting fatty acid synthesis [151].
Boosting lipid transporters
ApoE and ABCA1 are the major regulators of lipid transport in the brain. Multiple in vitro studies support the potential of using ApoE4 structure correctors to mitigate ApoE4-induced pathological effects [353,354]. In human iPSC-derived neurons, the compound PH002 is able to decrease ApoE4 degradative fragments, Aβ production, and tau hyperphosphorylation in a dose-dependent manner [353]. Future in vivo studies are needed to determine the efficacy of this ApoE4 structure-correcting approach in rescuing ApoE4-involving AD pathologies including the disrupted lipid transport.
Given the gain-of-toxic function of ApoE4 in AD, strategies aiming to reduce its expression are also being evaluated. At the RNA level, knockdown of ApoE4 in the APP/PS1 mice by antisense oligonucleotides (ASO) decreases neurite dystrophy independent of Aβ pathology, and this strategy is only effective if initiated during the seeding stage before fibril formation [355]. At the protein level, neutralizing antibodies that target nonlipidated and aggregated ApoE reduce Aβ deposition in the APP/PS1/ApoE4 mice [356]. A more direct approach to facilitate ApoE lipidation and lipid transport is to increase ABCA1 activity (see review [236]). In mice, intraperitoneal injection of an ABCA1 agonist, CS-6253, increases ApoE4 lipidation and decreases ApoE4-driven amyloid and tau pathologies [357]. While the primary outcome for these ApoE-or ABCA1-targeting studies is Aβ pathology, it would be interesting to investigate whether lipid metabolism and trafficking is involved.
Supplementing PUFAs
In distinction to approaches that intervene lipid metabolism by manipulating lipid enzymes or sensors, lipids that decline in AD and are prone to peroxidation such as PUFAs, have been assessed as AD treatment-or prevention strategies. Epidemiologically, increased intake of ω-3 PUFA, especially DHA and EPA, reduces AD risk, whereas lower ω-3 PUFA intake increases the risk [58,358].
Mechanistically, ω-3 PUFA affects multiple aspects of AD pathologies. DHA and EPA decrease Aβ production while increase Aβ degradation by reducing β- and γ-secretase activity and stimulating the activity of insulin-degrading enzyme (IDE), respectively [359,360]. ω-3 PUFA modulates brain inflammatory response to Aβ [361]. In addition to the direct effect of ω-3 PUFA on Aβ metabolism, some of the beneficial effects of ω-3 PUFA are contributed by its role as an activator of RXR and PPARs [362–364]. Clinically, small scale studies suggest potential beneficial effects of ω-3 PUFA on pathological outcomes in MCI or AD patients [365,366]. However, it should be noted that in many instances of these studies, ω-3 PUFA is supplemented in combination with other compounds such as antioxidants. It thus remains to be determined whether the observed effects are induced by ω-3 PUFA, by other ingredients, or by multiple ingredients combined. The last possibility pointing to a synergistic effect of ω-3 PUFA and antioxidants is supported by a report in which oxidized DHA, even at low levels, abrogates the protective effect of DHA on Aβ production [367].
In two larger scale, randomized clinical trials, DHA fails to delay the cognitive decline in mild to moderate AD subjects [368,369]. Nevertheless, subgroup analysis in these two trials detected positive effect of DHA in very mild AD cases [368] and in ApoE4 noncarriers [369], respectively. These interesting findings corroborate the critical role of disrupted lipid metabolism in early AD stages, as well as its close interaction with ApoE functionality. Notably, ω-3 fatty acid supplementation to MCI patients increases the levels of a SPM (RvD1) in their macrophage coupled with increased Aβ phagocytosis and decreased cytokine expression [365], suggesting some of the beneficial effect of ω-3 fatty acids might be mediated by the resolution of neuroinflammation via SPMs [365] (see section “Neuroinflammation” above).
Replenishing ketogenic fuels
Ketone bodies, such as acetoacetate and β-hydroxybutyrate, are mainly derived from fatty acid oxidation in liver upon starvation and represent the major non-glucose fuel source to the brain [370]. An adaptive metabolic switch to ketones in the brains of normal chronological aging, female reproductive aging, and AD models have been observed [371–375]. Since glucose hypometabolism is an early and persistent hallmark of AD, it is proposed that increased ketone body availability to the brain may promote its resistance to bioenergetic deficits [376]. Further, β-hydroxybutyrate exhibits antioxidative and anti-inflammatory effects in the brain [377]. How ketone body interventions affect brain lipid profiles and whether lipid metabolism is involved in the CNS effect of ketone bodies, warrant further investigation.
In both aging and AD animal models, ketogenic diets (high saturated fat and low carbohydrate) markedly attenuate Aβ pathology and improve brain metabolic and cognitive function [378,379]. However, despite initial indicators of efficacy for ketogenic diet/formula in human trials [380–382], daily administration of a ketogenic medium-chain TAG (AC-1204) fails to alter cognitive function in a phase III trial with mild-to-moderate AD subjects. Interestingly, subgroup analyses of two trials of ketogenic intervention suggest that its cognition-improving effect is more notable in early stage AD patients with Mini-Mental State Examination (MMSE) score ≤ 15 [383], or ApoE4 noncarriers [384,385]. These modifications of responsiveness by ApoE4 and disease stage resemble the findings in DHA interventions [368,369].
Taken together, while ketogenic interventions are effective in animal models and early phase clinical trials, its long-term effect in a broader AD population warrants further investigation. Moreover, the effectiveness of ketogenic strategies against AD is dependent on the individual metabolic status and lipid profile, which are markedly modified by ApoE genotype and disease stage as discussed in previous sections.
Restricting cholesterol levels
While retro/prospective, observational analyses suggest a protection of the use of cholesterol-lowering statins in reducing AD risk [386,387], randomized clinical trials find no benefit of statin treatment in AD patients [388–391]. In several reports, statin-associated short-term and reversible cognitive impairment are observed [392–394]. Schultz and colleagues ascribed such discrepancy to the independent effect of cholesterol in the central- and peripheral systems [395]. It is proposed that the risk-lowering effect of statins primarily comes from the reduced circulating cholesterol, which benefits cardiovascular and cerebrovascular systems and reduces vascular inflammation [395]. The negative impact of high-dose statins on cognition may stem from the decreased local cholesterol synthesis within the CNS (as seen in ApoE knockout mice [193]), where cholesterol participates in essential processes including synaptogenesis, myelination, and neurotransmission [395]. It is also plausible that the impact of statins on AD risk is in part contributed by its effect on vascular dementia, which is often misdiagnosed as AD [395]. Recent analyses of the UK Biobank database suggest that potential beneficial effect of statins is selective for ApoE4 carriers [386,396], which is in agreement with ApoE4-associated central- and systemic cholesterol dyshomeostasis. In addition to ApoE4 status, age [387] and sex [396] are also factors that modify AD risk profiles in statin users. These findings support the necessity of a precise and personized approach when evaluating the efficacy of cholesterol-lowering medicine for AD.
Besides the idea of reducing cholesterol biosynthesis by statins, studies in AD animal models indicate potential effect by modulating the levels of cholesterol derivatives. For example, boosted conversion of cholesterol to the brain-exportable 24S-hydroxycholesterol by expressing exogenous cholesterol 24-hydroxylase reduces Aβ pathology and improves spatial memory [397]. A pharmacological approach using Efavirenz, an FDA-approved anti-retroviral drug, to activate cholesterol 24-hydroxylase results in similar effect in the 5xFAD mice [398]. Alternatively, suppression of CE production by genetic knockdown or pharmacological inhibition of ACAT1 attenuates Aβ deposition and restores cognition in fAD mice [399–402].
Conclusions and perspectives
Large volumes of clinical evidence have clearly demonstrated the alterations in lipid species and lipid metabolism in the pathogenesis and progression of AD. It is noteworthy that many lipid-related changes emerge in early stages of the disease. This suggests that lipid dyshomeostasis may be an initiation mechanism of the disease, likely via interactions with amyloidogenesis, synaptogenesis and hypometabolism, thus highlighting the importance of early intervention for lipid-centric strategies towards improved efficacy.
Further, many of these lipid-related changes persist or exacerbate through later disease stages. These changes could be relevant to the critical interactions between lipid species and mechanisms such as bioenergetics, neuroinflammation, and oxidative stress, which potentially form a vicious cycle promoting neuronal loss and cognitive dysfunction. These lines of evidence indicate that in neurodegeneration, lipid metabolism could serve as a bidirectional link between bioenergetic decline and chronic neuroinflammation [403], both of which are hallmarks of AD [16]. Moreover, given the intimate and strong mechanistic connections among Aβ, ApoE and lipid trafficking, lipid metabolism could be the hub where Aβ-dependent and -independent mechanisms of ApoE-associated pathologies converge.
Several lessons are learned from the successful preclinical studies and failed human clinical trials of lipid-targeting AD therapies.
First, the efficacy of lipid-related therapeutics is substantially affected by genetic modifiers of AD risk, with ApoE status being the most significant one. It appears that fatty acid-centric therapeutics are more, and in some cases only, effective in ApoE4 noncarriers, such as PPAR agonists, DHA supplement and ketogenic intervention, whereas cholesterol targeting strategies such as statins are more selective for ApoE4 carriers. It is plausible that for ApoE4 carriers, fatty acid-related lipid dyshomeostasis is too severe to be corrected. In agreement with this speculation, diabetes worsens cognitive decline in ApoE3 and ApoE2 carriers, but not ApoE4 carriers [404]. Such an ApoE genotype-specific effect is in part mediated by vascular impairment, for which ApoE4 carriers have reached a plateau regardless of diabetic status [404]. A strong interaction with ApoE genotype also pertains to lifestyle-focusing strategies to reduce AD risk. It is estimated that improvements in modifiable risk factors can prevent or delay 30% to 40% of dementia cases [133,405]. However, a recent study suggests that the responsiveness to improvements to modifiable-risk factors (smoking, depression, diabetes, physical activity, social isolation, and healthy diet) is detectable in ApoE4 noncarriers only [406].
Second, emerging evidence suggests that many of the neuroprotective effect of lipid-modulating approaches are relevant to their indirect effect on peripheral metabolism. In addition to the ApoE status, metabolic- and vascular profile both modulate brain lipid metabolism and modify the responsiveness to lipid-modulating interventions. For example, PPARγ agonists are more effective in the AD population with metabolic disorders, whereas statins are more beneficial for those with hyperlipidemia, vascular abnormalities, and increased stroke risks. A personalized approach incorporating these interacting factors should be considered when intervening lipid metabolism against AD.
Third, developing therapeutics by targeting lipid metabolism should consider brain selectivity and peripheral side effects. For example, nuclear receptor (LXR/RXR/PPAR) agonists are known to elicit unwanted disruption to lipid synthesis and homeostasis in metabolic organs such as the liver and adipose tissue, which may complicate their direct effect on the brain.
Collectively, findings from large-scale populational studies and responsiveness analyses to investigational therapies in the past two decades emphasize the necessity to incorporate individual risk profile, especially genetic- and non-genetic factors that affect metabolic and vascular functions, when intervening lipid metabolism against AD. Further, successful lipid-targeting therapeutics should consider the dynamic changes and adaptations of lipid metabolism throughout the prodromal, early, and late phases of the disease.
Acknowledgements
This work has been supported by the National Institute on Aging (NIA) grants RF1AG068175 to FY, P01AG026572 (Project 1 and Analytic Core to FY), Arizona Alzheimer’s Consortium Pilot Project grants to FY, and the Packer-Wenz research endowment to FY. Figures were created with BioRender.com.
Abbreviations
- AA
arachidonic acid
- ABCA1/7
ATP-binding cassette subfamily A member 1/7
- ACAT1
acyl CoA:cholesterol acyltransferase 1
- ACC
acetyl-CoA carboxylase
- AD
alzheimer’s disease
- ALCAR
acetyl-l-carnitine
- ApoE
apolipoprotein E
- APP
amyloid precursor protein
- Aβ
β-amyloid
- BBB
blood-brain barrier
- CE
cholesteryl ester
- CLU
clusterin
- CNS
central nervous system
- COX-1/2
cyclooxygenase-1/2
- CPT
carnitine palmitoyltransferase
- CSF
cerebrospinal fluid
- DAG
diacylglycerol
- DHA
docosahexaenoic acid
- EPA
eicosapentaenoic acid
- ER
endoplasmic reticulum
- FFA
free fatty acid
- fAD
familial AD
- FAS
fatty acid synthase
- HDL
high-density lipoprotein
- LD
lipid droplet
- LOAD
late-onset AD
- LRP1
lipoprotein receptor-related protein 1
- LXR
liver X receptor
- MAG
monoacylglycerol
- MAM
mitochondria-associated membrane
- MCI
mild cognitive impairment
- MUFA
monounsaturated fatty acid
- NFT
neurofibrillary tangle
- PGE2
prostaglandin E2
- PICALM
phosphatidylinositol-binding clathrin assembly protein
- PLA2
phospholipase A2
- PPAR
peroxisome proliferator-activated receptor
- PSEN1/2
presenilin-1/2
- PUFA
polyunsaturated fatty acid
- ROS
reactive oxygen species
- RXR
Retinoid X receptor
- SNP
single nucleotide polymorphism
- SPM
specialized pro-resolution mediator
- SREBP
sterol regulatory-element binding protein
- T2D
type 2 diabetes
- TAG
triacylglycerol
- TREM2
triggering receptor expressed on myeloid cells 2
- TZD
thiazolidinedione
- VLCFA
very-long-chain fatty acid
Footnotes
Conflict of interest
The author declares no conflict of interest.
Data availability statement
Data sharing is not applicable to this review article as no new data were created or analyzed.
References
- 1.Alzheimers Disease International, Guerchet M, Prince M, Prina M. Numbers of people with dementia worldwide. 2020. [Google Scholar]
- 2.Knopman DS, Amieva H, Petersen RC, Chetelat G, Holtzman DM, Hyman BT, et al. Alzheimer disease. Nat Rev Dis Primers. 2021;7:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL. Alzheimer’s disease. Nat Rev Dis Primers. 2015;1:15056. [DOI] [PubMed] [Google Scholar]
- 4.Jack CR Jr, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA research framework: toward a biological definition of alzheimer’s disease. Alzheimers Dement. 2018;14:535–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vermunt L, Sikkes SAM, van den Hout A, Handels R, Bos I, van der Flier WM, et al. Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimers Dement. 2019;15:888–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van der Flier WM, Scheltens P. Epidemiology and risk factors of dementia. J Neurol Neurosurg Psychiatry. 2005;76(Suppl 5):2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nat Rev Neurol. 2011;7:137–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368:387–403. [DOI] [PubMed] [Google Scholar]
- 9.Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. 2019;179:312–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu PP, Xie Y, Meng XY, Kang JS. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct Target Ther. 2019;4:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knopman DS. Lowering of Amyloid-Beta by beta-Secretase Inhibitors - Some Informative Failures. N Engl J Med. 2019;380:1476–8. [DOI] [PubMed] [Google Scholar]
- 12.2021 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2021;17:327–406. [DOI] [PubMed] [Google Scholar]
- 13.Cummings J, Lee G, Zhong K, Fonseca J, Taghva K. Alzheimer’s disease drug development pipeline: 2021. Alzheimers Dement. 2021;7:e12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rafii MS, Aisen PS. The search for Alzheimer disease therapeutics - same targets, better trials? Nat Rev Neurol. 2020;16:597–8. [DOI] [PubMed] [Google Scholar]
- 15.The Lancet N. A contentious FDA ruling for Alzheimer’s disease. Lancet Neurol. 2021;20:585. [DOI] [PubMed] [Google Scholar]
- 16.Yin F, Sancheti H, Patil I, Cadenas E. Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic Biol Med. 2016;100:108–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Di Paolo G, Kim TW. Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat Rev Neurosci. 2011;12:284–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sarlus H, Heneka MT. Microglia in Alzheimer’s disease. J Clin Invest. 2017;127:3240–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swerdlow RH. Mitochondria and mitochondrial cascades in alzheimer’s disease. J Alzheimers Dis. 2018;62:1403–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mi Y, Qi G, Brinton RD, Yin F. Mitochondria-targeted therapeutics for alzheimer’s disease: the good, the bad, the potential. Antioxid Redox Signal. 2021;34:611–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cunnane SC, Trushina E, Morland C, Prigione A, Casadesus G, Andrews ZB, et al. Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat Rev Drug Discov. 2020;19:609–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mosconi L, Berti V, Quinn C, McHugh P, Petrongolo G, Varsavsky I, et al. Sex differences in Alzheimer risk: brain imaging of endocrine vs chronologic aging. Neurology. 2017;89:1382–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tan MS, Ji X, Li JQ, Xu W, Wang HF, Tan CC, et al. Longitudinal trajectories of Alzheimer’s ATN biomarkers in elderly persons without dementia. Alzheimers Res Ther. 2020;12:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin Anat. 1995;8:429–31. [DOI] [PubMed] [Google Scholar]
- 25.Naudi A, Cabre R, Jove M, Ayala V, Gonzalo H, Portero-Otin M, et al. Lipidomics of human brain aging and Alzheimer’s disease pathology. Int Rev Neurobiol. 2015;122:133–89. [DOI] [PubMed] [Google Scholar]
- 26.Han X. Neurolipidomics: challenges and developments. Front Biosci. 2007;12:2601–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sastry PS. Lipids of nervous tissue: composition and metabolism. Prog Lipid Res. 1985;24:69–176. [DOI] [PubMed] [Google Scholar]
- 28.Fantini J, Yahi N. Brain lipids in synaptic function and neurological disease : clues to innovative therapeutic strategies for brain disorders. London: Elsevier/Academic Press; 2015. [Google Scholar]
- 29.Schonfeld P, Reiser G. Brain energy metabolism spurns fatty acids as fuel due to their inherent mitotoxicity and potential capacity to unleash neurodegeneration. Neurochem Int. 2017;109:68–77. [DOI] [PubMed] [Google Scholar]
- 30.Li LO, Klett EL, Coleman RA. Acyl-CoA synthesis, lipid metabolism and lipotoxicity. Biochim Biophys Acta. 2010;1801:246–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fonteh AN, Cipolla M, Chiang J, Arakaki X, Harrington MG. Human cerebrospinal fluid fatty acid levels differ between supernatant fluid and brain-derived nanoparticle fractions, and are altered in Alzheimer’s disease. PLoS One. 2014;9:e100519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cunnane SC, Schneider JA, Tangney C, Tremblay-Mercier J, Fortier M, Bennett DA, et al. Plasma and brain fatty acid profiles in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis. 2012;29:691–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Snowden SG, Ebshiana AA, Hye A, An Y, Pletnikova O, O’Brien R, et al. Association between fatty acid metabolism in the brain and Alzheimer disease neuropathology and cognitive performance: a nontargeted metabolomic study. PLoS Med. 2017;14:e1002266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Belkouch M, Hachem M, Elgot A, Van Lo A, Picq M, Guichardant M, et al. The pleiotropic effects of omega-3 docosahexaenoic acid on the hallmarks of Alzheimer’s disease. J Nutr Biochem. 2016;38:1–11. [DOI] [PubMed] [Google Scholar]
- 35.Fonteh AN, Cipolla M, Chiang AJ, Edminster SP, Arakaki X, Harrington MG. Polyunsaturated fatty acid composition of cerebrospinal fluid fractions shows their contribution to cognitive resilience of a pre-symptomatic alzheimer’s disease cohort. Front Physiol. 2020;11:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prasad MR, Lovell MA, Yatin M, Dhillon H, Markesbery WR. Regional membrane phospholipid alterations in Alzheimer’s disease. Neurochem Res. 1998;23:81–8. [DOI] [PubMed] [Google Scholar]
- 37.Dennis EA. Diversity of group types, regulation, and function of phospholipase A2. J Biol Chem. 1994;269:13057–60. [PubMed] [Google Scholar]
- 38.Fonteh AN, Chiang AJ, Arakaki X, Edminster SP, Harrington MG. Accumulation of cerebrospinal fluid glycerophospholipids and sphingolipids in cognitively healthy participants with alzheimer’s biomarkers precedes lipolysis in the dementia stage. Front Neurosci. 2020;14:611393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stephenson DT, Lemere CA, Selkoe DJ, Clemens JA. Cytosolic phospholipase A2 (cPLA2) immunoreactivity is elevated in Alzheimer’s disease brain. Neurobiol Dis. 1996;3:51–63. [DOI] [PubMed] [Google Scholar]
- 40.Sebastiao AM, Colino-Oliveira M, Assaife-Lopes N, Dias RB, Ribeiro JA. Lipid rafts, synaptic transmission and plasticity: impact in age-related neurodegenerative diseases. Neuropharmacology. 2013;64:97–107. [DOI] [PubMed] [Google Scholar]
- 41.Sezgin E, Levental I, Mayor S, Eggeling C. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat Rev Mol Cell Biol. 2017;18:361–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin V, Fabelo N, Santpere G, Puig B, Marin R, Ferrer I, et al. Lipid alterations in lipid rafts from Alzheimer’s disease human brain cortex. J Alzheimers Dis. 2010;19:489–502. [DOI] [PubMed] [Google Scholar]
- 43.Marin R, Fabelo N, Fernandez-Echevarria C, Canerina-Amaro A, Rodriguez-Barreto D, Quinto-Alemany D, et al. Lipid raft alterations in aged-associated neuropathologies. Curr Alzheimer Res. 2016;13:973–84. [DOI] [PubMed] [Google Scholar]
- 44.Fabelo N, Martin V, Marin R, Moreno D, Ferrer I, Diaz M. Altered lipid composition in cortical lipid rafts occurs at early stages of sporadic Alzheimer’s disease and facilitates APP/BACE1 interactions. Neurobiol Aging. 2014;35:1801–12. [DOI] [PubMed] [Google Scholar]
- 45.Guan Z, Wang Y, Cairns NJ, Lantos PL, Dallner G, Sindelar PJ. Decrease and structural modifications of phosphatidylethanolamine plasmalogen in the brain with Alzheimer disease. J Neuropathol Exp Neurol. 1999;58:740–7. [DOI] [PubMed] [Google Scholar]
- 46.Nitsch RM, Blusztajn JK, Pittas AG, Slack BE, Growdon JH, Wurtman RJ. Evidence for a membrane defect in Alzheimer disease brain. Proc Natl Acad Sci USA. 1992;89:1671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stokes CE, Hawthorne JN. Reduced phosphoinositide concentrations in anterior temporal cortex of Alzheimer-diseased brains. J Neurochem. 1987;48:1018–21. [DOI] [PubMed] [Google Scholar]
- 48.Pettegrew JW, Panchalingam K, Hamilton RL, McClure RJ. Brain membrane phospholipid alterations in Alzheimer’s disease. Neurochem Res. 2001;26:771–82. [DOI] [PubMed] [Google Scholar]
- 49.Filippov V, Song MA, Zhang K, Vinters HV, Tung S, Kirsch WM, et al. Increased ceramide in brains with Alzheimer’s and other neurodegenerative diseases. J Alzheimers Dis. 2012;29:537–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, et al. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci USA. 2004;101:2070–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Han X, Holtzman M. D W. McKeel D, Kelley J, Morris JC. Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer’s disease: potential role in disease pathogenesis. J Neurochem. 2002;82:809–18. [DOI] [PubMed] [Google Scholar]
- 52.He X, Huang Y, Li B, Gong CX, Schuchman EH. Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol Aging. 2010;31:398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Soderberg M, Edlund C, Alafuzoff I, Kristensson K, Dallner G. Lipid composition in different regions of the brain in Alzheimer’s disease/senile dementia of Alzheimer’s type. J Neurochem. 1992;59:1646–53. [DOI] [PubMed] [Google Scholar]
- 54.Kosicek M, Zetterberg H, Andreasen N, Peter-Katalinic J, Hecimovic S. Elevated cerebrospinal fluid sphingomyelin levels in prodromal Alzheimer’s disease. Neurosci Lett. 2012;516:302–5. [DOI] [PubMed] [Google Scholar]
- 55.Gonzalez-Dominguez R, Garcia-Barrera T, Vitorica J, Gomez-Ariza JL. Region-specific metabolic alterations in the brain of the APP/PS1 transgenic mice of Alzheimer’s disease. Biochim Biophys Acta. 2014;1842:2395–402. [DOI] [PubMed] [Google Scholar]
- 56.Katsel P, Li C, Haroutunian V. Gene expression alterations in the sphingolipid metabolism pathways during progression of dementia and Alzheimer’s disease: a shift toward ceramide accumulation at the earliest recognizable stages of Alzheimer’s disease? Neurochem Res. 2007;32:845–56. [DOI] [PubMed] [Google Scholar]
- 57.Siskind LJ. Mitochondrial ceramide and the induction of apoptosis. J Bioenerg Biomembr. 2005;37:143–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morris MC, Evans DA, Bienias JL, Tangney CC, Bennett DA, Wilson RS, et al. Consumption of fish and n-3 fatty acids and risk of incident Alzheimer disease. Arch Neurol. 2003;60:940–6. [DOI] [PubMed] [Google Scholar]
- 59.Cheng H, Wang M, Li JL, Cairns NJ, Han X. Specific changes of sulfatide levels in individuals with pre-clinical Alzheimer’s disease: an early event in disease pathogenesis. J Neurochem. 2013;127:733–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Couttas TA, Kain N, Suchowerska AK, Quek LE, Turner N, Fath T, et al. Loss of ceramide synthase 2 activity, necessary for myelin biosynthesis, precedes tau pathology in the cortical pathogenesis of Alzheimer’s disease. Neurobiol Aging. 2016;43:89–100. [DOI] [PubMed] [Google Scholar]
- 61.Wood PL, Barnette BL, Kaye JA, Quinn JF, Woltjer RL. Non-targeted lipidomics of CSF and frontal cortex grey and white matter in control, mild cognitive impairment, and Alzheimer’s disease subjects. Acta Neuropsychiatr. 2015;27:270–8. [DOI] [PubMed] [Google Scholar]
- 62.Chan RB, Oliveira TG, Cortes EP, Honig LS, Duff KE, Small SA, et al. Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J Biol Chem. 2012;287:2678–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wood PL, Medicherla S, Sheikh N, Terry B, Phillipps A, Kaye JA, et al. Targeted lipidomics of fontal cortex and plasma diacylglycerols (DAG) in mild cognitive impairment and alzheimer’s disease: validation of DAG accumulation early in the pathophysiology of alzheimer’s disease. J Alzheimers Dis. 2015;48:537–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nguyen TB, Louie SM, Daniele JR, Tran Q, Dillin A, Zoncu R, et al. DGAT1-dependent lipid droplet biogenesis protects mitochondrial function during starvation-induced autophagy. Dev Cell. 2017;42:9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gomez-Ramos P, Asuncion Moran M. Ultrastructural localization of intraneuronal Abeta-peptide in Alzheimer disease brains. J Alzheimers Dis. 2007;11:53–9. [DOI] [PubMed] [Google Scholar]
- 66.Conte M, Medici V, Malagoli D, Chiariello A, Cirrincione A, Davin A, et al. Expression pattern of perilipins in human brain during aging and in Alzheimer’s disease. Neuropathol Appl Neurobiol. 2021;27:12756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pani A, Dessi S, Diaz G, La Colla P, Abete C, Mulas C, et al. Altered cholesterol ester cycle in skin fibroblasts from patients with Alzheimer’s disease. J Alzheimers Dis. 2009;18:829–41. [DOI] [PubMed] [Google Scholar]
- 68.Pani A, Mandas A, Diaz G, Abete C, Cocco PL, Angius F, et al. Accumulation of neutral lipids in peripheral blood mononuclear cells as a distinctive trait of Alzheimer patients and asymptomatic subjects at risk of disease. BMC Med. 2009;7:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ralhan I, Chang CL, Lippincott-Schwartz J, Ioannou MS. Lipid droplets in the nervous system. J Cell Biol. 2021;220;2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Farmer BC, Walsh AE, Kluemper JC, Johnson LA. Lipid droplets in neurodegenerative disorders. Front Neurosci. 2020;14:742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Feringa FM, van der Kant R. Cholesterol and alzheimer’s disease; from risk genes to pathological effects. Front Aging Neurosci. 2021;13:690372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vance JE, Hayashi H, Karten B. Cholesterol homeostasis in neurons and glial cells. Semin Cell Dev Biol. 2005;16:193–212. [DOI] [PubMed] [Google Scholar]
- 73.Heverin M, Bogdanovic N, Lutjohann D, Bayer T, Pikuleva I, Bretillon L, et al. Changes in the levels of cerebral and extracerebral sterols in the brain of patients with Alzheimer’s disease. J Lipid Res. 2004;45:186–93. [DOI] [PubMed] [Google Scholar]
- 74.Popp J, Meichsner S, Kolsch H, Lewczuk P, Maier W, Kornhuber J, et al. Cerebral and extracerebral cholesterol metabolism and CSF markers of Alzheimer’s disease. Biochem Pharmacol. 2013;86:37–42. [DOI] [PubMed] [Google Scholar]
- 75.Liu Y, Zhong X, Shen J, Jiao L, Tong J, Zhao W, et al. Elevated serum TC and LDL-C levels in Alzheimer’s disease and mild cognitive impairment: A meta-analysis study. Brain Res. 2020;1727:146554. [DOI] [PubMed] [Google Scholar]
- 76.Varma VR, Busra Luleci H, Oommen AM, Varma S, Blackshear CT, Griswold ME, et al. Abnormal brain cholesterol homeostasis in Alzheimer’s disease-a targeted metabolomic and transcriptomic study. NPJ Aging Mech Dis. 2021;7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hascalovici JR, Vaya J, Khatib S, Holcroft CA, Zukor H, Song W, et al. Brain sterol dysregulation in sporadic AD and MCI: relationship to heme oxygenase-1. J Neurochem. 2009;110:1241–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mori T, Paris D, Town T, Rojiani AM, Sparks DL, Delledonne A, et al. Cholesterol accumulates in senile plaques of Alzheimer disease patients and in transgenic APP(SW) mice. J Neuropathol Exp Neurol. 2001;60:778–85. [DOI] [PubMed] [Google Scholar]
- 79.Zhang J, Liu Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell. 2015;6:254–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tajima Y, Ishikawa M, Maekawa K, Murayama M, Senoo Y, Nishimaki-Mogami T, et al. Lipidomic analysis of brain tissues and plasma in a mouse model expressing mutated human amyloid precursor protein/tau for Alzheimer’s disease. Lipids Health Dis. 2013;12:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry. 2015;77:43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Picard C, Julien C, Frappier J, Miron J, Theroux L, Dea D. Alterations in cholesterol metabolism–related genes in sporadic Alzheimer’s disease. Neurobiol Aging. 2018;66:180. [DOI] [PubMed] [Google Scholar]
- 83.Hauser PS, Narayanaswami V, Ryan RO. Apolipoprotein E: from lipid transport to neurobiology. Prog Lipid Res. 2011;50:62–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chartier-Harlin MC, Parfitt M, Legrain S, Perez-Tur J, Brousseau T, Evans A, et al. Apolipoprotein E, epsilon 4 allele as a major risk factor for sporadic early and late-onset forms of Alzheimer’s disease: analysis of the 19q13.2 chromosomal region. Hum Mol Genet. 1994;3:569–74. [DOI] [PubMed] [Google Scholar]
- 85.Houlden H, Crook R, Backhovens H, Prihar G, Baker M, Hutton M, et al. ApoE genotype is a risk factor in nonpresenilin early-onset Alzheimer’s disease families. Am J Med Genet. 1998;81:117–21. [DOI] [PubMed] [Google Scholar]
- 86.Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC Jr, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7:180–4. [DOI] [PubMed] [Google Scholar]
- 87.Reiman EM, Arboleda-Velasquez JF, Quiroz YT, Huentelman MJ, Beach TG, Caselli RJ, et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat Commun. 2020;11:667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu C-C, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278:1349–56. [PubMed] [Google Scholar]
- 90.Neu SC, Pa J, Kukull W, Beekly D, Kuzma A, Gangadharan P, et al. Apolipoprotein E genotype and sex risk factors for alzheimer disease: a meta-analysis. JAMA Neurology. 2017;74:1178–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Huang Y, Mahley RW. Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol Dis. 2014;72:3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dallongeville J, Lussier-Cacan S, Davignon J. Modulation of plasma triglyceride levels by apoE phenotype: a meta-analysis. J Lipid Res. 1992;33:447–54. [PubMed] [Google Scholar]
- 93.Notkola IL, Sulkava R, Pekkanen J, Erkinjuntti T, Ehnholm C, Kivinen P, et al. Serum total cholesterol, apolipoprotein E epsilon 4 allele, and Alzheimer’s disease. Neuroepidemiology. 1998;17:14–20. [DOI] [PubMed] [Google Scholar]
- 94.Wang S, Li B, Solomon V, Fonteh A, Rapoport SI, Bennett DA, et al. Calcium-dependent cytosolic phospholipase A2 activation is implicated in neuroinflammation and oxidative stress associated with ApoE4. Mol Neurodegener. 2021;16:26. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 95.Grimm MOW, Michaelson DM, Hartmann T. Omega-3 fatty acids, lipids, and apoE lipidation in Alzheimer’s disease: a rationale for multi-nutrient dementia prevention. J Lipid Res. 2017;58:2083–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jay TR, von Saucken VE, Landreth GE. TREM2 in neurodegenerative diseases. Mol Neurodegener. 2017;12:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ulland TK, Colonna M. TREM2 - a key player in microglial biology and Alzheimer disease. Nat Rev Neurol. 2018;14:667–75. [DOI] [PubMed] [Google Scholar]
- 98.Kleinberger G, Yamanishi Y, Suarez-Calvet M, Czirr E, Lohmann E, Cuyvers E, et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014;6:243ra86. [DOI] [PubMed] [Google Scholar]
- 99.Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, Adolfsson R, et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet. 2002;71:656–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368:117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368:107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. 2015;160:1061–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron. 2016;91:328–40. [DOI] [PubMed] [Google Scholar]
- 104.Kober DL, Brett TJ. TREM2-Ligand Interactions in Health and Disease. J Mol Biol. 2017;429:1607–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–9. [DOI] [PubMed] [Google Scholar]
- 107.Nuutinen T, Suuronen T, Kauppinen A, Salminen A. Clusterin: a forgotten player in Alzheimer’s disease. Brain Res Rev. 2009;61:89–104. [DOI] [PubMed] [Google Scholar]
- 108.Xu W, Tan L, Yu JT. The role of PICALM in alzheimer’s disease. Mol Neurobiol. 2015;52:399–413. [DOI] [PubMed] [Google Scholar]
- 109.Zhao Z, Sagare AP, Ma Q, Halliday MR, Kong P, Kisler K, et al. Central role for PICALM in amyloid-beta blood-brain barrier transcytosis and clearance. Nat Neurosci. 2015;18:978–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Aikawa T, Holm ML, Kanekiyo T. ABCA7 and pathogenic pathways of alzheimer’s disease. Brain Sci. 2018;8:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nordestgaard LT, Tybjaerg-Hansen A, Nordestgaard BG, Frikke-Schmidt R. Loss-of-function mutation in ABCA1 and risk of Alzheimer’s disease and cerebrovascular disease. Alzheimers Dement. 2015;11:1430–8. [DOI] [PubMed] [Google Scholar]
- 112.Wahrle SE, Jiang H, Parsadanian M, Legleiter J, Han X, Fryer JD, et al. ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J Biol Chem. 2004;279:40987–93. [DOI] [PubMed] [Google Scholar]
- 113.Abe-Dohmae S, Ikeda Y, Matsuo M, Hayashi M, Okuhira K, Ueda K, et al. Human ABCA7 supports apolipoprotein-mediated release of cellular cholesterol and phospholipid to generate high density lipoprotein. J Biol Chem. 2004;279:604–11. [DOI] [PubMed] [Google Scholar]
- 114.De Roeck A, Van Broeckhoven C, Sleegers K. The role of ABCA7 in Alzheimer’s disease: evidence from genomics, transcriptomics and methylomics. Acta Neuropathol. 2019;138:201–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat Rev Endocrinol. 2017;13:710–30. [DOI] [PubMed] [Google Scholar]
- 116.Biessels GJ, Strachan MW, Visseren FL, Kappelle LJ, Whitmer RA. Dementia and cognitive decline in type 2 diabetes and prediabetic stages: towards targeted interventions. Lancet Diabetes Endocrinol. 2014;2:246–55. [DOI] [PubMed] [Google Scholar]
- 117.Knight EM, Martins IV, Gumusgoz S, Allan SM, Lawrence CB. High-fat diet-induced memory impairment in triple-transgenic Alzheimer’s disease (3xTgAD) mice is independent of changes in amyloid and tau pathology. Neurobiol Aging. 2014;35:1821–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Whitmer RA, Gustafson DR, Barrett-Connor E, Haan MN, Gunderson EP, Yaffe K. Central obesity and increased risk of dementia more than three decades later. Neurology. 2008;71:1057–64. [DOI] [PubMed] [Google Scholar]
- 119.Liu Z, Patil IY, Jiang T, Sancheti H, Walsh JP, Stiles BL, et al. High-fat diet induces hepatic insulin resistance and impairment of synaptic plasticity. PLoS One. 2015;10:e0128274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ma Y, Ajnakina O, Steptoe A, Cadar D. Higher risk of dementia in English older individuals who are overweight or obese. Int J Epidemiol. 2020;49:1353–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kivipelto M, Ngandu T, Fratiglioni L, Viitanen M, Kareholt I, Winblad B, et al. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch Neurol. 2005;62:1556–60. [DOI] [PubMed] [Google Scholar]
- 122.Anstey KJ, Cherbuin N, Budge M, Young J. Body mass index in midlife and late-life as a risk factor for dementia: a meta-analysis of prospective studies. Obes Rev. 2011;12:e426–37. [DOI] [PubMed] [Google Scholar]
- 123.Hassing LB, Dahl AK, Thorvaldsson V, Berg S, Gatz M, Pedersen NL, et al. Overweight in midlife and risk of dementia: a 40-year follow-up study. Int J Obes. 2009;33:893–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Willette AA, Johnson SC, Birdsill AC, Sager MA, Christian B, Baker LD, et al. Insulin resistance predicts brain amyloid deposition in late middle-aged adults. Alzheimers Dement. 2015;11:504–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kellar D, Craft S. Brain insulin resistance in Alzheimer’s disease and related disorders: mechanisms and therapeutic approaches. Lancet Neurol. 2020;19:758–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ekblad LL, Johansson J, Helin S, Viitanen M, Laine H, Puukka P, et al. Midlife insulin resistance, APOE genotype, and late-life brain amyloid accumulation. Neurology. 2018;90:e1150–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chornenkyy Y, Wang WX, Wei A, Nelson PT. Alzheimer’s disease and type 2 diabetes mellitus are distinct diseases with potential overlapping metabolic dysfunction upstream of observed cognitive decline. Brain Pathol. 2019;29:3–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Abner EL, Nelson PT, Kryscio RJ, Schmitt FA, Fardo DW, Woltjer RL, et al. Diabetes is associated with cerebrovascular but not Alzheimer’s disease neuropathology. Alzheimers Dement. 2016;12:882–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cristofano A, Sapere N, La Marca G, Angiolillo A, Vitale M, Corbi G, et al. Serum Levels of Acyl-Carnitines along the Continuum from Normal to Alzheimer’s Dementia. PLoS One. 2016;11:e0155694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ciavardelli D, Piras F, Consalvo A, Rossi C, Zucchelli M, Di Ilio C, et al. Medium-chain plasma acylcarnitines, ketone levels, cognition, and gray matter volumes in healthy elderly, mildly cognitively impaired, or Alzheimer’s disease subjects. Neurobiol Aging. 2016;43:1–12. [DOI] [PubMed] [Google Scholar]
- 131.Kivipelto M, Helkala EL, Laakso MP, Hanninen T, Hallikainen M, Alhainen K, et al. Midlife vascular risk factors and Alzheimer’s disease in later life: longitudinal, population based study. BMJ. 2001;322:1447–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zambon D, Quintana M, Mata P, Alonso R, Benavent J, Cruz-Sanchez F, et al. Higher incidence of mild cognitive impairment in familial hypercholesterolemia. Am J Med. 2010;123:267–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Livingston G, Huntley J, Sommerlad A, Ames D, Ballard C, Banerjee S, et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet. 2020;396:413–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Proitsi P, Lupton MK, Velayudhan L, Newhouse S, Fogh I, Tsolaki M, et al. Genetic predisposition to increased blood cholesterol and triglyceride lipid levels and risk of Alzheimer disease: a Mendelian randomization analysis. PLoS Med. 2014;11:e1001713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.McGrath ER, Beiser AS, DeCarli C, Plourde KL, Vasan RS, Greenberg SM, et al. Blood pressure from mid-to late life and risk of incident dementia. Neurology. 2017;89:2447–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Walker KA, Sharrett AR, Wu A, Schneider ALC, Albert M, Lutsey PL, et al. Association of midlife to late-life blood pressure patterns with incident dementia. JAMA. 2019;322:535–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Abell JG, Kivimaki M, Dugravot A, Tabak AG, Fayosse A, Shipley M, et al. Association between systolic blood pressure and dementia in the Whitehall II cohort study: role of age, duration, and threshold used to define hypertension. Eur Heart J. 2018;39:3119–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pase MP, Beiser A, Enserro D, Xanthakis V, Aparicio H, Satizabal CL, et al. Association of ideal cardiovascular health with vascular brain injury and incident dementia. Stroke. 2016;47:1201–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sabia S, Fayosse A, Dumurgier J, Schnitzler A, Empana JP, Ebmeier KP, et al. Association of ideal cardiovascular health at age 50 with incidence of dementia: 25 year follow-up of Whitehall II cohort study. BMJ. 2019;366:14414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Guttenplan KA, Weigel MK, Prakash P, Wijewardhane PR, Hasel P, Rufen-Blanchette U, et al. Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature. 2021;599:102–7. [DOI] [PubMed] [Google Scholar]
- 141.Ioannou MS, Jackson J, Sheu SH, Chang CL, Weigel AV, Liu H, et al. Neuron-astrocyte metabolic coupling protects against activity-induced fatty acid toxicity. Cell. 2019;177(1522–1535):e14. [DOI] [PubMed] [Google Scholar]
- 142.Pineau L, Colas J, Dupont S, Beney L, Fleurat-Lessard P, Berjeaud JM, et al. Lipid-induced ER stress: synergistic effects of sterols and saturated fatty acids. Traffic. 2009;10:673–90. [DOI] [PubMed] [Google Scholar]
- 143.Skulachev VP. Fatty acid circuit as a physiological mechanism of uncoupling of oxidative phosphorylation. FEBS Lett. 1991;294:158–62. [DOI] [PubMed] [Google Scholar]
- 144.Flores-Leon M, Perez-Dominguez M, Gonzalez-Barrios R, Arias C. Palmitic Acid-Induced NAD(+) Depletion is Associated with the Reduced Function of SIRT1 and Increased Expression of BACE1 in Hippocampal Neurons. Neurochem Res. 2019;44:1745–54. [DOI] [PubMed] [Google Scholar]
- 145.Schonfeld P, Reiser G. How the brain fights fatty acids’ toxicity. Neurochem Int. 2021;148:105050. [DOI] [PubMed] [Google Scholar]
- 146.Tracey TJ, Steyn FJ, Wolvetang EJ, Ngo ST. Neuronal lipid metabolism: multiple pathways driving functional outcomes in health and disease. Front Mol Neurosci. 2018; 11:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Du L, Hickey RW, Bayir H, Watkins SC, Tyurin VA, Guo F, et al. Starving neurons show sex difference in autophagy. J Biol Chem. 2009;284:2383–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Singh R, Cuervo AM. Lipophagy: connecting autophagy and lipid metabolism. Int J Cell Biol. 2012;2012:282041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ates G, Goldberg J, Currais A, Maher P. CMS121, a fatty acid synthase inhibitor, protects against excess lipid peroxidation and inflammation and alleviates cognitive loss in a transgenic mouse model of Alzheimer’s disease. Redox Biol. 2020;36:101648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Thangavel R, Kempuraj D, Zaheer S, Raikwar S, Ahmed ME, Selvakumar GP, et al. Glia maturation factor and mitochondrial uncoupling proteins 2 and 4 expression in the temporal cortex of alzheimer’s disease brain. Front Aging Neurosci. 2017;9:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Daugherty D, Goldberg J, Fischer W, Dargusch R, Maher P, Schubert D. A novel Alzheimer’s disease drug candidate targeting inflammation and fatty acid metabolism. Alzheimers Res Ther. 2017;9:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Barbero-Camps E, Fernandez A, Martinez L, Fernandez-Checa JC, Colell A. APP/PS1 mice overexpressing SREBP-2 exhibit combined Abeta accumulation and tau pathology underlying Alzheimer’s disease. Hum Mol Genet. 2013;22:3460–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wang H, Kulas JA, Wang C, Holtzman DM, Ferris HA, Hansen SB. Regulation of beta-amyloid production in neurons by astrocyte-derived cholesterol. Proc Natl Acad Sci USA. 2021;118:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wang C, Zhao F, Shen K, Wang W, Siedlak SL, Lee HG, et al. The sterol regulatory element-binding protein 2 is dysregulated by tau alterations in Alzheimer disease. Brain Pathol. 2019;29:530–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ebert D, Haller RG, Walton ME. Energy contribution of octanoate to intact rat brain metabolism measured by 13C nuclear magnetic resonance spectroscopy. J Neurosci. 2003;23:5928–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Baxter PS, Hardingham GE. Adaptive regulation of the brain’s antioxidant defences by neurons and astrocytes. Free Radic Biol Med. 2016;100:147–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Eraso-Pichot A, Braso-Vives M, Golbano A, Menacho C, Claro E, Galea E, et al. GSEA of mouse and human mitochondriomes reveals fatty acid oxidation in astrocytes. Glia. 2018;66:1724–35. [DOI] [PubMed] [Google Scholar]
- 158.Fecher C, Trovo L, Muller SA, Snaidero N, Wettmarshausen J, Heink S, et al. Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nat Neurosci. 2019;22:1731–42 [DOI] [PubMed] [Google Scholar]
- 159.Edmond J, Robbins RA, Bergstrom JD, Cole RA, de Vellis J. Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J Neurosci Res. 1987;18:551–61. [DOI] [PubMed] [Google Scholar]
- 160.Qi G, Mi Y, Shi X, Gu H, Brinton RD, Yin F. ApoE4 impairs neuron-astrocyte coupling of fatty acid metabolism. Cell Rep. 2021;34:108572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Rambold AS, Cohen S, Lippincott-Schwartz J. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell. 2015;32:678–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Farmer BC, Kluemper J, Johnson LA. Apolipoprotein E4 alters astrocyte fatty acid metabolism and lipid droplet formation. Cells. 2019;8:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sienski G, Narayan P, Bonner JM, Kory N, Boland S, Arczewska AA, et al. APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia. Sci Transl Med. 2021;13:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Qi G, Mi Y, Yin F. Characterizing brain metabolic function ex vivo with acute mouse slice punches. STAR Protoc. 2021;2:100559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Schulz JG, Laranjeira A, Van Huffel L, Gartner A, Vilain S, Bastianen J, et al. Glial beta-oxidation regulates Drosophila energy metabolism. Sci Rep. 2015;5:7805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.de la Monte SM, Wands JR. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J Alzheimers Dis. 2006;9:167–81. [DOI] [PubMed] [Google Scholar]
- 167.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Namgaladze D, Brune B. Macrophage fatty acid oxidation and its roles in macrophage polarization and fatty acid-induced inflammation. Biochim Biophys Acta. 2016;1861:1796–807. [DOI] [PubMed] [Google Scholar]
- 169.Malandrino MI, Fucho R, Weber M, Calderon-Dominguez M, Mir JF, Valcarcel L, et al. Enhanced fatty acid oxidation in adipocytes and macrophages reduces lipid-induced triglyceride accumulation and inflammation. Am J Physiol Endocrinol Metab. 2015;308:E756–69. [DOI] [PubMed] [Google Scholar]
- 170.Namgaladze D, Lips S, Leiker TJ, Murphy RC, Ekroos K, Ferreiros N, et al. Inhibition of macrophage fatty acid beta-oxidation exacerbates palmitate-induced inflammatory and endoplasmic reticulum stress responses. Diabetologia. 2014;57:1067–77. [DOI] [PubMed] [Google Scholar]
- 171.Mauerer R, Walczak Y, Langmann T. Comprehensive mRNA profiling of lipid-related genes in microglia and macrophages using taqman arrays. Methods Mol Biol. 2009;580:187–201. [DOI] [PubMed] [Google Scholar]
- 172.Song GJ, Suk K. Pharmacological modulation of functional phenotypes of microglia in neurodegenerative diseases. Front Aging Neurosci. 2017;9:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Qi G, Mi Y, Yin F. Cellular specificity and intercellular coordination in the brain bioenergetic system: implications for aging and neurodegeneration. Front Physiol. 2019; 10:1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Liu B, Huang B, Hu G, He D, Li Y, Ran X, et al. Isovitexin-mediated regulation of microglial polarization in lipopolysaccharide-induced neuroinflammation via activation of the CaMKKβ/AMPK-PGC-1α signaling axis. Front Immunol. 2019;10:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wang Y, Huang Y, Xu Y, Ruan W, Wang H, Zhang Y, et al. A dual AMPK/Nrf2 activator reduces brain inflammation after stroke by enhancing microglia M2 polarization. Antioxid Redox Signal. 2018;28:141–63. [DOI] [PubMed] [Google Scholar]
- 176.Kassmann CM, Lappe-Siefke C, Baes M, Brugger B, Mildner A, Werner HB, et al. Axonal loss and neuroinflammation caused by peroxisome-deficient oligodendrocytes. Nat Genet. 2007;39:969–76. [DOI] [PubMed] [Google Scholar]
- 177.Kou J, Kovacs GG, Hoftberger R, Kulik W, Brodde A, Forss-Petter S, et al. Peroxisomal alterations in Alzheimer’s disease. Acta Neuropathol. 2011;122:271–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Beckers L, Geric I, Stroobants S, Beel S, Van Damme P, D’Hooge R, et al. Microglia lacking a peroxisomal beta-oxidation enzyme chronically alter their inflammatory profile without evoking neuronal and behavioral deficits. J Neuroinflammation. 2019;16:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hamilton LK, Dufresne M, Joppe SE, Petryszyn S, Aumont A, Calon F, et al. Aberrant lipid metabolism in the forebrain niche suppresses adult neural stem cell proliferation in an animal model of alzheimer’s disease. Cell Stem Cell. 2015;17:397–411. [DOI] [PubMed] [Google Scholar]
- 180.Marschallinger J, Iram T, Zardeneta M, Lee SE, Lehallier B, Haney MS, et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat Neurosci. 2020;23:194–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.van der Kant R, Langness VF, Herrera CM, Williams DA, Fong LK, Leestemaker Y, et al. Cholesterol metabolism is a druggable axis that independently regulates tau and amyloid-beta in iPSC-derived alzheimer’s disease neurons. Cell Stem Cell. 2019;24 (363–375):e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Liu L, MacKenzie KR, Putluri N, Maletic-Savatic M, Bellen HJ. The Glia-neuron lactate shuttle and elevated ros promote lipid synthesis in neurons and lipid droplet accumulation in glia via APOE/D. Cell Metab. 2017;26:719–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, Sanz E, et al. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell. 2015;160:177–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Yin C, Ackermann S, Ma Z, Mohanta SK, Zhang C, Li Y, et al. ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat Med. 2019;25:496–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Moulton MJ, Barish S, Ralhan I, Chang J, Goodman LD, Harland JG, et al. Neuronal ROS-induced glial lipid droplet formation is altered by loss of alzheimers disease-associated genes. bioRxiv. 2021. [PREPRINT]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Björkhem I, Meaney S. Brain cholesterol: long secret life behind a barrier. Arterioscler Thromb Vasc Biol. 2004;24:806–15. [DOI] [PubMed] [Google Scholar]
- 187.Mauch DH. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–7. [DOI] [PubMed] [Google Scholar]
- 188.Bjorkhem I, Lutjohann D, Diczfalusy U, Stahle L, Ahlborg G, Wahren J. Cholesterol homeostasis in human brain: turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J Lipid Res. 1998;39:1594–600. [PubMed] [Google Scholar]
- 189.Meaney S, Bodin K, Diczfalusy U, Bjorkhem I. On the rate of translocation in vitro and kinetics in vivo of the major oxysterols in human circulation: critical importance of the position of the oxygen function. J Lipid Res. 2002;43:2130–5. [DOI] [PubMed] [Google Scholar]
- 190.Ferris HA, Perry RJ, Moreira GV, Shulman GI, Horton JD, Kahn CR. Loss of astrocyte cholesterol synthesis disrupts neuronal function and alters whole-body metabolism. Proc Natl Acad Sci. 2017; 114:1189–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Boyles JK, Zoellner CD, Anderson LJ, Kosik LM, Pitas RE, Weisgraber KH, et al. A role for apolipoprotein E, apolipoprotein A-I, and low density lipoprotein receptors in cholesterol transport during regeneration and remyelination of the rat sciatic nerve. J Clin Invest. 1989;83:1015–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Michikawa M, Fan QW, Isobe I, Yanagisawa K. Apolipoprotein E exhibits isoform-specific promotion of lipid efflux from astrocytes and neurons in culture. J Neurochem. 2000;74:1008–16. [DOI] [PubMed] [Google Scholar]
- 193.Nunes VS, Cazita PM, Catanozi S, Nakandakare ER, Quintao ECR. Decreased content, rate of synthesis and export of cholesterol in the brain of apoE knockout mice. J Bioenerg Biomembr. 2018;50:283–7. [DOI] [PubMed] [Google Scholar]
- 194.Fuentes D, Fernandez N, Garcia Y, Garcia T, Morales AR, Menendez R. Age-related changes in the behavior of Apolipoprotein E knockout mice. Behav Sci. 2018;8:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Liu Q, Trotter J, Zhang J, Peters MM, Cheng H, Bao J, et al. Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. J Neurosci. 2010;30:17068–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Li X, Zhang J, Li D, He C, He K, Xue T, et al. Astrocytic ApoE reprograms neuronal cholesterol metabolism and histone-acetylation-mediated memory. Neuron. 2021;109(957–970):e8. [DOI] [PubMed] [Google Scholar]
- 197.Mahley RW. Central nervous system lipoproteins: ApoE and regulation of cholesterol metabolism. Arterioscler Thromb Vasc Biol. 2016;36:1305–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Zhao J, Davis MD, Martens YA, Shinohara M, Graff-Radford NR, Younkin SG, et al. APOE epsilon4/epsilon4 diminishes neurotrophic function of human iPSC-derived astrocytes. Hum Mol Genet. 2017;26:2690–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Holtzman DM, Morris JC, Goate AM. Alzheimer’s disease: the challenge of the second century. Sci Transl Med. 2011;3:77sr1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Ehehalt R, Keller P, Haass C, Thiele C, Simons K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J Cell Biol. 2003;160:113–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Cheng H, Vetrivel KS, Gong P, Meckler X, Parent A, Thinakaran G. Mechanisms of disease: new therapeutic strategies for Alzheimer’s disease–targeting APP processing in lipid rafts. Nat Clin Pract Neurol. 2007;3:374–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Pike LJ. Rafts defined: a report on the Keystone Symposium on Lipid Rafts and Cell Function. J Lipid Res. 2006;47:1597–8. [DOI] [PubMed] [Google Scholar]
- 203.Marzolo MP, Bu G. Lipoprotein receptors and cholesterol in APP trafficking and proteolytic processing, implications for Alzheimer’s disease. Semin Cell Dev Biol. 2009;20:191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Puglielli L, Konopka G, Pack-Chung E, Ingano LA, Berezovska O, Hyman BT, et al. Acyl-coenzyme A: cholesterol acyltransferase modulates the generation of the amyloid beta-peptide. Nat Cell Biol. 2001;3:905–12. [DOI] [PubMed] [Google Scholar]
- 205.Rice HC, de Malmazet D, Schreurs A, Frere S, Van Molle I, Volkov AN, et al. Secreted amyloid-beta precursor protein functions as a GABABR1a ligand to modulate synaptic transmission. Science. 2019;363:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kojro E, Gimpl G, Lammich S, Marz W, Fahrenholz F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha -secretase ADAM 10. Proc Natl Acad Sci USA. 2001;98:5815–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Grimm MO, Grimm HS, Patzold AJ, Zinser EG, Halonen R, Duering M, et al. Regulation of cholesterol and sphingomyelin metabolism by amyloid-beta and presenilin. Nat Cell Biol. 2005;7:1118–23. [DOI] [PubMed] [Google Scholar]
- 208.Puglielli L, Ellis BC, Saunders AJ, Kovacs DM. Ceramide stabilizes beta-site amyloid precursor protein-cleaving enzyme 1 and promotes amyloid beta-peptide biogenesis. J Biol Chem. 2003;278:19777–83. [DOI] [PubMed] [Google Scholar]
- 209.Jana A, Pahan K. Fibrillar amyloid-beta peptides kill human primary neurons via NADPH oxidase-mediated activation of neutral sphingomyelinase. Implications for Alzheimer’s Disease. J Biol Chem. 2004;279:51451–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Malaplate-Armand C, Florent-Bechard S, Youssef I, Koziel V, Sponne I, Kriem B, et al. Soluble oligomers of amyloid-beta peptide induce neuronal apoptosis by activating a cPLA2-dependent sphingomyelinase-ceramide pathway. Neurobiol Dis. 2006;23:178–89. [DOI] [PubMed] [Google Scholar]
- 211.Jazvinscak Jembrek M, Hof PR, Simic G. Ceramides in Alzheimer’s disease: key mediators of neuronal apoptosis induced by oxidative stress and abeta accumulation. Oxid Med Cell Longev. 2015;2015:346783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Fong LK, Yang MM, Dos Santos Chaves R, Reyna SM, Langness VF, Woodruff G, et al. Full-length amyloid precursor protein regulates lipoprotein metabolism and amyloid-beta clearance in human astrocytes. J Biol Chem. 2018;293:11341–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Pierrot N, Tyteca D, D’Auria L, Dewachter I, Gailly P, Hendrickx A, et al. Amyloid precursor protein controls cholesterol turnover needed for neuronal activity. EMBO Mol Med. 2013;5:608–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Nguyen HN, Son DJ, Lee JW, Hwang DY, Kim YK, Cho JS, et al. Mutant presenilin 2 causes abnormality in the brain lipid profile in the development of Alzheimer’s disease. Arch Pharm Res. 2006;29:884–9. [DOI] [PubMed] [Google Scholar]
- 215.Gutierrez E, Lutjohann D, Kerksiek A, Fabiano M, Oikawa N, Kuerschner L, et al. Importance of gamma-secretase in the regulation of liver X receptor and cellular lipid metabolism. Life Sci Alliance. 2020;3:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Cadenas E Mitochondrial free radical production and cell signaling. Mol Aspects Med. 2004;25:17–26. [DOI] [PubMed] [Google Scholar]
- 217.Yin F, Sancheti H, Cadenas E. Mitochondrial thiols in the regulation of cell death pathways. Antioxid Redox Signal. 2012;17:1714–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Yin F, Boveris A, Cadenas E. Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid Redox Signal. 2014;20:353–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Yin F, Cadenas E. Mitochondria: the cellular hub of the dynamic coordinated network. Antioxid Redox Signal. 2015;22:961–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Yin F, Sancheti H, Liu Z, Cadenas E. Mitochondrial function in ageing: coordination with signalling and transcriptional pathways. J Physiol. 2016;594:2025–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Giorgi C, Missiroli S, Patergnani S, Duszynski J, Wieckowski MR, Pinton P. Mitochondria-associated membranes: composition, molecular mechanisms, and physiopathological implications. Antioxid Redox Signal. 2015;22:995–1019. [DOI] [PubMed] [Google Scholar]
- 222.Vance JE. MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim Biophys Acta. 2014;1841:595–609. [DOI] [PubMed] [Google Scholar]
- 223.Pera M, Larrea D, Guardia-Laguarta C, Montesinos J, Velasco KR, Agrawal RR, et al. Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. Embo J. 2017;36:3356–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Area-Gomez E, de Groof AJ, Boldogh I, Bird TD, Gibson GE, Koehler CM, et al. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am J Pathol. 2009;175:1810–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Area-Gomez E, de Groof A, Bonilla E, Montesinos J, Tanji K, Boldogh I, et al. A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis. 2018;9:335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Lee JT, Xu J, Lee JM, Ku G, Han X, Yang DI, et al. Amyloid-beta peptide induces oligodendrocyte death by activating the neutral sphingomyelinase-ceramide pathway. J Cell Biol. 2004;164:123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Bhattacharyya R, Black SE, Lotlikar MS, Fenn RH, Jorfi M, Kovacs DM, et al. Axonal generation of amyloid-beta from palmitoylated APP in mitochondria-associated endoplasmic reticulum membranes. Cell Rep. 2021;35:109134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Bhattacharyya R, Barren C, Kovacs DM. Palmitoylation of amyloid precursor protein regulates amyloidogenic processing in lipid rafts. J Neurosci. 2013;33:11169–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Area-Gomez E, Castillo DCL, Tambini MD, Guardia-Laguarta C, de Groof AJ, Madra M, et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. Embo J. 2012;31:4106–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Tambini MD, Pera M, Kanter E, Yang H, Guardia-Laguarta C, Holtzman D, et al. ApoE4 upregulates the activity of mitochondria-associated ER membranes. EMBO Rep. 2016;17:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Yu RK, Tsai YT, Ariga T, Yanagisawa M. Structures, biosynthesis, and functions of gangliosides–an overview. J Oleo Sci. 2011;60:537–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Yanagisawa K, Odaka A, Suzuki N, Ihara Y. GM1 ganglioside-bound amyloid beta-protein (A beta): a possible form of preamyloid in Alzheimer’s disease .Nat Med. 1995;1:1062–6. [DOI] [PubMed] [Google Scholar]
- 233.Matsuzaki K. Abeta-ganglioside interactions in the pathogenesis of Alzheimer’s disease, Biochim Biophys Acta. Biomembr 2020;1862:183233. [DOI] [PubMed] [Google Scholar]
- 234.Kanekiyo T, Xu H, Bu G. ApoE and Abeta in Alzheimer’s disease: accidental encounters or partners? Neuron. 2014;81:740–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Hu J, Liu CC, Chen XF, Zhang YW, Xu H, Bu G. Opposing effects of viral mediated brain expression of apolipoprotein E2 (apoE2) and apoE4 on apoE lipidation and Abeta metabolism in apoE4-targeted replacement mice. Mol Neurodegener. 2015;10:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Lanfranco MF, Ng CA, Rebeck GW. ApoE lipidation as a therapeutic target in alzheimer’s disease. Int J Mol Sci. 2020;21:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Heinsinger NM, Gachechiladze MA, Rebeck GW. Apolipoprotein E genotype affects size of ApoE complexes in cerebrospinal fluid. J Neuropathol Exp Neurol. 2016;75:918–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Hanson AJ, Bayer-Carter JL, Green PS, Montine TJ, Wilkinson CW, Baker LD, et al. Effect of apolipoprotein E genotype and diet on apolipoprotein E lipidation and amyloid peptides: randomized clinical trial. JAMA Neurol. 2013;70:972–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS, Frail DE. Isoform-specific binding of apolipoprotein E to beta-amyloid. J Biol Chem. 1994;269:23403–6. [PubMed] [Google Scholar]
- 240.Verghese PB, Castellano JM, Garai K, Wang Y, Jiang H, Shah A, et al. ApoE influences amyloid-beta (Abeta) clearance despite minimal apoE/Abeta association in physiological conditions. Proc Natl Acad Sci USA. 2013;110:E1807–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Glockner F, Ohm TG. Tau pathology induces intraneuronal cholesterol accumulation. J Neuropathol Exp Neurol. 2014;73:846–54. [DOI] [PubMed] [Google Scholar]
- 242.Glockner F, Meske V, Lutjohann D, Ohm TG. Dietary cholesterol and its effect on tau protein: a study in apolipoprotein E-deficient and P301L human tau mice. J Neuropathol Exp Neurol. 2011;70:292–301. [DOI] [PubMed] [Google Scholar]
- 243.Perreault S, Bousquet O, Lauzon M, Paiement J, Leclerc N. Increased association between rough endoplasmic reticulum membranes and mitochondria in transgenic mice that express P301L tau. J Neuropathol Exp Neurol. 2009;68:503–14. [DOI] [PubMed] [Google Scholar]
- 244.Motta C, Assogna M, Bonomi CG, Mascolo AP, De Lucia V, Semprini R, et al. Diabetes mellitus contributes to higher cerebrospinal fluid tau levels selectively in Alzheimer’s disease patients with the APOE4 genotype. Eur J Neurol. 2021;28:3965–71. [DOI] [PubMed] [Google Scholar]
- 245.Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol. 2021;17:157–72. [DOI] [PubMed] [Google Scholar]
- 246.Daynes RA, Jones DC. Emerging roles of PPARs in inflammation and immunity. Nat Rev Immunol. 2002;2:748–59. [DOI] [PubMed] [Google Scholar]
- 247.Basil MC, Levy BD. Specialized pro-resolving mediators: endogenous regulators of infection and inflammation. Nat Rev Immunol. 2016;16:51–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Whittington RA, Planel E, Terrando N. Impaired resolution of inflammation in Alzheimer’s disease: a review. Front Immunol. 2017;8:1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Emre C, Hjorth E, Bharani K, Carroll S, Granholm AC, Schultzberg M. Receptors for pro-resolving mediators are increased in Alzheimer’s disease brain. Brain Pathol. 2020;30:614–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Zhu M, Wang X, Hjorth E, Colas RA, Schroeder L, Granholm AC, et al. Pro-resolving lipid mediators improve neuronal survival and increase Abeta42 phagocytosis. Mol Neurobiol. 2016;53:2733–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Wang X, Zhu M, Hjorth E, Cortes-Toro V, Eyjolfsdottir H, Graff C, et al. Resolution of inflammation is altered in Alzheimer’s disease. Alzheimers Dement. 2015;11(40–50):e1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Kantarci A, Aytan N, Palaska I, Stephens D, Crabtree L, Benincasa C, et al. Combined administration of resolvin E1 and lipoxin A4 resolves inflammation in a murine model of Alzheimer’s disease. Exp Neurol. 2018;300:111–20. [DOI] [PubMed] [Google Scholar]
- 253.Dunn HC, Ager RR, Baglietto-Vargas D, Cheng D, Kitazawa M, Cribbs DH, et al. Restoration of lipoxin A4 signaling reduces Alzheimer’s disease-like pathology in the 3xTg-AD mouse model. J Alzheimers Dis. 2015;43:893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Kang J, Rivest S. Lipid metabolism and neuroinflammation in Alzheimer’s disease: a role for liver X receptors. Endocr Rev. 2012;33:715–46. [DOI] [PubMed] [Google Scholar]
- 255.Heneka MT, Reyes-Irisarri E, Hull M, Kummer MP. Impact and therapeutic potential of PPARs in alzheimer’s disease. Curr Neuropharmacol. 2011;9:643–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Montine TJ, Sidell KR, Crews BC, Markesbery WR, Marnett LJ, Roberts LJ 2nd, et al. Elevated CSF prostaglandin E2 levels in patients with probable AD. Neurology. 1999;53:1495–8. [DOI] [PubMed] [Google Scholar]
- 258.Combrinck M, Williams J, De Berardinis MA, Warden D, Puopolo M, Smith AD, et al. Levels of CSF prostaglandin E2, cognitive decline, and survival in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2006;77:85–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Shi J, Wang Q, Johansson JU, Liang X, Woodling NS, Priyam P, et al. Inflammatory prostaglandin E2 signaling in a mouse model of Alzheimer disease. Ann Neurol. 2012;72:788–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Liang X, Wang Q, Hand T, Wu L, Breyer RM, Montine TJ, et al. Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer’s disease. J Neurosci. 2005;25:10180–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Johansson JU, Woodling NS, Wang Q, Panchal M, Liang X, Trueba-Saiz A, et al. Prostaglandin signaling suppresses beneficial microglial function in Alzheimer’s disease models. J Clin Invest. 2015;125:350–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Minhas PS, Latif-Hernandez A, McReynolds MR, Durairaj AS, Wang Q, Rubin A, et al. Restoring metabolism of myeloid cells reverses cognitive decline in ageing. Nature. 2021;590:122–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Vlad SC, Miller DR, Kowall NW, Felson DT. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology. 2008;70:1672–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Dionisio-Santos DA, Olschowka JA, O’Banion MK. Exploiting microglial and peripheral immune cell crosstalk to treat Alzheimer’s disease. J Neuroinflammation. 2019;16:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Zhang C, Wang Y, Wang D, Zhang J, Zhang F. NSAID exposure and risk of alzheimer’s disease: an updated meta-analysis from cohort studies. Front Aging Neurosci. 2018;10:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.In’t Veld BA, Launer LJ, Hoes AW, Ott A, Hofman A, Breteler M, et al. NSAIDs and incident Alzheimer’s disease. Neurobiol Aging. 1998;19:607–11. [DOI] [PubMed] [Google Scholar]
- 267.Meyer PF, Tremblay-Mercier J, Leoutsakos J, Madjar C, Lafaille-Maignan ME, Savard M, et al. INTREPAD: A randomized trial of naproxen to slow progress of presymptomatic Alzheimer disease. Neurology. 2019;92:e2070–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Group, A. R., Martin BK, Szekely C, Brandt J, Piantadosi S, Breitner JC, et al. Cognitive function over time in the Alzheimer’s Disease Antiinflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch Neurol. 2008;65:896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Sanchez-Mejia RO, Newman JW, Toh S, Yu GQ, Zhou Y, Halabisky B, et al. Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer’s disease. Nat Neurosci. 2008;11:1311–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Miller YI, Navia-Pelaez JM, Corr M, Yaksh TL. Lipid rafts in glial cells: role in neuroinflammation and pain processing. J Lipid Res. 2020;61:655–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med. 2000;29:222–30. [DOI] [PubMed] [Google Scholar]
- 272.Benseny-Cases N, Klementieva O, Cotte M, Ferrer I, Cladera J. Microspectroscopy (muFTIR) reveals colocalization of lipid oxidation and amyloid plaques in human Alzheimer disease brains. Anal Chem. 2014;86:12047–54. [DOI] [PubMed] [Google Scholar]
- 273.Prasad VV, Nithipatikom K, Harder DR. Ceramide elevates 12-hydroxyeicosatetraenoic acid levels and upregulates 12-lipoxygenase in rat primary hippocampal cell cultures containing predominantly astrocytes. Neurochem Int. 2008;53:220–9. [DOI] [PubMed] [Google Scholar]
- 274.Sultana R, Perluigi M, Butterfield DA. Lipid peroxidation triggers neurodegeneration: a redox proteomics view into the Alzheimer disease brain. Free Radic Biol Med. 2013;62:157–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Markesbery WR, Kryscio RJ, Lovell MA, Morrow JD. Lipid peroxidation is an early event in the brain in amnestic mild cognitive impairment. Ann Neurol. 2005;58:730–5. [DOI] [PubMed] [Google Scholar]
- 276.Keller JN, Schmitt FA, Scheff SW, Ding Q, Chen Q, Butterfield DA, et al. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64:1152–6. [DOI] [PubMed] [Google Scholar]
- 277.Markesbery WR, Carney JM. Oxidative alterations in Alzheimer’s disease. Brain Pathol. 1999;9:133–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Ramassamy C, Averill D, Beffert U, Theroux L, Lussier-Cacan S, Cohn JS, et al. Oxidative insults are associated with apolipoprotein E genotype in Alzheimer’s disease brain. Neurobiol Dis. 2000;7:23–37. [DOI] [PubMed] [Google Scholar]
- 279.Dotson PP 2nd, Karakashian AA, Nikolova-Karakashian MN. Neutral sphingomyelinase-2 is a redox sensitive enzyme: role of catalytic cysteine residues in regulation of enzymatic activity through changes in oligomeric state. Biochem J. 2015;465:371–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Jayadev S, Linardic CM, Hannun YA. Identification of arachidonic acid as a mediator of sphingomyelin hydrolysis in response to tumor necrosis factor alpha. J Biol Chem. 1994;269:5757–63. [PubMed] [Google Scholar]
- 281.Panchal M, Gaudin M, Lazar AN, Salvati E, Rivals I, Ayciriex S, et al. Ceramides and sphingomyelinases in senile plaques. Neurobiol Dis. 2014;65:193–201. [DOI] [PubMed] [Google Scholar]
- 282.Lee JK, Jin HK, Park MH, Kim BR, Lee PH, Nakauchi H, et al. Acid sphingomyelinase modulates the autophagic process by controlling lysosomal biogenesis in Alzheimer’s disease. J Exp Med. 2014;211:1551–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Dinkins MB, Enasko J, Hernandez C, Wang G, Kong J, Helwa I, et al. Neutral sphingomyelinase-2 deficiency ameliorates alzheimer’s disease pathology and improves cognition in the 5XFAD mouse. J Neurosci. 2016;36:8653–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Snaidero N, Simons M. Myelination at a glance. J Cell Sci. 2014;127:2999–3004. [DOI] [PubMed] [Google Scholar]
- 285.Nave KA, Werner HB. Myelination of the nervous system: mechanisms and functions. Annu Rev Cell Dev Biol. 2014;30:503–33. [DOI] [PubMed] [Google Scholar]
- 286.O’Brien JS, Sampson EL. Lipid composition of the normal human brain: gray matter, white matter, and myelin. J Lipid Res. 1965;6:537–44. [PubMed] [Google Scholar]
- 287.Chrast R, Saher G, Nave KA, Verheijen MH. Lipid metabolism in myelinating glial cells: lessons from human inherited disorders and mouse models. J Lipid Res. 2011;52:419–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Nasrabady SE, Rizvi B, Goldman JE, Brickman AM. White matter changes in Alzheimer’s disease: a focus on myelin and oligodendrocytes. Acta Neuropathol Commun. 2018;6:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Lee S, Viqar F, Zimmerman ME, Narkhede A, Tosto G, Benzinger TL, et al. White matter hyperintensities are a core feature of Alzheimer’s disease: Evidence from the dominantly inherited Alzheimer network. Ann Neurol. 2016;79:929–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Tosto G, Zimmerman ME, Hamilton JL, Carmichael OT, Brickman AM, Neuroimaging AD, et al. The effect of white matter hyperintensities on neurodegeneration in mild cognitive impairment. Alzheimers Dement. 2015;11:1510–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Dimas P, Montani L, Pereira JA, Moreno D, Trotzmuller M, Gerber J, et al. CNS myelination and remyelination depend on fatty acid synthesis by oligodendrocytes. Elife. 2019;8:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Camargo N, Goudriaan A, van Deijk AF, Otte WM, Brouwers JF, Lodder H, et al. Oligodendroglial myelination requires astrocyte-derived lipids. PLoS Biol. 2017;15:e1002605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Zhao Y, Wu X, Li X, Jiang LL, Gui X, Liu Y, et al. TREM2 Is a receptor for beta-amyloid that mediates microglial Function. Neuron. 2018;97(1023–1031):e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Gouna G, Klose C, Bosch-Queralt M, Liu L, Gokce O, Schifferer M, et al. TREM2-dependent lipid droplet biogenesis in phagocytes is required for remyelination. J Exp Med. 2021;218:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Nugent AA, Lin K, van Lengerich B, Lianoglou S, Przybyla L, Davis SS, et al. TREM2 Regulates Microglial Cholesterol Metabolism upon Chronic Phagocytic Challenge. Neuron. 2020;105(837–854):e9. [DOI] [PubMed] [Google Scholar]
- 296.Segarra M, Aburto MR, Acker-Palmer A. Blood-brain barrier dynamics to maintain brain homeostasis. Trends Neurosci. 2021;44:393–405. [DOI] [PubMed] [Google Scholar]
- 297.Montagne A, Zhao Z, Zlokovic BV. Alzheimer’s disease: A matter of blood-brain barrier dysfunction? J Exp Med. 2017;214:3151–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Nation DA, Sweeney MD, Montagne A, Sagare AP, D’Orazio LM, Pachicano M, et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. 2019;25:270–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Ben-Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, Yan H, et al. Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature. 2014;509:507–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Andreone BJ, Chow BW, Tata A, Lacoste B, Ben-Zvi A, Bullock K, et al. Blood-brain barrier permeability is regulated by lipid transport-dependent suppression of caveolae-mediated transcytosis. Neuron. 2017;94 (581–594):e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Pan Y, Choy KHC, Marriott PJ, Chai SY, Scanlon MJ, Porter CJH, et al. Reduced blood-brain barrier expression of fatty acid-binding protein 5 is associated with increased vulnerability of APP/PS1 mice to cognitive deficits from low omega-3 fatty acid diets. J Neurochem. 2018;144:81–92. [DOI] [PubMed] [Google Scholar]
- 302.Rhea EM, Salameh TS, Logsdon AF, Hanson AJ, Erickson MA, Banks WA. Blood-brain barriers in obesity. AAPS J. 2017;19:921–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Ouyang S, Hsuchou H, Kastin AJ, Wang Y, Yu C, Pan W. Diet-induced obesity suppresses expression of many proteins at the blood-brain barrier. J Cereb Blood Flow Metab. 2014;34:43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Melo HM, Seixas da Silva GDS, Sant’Ana MR, Teixeira CVL, Clarke JR, Miya Coreixas VS, et al. Palmitate is increased in the cerebrospinal fluid of humans with obesity and induces memory impairment in mice via pro-inflammatory TNF-alpha. Cell Rep. 2020;30(2180–2194):e8. [DOI] [PubMed] [Google Scholar]
- 305.Ma L, Nelson ER. Oxysterols and nuclear receptors. Mol Cell Endocrinol. 2019;484:42–51. [DOI] [PubMed] [Google Scholar]
- 306.Olivares AM, Moreno-Ramos OA, Haider NB. Role of nuclear receptors in central nervous system development and associated diseases. J Exp Neurosci. 2015;9:93–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Zelcer N, Tontonoz P. Liver X receptors as integrators of metabolic and inflammatory signaling. J Clin Invest. 2006;116:607–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Gofflot F, Chartoire N, Vasseur L, Heikkinen S, Dembele D, Le Merrer J, et al. Systematic gene expression mapping clusters nuclear receptors according to their function in the brain. Cell. 2007;131:405–18. [DOI] [PubMed] [Google Scholar]
- 309.Andersson S, Gustafsson N, Warner M, Gustafsson JA. Inactivation of liver X receptor beta leads to adult-onset motor neuron degeneration in male mice. Proc Natl Acad Sci USA. 2005;102:3857–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Zelcer N, Khanlou N, Clare R, Jiang Q, Reed-Geaghan EG, Landreth GE, et al. Attenuation of neuroinflammation and Alzheimer’s disease pathology by liver x receptors. Proc Natl Acad Sci USA. 2007;104:10601–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Fitz NF, Nam KN, Koldamova R, Lefterov I. Therapeutic targeting of nuclear receptors, liver X and retinoid X receptors, for Alzheimer’s disease. Br J Pharmacol. 2019;176:3599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, et al. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Boehm-Cagan A, Michaelson DM. Reversal of apoE4-driven brain pathology and behavioral deficits by bexarotene. J Neurosci. 2014;34:7293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Ghosal K, Haag M, Verghese PB, West T, Veenstra T, Braunstein JB, et al. A randomized controlled study to evaluate the effect of bexarotene on amyloid-beta and apolipoprotein E metabolism in healthy subjects. Alzheimers Dement. 2016;2:110–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Cummings JL, Zhong K, Kinney JW, Heaney C, Moll-Tudla J, Joshi A, et al. Double-blind, placebo-controlled, proof-of-concept trial of bexarotene Xin moderate Alzheimer’s disease. Alzheimers Res Ther. 2016;8:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Pierrot N, Ris L, Stancu IC, Doshina A, Ribeiro F, Tyteca D, et al. Sex-regulated gene dosage effect of PPARalpha on synaptic plasticity. Life Sci Alliance. 2019;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Wang YX. PPARs: diverse regulators in energy metabolism and metabolic diseases. Cell Res. 2010;20:124–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–88. [DOI] [PubMed] [Google Scholar]
- 319.DiBattista AM, Dumanis SB, Newman J, Rebeck GW. Identification and modification of amyloid-independent phenotypes of APOE4 mice. Exp Neurol. 2016;280:97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Sastre M, Dewachter I, Landreth GE, Willson TM, Klockgether T, van Leuven F, et al. Nonsteroidal antiinflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J Neurosci. 2003;23:9796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Pedersen WA, McMillan PJ, Kulstad JJ, Leverenz JB, Craft S, Haynatzki GR. Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Exp Neurol. 2006;199:265–73. [DOI] [PubMed] [Google Scholar]
- 322.Mandrekar-Colucci S, Karlo JC, Landreth GE. Mechanisms underlying the rapid peroxisome proliferator-activated receptor-gamma-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer’s disease. J Neurosci. 2012;32:10117–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Yamanaka M, Ishikawa T, Griep A, Axt D, Kummer MP, Heneka MT. PPARgamma/RXRalpha-induced and CD36-mediated microglial amyloid-beta phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J Neurosci. 2012;32:17321–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.O’Reilly JA, Lynch M. Rosiglitazone improves spatial memory and decreases insoluble Abeta(1–42) in APP/PS1 mice. J Neuroimmune Pharmacol. 2012;7:140–4. [DOI] [PubMed] [Google Scholar]
- 325.Toledo EM, Inestrosa NC. Activation of Wnt signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1DeltaE9 mouse model of Alzheimer’s disease. Mol Psychiatry. 2010;15(272–85):228. [DOI] [PubMed] [Google Scholar]
- 326.Yu Y, Li X, Blanchard J, Li Y, Iqbal K, Liu F, et al. Insulin sensitizers improve learning and attenuate tau hyperphosphorylation and neuroinflammation in 3xTg-AD mice. J Neural Transm (Vienna). 2015;122:593–606. [DOI] [PubMed] [Google Scholar]
- 327.Hanyu H, Sato T, Kiuchi A, Sakurai H, Iwamoto T. Pioglitazone improved cognition in a pilot study on patients with Alzheimer’s disease and mild cognitive impairment with diabetes mellitus. J Am Geriatr Soc. 2009;57:177–9. [DOI] [PubMed] [Google Scholar]
- 328.Sato T, Hanyu H, Hirao K, Kanetaka H, Sakurai H, Iwamoto T. Efficacy of PPAR-gamma agonist pioglitazone in mild Alzheimer disease. Neurobiol Aging. 2011;32:1626–33. [DOI] [PubMed] [Google Scholar]
- 329.Burns DK, Alexander RC, Welsh-Bohmer KA, Culp M, Chiang C, O’Neil J, et al. Safety and efficacy of pioglitazone for the delay of cognitive impairment in people at risk of Alzheimer’s disease (TOMMORROW): a prognostic biomarker study and a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2021;20:537–47. [DOI] [PubMed] [Google Scholar]
- 330.Gold M, Alderton C, Zvartau-Hind M, Egginton S, Saunders AM, Irizarry M, et al. Rosiglitazone monotherapy in mild-to-moderate Alzheimer’s disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cogn Disord. 2010;30:131–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Watson GS, Cholerton BA, Reger MA, Baker LD, Plymate SR, Asthana S, et al. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: a preliminary study. Am J Geriatr Psychiatry. 2005;13:950–8. [DOI] [PubMed] [Google Scholar]
- 332.Tzimopoulou S, Cunningham VJ, Nichols TE, Searle G, Bird NP, Mistry P, et al. A multi-center randomized proof-of-concept clinical trial applying [(1)(8)F]FDG-PET for evaluation of metabolic therapy with rosiglitazone XR in mild to moderate Alzheimer’s disease. J Alzheimers Dis. 2010;22:1241–56. [DOI] [PubMed] [Google Scholar]
- 333.Harrington C, Sawchak S, Chiang C, Davies J, Donovan C, Saunders AM, et al. Rosiglitazone does not improve cognition or global function when used as adjunctive therapy to AChE inhibitors in mild-to-moderate Alzheimer’s disease: two phase 3 studies. Curr Alzheimer Res. 2011;8:592–606. [DOI] [PubMed] [Google Scholar]
- 334.Risner ME, Saunders AM, Altman JF, Ormandy GC, Craft S, Foley IM, et al. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics J. 2006;6:246–54. [DOI] [PubMed] [Google Scholar]
- 335.Feinstein DL, Spagnolo A, Akar C, Weinberg G, Murphy P, Gavrilyuk V, et al. Receptor-independent actions of PPAR thiazolidinedione agonists: is mitochondrial function the key? Biochem Pharmacol. 2005;70:177–88. [DOI] [PubMed] [Google Scholar]
- 336.Reddy JK, Hashimoto T. Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: an adaptive metabolic system. Annu Rev Nutr. 2001;21:193–230. [DOI] [PubMed] [Google Scholar]
- 337.Basu-Modak S, Braissant O, Escher P, Desvergne B, Honegger P, Wahli W. Peroxisome proliferator-activated receptor beta regulates acyl-CoA synthetase 2 in reaggregated rat brain cell cultures. J Biol Chem. 1999;274:35881–8. [DOI] [PubMed] [Google Scholar]
- 338.Roy A, Jana M, Corbett GT, Ramaswamy S, Kordower JH, Gonzalez FJ, et al. Regulation of cyclic AMP response element binding and hippocampal plasticity-related genes by peroxisome proliferator-activated receptor alpha. Cell Rep. 2013;4:724–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Zhang H, Gao Y, Qiao PF, Zhao FL, Yan Y. PPAR-alpha agonist regulates amyloid-beta generation via inhibiting BACE-1 activity in human neuroblastoma SH-SY5Y cells transfected with APPswe gene. Mol Cell Biochem. 2015;408:37–46. [DOI] [PubMed] [Google Scholar]
- 340.Corbett GT, Gonzalez FJ, Pahan K. Activation of peroxisome proliferator-activated receptor alpha stimulates ADAM10-mediated proteolysis of APP. Proc Natl Acad Sci USA. 2015;112:8445–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Santos MJ, Quintanilla RA, Toro A, Grandy R, Dinamarca MC, Godoy JA, et al. Peroxisomal proliferation protects from beta-amyloid neurodegeneration. J Biol Chem. 2005;280:41057–68. [DOI] [PubMed] [Google Scholar]
- 342.Roy A, Kundu M, Jana M, Mishra RK, Yung Y, Luan CH, et al. Identification and characterization of PPARalpha ligands in the hippocampus. Nat Chem Biol. 2016;12:1075–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Inestrosa NC, Carvajal FJ, Zolezzi JM, Tapia-Rojas C, Serrano F, Karmelic D, et al. Peroxisome proliferators reduce spatial memory impairment, synaptic failure, and neurodegeneration in brains of a double transgenic mice model of Alzheimer’s disease. J Alzheimers Dis. 2013;33:941–59. [DOI] [PubMed] [Google Scholar]
- 344.Luo R, Su LY, Li G, Yang J, Liu Q, Yang LX, et al. Activation of PPARA-mediated autophagy reduces Alzheimer disease-like pathology and cognitive decline in a murine model. Autophagy. 2020;16:52–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Esmaeili MA, Yadav S, Gupta RK, Waggoner GR, Deloach A, Calingasan NY, et al. Preferential PPAR-alpha activation reduces neuroinflammation, and blocks neurodegeneration in vivo. Hum Mol Genet. 2016;25:317–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Pettegrew JW, Levine J, McClure RJ. Acetyl-L-carnitine physical-chemical, metabolic, and therapeutic properties: relevance for its mode of action in Alzheimer’s disease and geriatric depression. Mol Psychiatry. 2000;5:616–32. [DOI] [PubMed] [Google Scholar]
- 347.Lodeiro M, Ibanez C, Cifuentes A, Simo C, Cedazo-Minguez A. Decreased cerebrospinal fluid levels of L-carnitine in non-apolipoprotein E4 carriers at early stages of Alzheimer’s disease. J Alzheimers Dis. 2014;41:223–32. [DOI] [PubMed] [Google Scholar]
- 348.Mota SI, Pita I, Aguas R, Tagorti S, Virmani A, Pereira FC, et al. Mechanistic perspectives on differential mitochondrial-based neuroprotective effects of several carnitine forms in Alzheimer’s disease in vitro model. Arch Toxicol. 2021;95:2769–84. [DOI] [PubMed] [Google Scholar]
- 349.Hagen TM, Ingersoll RT, Wehr CM, Lykkesfeldt J, Vinarsky V, Bartholomew JC, et al. Acetyl-L-carnitine fed to old rats partially restores mitochondrial function and ambulatory activity. Proc Natl Acad Sci USA. 1998;95:9562–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Barnes CA, Markowska AL, Ingram DK, Kametani H, Spangler EL, Lemken VJ, et al. Acetyl-1-carnitine. 2: Effects on learning and memory performance of aged rats in simple and complex mazes. Neurobiol Aging. 1990;11:499–506. [DOI] [PubMed] [Google Scholar]
- 351.Hudson S, Tabet N. Acetyl-L-carnitine for dementia. Cochrane Database Syst Rev. 2003;58:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Montgomery SA, Thal LJ, Amrein R. Meta-analysis of double blind randomized controlled clinical trials of acetyl-L-carnitine versus placebo in the treatment of mild cognitive impairment and mild Alzheimer’s disease. Int Clin Psychopharmacol. 2003;18:61–71. [DOI] [PubMed] [Google Scholar]
- 353.Wang C, Najm R, Xu Q, Jeong DE, Walker D, Balestra ME, et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat Med. 2018;24:647–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Mahley RW, Huang Y. Small-molecule structure correctors target abnormal protein structure and function: structure corrector rescue of apolipoprotein E4-associated neuropathology. J Med Chem. 2012;55:8997–9008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Huynh TV, Liao F, Francis CM, Robinson GO, Serrano JR, Jiang H, et al. Age-dependent effects of apoe reduction using antisense oligonucleotides in a model of beta-amyloidosis. Neuron. 2017;96(1013–1023):e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Liao F, Li A, Xiong M, Bien-Ly N, Jiang H, Zhang Y, et al. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J Clin Invest. 2018;128:2144–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Boehm-Cagan A, Bar R, Liraz O, Bielicki JK, Johansson JO, Michaelson DM. ABCA1 agonist reverses the ApoE4-driven cognitive and brain pathologies. J Alzheimers Dis. 2016;54:1219–33. [DOI] [PubMed] [Google Scholar]
- 358.Zhang Y, Chen J, Qiu J, Li Y, Wang J, Jiao J. Intakes of fish and polyunsaturated fatty acids and mild-to-severe cognitive impairment risks: a dose-response meta-analysis of 21 cohort studies. Am J Clin Nutr. 2016;103:330–40. [DOI] [PubMed] [Google Scholar]
- 359.Grimm MO, Mett J, Stahlmann CP, Haupenthal VJ, Blumel T, Stotzel H, et al. Eicosapentaenoic acid and docosahexaenoic acid increase the degradation of amyloid-beta by affecting insulin-degrading enzyme. Biochem Cell Biol. 2016;94:534–42. [DOI] [PubMed] [Google Scholar]
- 360.Grimm MO, Kuchenbecker J, Grosgen S, Burg VK, Hundsdorfer B, Rothhaar TL, et al. Docosahexaenoic acid reduces amyloid beta production via multiple pleiotropic mechanisms. J Biol Chem. 2011;286:14028–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Hopperton KE, Trepanier MO, Giuliano V, Bazinet RP. Brain omega-3 polyunsaturated fatty acids modulate microglia cell number and morphology in response to intracerebroventricular amyloid-beta 1–40 in mice. J Neuroinflammation. 2016;13:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.de Urquiza AM, Liu S, Sjoberg M, Zetterstrom RH, Griffiths W, Sjovall J, et al. Docosahexaenoic acid, a ligand for the retinoid X receptor in mouse brain. Science. 2000;290:2140–4. [DOI] [PubMed] [Google Scholar]
- 363.Forman BM, Chen J, Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc Natl Acad Sci USA. 1997;94:4312–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Casali BT, Corona AW, Mariani MM, Karlo JC, Ghosal K, Landreth GE. Omega-3 fatty acids augment the actions of nuclear receptor agonists in a mouse model of alzheimer’s disease. J Neurosci. 2015;35:9173–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Fiala M, Halder RC, Sagong B, Ross O, Sayre J, Porter V, et al. omega-3 Supplementation increases amyloid-beta phagocytosis and resolvin D1 in patients with minor cognitive impairment. FASEB J. 2015;29:2681–9. [DOI] [PubMed] [Google Scholar]
- 366.Famenini S, Rigali EA, Olivera-Perez HM, Dang J, Chang MT, Halder R, et al. Increased intermediate M1–M2 macrophage polarization and improved cognition in mild cognitive impairment patients on omega-3 supplementation. FASEB J. 2017;31:148–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Grimm MO, Haupenthal VJ, Mett J, Stahlmann CP, Blumel T, Mylonas NT, et al. Oxidized docosahexaenoic acid species and lipid peroxidation products increase amyloidogenic amyloid precursor protein processing. Neurodegener Dis. 2016;16:44–54. [DOI] [PubMed] [Google Scholar]
- 368.Freund-Levi Y, Eriksdotter-Jonhagen M, Cederholm T, Basun H, Faxen-Irving G, Garlind A, et al. Omega-3 fatty acid treatment in 174 patients with mild to moderate Alzheimer disease: OmegAD study: a randomized double-blind trial. Arch Neurol. 2006;63:1402–8. [DOI] [PubMed] [Google Scholar]
- 369.Quinn JF, Raman R, Thomas RG, Yurko-Mauro K, Nelson EB, Van Dyck C, et al. Docosahexaenoic acid supplementation and cognitive decline in Alzheimer disease: a randomized trial. JAMA. 2010;304:1903–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Cunnane S, Nugent S, Roy M, Courchesne-Loyer A, Croteau E, Tremblay S, et al. Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition. 2011;27:3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Yin F, Yao J, Sancheti H, Feng T, Melcangi RC, Morgan TE, et al. The perimenopausal aging transition in the female rat brain: decline in bioenergetic systems and synaptic plasticity. Neurobiol Aging. 2015;36:2282–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Ding F, Yao J, Rettberg JR, Chen S, Brinton RD. Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer’s mouse brain: implication for bioenergetic intervention. PLoS One. 2013;8:e79977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Klosinski LP, Yao J, Yin F, Fonteh AN, Harrington MG, Christensen TA, et al. White matter lipids as a ketogenic fuel supply in aging female brain: implications for alzheimer’s disease. EBioMedicine. 2015;2:1888–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Wang Y, Shang Y, Mishra A, Bacon E, Yin F, Brinton R. Midlife chronological and endocrinological transitions in brain metabolism: system biology basis for increased alzheimer’s risk in female brain. Sci Rep. 2020;10:8528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Brinton RD, Yao J, Yin F, Mack WJ, Cadenas E. Perimenopause as a neurological transition state. Nat Rev Endocrinol. 2015;11:393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Mattson MP, Moehl K, Ghena N, Schmaedick M, Cheng A. Intermittent metabolic switching, neuroplasticity and brain health. Nat Rev Neurosci. 2018;19:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Yang H, Shan W, Zhu F, Wu J, Wang Q. Ketone bodies in neurological diseases: focus on neuroprotection and underlying mechanisms. Front Neurol. 2019;10:585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Van der Auwera I, Wera S, Van Leuven F, Henderson ST. A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer’s disease. Nutrition & Metabolism. 2005;2:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Newman JC, Covarrubias AJ, Zhao M, Yu X, Gut P, Ng C-P, et al. Ketogenic diet reduces midlife mortality and improves memory in aging mice. Cell Metab. 2017;26(547–557):e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Neth BJ, Mintz A, Whitlow C, Jung Y, Solingapuram Sai K, Register TC, et al. Modified ketogenic diet is associated with improved cerebrospinal fluid biomarker profile, cerebral perfusion, and cerebral ketone body uptake in older adults at risk for Alzheimer’s disease: a pilot study. Neurobiol Aging. 2020;86:54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Ota M, Matsuo J, Ishida I, Takano H, Yokoi Y, Hori H, et al. Effects of a medium-chain triglyceride-based ketogenic formula on cognitive function in patients with mild-to-moderate Alzheimer’s disease. Neurosci Lett. 2019;690:232–6. [DOI] [PubMed] [Google Scholar]
- 382.Taylor MK, Sullivan DK, Mahnken JD, Burns JM, Swerdlow RH. Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer’s disease. Alzheimers Dement (N Y). 2018;4:28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Kimoto A, Ohnuma T, Toda A, Takebayashi Y, Higashiyama R, Tagata Y, et al. Medium-chain triglycerides given in the early stage of mild-to-moderate Alzheimer’s disease enhance memory function. Psychogeriatrics. 2017;17:520–1. [DOI] [PubMed] [Google Scholar]
- 384.Henderson ST, Poirier J. Pharmacogenetic analysis of the effects of polymorphisms in APOE, IDE and IL1B on a ketone body based therapeutic on cognition in mild to moderate Alzheimer’s disease; a randomized, double-blind, placebo-controlled study. BMC Med Genet. 2011;12:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Henderson ST, Vogel JL, Barr LJ, Garvin F, Jones JJ, Costantini LC. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutrition & Metabolism. 2009;6:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Geifman N, Brinton RD, Kennedy RE, Schneider LS, Butte AJ. Evidence for benefit of statins to modify cognitive decline and risk in Alzheimer’s disease. Alzheimers Res Ther. 2017;9:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Li G, Shofer JB, Rhew IC, Kukull WA, Peskind ER, McCormick W, et al. Age-varying association between statin use and incident Alzheimer’s disease. J Am Geriatr Soc. 2010;58:1311–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Sano M, Bell KL, Galasko D, Galvin JE, Thomas RG, van Dyck CH, et al. A randomized, doubleblind, placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology. 2011;77:556–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Feldman HH, Doody RS, Kivipelto M, Sparks DL, Waters DD, Jones RW, et al. Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease: LEADe. Neurology. 2010;74:956–64. [DOI] [PubMed] [Google Scholar]
- 390.McGuinness B, Craig D, Bullock R, Passmore P. Statins for the prevention of dementia. Cochrane Database Sys Rev. 2016;146:3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Li G, Mayer CL, Morelli D, Millard SP, Raskind WH, Petrie EC, et al. Effect of simvastatin on CSF Alzheimer disease biomarkers in cognitively normal adults. Neurology. 2017;89:1251–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Muldoon MF, Ryan CM, Sereika SM, Flory JD, Manuck SB. Randomized trial of the effects of simvastatin on cognitive functioning in hypercholesterolemic adults. Am J Med. 2004;117:823–9. [DOI] [PubMed] [Google Scholar]
- 393.Muldoon MF, Barger SD, Ryan CM, Flory JD, Lehoczky JP, Matthews KA, et al. Effects of lovastatin on cognitive function and psychological well-being. Am J Med. 2000;108:538–46. [DOI] [PubMed] [Google Scholar]
- 394.Wagstaff LR, Mitton MW, Arvik BM, Doraiswamy PM. Statin-associated memory loss: analysis of 60 case reports and review of the literature. Pharmacotherapy. 2003;23:871–80. [DOI] [PubMed] [Google Scholar]
- 395.Schultz BG, Patten DK, Berlau DJ. The role of statins in both cognitive impairment and protection against dementia: a tale of two mechanisms. Transl Neurodegener. 2018;7:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Dagliati A, Peek N, Brinton RD, Geifman N. Sex and APOE genotype differences related to statin use in the aging population. Alzheimers Dement. 2021;7:e12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Hudry E, Van Dam D, Kulik W, De Deyn PP, Stet FS, Ahouansou O, et al. Adeno-associated virus gene therapy with cholesterol 24-hydroxylase reduces the amyloid pathology before or after the onset of amyloid plaques in mouse models of Alzheimer’s disease. Mol Ther. 2010;18:44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Petrov AM, Lam M, Mast N, Moon J, Li Y, Maxfield E, et al. CYP46A1 activation by efavirenz leads to behavioral improvement without significant changes in amyloid plaque load in the brain of 5XFAD Mice. Neurotherapeutics. 2019;16:710–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Hutter-Paier B, Huttunen HJ, Puglielli L, Eckman CB, Kim DY, Hofmeister A, et al. The ACAT inhibitor CP-113,818 markedly reduces amyloid pathology in a mouse model of Alzheimer’s disease. Neuron. 2004;44:227–38. [DOI] [PubMed] [Google Scholar]
- 400.Bryleva EY, Rogers MA, Chang CC, Buen F, Harris BT, Rousselet E, et al. ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proc Natl Acad Sci USA. 2010;107:3081–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Murphy SR, Chang CC, Dogbevia G, Bryleva EY, Bowen Z, Hasan MT, et al. Acat1 knockdown gene therapy decreases amyloid-beta in a mouse model of Alzheimer’s disease. Mol Ther. 2013;21:1497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Shibuya Y, Chang CC, Chang TY. ACAT1/SOAT1 as a therapeutic target for Alzheimer’s disease. Future Med Chem. 2015;7:2451–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Yin F, Yao J, Brinton RD, Cadenas E. Editorial: The Metabolic-Inflammatory Axis in Brain Aging and Neurodegeneration. Front Aging Neurosci. 2017;9:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Shinohara M, Tashiro Y, Suzuki K, Fukumori A, Bu G, Sato N. Interaction between APOE genotype and diabetes in cognitive decline. Alzheimers Dement (Amst). 2020;12:e12006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.de Bruijn RF, Bos MJ, Portegies ML, Hofman A, Franco OH, Koudstaal PJ, et al. The potential for prevention of dementia across two decades: the prospective, population-based Rotterdam Study. BMC Med. 2015;13:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Licher S, Ahmad S, Karamujić-Čomić H, Voortman T, Leening MJ, Ikram MA, et al. Genetic predisposition, modifiable-risk-factor profile and longterm dementia risk in the general population. Nat Med. 2019;25:1364–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this review article as no new data were created or analyzed.