

Abstract
The eukaryotic cell is compartmentalized into subcellular niches, including membrane-bound and membrane-less organelles. Proteins localize to these niches to fulfil their function, enabling discreet biological processes to occur in synchrony. Dynamic movement of proteins between niches is essential for cellular processes such as signalling, growth, proliferation, motility and programmed cell death, and mutations causing aberrant protein localization are associated with a wide range of diseases. Determining the location of proteins in different cell states and cell types and how proteins relocalize following perturbation is important for understanding their functions, related cellular processes and pathologies associated with their mislocalization. In this Primer, we cover the major spatial proteomics methods for determining the location, distribution and abundance of proteins within subcellular structures. These technologies include fluorescent imaging, protein proximity labelling, organelle purification and cell-wide biochemical fractionation. We describe their workflows, data outputs and applications in exploring different cell biological scenarios, and discuss their main limitations. Finally, we describe emerging technologies and identify areas that require technological innovation to allow better characterization of the spatial proteome.
Compartmentalization of the eukaryotic cell into membrane-bound and membrane-less organelles and other subcellular niches allows biological processes to occur synchronously1. Proteins often localize to specific subcellular niches to fulfil their function and dynamic movement of proteins between compartments is essential for cellular processes including signalling, growth, proliferation, motility and programmed cell death; indeed, cells employ dedicated mechanisms to ensure the correct trafficking of proteins and mislocalization of proteins has been implicated in various different pathological states2,3. Mutations causing aberrant protein localization underpin some forms of obesity4, cancers5, laminopathies6 and lung and liver disease7, and translation at inappropriate subcellular locations has been linked to cancer8 and dementia9.
Determining the subcellular location of a protein and how it changes upon perturbation or varies between different cell types is essential for understanding the protein’s biochemical function. This is complicated in the case of multi-localized proteins (MLPs), which reside in multiple subcellular locations because trafficking between locations is part of their cellular function or enables them to adopt different functions in the cell in a context-specific manner10,11. Up to 50% of the proteome is estimated to be composed of MLPs11. Recently, community-led spatial proteomics approaches and the refinement of experimental techniques have made substantial progress in determining and understanding the subcellular localization of proteins and assembling subcellular protein atlases11–18. These experimental methods range from single-cell approaches to those giving information on bulk steady-state protein location in multiple cells, tissues or even whole organisms. The application of these techniques to dynamic systems has detailed protein relocalization events associated with pathologies, cellular stresses and exposure to therapeutic agents. Together, these studies have uncovered details of the spatial proteome and revealed the context-specific properties of its components19,20.
In this Primer, we cover the major spatial proteomics approaches for determining the localization and abundance of proteins within intricate subcellular structures, rather than whole cell protein abundance in tissue-specific cell types. These technologies include fluorescent imaging approaches and protein proximity labelling, organelle purification or cell-wide biochemical fractionation coupled to mass spectrometry (MS), summarized in Fig. 1. We discuss the experimental procedures and data analysis principles for these techniques and cover examples of their applications. Irrespective of the approach taken, the importance of rigorous data analysis and raw data accessibility is of paramount importance and is described along with emerging high-throughput methods for the identification, quantification and subcellular mapping of proteins within the cell and at the cell surface.
Fig. 1 |. Overview of spatial proteomics approaches.
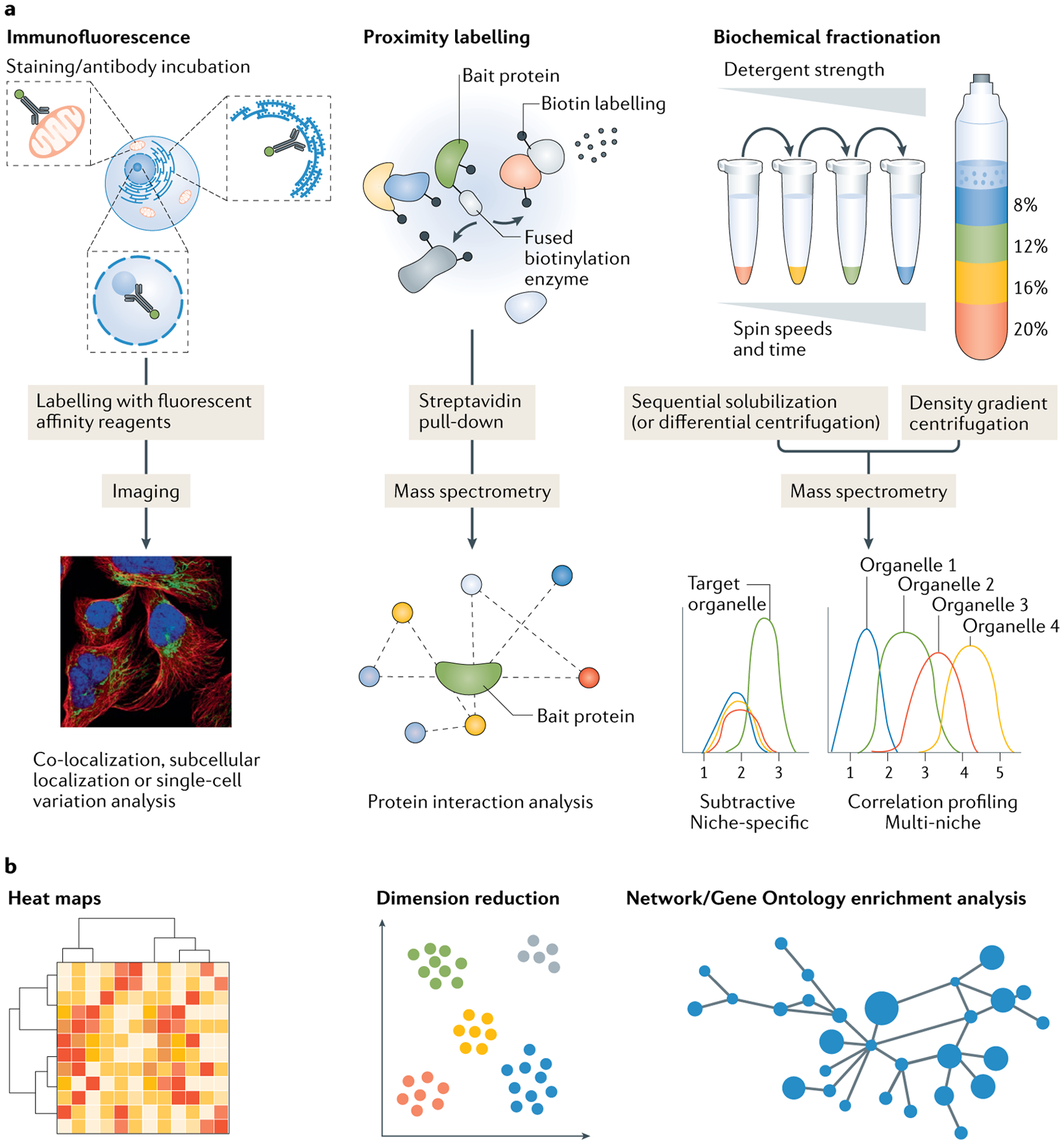
Spatial proteomics approaches include fluorescence imaging approaches and proximity labelling or biochemical fractionation techniques coupled with mass spectrometry (MS). a | Imaging of cells and tissues stained with fluorescently labelled antibodies (or other affinity reagents) allows for subcellular protein localization in situ. Proximity labelling strategies permit in vivo biotin labelling of proteins in close proximity to a chosen bait protein that has been genetically fused to a biotinylating enzyme. Following labelling, samples can be processed using MS proteomics protocols. Biochemical fractionation methods can produce cell fractions that are enriched for organelles of interest based on the different biophysical and chemical properties of different subcellular niches. These fractions are then subject to MS analysis. Typically, organellar separation is achieved using density gradient or differential/sedimentation centrifugation, or sequential solubilization using detergents190,191,303–305,315. b | All of these methods produce data-rich outputs that require computational analysis using techniques such as hierarchical clustering, dimension reduction or network analysis to visually represent and calculate statistical information. Machine learning techniques can also be used (not pictured). Correlation profiling plot in part a and dimension reduction plot in part b adapted from REF.21, Springer Nature Limited.
Experimentation
Workflows required to interrogate the spatial proteome are extremely varied and the choice of workflow depends on the system and scale of spatial information required21. For simplicity, we divide methods into those that use quantitative MS or fluorescent imaging.
Mass spectrometry-based methods
MS approaches offer accurate proteome-wide identification and quantification of proteins and proteoforms. MS-based workflows for subcellular proteomics use biochemical fractionation or proximity labelling methods to separate or discriminate specific subcellular compartments before MS analysis. We describe specific strategies for producing spatially informative samples for MS analysis and common quantitative MS techniques below. Note that we focus on centrifugation-based and detergent-based fractionation methods, although electrophoresis and affinity purification strategies have also been employed for organellar fractionation22–28.
Proteoforms.
Different molecular forms in which the protein product of a single gene can be found.
Biochemical fractionation and enrichment.
Biochemical fractionation separates organelles based on size, density, membrane solubility or charge prior to their analysis by MS. Methods that produce fractions enriched for specific organelles typically achieve very high sensitivity and proteome coverage in MS-based analysis. A common strategy uses differential centrifugation to pellet a crude organelle preparation, followed by density centrifugation using sucrose, Nycodenz, iodixanol or Percoll to produce discrete organelle fractions29–38. Bona fide target organelle components can be differentiated from co-purifying contaminants in MS data using subtractive proteomics39; in this experimental workflow, a single fraction containing contaminant organelles serves as a negative control. The control can also consist of several subcellular fractions collected during purification of the target organelle, which are then analysed using protein correlation profiling approaches to differentiate the target niche from the control. Fractions from density centrifugation will inevitably contain contaminant proteins and these may account for the majority of protein identifications29; it is therefore important to achieve adequate enrichment of the target organelle or organelles in the purified sample over the control fraction(s). A typical amount of starting sample for the purification of a target organelle such as the mitochondria from human cell lines (for example, HeLa or HEK293) is 2 × 108 cells, although the required starting material varies between model systems and the organelle of interest21.
Protein correlation profiling.
Using distributions profiles of proteins unique to different organelles and protein complexes across subcellular biochemical fractions to determine the subcellular location or complex association of uncharacterized proteins.
In correlation profiling methods such as protein correlation profiling30,40, localization of organelle proteins using isotope tagging (LOPIT)10,41–43, dynamic organellar maps44, Prolocate45, COLA46 and SubCellBarCode47, fractions are collected across a separation gradient using either density or differential centrifugation and analysed using MS and multivariate statistics, and machine learning methods are used to compare the abundance distribution of every protein with known organelle markers in order to determine the probable locations of the proteins10,42,44,48–53 and make inferences regarding protein trafficking44,47,54,55. These techniques can identify organelle protein distribution trends even in the presence of structural alterations, which may not be captured by traditional fractionation methods that focus on enriching a specific organelle53,56. They are based on de Duve’s principle57, which states that proteins from the same subcellular niche will share a distinct abundance profile across a separation gradient. Typically, these approaches require a minimum of ~1 × 107 cells or ~1.3 g of tissue40,44, although this is highly dependent on cell size and type, the number of organellar fractions required, the organelles of interest and the homogenization technique used.
de Duve’s principle.
Comparing the distribution pattern across subcellular fractions of proteins known to be resident within a specific organelle of interest allows for inference of other proteins with similar distribution profiles that must also reside in the same compartment.
Detergent-based subcellular fractionation methods separate subcellular compartments based on their solubilization. This strategy allows the capture of a broader distribution of proteins in individual fractions than density centrifugation methods, for example comparing cytosolic, membrane and nuclear distributions. The required starting material is 106–107 cells or 25–50 mg of tissue; this approach is therefore useful where starting material is limited, for example when using primary cell cultures. Frequently used detergents for these methods and resultant subcellular fractions are summarized in TABLE 1 (REF.47).
Table 1 |.
Summary of detergents for subcellular fractionation
| Detergent | Properties | Recommended concentration (%) | Targeted subcellular proteins | Critical micelle concentrationa (mM at 20–25 °C) |
|---|---|---|---|---|
| Digitonin | Mild, non-ionic | 0.0045–0.01 (refs303–305,306) | Cytosolic and soluble cytoskeletal proteins | <0.5 |
| Triton X-100 | Non-ionic | 0.5 (REFs303,306) | Membrane and organellar proteins | 0.2–0.9 |
| NP-40 | Non-ionic | 1 (REFs304,305) | Membrane and organellar proteins | 0.05–0.3 |
| Tween-40/deoxycholate (DOC) buffer | Non-ionic/anionic | 0.5–1 (REFs303,306) | Soluble nuclear proteins | 0.027–6 |
| Sodium dodecyl sulfate (SDS) | Anionic | 0.1–4 (REFs304,305) | Nuclear matrix proteins and more insoluble proteins | 7–10 |
The minimum concentration at which the surfactant produces micelles. This information may be important for optimizing the isolation of organellar compartments of interest in the context of detergent-based organellar isolation methods.
During any biochemical fractionation experiment, samples should be maintained at 4 °C or on ice throughout organellar enrichment. For centrifugation-based methods, disruption and breakage of the compartments should be avoided during cell lysis; this can be minimized by performing sonication, hypotonic shock, Dounce homogenization or ball bearing homogenization in detergent-free lysis buffer58. Quality control checks can be performed following organellar enrichment to ensure successful fractionation by measuring organelle-specific protein markers using immunoblotting or targeted MS, or by assessing the biophysical homogeneity of fractions using electron microscopy or fluorescence microscopy59. Independent replicates should be generated at this stage.
Proximity labelling.
In proximity labelling techniques, a ‘bait’ protein is fused to a labelling enzyme that can covalently label neighbouring ‘prey’ proteins in the cell. Labelled proteins can then be purified and characterized by MS to generate spatially resolved proteomic data. Two main labelling approaches are used in spatial proteomics. The first uses an engineered ascorbate peroxidase — either APEX or the derivative APEX2 (REFS60,61) — which biotinylates the tyrosine residues of proteins within a radius of ~20 nm upon stimulation with peroxide. The second approach, BioID, uses a mutated bacterial biotin ligase, which creates a ~10 nm cloud of activated and reactive biotin-AMP62 that can covalently biotinylate lysine ε-amines on proximal proteins. Labelling enzymes for BioID include a mutant form of the Escherichia coli BirA protein known as BirA*63, the more active miniTurbo or TurboID variants64 and biotin ligases from other species such as BioID2 (REF.65) and BASU66. Biotinylated proteins are recovered from the lysate using streptavidin–agarose or magnetic beads before MS analysis (FIG. 2). Unlike the fractionation approaches mentioned above, organelles and protein–protein interactions do not need to be intact during cell lysis, and detergents and chaotropic agents — such as SDS or urea, respectively — can be employed to efficiently solubilize subcellular compartments.
Fig. 2 |. Proximity labelling proteomics.
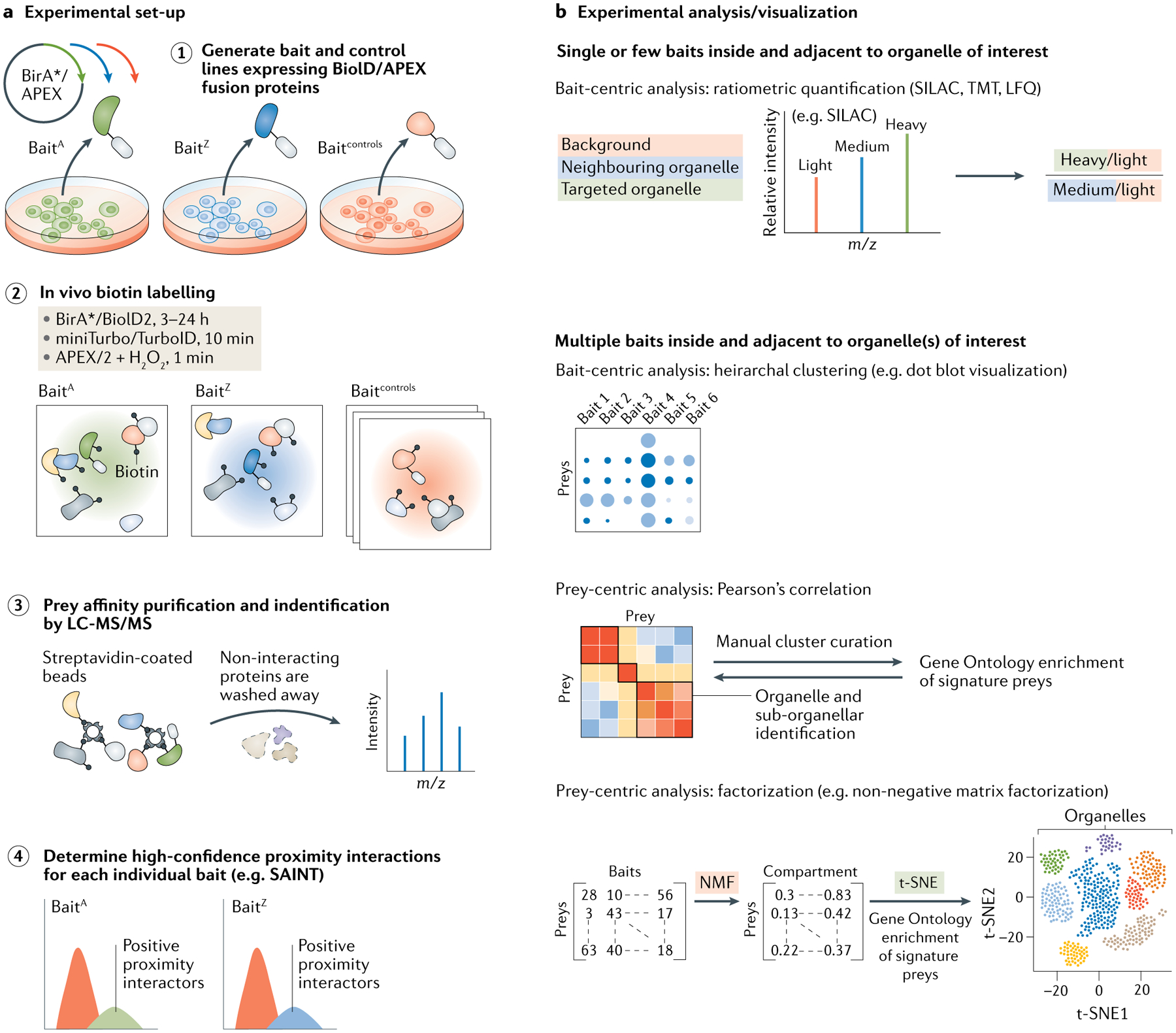
Proximity labelling strategies permit biotin labelling of proteins in immediate proximity to the chosen bait proteins in living cells. a | Baits of interest (BaitA, BaitZ) are genetically fused with an enzyme such as APEX/APEX2 or BirA*, BioID2, miniTurbo or TurboID for BioID (step 1), which biotinylate nearby proteins upon incubation of engineered cells with biotin in culture (step 2). Control lines can express the labelling enzyme fused to a non-specifically localized control bait such as green fluorescent protein or a localization signal specific for a non-target organelle. In the case of APEX, the addition of H2O2 generates short-lived biotin-phenol free radicals that react with nearby biomolecules. Following labelling, a streptavidin pull-down step enriches for labelled proteins, which can then be identified by mass spectrometry (MS) (step 3). High-confidence proximity interactors are determined by comparing preys with proteins isolated to control lines using such tools as SAINT (Significance Analysis of INTeractome) (step 4). b | Organellar components can be elucidated using bait-centric or prey-centric analyses. Ratiometric quantification of baits, for example using isotopic labelling approaches with baits in and outside the organelle, or hierarchical clustering of multiple baits can be used to identify enrichment of proteins within an organelle of interest in a bait-centric manner. Alternatively, extensive proximity interaction networks can be elucidated using multiple baits and prey-centric analyses including Pearson’s correlation and factorization approaches such as non-negative matrix factorization (NMF). LC-MS/MS, liquid chromatography with tandem mass spectrometry; m/z, mass to charge ratio; SILAC, stable isotope labelling by amino acids in cell culture; TMT, tandem mass tagging; t-SNE, t-distributed stochastic embedding; LFQ, label-free quantitation.
The range of labelling proteins is far shorter than the size of membrane-bound and membrane-less organelles, which are generally hundreds of nanometres to a few micrometres in diameter. Therefore, proximity labelling can obtain detailed information about the spatial organization of proteins within organelles. Quantitative proteomics comparisons can be made between prey proteins from baits in an organelle of interest and those from control baits labelling adjacent compartments; this is exemplified in an early study of the mitochondria inner membrane space67. Sub-organellar organization of proteins can be inferred when an experimental set-up involves multiple baits both inside and outside an organelle, as demonstrated in the characterization of different modules within the centrosome–cilium compartment68. Large data sets can also reveal the organization of subcellular proteomes in a prey-centric fashion, as preys in close proximity should be co-labelled by the same set of baits. Finally, correlation of prey profiles can be used to reveal clusters of preys defined by their organellar association69,70. For further information on proximity labelling methods, we refer readers to recent reviews71,72.
Mass spectrometry techniques.
In a typical bottom-up or shotgun spatial proteomics workflow, proteins isolated from the above biochemical fractionation or proximity labelling techniques are enzymatically digested to produce peptides that are then characterized using liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS). The initial liquid chromatography step separates peptides to reduce sample complexity (improving proteome coverage) before aerosolization and direct injection of the samples into the MS using electrospray ionization. Matrix-assisted laser desorption/ionization is an alternative to electrospray ionization, but has a smaller mass range of detection that is not as suitable for peptide samples with diverse molecular masses. In commonly used data-dependent acquisition (DDA) approaches using MS/MS, an initial MS scan (MS1) detects peptide ions of which the most highly abundant are then isolated and fragmented before a subsequent MS scan (MS2) to determine their specific amino acid sequence. Mass spectra are compared against a reference database of canonical sequences to infer peptide identity73. In emerging data-independent acquisition (DIA) approaches, large overlapping isolation windows are used at the MS1 stage to allow more peptide ions to be measured in MS2. DIA can improve peptide identification, reproducibility and detection of low-abundance proteins74, although the complex MS2 spectra from DIA can be difficult to interpret. Experimental considerations of DDA and DIA are covered in a recent review75. Note that spatial proteomics strategies are not restricted to shotgun proteomics and can include other analytical methodologies; for example, SDS-PAGE and western blotting can assess a small, targeted group of proteins. MS technology can also be used for cell imaging, which is briefly covered in BOX 1.
Box 1 |. Imaging mass cytometry and multiplexed ion beam imaging.
New combinatorial methods using both imaging and mass spectrometry (MS) or conceptually similar techniques include imaging mass cytometry (IMC)316, multiplexed ion beam imaging (MIBI)317 and mass spectrometry imaging (MSI)318,319. These techniques allow highly multiplexed imaging of proteins. In IMC, rare metal ions with distinct valences and masses — such as ytterbium or neodymium — are conjugated to antibodies and detected using a mass cytometer, which ionizes the sample material by ablating it with a laser. The number of protein targets detected is theoretically limited by the number of available metals (close to 100); however, only ~40 parameters can be measured in practice owing to restrictions in conjugating isotopes to antibodies and isotope purification316,320. Spatial resolution is limited by the diameter of the laser beam, which is around 1 μm316, and the technique is therefore constrained to imaging large subcellular organelles. MIBI, a very similar method to IMC, has reported resolutions of 5–30 nm using an atomic ion beam for more precise ionization of antibody–metal tags316,317. A general limitation of these approaches is the lack of commercially available probes. For IMC and MIBI, instrument accessibility, cost and low acquisition speeds are limiting factors. In MSI — typically performed with a matrix-assisted laser desorption/ionization (MALDI) instrument — each ‘pixel’ of an ionized sample produces a corresponding label-free mass spectrum. MSI preparations and related techniques such as secondary ion MS enable resolution at the nanometre scale321, but can struggle to ionize peptides and proteins.
Label-free MS approaches are popular amongst most groups owing to their ease of application, although proteins can be labelled with stable isotopes before DDA to reduce missing values caused by and improve reproducibility over label-free approaches76–80 (FIG. 3). Missing values can be caused by biological variation or technical issues arising from sample storage, protein extraction, the stochastic sampling of peptide ions during MS acquisition or signal-to-noise thresholding81. Labelling allows for multiplexing of samples, which minimizes some of these technical issues. In stable isotope labelling by amino acids in cell culture (SILAC)76, non-radioactive, stable isotope-labelled amino acids — typically Arg and Lys — are incorporated into the proteome during cell culture. This is arguably the most robust labelling approach and allows up to three samples to be combined immediately after cell culture and analysed simultaneously (FIG. 3b). Higher multiplexing capacity is theoretically possible with SILAC, although only a limited set of reagents are currently available82. In tandem mass tagging (TMT)83–85 (FIG. 3c), peptides generated after cell lysis and protein digestion are chemically labelled with an isobaric compound containing an identifiable mass reporter ion. During fragmentation, these reporter ions are cleaved and can be quantified using their MS2 spectra. This technique allows for multiplexing of up to 16 samples and has applications beyond in vivo incorporation of labels. Isobaric tags suffer from more experimental variation than SILAC approaches, owing to their incorporation and multiplexing later in the sample preparation workflow. TMT also suffers from precursor co-isolation issues (see REFS84,86). To achieve higher proteome coverage and include low abundance and low molecular mass proteins when performing bottom-up techniques, the use of alternative proteases or orthogonal peptide fractionation techniques such as high-pH reversed-phase chromatography is advised29,87. In most spatial proteomics applications, it is common practice to scale intensities of each protein to values between zero and one (REFS44,88,89). Normalization at the sample preparation stage is also important to consider, such as creating balanced experiments to minimize batch effects and normalizing protein amounts when using TMT labelling. These are just a couple of examples and these considerations are extensively reviewed in REF.90.
Fig. 3 |. Generic data-dependent acquisition workflows in quantitative proteomics.
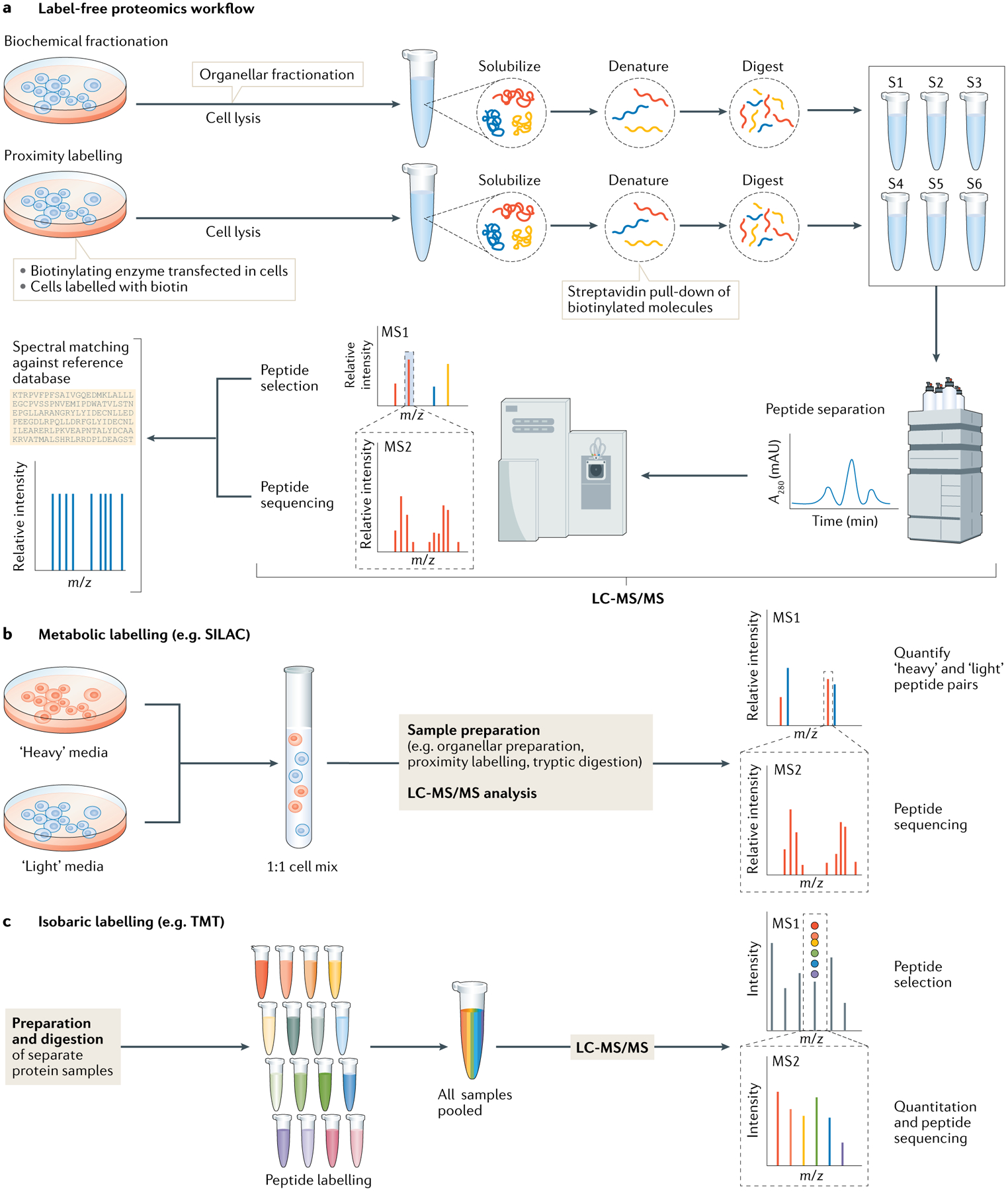
a | Two sample preparation workflows incorporating biochemical fractionation and proximity labelling techniques are shown in the context of a standard data-dependent acquisition (DDA) proteomics workflow. In DDA workflows, proteins are solubilized and denatured using buffers containing chaotropic agents and detergents, such as urea and SDS. Reduction of disulfide bonds and alkylation of free cysteine thiols allows for efficient digestion of the proteins to peptides using proteolytic enzymes, typically trypsin. Samples are then acidified and analysed by liquid chromatography with tandem mass spectrometry (LC-MS/MS). MS/MS consists of an initial MS1 scan, which detects charged peptides (known as precursor ions), followed by isolation, fragmentation and detection of these ions in a subsequent MS2 scan to determine the amino acid sequence of the precursor ions. The complex spectra can then be deconvoluted using in silico reference databases, using algorithms that account for experimental parameters and sample preparations. b | Labelling-based proteomics methods use the above strategy, although additional steps are added to the workflow. For metabolic labelling methods such as stable isotope labelling by amino acid in culture (SILAC), a light and heavy isotopic version of an amino acid are added to the cell culture growth media, allowing metabolic incorporation of stable isotope-labelled amino acids into newly synthesized proteins. For complete incorporation, approximately six cell doublings are needed. Labelling enables sample pooling after cell harvest to minimize downstream technical variability. c | Isobaric labelling methods such as tandem mass tagging (TMT) also reduce technical variability. Labelling occurs post digestion and up to 16 samples can be multiplexed and measure in one MS run using TMT labelling. Although each tag has the same mass when bound to the peptides, upon fragmentation by higher-energy collisional dissociation during MS, their ion reporter components — which have distinguishing mass — are displaced from the peptide and can be observed in the low mass region of the MS2 spectra to determine the relative quantities of the same peptide across multiple samples. m/z, mass to charge ratio. S1–S6, sample 1–sample 6.
Fluorescent imaging methods
Imaging approaches give subcellular information of protein distribution in intact cells, and microscopy has been used for centuries to explore the cell interior. Recent advances in genome-wide RNA interference technologies and CRISPR–Cas9 (CRISPR-associated protein 9) have further contributed to imaging being a powerful approach for linking phenotype — including changes in subcellular location — to genotype91,92. Two main fluorescent imaging approaches can be employed to capture subcellular information: live-cell imaging, where the protein of interest is modified to express a fluorescent tag to enable detection; and immunocytochemistry, where fluorescently tagged affinity reagents are applied to fixed material to bind specific proteins and allow their visualization. Antibodies are the most commonly used affinity reagents and can be labelled directly with fluorophores for detection of the bound protein (primary antibodies), or secondary antibodies conjugated to fluorophores can be used, which bind to unlabelled primary antibodies in turn. Alternatives to antibodies include nanobodies, affimers and aptamers93–95. Several chemical dyes are available for live and fixed material to counterstain cellular structures such as the nucleus, cell membranes, mitochondria and actin filaments95,96. We refer readers to a review on the range of available counterstains97.
Nanobodies.
Antibody fragments consisting of a single monomeric variable antibody domain.
Affimers.
Small proteins that bind to target molecules with a similar specificity and affinity to antibodies.
Aptamers.
Oligonucleotides or peptide molecules that bind to a specific target molecule.
Use of live or fixed cells.
The biological context, accessibility of samples, instrumentation, technological expertise and number of studied proteins can determine the method of choice. Studying transient protein dynamics in real time requires live-cell, time-lapse microscopy, with instrumentation that enables temperature and CO2 control to promote cell survival throughout several cell cycles. Using fixed material and antibody labelling is useful in cases where the protein of interest is difficult to tag or express endogenously, in cases where the cell type of relevance is difficult to genetically modify or for large-scale experiments of hundreds of proteins. Further, once affinity reagents are generated, they can be validated and applied to detect the same protein in different cell types under various conditions, allowing for comparative studies. For these reasons, we focus on immunocytochemistry (FIG. 4); however, most global microscopy studies so far have been performed with protein tagging approaches and we refer readers interested in these methodologies for subcellular profiling to other studies98–101. These efforts and methodologies serve as complementary methods for the validation of data obtained from fixed material102.
Fig. 4 |. Generic fluorescence immunocytochemistry proteomics workflow.
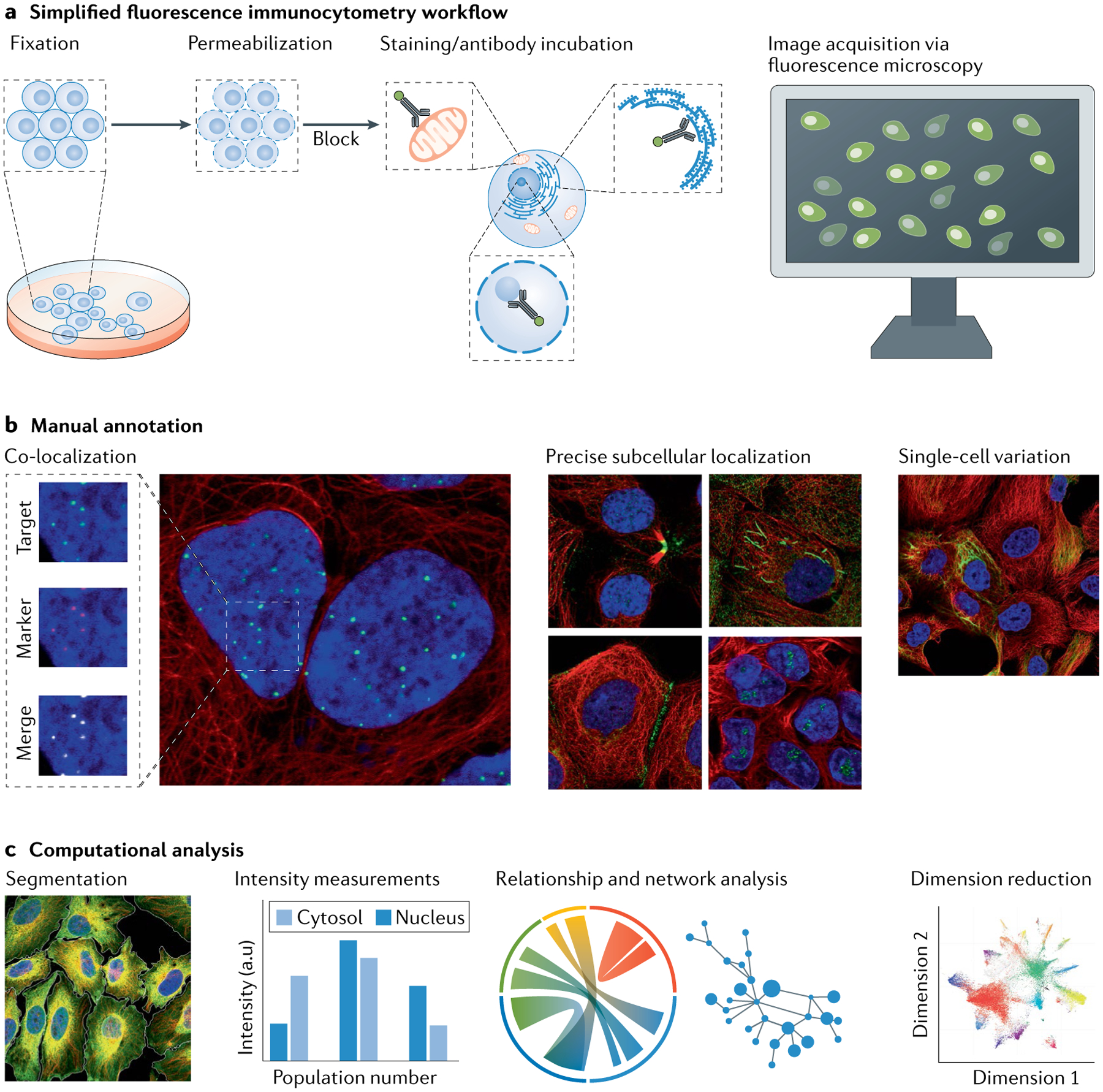
a | Cells are fixed using either cross-linking agents or organic solvents, such as aldehydes or alcohols. Cross-linkers generally outperform organic solvents for preserving subcellular structures but may reduce antigen retrieval for certain proteins. Permeabilization of cells with detergents such as digitonin or Triton X-100 extracts lipids from the cell membrane and allows for penetration of affinity agents such as antibodies into the cells. Blocking buffer containing serum or albumin helps reduce unspecific labelling before addition of affinity reagents. Cells are then counterstained with different organelle probes before imaging. Image acquisition is typically performed using confocal microscopy and an optional embedding step can be used for archiving of the samples. b,c | After image acquisition, the image data are annotated to assign protein location to subcellular structures. b | Manual inspection of images can be used to acquire qualitative data. For obvious staining patterns easily interpreted by eye, qualitative annotation might be sufficient if the data sets are small. c | For large image data sets, computational analysis is required. Computational strategies enable high-throughput collection of quantitative, morphological and comparative information, such as algorithms for segmentation and intensity measurements. Such quantitative data can then be used to generate networks and dimension reduction plots. Multi-label patterns and fine structures stained in a subset of cells may require manual annotation or more advanced computational analysis. Relationship and network analysis in part c adapted from REF.11, AAAS. Dimension reduction plot in part c adapted from REF.180, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/).
Sample preparation.
Imaging-based spatial proteomics techniques can be applied to cell cultures, tissue sections and organoids. Cultured cells are most often used owing to their accessibility and flexibility. Cell culture samples are often fixed using paraformaldehyde (PFA)103 to preserve subcellular structures and permeabilized using detergents to allow penetration of affinity reagents; this can be done using non-ionic detergents such as Triton X-100, Tween-20 and NP-40, or saponin glycosides, which better preserve membrane morphology than detergents104,105. Alternatively, alcohols or other organic solvents can fix and permeabilize cells in a single step106 and could be an option when more rigid structures of the cells, such as cytoskeletal components or nuclear structures, are to be visualized107. Fixation and permeabilization protocols should be optimized for the specific target protein and associated affinity reagents107–111. Tissue samples should be frozen immediately and stored at −20 °C112 or preserved in formalin-fixed paraffin-embedded blocks113 before sectioning using a cryostat or microtome. To resolve fine subcellular structures, the sample thickness should be <10 μm to reduce scatter from out of focus light. Frozen sections can be stained directly after sectioning, whereas formalin-fixed paraffin-embedded sections require de-paraffinization and an antigen retrieval step to allow for affinity reagent penetration and binding of the protein target114,115.
Microscope instrumentation.
The use of different fluorophores can allow for simultaneous detection of signals from different proteins. Choosing fluorophores with narrow excitation and emission peaks or microscope systems that detect broad spectral ranges can increase the number of proteins that can be imaged at once. Spectral overlap when introducing additional fluorophores can limit the number of proteins that can be detected simultaneously, although approaches are being developed based on the cyclic detection of a few proteins at a time that enable more proteins to be detected in a single sample without introducing additional fluorophores101,116–118.
Instrumentation for image capture depends on several factors and is usually a trade-off between resolution, sample size and speed. For example, high-resolution imaging requires an objective with a high numerical aperture and short working distance that captures a smaller field of view, requiring many images to be taken to collect data from a large area. By contrast, in high-throughput screening techniques — used extensively by the pharmaceutical industry owing to the information-rich data being generated from imaging119 — throughput is more important than achieving the best possible resolution, as these approaches require a large number of replicates to gain statistical power.
Confocal microscopy can block out of focus light and has an improved signal-to-noise ratio compared with conventional fluorescence microscopy, allowing the localization of proteins to fine structures such as fibrillar centres, microtubule ends and substructures of the cytokinetic bridge120. Confocal microscopy is limited by the diffraction limit of light according to abbe’s law and minimum resolution is therefore around 200 nm121. Super-resolution microscopy techniques, such as stimulated emission depletion microscopy122, photoactivated localization microscopy123 and stochastic optical reconstruction microscopy124, can resolve distances as low as 20 nm and are therefore able to localize proteins to smaller structures or sub-organelle domains. We refer readers to a review of these techniques121.
Abbe’s law.
The approximate diffraction limit of a microscope determined using the wavelength of light (λ), the refraction index of the medium the imaged object is in (n) and the numerical aperture (θ).
Results
Subcellular proteomics approaches give rise to diverse and complex data sets. Here, we discuss the expected data outputs from these approaches and the recommended manual and computational data analysis strategies for these methods.
Shotgun MS proteomics data processing
The raw output of organellar fractionation and proximity labelling approaches consists of thousands of tandem mass spectra and it is therefore impractical to manually sequence peptides from spectra. Complex algorithmic database searching tools have been developed for DDA spectra, including Andromeda125, Sequest126, Mascot127 and X!Tandem128. These compare experimental peptide spectra with theoretical in silico spectra and generate peptide spectral match scores, which reflect the likelihood that the corresponding peptide was present in the sample. To ensure that peptide spectral match scores are reliable, spectra are searched against the target species peptide database and a decoy database that consists of a reversed or randomized version of the target database129,130. This allows for the calculation of the percentage of peptide spectral match hits that are false positives, enabling estimation of the false discovery rate131. Database searching tools are often packaged into user-friendly software such as Proteome Discoverer and MaxQuant that allow for customizable and experiment-specific pipeline design132. For DIA spectra deconvolution, alternative software is required, such as OpenSWATH133, Skyline134 and DIA-NN135, which are reviewed elsewhere136. The output of these packages is a list of peptides and proteins alongside their quantitation across all samples, as well as other statistical and descriptive data. Statistical and visual analysis of this output can be performed using Perseus137, Jupyter138, Python139 or R programming packages. We direct interested readers to a repository of open-source Python tools for proteomics analysis.
Analysing fractionation experiments
All organellar fractionation methods coupled with MS use ion intensities as a proxy for protein abundance. Proteins with similar abundance profiles or characteristics of defined organellar proteins are assigned to their respective organelles. As proteins are dynamic and can simultaneously reside in multiple cellular compartments140, validation of organelle markers within different biological contexts is critical. Subtractive proteomics data must include an accurate reference set of known protein components of the target organelle and reference lists for protein contaminants from other subcellular compartments. These can be selected using Gene Ontology141,142, although Gene Ontology is frequently not a robust way to define marker sets as terms can be broadly applied. Other databases can be used in conjunction with Gene Ontology that contain evidence-based information, such as COMPARTMENTS143 and UniProt144. Calculation of the abundance ratios of reference proteins against contaminant proteins allows proteins with enrichment comparable with the reference markers to be assigned to the target organelle and visualized in a ratio versus intensity plot (FIG. 5a). This analysis accounts for impure isolations and enables high-confidence assignment; however, proteins that also reside outside the target organelle can easily elude identification.
Fig. 5 |. Subtractive versus correlation profiling analysis.
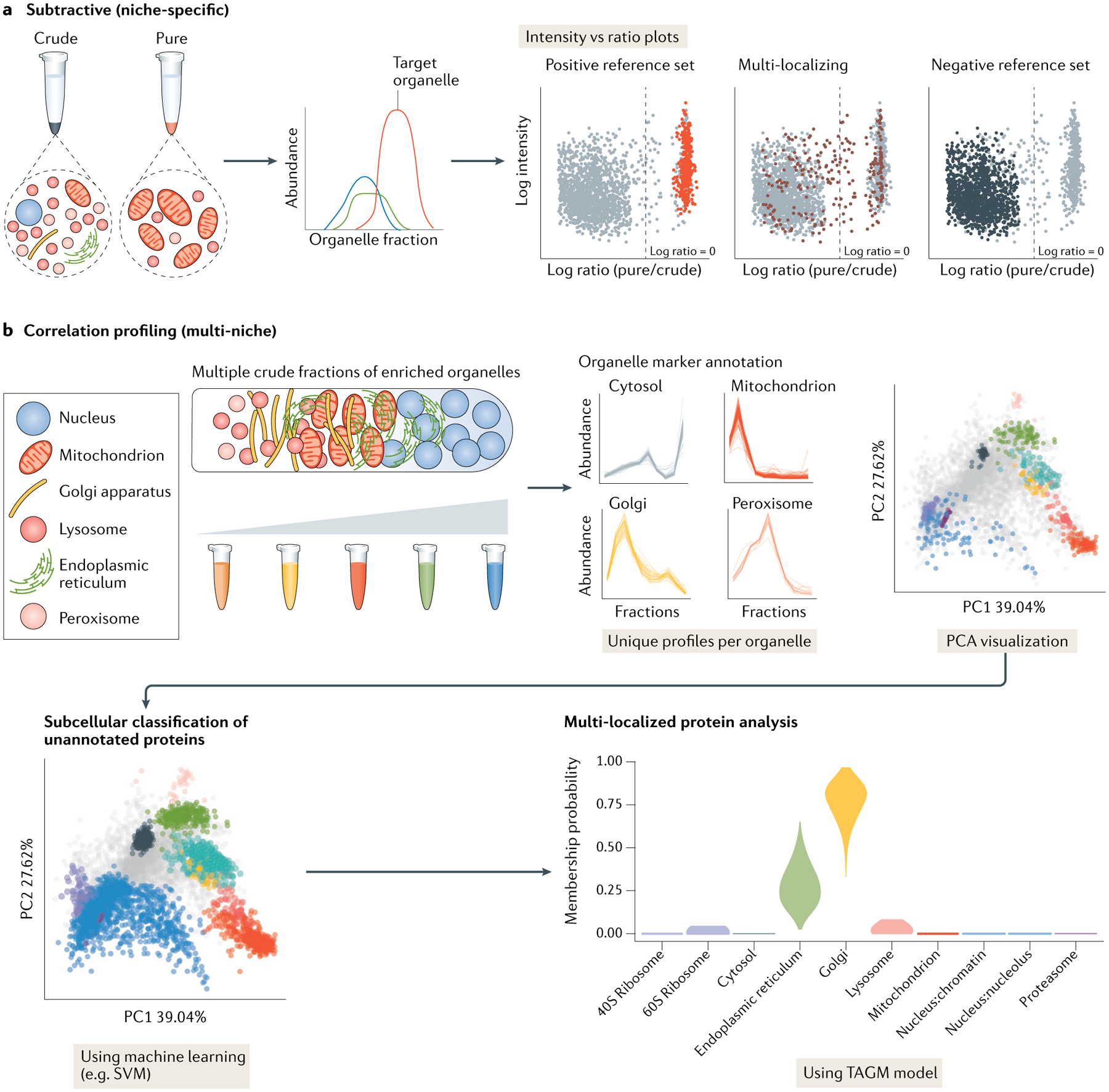
a | Subtractive proteomics techniques involve purifying an organelle of interest — usually using a combination of sedimentation and density centrifugation — alongside one or more crude fractions containing ‘contaminant’ components. By quantifying protein markers for the organelle of interest and proteins from the crude fractions using quantitative liquid chromatography with tandem mass spectrometry (LC-MS/MS), intensity versus ratio plots can be produced and used to distinguish true residents of the organelle of interest versus contaminants. b | In cell-wide correlation profiling experiments, multiple organelle-enriched fractions are collected across a sedimentation or density gradient. This allows for the unique profiles of multiple subcellular niches to be generated by annotation with organellar marker proteins. The variance of each organellar niche can be represented using dimension reduction methods such as principle component analysis (PCA). Markers can be used to train machine learning algorithms such as support vector machines (SVMs) or T-augmented Gaussian mixture models (TAGMs)52 to assign unannotated proteins, and find posterior probabilities of proteins belonging to multiple organelles in order to elucidate the locations of MLPs. Intensity versus ratio plots in part a adapted with permission from REF.29. Elements of part b adapted from REF.43, Springer Nature Limited, apart from TAGM plot adapted from REF.156, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/).
Correlation profiling data do not require a contaminant reference and consist of multiple organelle profile plots, where the abundance of the protein is plotted across the biochemical fractionation gradient (FIG. 5b). To reduce complexity and improve visualization, dimension reduction methods such as principle component analysis (PCA) can assess subcellular resolution as proteins associated with the same subcellular niche should form defined clusters in PCA plots145 (discussed further in BOX 2). Supervised, semi-supervised or unsupervised machine learning algorithms can also be used for correlation profiling; supervised and semi-supervised algorithms require the spatial profile of known organelle marker proteins to assign proteins with unknown localizations to subcellular niches44,146–149 and include support vector machines, neural networks, random forest models and naive Bayes algorithms. The choice of machine learning algorithm is dependent on the available computation power, processing time frames, experience and what assumptions about the data are appropriate to make. We refer readers to a review of common machine learning algorithms in proteomics150.
Box 2 |. Dimension reduction methods.
Dimension reduction is a technique for reducing the dimensional space of multidimensional data to aid data interpretation. Dimension reduction is particularly useful for visualizing complex omics data sets and often used for highlighting variable features across samples, such as expression levels between different treatment groups. These techniques can highlight variation between different populations of cells or subcellular fractions in spatial proteomics data. The most common linear method is principle components analysis (PCA), which projects data onto principle components that help to describe key features in the data (see REF.322 for a thorough explanation). Principle components cannot always detect patterns such as polynomial relationships and non-linear methods are better for detecting such patterns, although usually at the cost of losing characteristics of the original data and producing artefacts. An example of a non-linear dimension reduction approach is t-distributed stochastic neighbour embedding (t-SNE), which calculates and redefines local relationships between data points using Student t distributions to expose non-linear features163. This is a powerful technique for data exploration, although limitations include its stochasticity, artefact clustering and the loss of data structure. Other dimension reduction methods such as uniform manifold approximation and projection (UMAP)323 are available, and unsupervised clustering techniques such as DBSCAN and hierarchical clustering can aid marker curation for challenging or poorly researched systems152 (reviewed in REF.324).
Unsupervised clustering algorithms, such as k-means clustering or DBSCAN, are useful when training data are scarce, for example when investigating non-model organisms with limited marker proteins45,151,152. These methods are most suited to static and single locale assignment of proteins. Dynamic classification of proteins and classification of MLPs using these methods is challenging, but has been performed in previous studies44,47,56,89,153–155. Bespoke pipelines that use training data have recently facilitated dynamic classifications and MLP classifications, including T-augmented Gaussian mixture model approaches able to quantify the posterior probabilities of proteins residing in multiple organelles and TRANSPIRE (Translocation Analysis of Spatial pRotEomics), which models synthetic translocations from experimental organelle marker profiles to train a classifier to identify true protein dynamic events52,54,149,156,157.
Posterior probabilities.
In bayesian statistics, the revised or updated probability of an event after incorporating prior knowledge with observed data.
Proximity labelling data
The selection of benchmarking baits as organellar markers and appropriate controls informs downstream analytical pipelines. Negative controls must identify likely contaminants71, which include endogenously biotinylated proteins and proteins that become biotinylated by BirA* or APEX2 in a dose-dependent manner irrespective of the bait used71. These controls may include samples omitting the substrate or activator (for example, biotin or biotin-phenol/peroxide) or the fused enzyme, or samples where the labelling enzyme is fused to an irrelevant protein and this fusion protein is expressed at the same level as the bait fusions in the experimental samples60,71. Input of bait and control data into computational tools such as SAINT (Significance Analysis of INTeractome) allows the probability of bona fide protein–protein interactions to be derived158. Depending on the questions being asked, inclusion of additional controls assists in organellar proteome definition on a case-by-case basis and can help to discriminate between adjacent compartments. For example, to define the proteome of the mitochondrial intermembrane space, a ratiometric strategy based on SILAC was used to distinguish intermembrane space resident proteins from those in the cytoplasm67.
When analysing compartments that are not separated by membranes, using components of adjacent organelles or structures as controls can also help compartment annotation. This was exemplified by use of the SNARE VAMP8 as an adjacent-compartment bait to identify proteins enriched with the endolysosome-specific VAMP7 bait159. The use of multiple baits — labelling both the desired organelle and its neighbours — has been exploited to define the composition of membrane-less organelles such as the centrosome68, RNA granules and processing bodies69. Sub-organellar protein organization and contact sites between organelles and other structures can be resolved by interrogating multiple organellar baits, as was recently demonstrated within the mitochondria70. For these data sets, which typically contain data from at least 20 baits, prey-wise analysis using factorization or correlation approaches aids in organellar inference69,70,160. Factorization approaches are most useful for the definition of distinct groups of proteins associated with sets of structures, whereas correlation approaches produce continuums of scores displaying the relationships between pairs of tested proteins. In either case, the integration of these scoring tools into visualization and analysis schemes helps to explore localization assignments and relationships between proteins predicted to co-localize161–163.
Processing imaging data
Imaging data give spatial information regarding target proteins in intact cells. A trained human eye can distinguish between more than 20 subcellular structures using no more than three standard markers11; however, reference proteins or dyes are needed to stain and distinguish between organelles with similar distributions and size, for example cytosolic bodies and vesicles. Adequate controls and replicates are crucial to distinguish true localization from false staining patterns caused by non-specific binding or artefacts from sample preparation. Negative controls can include a sample lacking the affinity reagent towards the protein target, or a sample lacking the expression of the protein target, such as a knockout cell line to allow for background subtraction164,165. Affinity reagents targeting specific cellular structures, such as cytoskeletal components or the intranuclear domain, should be included to evaluate reproducibility, fixation and sufficient permeabilization. Affinity reagents can be validated by evaluating signal loss following partial or complete knockdown of the target protein, comparing the staining pattern with the same, endogenously tagged protein or examining correlations between signal intensity and target protein expression levels according to RNA sequencing data across samples. To date, manual pattern recognition has been the primary approach to assign protein subcellular localization simply by inspecting images using the microscope software or any software supporting the image file format. Popular open-source software for image analysis include ImageJ, CellProfiler, QuPath, ilastik and Orbit166–170.
A significant proportion of proteins localize to multiple cellular structures11 and populations of genetically identical cells show variability in their protein expression levels and localization11,171–173. Quantitative analysis techniques are needed to reveal subtle cell to cell variations and partial translocations between organelles across cellular states. This requires cells, and sometimes also subcellular structures, to be segmented by measuring the signal intensity for a structure-specific marker and determining the boundaries of the structure with respect to a threshold for defining a positive signal from the background. Once the objects are defined, fluorescence intensities from any stained marker can be measured and compared across segmented objects.
Machine learning and especially deep learning methods are becoming increasingly popular for image analysis owing to their superior performance and scalability, which make them attractive for analysing large data sets174,175. These involve training a prediction model on an existing data set of images representing known protein localization patterns defined by the user — referred to as the ‘ground truth’. The model extracts features from this training data set and through iterative cycles of model fitting and improvement can predict the subcellular localization of a protein based on the staining pattern in sample data. Machine learning techniques such as k-nearest neighbour classifiers, artificial neural networks and support vector machines use feature sets to capture protein location in relation to cellular morphology, as displayed by reference markers for certain organelle structures176–179. As many proteins localize to more than one organelle, models must accurately assign complex staining patterns to several fluorescent labels representing different organelles. This challenge was addressed in the Kaggle challenge for multi-label classification of cell organelles using the proteome scale collection of immunofluorescence images from the Human Protein Atlas (HPA) to generate a model that could assign subcellular locations for proteins in various cell lines displaying different morphologies180. Machine learning techniques have also been applied as high-content screening approaches for identifying changes in phenotypes in response to small molecules176,180,181. Model generation and training analysis software include WEKA182 and Scikit-learn183.
Applications
Pinpointing the subcellular localization of proteins provides insights into their biochemical function, and spatial proteomics has become an invaluable tool for characterizing the proteomes of individual organelles across cell types, organisms and conditions. Improved accessibility of technologies has enabled researchers from various backgrounds to conduct subcellular studies in a broad range of biological settings. We discuss some of these below.
Characterizing organelle proteomes
Early organelle-centric purification methods generally isolated target organelles at a high purity before systematically identifying organellar proteins using LC-MS184–189. These approaches established the protein inventories of organelles, although they could not discriminate between targeted organellar constituents and co-purified proteins originating from non-target subcellular compartments. Typically, fewer than 1,000 proteins were measured over as few as four fractions in these early studies190,191 — a decade later, more than 8,000 proteins were identified in four distinct fractions from mouse lungs following the rapid development of liquid chromatography and MS technology192.
Subtractive proteomics methodologies using differential separation technologies were first applied to define the nuclear envelope proteome from mouse liver39. Subsequently, the quantitative comparison of a crude preparation of mitochondria against purified mitochondria from ten different mouse tissues greatly contributed to the mitochondrial compendium, MitoCarta36. Subtractive proteomics has helped establish the protein inventories of peroxisomes193–196, autophagosomes197 and lysosomes198. A landmark single-organelle spatial proteomics paper described the systematic study of the human centrosome using intensity-based, label-free protein correlation profiling of consecutive density gradient fractions30. Several adaptations of subtractive proteomics have used SILAC ratios produced by spiking a differentially labelled internal standard into each fraction33,34,199,200; this approach, with or without stable isotope labelling, has been used to characterize the proteome of peroxisomes38,201, nuclei202, autophagosomes33,199, lipid droplets34, vesicles200 and mitochondria35,203.
For proximity labelling approaches, the initial BioID study in which BioID was fused to lamin A identified new components of the nuclear lamina63. The first APEX study targeted the peroxidase to the mitochondria to define the proteome of the mitochondrial matrix and inner mitochondrial space67. Proximity labelling has now contributed to the characterization of various organelles and some of the more recent studies have employed multiple baits to provide extensive coverage of specific structures, in some cases revealing their organization68–70,204–212. Proximity labelling is typically performed on whole cell lysates; upstream fractionation steps can also be coupled with the capture of biotinylated proteins to assist in defining specific proteomes, although these approaches can have issues with post-lysis artefacts and imperfect fractionation209,211.
Imaging approaches have helped characterize the proteome of many organelles, such as the centrosome and nucleolus213, and revealed novel structures including cytoplasmic rods and rings11,214 and the nucleoli rim213. Complete characterization of the organelle proteome using affinity reagents or fluorescent protein fusions is both expensive and laborious; these approaches are complementary to MS-based approaches and valuable for validation of subsets of proteins identified in MS data.
Cell-wide correlation profiling
Cell-wide mapping of proteins using correlation profile approaches has been achieved in many cell types10,44,47, tissues and even whole organisms. Models include Arabidopsis root-derived cells42, chicken DT-40 cells13, mouse neuronal cells14, rat liver40,45, Saccharomyces cerevisiae15, Drosophila embryos16 and the photo-synthetic bacterium Synechocystis18. A recent study achieved the first subcellular-resolution proteome of an apicomplexan parasite, Toxoplasma gondii17; T. gondii is a widespread parasite that infects 30% of the human population, leading to various pathologies in infected individuals215. Using a variant of the LOPIT workflow known as hyperplexed LOPIT (or hyperLOPIT) along with image-based validation10,216, this study mapped two-thirds of the predicted proteome to 26 subcellular and sub-organellar niches in the tachyzoite form of the parasite. High-resolution isoelectric focusing LC-MS (HiRIEF-LC-MS)217 coupled with differential detergent solubility and TMT 10-plex labelling was recently used to generate comprehensive subcellular maps with an 8,140-protein overlap between replicates across five human cell lines47. Using support vector machines, 9,594 proteins were classified into 15 specific compartments and 12,125 proteins were classified into 4 subcellullar ‘neighbourhoods’ — groups of functionally related subcellular compartments such as those involved in the secretory pathway. Cell-wide correlation profiling can also be used to characterize protein complexes (BOX 3).
Box 3 |. Interrogating protein complexes.
Correlation profiling can investigate protein complexes globally in several systems325–327. Chromatography or electrophoresis is preferred for protein complex separation as ultracentrifugation methods can only resolve large complexes. Ion exchange is widely used328, although samples are exposed to high salt concentrations; techniques such as size exclusion chromatography can be conducted under native conditions to better preserve protein complexes during separation. Peptide cross-linkers can preserve weakly interacting proteins for separation under denaturing conditions329,330, although optimizing cross-linking conditions for a broad range of complexes is challenging. In these experiments, cross-linked proteins that co-migrate are considered part of the same complex. Blue native polyacrylamide gel electrophoresis (BN-PAGE) coupled with semi-quantitative mass spectrometry (MS) has been used to assess interactions in multi-protein complexes331. It is possible to measure subcomplexes, different assembly states or how protein–protein interactions change in response to stimulus through coupling co-migration methods with stable isotope labelling by amino acids in cell culture (SILAC)325.
Thermal proximity co-aggregation (TPCA) exploits the propensity of proteins to co-aggregate during thermal denaturing332,333. The principle of TPCA is that interacting proteins exhibit similar co-aggregation behaviours and therefore have similar melting curves. The assay involves briefly heating cells or cell lysates along a gradient of increasing temperatures. Changes in the soluble proteins at each temperature are then quantified using MS, allowing for comparisons of protein complex formation and dissolution across experimental conditions — for example, drug treatment or infection. Although TCPA has a lower resolution than other biochemical fractionation techniques, it has been applied to characterize and monitor complex dynamics in disease models and biological processes332–334. TPCA can also provide information about a protein’s subcellular localization or movement between organelles during a biological process through inferring associations between organellar resident proteins334.
The imaging equivalent of these cell-wide organellar fractionation methods to achieve cell-wide distribution of proteins is the work by the HPA project. This is the largest collection of subcellular imaging data to date, with 12,813 human proteins mapped to 30 different subcellular structures on a panel of 22 different human cell lines using immunofluorescence and confocal microscopy as of January 2021. This project has predicted that up to 50% of human proteins localize to multiple compartments, with 17% showing single-cell variations11. Several studies have investigated the sources of this single-cell heterogeneity11,218,219. Characterizing single-cell heterogeneity has implications in health and disease; for example, many proteins are expressed heterogeneously across tumours, which has presented a challenge for developing treatments220,221. Imaging further enables analysis of spatio-temporal human proteome reorganization over the course of the cell cycle or as a response to metabolic change222,223.
Sub-organellar resolution
The application of tailored and combinational biochemical separation approaches, such as centrifugation and affinity purification, has enabled the definition of the sub-organellar proteomes of lysosomes32, nuclear envelopes37,39, chloroplast envelopes31 and sub-compartments of mitochondria29,35,224,225. For example, the incorporation of organelle purification, proximity labelling and subtractive methods effectively achieved sub-organellar resolution of the nuclear envelope, lipid droplets and centrosomes in Arabidopsis37,68,211. To map the nuclear envelope, subtractive proteomics was used to distinguish proteins associated with the nuclear envelope from microsomal membrane proteins, followed by BioID2 with several bait proteins to provide complementary data for the characterization of the plant nuclear membrane proteome and nuclear pore complex. Subsequent studies used LOPIT in a targeted manner alongside electrophoresis-based protein separation to resolve and identify previously unknown endoplasmic reticulum proteins and cis, medial and trans-Golgi proteins in Arabidopsis41,51,226; these findings have implications for understanding the role of Golgi proteins in the cell wall synthesis pathway and the development of biofuels227. A recent large-scale subtractive proteomics study used a combination of label-free and SILAC-based MS profiling to profile the sub-organellar compartments of mitochondria in S. cerevisiae cultures grown in different carbon sources. Integration of these multiple MS data sets resulted in the high-confidence identification of 901 mitochondrial proteins, including numerous proteins of previously unknown mitochondrial residency and some with multiple cellular locations29.
As mentioned above, the breadth and precision of organellar and sub-organellar proteomics by proximity labelling can be improved by selecting multiple baits for organelle profiling. In a ground-breaking 2015 study, 58 baits mapping to the centrosome and primary cilia were systematically profiled to define the composition of these structures using bait–prey hierarchical clustering, revealing different groups of proteins that performed distinct functions68. This study set the stage for mapping organelles with more extensive sets of baits. Recently, the steady-state composition and organization of RNA stress granules, processing bodies69 and mitochondria70 were defined using more than 100 baits. In each of these studies, clustering of preys rather than baits provided a high-resolution map of these structures, in some cases suggesting direct protein–protein interactions within the structures that could be experimentally validated using immunoblotting and microscopy69. A comprehensive map of the human cell made using ~190 baits spanning most organelles160 provided a global reference map. As these resources become increasingly available, selection of ideal baits to profile an organelle across different cell types or following different treatments will permit more in-depth investigation of spatio-temporal changes in organellar composition and organization. As imaging allows the study of proteins in their native cellular environment without the need for cell lysis, fine sub-organellar structures can be preserved in situ to enable detection of dynamic and MLP behaviour across the sub-organellar proteome11. This trait of immunofluorescence has enabled the spatio-temporal characterization of the nucleolar proteome and its relocalization to the mitotic chromosomes during mitosis228. Further, MS analysis has exposed diverse extracellular vesicle subpopulations with roles in important biological functions and disease, such as angiogenesis and cancers229–232. Multiple strategies to isolate extracellular vesicles can be used; some are similar to subtractive proteomics or correlation profile strategies, such as density or differential centrifugation, and others use alternative biochemical isolation strategies, such as size exclusion, microfluidics and immune purification. These methods are detailed and compared in a recent review233.
Multi-localized proteins
Owing to the dynamic nature of proteins during signalling and heterogeneity between different cell types, there is no consensus on exactly what proportion of the proteome is multi-localized. Predictions of MLPs vary and have been reported as 50% of the human proteome by hyperLOPIT studies and the HPA11 but as little as 10% or less by other correlation profiling studies47,53. This discrepancy could be owing to low confidence measurements, the type of classification tool used or biological variation47,53 and demonstrates the difficulty of accurately measuring MLPs.
There is a growing repertoire of tools for describing MLPs. An unorthodox citizen science-based deep learning approach was able to create a robust tool for overcoming the challenges of automated subcellular allocation of MLPs for the analysis of the HPA12. This approach robustly classified proteins into 29 subcellular compartments across 17 human cell lines. Despite inaccurate annotation of structurally similar subcellular features such as centrosomes and microtubule organization centres, unusual features such as blebs and condensed chromosomes were successfully profiled using this approach12. The identification of MLPs in T. gondii using T-augmented Gaussian mixture model–Markov-chain Monte Carlo models demonstrated that these proteins are primarily associated with organelles associated with protein transportation, such as the plasma membrane and Golgi. Organelles known to be relatively static such as rhoptries, micronemes and dense granules showed more proteins with single assignments17.
Dynamic studies
A study to chart the mitochondrial importome in Trypanosoma brucei employed an approach known as importomics to identify substrates of glycosomal and mitochondrial import pathways155,234. Importomics approaches can assess changes in protein abundance in a target organelle following RNAi-mediated knockdown of a central component of the protein import machinery, to determine potential substrate proteins that are trafficked to the organelle and imported via this machinery155,234. Importomics is not limited to use with organelle-enriched fractions and can be used to globally study organellar protein import using cell lysates155,235. A more simplistic importomics study design for investigating the nucleus used nucleocytoplasmic fractionation coupled with quantitative MS to study mechanisms underlying nucleocytoplasmic partitioning in Xenopus laevis eggs202. Knockdown of the nuclear export receptor exportin 1 largely did not affect nucleocytoplasmic partitioning, suggesting that protein distribution between the nucleus and cytoplasm is largely maintained through passive processes rather than active trafficking202.
Cell-wide dynamic studies studying trafficking proteins on the proteome scale using correlation profiling have given unprecedented insight into EGFR-related protein trafficking events47. The combination of CRISPR–Cas9-based gene knockouts with dynamic organellar maps has been used to assess AP-4 vesicles153 and AP-5 cargo154 and perform the first cell-wide, dynamic correlation profiling experiment intended to define drug mechanisms236. Recently, LOPIT was coupled with a technique where selective capturing of vesicles destined for the Golgi were relocated to mitochondria by replacing the Golgi targeting domains of golgins with a mitochondrial transmembrane domain55. This enabled characterization of vesicle cargo and regulatory proteins by redirecting them to the mitochondria, bypassing technical issues that arise owing to the genetic redundancy of golgin proteins and their transient interaction with vesicles.
Golgins.
A family of proteins that selectively tether vesicles at the golgi apparatus and mediate transport of vesicles as part of the secretory pathway.
Several studies have been published assessing organelle remodelling during viral infection and the distribution of virus and host proteins. Organellar fractionation followed by MS has been used to quantify temporal changes in plasma membrane and organelle proteomes during human cytomegalovirus infection53,237,238. Analysis of organelle remodelling during the virus replication cycle demonstrated that alterations in organelle composition can reflect changes in the structure and function of an organelle, as peroxisomes shift between antiviral and proviral functions239,240. Coupling of density and differential centrifugation has also been utilized to explore the mitochondrial-associated endoplasmic reticulum membrane, the endoplasmic reticulum and cytosol proteomes during infection with hepatitis C and Sendai virus241. The above studies have described virus manipulation of host protein subcellular localization, allowing for the increased understanding of virus–host interactions and identification of potential therapeutic targets.
Generally, MS techniques are only able to capture averaged information for heterogeneous populations in samples, losing dynamic events occurring in subgroups or single cells. Imaging studies have reported that heterogeneity in cell samples could be the result of sudden stresses, cell crowding, cell–cell contacts, cell differentiation and different metabolic states218,242–245. Several imaging studies have reported a spatio-temporal resolved map of the human proteome over the course of a cell cycle using imaging227,245,246 and similar techniques have been applied to disease settings. Indeed, mislocalization of proteins and variations in protein expression levels have been shown to be associated with diseases such as cancer, neurodegenerative diseases and cardiac diseases247–249. Combining imaging with computational analysis enables the identification of proteins with similar relocalization and changes in expression levels, which implies functional connectivity for such groups of proteins101,250,251. Imaging-based approaches allow quantification of intensity levels in different cellular compartments that can better indicate whether a protein translocates from one compartment to the other, or merely changes in abundance.
Reproducibility and data deposition
Spatial proteomics approaches tend to vary from laboratory to laboratory. Therefore, careful documentation of workflows and data deposition is important for ensuring reproducibility. Here, we cover these aspects, plus other method-dependent considerations.
Mass spectrometry-based methods
As mentioned above, MS-based proteomics data are prone to missing values owing to stochastic extraction and solubilization of peptides in sample preparation and during MS acquisition. Despite these issues, BioID and affinity purification MS can typically achieve high reproducibility (R2 > 0.9) as the complexity of MS samples in these methods is low compared with the complexity of whole cell lysates252. Reproducibility and coverage is usually more achievable when performing LC-MS/MS with less complex peptide samples, as in these samples there are fewer precursor ions per isolation window in the MS1 step and fewer ions are therefore omitted for subsequent sequencing in MS2. Labelling methods can reduce missing values, although cell-wide correlation profiling methods that allow for multiplexed characterization of many organellar fractions in each replicate still suffer from losses in protein identification of ~40–50% across three biological replicates. Unlabelled methods, by contrast, have attrition rates of ~60%14,43. There are no tools to assess correlation profile reproducibility, only those to assess and compare the quality of organellar separation between cell-wide spatial proteomics experiments88. DIA provides improved protein identification and quantification as it involves less stochastic ion sampling during MS acquisition than DDA.
For analyses of data collected after using biochemical separation techniques such as density centrifugation, losses in protein identification are proportional to the number of organellar fractions and replicates analysed. Performing reproducible subcellular fractionation is partly dependent on the simplicity of the fractionation procedure. Differential centrifugation and sequential detergent preparations are more reproducible than density equilibrium centrifugation owing to the intricacy of forming and collecting density equilibrium gradients, although spatio-temporal analysis using density gradients has proven to be effective17,18,45,53,89.
For any MS-based proteomics experiment, the Minimum Information About a Proteomics Experiment (MIAPE) guidelines should be adhered to when publishing. The principles of MIAPE and the basic criteria are shown in BOX 4. For MIAPE compliance, the location of raw mass spectra files should be published; files can be stored in a public repository, such as the PRoteomics IDEntifications Database (PRIDE). There is no overarching deposition resource for data from all subcellular MS methodologies, although bespoke platforms and pipelines are available47,156,253 (TABLE 2). The creation of guidelines to ensure efficient capture and reporting of crucial metadata associated with spatial proteomics data will be essential to enable data reanalysis and align the field with Findability, Accessibility, Interoperability, and Reusability (FAIR) principles254.
Box 4 |. Minimum Information About a Proteomics Experiment (MIAPE) guidelines.
Proteomics protocols in the field are heterogeneous in nature and data output is context-specific. This led the Human Proteome Organization’s Proteomics Standards Initiative to establish guidelines for the minimal information required to report proteomics data. The MIAPE guidelines facilitate the sharing of metadata, allowing for transparency and reducing ambiguity for those who wish to review, reproduce or reanalyse the data335. Currently, the following information should be shared for any mass spectrometry (MS) experiment to be MIAPE compliant336:
General details such as the name of the person responsible for the data, the instrument model and manufacturer, and any customization of the instrumentation.
The ion source, for example electrospray ionization or matrix-assisted laser-desorption/ionization, including the manufacturer, make and model.
Information on post-source components such as Orbitrap or time of flight, including the manufacturer, whether activation or dissociation was used, the component where dissociation occurs and what collision gas was used, if applicable.
Data acquisition and analysis software and parameters, the resulting data (for example, chromatograms and spectra) with their corresponding annotations (such as MS level or ion mode) and information on where to find the raw data. Raw data are almost always located in the PRoteomics IDEntifications Database (PRIDE) repository as this is often a prerequisite for publication.
Further MIAPE criteria apply to MS quantitation and informatics, chromatography, and gel electrophoresis-based and molecular interaction methods.
Table 2 |.
Recommended resources for subcellular proteomics data
| Data resource | Description | URL |
|---|---|---|
| Mass spectrometry-based proteomics | ||
| PRoteomics IDEntifications Database (PRIDE)307 | World’s largest data repository for a diverse range of MS workflows Mandatory submission requirements for each experiment include raw MS files and processed results showing protein identification Part of the ProteomeXchange Consortium |
https://www.ebi.ac.uk/pride |
| PeptideAtlas308 | A resource that provides both a uniform analysis pipeline and repository for genome annotation of raw tandem MS data | http://www.peptideatlas.org |
| Mass Spectrometry Interactive Virtual Environment (MassIVE)309 | Part of the ProteomeXchange Consortium A resource to allow the free exchange of MS data Includes proximity-dependent biotinylation data sets |
https://massive.ucsd.edu/ |
| Bespoke subcellular MS repositories and pipelines | ||
| ProHits-web | Repository for storing, visualizing and disseminating affinity purification MS and proximity labelling data Designed to complement MassIVE, BioGRID and IntAct, which also store interaction data |
https://prohits-web.lunenfeld.ca/ |
| The Cell Map160 | A large-scale project aiming to map the entire interaction network of the human embryonic kidney cell line, HEK293, as an open-access resource | https://humancellmap.org/ |
| pRoloc and pRolocdata253 | A series of R packages with user-friendly pipelines for data analysis of biochemical fractionation experiments The pRolocdata package contains existing spatial proteomics data sets from LOPIT and protein correlation profiling |
https://lgatto.github.io/pRoloc/ |
| SubCellBarCode47 | Exploratory web interface for localization data across five different human cancer cell lines collected from biochemical fractionation experiments | https://www.subcellbarcode.org/ |
| HeLa Spatial Proteome database44 | Interactive subcellular biochemical fractionation maps of the human cervical cancer cell line, HeLa | www.mapofthecell.org |
| MitoCarta310 | An inventory of highly confident human and mouse mitochondrial proteins annotated using multiple microscopy and genome-scale data sets | https://www.broadinstitute.org/mitocarta/ |
| MitoMiner | A database of mitochondrial localization experiments in mammals, zebrafish and yeasts | https://mitominer.mrc-mbu.cam.ac.uk/ |
| PeroxisomeDB311 | A curated resource collating identified peroxisomal proteins from 38 organisms | http://www.peroxisomedb.org/ |
| RareLSD312 | A manually curated inventory of lysosomal enzymes | https://webs.iiitd.edu.in/raghava/rarelsd/ |
| Imaging resources | ||
| Cell Image Library (CIL)313 | A public repository providing a resource of thousands of imaging data sets, including videos and animations, from various organisms | http://www.cellimagelibrary.org |
| Image Data Resource (IDR)314 | Publicly accessible light microscopy data alongside extensive metadata for the purpose of improving accessibility and reanalysis | https://idr.openmicroscopy.org |
| BioImage Archive | A central archive for data from the IDR and the Electron Microscopy Public Image Archive (EMPIAR) | https://www.ebi.ac.uk/bioimage-archive/ |
| Multidisciplinary repositories | ||
| Human Protein Atlas (HPA)11 | A large-scale project with open-source data that aims to map all human proteins in cells, tissues and organs, including subcellular information Data types include antibody-based imaging and systems biology data |
https://www.proteinatlas.org/ |
| Universal Protein Resource (UniProt)144 | A consortium of multiple databases providing protein sequence and annotation data Annotations are performed through automated and manual curation of information extracted from the literature and computational analyses such as Gene Ontology |
https://www.uniprot.org/ |
LOPIT, localization of organelle proteins using isotope tagging; MS, mass spectrometry.
Imaging
Immuno-imaging data can be difficult to reproduce because of biological variation — for example, variation between cell types — and technical variation introduced by different sample preparation procedures or affinity reagents. Both the HPA and the independent antibody validation initiative antibodies-online found that less than 50% of commercially available antibodies are appropriate for studying protein distribution, owing to specificity issues in the context of intact tissues and cells255,256. Major research and awareness efforts are improving this situation by using alternative antibody production and product validation procedures (see the Limitations and optimizations section).
As each microscopy vendor has their own internal imaging format and software for analysing and visualizing data, exporting files into standardized formats is important to disseminate scientific discoveries. The Open Microscopy Environment is an open-source informatics framework for biological microscopy experiments, designed to support various imaging applications. Most imaging platforms and open-source software are compatible with the Open Microscopy Environment166,170,257,258. Publishing imaging data in a suitable data repository for data collection and mining is encouraged and is a prerequisite for publishing in many journals. Providing the original raw images is crucial, along with metadata accompanying each individual image containing all hardware and software settings applied upon image acquisition. When raw images are processed — for example, to increase intensity or contrast — these processes must be described. Upon submission of images to most journals there are clear guidelines regarding what image processing is accepted and part of the review process is sometimes dedicated to tracking image manipulations. Public repositories for imaging experiments are noted in TABLE 2 (REF.259).
Limitations and optimizations
Below, we discuss technical limitations to consider in MS-based and imaging-based subcellular proteomics methods and existing optimizations to address these issues and ensure accurate interpretation of results.
Mass spectrometry-based methods
Organelle fractionation.
Limitations of organelle fractionation approaches are largely data analysis-centric and arise from the significant portion of MLPs in the proteome, uncertainty in current database annotations and classifier overfitting from using small reference sets. Although curated marker sets for humans and some model organisms are becoming larger and more established145, non-model organisms are often poorly curated; however, orthologue identification and unsupervised clustering tools can make their analysis possible260. Other semi-supervised tools can help with marker curation149,261. As proteomics data represent an average location for each protein, computationally identifying primary locations is relatively simple, although when using subtractive proteomics or other similar methods, information on MLPs is lost as limited fractions are assessed rather than complete protein distributions. Similarly, dynamic analysis does not distinguish between genuine translocation and synthesis or degradation of proteins within subcellular compartments. Recent advances in computation have begun to address these questions, for example Bayesian methodologies for the localization assignment of proteins exhibiting multiple localization52 and pipelines to identify changes in protein distribution, such as the T-augmented Gaussian mixture model, protein translocation magnitude and translocation reproducibility scoring and TRANSPIRE44,54,55. For correlation profiling experiments, the quantitative approach taken — for example, labelling or label-free approaches — can affect the sensitivity, resolution and coverage of the experiment146,262–264.
Established cell lysis protocols for organelle fractionation methods may be inappropriate for non-model organisms, especially when using species with cell walls. Regardless of the model system, the lysis method and buffer conditions must be optimized15,17. A full lysis of the cell can be performed before centrifugation to show the absence of organellar profiles as a negative control. Also, when performing dynamic studies, it is preferred to perform sample preparation and analysis of conditions simultaneously for each replicate, so that the changes seen between conditions are likely to be biological and not caused by technical factors.
Proximity labelling.
A major limitation of proximity labelling-based spatial proteomics approaches is the time investment required for selection and validation of appropriate baits. Ideally, it should be confirmed that addition of the fusion protein does not disrupt the function of the protein or result in gross mislocalization160, either using microscopy or by systematically tagging both termini69. The bait fusion protein should be expressed at close to physiologically relevant levels, which is not always straightforward when using transient or stable expression from generic promoters. The inaccessibility of amino acid residues to biotinylation can lead to unidentified prey proteins. Further, interpretation of proximity profiles is often complicated by the dynamic nature of protein localizations and interactions and the fact that many proteins localize to multiple compartments71. Careful bait selection, systematic profiling and prey-centric analyses, although labour intensive, can overcome such complications.
General limitations of subcellular proteomics techniques.
Spatial proteomics approaches often miss information on protein post-translational modifications (PTMs) and isoforms, in part owing to the gene-centric focus of downstream analytical pipelines. There is currently no PTM-level imaging equivalent to the HPA as there are insufficient site-specific PTM-targeting antibodies265. Similarly, generating isoform-specific antibodies, although possible, can be very challenging266. MS techniques for analysing PTM sites in a high-throughput manner are emerging — particularly for phosphorylation and acetylation — and can be aided by enrichment techniques, such as those that use metal cation affinity or antibody purification. These are covered in recent papers267–269. These experiments typically require large amounts of starting material (>1 mg) owing to the sub-stoichiometric nature of PTMs and the low recovery and inherent losses in proteomics and enrichment protocols270, which may be limiting when using primary cell cultures or organellar fractions. However, streamlining workflows and improved enrichment methods will aid analysis of phosphorylation and acetylation in such experimental scenarios89,271–273. Other modifications, such as glycosylation, are difficult to analyse owing to their complexity, difficulty in distinguishing their mass shift, low stoichiometry, instability and poor ionizing efficiency274. Sample preparations or MS acquisitions must be adjusted for the PTM of interest270,271,275.
Imaging
Although use of a fixative allows cells to be preserved at precise time points, it can cause high background and signal loss, or disruption of subcellular structures and protein macromolecules if the fixation protocol used is inappropriate for the protein of interest276. An optimized single fixation protocol can be used for proteome-wide compatibility for studies such as the HPA107; although this protocol has proven to work for a large variety of proteins representing >30 cellular structures, the best staining conditions for each protein depend on the sample context and the combination of the target protein and antibody used for detection.
Antibodies used in imaging studies are subject to biological batch-to-batch variability, with inconsistent binding properties between batches. Every antibody batch needs careful validation for their prospective application and sample type prior to use. More commercial suppliers are using recombinant means to manufacture their antibodies over traditional immunization, providing more sustainable and reproducible products. Awareness of careful antibody selection, validation and optimization has improved significantly over recent years. In subcellular analysis, the potential for high local concentrations of any cross-reacting proteins increase the need for thorough validation.
Outlook
Substantial progress has been made in both MS-based and imaging-based spatial proteomics over the past decade, with the development of highly multiplexed protein detection methods coupled with subcellular-resolution imaging techniques277,278. This highly multiplexed imaging has also been shown to have huge potential for healthcare by allowing deep profiling of cell phenotypes in clinical samples such as tumours. Advances in instrumentation, such as the development of laser microdissection microscopes, now allow for precise isolation of single cells from tissue sections that can then be explored with ultra-high-sensitivity MS, a concept recently introduced as deep visual proteomics279. Deep visual proteomics and similar approaches combine the best of two powerful technologies and overcome their individual limitations. New technologies also include artificial intelligence-driven image analysis methods, which could potentially achieve the unbiased identification of subcellular characteristics in specific cellular phenotypes.
Several challenges remain for the holistic study of subcellular biology. These include determining the copy number of proteins in different parts of the cell, organelle-specific turnover rates, organellar interfaces and the extent of organelle crosstalk. The continual developmental of new tools to access difficult to sample subcellular regions, such as mitochondrial–endoplasmic reticulum contact sites280,281, and efficient ways to detect newly synthesized proteins282–286 promise to further our ability to elucidate the dynamic processes that occur within a cell. Crosstalk between PTM regulatory mechanisms and its role in subcellular protein movement remains understudied and the general patterns governing proteoform-related subcellular protein location are yet to be discovered47,89,287. New spatial proteomics methodologies will undoubtedly allow the mapping of individual proteoforms.
Single-cell transcriptomics data on the composition and variation of cellular mRNA populations288–290 are often used as a proxy for protein localization within different cell populations; however, this makes the misleading assumption that proteins and their corresponding transcript co-locate291–293. Gene products vary substantially in their translation efficiency, post-translational processing, trafficking and stability, confounding the accurate prediction of protein abundance and subcellular localization from mRNA reads alone. Genomic profiles do not readily identify causal mechanisms, clinical markers, physical associations or drug targets. For these reasons, a single-cell, multi-omics approach incorporating single-cell spatial proteomics techniques is needed to fully understand cell functionality by determining subcellular protein levels, PTMs and protein folding, binding and turnover. There are several challenges to overcome beyond improving instrument sensitivity and reducing loss of material during sample preparation before the acquisition of meaningful single-cell spatial proteomics data will be possible; first, significant compositional heterogeneity is seen across even genetically and phenotypically identical cells294, presumably stemming from differences in cellular proteostasis machinery, the impact of environmental cues and stochastic variations in protein expression programmes. Second, the regulation of protein turnover rate is likely related to subcellular location through compartment-dependent PTM modification patterns and protein complex formation. Indeed, compared with single-cell genomics workflows, single-cell proteomics methods — spatial or otherwise — remain in their infancy. Priorities over the next 5 years will be centred on measuring larger numbers of proteins accurately in single cells295. Although the field currently lacks robust methods for identifying, quantifying and localizing the myriad of proteins present in individual cells, recent developments such as single-cell MS-based methods and newer imaging-based protein sequencing modalities296 suggest we are on the cusp of a new era of single-cell spatial proteomics297,298.
Super-resolution fluorescence microscopy techniques can measure individual protein molecules within single cells, and although most fluorescent imaging methods are typically limited to resolving 1–5 protein targets at a given time, recent innovations with cyclic fluorescent microscopy approaches allow for the analysis of >50 proteins278. Mass cytometry (BOX 1) and cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq)299 technologies are able to probe upwards of 30 cell surface markers for single-cell proteomics applications. Similar to other fluorescent imaging techniques, these methods depend on access to highly validated antibodies. The future of single-cell proteomics is likely to be driven by the development and application of optical imaging modalities that do not depend on antibodies300. Promising technologies include a microscopy-based imaging workflow that exploits edman degradation for the parallel partial sequencing of thousands of individual fluorescently labelled peptides arrayed on the surface of a glass slide296,301. Indeed, the companies that brought single-molecule nucleic acid sequencing to the market are moving to develop devices capable of generating partial sequence information for polypeptides in a similarly high-throughput manner. As single-molecule measurements are inherently noisy, such devices are likely to depend on sophisticated data analysis tools to ensure that reliable and statistically meaningful results are generated. No technology has yet been reported that allows for de novo spatial sequencing of proteins within a native, single-cell or subcellular context. Additionally, data-driven approaches will be increasingly used for predicting protein localization directly from unlabelled images and for generating whole cell models that allow for predictions of cell morphology and protein localization in normal and perturbed cellular states302.
Edman degradation.
A cyclic peptide sequencing technique where amino-terminal amino acid groups are sequentially cleaved and identified using chromatography or electrophoresis.
A ‘Global Positioning System’ for proteins is required that can simultaneously monitor the identity, location and physical neighbourhood of all protein molecules associated with organellar compartments across numerous individual cells, with high confidence and quantitative precision. Progress will result from collaborative thinking and iterative technology optimization supported by sizeable investment from public and commercial sources. We anticipate that pioneering breakthroughs in this domain are lurking around the corner and have the potential to transform biomedical research to an even greater extent than even single-cell genomics approaches. Advances in single-cell spatial proteomics could reveal localizations of pathway components and protein assemblies that drive the early stages of major diseases such as diabetes, cancer, Alzheimer disease, heart failure and infection. Finally, the development and application of machine learning tools promises to allow the interrogation of increasingly complex spatial proteomics data, delivering unprecedented insights into the dynamics of the subcellular proteome.
Acknowledgements
J.A.C. is funded through a BBSRC iCASE award with Astra Zeneca. D.M. is funded by the Knut and Alice Wallenberg Foundation (2016.0204) and the Swedish Research Council (2017-05327). C.S. is funded by Science for Life (SciLifeLab) national funding, the National Microscopy Infrastructure (VR-RFI 2019-00217), the European Proteomics Infrastructure Consortium EPIC-XS (project number 823839) and the EU Horizon 2020 programme. A.-C.G. is the Tier 1 Canada Research Chair in Functional Proteomics and is supported by the Canadian Institutes of Health Research (FDN143301). C.E.M. is supported by a KRESCENT Post-Doctoral Fellowship and Canadian Institutes of Health Research Fellowship. B.W. is supported by the Deutsche Forschungsgemeinschaft (Project IDs 403222702/SFB 1381, FOR 1905, FOR 2743), Germany’s Excellence Strategy (CIBSS — EXC-2189 — Project ID 390939984), European Research Council Consolidator Grant No. 648235 and the European Union Marie Curie Initial Training Networks program PerICo (Grant Agreement Number 812968). Work included in this study has also been performed in partial fulfilment of the requirements for the doctoral thesis of M.M. at the University of Freiburg. L.J.F. is supported by Genome Canada/Genome British Columbia (Project 264PRO). I.M.C. is funded by the National Institute of General Medical Sciences (GM114141), the National Institute of Child Health and Human Development (HD089275) and the Edward Mallinckrodt Jr. foundation. C.N.B. is funded by the National Institute of General Medical Sciences (T32GM007388). Y.P. is funded through the Swedish Cancer Society. J.L. is funded though the Erling-Persson Family Foundation, the Swedish Cancer Society, the Swedish Childhood Cancer Foundation, the Swedish Foundation for Strategic Research, the Swedish Research Council and the EU Horizon 2020 project (RESCUER and OncoBiome). A.E. acknowledges previous and ongoing grant support from the National Institutes of Health (NIH) (1UL1TR001430, R01AG064932, R01AG061706, R01DK110520).
Footnotes
Competing interests
The authors declare no competing interests.
RELATED LINKS
COMPARTMENTS: https://compartments.jensenlab.org/
Gene Ontology: http://geneontology.org/
Human Protein Atlas: https://www.proteinatlas.org/humanproteome/cell
Kaggle challenge for multi-label classification of cell organelles: https://www.kaggle.com/c/human-protein-atlas-image-classification
miAPe guidelines: http://www.psidev.info/miape
Open microscopy environment: https://www.openmicroscopy.org/
Open-source Python tools for proteomics analysis: https://github.com/Roestlab/PythonProteomics
R programming packages: https://www.R-project.org/
UniProt: https://www.uniprot.org/
References
- 1.Gibson TJ Cell regulation: determined to signal discrete cooperation. Trends Biochem. Sci 34, 471–482 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Hung M-C & Link W Protein localization in disease and therapy. J. Cell Sci 124, 3381 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Pankow S, Martínez-Bartolomé S, Bamberger C & Yates JR Understanding molecular mechanisms of disease through spatial proteomics. Curr. Opin. Chem. Biol 48, 19–25 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siljee JE et al. Subcellular localization of MC4R with ADCY3 at neuronal primary cilia underlies a common pathway for genetic predisposition to obesity. Nat. Genet 50, 180–185 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neel DS et al. Differential subcellular localization regulates oncogenic signaling by ROS1 kinase fusion proteins. Cancer Res. 79, 546 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hübner S, Eam JE, Hübner A & Jans DA Laminopathy-inducing lamin A mutants can induce redistribution of lamin binding proteins into nuclear aggregates. Exp. Cell Res 312, 171–183 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Valastyan JS & Lindquist S Mechanisms of protein-folding diseases at a glance. Dis. Model. Mech 7, 9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shin SJ et al. Unexpected gain of function for the scaffolding protein plectin due to mislocalization in pancreatic cancer. Proc. Natl Acad. Sci. USA 110, 19414–19419 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thelen MP & Kye MJ The role of RNA binding proteins for local mRNA translation: implications in neurological disorders. Front. Mol. Biosci 10.3389/fmolb.2019.00161 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christoforou A et al. A draft map of the mouse pluripotent stem cell spatial proteome. Nat. Commun 7, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thul PJ et al. A subcellular map of the human proteome. Science 10.1126/science.aal3321 (2017). [DOI] [PubMed] [Google Scholar]; This ambitious work performs immunofluorescence and confocal microscopy to systematically assess the subcellular localization of more than 12,000 human proteins in several human cell lines, published in the HPA database.
- 12.Sullivan DP et al. Deep learning is combined with massive-scale citizen science to improve large-scale image classification. Nat. Biotechnol 36, 820–828 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Hall SL, Hester S, Griffin JL, Lilley KS & Jackson AP The organelle proteome of the DT40 lymphocyte cell line. Mol. Cell Proteom 8, 1295–1305 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Itzhak DN et al. A mass spectrometry-based approach for mapping protein subcellular localization reveals the spatial proteome of mouse primary neurons. Cell Rep. 20, 2706–2718 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nightingale DJ, Geladaki A, Breckels LM, Oliver SG & Lilley KS The subcellular organisation of Saccharomyces cerevisiae. Curr. Opin. Chem. Biol 48, 86–95 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan DJL et al. Mapping organelle proteins and protein complexes in Drosophila melanogaster. J. Proteome Res 8, 2667–2678 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Barylyuk K et al. A subcellular atlas of Toxoplasma reveals the functional context of the proteome. Cell Host Microbe 28, 752–766.e9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baers LL et al. Proteome mapping of a cyanobacterium reveals distinct compartment organization and cell-dispersed metabolism. Plant. Physiol 181, 1721–1738 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jeffery CJ Protein moonlighting: what is it, and why is it important? Philos. Trans. R. Soc. B: Biol. Sci 373, 20160523 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gancedo C, Flores C-L & Gancedo JM The expanding landscape of moonlighting proteins in yeasts. Microbiol. Mol. Biol. Rev 80, 765 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lundberg E & Borner GHH Spatial proteomics: a powerful discovery tool for cell biology. Nat. Rev. Mol. Cell Biol 20, 285–302 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Pasquali C, Fialka I & Huber LA Subcellular fractionation, electromigration analysis and mapping of organelles. J. Chromatogr. B Biomed. Sci. Appl 722, 89–102 (1999). [DOI] [PubMed] [Google Scholar]
- 23.Parsons HT Preparation of highly enriched ER membranes using free-flow electrophoresis. Methods Mol. Biol 1691, 103–115 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Moon MH Flow field-flow fractionation: recent applications for lipidomic and proteomic analysis. TrAC 118, 19–28 (2019). [Google Scholar]
- 25.Oeyen E et al. Ultrafiltration and size exclusion chromatography combined with asymmetrical-flow field-flow fractionation for the isolation and characterisation of extracellular vesicles from urine. J. Extracell. Vesicles 7, 1490143 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen WW, Freinkman E & Sabatini DM Rapid immunopurification of mitochondria for metabolite profiling and absolute quantification of matrix metabolites. Nat. Protoc 12, 2215–2231 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiong J et al. Rapid affinity purification of intracellular organelles using twin strep tag. J. Cell Sci 132, jcs235390 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ito Y, Grison M, Esnay N, Fouillen L & Boutté Yin Plant Endosomes: Methods and Protocols (ed Otegui MS) 119–141 (Springer, 2020). [DOI] [PubMed] [Google Scholar]
- 29.Morgenstern M et al. Definition of a high-confidence mitochondrial proteome at quantitative scale. Cell Rep. 19, 2836–2852 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andersen JS et al. Proteomic characterization of the human centrosome by protein correlation profiling. Nature 426, 570–574 (2003). [DOI] [PubMed] [Google Scholar]; This article presents the first protein correlation profiling experiment, which coupled de Duve’s principle with MS to characterize the human centrosome.
- 31.Bouchnak I, Brugire S & Moyet LA Unraveling hidden components of the chloroplast envelope proteome: opportunities and limits of better MS sensitivity. Mol. Cell. Proteomics 18, 1285–1306 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chapel A, Kieffer-Jaquinod S & Sagn. An extended proteome map of the lysosomal membrane reveals novel potential transporters. Mol. Cell. Proteomics 12, 1572–1588 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dengjel J et al. Identification of autophagosome-associated proteins and regulators by quantitative proteomic analysis and genetic screens. Mol. Cell. Proteomics 10.1074/mcp.M111.014035 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krahmer N et al. Protein correlation profiles identify lipid droplet proteins with high confidence. Mol. Cell. Proteom 12, 1115–1126 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niemann M et al. Mitochondrial outer membrane proteome of Trypanosoma brucei reveals novel factors required to maintain mitochondrial morphology. Mol. Cell. Proteomics 12, 515–528 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pagliarini DJ et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 134, 112–123 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang Y, Huang A & Gu Y Global profiling of plant nuclear membrane proteome in Arabidopsis. Nat. Plants 6, 838–847 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Wiese S et al. Proteomics characterization of mouse kidney peroxisomes by tandem mass spectrometry and protein correlation profiling. Mol. Cell. Proteom 6, 2045–2057 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Schirmer EC, Florens L, Guan T, Yates JR & Gerace L Nuclear membrane proteins with potential disease links found by subtractive proteomics. Science 301, 1380–1382 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Foster LJ et al. A mammalian organelle map by protein correlation profiling. Cell 125, 187–199 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Dunkley TPJ, Watson R, Griffin JL, Dupree P & Lilley KS Localization of organelle proteins by isotope tagging (LOPIT). Mol. Cell. Proteom 3, 1128–1134 (2004). [DOI] [PubMed] [Google Scholar]; This article is the first published LOPIT experiment and multi-organellar mapping of protein endoplasmic reticulum and Golgi proteins in Arabidopsis using MS-based proteomics.
- 42.Dunkley TPJ et al. Mapping the Arabidopsis organelle proteome. Proc. Natl Acad. Sci. USA 103, 6518–6523 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Geladaki A et al. Combining LOPIT with differential ultracentrifugation for high-resolution spatial proteomics. Nat. Commun 10, 331 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Itzhak DN, Tyanova S, Cox J & Borner GHH Global, quantitative and dynamic mapping of protein subcellular localization. eLife 5, e16950 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jadot M et al. Accounting for protein subcellular localization: a compartmental map of the rat liver proteome. Mol. Cell. Proteom 16, 194–212 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mardakheh FK et al. Proteomics profiling of interactome dynamics by colocalisation analysis (COLA). Mol. Biosyst 13, 92–105 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Orre LM et al. SubCellBarCode: proteome-wide mapping of protein localization and relocalization. Mol. Cell 73, 166–182 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Nikolovski N et al. Putative glycosyltransferases and other plant Golgi apparatus proteins are revealed by LOPIT proteomics. Plant. Physiol 160, 1037–1051 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tardif M et al. PredAlgo: a new subcellular localization prediction tool dedicated to green algae. Mol. Biol. Evol 29, 3625–3639 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Ohta S et al. The protein composition of mitotic chromosomes determined using multiclassifier combinatorial proteomics. Cell 142, 810–821 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Groen AJ et al. Identification of trans-Golgi network proteins in Arabidopsis thaliana root tissue. J. Proteome Res 13, 763–776 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Crook OM, Mulvey CM, Kirk PDW, Lilley KS & Gatto L A Bayesian mixture modelling approach for spatial proteomics. PLoS Comput. Biol 14, e1006516 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jean Beltran PM, Mathias RA & Cristea IM A portrait of the human organelle proteome in space and time during cytomegalovirus infection. Cell Syst. 3, 361–373 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kennedy MA, Hofstadter WA & Cristea IM TRANSPIRE: a computational pipeline to elucidate intracellular protein movements from spatial proteomics data sets. J. Am. Soc. Mass. Spectrom 31, 1422–1439 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shin JJH et al. Spatial proteomics defines the content of trafficking vesicles captured by golgin tethers. Nat. Commun 11, 5987 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article presents a general strategy for analysing intracellular sub-proteomes by combining acute cellular rewiring with high-resolution spatial proteomics.
- 56.Jean Beltran PM, Cook KC & Cristea IM Exploring and exploiting proteome organization during viral infection. J. Virol 91, e00268–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Duve C, Pressman BC, Gianetto R, Wattiaux R & Appelmans F Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J 60, 604–617 (1955). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study forms the basis for most biochemical fractionation strategies and demonstrates the importance of capturing quantitative data, as opposed to achieving ultra-pure organellar samples.
- 58.Shehadul Islam M, Aryasomayajula A & Selvaganapathy PR A review on macroscale and microscale cell lysis methods. Micromachines 8, 83 (2017). [Google Scholar]
- 59.Drissi R, Dubois M-L & Boisvert F-M Proteomics methods for subcellular proteome analysis. FEBS J. 280, 5626–5634 (2013). [DOI] [PubMed] [Google Scholar]
- 60.Rhee HW et al. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339, 1328–1331 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article is the first example of combining APEX with MS, capturing spatial and temporal information for the human mitochondria matrix proteome, including 31 proteins not previously associated with this compartment.
- 61.Lam SS et al. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 12, 51–54 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim DI et al. Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc. Natl Acad. Sci. USA 111, 2453–2461 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roux KJ, Kim DI, Raida M & Burke B A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol 196, 801–810 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article is the first description of BioID technology, identifying known and new components of the nuclear envelope using the well-characterized nuclear filament protein lamin A.
- 64.Branon TC et al. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol 36, 880–887 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim DI et al. An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell 27, 1188–1196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ramanathan M et al. RNA–protein interaction detection in living cells. Nat. Methods 15, 207–212 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hung V et al. Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol. Cell 55, 332–341 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gupta GD et al. A dynamic protein interaction landscape of the human centrosome–cilium interface. Cell 163, 1484–1499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Youn JY et al. High-density proximity mapping reveals the subcellular organization of mRNA-associated granules and bodies. Mol. Cell 69, 517–532 (2018). [DOI] [PubMed] [Google Scholar]; This article is an extensive BioID study using 119 baits to conduct prey–prey analysis of the proteomes of stress granules and processing bodies to investigate mRNA biology.
- 70.Antonicka H et al. A high-density human mitochondrial proximity interaction network. Cell Metab. 32, 479–497 (2020). [DOI] [PubMed] [Google Scholar]
- 71.Gingras AC, Abe KT & Raught B Getting to know the neighborhood: using proximity-dependent biotinylation to characterize protein complexes and map organelles. Curr. Opin. Chem. Biol 48, 44–54 (2019). [DOI] [PubMed] [Google Scholar]
- 72.Qin W, Cho KF, Cavanagh PE & Ting AY Deciphering molecular interactions by proximity labeling. Nat. Methods 18, 133–143 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Weston LA, Bauer KM & Hummon AB Comparison of bottom-up proteomic approaches for LC-MS analysis of complex proteomes. Anal. Methods 5, 4615–4621 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lambert J-P et al. Interactome rewiring following pharmacological targeting of BET bromodomains. Mol. Cell 73, 621–638 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ludwig C et al. Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Mol. Syst. Biol 14, e8126 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ong S-E et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom 1, 376–386 (2002). [DOI] [PubMed] [Google Scholar]
- 77.Rauniyar N & Yates JR Isobaric labeling-based relative quantification in shotgun proteomics. J. Proteome Res 13, 5293–5309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Boersema PJ, Raijmakers R, Lemeer S, Mohammed S & Heck AJR Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat. Protoc 4, 484–494 (2009). [DOI] [PubMed] [Google Scholar]
- 79.Ross PL et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteom 3, 1154–1169 (2004). [DOI] [PubMed] [Google Scholar]
- 80.Ankney JA, Muneer A & Chen X Relative and absolute quantitation in mass spectrometry–based proteomics. Annu. Rev. Anal. Chem 11, 49–77 (2018). [DOI] [PubMed] [Google Scholar]
- 81.Fernández-Costa C et al. Impact of the identification strategy on the reproducibility of the DDA and DIA results. J. Proteome Res 19, 3153–3161 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Merrill AE et al. NeuCode labels for relative protein quantification. Mol. Cell. Proteom 13, 2503–2512 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Erickson BK et al. Evaluating multiplexed quantitative phosphopeptide analysis on a hybrid quadrupole mass filter/linear ion trap/orbitrap mass spectrometer. Anal. Chem 87, 1241–1249 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Altelaar AF et al. Benchmarking stable isotope labeling based quantitative proteomics. J. Proteom 88, 14–26 (2013). [DOI] [PubMed] [Google Scholar]
- 85.Thompson A et al. TMTpro: design, synthesis, and initial evaluation of a proline-based isobaric 16-plex tandem mass tag reagent set. Anal. Chem 91, 15941–15950 (2019). [DOI] [PubMed] [Google Scholar]
- 86.McAlister GC et al. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem 86, 7150–7158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang Y et al. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 11, 2019–2026 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gatto L, Breckels LM & Lilley KS Assessing subcellular resolution in spatial proteomics experiments. Curr. Opin. Chem. Biol 48, 123–149 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Krahmer N et al. Organellar proteomics and phosphor-proteomics reveal subcellular reorganization in diet-induced hepatic steatosis. Dev. Cell 47, 205–221 (2018). [DOI] [PubMed] [Google Scholar]
- 90.O’Rourke MB et al. What is normalization? The strategies employed in top-down and bottom-up proteome analysis workflows. Proteomes 7, 29 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stertz S & Shaw ML Uncovering the global host cell requirements for influenza virus replication via RNAi screening. Microbes Infect. 13, 516–525 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.de Groot R, Lüthi J, Lindsay H, Holtackers R & Pelkmans L Large-scale image-based profiling of single-cell phenotypes in arrayed CRISPR–Cas9 gene perturbation screens. Mol. Syst. Biol 14, e8064 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Marx V Calling the next generation of affinity reagents. Nat. Methods 10, 829–833 (2013). [DOI] [PubMed] [Google Scholar]
- 94.Tiede C et al. Affimer proteins are versatile and renewable affinity reagents. eLife 6, e24903 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Alamudi SH & Chang Y-T Advances in the design of cell-permeable fluorescent probes for applications in live cell imaging. Chem. Commun 54, 13641–13653 (2018). [DOI] [PubMed] [Google Scholar]
- 96.Chazotte B Labeling mitochondria with mitotracker dyes. Cold Spring Harb. Protoc 2011, 990–992 (2011). [DOI] [PubMed] [Google Scholar]
- 97.Giepmans BN, Adams SR, Ellisman MH & Tsien RY The fluorescent toolbox for assessing protein location and function. Science 312, 217–224 (2006). [DOI] [PubMed] [Google Scholar]
- 98.Neumann B et al. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 464, 721–727 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study uses time-lapse microscopy and genome-wide small interfering RNA silencing of green fluorescent protein tagged cell lines to identify 592 essential genes for mitosis; the majority had previously not been annotated with cellular processes consistent with a function in mitosis.
- 99.Leonetti MD, Sekine S, Kamiyama D, Weissman JS & Huang B A scalable strategy for high-throughput GFP tagging of endogenous human proteins. Proc. Natl Acad. Sci. USA 113, E3501–E3508 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sarov M et al. A genome-scale resource for in vivo tag-based protein function exploration in C. elegans. Cell 150, 855–866 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chong YT et al. Yeast proteome dynamics from single cell imaging and automated analysis. Cell 161, 1413–1424 (2015). [DOI] [PubMed] [Google Scholar]
- 102.Stadler C et al. Immunofluorescence and fluorescent-protein tagging show high correlation for protein localization in mammalian cells. Nat. Methods 10, 315–323 (2013). [DOI] [PubMed] [Google Scholar]
- 103.Cheng R et al. Influence of fixation and permeabilization on the mass density of single cells: a surface plasmon resonance imaging study. Front. Chem 7, 58 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Amidzadeh Z et al. Assessment of different permeabilization methods of minimizing damage to the adherent cells for detection of intracellular RNA by flow cytometry. Avicenna J. Med. Biotechnol 6, 38–46 (2014). [PMC free article] [PubMed] [Google Scholar]
- 105.Jamur MC & Oliver C in Immunocytochemical Methods and Protocols (eds Oliver C & Jamur M) 63–66 (Humana, 2010). [DOI] [PubMed] [Google Scholar]
- 106.Hobro AJ & Smith NI An evaluation of fixation methods: spatial and compositional cellular changes observed by Raman imaging. Vib. Spectrosc 91, 31–45 (2017). [Google Scholar]
- 107.Stadler C, Skogs M, Brismar H, Uhlen M & Lundberg E A single fixation protocol for proteome-wide immunofluorescence localization studies. J. Proteom 73, 1067–1078 (2010). [DOI] [PubMed] [Google Scholar]
- 108.Nakagawa T et al. Optimum immunohistochemical procedures for analysis of macrophages in human and mouse formalin fixed paraffin-embedded tissue samples. J. Clin. Exp. Hematop 57, 31–36 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Syrbu SI & Cohen MB An enhanced antigen-retrieval protocol for immunohistochemical staining of formalin-fixed, paraffin-embedded tissues. Methods Mol. Biol 717, 101–110 (2011). [DOI] [PubMed] [Google Scholar]
- 110.Cohen M, Varki NM, Jankowski MD & Gagneux P Using unfixed, frozen tissues to study natural mucin distribution. J. Vis. Exp 10.3791/3928 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Scheffler JM, Schiefermeier N & Huber LA Mild fixation and permeabilization protocol for preserving structures of endosomes, focal adhesions, and actin filaments during immunofluorescence analysis. Methods Enzymol. 535, 93–102 (2014). [DOI] [PubMed] [Google Scholar]
- 112.Micke P et al. Biobanking of fresh frozen tissue: RNA is stable in nonfixed surgical specimens. Lab. Invest 86, 202–211 (2006). [DOI] [PubMed] [Google Scholar]
- 113.Magdeldin S & Yamamoto T Toward deciphering proteomes of formalin-fixed paraffin-embedded (FFPE) tissues. Proteomics 12, 1045–1058 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Robertson D, Savage K, Reis-Filho JS & Isacke CM Multiple immunofluorescence labelling of formalin-fixed paraffin-embedded (FFPE) tissue. BMC Cell Biol. 9, 13 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pan J, Thoeni C, Muise A, Yeger H & Cutz E Multilabel immunofluorescence and antigen reprobing on formalin-fixed paraffin-embedded sections: novel applications for precision pathology diagnosis. Mod. Pathol 29, 557–569 (2016). [DOI] [PubMed] [Google Scholar]
- 116.Goltsev Y et al. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell 174, 968–981 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lin JR, Fallahi-Sichani M & Sorger PK Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nat. Commun 6, 8390 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gut G, Herrmann MD & Pelkmans L Multiplexed protein maps link subcellular organization to cellular states. Science 361, eaar7042 (2018). [DOI] [PubMed] [Google Scholar]; This work describes a protocol that achieves 40-plex protein staining in the same biological sample using off-the-shelf antibodies for immunofluorescence in an iterative manner.
- 119.Swinney DC & Anthony J How were new medicines discovered? Nat. Rev. Drug Discov 10, 507–519 (2011). [DOI] [PubMed] [Google Scholar]
- 120.Rathbun LI et al. Cytokinetic bridge triggers de novo lumen formation in vivo. Nat. Commun 11, 1269 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Huang B, Babcock H & Zhuang X Breaking the diffraction barrier: super-resolution imaging of cells. Cell 143, 1047–1058 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hell SW & Wichmann J Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt. Lett 19, 780–782 (1994). [DOI] [PubMed] [Google Scholar]
- 123.Betzig E et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–1645 (2006). [DOI] [PubMed] [Google Scholar]
- 124.Rust MJ, Bates M & Zhuang X Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3, 793–795 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cox J et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res 10, 1794–1805 (2011). [DOI] [PubMed] [Google Scholar]
- 126.Eng JK, McCormack AL & Yates JR An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass. Spectrom 5, 976–989 (1994). [DOI] [PubMed] [Google Scholar]
- 127.Perkins DN, Pappin DJC, Creasy DM & Cottrell JS Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 (1999). [DOI] [PubMed] [Google Scholar]
- 128.Fenyö D & Beavis RC A method for assessing the statistical significance of mass spectrometry-based protein identifications using general scoring schemes. Anal. Chem 75, 768–774 (2003). [DOI] [PubMed] [Google Scholar]
- 129.Moore RE, Young MK & Lee TD Qscore: an algorithm for evaluating SEQUEST database search results. J. Am. Soc. Mass. Spectrom 13, 378–386 (2002). [DOI] [PubMed] [Google Scholar]
- 130.Colinge J, Masselot A, Giron M, Dessingy T & Magnin J OLAV: towards high-throughput tandem mass spectrometry data identification. Proteomics 3, 1454–1463 (2003). [DOI] [PubMed] [Google Scholar]
- 131.Benjamini Y & Hochberg Y Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Society B 57, 289–300 (1995). [Google Scholar]
- 132.Tyanova S, Temu T & Cox J The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc 11, 2301–2319 (2016). [DOI] [PubMed] [Google Scholar]
- 133.Röst HL et al. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat. Biotechnol 32, 219–223 (2014). [DOI] [PubMed] [Google Scholar]
- 134.MacLean B et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Demichev V, Messner CB, Vernardis SI, Lilley KS & Ralser M DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 17, 41–44 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhang F, Ge W, Ruan G, Cai X & Guo T Data-independent acquisition mass spectrometry-based proteomics and software tools: a glimpse in 2020. Proteomics 20, 1900276 (2020). [DOI] [PubMed] [Google Scholar]
- 137.Tyanova S et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016). [DOI] [PubMed] [Google Scholar]
- 138.Malmström L Computational proteomics with Jupyter and Python. Methods Mol. Biol 1977, 237–248 (2019). [DOI] [PubMed] [Google Scholar]
- 139.Levitsky LI, Klein JA, Ivanov MV & Gorshkov MV Pyteomics 4.0: five years of development of a python proteomics framework. J. Proteome Res 18, 709–714 (2019). [DOI] [PubMed] [Google Scholar]
- 140.Mendik P et al. Translocatome: a novel resource for the analysis of protein translocation between cellular organelles. Nucleic Acids Res. 47, D495–D505 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ashburner M et al. Gene ontology: tool for the unification of biology. the gene ontology consortium. Nat. Genet 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chibucos MC, Siegele DA, Hu JC & Giglio M The Evidence and Conclusion Ontology (ECO): supporting GO annotations. Methods Mol. Biol 1446, 245–259 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Binder JX et al. COMPARTMENTS: unification and visualization of protein subcellular localization evidence. Database 10.1093/database/bau012 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Borner GHH Organellar maps through proteomic profiling — a conceptual guide. Mol. Cell. Proteom 19, 1076–1087 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gatto L et al. A foundation for reliable spatial proteomics data analysis. Mol. Cell. Proteom 13, 1937–1952 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gatto L, Breckels LM, Naake T & Gibb S Visualization of proteomics data using R and Bioconductor. Proteomics 15, 1375–1389 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Crook OM, Smith T, Elzek M & Lilley KS Moving profiling spatial proteomics beyond discrete classification. Proteomics 20, 1900392 (2020). [DOI] [PubMed] [Google Scholar]
- 149.Crook OM et al. A semi-supervised Bayesian approach for simultaneous protein subcellular localisation assignment and novelty detection. PLoS Comput. Biol 16, e1008288 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Swan AL, Mobasheri A, Allaway D, Liddell S & Bacardit J Application of machine learning to proteomics data: classification and biomarker identification in postgenomics biology. OMICS 17, 595–610 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.MacQueen J in Proc. Fifth Berkeley Symp. Math. Stat. Prob, Vol. 1: Statistics 281–297 (Univ. of California Press, 1967). [Google Scholar]
- 152.Ester M, Kriegel HP, Sander J & Xu X A density-based algorithm for discovering clusters in large spatial databases with noise. KDD 96, 226–231 (1996). [Google Scholar]
- 153.Davies AK et al. AP-4 vesicles contribute to spatial control of autophagy via RUSC-dependent peripheral delivery of ATG9A. Nat. Commun 9, 3958 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hirst J, Itzhak DN, Antrobus R, Borner GHH & Robinson MS Role of the AP-5 adaptor protein complex in late endosome-to-Golgi retrieval. PLoS Biol. 16, e2004411 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Peikert CD et al. Charting organellar importomes by quantitative mass spectrometry. Nat. Commun 8, 1–14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Crook OM, Breckels LM, Lilley KS, Kirk PDW & Gatto L A Bioconductor workflow for the Bayesian analysis of spatial proteomics. F1000Research 8, 446 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Crook OM, Davies CTR, Gatto L, Kirk PDW & Lilley KS Inferring differential subcellular localisation in comparative spatial proteomics using BANDLE. Preprint at https://www.biorxiv.org/content/10.1101/2021.01.04.425239v2 (2021). [DOI] [PMC free article] [PubMed]
- 158.Choi H et al. SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods 8, 70–73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hesketh GG et al. The GATOR–Rag GTPase pathway inhibits mTORC1 activation by lysosome-derived amino acids. Science 370, 351–356 (2020). [DOI] [PubMed] [Google Scholar]
- 160.Go CD et al. A proximity biotinylation map of a human cell. Preprint atbioRxiv https://www.biorxiv.org/content/10.1101/796391v1 (2019). [Google Scholar]
- 161.Knight JDR et al. ProHits-viz: a suite of web tools for visualizing interaction proteomics data. Nat. Methods 14, 645–646 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Omasits U, Ahrens CH, Müller S & Wollscheid B Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 30, 884–886 (2014). [DOI] [PubMed] [Google Scholar]
- 163.Maarten LVD & Hinton GE Visualizing data using t-SNE. J. Mach. Learn. Res 9, 2579–2605 (2008). [Google Scholar]
- 164.Burry RW Controls for immunocytochemistry: an update. J. Histochem. Cytochem 59, 6–12 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Uhlen M et al. A proposal for validation of antibodies. Nat. Methods 13, 823–827 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Schneider CA, Rasband WS & Eliceiri KW NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lamprecht MR, Sabatini DM & Carpenter AE CellProfiler™: free, versatile software for automated biological image analysis. BioTechniques 42, 71–75 (2007). [DOI] [PubMed] [Google Scholar]
- 168.Bankhead P et al. QuPath: open source software for digital pathology image analysis. Sci. Rep 7, 16878 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Sommer C, Straehle C, Köthe U & Hamprecht FA in 2011 IEEE Int. Symp. Biomed. Imaging: From Nano to Macro 10.1109/ISBI.2011.5872394 (IEEE, 2011). [DOI] [Google Scholar]
- 170.Goldberg IG et al. The Open Microscopy Environment (OME) data model and XML file: open tools for informatics and quantitative analysis in biological imaging. Genome Biol. 6, R47 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Sigal A et al. Variability and memory of protein levels in human cells. Nature 444, 643–646 (2006). [DOI] [PubMed] [Google Scholar]
- 172.Breker M, Gymrek M & Schuldiner M A novel single-cell screening platform reveals proteome plasticity during yeast stress responses. J. Cell Biol 200, 839–850 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lu AX et al. Integrating images from multiple microscopy screens reveals diverse patterns of change in the subcellular localization of proteins. eLife 7, e31892 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.LeCun Y, Bengio Y & Hinton G Deep learning. Nature 521, 436–444 (2015). [DOI] [PubMed] [Google Scholar]
- 175.Lundervold AS & Lundervold A An overview of deep learning in medical imaging focusing on MRI. Z. für Medizinische Phys 29, 102–127 (2019). [DOI] [PubMed] [Google Scholar]
- 176.Caicedo JC et al. Data-analysis strategies for image-based cell profiling. Nat. Methods 14, 849–863 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Coelho LP et al. Determining the subcellular location of new proteins from microscope images using local features. Bioinformatics 29, 2343–2349 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Li J, Newberg JY, Uhlen M, Lundberg E & Murphy RF Automated analysis and reannotation of subcellular locations in confocal images from the Human Protein Atlas. PLoS ONE 7, e50514 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Li J, Xiong L, Schneider J & Murphy RF Protein subcellular location pattern classification in cellular images using latent discriminative models. Bioinformatics 28, i32–i39 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Ouyang W et al. Analysis of the Human Protein Atlas image classification competition. Nat. Methods 16, 1254–1261 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kraus OZ, Ba JL & Frey BJ Classifying and segmenting microscopy images with deep multiple instance learning. Bioinformatics 32, i52–i59 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Witten IH, Frank E, Hall MA & Pal CJ Data Mining: Practical Machine Learning Tools and Techniques 4th edn (Morgan Kaufmann, 2016). [Google Scholar]
- 183.Pedregosa F et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res 12, 2825–2830 (2011). [Google Scholar]
- 184.Bagshaw RD, Mahuran DJ & Callahan JW A proteomic analysis of lysosomal integral membrane proteins reveals the diverse composition of the organelle. Mol. Cell. Proteom 4, 133–143 (2005). [DOI] [PubMed] [Google Scholar]
- 185.Kikuchi M et al. Proteomic analysis of rat liver peroxisome: presence of peroxisome-specific isozyme of Lon protease. J. Biol. Chem 279, 421–428 (2004). [DOI] [PubMed] [Google Scholar]
- 186.Kleffmann T & Russenberger DA The Arabidopsis thaliana chloroplast proteome reveals pathway abundance and novel protein functions. Curr. Biol 14, 354–362 (2004). [DOI] [PubMed] [Google Scholar]
- 187.Sickmann A et al. The proteome of Saccharomyces cerevisiae mitochondria. Proc. Natl. Acad. Sci. USA 100, 13207–13212 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Taylor SW et al. Characterization of the human heart mitochondrial proteome. Nat. Biotechnol 21, 281–286 (2003). [DOI] [PubMed] [Google Scholar]
- 189.Zhang L et al. Proteomic analysis of mouse liver plasma membrane: use of differential extraction to enrich hydrophobic membrane proteins. Proteomics 5, 4510–4524 (2005). [DOI] [PubMed] [Google Scholar]
- 190.van den Berg BH, Harris T, McCarthy FM, Lamont SJ & Burgess SC Non-electrophoretic differential detergent fractionation proteomics using frozen whole organs. RCM 21, 3905–3909 (2007). [DOI] [PubMed] [Google Scholar]
- 191.McCarthy FM, Burgess SC, van den Berg BHJ, Koter MD & Pharr GT Differential detergent fractionation for non-electrophoretic eukaryote cell proteomics. J. Proteome Res 4, 316–324 (2005). [DOI] [PubMed] [Google Scholar]
- 192.Schiller HB et al. Time- and compartment-resolved proteome profiling of the extracellular niche in lung injury and repair. Mol. Syst. Biol 11, 819 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Guther MLS, Urbaniak MD, Tavendale A, Prescott A & Ferguson MAJ High-confidence glycosome proteome for procyclic form Trypanosoma brucei by epitope-tag organelle enrichment and SILAC proteomics. J. Proteome Res 13, 2796–2806 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Islinger M, Lers GH, Li KW, Loos M & Vlkl A Rat liver peroxisomes after fibrate treatment. A survey using quantitative mass spectrometry. J. Biol. Chem 282, 23055–23069 (2007). [DOI] [PubMed] [Google Scholar]
- 195.Marelli M et al. Quantitative mass spectrometry reveals a role for the GTPase Rho1p in actin organization on the peroxisome membrane. J. Cell Biol 167, 1099–1112 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Ray GJ et al. A PEROXO-tag enables rapid isolation of peroxisomes from human cells. iScience 23, 101109 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Goebel T et al. Proteaphagy in mammalian cells can function independent of ATG5/ATG7. Mol. Cell. Proteom 19, 1120–1131 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Schmidtke C, Tiede S, Thelen M & Kkel Lysosomal proteome analysis reveals that CLN3-defective cells have multiple enzyme deficiencies associated with changes in intracellular trafficking. J. Biol. Chem 294, 9592–9604 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Becker AC & Gannag Influenza a virus induces autophagosomal targeting of ribosomal proteins. Mol. Cell. Proteom 17, 1909–1921 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Borner GHH et al. Fractionation profiling: a fast and versatile approach for mapping vesicle proteomes and protein–protein interactions. Mol. Biol. Cell 25, 3178–3194 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Gronemeyer T et al. The proteome of human liver peroxisomes: identification of five new peroxisomal constituents by a label-free quantitative proteomics survey. PLoS ONE 8, e57395 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Wühr M et al. The nuclear proteome of a vertebrate. Curr. Biol 25, 2663–2671 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Kislinger T et al. Global survey of organ and organelle protein expression in mouse: combined proteomic and transcriptomic profiling. Cell 125, 173–186 (2006). [DOI] [PubMed] [Google Scholar]
- 204.Loh KH et al. Proteomic analysis of unbounded cellular compartments: synaptic clefts. Cell 166, 1295–1307 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Xie W et al. A-type lamins form distinct filamentous networks with differential nuclear pore complex associations. Curr. Biol 26, 2651–2658 (2016). [DOI] [PubMed] [Google Scholar]
- 206.Dong JM et al. Proximity biotinylation provides insight into the molecular composition of focal adhesions at the nanometer scale. Sci. Signal 9, rs4 (2016). [DOI] [PubMed] [Google Scholar]
- 207.Guo Z et al. E-cadherin interactome complexity and robustness resolved by quantitative proteomics. Sci. Signal 7, rs7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Markmiller S et al. Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell 172, 590–604 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Firat-Karalar EN, Rauniyar N, Yates JR III & Stearns T Proximity interactions among centrosome components identify regulators of centriole duplication. Curr. Biol 24, 664–670 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Liu X et al. An AP-MS- and BioID-compatible MAC-tag enables comprehensive mapping of protein interactions and subcellular localizations. Nat. Commun 9, 1188 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Bersuker K et al. A proximity labeling strategy provides insights into the composition and dynamics of lipid droplet proteomes. Dev. Cell 44, 97–112 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Chastney MR et al. Topological features of integrin adhesion complexes revealed by multiplexed proximity biotinylation. J. Cell Biol 219, e202003038 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Stenström L et al. Mapping the nucleolar proteome reveals a spatiotemporal organization related to intrinsic protein disorder. Mol. Syst. Biol 16, e9469–e9469 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Carcamo WC et al. Induction of cytoplasmic rods and rings structures by inhibition of the CTP and GTP synthetic pathway in mammalian cells. PLoS ONE 6, e29690 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Havelaar AH et al. World Health Organization global estimates and regional comparisons of the burden of foodborne disease in 2010. PLoS Med. 12, e1001923 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Mulvey CM et al. Subcellular proteomics reveals a role for nucleo-cytoplasmic trafficking at the DNA replication origin activation checkpoint. J. Proteome Res 12, 1436–1453 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Branca RMM et al. HiRIEF LC-MS enables deep proteome coverage and unbiased proteogenomics. Nat. Methods 11, 59–62 (2014). [DOI] [PubMed] [Google Scholar]
- 218.Snijder B & Pelkmans L Origins of regulated cell-to-cell variability. Nat. Rev. Mol. Cell Biol 12, 119–125 (2011). [DOI] [PubMed] [Google Scholar]
- 219.Dueck H, Eberwine J & Kim J Variation is function: are single cell differences functionally important?: testing the hypothesis that single cell variation is required for aggregate function. BioEssays 38, 172–180 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.[No authors listed].The global challenge of cancer. Nature Cancer 1, 1–2 (2020). [DOI] [PubMed] [Google Scholar]; This paper emphasizes the importance of understanding cell to cell heterogeneity to understand disease development, resistance to therapy and disease recurrence.
- 221.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 222.Mahdessian D et al. Spatiotemporal dissection of the cell cycle with single-cell proteogenomics. Nature 590, 649–654 (2021). [DOI] [PubMed] [Google Scholar]; This article presents a comprehensive spatio-temporal map of proteomics heterogeneity integrating immunofluorescence imaging with single-cell transcriptomics and precise measurements of the cell cycle in individual cells.
- 223.Nagao Y, Sakamoto M, Chinen T, Okada Y & Takao D Robust classification of cell cycle phase and biological feature extraction by image-based deep learning. Mol. Biol. Cell 31, 1346–1354 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Vögtle FN et al. Landscape of submitochondrial protein distribution. Nat. Commun 8, 290 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Vögtle FN et al. Intermembrane space proteome of yeast mitochondria. Mol. Cell. Proteom 11, 1840–1852 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Parsons HT et al. Separating golgi proteins from cis to trans reveals underlying properties of cisternal localization. Plant. Cell 31, 2010–2034 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Parsons HT et al. Isolation and proteomic characterization of the Arabidopsis Golgi defines functional and novel components involved in plant cell wall biosynthesis. Plant. Physiol 159, 12–26 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Willms E et al. Cells release subpopulations of exosomes with distinct molecular and biological properties. Sci. Rep 6, 22519 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Bobrie A, Colombo M, Krumeich S, Raposo G & Théry C Diverse subpopulations of vesicles secreted by different intracellular mechanisms are present in exosome preparations obtained by differential ultracentrifugation. J. Extracell. Vesicles 1, 18397 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Anderson JD et al. Comprehensive proteomic analysis of mesenchymal stem cell exosomes reveals modulation of angiogenesis via nuclear factor-κB signaling. Stem Cells 34, 601–613 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Bandu R, Oh JW & Kim KP Mass spectrometry-based proteome profiling of extracellular vesicles and their roles in cancer biology. Exp. Mol. Med 51, 1–10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Rontogianni S et al. Proteomic profiling of extracellular vesicles allows for human breast cancer subtyping. Commun. Biol 2, 325 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Li J, He X, Deng Y & Yang C An update on isolation methods for proteomic studies of extracellular vesicles in biofluids. Molecules 24, 3516 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Gomkale R et al. Defining the substrate spectrum of the TIM22 complex identifies pyruvate carrier subunits as unconventional cargos. Curr. Biol 30, 1119–1127 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Nguyen D et al. Proteomics reveals signal peptide features determining the client specificity in human TRAP-dependent ER protein import. Nat. Commun 9, 3765 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Kozik P et al. Small molecule enhancers of endosome-to-cytosol import augment anti-tumor immunity. Cell Rep. 32, 107905 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Weekes MP et al. Quantitative temporal viromics: an approach to investigate host–pathogen interaction. Cell 157, 1460–1472 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Cook KC & Cristea IM Location is everything: protein translocations as a viral infection strategy. Curr. Opin. Chem. Biol 48, 34–43 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Jean Beltran PM et al. Infection-induced peroxisome biogenesis is a metabolic strategy for herpesvirus replication. Cell Host Microbe 24, 526–541 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Federspiel JD et al. Mitochondria and peroxisome remodeling across cytomegalovirus infection time viewed through the lens of inter-ViSTA. Cell Rep. 32, 107943 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Horner SM, Wilkins C, Badil S, Iskarpatyoti J & Gale M Proteomic analysis of mitochondrial-associated ER membranes (MAM) during RNA virus infection reveals dynamic changes in protein and organelle trafficking. PLoS ONE 10, e0117963 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Dehmelt L & Bastiaens PI Spatial organization of intracellular communication: insights from imaging. Nat. Rev. Mol. Cell Biol 11, 440–452 (2010). [DOI] [PubMed] [Google Scholar]; This review discusses how changes in subcellular localization and regulation of proteins can contribute to drastic consequences in the cell.
- 243.Smith ZD, Nachman I, Regev A & Meissner A Dynamic single-cell imaging of direct reprogramming reveals an early specifying event. Nat. Biotechnol 28, 521–526 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Bar-Even A et al. Noise in protein expression scales with natural protein abundance. Nat. Genet 38, 636–643 (2006). [DOI] [PubMed] [Google Scholar]
- 245.Newman JR et al. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature 441, 840–846 (2006). [DOI] [PubMed] [Google Scholar]
- 246.Landhuis E Deep learning takes on tumours. Nature 580, 551–553 (2020). [DOI] [PubMed] [Google Scholar]; This study discusses how artificial intelligence methods combined with imaging tools for subcellular proteomics could be a useful advance for cancer research.
- 247.Guardia CM, De Pace R, Mattera R & Bonifacino JS Neuronal functions of adaptor complexes involved in protein sorting. Curr. Opin. Neurobiol 51, 103–110 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Hanash S Disease proteomics. Nature 422, 226–232 (2003). [DOI] [PubMed] [Google Scholar]
- 249.Kavallaris M & Marshall GM Proteomics and disease: opportunities and challenges. Med. J. Aust 182, 575–579 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Dénervaud N et al. A chemostat array enables the spatio-temporal analysis of the yeast proteome. Proc. Natl Acad. Sci. USA 110, 15842–15847 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Tkach JM et al. Dissecting DNA damage response pathways by analysing protein localization and abundance changes during DNA replication stress. Nat. Cell Biol 14, 966–976 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Samavarchi-Tehrani P, Abdouni H, Samson R & Gingras A-C A versatile lentiviral delivery toolkit for proximity-dependent biotinylation in diverse cell types. Mol. Cell. Proteom 17, 2256 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Gatto L, Breckels LM, Wieczorek S, Burger T & Lilley KS Mass-spectrometry-based spatial proteomics data analysis using pRoloc and pRolocdata. Bioinformatics 30, 1322–1324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Wilkinson MD et al. The FAIR guiding principles for scientific data management and stewardship. Sci. Data 3, 1–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Berglund L et al. A genecentric Human Protein Atlas for expression profiles based on antibodies. Mol. Cell Proteom 7, 2019–2027 (2008). [DOI] [PubMed] [Google Scholar]
- 256.Baker M Reproducibility crisis: blame it on the antibodies. Nature 521, 274–276 (2015). [DOI] [PubMed] [Google Scholar]
- 257.Linkert M et al. Metadata matters: access to image data in the real world. J. Cell Biol 189, 777–782 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Carpenter AE et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7, R100 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Vizcaíno JA et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol 32, 223–226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Hahsler M, Piekenbrock M & Doran D DBCSCAN: fast density-based clustering with R. J. Stat. Soft 10.18637/jss.v091.i01 (2019). [DOI] [Google Scholar]
- 261.Lund-Johansen F et al. MetaMass, a tool for meta-analysis of subcellular proteomics data. Nat. Methods 13, 837–840 (2016). [DOI] [PubMed] [Google Scholar]
- 262.Thompson A et al. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem 75, 1895–1904 (2003). [DOI] [PubMed] [Google Scholar]
- 263.Plubell DL et al. Extended multiplexing of tandem mass tags (TMT) labeling reveals age and high fat diet specific proteome changes in mouse epididymal adipose tissue. Mol. Cell. Proteom 16, 873–890 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.O’Brien JJ et al. The effects of nonignorable missing data on label-free mass spectrometry proteomics experiments. Ann. Appl. Stat 12, 2075–2095 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Kurosawa N et al. Novel method for the high-throughput production of phosphorylation site-specific monoclonal antibodies. Sci. Rep 6, 25174 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Smith TC, Saul RG, Barton ER & Luna EJ Generation and characterization of monoclonal antibodies that recognize human and murine supervillin protein isoforms. PLoS ONE 13, e0205910 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Li X-S, Yuan B-F & Feng Y-Q Recent advances in phosphopeptide enrichment: strategies and techniques. Trends Anal. Chem 78, 70–83 (2016). [Google Scholar]
- 268.Svinkina T et al. Deep, quantitative coverage of the lysine acetylome using novel anti-acetyl-lysine antibodies and an optimized proteomic workflow. Mol. Cell Proteom 14, 2429–2440 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Weinert BT et al. Time-resolved analysis reveals rapid dynamics and broad scope of the CBP/p300 acetylome. Cell 174, 231–244.e212 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Bekker-Jensen DB et al. Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nat. Commun 11, 787 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Humphrey SJ, Azimifar SB & Mann M High-throughput phosphoproteomics reveals in vivo insulin signaling dynamics. Nat. Biotechnol 33, 990–995 (2015). [DOI] [PubMed] [Google Scholar]
- 272.Masuda T, Sugiyama N, Tomita M, Ohtsuki S & Ishihama Y Mass spectrometry-compatible subcellular fractionation for proteomics. J. Proteome Res 19, 75–84 (2020). [DOI] [PubMed] [Google Scholar]
- 273.Murray LA, Sheng X & Cristea IM Orchestration of protein acetylation as a toggle for cellular defense and virus replication. Nat. Commun 9, 4967 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Parker CE, Mocanu V, Mocanu M, Dicheva N & Warren MR in Neuroproteomics Ch. 6 (CRC Press/Taylor & Francis, 2010). [PubMed] [Google Scholar]
- 275.Virág D et al. Current trends in the analysis of post-translational modifications. Chromatographia 83, 1–10 (2020). [Google Scholar]
- 276.Lundberg E & Uhlén M Creation of an antibody-based subcellular protein atlas. Proteomics 10, 3984–3996 (2010). [DOI] [PubMed] [Google Scholar]
- 277.Jackson HW et al. The single-cell pathology landscape of breast cancer. Nature 578, 615–620 (2020). [DOI] [PubMed] [Google Scholar]
- 278.Schürch CM et al. Coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive front. Cell 182, 1341–1359.e19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Mund A et al. AI-driven deep visual proteomics defines cell identity and heterogeneity. Preprint at https://www.biorxiv.org/content/10.1101/2021.01.25.427969v1.abstract (2021). [DOI] [PMC free article] [PubMed]
- 280.Kwak C et al. Contact-ID, a tool for profiling organelle contact sites, reveals regulatory proteins of mitochondrial-associated membrane formation. Proc. Natl Acad. Sci. USA 117, 12109 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Cho KF et al. Split-TurboID enables contact-dependent proximity labeling in cells. Proc. Natl Acad. Sci. USA 117, 12143–12154 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Ma Y, McClatchy DB, Barkallah S, Wood WW & Yates JR Quantitative analysis of newly synthesized proteins. Nat. Protoc 13, 1744–1762 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Fornasiero EF et al. Precisely measured protein lifetimes in the mouse brain reveal differences across tissues and subcellular fractions. Nat. Commun 9, 4230 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Zecha J et al. Peptide level turnover measurements enable the study of proteoform dynamics. Mol. Cell. Proteom 17, 974 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Kleinpenning F, Steigenberger B, Wu W & Heck AJR Fishing for newly synthesized proteins with phosphonate-handles. Nat. Commun 11, 3244 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Bogenhagen DF & Haley JD Pulse-chase SILAC-based analyses reveal selective oversynthesis and rapid turnover of mitochondrial protein components of respiratory complexes. J. Biol. Chem 295, 2544–2554 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Duan J et al. Stochiometric quantification of the thiol redox proteome of macrophages reveals subcellular compartmentalization and susceptibility to oxidative perturbations. Redox Biol. 36, 101649 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Klein AM & Macosko E InDrops and Drop-seq technologies for single-cell sequencing. Lab. Chip 17, 2540–2541 (2017). [DOI] [PubMed] [Google Scholar]
- 289.Chen KH, Boettiger AN, Moffitt JR, Wang S & Zhuang X RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Lee JH et al. Highly multiplexed subcellular RNA sequencing in situ. Science 343, 1360–1363 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Mardakheh FK et al. Global analysis of mRNA, translation, and protein localization: local translation is a key regulator of cell protrusions. Dev. Cell 35, 344–357 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Adekunle DA & Wang ET Transcriptome-wide organization of subcellular microenvironments revealed by ATLAS-seq. Nucleic Acids Res. 48, 5859–5872 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Lefebvre FA et al. CeFra-seq: systematic mapping of RNA subcellular distribution properties through cell fractionation coupled to deep-sequencing. Methods 126, 138–148 (2017). [DOI] [PubMed] [Google Scholar]
- 294.Taniguchi Y et al. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science 329, 533–538 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Slavov N Unpicking the proteome in single cells. Science 367, 512–513 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Swaminathan J et al. Highly parallel single-molecule identification of proteins in zeptomole-scale mixtures. Nat. Biotechnol 36, 1076–1082 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Budnik B, Levy E, Harmange G & Slavov N SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 19, 161 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports a tandem mass tag carrier-based method to increase protein detection sensitivity sufficiently to allow for single-cell proteomics.
- 298.Kelly RT Single-cell proteomics: progress and prospects. Mol. Cell Proteom 19, 1739–1748 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Stoeckius M et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports the use of DNA-barcoded antibodies to convert the detection of surface proteins into a quantitative read-out jointly with RNA sequencing of single cells.
- 300.Paul I, White C, Turcinovic I & Emili A Imaging the future: the emerging era of single-cell spatial proteomics. FEBS J. 10.1111/febs.15685 (2020). [DOI] [PubMed] [Google Scholar]
- 301.Yao Y, Docter M, van Ginkel J, de Ridder D & Joo C Single-molecule protein sequencing through fingerprinting: computational assessment. Phys. Biol 12, 055003 (2015). [DOI] [PubMed] [Google Scholar]
- 302.Christiansen EM et al. In silico labeling: predicting fluorescent labels in unlabeled images. Cell 173, 792–803 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Bernocco S et al. Sequential detergent fractionation of primary neurons for proteomics studies. Proteomics 8, 930–938 (2008). [DOI] [PubMed] [Google Scholar]
- 304.Holden P & Horton WA Crude subcellular fractionation of cultured mammalian cell lines. BMC Res. Notes 2, 243 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Baghirova S, Hughes BG, Hendzel MJ & Schulz R Sequential fractionation and isolation of subcellular proteins from tissue or cultured cells. MethodsX 2, 440–445 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Ramsby ML, Makowski GS & Khairallah EA Differential detergent fractionation of isolated hepatocytes: biochemical, immunochemical and two-dimensional gel electrophoresis characterization of cytoskeletal and noncytoskeletal compartments. Electrophoresis 15, 265–277 (1994). [DOI] [PubMed] [Google Scholar]
- 307.Perez-Riverol Y et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Deutsch EW, Lam H & Aebersold R PeptideAtlas: a resource for target selection for emerging targeted proteomics workflows. EMBO Rep. 9, 429–434 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Wang M et al. Assembling the community-scale discoverable human proteome. Cell Syst. 7, 412–421 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Calvo SE, Clauser KR & Mootha VK MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 44, D1251–D1257 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Schlüter A, Real-Chicharro A, Gabaldón T, Sánchez-Jiménez F & Pujol A PeroxisomeDB 2.0: an integrative view of the global peroxisomal metabolome. Nucleic Acids Res. 38, D800–D805 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Akhter S, Kaur H, Agrawal P & Raghava GP S. RareLSD: a manually curated database of lysosomal enzymes associated with rare diseases. Database 10.1093/database/baz112 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Orloff DN, Iwasa JH, Martone ME, Ellisman MH & Kane CM The cell: an image library-CCDB: a curated repository of microscopy data. Nucleic Acids Res. 41, D1241–D1250, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Williams E et al. Image Data Resource: a bioimage data integration and publication platform. Nat. Methods 14, 775–781 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Forsberg L et al. Pre-fractionation of archival frozen tumours for proteomics applications. J. Biotechnol 126, 582–586 (2006). [DOI] [PubMed] [Google Scholar]
- 316.Giesen C et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 11, 417–422 (2014). [DOI] [PubMed] [Google Scholar]
- 317.Angelo M et al. Multiplexed ion beam imaging of human breast tumors. Nat. Med 20, 436–442 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Aichler M & Walch A MALDI imaging mass spectrometry: current frontiers and perspectives in pathology research and practice. Lab. Invest 95, 422–431 (2015). [DOI] [PubMed] [Google Scholar]
- 319.Buchberger AR, DeLaney K, Johnson J & Li L Mass spectrometry imaging: a review of emerging advancements and future insights. Anal. Chem 90, 240–265 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Bendall SC, Nolan GP, Roederer M & Chattopadhyay PK A deep profiler’s guide to cytometry. Trends Immunol. 33, 323–332 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Gorman BL & Kraft ML High-resolution secondary ion mass spectrometry analysis of cell membranes. Anal. Chem 92, 1645–1652 (2020). [DOI] [PubMed] [Google Scholar]
- 322.Lever J, Krzywinski M & Altman N Principal component analysis. Nat. Methods 14, 641–642 (2017). [Google Scholar]
- 323.Becht E et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol 37, 38–44 (2019). [DOI] [PubMed] [Google Scholar]
- 324.Hansen P & Jaumard B Cluster analysis and mathematical programming. Math. Program 79, 191–215 (1997). [Google Scholar]
- 325.Kristensen AR, Gsponer J & Foster LJ A high-throughput approach for measuring temporal changes in the interactome. Nat. Methods 9, 907–909 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Havugimana PC et al. A census of human soluble protein complexes. Cell 150, 1068–1081 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Guerrero-Castillo S et al. The assembly pathway of mitochondrial respiratory chain complex I. Cell Metab. 25, 128–139 (2017). [DOI] [PubMed] [Google Scholar]
- 328.Tackett AJ et al. Proteomic and genomic characterization of chromatin complexes at a boundary. J. Cell Biol 169, 35–47 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Larance M et al. Global membrane protein interactome analysis using in vivo crosslinking and mass spectrometry-based protein correlation profiling. Mol. Cell. Proteom 15, 2476 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Kastritis PL et al. Capturing protein communities by structural proteomics in a thermophilic eukaryote. Mol. Syst. Biol 13, 936 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Wessels HJCT et al. LC-MS/MS as an alternative for SDS-PAGE in blue native analysis of protein complexes. Proteomics 9, 4221–4228 (2009). [DOI] [PubMed] [Google Scholar]
- 332.Savitski MM et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 346, 1255784 (2014). [DOI] [PubMed] [Google Scholar]
- 333.Tan CSH et al. Thermal proximity coaggregation for system-wide profiling of protein complex dynamics in cells. Science 359, 1170–1177 (2018). [DOI] [PubMed] [Google Scholar]
- 334.Hashimoto Y, Sheng X, Murray-Nerger LA & Cristea IM Temporal dynamics of protein complex formation and dissociation during human cytomegalovirus infection. Nat. Commun 11, 1–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Taylor CF et al. The Minimum Information About a Proteomics Experiment (MIAPE). Nat. Biotechnol 25, 887–893 (2007). [DOI] [PubMed] [Google Scholar]
- 336.Taylor CF et al. Guidelines for reporting the use of mass spectrometry in proteomics. Nat. Biotechnol 26, 860–861 (2008). [DOI] [PubMed] [Google Scholar]