

Abstract
Cultured cell lines are the workhorse of cancer research, but it is unclear to what extent they recapitulate the cellular heterogeneity observed among malignant cells in tumors. To address this, we used multiplexed single cell RNA-seq to profile ~200 cancer cell lines from 22 cancer types. We uncovered 12 expression programs that are recurrently heterogeneous within many cancer cell lines. These programs are associated with diverse biological processes including cell cycle, senescence, stress and interferon responses, epithelial-mesenchymal transition, and protein maturation and degradation. Notably, most of these recurrent programs of heterogeneity recapitulate those recently observed within human tumors. The similarity to tumors allowed us to prioritize specific cell lines as model systems of cellular heterogeneity. We used two such models to demonstrate the regulation and dynamics of an epithelial senescence-related program that is observed in subpopulations of cells within cell lines and tumors. We further demonstrate unique drug responses of these subpopulations, highlighting their potential clinical significance. Our work describes the landscape of cellular heterogeneity within diverse cancer cell lines, and identifies recurrent patterns of heterogeneity that are shared between tumors and specific cell lines.
Cellular plasticity and heterogeneity are fundamental features of human tumors that play a major role in disease progression and treatment failure1,2. For example, rare subpopulations of tumor cells may underlie resistance to treatments or facilitate metastasis. Single-cell RNA sequencing (scRNA-seq) has emerged as a valuable tool to study the heterogeneity within tumors3–12. Initial scRNA-seq studies defined the expression patterns of intra-tumoral heterogeneity (ITH), yet their mechanisms and functional implications were difficult to resolve, calling for extensive follow up studies in model systems.
In principle, genetic diversity, epigenetic plasticity, and interactions within the tumor microenvironment all contribute to the heterogeneity observed across malignant cells. However, we hypothesize that a considerable fraction of the ITH expression patterns reflect intrinsic cellular plasticity that exists even in the absence of genetic diversity and a native microenvironment. For example, we previously reported an epithelial-to-mesenchymal transition (EMT)-like program in head and neck squamous cell carcinoma (HNSCC) that was partially preserved in one of a few tested cell lines5. Similarly, drug resistance programs identified in tumors were recapitulated and studied in melanoma cell lines6,13,14. Additionally, the existence of phenotypic diversity within cancer cell lines has been established for many years, but often in a highly context-specific manner and without a direct link back to in vivo patterns of diversity15–18. To further examine the ability of cancer cell lines to recapitulate ITH programs, we sought to define the landscape of cellular diversity within a large number of cell lines from the Cancer Cell Line Encyclopedia (CCLE) collection19,20.
Pan-cancer scRNA-seq of human cell lines
We developed and applied a multiplexing strategy where cells from different cell lines are profiled in pools by scRNA-seq and then computationally assigned to the corresponding cell line (Fig. 1A). We utilized existing pools that were previously generated from the CCLE collection19,21. Each pool consisted of 24-27 cell lines from diverse lineages but with comparable proliferation rates, and was profiled by scRNA-seq with the 10x Genomics Chromium system, for an average of 280 cells per cell line (Methods). We profiled eight CCLE pools, along with one smaller custom pool that included HNSCC cell lines.
Figure 1. Characterizing intra-cell line expression heterogeneity by multiplexed scRNA-seq.
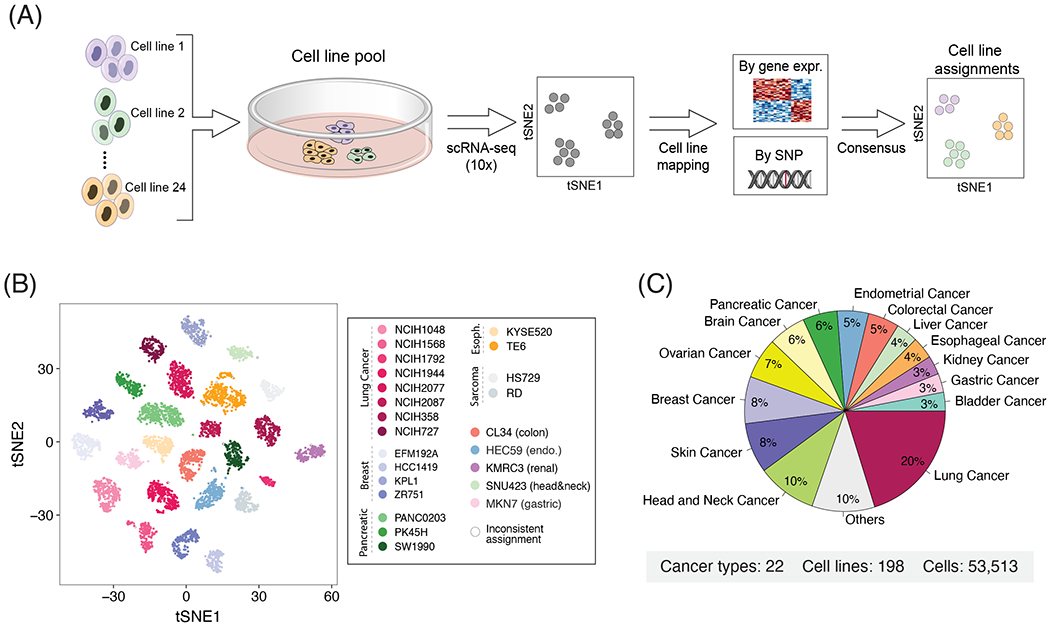
(A) Workflow of the multiplexing strategy used to profile multiple cell lines simultaneously. Cell lines were pooled and profiled by droplet-based scRNA-seq. We used reference CCLE data to assign cells to the most similar cell line based on their overall gene expression and SNP pattern. (B) t-SNE plot of a representative pool demonstrating the robustness of cells’ assignments to cell lines. Cells with inconsistent assignments (by gene expression and SNPs) are denoted and these were excluded from further analyses. (C) Distribution of cancer types profiled.
We assigned profiled cells to cell lines based on consensus between two complementary approaches, using genetic and expression profiles (Fig. 1A). First, cells were clustered by their global expression profile, and each cluster was mapped to the cell line with the most similar bulk RNA-seq profile20. Second, by detection of single nucleotide polymorphisms (SNPs) in the scRNA-seq reads, we assigned cells to the cell line with highest similarity by SNP profiles derived from bulk RNA-seq20,22. Cell line assignments based on gene expression and SNPs were consistent for 98% of the cells, which were retained for further analysis (e.g. Fig. 1B). The few inconsistent assignments were observed primarily in cells with low data quality, resulting in low SNP coverage, which were therefore excluded. Cell lines with less than 50 assigned cells were also excluded from further analyses, as were low-quality cells and suspected doublets. Overall, following assignment and quality control filters, we studied the expression profiles of 53,513 cells, from 198 cell lines (56-1,990 cells per cell line; fig. S1A), reflecting 22 cancer types (Fig. 1C; Table S1). We detected an average of 19,264 UMIs and 3,802 genes per cell, underscoring the high quality of our dataset (fig. S1A).
A potential caveat of our multiplexing approach is that in the previously generated CCLE pools, but not in the custom HNSCC pool, cell lines were co-cultured for 3 days prior to profiling by scRNA-seq and hence their expression patterns may have been affected. However, our analyses suggest a limited effect of co-culturing, particularly when considering the heterogeneity within each cell line, which is our focus in this work. First, the patterns of heterogeneity were as similar between cell lines from the same pool as between cell lines of different pools (fig. S1B). Second, we performed a control experiment in which six cell lines were profiled with and without co-culturing for 3 days. Co-culturing had a modest effect on average gene expression in each cell line, while patterns of heterogeneity were highly consistent between the two conditions (fig. S1C–F).
Discrete and continuous patterns of expression heterogeneity within cell lines
Extensive variability of gene expression was identified across cells within individual cell lines, including discrete subpopulations of cells, as well as continuous patterns that reflect spectra of cellular states (Fig. 2A). To identify discrete subpopulations we used dimensionality reduction with t-Distributed Stochastic Neighbor Embedding (t-SNE) followed by density-based clustering (DBSCAN; fig. S2A; Methods). Discrete clusters of cells within a cell line were found only for a minority (11%) of the cell lines: three cell lines had three or more clusters, three had two clusters of comparable sizes, and 16 had one major and one minor cluster (Fig. 2B and S2B). For each such cluster, we identified the top 50 upregulated genes compared to all other cells from the same cell line (Table S2). These expression programs showed limited similarities to one another, both within cell lines of the same cancer type and across different cancer types, indicating that discrete subpopulations are typically unique and cell line-specific (Fig. 2C). The main exceptions were seven subpopulations that commonly upregulated cell cycle-related genes, and six subpopulations that commonly upregulated stress response genes. Similar results were obtained using DBSCAN with different parameters (fig. S2C–D).
Figure 2. Discrete and continuous patterns of heterogeneity within cell lines.
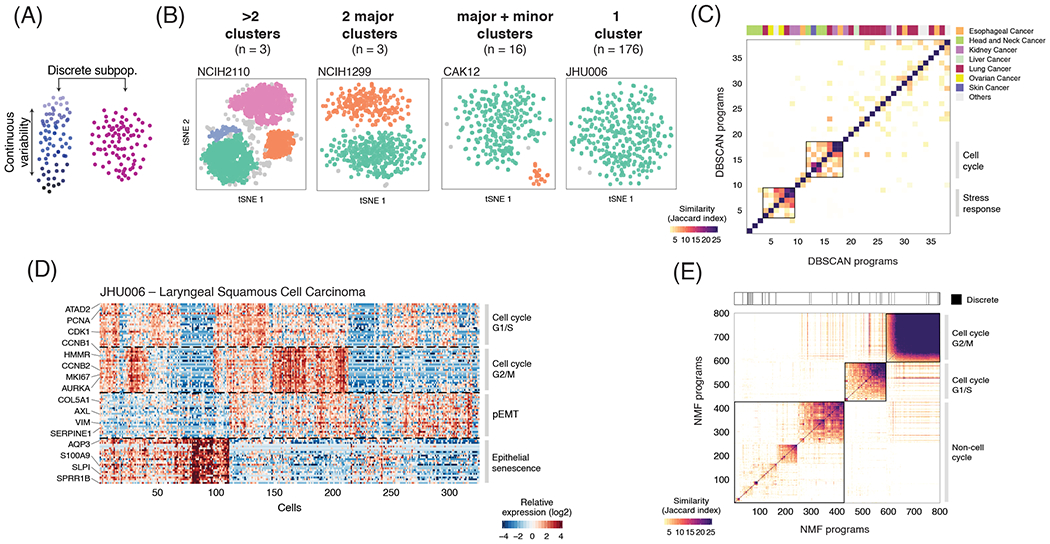
(A) Illustration of the two types of expression variability investigated. (B) t-SNE plots show exemplary cell lines for the four classes defined by the presence and number of discrete subpopulations identified using DBSCAN. The description of each class and number of cell lines is indicated above the t-SNE plots. (C) Heatmap depicts pairwise similarities between gene expression programs defined for each of the cell clusters derived from the 22 cell lines identified as having one or more discrete subpopulations. Hierarchical clustering identifies only two groups of similar programs (metaprograms). Top panel shows assignment to cancer types. (D) Continuous programs of heterogeneity identified using NMF in a representative cell line that lacks discrete subpopulations (JHU006; see B). Heatmap shows relative expression of genes from four programs, across all cells ordered by hierarchical clustering. NMF programs are annotated (right) and selected genes are indicated (left). (E) Pairwise similarities between NMF programs identified across all the cell lines analyzed and ordered by hierarchical clustering. Programs with limited similarity to all other programs as well as those associated with technical confounders were excluded. Top panel indicates the 4% of NMF programs that were consistent with discrete subpopulations identified by DBSCAN (P<0.001, Fisher’s exact test).
To also identify continuous variability of cellular states, we applied non-negative matrix factorization (NMF) to each cancer cell line5. We repeated the NMF analysis with distinct parameters, to identify robust expression programs (i.e. consistently observed as variable using different parameters), each defined by the top 50 genes based on NMF scores (e.g., Fig. 2D; Methods). This procedure captures both continuous and discrete programs. Overall, we detected 1,445 robust expression programs across all cell lines, with 4-9 such programs in individual cell lines (fig. S2E; Table S3). To identify common expression programs varying within multiple cell lines, we first excluded those with limited similarity to all other programs as well as those associated with the technical confounder of low data quality (fig. S2F), retaining 800 programs (0-8 per cell line, fig. S2E). Of these programs, only 4.75% corresponded to the discrete subpopulations above (Fig. 2E).
Hierarchical clustering of the NMF programs based on their shared genes emphasized multiple recurrent heterogeneous programs (RHPs) of gene expression, which are present in multiple cell lines. As expected, the two most prominent RHPs reflected the cell cycle, and 10 additional RHPs were associated with other cellular processes but were not positively associated with the cell cycle (Fig. 2E; Table S4). The cell cycle RHPs corresponded to the G1/S and the G2/M phases (Fig. 2E), as was also observed in clinical tumor samples (fig. S3A). G2/M programs were highly similar across cell lines, as well as between cell lines and tumors, thus defining a generic mitotic program (fig. S3B). In contrast, G1/S programs differed more both across cell lines and between cell lines and tumors (fig. S3B), indicating that expression programs associated with genome replication are more context-dependent. A central difference in G1/S programs involved the MCM complex genes (MCM2-7) and the linker histone H1 family genes (HIST1H1B-E), which were robustly upregulated only in tumors or cell lines, respectively (fig. S3B,D). This may reflect an in vitro adaptation to rapid growth and loss of the G1 checkpoint in cell lines. Consistent with this possibility, while tumors have a high percentage of apparent G0 cells (i.e., lacking both G1/S and G2/M expression programs), such cells are much less prevalent in cell lines (fig. S3E).
RHPs reflect distinct biological processes and mirror in vivo states
The ten additional RHPs reflect diverse biological processes, and are each described in detail in the next sections (Fig. 3 and Table S4). These RHPs were either largely independent of cell cycle status or preferentially expressed by non-cycling cells (fig S4A,B). Importantly, each of the RHPs were detected across at least 8 different cell lines and from at least four different pools, highlighting their robustness (fig. S4C). We characterized these 10 RHPs by functional enrichment of their signature genes (Fig. 3D), by the cell lines in which they are observed (fig. S4D,E), and by their potential regulators23 (Table S5–6).
Figure 3. Functional annotation of RHPs.
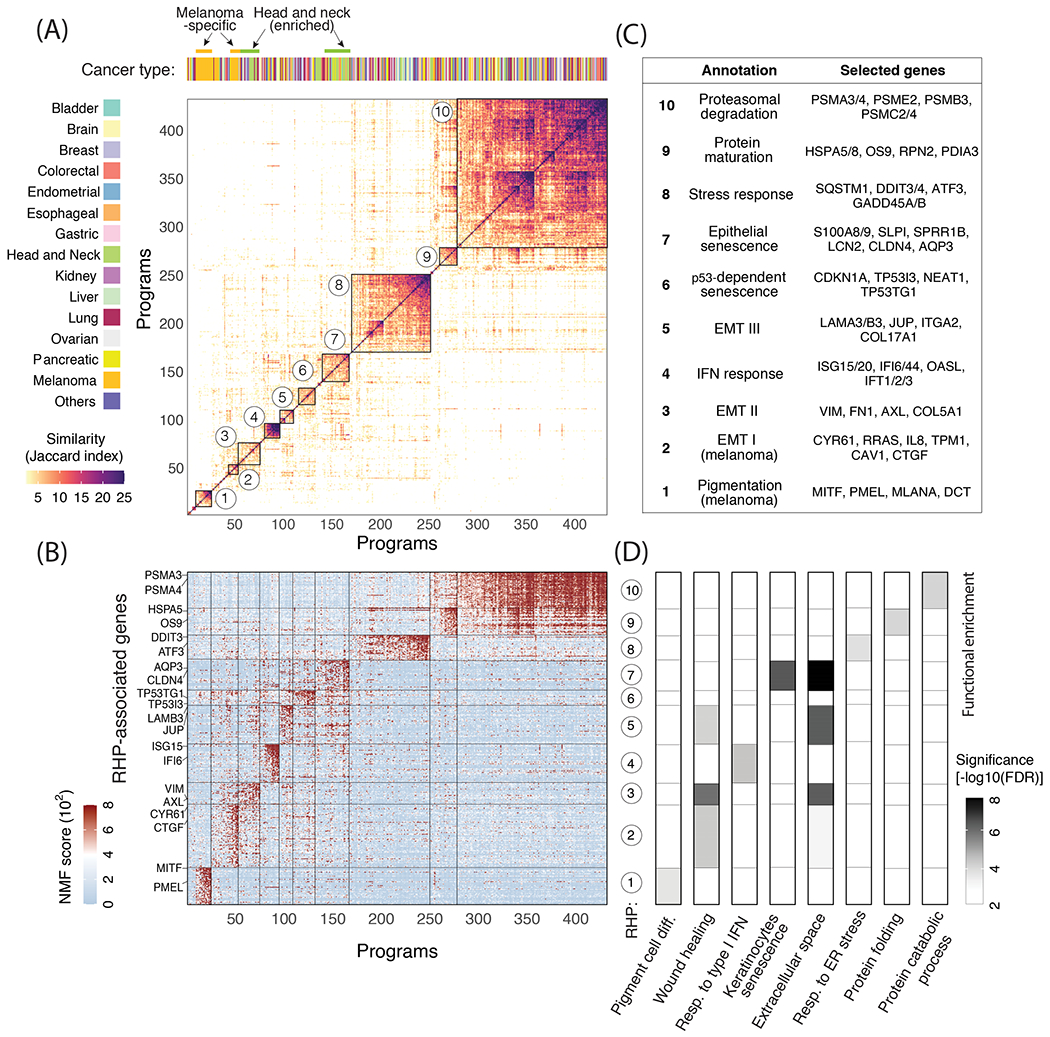
(A) The main heatmap depicts pairwise similarities between all NMF programs (except for those linked to the cell cycle, see Fig. 2E), ordered by hierarchical clustering. Ten clusters (RHPs) are indicated by squares and numbers. Top panel shows assignment to cancer types, highlighting significant enrichment (P<0.05, hypergeometric test) of melanoma and HNSCC cell lines. (B) NMF scores of signature genes of each RHP (rows), with selected genes labeled. Cells (columns) are ordered as in (A). (C) Annotation and selected top genes for each of the 10 RHPs. (D) Functional enrichment (−log10 of FDR-adjusted p-value, hypergeometric test) of RHP genes with eight annotated gene-sets.
In addition, we examined the similarity of these in vitro RHPs with recurrent in vivo expression programs that vary across cells within patient tumor samples. In vivo RHPs were defined previously in HNSCC5, melanoma6, glioblastoma24, and ovarian cancer25, and we defined additional RHPs by NMF analysis of scRNA-seq datasets in HNSCC5, melanoma6, breast cancer9 and lung cancer12 samples (fig. S3A and Table S7). Strikingly, 7 out of the 10 cell line RHPs are highly similar to the in vivo RHPs, as defined by highly significant overlap of signature genes (Fig. 4A, FDR-adjusted p<10−9 by hypergeometric test), as well as by high correlation of cell scores (Fig. 4B). The in vivo relevance of cell line RHPs was further demonstrated in melanoma and in HNSCC by a combined analysis of cells from cell lines and tumors, demonstrating their common patterns of variation as described below (Fig. 4C–F and S5).
Figure 4. In vitro RHPs recapitulate in vivo programs of heterogeneity.
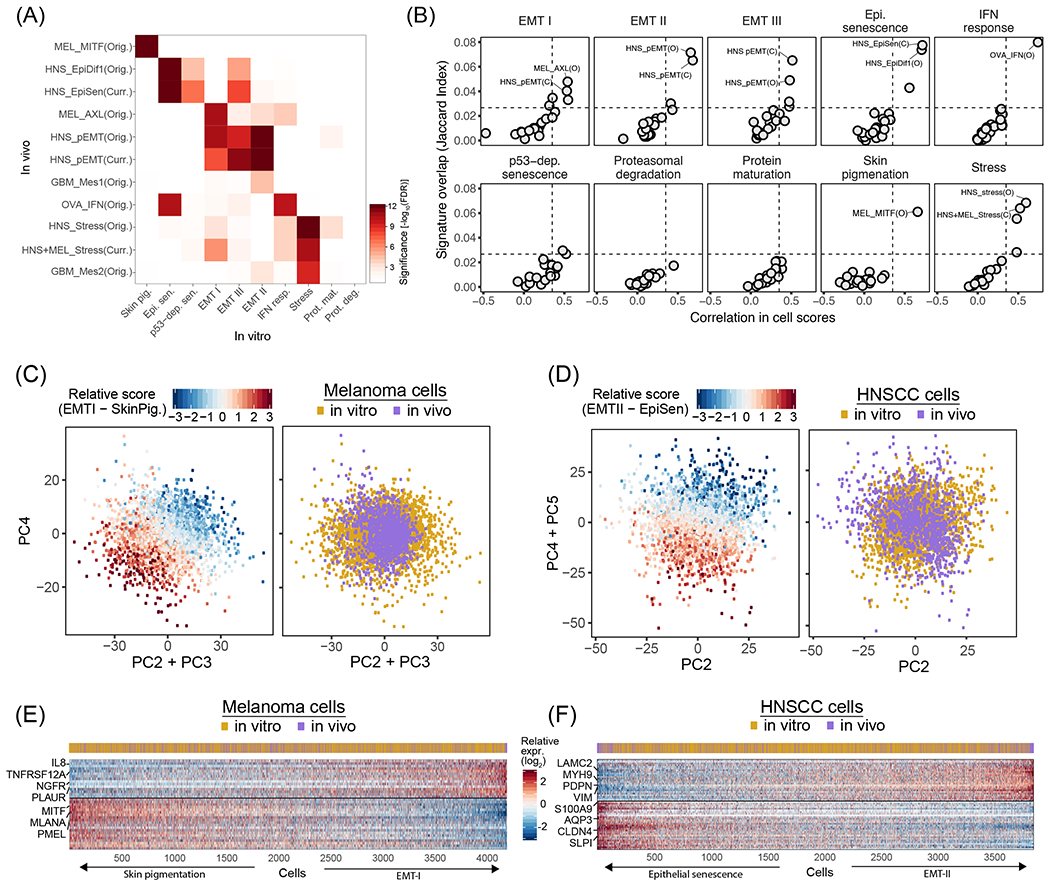
(A) Significance of the overlap (−log10(P) for FDR-adjusted hypergeometric test), between RHP gene-sets defined in cell lines (in vitro, X-axis) and in tumors (in vivo, Y-axis). In vivo RHPs are named by a cancer type abbreviation (MEL: melanoma, HNS: HNSCC, GBM: glioblastoma, OVA: ovarian cancer), followed by an associated functional annotation, and whether it was defined by the original study (Orig.) or the current study (Curr., see Fig. S3). (B) Each panel shows the mean Jaccard index (Y-axis) and mean correlation of single cell scores (X-axis) between the NMF programs constituting a specific In vitro RHP (as noted at the top) and all In vivo RHPs. The most similar In vivo RHPs are labeled as in (A). Dashed lines indicate a 99.9% confidence threshold determined by permutations of NMF programs. (C) Scatterplots of melanoma cells based on PC2+PC3 (X-axis) and PC4 (Y-axis). Cells are colored by the relative score for EMT-I and SkinPig genes shared between cell lines and tumors RHPs (left panel) and by whether the cells are from tumors or cell lines (right panel). (D) Scatterplots of HNSCC cells based on PC2 (X-axis) and PC4+PC5 (Y-axis). Cells are colored by the relative score for EMT-II and EpiSen genes shared between cell lines and tumors RHPs (left panel) and by whether the cells are from tumors or cell lines (right panel). (E) Heatmap showing the relative expression of shared EMT-I and SkinPig RHP genes (rows) across melanoma cells (columns), sorted by the relative RHP scores. The cells’ origin from tumors or cell lines is shown by the top panel. (F) Heatmap showing the relative expression of shared EMT-II and EpiSen RHP genes (rows) across HNSCC cells (columns), sorted by the relative RHP scores. The cells’ origin from tumors or cell lines is shown by the top panel.
RHPs are associated with multiple types of stress responses
One of the RHPs (#8) reflected a stress response, including DNA damage-induced and immediate early genes (e.g. DDIT3-4 and ATF3). This RHP resembles programs of heterogeneity previously observed in melanoma and HNSCC tumors5,6 (Fig. 4A,B), and may reflect the response to various cellular insults. Another RHP (#4) contained interferon (IFN) response genes (e.g., IFT1-3 and ISG15,20), highly resembling a program of heterogeneity observed within ascites samples of ovarian cancer patients25. Recent studies revealed that IFN response may be triggered by genomic instability through the cGAS-STING pathway26. Accordingly, the IFN-response program was depleted in cell lines with mutations in MRE11A (fig. S6A), which recognizes cytosolic dsDNA and activates STING27.
Two other RHPs (#9 and #10) consisted of genes related to protein folding and maturation (e.g. HSPA1A, RPN2) and to proteasomal degradation (e.g. PSMA3-4), respectively. These RHPs were the only ones that did not seem to resemble any of the in vivo programs of heterogeneity observed previously among tumor cells. However, it is possible that such programs exist in vivo and have not been detected yet due to the limited scRNA-seq data in tumors.
RHPs recapitulate in vivo EMT programs, and are associated with specific cancer types and NOTCH mutations
Three distinct RHPs were related to EMT: two shared across cancer types, and one unique to melanoma cell lines (fig. S4D). The melanoma-specific EMT (RHP #2; EMT-I) was negatively correlated with another melanoma-specific RHP (#1) that was enriched with skin pigmentation genes (e.g., MITF and PMEL). Both of these melanoma-specific RHPs, and their negative correlation, recapitulated the patterns of variability previously observed in melanoma tumors (Fig. 4A,B), in which they were linked to drug resistance6,13. Accordingly, these two RHPs were associated with three of the top five principle components, and with a range of cellular states, in a combined analysis of in vitro and in vivo melanoma cells (Figs. 4C,E, S5A,B,E). Notably, as observed in patient samples, many of the melanoma cell lines (50%; Table S3) harbored cells in both of these alternate cellular states, yet our data highlight certain melanoma cell lines as more faithful model systems for these in vivo-related RHPs (fig. S5E).
Two other RHPs, EMT-II (#3) and EMT-III (#5), also reflected EMT-like processes in distinct cell lines. EMT-II was mainly observed in HNSCC cell lines, although across 7 distinct pools (fig. S4C,D). It included vimentin (VIM), fibronectin (FN1), the AXL receptor tyrosine kinase, and other genes, closely mirroring the partial EMT state we previously observed in HNSCC tumors (Figs. 4A,B,D,F, S5) , where it was linked to metastasis5. Cell lines harboring EMT-II were depleted of NOTCH4 mutations (fig. S6A) and were more sensitive to inhibitors of NOTCH signaling (fig. S6B), suggesting a potential role of the NOTCH pathway in enabling EMT-II variability. This is similar to the association we found in glioblastoma between specific mutations and patterns of intra-tumoral heterogeneity24. In contrast, EMT-III was enriched among non-cycling cells (fig. S4A,B) and contained genes involved in cell junction organization such as laminin A3, B3 and C3, and plakoglobin (JUP). Interestingly, JUP was shown to promote collective migration of circulating tumor cells with increased metastatic potential28. The identification of three distinct EMT programs, two of which are enriched in specific cancer types, highlights EMT as a common, yet context-specific, pattern of cellular heterogeneity, which may have important implications for metastasis and drug responses.
RHPs related to classical and epithelial senescence programs
RHPs #6 and #7 were preferentially observed in G0 cells (fig. S4A,B) and seem to reflect different expression programs related to cellular senescence. RHP #6 was enriched in p53-wild type cell lines and in those sensitive to the pharmacological activation of p53 by the MDM2 inhibitor Nutlin-3a (fig. S6). Moreover, it included the senescence mediator p21 (CDKN1A) and other p53-target genes. Thus, we annotated it as “classical” p53-dependent senescence. In contrast, RHP #7 was not enriched in p53-wild type cell lines, but was enriched in HNSCC cell lines (fig. S4D,E).
RHP #7 was highly similar to the senescence program of keratinocytes and had similarity to other published senescence programs29–32 (Figs. 5A, S7A,B,Table S8). To further examine the similarity of RHP #7 with senescence-related programs of epithelial cells, we profiled primary lung bronchial cells by bulk RNA-seq after induction of senescence by etoposide. The etoposide-treated cells stained for the senescence marker SA-β-GAL and strongly downregulated the expression of cell cycle genes, indicating a bona fide senescence phenotype (Fig. 5B). Both of the senescence-associated RHPs (#6 and #7) were upregulated, although the effect was stronger for RHP #7 genes, which were among the top upregulated genes (Fig. 5B, S7C). RHP #7 also contained many secreted factors, consistent with a Senescence-Associated Secretory Phenotype (SASP). These include S100A8, S100A9, SAA1, SAA2, LCN2, CXCL1 and SLPI, which are involved in inflammatory responses and may influence cancer, stromal and immune cells in the tumor microenvironment. While most of these factors are not traditionally considered as classical SASP genes33, we found a significant overlap (P<0.01, hypergeometric test) with secreted factors from multiple other senescence-related programs, including from the in vivo counterpart in HNSCC tumors (fig. S7D,E).
Figure 5. Interrogating the EpiSen RHP in primary cells and model cell lines.
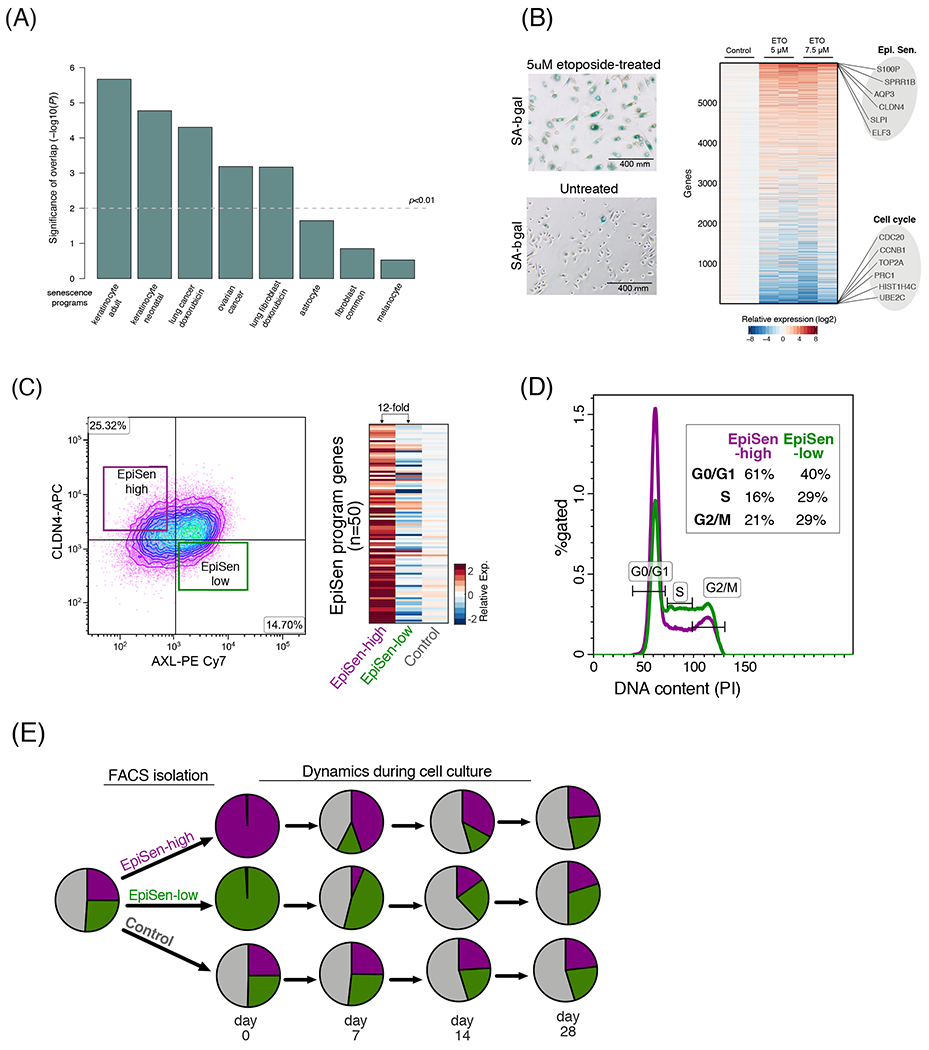
(A) Significance of the overlap (−log10(P), hypergeometric test) between the EpiSen RHP and eight previously reported senescence programs. (B) Left: induction of senescence by etoposide in primary lung bronchial cells confirmed by SA-β-gal staining. Right: heatmap depicts the relative expression of 6,000 genes (rows) in primary lung bronchial cells, 9 days after induction of senescence by etoposide treatment for 48h at two concentrations with 2 biological replicates. EpiSen and cell cycle programs were the most upregulated and downregulated programs, respectively (see fig. S5D); selected genes from these programs are labeled. (C) Isolation of the EpiSen-high (AXL+ CLDN4−) and EpiSen-low (AXL+CLDN4−) populations by FACS in JHU006. Heatmap shows relative expression of the EpiSen program genes in three sorted subpopulations: two as shown at the right panel and a third control population. (D) FACS analysis of cell cycle by the DNA binding dye propidium iodide (PI) on sorted EpiSen-high and EpiSen-low cells in JHU006. (E) Pie charts depict relative proportions of the EpiSen-high and EpiSen-low subpopulations in SCC47, for an unsorted sample (left, initial distribution), and for sorted subpopulations that were analyzed immediately after sorting (day 0) and at three additional time points (at days 7, 14 and 28 in culture).
Taken together, RHP #7 is associated with low proliferation and a secretory phenotype, and highly resembles the senescence response of keratinocytes, lung bronchial cells, and other epithelial cells. It lacks classical senescence markers (e.g., p16 and p21) and differs from published senescence signatures of fibroblasts and melanocytes29, underscoring the context-specificity of senescence expression programs. We therefore denote it as an epithelial senescence-associated (EpiSen) program. We note that although the EpiSen program is induced in senescent cells, its expression does not necessarily imply a complete senescent phenotype.
Notably, EpiSen recapitulates a program we previously observed in HNSCC tumors, “Epi-Difl” (Figs. 4A,B, 5A), which was negatively associated with the cell cycle and spatially restricted to the hypoxic tumor core5. This program was negatively correlated with the EMT-II program, defining a spectrum of cellular states that is shared by multiple HNSCC cell lines and tumors. Accordingly, the two RHPs were associated with three of the top five principle components, and with a range of cellular states, in a combined analysis of in vitro and in vivo HNSCC cells (Fig. 4D,F, S5C–E).
Proliferation and dynamics of EpiSen subpopulations in HNSCC
We selected two HNSCC cell lines (JHU006 and SCC47) with high variability of the EpiSen and EMT-II RHPs for further analysis. EpiSen-high and EpiSen-low subpopulations of cells could be prospectively isolated by FACS (as AXL−/CLDN4+ and AXL+/CLDN4−, respectively, fig. S8A), with ~12-fold difference in the expression of the EpiSen program (Fig. 5C). We note that EpiSen-low cells are only minimally enriched for the EMT-II RHP, and are used here as a negative control for the EpiSen RHP. The EpiSen-high subpopulation was enriched for G0/G1 phases, consistent with lower proliferation (Fig. 5D, S8B). Nevertheless, it still contained cells in the S and G2/M phases, similar to its in vivo counterpart (fig. S8C) and did not stain for the classical senescence marker SA-β-gal (data not shown). These results suggest that the EpiSen program represent an incomplete or reversible cell cycle arrest, consistent with previous studies in cancer cells34.
Interestingly, the EpiSen-high and EpiSen-low sorted subpopulations began to shift by one week in culture and each of them returned to the pre-sorting distribution of cellular states by four weeks, suggesting cellular transitions (Figs. 5E, S8D). This distribution of cellular states was stably maintained in culture, suggesting a steady-state which is maintained through a balance between proliferation (favoring EpiSen-low cells) and cellular transitions (favoring EpiSen-high cells). These results indicate that the EpiSen program is dynamically regulated, although we cannot determine if cellular transitions occur only from EpiSen-low to EpiSen-high or in both directions.
RHP regulation by genetics and tumor microenvironment
Expression heterogeneity could be driven by either genetic or non-genetic mechanisms. To search for the contribution of genetic heterogeneity, we identified large-scale copy number aberrations (CNAs) in each cell, based on average expression levels in windows of 100 genes around each locus3–8 (fig. S9). CNA patterns allowed the robust identification of multiple genetic subclones in 26% (58/198) of the cell lines, based on the gain or loss of chromosomes (or chromosome arms) that was restricted to subsets of cells (Methods). Co-existence of genetic subclones is consistent with ongoing evolutionary dynamics within cell lines, as demonstrated recently15. Next, we compared the assignment of cells to CNA-based genetic subclones with their patterns of expression heterogeneity. Among the discrete expression-based clusters, 39% were significantly associated with specific genetic subclones, suggesting a genetic basis for these cases of expression heterogeneity (Fig. 6A–B; P<0.001, Fisher’s exact test). In contrast, only 8% of the continuous NMF programs were significantly associated with genetic subclones (Fig. 6B, P<0.001, t-test). This analysis likely underestimates the contribution of genetic heterogeneity, as it relies on CNAs for subclone identification. However, it suggests that genetic heterogeneity contributes primarily to discrete clusters, while the continuous programs of heterogeneity may primarily reflect cellular plasticity, consistent with the established plasticity of EMT and the dynamics of the EpiSen program described above.
Figure 6. Genetic and microenvironmental factors partially explain expression heterogeneity.
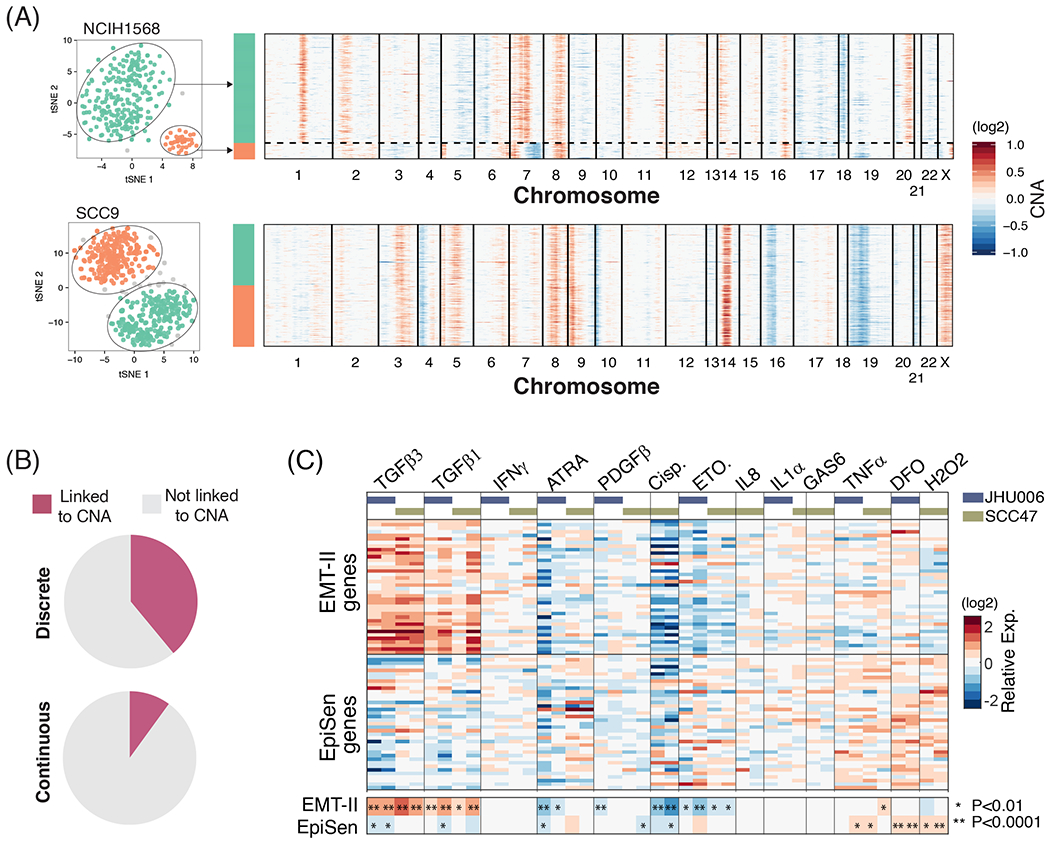
(A) Representative cell lines showing the association (top panel) or lack thereof (bottom panel) between discrete subpopulations and CNA-based subclones. t-SNE plots on the left show discrete subpopulations identified using DBSCAN (as in Fig. 2B and S1C). Heatmaps on the right depict inferred CNAs ordered according to the expression-based clusters. (B) Percentage of discrete (left) and continuous (right) heterogeneity programs that are associated with genetic subclones. For discrete programs, associations were assessed by comparing the assignment of cells to CNA subclones and to expression-based subpopulations (P<0.001, Fisher’s exact test); for continuous programs, we compared NMF cell scores between different clones (P<0,001, t-test). (C) Main heatmap depicts relative expression of EpiSen program genes and EMT-II program genes following multiple perturbations in SCC47 and JHU006. Smaller heatmap at the bottom shows the average values for the EMT-II genes and EpiSen genes, and asterisks denote significant up or down-regulation (by t-test, P value indicated in figure).
Next, we examined the induction of these programs by soluble factors that are secreted by components of the tumor microenvironment and by related perturbations. The most dynamic programs were EMT-II and EpiSen, which responded in opposite ways to several of the perturbations (Fig. 6C, fig. S10A). As expected, TGF-β1 and TGF-β3 upregulated the expression of the EMT-II genes and increased migration in a wound healing assay (fig. S10B–C). Interestingly, TGF-β treatments also downregulated the expression of EpiSen genes, underscoring the potential interplay between EpiSen and EMT. A negative association between these two programs was further supported by the single cell profiles of HNSCC cell lines and tumors (Fig. 4D,F, S5E) and by our prior findings that EMT-high cells were enriched at the invasive edge, while EpiSen-high cells were enriched at the core of tumors5. Tumor cores are often associated with increased hypoxia, suggesting a potential mechanism for the spatial enrichment of senescent cells. In accordance with this possibility, the hypoxia mimetic desferrioxamine (DFO) induced the expression of the EpiSen program. A similar effect was observed upon hydrogen peroxide treatment, consistent with oxidative stress as a potent inducer of senescence35 (Fig. 6C). Taken together, the EpiSen and EMT-II programs reflect cellular plasticity that exists in certain cell lines even in the absence of perturbations and the native tumor microenvironment, but they are further induced by stresses (e.g. hypoxia and oxidative stress) and by secreted factors (e.g. TGF-β).
Co-existing subpopulations differ in drug sensitivity
An important implication of cellular diversity in cancer is the possibility that distinct subpopulations of cells respond differently to treatments and thereby facilitate treatment failure and recurrence. Thus, we compared the sensitivities of EpiSen-high and EpiSen-low subpopulations sorted from each of the two model cell lines selected (Fig. 7A). We initially screened 2,198 bioactive compounds using a CTG-based viability assay (fig. S11A–C). Putative hits (n=248) defined based on differential sensitivity or the ability to kill both subpopulations (≤10% viability) were selected for a secondary screen performed in duplicates in each cell line (Fig. 7B). Compounds that killed both subpopulations in the primary screen were tested at reduced concentration in the secondary screen. The secondary screen identified 113 compounds with differential killing of the subpopulations in at least one cell line (Table S9).
Figure 7. Co-existing cellular states differ in drug sensitivity.
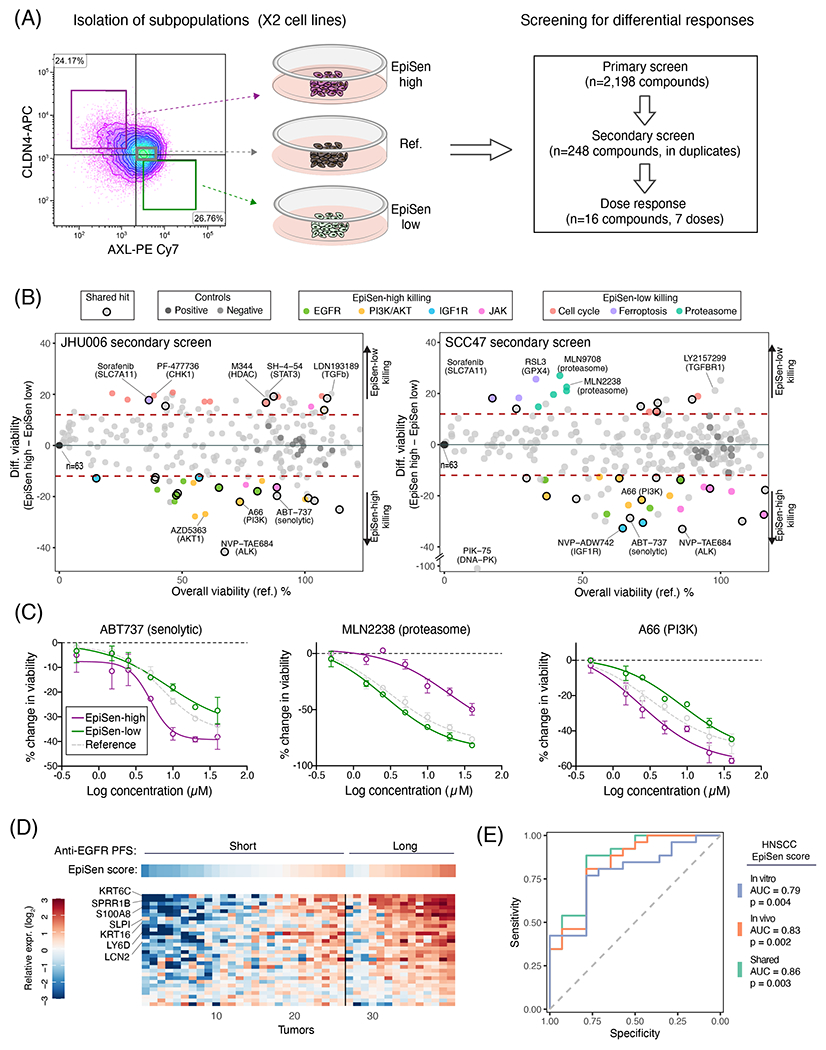
(A) Experimental scheme for drug screening: three subpopulations were isolated by FACS and subjected to primary screen, secondary screen and dose response analysis of selected hits. (B) Viability of the reference population (X-axis) and differential viability of the EpiSen-high vs. EpiSen-low populations (Y-axis) upon treatment with 248 compounds tested in the secondary screen, in JHU006 (left) and SCC47 (right), averaged over 2 replicates. Dotted lines represent thresholds for differential sensitivity (as described in Methods). Selected hits and controls are colored by target as specified in the top legends. (C) Dose response curves of three selected compounds in SCC47 measured in duplicate at each concentration, presented by the change in viability relative to vehicle controls. Error bars represent standard deviation. (D) Heatmap showing the expression of EpiSen genes shared between the HNSCC cell lines (in vitro) and tumors (in vivo) programs (see fig. S12) in bulk pre-treatment samples of 40 recurrent or metastatic HNSCC patients, stratified into short and long PFS following treatment with Cetuximab plus platinum-based chemotherapy. Top panel shows the corresponding EpiSen scores. Genes are ordered by differential expression (log2(fold change)) comparing short and long PFS patients, and tumors are ordered within each group according to the EpiSen score. Selected genes are labeled. (E) Receiver operating characteristic (ROC) curves for prediction of long vs. short PFS patients following Cetuximab treatment. Curves depict the predictive power of three potential HNSCC EpiSen signatures (in vitro, in vivo and shared). P values were calculated for each signature separately using multivariate logistic regression correcting for relevant clinicopathological features.
Of the hits with preferential sensitivity of EpiSen-high cells, >40% were shared between both cell lines. This fraction of shared hits further increases to 71.4% (for both cell lines) when considering the targets of compounds rather than the exact compounds, highlighting consistent vulnerabilities of EpiSen-high cells. Fourteen compounds with differential sensitivities, including five shared hits and nine that were specific to one cell line, were analyzed by a full dose response (Figs. 7C, S11D, Table S10). All five of the shared compounds, and five of the nine cell line-specific compounds (56%), displayed significant differential sensitivity as in the secondary screen (P<0,05, paired t-test).
As expected, EpiSen-high cells were more sensitive to the senolytic compound ABT-73736 while EpiSen-low cells were preferentially sensitive to inhibitors of cell cycle regulators (CDKs, CHK1 and topoisomerase), consistent with their increased proliferation. Additional EpiSen-high sensitivities included multiple inhibitors of EGFR, AKT, PI3K, DNA-PK, IGF1R, and JAK (Fig. 7B). Several of these targets (DNA-PK, IGF1R and AKT) converge on repair of double-strand breaks as part of the DNA repair machinery37,38. Together with the observation that hydrogen peroxide induces the expression of EpiSen genes (Fig. 6C), these results reinforce the role of DNA damage as a potential inducer of this RHP. The PI3K/AKT axis is hyper-activated in HNSCC, and resistance to PI3K inhibition in HNSCC is AXL-dependent39. Accordingly, EpiSen-high cells (which are defined by low AXL expression) were more sensitive to inhibitors of PI3K and AKT, as well as those of EGFR and IGF1R that signal via the PI3K/AKT axis.
Apart from cell cycle inhibitors, EpiSen-low cells in SCC47 (HPV+, p53 WT) also had increased sensitivity to multiple proteasome inhibitors. In contrast, all subpopulations of JHU006 (HPV−) were sensitive to proteasomal inhibition. The differential sensitivity to proteasomal inhibitors between different HNSCC cell lines is consistent with previous studies, which ascribed those differences to HPV and p53 status40 and to activation of NFkB41. Our analysis further highlights differential sensitivity to such inhibitors within the same cell line (SCC47) and a connection to the EpiSen program. EpiSen-low cells in SCC47 also had increased sensitivity to drugs that induce cell death by sensitizing cells to ferroptosis (the GPX4 inhibitor RSL3, Erastin, and the SLC7A11 inhibitor Sorafenib) (Fig. 7B). Recent work demonstrated that mesenchymal cells are particularly sensitive to ferroptosis-inducing compounds42,43. Thus, sensitivity to ferroptosis-inducing compounds may be increased in cells with mesenchymal features, consistent with previous work, and decreased in cells with features of senescence.
Taken together, EpiSen-high and EpiSen-low cells are associated with differential vulnerabilities that are largely consistent across two model cell lines and potentially across human tumors. In addition to these differential sensitivities, we also observed consistent sensitivities between EpiSen-high and EpiSen-low cells that were shared between cell lines. Nine compounds killed both subpopulations (viability ≥ 10%) in both of the cell lines (Table S9), including disulfiram (Antabuse), which was proposed recently as a potential HNSCC therapy44,45.
EpiSen subpopulations are predictive of clinical drug response
Of the multiple differential vulnerabilities described above, the increased sensitivity of EpiSen-high cells to multiple EGFR inhibitors captured our interest due to its potential clinical relevance. Cetuximab is an EGFR inhibitor routinely used for the treatment of HNSCC patients46. Most patients with recurrent or metastatic HNSCC progress shortly after Cetuximab treatment, combined with platinum-based chemotherapy, but a minority of patients have long progression-free survival (PFS). To examine the potential relevance of EpiSen in clinical response to Cetuximab, we examined bulk pre-treatment transcriptome data of 40 recurrent or metastatic HNSCC patients, stratified by PFS following Cetuximab treatment46. Twenty six patients had short PFS (PFS<5.6 months) while fourteen patients had long PFS (PFS>12 months).
Consistent with our in vitro observations, patients with long PFS had significantly higher EpiSen scores compared to those with short PFS (Fig. 7D–E, fig. S12). Accordingly, bulk EpiSen scores, a proxy for the abundance of EpiSen cells, were predictive of patients’ responses, with an area under curve (AUC) of 0.86. For example, a potential EpiSen threshold identifies 79% (11/14) of the long PFS but only 23% (6/26) of the short PFS patients, corresponding to sensitivity of 79% and specificity of 77%. While this predictive power may not be sufficient for clinical use, future work and larger cohorts may consider a combination of EpiSen with other features for improved prediction. Notably, the predictive power of EpiSen was comparable between the HNSCC in vitro RHP defined in this work and the in vivo program defined previously, and was slightly higher for the shared genes among the two programs (Fig. 7E).
Discussion
Our analysis of cell lines identified 12 RHPs (2 cell cycle and 10 others), 9 of which were highly similar to programs of heterogeneity observed within tumors, indicating that they are retained in the absence of a native microenvironment. The continuous pattern of such RHPs contrasts with the discrete nature of genetic heterogeneity. Accordingly, we observed dynamic plasticity of the EpiSen program and found only limited associations with genetic subclones (albeit only by inferred CNAs). Thus, cancer cells may harbor variability through two largely distinct processes of genetic and nongenetic mechanisms, both of which may contribute to drug resistance and tumor progression. We speculate that by focusing on recurrent patterns of heterogeneity our analysis highlights nongenetic plasticity, as this form of variability tends to be shared across cell lines, while genetic forms of variability may be more unique to each cell line.
We suggest that the significance of RHPs is derived directly from their definition as recurrent. A central observation from scRNA-seq studies of tumors is that although each tumor is unique, the diversity of cancer cells within individual tumors highlights few programs that recur as heterogeneous across many tumors47. Here we show that some of these programs also recur as heterogeneous in specific cell lines, sometimes across multiple cancer types, paving the way for mechanistic and functional studies. Just as cancer genetics has focused on recurrent mutations, based on the premise that they are drivers of tumorigenesis, we propose that cancer transcriptomics should focus on recurrent programs, as they may drive important cancer phenotypes, such as drug resistance and metastasis. Extending the analogy to therapeutics, we envisage that while current targeted therapies attempt to reverse the action of recurrent oncogenic mutations, future targeted therapies may also be targeted at recurrent programs associated with proliferation, drug resistance or metastasis.
Careful examination of the recurrent in vitro programs highlights their consistency with in vivo tumor programs, but also the divergence from their developmental “normal” counterparts. During development and wound healing, both EMT and senescence are associated with precise phenotypes and well-defined regulators. Yet in the context of tumors and cancer cell lines, we observe only partial phenotypes and limited dependence on these regulators. The EMT-like profiles we observe include many EMT-related genes and are associated with increased migration, but do not involve other EMT hallmarks such as the loss of epithelial markers, a drastic change in morphology, and high expression of EMT transcription factors. Similarly, EpiSen-high cells resemble the senescence response of keratinocytes and lung bronchial cells, are associated with reduced proliferation, and possess markers of SASP, yet they retain some proliferative capacity, do not express high levels of p16 and p21, and do not stain with SA-β-GAL. This is consistent with studies showing evidence for incomplete and reversible senescence programs in cancer: Low levels of p16 at induction of senescence may confer cell cycle re-entry upon p53 inactivation or RAS expression48, a program coined “light senescence”34 and likewise, loss of Rb in senescent cells may lead to renewed proliferation49. We hypothesize that cancer cells often activate partial or distorted programs, possibly not through the canonical developmental mechanisms, and in a context-dependent manner. This could contribute to the difficulties in resolving long-standing debates in the cancer field about the role of EMT and senescence, which are often evaluated through the activity of developmental regulators and markers that may fail to detect certain partial programs. Comprehensive single cell profiling helps to detect such partial programs that vary in their magnitude across the cells.
Multiple RHPs are of potential clinical relevance. First, the EpiSen program, which mirrors subpopulations of cells detected in HNSCC tumors, is associated with distinct responses to several drugs. Notably, the sensitivity of EpiSen-high cells to EGFR inhibitors appears to extend from cell lines to patients, underscoring its significance. These results provide a rationale for the combination of EpiSen-killing drugs (e.g. Cetuximab) with chemotherapies that target the more proliferative subpopulations. Second, for two EMT-like RHPs (EMT-I and EMT-II), clinical relevance is strongly supported by previous studies of tumor samples. EMT-II is highly consistent with a heterogeneity program we previously described in HNSCC tumors5, where it was shown to be localized to the invasive edge of the tumor and predictive of nodal metastasis. Recent work further evaluated the quantification of this program as a tool for clinical decision making50.
Similarly, EMT-I, seen only in melanoma cell lines, is highly consistent with a heterogeneity program we previously described in melanoma tumors6. Moreover, other studies defined different versions of this program, both in tumors and in cell lines, variably naming it as an ‘invasive’51,52, ‘AXL-high’/‘MITF-low’53,54, or ’resistance’13 program, but invariably involving upregulation of EMT-related genes, downregulation of skin pigmentation genes (e.g. MITF) and resistance to targeted therapies. While the specific definition of this program varies between studies, this overall convergence indicates a robust phenomenon in melanoma whereby tumors often consist of drug-sensitive and drug-resistant subpopulations that differ in levels of pigmentation and EMT-related genes, which are recapitulated here by the Skin pigmentation and EMT-I RHPs.
For two other RHPs, our observations in cell lines suggest a potential for clinical relevance, although further studies would be needed to evaluate this. The p53-dependent senescence program (RHP #6) is significantly correlated with response to the p53 activating drug, Nutlin-3a. Notably, in Nutlin-sensitive cell lines, only a minority of cells express this program before treatment, while most or all cells appear to express it after treatment55, suggesting that rare senescent cells may serve as a biomarker for p53 activity that implies sensitivity to Nutlin-3a or similar treatments. Although p53 mutations are routinely tested, the multitude of functionally distinct p53 mutations and of other mechanisms that influence p53 activity warrant a direct functional readout in case p53-based treatments will be incorporated in clinical practice. Third, expression of the IFN response program (RHP #4) by subpopulations of cancer cells, as recently described in ovarian cancer25, may influence immune cells in the tumor microenvironment and the response to immunotherapies. For example, recent work demonstrated opposing functions of the IFN response by cancer and immune cells through complex cancer-immune crosstalk56.
With the advent of single cell genomics, cellular heterogeneity is now being characterized in various clinical contexts. However, the ability to model ITH is a prerequisite for deeper understanding of the mechanisms that govern such heterogeneity. Here we described the landscape of cellular diversity across ~200 cell lines, highlighting particular models that recapitulate programs of heterogeneity observed in human tumors. Further studies of these programs and model systems will provide a better understanding of ITH, and may help to transform this understanding to novel treatment strategies that exploit ITH.
Supplementary Material
Acknowledgements
This work was supported by funding from the Israel Science Foundation (I.T.), the Zuckerman STEM leadership program (I.T.), the Mexican Friends New Generation grant (I.T.), the Rising Tide Foundation (I.T.), the A.M.N. Fund for the Promotion of Science, Culture and Arts in Israel (I.T.), the Estate of Dr. David Levinson, the Dr. Celia Zwillenberg-Fridman and Dr. Lutz Zwillenberg Career Development Chair (I.T.), the Sao Paulo Research Foundation (FAPESP) Fellowship 2014/27287-0 and 2017/24287-8 (G.S.K.), the Clore Foundation Postdoctoral Fellowship (A.C.G.), the Klarman Cell Observatory (A.R.) and HHMI (A.R.).
Footnotes
Code availability
R code for performing the analyses described here is available at https://github.com/gabrielakinker/CCLE_heterogeneity and upon request from the corresponding author.
Competing interests
A.R. is a founder and equity holder of Celsius Therapeutics and a SAB member of Neogene Therapeutics, ThermoFisher Scientific and Syros Pharmaceuticals.
Data availability
Raw and processed scRNA-seq data is available through the Broad Institute’s single cell portal (SCP542).
References
- 1.McGranahan N & Swanton C Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 27, 15–26 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Chaffer CL, San Juan BP, Lim E & Weinberg RA EMT, cell plasticity and metastasis. Cancer Metastasis Rev 35, 645–654 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Filbin MG et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 360, 331–335 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel AP et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Puram S et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tirosh I et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–96 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tirosh I et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Venteicher AS et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chung W et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun 8, 15081 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim KT et al. Application of single-cell RNA sequencing in optimizing a combinatorial therapeutic strategy in metastatic renal cell carcinoma. Genome Biol 17, 80 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat Genet 49, 708–718 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Lambrechts D et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med 24, 1277–1289 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Shaffer SM et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 546, 431–435 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jerby-Arnon L et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175, 984–997 e24 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ben-David U et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 560, 325–330 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fillmore CM & Kuperwasser C Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res 10, R25 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gupta PB et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 146, 633–44 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Stackpole CW Generation of phenotypic diversity in the B16 mouse melanoma relative to spontaneous metastasis. Cancer Res 43, 3057–65 (1983). [PubMed] [Google Scholar]
- 19.Barretina J et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghandi M et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu C et al. High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat Biotechnol 34, 419–23 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang HM et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat Biotechnol 36, 89–94 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aibar S et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods 14, 1083–1086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neftel C et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 178, 835–849 e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Izar B et al. A single-cell landscape of high-grade serous ovarian cancer. Nature Medicine (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Q, Sun L & Chen ZJ Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol 17, 1142–9 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Kondo T et al. DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc Natl Acad Sci U S A 110, 2969–74 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aceto N et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158, 1110–1122 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hernandez-Segura A et al. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr Biol 27, 2652–2660 e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jang DH et al. A transcriptional roadmap to the senescence and differentiation of human oral keratinocytes. J Gerontol A Biol Sci Med Sci 70, 20–32 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Musiani D et al. PRMT1 Is Recruited via DNA-PK to Chromatin Where It Sustains the Senescence-Associated Secretory Phenotype in Response to Cisplatin. Cell Rep 30, 1208–1222 e9 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Yang L, Fang J & Chen J Tumor cell senescence response produces aggressive variants. Cell Death Discov 3, 17049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coppe JP, Desprez PY, Krtolica A & Campisi J The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5, 99–118 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee S & Schmitt CA The dynamic nature of senescence in cancer. Nat Cell Biol 21, 94–101 (2019). [DOI] [PubMed] [Google Scholar]
- 35.te Poele RH, Okorokov AL, Jardine L, Cummings J & Joel SP DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res 62, 1876–83 (2002). [PubMed] [Google Scholar]
- 36.Yosef R et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun 7, 11190 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bozulic L, Surucu B, Hynx D & Hemmings BA PKBalpha/Aktl acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol Cell 30, 203–13 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Wong RH et al. A role of DNA-PK for the metabolic gene regulation in response to insulin. Cell 136, 1056–72 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elkabets M et al. AXL mediates resistance to PI3Kalpha inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell 27, 533–46 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li C & Johnson DE Liberation of functional p53 by proteasome inhibition in human papilloma virus-positive head and neck squamous cell carcinoma cells promotes apoptosis and cell cycle arrest. Cell Cycle 12, 923–34 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Z et al. Differential bortezomib sensitivity in head and neck cancer lines corresponds to proteasome, nuclear factor-kappaB and activator protein-1 related mechanisms. Mol Cancer Ther 7, 1949–60 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hangauer MJ et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Viswanathan VS et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park YM et al. Anti-cancer effects of disulfiram in head and neck squamous cell carcinoma via autophagic cell death. PLoS One 13, e0203069 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shah O’Brien P et al. Disulfiram (Antabuse) Activates ROS-Dependent ER Stress and Apoptosis in Oral Cavity Squamous Cell Carcinoma. J Clin Med 8(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bossi P et al. Functional Genomics Uncover the Biology behind the Responsiveness of Head and Neck Squamous Cell Cancer Patients to Cetuximab. Clin Cancer Res 22, 3961–70 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Suva ML & Tirosh I Single-Cell RNA Sequencing in Cancer: Lessons Learned and Emerging Challenges. Mol Cell 75, 7–12 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Beausejour CM et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 22, 4212–22 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sage J, Miller AL, Perez-Mancera PA, Wysocki JM & Jacks T Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature 424, 223–8 (2003). [DOI] [PubMed] [Google Scholar]
- 50.Parikh AS et al. Immunohistochemical quantification of partial-EMT in oral cavity squamous cell carcinoma primary tumors is associated with nodal metastasis. Oral Oncol 99, 104458 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hoek KS et al. Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Res 19, 290–302 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Verfaillie A et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat Commun 6, 6683 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Konieczkowski DJ et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov 4, 816–27 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Muller J et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat Commun 5, 5712 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McFarland JM et al. Multiplexed single-cell profiling of post-perturbation transcriptional responses to define cancer vulnerabilities and therapeutic mechanism of action. bioRxiv, 868752 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Benci JL et al. Opposing Functions of Interferon Coordinate Adaptive and Innate Immune Responses to Cancer Immune Checkpoint Blockade. Cell 178, 933–948 e14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed scRNA-seq data is available through the Broad Institute’s single cell portal (SCP542).