

ABSTRACT
The precise degradation of dysfunctional mitochondria by mitophagy is essential for maintaining neuronal homeostasis. HTT (huntingtin) can interact with numerous other proteins and thereby perform multiple biological functions within the cell. In this study, we investigated the role of HTT during mitophagy and analyzed the impact of the expansion of its polyglutamine (polyQ) tract. HTT is involved in different mitophagy steps, promoting the physical proximity of different protein complexes during the initiation of mitophagy and recruiting mitophagy receptors essential for promoting the interaction between damaged mitochondria and the nascent autophagosome. The presence of the polyQ tract in mutant HTT affects the formation of these protein complexes and determines the negative consequences of mutant HTT on mitophagy, leading to the accumulation of damaged mitochondria and an increase in oxidative stress. These outcomes contribute to general mitochondrial dysfunction and neurodegeneration in Huntington disease.
Abbreviations: AMPK: AMP-activated protein kinase; ATG13: autophagy related 13; BECN1: beclin 1, autophagy related; BNIP3: BCL2/adenovirus E1B interacting protein 3; BNIP3L/Nix: BCL2/adenovirus E1B interacting protein 3-like; CCCP: carbonyl cyanide 3-chlorophenyl hydrazone; DMEM: Dulbecco’s modified eagle medium; EDTA: ethylene-diamine-tetra-acetic acid; EGFP: enhanced green fluorescent protein; EGTA: ethylene glycol bis(2-aminoethyl ether)tetraacetic acid; FUNDC1: FUN14 domain containing 1; HD: Huntington disease; HRP: horseradish peroxidase; HTT: huntingtin; LC3-II: lipidated form of MAP1LC3/LC3; mtDNA: mitochondrial deoxyribonucleic acid; MTDR: MitoTracker Deep Red; MTOR: mechanistic target of rapamycin kinase; MTORC1: mechanistic target of rapamycin kinase complex 1; NBR1: NBR1, autophagy cargo receptor; CALCOCO2/NDP52: calcium binding and coiled-coil domain 2; OCR: oxygen consumption rate; OPTN: optineurin; OXPHOS: oxidative phosphorylation; PIK3C3/VPS34: phosphatidylinositol 3-kinase catalytic subunit type 3; PIK3R4/VPS15: phosphoinositide-3-kinase regulatory subunit 4; PINK1: PTEN induced putative kinase 1; PLA: proximity ligation assay; PMSF: phenylmethylsulfonyl fluoride; polyQ: polyglutamine; PtdIns3K: phosphatidylinositol 3-kinase; ROS: reactive oxygen species; Rot: rotenone; SDS-PAGE: sodium dodecyl sulfate-polyacrylamide gel electrophoresis; SEM: standard error of the mean; SQSTM1/p62: sequestosome 1; TMRM: tetramethylrhodamine methyl ester; UB: ubiquitin; ULK1: unc-51 like kinase 1.
KEYWORDS: Autophagy, huntingtin, Huntington disease, mitochondria, mitophagy
Introduction
Cellular homeostasis depends on the proper functioning of different quality control systems that ensure the continuous turnover, degradation, and recycling of intracellular components such as proteins, organelles, and aggregates. Autophagy is the catabolic mechanism by which intracellular components are delivered to lysosomes for degradation [1]. This mechanism plays an essential role in neuronal homeostasis and survival since neurons, in contrast to other non-postmitotic cells, require a constitutive basal autophagic activity to prevent the accumulation of misfolded and aggregated proteins, eliminate unwanted organelles, and keep the cell “clean” [2,3].
Mitophagy is a form of selective macroautophagy/autophagy in which damaged mitochondria are targeted for degradation in the lysosome. While basal mitophagy is responsible for the constant turnover of the mitochondrial pool, mitophagy can also be induced during development in certain cell types or as a stress-response mechanism following mitochondrial membrane depolarization, hypoxia, or metabolic stress [4–6]. Despite the potential for different molecules to be involved, the various forms of mitophagy follow a similar mechanism whereby a receptor molecule acts as a bridge between the mitochondria targeted for elimination and the lipidated form of MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3), termed LC3-II, present in the phagophore membrane. This interaction permits the targeted mitochondrion to be engulfed by the phagophore and be subsequently degraded after fusion of the completed autophagosome with a lysosome.
Huntington disease (HD) is an autosomal-dominant neurodegenerative disorder caused by an expansion of the CAG trinucleotide repeat encoding a polyglutamine (polyQ) tract in the amino-terminal region of HTT (huntingtin) protein [7]. Despite the ubiquitous expression of mutant HTT (mHTT), this disorder is characterized by preferential atrophy of the striatum caused by the loss of GABAergic medium spiny neurons in the caudate nucleus and putamen. However, other brain areas such as the cerebral cortex are also affected [8,9]. The neuropathological hallmark of HD is the formation of inclusion bodies within the affected neurons due to the presence of an expanded polyQ tract that renders mHTT prone to aggregation [10]. HTT is a particularly large protein (347.603 kDa) with multiple protein-protein interaction regions that allow the protein to interact physically with several partners [11,12], thereby providing structural stabilization to different protein complexes. To date, HTT has been proposed to act as a scaffold protein stabilizing different protein complexes involved in transcriptional regulation [13], axonal trafficking of vesicles, autophagosomes, and mitochondria [14–16], nuclear export/import, and ubiquitin (UB)-mediated proteolysis [17]. Recently it was shown that HTT could also act as a scaffold protein during the initiation of stress-induced selective autophagy. This is done by the interaction of HTT with ULK1 (unc-51 like kinase 1), required to initiate macroautophagy [18,19], and with SQSTM1/p62 (sequestosome 1), one of the selective macroautophagy receptors that bind simultaneously to autophagosomes via the interaction with LC3-II and to the substrate through the direct interaction with polyubiquitin (polyUB) chains tagged to the substrate [18,19]. In fact, the dysfunction of cellular proteolytic systems, including mostly autophagic pathways but also the proteasome system, has been extensively linked to HD [20–22]. Simultaneously with the proteolytic dysfunction, and similar to other neurodegenerative diseases, mitochondrial dysfunction has also been described in neurons affected by HD [23–25]. We believe that the mitochondrial dysfunction is caused in part by impairment of the selective degradation of mitochondria by autophagy. Indeed, impairment of neuronal mitophagy leads to the accumulation of damaged mitochondria, leading to an increase in reactive oxygen species (ROS) production, and consequently contributes to neurodegeneration.
Here, we focus attention on the proposed role of HTT in mitophagy, not only through its interaction with ULK1 and SQSTM1/p62 as previously reported but also through its interaction with other essential components of the mitophagy pathway (BECN1, NBR1, OPTN, and CALCOCO2/NDP52). We also analyze the consequences of the presence of mHTT in the stabilization of the different protein-protein interactions involved in mitophagy. In contrast to previous works, we use differentiated striatal neurons to determine the role of mHTT in mitophagy without artificially modifying the endogenous levels of the molecular components involved in this process. Furthermore, we induce severe mitochondrial depolarization with the mitochondrial uncoupler CCCP (carbonyl cyanide 3-chlorophenyl hydrazone), and also study mitophagy in response to mild depolarization, which closely mimics the moderate mitochondrial damage that occurs during neurodegeneration.
Results
Mitochondrial dysfunction in ST-Q111 cells
Mitochondrial dysfunction and impairment of the proteolytic systems have been extensively linked to neurodegenerative diseases, including HD [4]. Neurons are particularly vulnerable to autophagic impairment as well as to mitochondrial dysfunction, mostly due to their particularly high-energy dependence and to their post-mitotic nature. The precise and proper degradation of dysfunctional mitochondria by mitophagy is essential for maintaining control over mitochondrial quality and quantity in neurons. Here, we focus on the role of HTT protein in the mechanism of mitophagy and the consequences of the polyQ tract expansion. To this end, we used differentiated immortalized striatal neurons derived from a knock-in mouse expressing human exon 1 carrying a polyQ tract with 7 glutamines STHdh-Q7 (ST-Q7, control) or 111 glutamines STHdh-Q111 (ST-Q111, mutant) inserted into the endogenous mouse Htt gene. To validate our model, we first verified the presence of mitochondrial dysfunction with different approaches. The ratio between non-depolarized mitochondria, detected with a mitochondrial probe that is sensitive to the mitochondrial membrane potential (TMRM), and total mitochondria, detected with a mitochondrial membrane potential-insensitive mitochondrial probe (MitoTracker Deep Red, MTDR), indicates the number of healthy mitochondria remaining after depolarization. After 24 h of mild (rotenone 1 μM for 4 h) or acute depolarization (CCCP 10 μM for 24 h), this ratio was higher in control cells compared to mutant cells (Figure 1A). This finding is compatible with a reduced capacity for the turnover of damaged mitochondria in mutant cells and; therefore, further analysis is required. We evaluated mitochondrial function by measuring the oxygen consumption rate (OCR) of differentiated striatal cells under basal conditions and in response to cellular respiration stress. Under basal conditions, both differentiated striatal cell lines had similar respiration levels (Figure 1B and C). Inhibition of adenosine triphosphate (ATP) synthase with oligomycin caused an expected decrease in respiration in both ST-Q7 and ST-Q111 cells, with no significant difference between the two cell types. The addition of the uncoupling agent CCCP collapsed the proton gradient and caused an increase in the OCR until the maximum respiration rate was achieved. ST-Q111 cells clearly showed a lower rate of maximal respiration and a lower spare respiratory capacity compared with control cells (Figure 1B and C), indicating that mitochondria from ST-Q111 cells are bioenergetically less-active and have a decreased ability to respond to increased energy demand. When cells were initially subjected to mild depolarization by exposure to rotenone for 4 h and then left to recover for 24 h, mitochondria from mutant ST-Q111 cells exhibited lower levels of basal respiration. After the addition of the different inhibitors and uncoupling agents, mutant cells showed reductions in ATP production, maximal respiration, and spare respiratory capacity compared to control cells (Figure 1D), suggesting that inefficient mitophagy triggered in response to cellular stress leads to the accumulation of damaged mitochondria. Consequently, the remaining pool of mitochondria present in the cell is not fully healthy, thus, contributing to the general mitochondrial dysfunction observed in HD. One of the main consequences of the cell’s inability to properly eliminate damaged mitochondria is the increase in ROS. These can be detected by the fluorescent probes CellROX Green (Figure 1E and F) and CM-H2DCFDA (Figure 1G). Our data showed increased ROS levels in ST-Q111 cells compared with ST-Q7 cells, in part, due to the higher levels of damaged mitochondria present in mutant cells. In conclusion, differentiated ST-Q111 cells after depolarization are more prone to accumulate damaged mitochondria and to display altered mitochondrial respiration and higher levels of ROS.
Figure 1.
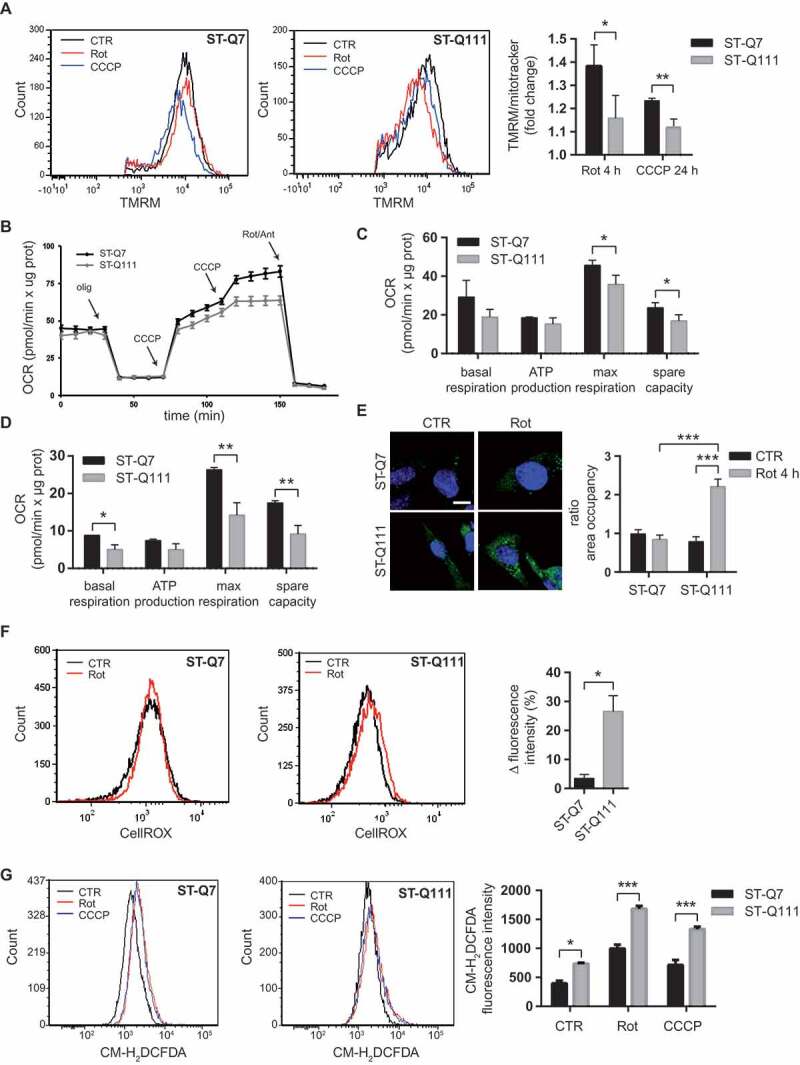
Impaired mitophagy leads to the accumulation of damaged mitochondria in ST-Q111 cells. (a) Flow cytometry was used to determine TMRM and MitoTracker DeepRed staining in control ST-Q7 and ST-Q111 cells (vehicle condition, DMSO 0.1%, CTR) or treated with rotenone (Rot, 1 μM, 4 h) or CCCP (10 μM, 4 h). Chloroquine was added with the treatments (60 μM; n = 3 independent experiments). Quantification is represented as fold-change compared with the corresponding CTR condition. (b) Seahorse XF-24 analysis of oxygen consumption rate (OCR) detected in ST-Q7 and ST-Q111 cells under basal conditions or following the addition of oligomycin (olig, 1 μM), CCCP (CCCP, 0.5 μM each time) and rotenone and antimycin (Rot/Ant, 1 μM) at the indicated times (n = 3 independent experiments). (c and d) Rates of basal respiration, ATP production, maximal respiration and spare capacity were quantified by normalization of OCR level to the total protein OD values (n = 3 independent experiments) in ST-Q7 and ST-Q111 (c) CTR cells or (d) after 4 h of rotenone treatment and 24 h of recovery. (e) Representative images of CellROX Green immunostained ST-Q7 and ST-Q111 cells CTR or treated with Rot (1 μM, 4 h). Scale bar: 10 μm. Intracellular ROS levels were quantified as a measure of the area fraction occupied by CellROX staining and represented as the fold-change compared with ST-Q7 CTR. A minimum of 40 cells was analyzed per condition. (f) Flow cytometry analysis of CellROX intensity in ST-Q7 and ST-Q111 cells CTR or treated with Rot (1 μM, 4 h). Intracellular ROS levels were quantified as a measure of CellROX intensity and represented as the ratio between rotenone and control (vehicle, CTR) conditions (n = 3 independent experiments). (g) Flow cytometry analysis of CM-H2DCFDA staining in ST-Q7 and ST-Q111 cells CTR or treated with Rot (1 μM, 4 h). Intracellular ROS levels were quantified as a measure of CM-H2DCFDA intensity and represented as mean fluorescence intensity (n = 3 independent experiments)
Mitophagy is affected in ST-Q111 cells
Following the confirmation that our model, in accordance with other HD models [26–30], presents mitochondrial dysfunction, we next sought to verify if the mitophagy was indeed affected in differentiated striatal ST-Q111 cells and could at least in part, explain the mitochondrial dysfunction observed in HD. Altered autophagy has been widely described in vivo and in vitro models of HD, as well as in postmortem brain samples from patients [21,31–36]. Indeed, HTT protein has been shown to be implicated in the cargo recognition step during selective autophagy [18,19,21]. Due to its ability to act as a scaffold, HTT brings together different proteins required for autophagy to take place. However, the presence of the polyQ tract can alter these interactions and destabilize different protein complexes, thus affecting the efficiency of selective autophagy processes, including mitophagy. To study the consequences of glutamine expansion in HTT’s role in mitophagy, we used different approaches to monitor the autophagy-dependent degradation of mitochondria. First, we studied mitochondrial clearance by quantifying the colocalization of mitochondria within lysosomes, as the last step of the mitophagy process. Under basal conditions, the number of lysosomes loaded with mitochondrial material in the presence of lysosomal inhibitors was similar between control and mutant striatal cells (Figure 2A). However, after mild and acute mitochondrial depolarization, control cells showed a clear increase in the number of lysosomes colocalizing with mitochondria, but we observed no increase in the mutant ST-Q111 cells (Figure 2A). This finding indicates that mHTT cells are less efficient at engulfing and digesting damaged mitochondria following depolarization. To analyze mitochondrial clearance in an alternative way, we quantified mitophagy flux by detecting the mitochondrial pool with the MTDR probe under basal conditions and after induction of mitophagy by rotenone and CCCP, in the presence and absence of a lysosomal inhibitor (60 μM chloroquine) to avoid mitochondrial degradation within lysosomes. The increase of the MTDR signal in each condition due to the presence of chloroquine is representative of the number of mitochondria degraded by mitophagy (Figure 2B). ST-Q7 cells showed a significant increase of MTDR signal in the presence of lysosomal inhibitors (chloroquine) in all three conditions, indicating that mitophagy flux is active. However, in QT-111 cells, the increase of the MTDR signal in the presence of chloroquine is lower and, after induction of mitophagy with rotenone or CCCP, is not statistically significant, indicating that mitophagy flux is deficient in these cells.
Figure 2.
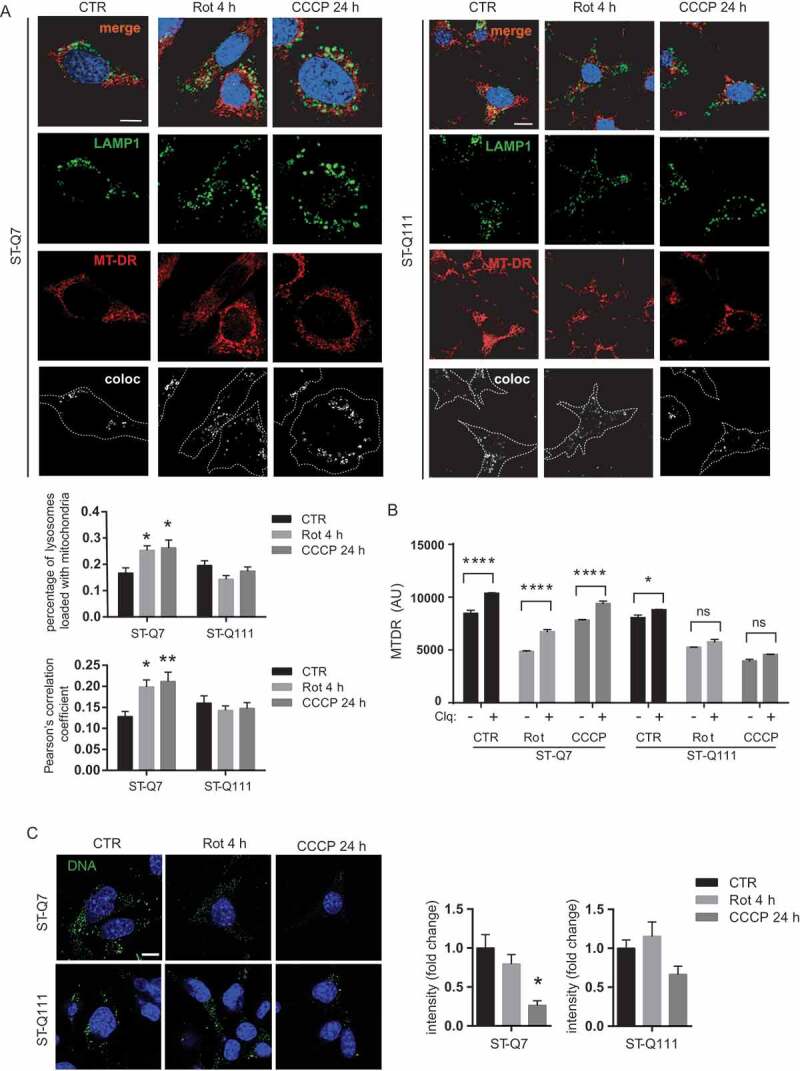
Mitophagy is affected in ST-Q111 cells. (a) Representative images of LAMP1 and MitoTracker DeepRed immunostained ST-Q7 and ST-Q111 control cells (vehicle, CTR) or treated with rotenone (Rot, 1 μM, 4 h) or CCCP (10 μM, 24 h). Insets show higher magnification images. All conditions were treated with the lysosomal inhibitor bafilomycin. Colocalization of both channels is indicated in bottom panels as a white signal and was quantified using Pearson’s correlation coefficient and as the ratio of LAMP1-stained lysosomes loaded with MitoTracker DeepRed-stained mitochondria. A minimum of 30 cells was analyzed per condition. (b) Mitophagy flux measured as mean fluorescence intensity (MFI) of MTDR probe in ST-Q7 and ST-Q111 control (CTR) cells or treated 4 h with 1 μM, rotenone (Rot) or 24 h with 10 μM, CCCP (CCCP) in the presence and absence of lysosomal inhibitor (60 μM chloroquine). (c) Representative images of ST-Q7 and ST-Q111 cells immunostained with anti-DNA CTR or treated with Rot (1 μM, 4 h) or CCCP (10 μM, 24 h). Quantification is represented as the fold-change in DNA intensity compared with CTR conditions. 15 to 50 cells were analyzed per condition. Scale bar: 10 μm
As another measure of mitochondrial degradation, we quantified mitochondrial (mt)DNA as a reporter of mitochondrial mass using an anti-DNA antibody that preferentially binds to mtDNA [37]. ST-Q7 cells displayed a loss of DNA intensity upon rotenone and CCCP treatment, which is indicative of mitochondrial removal in response to damage. However, we observed no significant changes in ST-Q111 cells, further implying that mitochondria were not being degraded with the same efficiency as ST-Q7 cells upon stress-induction (Figure 2C). These results indicate that induced mitophagy is impaired in differentiated ST-Q111 cells, and suggest that mHTT can directly or indirectly affect the efficiency of this process.
The polyQ tract in mHTT affects the initiation of mitophagy
To gain insight into the mechanism by which mHTT affects mitophagy, we analyzed the different steps of the mitophagy process, on the one hand, the induction of autophagy and, on the other, the labeling and subsequent recognition of the targeted mitochondria. First, by focusing on the parallel activation of the ULK1 and BECN1-containing class III phosphatidylinositol 3-kinase (PtdIns3K) complexes, we studied if mHTT could affect the initiation of the autophagy machinery. The initiation process is coordinated by ULK1, which forms a complex with ATG13 and RB1CC1/FIP200 to regulate the initial step of autophagy induction in mammalian cells [38–40]. To induce autophagy, ULK1 must be activated and released from the MTOR complex 1 (MTORC1), losing the MTOR-mediated inhibitory phosphorylation at Ser757 [41]. Simultaneously, AMP-activated protein kinase (AMPK) activates ULK1 through physical interaction [42] and via phosphorylation of the Ser555, Ser777, and Ser317 residues [41,43]. Previous studies showed that HTT plays a key role as a scaffold protein by binding active ULK1 at its C-terminal domain, competing with MTORC1 for ULK1 binding, and promoting autophagy initiation [18,19]. To understand how the presence of the mutant polyQ tract might affect this step, we analyzed the interaction of ULK1 with MTOR and HTT. To this end, we used the proximity ligation assay (PLA) since immunoprecipitation with the endogenous proteins was technically not feasible due to the low levels of these proteins. PLA allows a highly sensitive and highly specific quantification of protein-protein interaction under different conditions. PLA methodology was initially validated by quantifying a well-known HTT-SYVN1/HRD1 interaction [44] (Fig. S1). We next studied the interactions of ULK1 by PLA and observed that in differentiated ST-Q111 cells, the ULK1-MTOR interaction was more stable after depolarization compared to that seen in ST-Q7 cells, where the ULK1-MTOR interaction decreased upon depolarization to permit ULK1 activation (Figure 3A and negative controls in Fig. S2). These results suggest that, in mutant cells, ULK1 remains more inactive and bound to MTOR than in control cells. Similarly, the ULK1-HTT interaction increased in ST-Q7 cells after depolarization, but this interaction was not induced in ST-Q111 cells after depolarization, confirming that ULK1 has a reduced affinity for mHTT (Figure 3B). Using PLA, we also detected a lower amount of active ULK1 (phosphorylated at S317) bound to HTT in ST-Q111 cells after depolarization (Figure 3C). In order to determine if the changes in ULK1-MTOR and ULK1-HTT interactions could affect ULK1’s phosphorylation state, we analyzed MTOR-dependent and AMPK-dependent phosphorylations. MTOR-dependent Ser757 inactivating phosphorylation showed a tendency to decrease after mitochondrial depolarization that, although was non-significant, would agree with ULK1 activation and autophagy induction. However, ST-Q111 cells maintained high levels of Ser757 phosphorylation upon depolarization, indicating that ULK1 remains inactivated(Figure 3D). We also analyzed ULK1’s AMPK-dependent phosphorylations at Ser555 and Ser317, both of which are more abundant after mitochondrial depolarization in both ST-Q7 and ST-Q111 cell lines, with no significant changes between cell lines (Figure 3D), indicating that AMPK-dependent phosphorylations are not negatively affected by mHTT. Together, these results suggest that the extended polyQ tract in mHTT might be interfering with ULK1 activation and the shift from the MTORC1 to the HTT scaffolding complex, a necessary step to initiate autophagy and consequently that of mitophagy as well.
Figure 3.
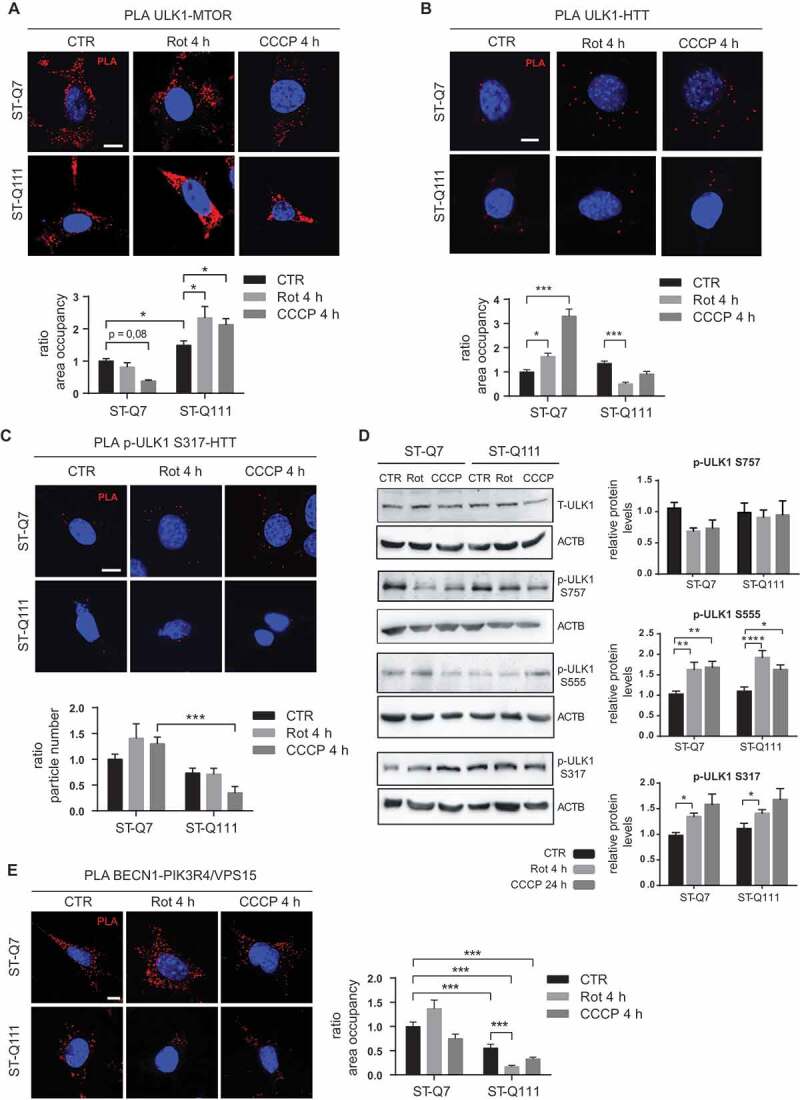
Mutant HTT alters the autophagy initiation step. (a) ULK1-MTOR interaction detected by PLA in ST-Q7 and ST-Q111 control cells (vehicle, CTR) or treated with rotenone (Rot, 1 μM, 4 h) or CCCP (10 μM, 4 h). (b) ULK1-HTT interaction detected by PLA in ST-Q7 and ST-Q111 cells CTR or treated with Rot (1 μM, 4 h) or CCCP (10 μM, 4 h). (c) Phospho-ULK1 S317-HTT interaction detected by PLA in ST-Q7 and ST-Q111 cells CTR or treated with Rot (1 μM, 4 h) and CCCP (10 μM, 4 h). (d) Representative immunoblots of total ULK1 (T-ULK1), Phospho-ULK1-S757, Phospho-ULK1-S555, Phospho-ULK1-S317 and ACTB protein levels in total homogenates from ST-Q7 and ST-Q111 CTR cells or treated with Rot (1 μM, 4 h) or CCCP (10 μM, 24 h). (e) BECN1-PIK3R4/VPS15 interaction detected by PLA in ST-Q7 and ST-Q111 control cells (vehicle, CTR) or treated with Rot (1 μM, 4 h) and CCCP (10 μM, 4 h). All PLA interactions were quantified as a ratio of area occupancy by the PLA signal or number of particles represented as the fold-change compared with the ST-Q7 CTR condition. A minimum of 30 cells was analyzed per condition. Scale bars: 10 μm
Following its activation, ULK1 phosphorylates BECN1, a component of the autophagy initiation complex or PtdIns3K complex formed by BECN1, PIK3R4/VPS15, PIK3C3/VPS34, and other proteins and responsible for autophagosome formation [45]. Recently it was shown that the deubiquitinase ATXN3 (ataxin 3), which is altered in spinocerebellar ataxia type 3, interacts through its wild-type (WT) polyQ domain with BECN1 and protects BECN1 from proteasomal degradation. However, BECN1 binds not only to ATXN3 but also to a greater extent to expanded polyQ tracts such as the ones present on mHTT, thus, competing with the physiological binding of ATXN3 and promoting the degradation of BECN1 [46]. Lower levels of BECN1 are related to a reduced initiation of complex assembly and lower macroautophagy rates [46]. Using PLA, we investigated whether mHTT could affect mitophagy at this step by impairing the formation of the PtdIns3K complex. At basal levels, the presence of mHTT downregulated the BECN1-PIK3R4/VPS15 interaction. Upon depolarization, differentiated ST-Q7 cells exhibited high levels of BECN1-PIK3R4/VPS15 interaction, which is indicative of assembly of the PtdIns3K initiation complex, while differentiated ST-Q111 cells displayed downregulation of the formation of this complex (Figure 3E and S2). These results further confirm that expanded polyQ tracts present in mHTT impair the formation of the autophagy initiation complex in striatal cells.
Mitophagy in differentiated striatal cells does not affect the polyUB labeling of mitochondria
Mitophagy can degrade mitochondria through various pathways depending on the cell type and conditions [4]. Some of these pathways may be alternative, but others could be redundant and might compensate for each other under certain physiologic or pathologic conditions [4].
Mitophagy can be achieved by a UB-mediated process with the collaboration of E3 ubiquitin ligases and the presence of non-mitochondrial receptors like SQSTM1/p62, NBR1, OPTN, and CALCOCO2 containing an UB-associated (UBA) and an LC3-interacting region (LIR) domain in their structure. Alternatively, UB-independent mitophagy can take place with the participation of mitochondrial receptors like BNIP3 (BCL2/adenovirus E1B 19 kDa protein-interacting protein), BNIP3L (BCL2/adenovirus E1B 19 kDa protein-interacting protein 3-like, also known as NIX) and FUNDC1 (FUN14 domain-containing protein 1). These receptors can bind directly to LC3 through their LIR domain in response to different conditions like hypoxia or metabolic stress [4].
We further investigated the different alternative pathways to elucidate the effect of mHTT in mitophagy in differentiated striatal cells. First, we analyzed the effect of mHTT in the best-characterized mitophagy mechanism, mediated by the action of PINK1 (PTEN induced kinase 1) protein. Depolarized mitochondria are labeled with phosphorylated polyUB at serine 65 (polyUB-p-S65) tagged on a variety of substrates present on the outer mitochondrial membrane, and these tags act as the signal to be recognized by the mitophagy machinery. E3 UB ligases mediate this tagging process, along with PRKN/PARKIN, the most well-characterized UB ligase protein involved in this step. However, PRKN requires activation by the kinase PINK1, which phosphorylates both PRKN and also UB at their serine 65. Thus, and according to many studies, the first step of this mitochondrial labeling is the retention of full-length PINK1 protein on the mitochondrial surface and the subsequent recruitment and activation of PRKN protein [47–50]. Active PRKN ubiquitinates the substrates on the outer mitochondrial membrane, which feeds a positive feedback loop, amplifying the “eat-me” signals [37]. However, recent studies have shown that this scenario might not be common during neuronal mitophagy since PRKN cannot be detected in mitochondria in several neuronal models [51–54], indicating that other alternative E3 UB ligases might take the role of PRKN [55]. Consequently, neuronal mitophagy might take place in a slower process compared to that in other cell types or models that exhibit high levels of PRKN translocated to the mitochondria [56].
According to this situation, we first investigated the possible role of PINK1 and PRKN upon mitophagy induction in our cellular model. We observed that PINK1 protein levels remained steady in differentiated ST-Q7 cells upon mild or acute depolarization. In differentiated ST-Q111 cells, basal PINK1 protein levels slightly increased, but again no changes were observed upon rotenone- or CCCP-treatment (Figure 4A). Depolarization induced by various effectors did not alter PRKN protein levels in total cellular homogenates. Importantly, we were unable to detect PRKN in isolated mitochondrial fractions (Figure 4B). PRKN translocation to mitochondria was also not detected by confocal immunofluorescence in the presence of basal or depolarization-induced conditions in differentiated ST-Q7 and ST-Q111 cells (Figure 4C). This scenario is similar to that observed in neuronal cell lines where endogenous PRKN translocation to the mitochondria was not detectable [51–54]. To test if the lack of mitochondrial-associated PRKN after depolarization was due to the lower levels of PRKN protein, we artificially overexpressed PRKN protein tagged to a fluorescent protein (EGFP). Even with the overexpression of EGFP-PRKN protein, we did not observe a clear translocation of EGFP-PRKN within a short time with mild depolarization (1 μM Rot 4 h). Only after 24 h with a strong depolarizing agent (10 μM CCCP) was it possible to observe a partial translocation of EGFP-PRKN from a diffuse cytosolic distribution to a mitochondrial localization (Figure 4D).
Figure 4.
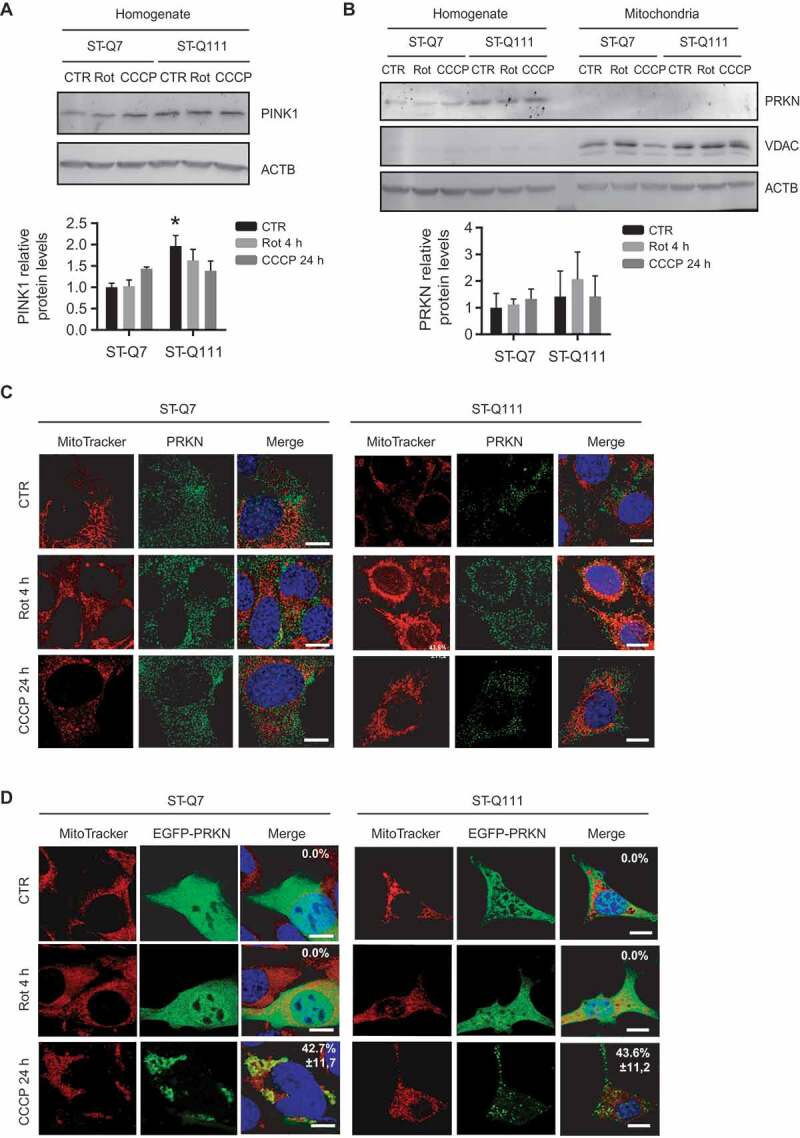
Mitophagy in differentiated striatal cells is not mediated by PRKN translocation to mitochondria. (a) Representative immunoblots of PINK1 protein levels in total homogenates from ST-Q7 and ST-Q111 control cells (vehicle, CTR) or treated with rotenone (Rot, 1 μM, 4 h) or CCCP (10 μM, 24 h). Protein levels were normalized relative to ACTB and quantification is depicted as fold-change to control ST-Q7. (b) Representative immunoblots of PRKN protein levels in total homogenates and isolated mitochondria from ST-Q7 and ST-Q111 control cells (vehicle, CTR) or treated with Rot (1 μM, 4 h) or CCCP (10 μM, 24 h). Protein levels were normalized relative to ACTB and quantification is depicted as fold-change to control ST-Q7. (c) Representative images of MitoTracker DeepRed and PRKN immunostained ST-Q7 and ST-Q111 cells CTR or treated with Rot (1 μM, 4 h) or CCCP (10 μM, 24 h). (d) Representative images of MitoTracker Deep Red and EGFP-PRKN immunostained ST-Q7 and ST-Q111 cells CTR or treated with Rot (1 μM, 4 h) or CCCP (10 μM, 24 h). A minimum of 30 cells was analyzed per condition. Scale bar: 10 μm
Nevertheless, EGFP-PRKN mitochondrial translocation was only detectable in less than 50% of ST-Q7 (42.7% ± 11.8), and ST-Q111 (46.3% ± 11.2) differentiated striatal cells (Figure 4D), as previously reported [57]. This result contrasts with other studies where PRKN overexpression led to a clear mitochondrial localization in other cell lines, including neuronal cells [51,52,58,59]. Based on these findings, we analyzed endogenous and EGFP-PRKN translocation to mitochondria in two human immortalized neuroblastoma cell lines, BE(2)-M17 and SH-SY5Y. Similarly, in differentiated striatal cells, endogenous PRKN did not translocate to mitochondria in either cell line upon mild or acute depolarization (Fig. S4A). In contrast, artificial overexpression of EFGP-PRKN under strong depolarizing conditions led to nearly all cells showing EGFP-PRKN translocation to the mitochondria (Fig. S4B). According to these results, and in agreement with other studies focused on neuronal mitophagy, endogenous PRKN levels in our differentiated cellular model seemed too low for clear detection of PRKN translocation to the mitochondria. Nevertheless, mitophagy still takes place since we still detected mitochondria engulfed by autophagosomes and fused with lysosomes (Figure 2A).
Next, we analyzed if, despite the absence of PRKN in mitochondria upon depolarization, the labeling of mitochondria with polyUB chains could still take place, probably by the action of alternative E3 UB ligases. To this end, we analyzed the polyUB labeling of total homogenate and isolated mitochondrial fractions detected with anti-polyUB antibody (Figure 5A). We observed no significant differences between control and mHTT samples for both homogenate and isolated mitochondrial fractions after depolarization. When studying only polyUB-p-S65 both in total homogenate and isolated mitochondria, we observed a tendency toward increased levels of this mitophagy-related labeling (Figure 5A) in both control and mHTT cells upon depolarization. These results indicate that although we could not detect PRKN translocation to mitochondria, ubiquitin chains are phosphorylated at Ser65 in the presence of both control and mHTT. Therefore, mitophagy is taking place in differentiated striatal cells through a UB-dependent mechanism, although this UB-labeling step seems to be PRKN-independent and HTT-independent. However, since other forms of mitophagy independent of ubiquitin labeling [4,60] could also be participating, we analyzed UB-independent mitophagy mediated by the receptors BNIP3, BNIP3L, and FUNDC1 to determine if they could also be assisting in the induced mitophagy in our cellular model. We analyzed the levels of these three receptors in mitochondria before and after depolarization. FUNDC1 protein could not be detected in striatal cell lines under basal conditions or after depolarization treatment (data not shown). We analyzed levels of BNIP3 and BNIP3L proteins in the isolated mitochondrial fractions (Figure 5B). We detected both proteins, as members of the BH3-only protein family, as monomers and oligomers. Quantification of total levels of these proteins did not increase after mitophagy induction by depolarization, suggesting that, under these conditions, these proteins are probably not participating in mitophagy. These findings are in accordance with other studies, where other stress-associated conditions like hypoxia and metabolic stress trigger BNIP3/BNIP3L/FUNDC1-mediated mitophagy, but not depolarization [4,61]. In conclusion, in striatal cells, mitophagy after depolarization seems to be UB-dependent (although without the intervention of PRKN); however, none of these steps seem to be impaired by the presence of mHTT.
Figure 5.
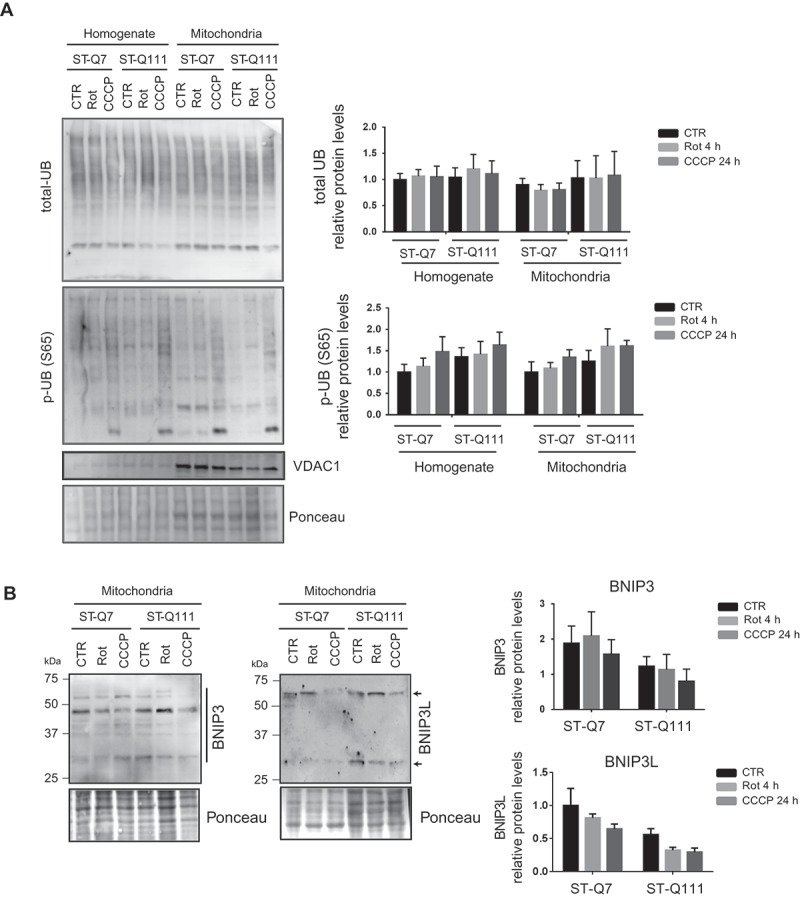
PolyQ tract in mutant HTT does not affect the polyUB labeling of mitochondria. (a) Representative immunoblots of total polyUB (total-UB) and phosphorylated ubiquitin (Ser65; p-UB-S65) in total homogenate and isolated mitochondria from ST-Q7 and ST-Q111 control cells (vehicle, CTR) or treated with rotenone (Rot, 1 μM, 4 h) or CCCP (10 μM, 24 h). Protein levels were normalized relative to Ponceau staining and quantification is depicted as fold-change to control ST-Q7 cells of at least three independent experiments. (b) Representative immunoblots of BNIP3 and BNIP3L in isolated mitochondria from ST-Q7 and ST-Q111 control cells (vehicle, CTR) or treated with rotenone (Rot, 1 μM, 4 h) or CCCP (10 μM, 24 h). Protein levels were quantified using all bands present in each lane independently of the molecular weight or oligomerization state (indicated with line and arrows) and were normalized relative to Ponceau staining and quantification is depicted as fold-change to control ST-Q7 cells. Data are presented as mean ± s.e.m of at least 3 independent experiments and no significant changes were observed after statistical analysis
The polyQ tract in mHTT affects the recruitment of mitophagy receptors during mitophagy
Once we had shown that despite the absence of PRKN, mitochondria are still labeled with the polyUB-p-S65, we analyzed the next step in mitophagy mediated by the UB tag. After the labeling of damaged mitochondria with polyUB-p-S65 chains, a set of receptors are able to recognize and bind the mitochondria and simultaneously recruit LC3-II to form an autophagosome around the mitochondria.
Previously, we and others showed that HTT could also interact with the selective autophagy receptor protein SQSTM1/p62 [18,19,21]. In addition to SQSTM1/p62, different protein receptors for selective mitophagy like CALCOCO2 (calcium binding and coiled-coil domain), OPTN (optineurin), NBR1 (NBR1, autophagy cargo receptor) and TAX1BP1 (Tax1 binding protein 1) have been identified [62]. In a recent study using the combined knockout of all five mitophagy receptors, it was shown that, although all of the receptors can intervene during mitophagy, only OPTN and CALCOCO2 are essential [37], with the others probably having redundant roles. First, and in agreement with previous data [18,19,21] and with the computer-based predictions, we used immunoprecipitation to confirm the interaction of HTT with the mitophagy receptors SQSTM1/p62, NBR1, OPTN, and CALCOCO2 (Fig. S3). Once we validated the interactions, we used PLA to investigate and quantify the efficiency of the interaction of HTT with the essential mitophagy receptors OPTN and CALCOCO2 (Figure 6A and B). The OPTN-HTT interaction became reduced after depolarization only when HTT was carrying the mutant polyQ tract (Figure 6A and S2). Moreover, we could not detect the CALCOCO2-HTT interaction in the presence of the mutant polyQ tract (Figure 6B). The lack of interaction between CALCOCO2 and mHTT was not due to the absence of CALCOCO2 protein but rather due to the total loss of interaction since CALCOCO2 protein could still be detected by immunofluorescence in mHTT cells before and after depolarization (Fig. S5). These results show that the interaction between the two essential mitophagy receptors and HTT is strongly influenced by the number of glutamines in the polyQ tract. In the case of CALCOCO2, the interaction is totally abolished when mHTT is present.
Figure 6.
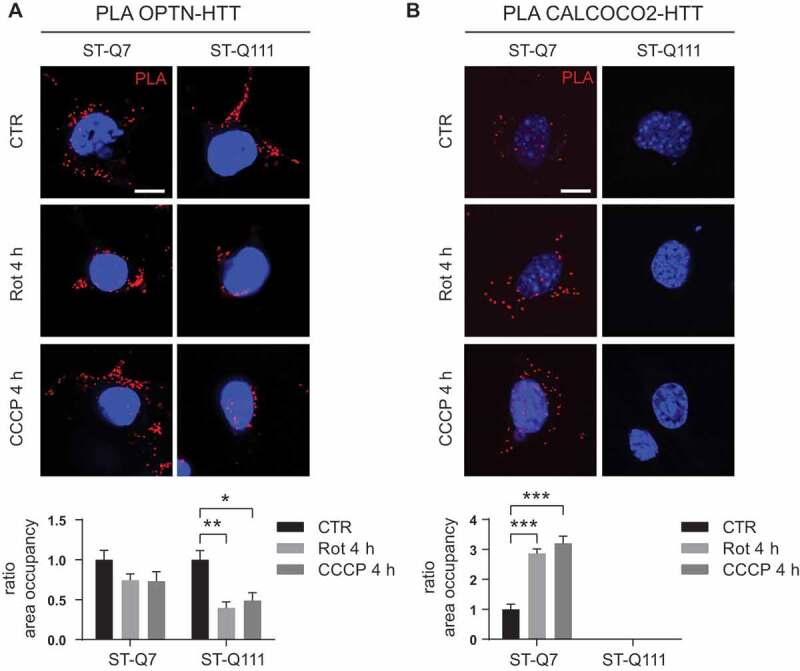
PolyQ tract in mutant HTT affects the interactions with mitophagy receptors. (a) OPTN-HTT interaction detected by PLA in ST-Q7 and ST-Q111 control cells (vehicle, CTR) or treated with rotenone (Rot, 1 μM, 4 h) or CCCP (10 μM, 4 h). (b) CALCOCO2-HTT interaction detected by PLA in ST-Q7 and ST-Q111 cells CTR or treated with Rot (1 μM, 4 h) or CCCP (10 μM, 4 h). A minimum of 40 cells was analyzed per condition. Scale bars: 10 μm
The polyQ tract in mHTT affects the interaction of LC3-II with mitophagy receptors
As the last step of substrate recognition during mitophagy, receptors interact with and recruit LC3-II, thereby allowing elongation of the autophagosome membrane around mitochondria. We used PLA to analyze the interaction between the two essential mitophagy receptors (OPTN and CALCOCO2) with LC3 and observed that, upon depolarization, the OPTN-LC3 and CALCOCO2-LC3 interactions increased in differentiated ST-Q7 cells (Figure 7A and 7B). Conversely, the presence of the mutant polyQ tract impaired the OPTN-LC3 interaction and decreased the CALCOCO2-LC3 interaction, probably as a consequence of the failure recruitment of receptors to the mitophagy site (Figure 6A and B). We also used PLA to examine interactions between the SQSTM1/p62 and NBR1 autophagy receptors with LC3 and observed the same effect; mHTT dramatically impaired SQSTM1/p62 and NBR1 interactions with LC3 (Figure 7C and D). These results show the impact of the presence of mHTT on the recruitment of LC3-II to the mitophagy initiation site, a key step to promote the elongation of the autophagosome membrane to engulf tagged mitochondria.
Figure 7.
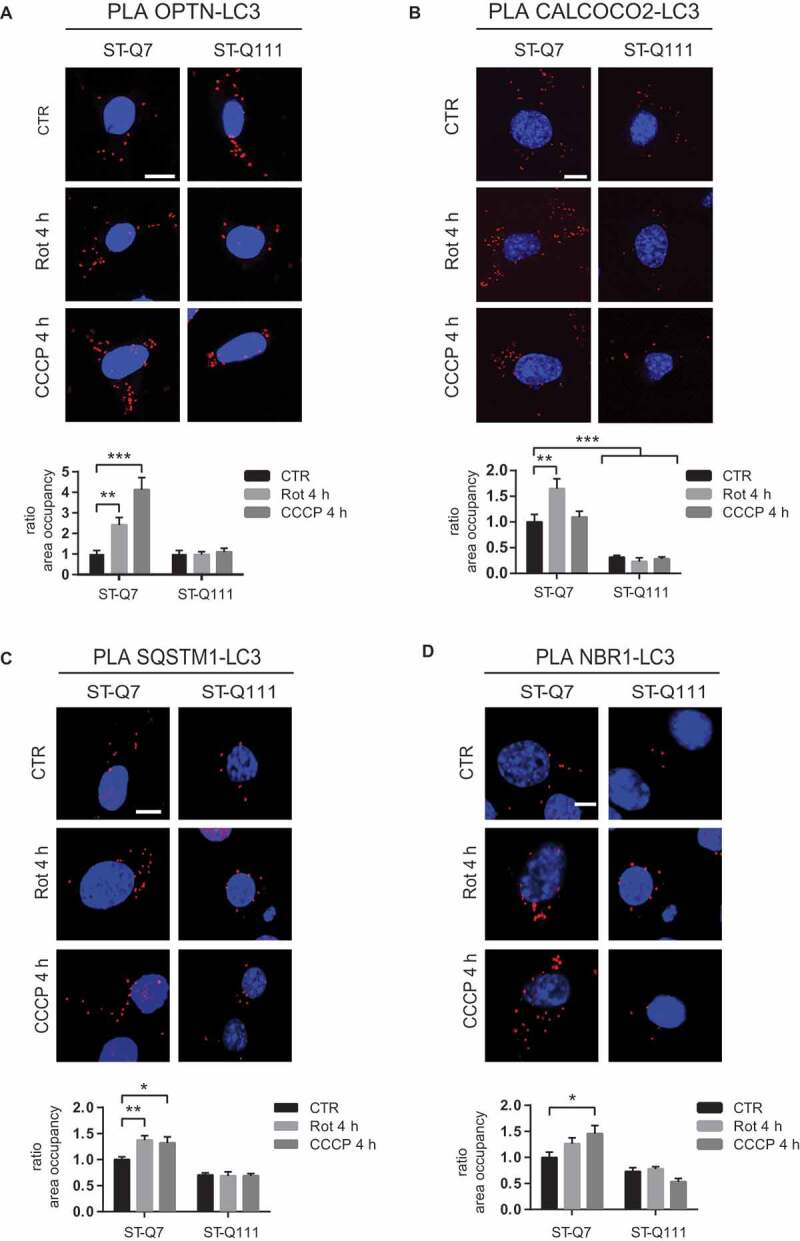
PolyQ tract in mutant HTT affects recruitment of LC3-II (a) OPTN-LC3 interaction detected by PLA in ST-Q7 and ST-Q111 control cells (vehicle, CTR) or treated with rotenone (Rot, 1 μM, 4 h) or CCCP (10 μM, 4 h). (b) CALCOCO2-LC3 interaction detected by PLA in ST-Q7 and ST-Q111 cells CTR or treated with Rot (1 μM, 4 h) or CCCP (10 μM, 4 h). (c) SQSTM1/p62-LC3 interaction detected by PLA in ST-Q7 and ST-Q111 control cells (vehicle, CTR) or treated with rotenone (Rot, 1 μM, 4 h) or CCCP (10 μM, 4 h). (d) NBR1-LC3 interaction detected by PLA in ST-Q7 and ST-Q111 cells CTR or treated with Rot (1 μM, 4 h) or CCCP (10 μM, 4 h). PLA interaction was quantified as a ratio of area occupancy by the PLA signal represented as the fold-change compared with CTR ST-Q7 condition. A minimum of 20 cells was analyzed per condition Scale bars: 10 μm
Discussion
This work has analyzed in-depth the steps by which HTT modulates mitophagy under conditions mimicking different physiological and pathogenic scenarios, providing insights to identifying the consequences accompanying the presence of the polyQ tract expansion. To this end, we worked with differentiated striatal cells expressing endogenous levels of mitophagy-related proteins. We have used two alternative treatments to induce mitochondrial depolarization, the first being the most widely used method and consisting of treatment with CCCP 10 μM for 24 h to induce a fast and substantial mitochondrial depolarization. However, this treatment could be too aggressive, as CCCP has been shown to also interfere directly in the initiation of autophagy and lysosomal degradation in some cell types like yeast and HeLa cells, in addition to its role as a mitochondrial uncoupler [63]. Given this possible dual role as an autophagy inhibitor, as well as a mitochondrial uncoupler, we also included a second type of mitochondrial depolarization using a low concentration of rotenone (1 μM for 4 h), which induces a milder depolarization closer to that which may occur during the evolution of HD [64]. By promoting the activation of mitophagy and the recruitment of different elements participating in this mechanism, our studies demonstrated the key role of HTT protein in mitophagy (Fig. S6). The presence of mHTT affects the ability of the protein to execute its role and consequently impairs the efficiency of the mitophagy process, leading to the accumulation of damaged mitochondria and the generation of ROS, thus arguing in favor of the presence of loss-of-function mechanisms in HD pathology. Indeed, we have previously described the presence of “empty” autophagosomes in HD striatal cells as a consequence of inefficient mitochondrial loading during selective macroautophagy [21]. Other authors have also described reduced levels of mitophagy in an HD mouse model expressing mt-keima in vivo, confirming that this mechanism is also altered in vivo [65].
In this work, we analyzed how the presence of the mutant polyQ tract impairs the mitophagy mechanism. WT HTT participates as a scaffold protein in different steps of the mitophagy mechanism [18,19]. The presence of the mutant polyQ tract in mHTT impairs several crucial interactions, with our results showing the impaired formation of the autophagy initiation complexes by preventing both the release of ULK1 from MTORC1 to form the ULK1 complex and the interaction between BECN1 and PIK3R4/VPS15 to form the PtdIns3K complex. The latter is in agreement with a previously described reduction of BECN1 levels in different cellular and animal models with expanded polyQ forms [46]. Therefore, mHTT affects the formation and stability of two complexes that are essential for autophagosome formation during autophagy initiation. This scenario might affect all forms of autophagy initiation, among them mitophagy, due to the reduced assembly of these complexes. While we detected lower levels of the MTOR-mediated inhibitory phosphorylation of ULK1 at S757 upon mitophagy induction using both rotenone and CCCP treatments, mutant cells displayed no changes. This observation correlates with the fact that ULK1 remains more bound to MTOR in ST-Q111 cells upon mitophagy induction, suggesting that the presence of mHTT impairs the release of ULK1 from MTOR inhibitory interaction. Importantly, AMPK-mediated phosphorylation on S555 and S317 are not affected by the presence of mHTT, in agreement with Rui et al. (2015) that also show that the absence of HTT had no significant impact on ULK1 activation by AMPK-phosphorylation.
Most studies on mitophagy employ the artificial introduction, overexpression or deletion, of different proteins involved in mitophagy. However, the reproduction of these studies under endogenous conditions in neuronal cell lines resulted in no detection of endogenous PRKN translocation to the mitochondria [51–54], and that other types of mitophagy might be taking place [64,66]. We have also confirmed these results in our striatal cell line. Considering the latest model of PINK1/PRKN-dependent mitophagy [37], the role of PRKN in mitophagy is likely to be important as an amplifier of the mitophagy signal, but it is unlikely to be essential for mitophagy to take place. When PRKN levels in mitochondria are low, mitophagy can still take place through a PINK1-dependent process, but at a lower rate [37]. According to our results, we detected mitochondria labeled with polyUB-p-S65 chains after acute depolarization, which suggests that alternative E3 UB ligases might be involved in this step. Indeed, the observed PRKN-independent mitophagy seems to be related to the nature of the differentiated immortalized striatal cells themselves, since we observed the same effect in both control and mHTT cells. We can, therefore, postulate that PRKN translocation does not occur in this cell type and that mitophagy is taking place at a low level. The observed increase in basal PINK1 levels could be related to alternative roles of PINK1 in other mitochondrial quality control mechanisms [4,60,67] since PINK1 is involved in many other mitophagy-independent mechanisms in response to mitochondrial damage. Given that UB-labeling of the mitochondria does not seem to be affected in mHTT, we can confirm that this step in the mitophagy process does not contribute to the global mitophagy impairment observed in HD. We also ruled out the involvement of UB-independent mitochondrial receptors like BNIP3 and BNIP3L that do not seem to be activated under these stress conditions and do not show alterations in the presence of mHTT protein.
Finally, we found that mutant polyQ tracts affect the interaction of OPTN and CALCOCO2 with HTT and consequently affect the recruitment of LC3 to the mitochondrial site. We also observed this effect with other alternative receptors like SQSTM1/p62 and NBR1. Altogether, the deficient recruitment of LC3 may affect the ability of the cell to engulf tagged mitochondria efficiently.
As a consequence of the hampered mitophagy, damaged mitochondria accumulated, leading to deficits in mitochondrial respiration and increased ROS production. One previous study also reported the presence of decreased spare respiratory capacity in mHTT striatal cells [30], while others observed respiratory deficits only when respiration demand was high [35,68]. Interestingly, the only observation of dysfunctional respiration occurs in striatal neurons but not in cortical neurons [35], which degenerate later in the course of HD. In the same way, several studies have also described dysregulated glycolysis in the context of HD [69–71]. There are also reports of higher basal levels of mitochondria-generated ROS, and mtDNA lesions in mHTT striatal cells [30], which correlates with the increased presence of oxidative damage in proteins and lipids in the striatum and cortex of human HD brains [72–75].
We can conclude that the effect of mHTT on mitophagy is not only a consequence of the failure of autophagy initiation due to the abnormal interaction of mHTT with ULK1 and BECN1 but also due to the loss of interaction of HTT with two essential receptors (OPTN and CALCOCO2), as well as other alternative receptors, such as SQSTM1/p62 and NBR1. These occurrences led to reduced LC3-II recruitment to the damaged mitochondria. Since the receptor recruitment step by HTT is independent of ULK1 and BECN1 and is clearly impaired when the polyQ tract is expanded, we can conclude that the detrimental effect of mHTT occurs in parallel at different steps in the mitophagy process (Fig. S6).
In addition to these effects on mitophagy, there are also reports of numerous other alterations to mitochondrial homeostasis in HD models and tissue, including bioenergetic alterations [76,77], reduced activity of OXPHOS complex I, II and IV [26,27,78], reduced ATP levels [79,80], increased oxidative stress [72–75,81], and defects in mitochondrial dynamics [82–84], biogenesis [85–87], and trafficking [15,16,84,88–91]. Obviously, not all of these mitochondria-associated alterations are likely to be due to the role of HTT in mitophagy, but they could occur as a consequence of alternative HTT functions [25]. As the events occurring in HD neurons would likely accumulate over the course of the disease, and since mitophagy is one of the main mitochondrial quality control mechanisms that guarantees a healthy mitochondrial pool, the impairment of mitophagy described here must play an important role in the pathogenesis of the disease and contribute to the process of neurodegeneration.
Materials and methods
Reagents
Rotenone (R-8875), CCCP (carbonyl cyanide 3-chlorophenyl hydrazone, C2759), oligomycin (75351), antimycin A (A8674) and the lysosomal inhibitors chloroquine (C6628) and bafilomycin A1 (B1793) were from Sigma-Aldrich. MitoTracker™ Deep Red FM (Molecular Probes, M22426), CM-H2DCFDA (Life Technologies, C6827), Cell Rox Green Reagent (Thermo Fisher Scientific, C10444) and TMRM (Tetramethylrhodamine methyl ester, Sigma-Aldrich, T668). Sodium ortho-vanadate (Sigma-Aldrich, S6508), NaF (Sigma-Aldrich, 201154), sodium pyrophosphate (Sigma-Aldrich, 221368), sodium 2-glycerophosphate (Sigma-Aldrich, G9422), benzamidine (Sigma-Aldrich, 12072), PMSF (Sigma-Aldrich, P7626), iodoacetamide (Sigma-Aldrich, I1149), MG132 (Enzo BML, PI102).
Antibodies
LC3 (NB-2220), PINK1 (BC100-494SS), ULK1 (NBP2-24738S) and PIK3R4/VPS15 (NBP1-30463) from Novus, LAMP1 (sc-19992), CALCOCO2 (sc-376540) and BECN1/Beclin 1 (sc-48341) from Santa Cruz Biotechnology. PRKN (ab15954), PRKN (ab77924) and VDAC1/Porin (ab15895) from Abcam. ULK1 Ser555 (5869P), ULK1 Ser317 (12753S) and ULK1 Ser757 (6888S) from Cell Signaling Technology. OPTN (10837-1-AP) from Proteintech Group, HTT (MAB2166) from Chemicon, SQSTM1/p62 (GP62-C) and NBR1 (H00004077-M01) from Abnova, anti-DNA (AC-30-10) from Progen, MTOR (GT649) mouse (GTX630198) from Genetex, total-UB (U5379) and ACTB/β-actin (A5441) from Sigma-Aldrich, BNIP3 (M01469), BNIP3L (M03107) and FUNDC1 (A08688) from Boster. Secondary antibodies goat anti-mouse HRP, donkey anti-rabbit HRP, donkey anti-rat HRP from GE Healthcare, goat anti-guinea pig HRP from Santa Cruz Biotechnology, Alexa Fluor 488 goat anti-mouse, Alexa Fluor 488 goat anti-rabbit, Alexa Fluor 488 donkey anti-rat, Alexa Fluor 594 goat anti-mouse and Alexa Fluor 594 goat anti-rabbit from Thermo Fisher Scientific.
Cell culture and treatments
ST-Q7 and ST-Q111 cell lines
STHdhQ7/Q7 (ST-Q7) and STHdhQ111/Q111 (ST-Q111) were obtained from the Coriell Institute (CH00097 and CH00095, respectively). ST-Q7 and ST-Q111 cells were grown in Dulbecco’s Modified Eagle Medium (DMEM, Gibco, Thermo Fisher Scientific, 41966–029) supplemented with 10% inactivated fetal bovine serum (Thermo Fisher Scientific, 26,140–079), 1% penicillin-streptomycin (Sigma-Aldrich, P4333) and 0.4 mg/mL active geneticin (Thermo Fisher Scientific, 11558616) and maintained at 33°C in humidified 95% air/5% CO2 incubator. For differentiation, cells were supplemented with dopamine (DOPA) cocktail consisting of 10 ng/mL α-FGF-acidic mouse recombinant (Fibroblast growth factor, Sigma-Aldrich, SRP3197-50U), 250 μM IBMx (3-isobutyl-1-methylxanthine, Sigma-Aldrich, I5879), 200 nM TPA/PMA (Phorbol 12-myristate 13-acetate, Sigma-Aldrich, P1585), 50 μM forskolin (Sigma-Aldrich, F6886) and 20 μM dopamine hydrochloride (Sigma-Aldrich, H8502) during at least 24 h.
BE(2)-M17
Human neuroblastoma dopaminergic cell line BE(2)-M17 (Sigma-Aldrich, 95011816) was grown in Minimal Essential Medium optimized (Opti-MEM, Gibco, Thermo Fisher Scientific, 31985062) supplemented with 10% inactivated fetal bovine serum, 1% penicillin-streptomycin and 0.5 mg/mL active geneticin and maintained at 37°C in humidified 95% air/5% CO2 incubator.
SH-SY5Y
Human neuroblastoma SH-SY5Y cell line was obtained from ATCC (CRL-2266TM) and grown in DMEM supplemented with 10% inactivated fetal bovine serum and 1% penicillin-streptomycin and maintained at 37°C in humidified 95% air/5% CO2 incubator.
Plasmid transfection
For plasmid transfection, Lipofectamine 3000 (Thermo Fisher Scientific, L3000015) was used. Cells were transfected with 0.8 µg (EGFP-PRKN) of DNA following the manufacturer’s instructions.
Immunofluorescence microscopy
Cells were fixed in 4% paraformaldehyde for 30 min at RT. Following the incubation with a blocking solution containing normal goat serum 3% (Vector Laboratories, S100), Triton X-100 0.1% (Sigma, T9284) in PBS (Thermo Fisher, 70013065) for 1 h at RT, the corresponding primary antibodies were used diluted in the same blocking solution overnight at 4°C. Then, cells were incubated 1 h at RT with the corresponding Alexa secondary antibodies, diluted in blocking solution. Nuclei were stained with 10 µM Hoechst 33342 (Invitrogen, H3570) in PBS. When indicated, 100 nM MitoTracker™ Deep Red FM prepared in Opti-MEM medium was added before proceeding with the protocol. Coverslips were mounted onto superfrost ultra plus slide (Menzel-Gläser, Ref.10417002) using DAKO Fluorescent Mounting Medium (DAKO, S3023). Images were acquired using an Olympus FV1000 confocal microscope (Tokyo, Japan). Intensity analysis was performed by drawing a region of interest to each cell and measuring its intensity using ImageJ 1.50a (NIH, USA).
Immunoblot detection
For biochemical studies, cells were detached and centrifuged at 800 g for 5 min at 4°C. The pellet was resuspended in PBS, centrifuged two more times, and resuspended in radioimmunoprecipitation assay buffer or RIPA buffer supplemented with proteases inhibitors (HALT Protease inhibitor cocktail EDTA free, Thermo Scientific, 1862209) and 1 mM PMSF (Sigma-Aldrich, P7626). For ubiquitin detection, cells where resuspended in UB-buffer (50 mM Tris/HCl pH 7.5, 1% Triton X-100, 1 mM sodium ortho-vanadate, 50 mM NaF, 5 mM sodium pyrophosphate, 0.27 M sucrose, 10 mM sodium 2-glycerophosphate, 0.2 mM PMSF, 1 mM benzamidine, 1 mM EGTA, 1 mM EDTA, 100 mM iodoacetamide, 25 mM MG132) in order to avoid deubiquitination. Homogenates were incubated on ice for 15 min and sonicated. Total protein concentration was determined by the bicinchoninic assay method with BSA as a standard protein. Cellular protein extracts were heated at 95°C for 5 min after adding loading buffer 6x. Proteins were resolved by SDS-PAGE on different percentages of polyacrylamide gels, ranging from 7% to 15%. Protein All Blue Standards ladder (Biorad, 161–0373) was used as a ladder. Resolved proteins were transferred onto nitrocellulose membranes (GE Healthcare, 10600002) or PVDF membranes (Whatman, 10485289) in the case of polyUB immunodetection, and blocked with 5% nonfat milk powder (Sigma-Aldrich, 70166) in PBS for 1 h at RT. Membranes were incubated with the corresponding primary antibodies diluted in 4% bovine serum albumin (Sigma-Aldrich, A4503) in PBS overnight at 4°C. Then, we proceeded to the incubation with the corresponding secondary antibodies coupled with horseradish peroxidase and diluted in 5% milk in PBS for 1 h at RT. Finally, proteins were visualized using either West Pico SuperSignal Substrate or SuperSignal West Femto (Thermo Fisher Scientific, 34080 and 34095, respectively) on an ImageQuant RT ECL imaging system (GE Healthcare). Immunoblots were quantified by densitometry using ImageJ 1.50a.
Mitochondrial enrichment
Mitochondria were isolated from striatal cells, as previously described on Frezza et al. [92]. Briefly, cells were washed with PBS, detached, and centrifuged at 600 g at 4°C for 5 min. The pellet was resuspended in ice-cold IBc buffer (10 mM Tris-MOPS [Sigma-Adrich, M5162], 1mM EGTA/Tris [Sigma-Aldrich, E4378] and 0.2 M sucrose [Sigma-Aldrich, S9378] in H2O at pH 7.4). To homogenize, we used a Teflon pestle operated at 1600 rpm and stroked the cell suspension placed in a glass potter during 30–40 times. The cell suspension was centrifuged at 600 g for 10 min at 4°C, and the supernatant was further centrifuged at 7000 g for 10 min at 4°C. Pellet was washed with ice-cold IBc buffer and centrifuged again at 7000 g for 10 min at 4°C. Finally, the pellet containing mitochondria was resuspended in an adequate volume of IBc buffer, and mitochondrial protein concentration was determined by the bicinchoninic assay method with BSA as a standard protein.
Mitochondrial membrane potential
Differentiated ST-Q7 and ST-Q111 cells were incubated with 50 nM TMRM or 10 nM MitoTracker™ Deep Red FM probes for 30 min in a cell incubator. Cells were washed twice with pre-warmed PBS and recollected with a cell scraper in the medium. Using a FACSAria flow cytometer (BD Biosciences), 10,000 cells were acquired, and fluorescence detected with the indicated lasers and detectors. Membrane potential was analyzed as mean fluorescence intensity (MFI) and represented as fold-change compared to the corresponding controls using the FCS Express (v3 De Novo SoftwareTM).
Seahorse XF24 mitochondrial respiration analysis
Oxygen consumption rate (OCR) was measured using the Seahorse XF24 equipment (Seahorse bioscience Inc., North Billerica, MA, USA). ST-Q7 and ST-Q111 cells were seeded in Seahorse XF24 Cell Culture Microplate (Seahorse Biosciences, 100777–004), differentiated and treated with the corresponding treatments. Cells were rinsed once and incubated in 700 μl of XF assay buffer (DMEM without NaHCO3, 2 mM glutamax; pH 7.4, 5 mM glucose, 1 mM sodium pyruvate), then equilibrated for 1 h at 37°C in a non-CO2 incubator. All medium and solutions of mitochondrial complex inhibitors were adjusted to pH 7.4 on the day of assay. Following three baseline measurements of OCR, mitochondrial complex inhibitors were sequentially injected into each well. Three OCR readings were taken after the addition of each inhibitor and before the automated injection of the subsequent inhibitor. Mitochondrial complex inhibitors, in order of injection, included oligomycin (1 μM), CCCP (0.5 μM each time), antimycin A (1 μM), and rotenone (1 μM). OCR was automatically calculated, recorded, and plotted by Seahorse XF24 software version 1.8 (Seahorse Bioscience, Billerica, MA, USA). At the end of each assay, cells were lysed with ice-cold RIPA buffer and the protein content estimated by the bicinchoninic assay method with BSA as a standard protein.
Quantification of reactive oxygen species (ROS) by fluorescent probes
Cells were washed twice with pre-warmed PBS and either 50 μM of CM-H2DCFDA probe or 50 μM CellROX was added in medium and incubated for 30 min in a cell incubator. Using a FACSAria flow cytometer, 10,000 cells were acquired, and fluorescence was read at 488 nm laser with a 530/30 emission filter. ROS was analyzed as mean fluorescence intensity (MFI) and represented either as MFI or as a percentage compared to the corresponding controls using the FCS Express v3 software. For immunofluorescence analysis, 5 μM of CellROX was added to the medium and incubated for 30 min in a cell incubator, before proceeding with the immunofluorescence protocol. Images were acquired using an Olympus FV1000 confocal microscope.
Mitophagy flux assay
Differentiated ST-Q7 and ST-Q111 cells were treated with the corresponding reagents (vehicle, rotenone, CCCP, chloroquine). Cells were incubated with 10 nM MitoTracker™ Deep Red FM probe for 30 min in cell incubator before harvesting. Cells were washed twice with pre-warmed PBS and collected with a cell scraper in the medium. Using a FACSAria flow cytometer, 10.000 cells were acquired and fluorescence was read at 640 nm red laser with a 660/20 emission filter. Mitophagy flux was analyzed as previously described [93] as a mean fluorescence intensity (MFI) in the presence and absence of 60 μM chloroquine using the FCS Express v3 software.
Immunoprecipitation
Differentiated ST-Q7 and ST-Q111 cells were harvested in PBS using and centrifuged at 800 g at 4ºC for 5 min. Pellet was resuspended in IP buffer containing 0.01% Triton in PBS and protease inhibitors. 1:2 mixture of Dynabeads protein A and protein G (Thermo Scientific, 10,001 and 10,003, respectively) was incubated with the corresponding antibody, together with 5 mM BS3 (bis[sulfosuccinimidyl]suberate, Sigma-Aldrich, S5799), to crosslink the antibody to the Dynabeads, in IP buffer and let rocking 30 min at RT. The supernatant was discarded using a magnetic particle concentrator (DynaMagTM-2, Invitrogen, 4211) and BS3 was inactivated with 50 mM Tris-HCl pH 7.4 for 15 min. Dynabeads were washed three times with IP buffer and incubated with the corresponding cell lysate for 90 min at 4ºC. After collecting the flow-through, Dynabeads were washed three times with IP buffer, and samples were incubated in elution buffer containing 0.2 M glycine pH 2.5, LB6x and 16 mM dithiothreitol (DTT) 10 min at 70ºC and immunoprecipitated samples were collected. Immunoprecipitates (IP), whole-cell extracts (input), and flow-through were analyzed by standard immunoblotting.
Proximity ligation assay (PLA)
PLA was performed in 3% formaldehyde-fixed cells (Sigma-Aldrich, F1635) following the manufacturer’s instructions. Briefly, samples were blocked for 30 min at RT and incubated with specific primary antibodies overnight. Secondary antibodies anti-mouse PLUS and anti-rabbit MINUS conjugated with oligonucleotides were added to the reaction (Duolink PLA Probe anti-mouse PLUS and anti-rabbit MINUS, Duolink, Sigma-Aldrich, DUO92001 and DUO92005, respectively) and incubated for 1 h at 37°C. Ligation reagents were added and incubated for 30 min at 37°C. Amplification solution containing nucleotides and fluorescently-labeled oligonucleotides, and polymerase was added and incubated for 100 min at 37°C (Detection Reagent RED, Duolink; Sigma-Aldrich, DUO92008). The proximity ligation signal is observed as fluorescence spots and was analyzed using an Olympus FV1000 confocal microscope. The corresponding negative control experiments were performed under identical experimental conditions. Quantification was performed by analyzing the number of dots per cell and the area fraction that these dots occupy per cell using ImageJ 1.50a.
Statistical analysis
The values were expressed as the mean ± standard error of the mean (SEM). The significant differences (*p < 0.05, **p < 0.01, ***p < 0.001) when comparing two groups were determined by a two-tailed unpaired Student’s t-test. When comparing more than two groups, significant differences were determined by one-way or two-way ANOVA followed by Tukey’s post hoc test. Statistical analyzes were performed using GraphPad Prism 6.
Supplementary Material
Funding Statement
This work was supported by the Instituto de Salud Carlos III [PI14/01529]; Instituto de Salud Carlos III [PI17/00496]; Instituto de Salud Carlos III [CP09/00184]; Instituto de Salud Carlos III [MSII15/00007]; Spanish Ministry of Science and Innovation[SAF2009-08374]; Spanish Ministry of Science and Innovation[FPU13/01339]; Spanish Ministry of Science and Innovation [JCI-2012-13544].
Aknowledgements
The authors wish to thank C. Malagelada (University of Barcelona) for the EGFP-PRKN plasmid and C. Perier for her insightful comments. This work was supported by Fondo de Investigación Sanitaria-Instituto de Salud Carlos III (Spain)-FEDER (CP09/00184, PI14/01529, PI17/00496, MSII15/00007), Spanish Ministry of Science and Innovation SAF2009-08374, and CIBERNED. S.F.I. was supported by an FPU doctoral fellowship (FPU13/01339) from Spanish Ministry of Science and Innovation. A.P.Z. was supported by a Juan de la Cierva post-doctoral fellowship (JCI-2012-13,544) from Spanish Ministry of Science and Innovation .
Disclosure statement
The authors declare that they have no competing interests.
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].He C, Klionsky DJ.. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;15:1–5. [DOI] [PubMed] [Google Scholar]
- [3].Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. [DOI] [PubMed] [Google Scholar]
- [4].Martinez-vicente M. Neuronal mitophagy in neurodegenerative diseases. Front Mol Neurosci. 2017;10:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Evans CS, Holzbaur ELF. Quality control in neurons: mitophagy and other selective autophagy mechanisms. J Mol Biol. 2019;432(1):240–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Stavoe AKH, Holzbaur ELF. Autophagy in neurons. Annu Rev Cell Dev Biol. 2019;35:477–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83–98. [DOI] [PubMed] [Google Scholar]
- [8].Bates GP, Dorsey R, Gusella JF, et al. Huntington disease. Nat Rev Dis Prim. 2015;1:15005. [DOI] [PubMed] [Google Scholar]
- [9].Rosas HD, Koroshetz WJ, Chen YI, et al. Evidence for more widespread cerebral pathology in early HD: an MRI-based morphometric analysis. Neurology. 2003;60:1615–1620. [DOI] [PubMed] [Google Scholar]
- [10].DiFiglia M, Sapp E, Chase KO, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. [DOI] [PubMed] [Google Scholar]
- [11].Shirasaki DI, Greiner ER, Al-Ramahi I, et al. Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron. 2012;75:41–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Li S-H, Li X-J. Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 2004;20:146–154. [DOI] [PubMed] [Google Scholar]
- [13].Luthi-Carter R, Cha JHJ. Mechanisms of transcriptional dysregulation in Huntington’s disease. Clin Neurosci Res. 2003;3:165–177. [Google Scholar]
- [14].Gauthier LR, Charrin BC, Borrell-Pagès M, et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. [DOI] [PubMed] [Google Scholar]
- [15].Trushina E, Dyer RB, Badger JD, et al. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol Cell Biol. 2004;24:8195–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wong YC, Holzbaur ELF. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J Neurosci. 2014;34:1293–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Martin DDO, Ladha S, Ehrnhoefer DE, et al. Autophagy in huntington disease and huntingtin in autophagy. Trends Neurosci. 2014;38:1–10. [DOI] [PubMed] [Google Scholar]
- [18].Rui Y-N, Xu Z, Patel B, et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat Cell Biol. 2015;17:262–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ochaba J, Lukacsovich T, Csikos G, et al. Potential function for the huntingtin protein as a scaffold for selective autophagy. Proc Natl Acad Sci USA. 2014;111:16889–16894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Diaz-Hernandez M, Valera AG, Moran MA, et al. Inhibition of 26S proteasome activity by huntingtin filaments but not inclusion bodies isolated from mouse and human brain. J Neurochem. 2006;98:1585–1596. [DOI] [PubMed] [Google Scholar]
- [21].Martinez-Vicente M, Talloczy Z, Wong E, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci. 2010;13:567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Heng MY, Duong DK, Albin RL, et al. Early autophagic response in a novel knock-in model of Huntington disease. Hum Mol Genet. 2010;19:3702–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Franco-Iborra S, Vila M, Perier C. Mitochondrial quality control in neurodegenerative diseases: focus on parkinson’s disease and Huntington’s disease. Front Neurosci. 2018;12:342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Johri A, Chandra A, Flint Beal M. PGC-1α, mitochondrial dysfunction, and Huntington’s disease. Free Radic Biol Med. 2013;62:37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Guedes-Dias P, Pinho BR, Soares TR, et al. Mitochondrial dynamics and quality control in Huntington’s disease. Neurobiol Dis. 2016;90:51–57 [DOI] [PubMed] [Google Scholar]
- [26].Damiano M, Diguet E, Malgorn C, et al. A role of mitochondrial complex II defects in genetic models of Huntington’s disease expressing N-terminal fragments of mutant huntingtin. Hum Mol Genet. 2013;22:3869–3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gu M, Gash MT, Mann VM, et al. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann Neurol. 1996;39:385–389. [DOI] [PubMed] [Google Scholar]
- [28].Guo X, Sun X, Hu D, et al. VCP recruitment to mitochondria causes mitophagy impairment and neurodegeneration in models of Huntington’s disease. Nat Commun. 2016;7:12646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Panov AV, Gutekunst C-A, Leavitt BR, et al. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5:731–736. [DOI] [PubMed] [Google Scholar]
- [30].Siddiqui A, Rivera-Sánchez S, Castro MDR, et al. Mitochondrial DNA damage is associated with reduced mitochondrial bioenergetics in Huntington’s disease. Free Radic Biol Med. 2012;53:1478–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Pal A, Severin F, Lommer B, et al. Huntingtin-HAP40 complex is a novel Rab5 effector that regulates early endosome motility and is up-regulated in Huntington’s disease. J Cell Biol. 2006;172:605–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Caviston JP, Zajac AL, Tokito M, et al. Huntingtin coordinates the dynein-mediated dynamic positioning of endosomes and lysosomes. Mol Biol Cell. 2011;22:478–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Koga H, Martinez-Vicente M, Arias E, et al. Constitutive upregulation of chaperone-mediated autophagy in Huntington’s disease. J Neurosci. 2011;31:18492–18505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Costa V, Scorrano L. Shaping the role of mitochondria in the pathogenesis of Huntington’s disease. Embo J. 2012;31:1853–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gouarné C, Tardif G, Tracz J, et al. Early deficits in glycolysis are specific to striatal neurons from a rat model of huntington disease. PLoS One. 2013;8:e81528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Guedes-Dias P, de Proença J, Soares TR, et al. HDAC6 inhibition induces mitochondrial fusion, autophagic flux and reduces diffuse mutant huntingtin in striatal neurons. Biochim Biophys Acta. 2015;1852:2484–2493. [DOI] [PubMed] [Google Scholar]
- [37].Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ganley IG, Lam DH, Wang J, et al. ULK1·ATG13·FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284:12297–12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Jung CH, Jun CB, Ro S-H, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hosokawa N, Hara T, Kaizuka T, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lee JW, Park S, Takahashi Y, et al. The association of AMPK with ULK1 regulates autophagy. PLoS One. 2010;5:e15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yang H, Zhong X, Ballar P, et al. Ubiquitin ligase Hrd1 enhances the degradation and suppresses the toxicity of polyglutamine-expanded huntingtin. Exp Cell Res. 2007;313:538–550. [DOI] [PubMed] [Google Scholar]
- [45].Matsunaga K, Morita E, Saitoh T, et al. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol. 2010;190:511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ashkenazi A, Bento CF, Ricketts T, et al. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature. 2017;48:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Narendra DP, Jin SM, Tanaka A, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Vives-Bauza C, Zhou C, Huang Y, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A. 2010;107:378–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kazlauskaite A, Martínez-torres RJ, Wilkie S, et al. Binding to serine 65-phosphorylated ubiquitin primes Parkin for optimal PINK 1 -dependent phosphorylation and activation. EMBO Rep. 2015;16:939–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Koyano F, Okatsu K, Kosako H, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–166. [DOI] [PubMed] [Google Scholar]
- [51].Cai Q, Zakaria HM, Simone A, et al. Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr Biol. 2012;22:545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Rakovic A, Shurkewitsch K, Seibler P, et al. Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J Biol Chem. 2013;288:2223–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Sterky FH, Lee S, Wibom R, et al. Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc Natl Acad Sci U S A. 2011;108:12937–12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Van Laar VS, Arnold B, Cassady SJ, et al. Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Hum Mol Genet. 2011;20:927–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Szargel R, Shani V, Abd Elghani F, et al. The PINK1, synphilin-1 and SIAH-1 complex constitutes a novel mitophagy pathway. Hum Mol Genet. 2016;25(16):3476–3490. [DOI] [PubMed] [Google Scholar]
- [56].Villa E, Marchetti S, Ricci JE. No Parkin zone: mitophagy without Parkin. Trends Cell Biol. 2018;S0962-8924:30125–30129. [DOI] [PubMed] [Google Scholar]
- [57].Khalil B, El Fissi N, Aouane A, et al. PINK1-induced mitophagy promotes neuroprotection in Huntington’s disease. Cell Death Dis. 2015;6:e1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ashrafi G, Schwarz TL. PINK1- and PARK2-mediated local mitophagy in distal neuronal axons. Autophagy. 2015;11:187–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Joselin AP, Hewitt SJ, Callaghan SM, et al. ROS-dependent regulation of parkin and DJ-1 localization during oxidative stress in neurons. Hum Mol Genet. 2012;21:4888–4903. [DOI] [PubMed] [Google Scholar]
- [60].Chu CT. Multiple pathways for mitophagy: A neurodegenerative conundrum for Parkinson’s disease. Neurosci Lett. 2019;697:66–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Chu CT. Mechanisms of selective autophagy and mitophagy: implications for neurodegenerative diseases. Neurobiol Dis. 2019;122:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Yoshii SR, Mizushima N. Autophagy machinery in the context of mammalian mitophagy. Biochim Biophys Acta. 2015;1853:2797–2801. [DOI] [PubMed] [Google Scholar]
- [63].Padman BS, Bach M, Lucarelli G, et al. The protonophore CCCP interferes with lysosomal degradation of autophagic cargo in yeast and mammalian cells. Autophagy. 2013;9:1862–1875. [DOI] [PubMed] [Google Scholar]
- [64].Chu CT, Ji J, Dagda RK, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15:1197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Sun N, Yun J, Liu J, et al. Measuring in vivo mitophagy. Mol Cell. 2015;60:685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Shi RY, Zhu SH, Li V, et al. BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci Ther. 2014;20:1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Scarffe LA, Stevens DA, Dawson VL, et al. Parkin and PINK1: much more than mitophagy. Trends Neurosci. 2014;37:315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Oliveira JM, Jekabsons MB, Chen S, et al. Mitochondrial dysfunction in Huntington’s disease: the bioenergetics of isolated and in situ mitochondria from transgenic mice. J Neurochem. 2007;101:241–249. [DOI] [PubMed] [Google Scholar]
- [69].Ferreira IL, Cunha-Oliveira T, Nascimento MV, et al. Bioenergetic dysfunction in Huntington’s disease human cybrids. Exp Neurol. 2011;231:127–134. [DOI] [PubMed] [Google Scholar]
- [70].Oláh J, Klivényi P, Gardián G, et al. Increased glucose metabolism and ATP level in brain tissue of Huntington’s disease transgenic mice. Febs J. 2008;275:4740–4755. [DOI] [PubMed] [Google Scholar]
- [71].Jin YN, Hwang WY, Jo C, et al. Metabolic state determines sensitivity to cellular stress in Huntington disease: normalization by activation of PPARγ. PLoS One. 2012;7(1):e30406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Browne SE, Bowling AC, Macgarvey U, et al. Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann Neurol. 1997;41:646–653. [DOI] [PubMed] [Google Scholar]
- [73].Perluigi M, Poon HF, Maragos W, et al. Proteomic analysis of protein expression and oxidative modification in R6/2 transgenic mice: a model of Huntington disease. Mol Cell Proteomics. 2005;4:1849–1861. [DOI] [PubMed] [Google Scholar]
- [74].Sorolla MA, Rodríguez-Colman MJ, Tamarit J, et al. Protein oxidation in Huntington disease affects energy production and vitamin B6 metabolism. Free Radic Biol Med. 2010;49:612–621. [DOI] [PubMed] [Google Scholar]
- [75].Tabrizi SJ, Cleeter MWJ, Xuereb J, et al. Biochemical abnormalities and excitotoxicity in Huntington’s disease brain. Ann Neurol. 1999;45:25–32. [DOI] [PubMed] [Google Scholar]
- [76].Grafton ST, Mazziotta JC, Pahl JJ, et al. Serial changes of cerebral glucose metabolism and caudate size in persons at risk for Huntington’s disease. Arch Neurol. 1992;49:1161–1167. [DOI] [PubMed] [Google Scholar]
- [77].Jenkins BG, Koroshetz WJ, Beal MF, et al. Evidence for impairment of energy metabolism in vivo in Huntington’s disease using localized 1H NMR spectroscopy. Neurology. 1993;43:2689–2695. [DOI] [PubMed] [Google Scholar]
- [78].Brennan WA, Bird ED, Aprille JR. Regional mitochondrial respiratory activity in Huntington’s disease brain. J Neurochem. 1985;44:1948–1950. [DOI] [PubMed] [Google Scholar]
- [79].Mochel F, Durant B, Meng X, et al. Early alterations of brain cellular energy homeostasis in huntington disease models. J Biol Chem. 2012;287:1361–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Naia L, Ferreira IL, Cunha-Oliveira T, et al. Activation of IGF-1 and insulin signaling pathways ameliorate mitochondrial function and energy metabolism in huntington’s disease human lymphoblasts. Mol Neurobiol. 2014;51:331–348. [DOI] [PubMed] [Google Scholar]
- [81].Stack EC, Matson WR, Ferrante RJ. Evidence of oxidant damage in Huntington’s disease: translational strategies using antioxidants. Ann N Y Acad Sci. 2008;1147:79–92. [DOI] [PubMed] [Google Scholar]
- [82].Kim J, Moody JP, Edgerly CK, et al. Mitochondrial loss, dysfunction and altered dynamics in Huntington’s disease. Hum Mol Genet. 2010;19:3919–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Shirendeb U, Reddy AP, Manczak M, et al. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Hum Mol Genet. 2011;20:1438–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Shirendeb UP, Calkins MJ, Manczak M, et al. Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum Mol Genet. 2012;21:406–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Cui L, Jeong H, Borovecki F, et al. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. [DOI] [PubMed] [Google Scholar]
- [86].Dickey AS, Pineda VV, Tsunemi T, et al. PPAR-δ is repressed in Huntington’s disease, is required for normal neuronal function and can be targeted therapeutically. Nat Med. 2016;22:37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Weydt P, Pineda VV, Torrence AE, et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metab. 2006;4:349–362. [DOI] [PubMed] [Google Scholar]
- [88].Chang DTW, Rintoul GL, Pandipati S, et al. Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol Dis. 2006;22:388–400. [DOI] [PubMed] [Google Scholar]
- [89].Gunawardena S, Her L-S, Brusch RG, et al. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40:25–40. [DOI] [PubMed] [Google Scholar]
- [90].Orr AL, Li S, Wang CE, et al. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J Neurosci. 2008;28:2783–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Song W, Chen J, Petrilli A, et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat Med. 2011;17:377–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured filroblasts. Nat Protoc. 2007;2:287–295. [DOI] [PubMed] [Google Scholar]
- [93].Mauro-Lizcano M, Esteban-Martínez L, Seco E, et al. New method to assess mitophagy flux by flow cytometry. Autophagy. 2015;11:833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.