

Abstract
Prion diseases are progressive neurodegenerative disorders affecting humans and other mammalian species. The term prion, originally put forward to propose the concept that a protein could be infectious, refers to PrPSc, a misfolded isoform of the cellular prion protein (PrPC) that represents the pathogenetic hallmark of these disorders. The discovery that other proteins characterized by misfolding and seeded aggregation can spread from cell to cell, similarly to PrPSc, has increased interest in prion diseases. Among neurodegenerative disorders, however, prion diseases distinguish themselves for the broader phenotypic spectrum, the fastest disease progression and the existence of infectious forms that can be transmitted through the exposure to diseased tissues via ingestion, injection or transplantation. The main clinicopathological phenotypes of human prion disease include Creutzfeldt–Jakob disease, by far the most common, fatal insomnia, variably protease‐sensitive prionopathy, and Gerstmann–Sträussler–Scheinker disease. However, clinicopathological manifestations extend even beyond those predicted by this classification. Because of their transmissibility, the phenotypic diversity of prion diseases can also be propagated into syngenic hosts as prion strains with distinct characteristics, such as incubation period, pattern of PrPSc distribution and regional severity of histopathological changes in the brain. Increasing evidence indicates that different PrPSc conformers, forming distinct ordered aggregates, encipher the phenotypic variants related to prion strains. In this review, we summarize the most recent advances concerning the histo‐molecular pathology of human prion disease focusing on the phenotypic spectrum of the disease including co‐pathologies, the characterization of prion strains by experimental transmission and their correlation with the physicochemical properties of PrPSc aggregates.
Keywords: amyloidosis, Creutzfeldt–Jakob disease, fatal insomnia, Gerstmann–Sträussler–Scheinker disease, human prions, neurodegenerative dementia, prion strains
Introduction
Prion diseases are neurodegenerative disorders of humans and other mammals, characterized by tissue deposition of a structurally modified isoform of the cellular prion protein (PrPC). In prion disease, PrPC converts into a misfolded isoform commonly identified as scrapie prion protein (PrPSc), which is enriched in β‐sheet secondary structure and prone to form aggregates that are partially resistant to protease digestion 137. PrPSc is generated by a seeded‐conversion mechanism where PrPSc binds PrPC and mediates its conversion to PrPSc 31. Newly converted PrPSc then propagates and accumulates preferentially, but not exclusively, in the central nervous system (CNS). Notably, a similar prion‐like seeded‐polymerization mechanism appears to be responsible for the formation of protein aggregates involving α‐synuclein, amyloid‐beta (Aβ), and tau in Parkinson’s disease, Alzheimer’s disease (AD) and primary tauopathies 173. These findings have increased the interest in prion disease, and there is hope that there might be a shared benefit from future therapies between prion and prion‐like diseases.
Among protein misfolding neurodegenerative disorders, prion diseases manifest the broader phenotypic spectrum and the fastest disease progression. Moreover, to date, they uniquely include disease subtypes proven to be transmissible between humans by exposure to affected tissues via ingestion, injection or transplantation.
The experimental transmission of animal and human prions led to the demonstration of prion strains, a term which has been borrowed from microbiology to underline the similarities between viral and prion strains. These are defined as animal or human “isolates” that, after inoculation into syngenic hosts, cause diseases with distinct characteristics, such as the incubation period, the pattern of PrPSc deposition and the regional severity of neuropathological changes 2, 22. The existence of prion strains has been difficult to accommodate within a protein‐only model of prion propagation, and the understanding of how a protein‐only infectious agent can encode distinct disease phenotypes remains of considerable scientific interest. A wealth of experimental evidence now suggests that prion strain diversity is enciphered within PrP itself and that phenotypic heterogeneity in human prion disease relates to differing physicochemical properties of PrPSc isoforms [15, 116, 120, 163, for additional studies, see 126].
Human prion disease comprises several major phenotypes, namely Creutzfeldt–Jakob disease (CJD), fatal insomnia (FI), variably protease‐sensitive prionopathy (VPSPr) and the inherited PrP‐amyloidoses [ie, Gerstmann–Sträussler–Scheinker disease (GSS), PrP‐cerebral amyloid angiopathy (PrP‐CAA) and PrP‐systemic amyloidosis (PrP‐SA)].
While CJD and FI present as rapidly progressive neurological syndromes, the inherited PrP‐amyloidoses manifest a slower clinical course and are mainly characterized by amyloid deposition rather than spongiform change in the affected brain tissue 48. Interestingly, this major difference in the distinctive histopathology strongly correlates with the composition and topology of PrPSc aggregates. Indeed, while in CJD and FI they are formed by PrP27‐30, namely full‐length PrPSc generating proteinase K(PK)‐resistant N‐terminally truncated fragments retaining the glycophosphatidylinositol (GPI) anchor and the attachment to the cell membrane, in the PrP‐amyloidoses the pathological protein aggregates mainly deposit in the extracellular space as unglycosylated anchorless fragments 118, 131.
In humans, prion diseases are also classified into sporadic (or idiopathic), genetic and acquired forms based on the presumed etiology (Figure 1).
Figure 1.
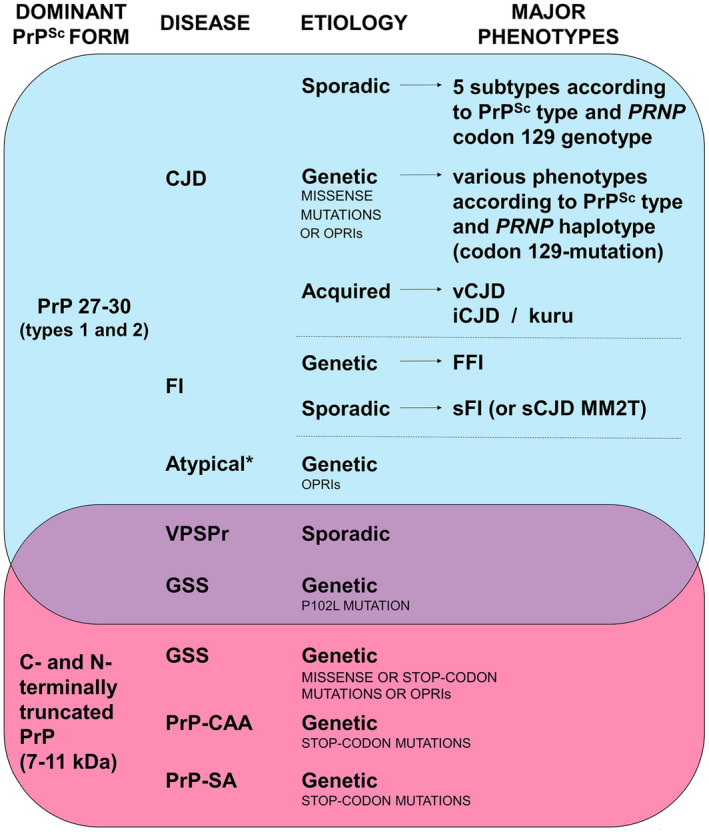
Classification of human prion diseases according to the dominant PrPSc form, etiology and phenotypic features. *Atypical refers to disease lacking distinctive histopathological features. List of abbreviations: CJD, Creutzfeldt–Jakob disease; vCJD, variant CJD; iCJD, iatrogenic CJD; FI, fatal insomnia; FFI, fatal familial insomnia; sFI, sporadic FI; VPSPr, variably protease‐sensitive prionopathy; GSS, Gerstmann–Sträussler–Scheinker disease; PrP‐CAA, PrP‐cerebral amyloid angiopathy; PrP‐SA, PrP‐systemic amyloidosis; OPRIs, octapeptide repeat insertions.
In this review, we provide an update on current knowledge on human prion disease focusing on the most recent studies addressing the pathological spectrum of disease, the isolation and characterization of strains by experimental transmissions and the molecular basis of strains. Recent studies exploring the prevalence and significance of the co‐occurrence of CJD and prion‐like disorders are also reviewed.
Phenotypic Spectrum and Classification of Disease Subtypes
Creutzfeldt–Jakob disease and fatal insomnia
Sporadic form
Sporadic CJD (sCJD), by far the most common human prion disease, is characterized by a widespread brain deposition of PrPSc leading to spongiform change, microglial activation, synaptic and neuronal loss, and astrocytic gliosis of variable severity and regional distribution. Moreover, PrP‐amyloid plaques occur in about 10% of cases.
Current sCJD classification recognizes six major clinicopathological phenotypes that largely correlate at the molecular level with the genotype (methionine, M or valine, V) at the polymorphic codon 129 of PRNP and two PrPSc forms (types 1 and 2) with a distinctive size (21 and 19 kDa) of their PK‐resistant core 120, 123, 126. Specifically, each phenotypic variant or “subtype” results from a specific codon 129 genotype/PrPSc type combination, with only two exceptions (Table 1 and Figure 2). The MM(V)1 subtype includes both MM1 and MV1 cases given that they are phenotypically indistinguishable, whereas the MM2 group comprises two subtypes with distinctive histopathological features and topographical distribution of lesions mainly affecting the cerebral cortex (MM2‐Cortical or MM2C) or the thalamus (MM2‐Thalamic or MM2T). The latter subtype is also known as the sporadic form of FI 1. It is also noteworthy that about 35% of sCJD cases show the co‐occurrence of PrPSc types 1 and 2, an intriguing and still unexplained phenomenon that is more often observed in subjects carrying MM at codon 129 than in the other genotypes 123. In these “mixed” subtypes, the pathological features are the same described for the “pure” subtypes but with a predominance of the traits reflecting the most prevalent PrPSc type accumulating in the brain 123.
Table 1.
Distinctive pathological features of sCJD subtypes.
| Disease subtype | Mean duration, months | Regional pattern of distribution | Distinctive histopathological features | |
|---|---|---|---|---|
| Frequent (overall about 90% of all sCJD cases) | ||||
| 65% | MM(V)1 (typical CJD) | 4 | Neocortex (especially in occipital lobe), striatum, thalamus and cerebellum constantly affected | Small vacuoles and “synaptic type” PrP staining |
| 15%–20% | VV2 (ataxic/cerebellar variant) | 6.5 | Prominent subcortical and cerebellar pathology, late cortical involvement (occipital lobe least affected) | Laminar intracortical distribution of spongiform change (ie, predominant involvement of deep layers), plaque‐like and perineuronal PrP staining |
| 10% | MV2K (kuru‐plaque variant) | 17 | Similar to VV2 with more consistent involvement of the cerebral cortex | Amyloid‐kuru plaques in the cerebellum, and consistent plaque‐like and focal PrP deposits |
| Rare (overall about 10% of all sCJD cases) | ||||
| MM2T (thalamic variant or sFI) | 26 | Atrophy of the thalamus and inferior olive, patchy spongiosis limited to the cerebral cortex and limbic regions | ||
| MM2C (cortical variant) | 16 | Spongiosis in all cortices with sparing of the cerebellum | Large confluent vacuoles and coarse/perivacuolar PrP staining | |
| VV1 (cortico‐striatal variant) | 21 | Severe pathology in the cerebral cortex and striatum with sparing of the brainstem nuclei and cerebellum | Intermediate vacuoles | |
| p‐MM1 | 22 | Distribution profile MM(V)1‐like | Widespread PrP‐amyloid plaques in subcortical and deep nuclei white matter | |
Figure 2.
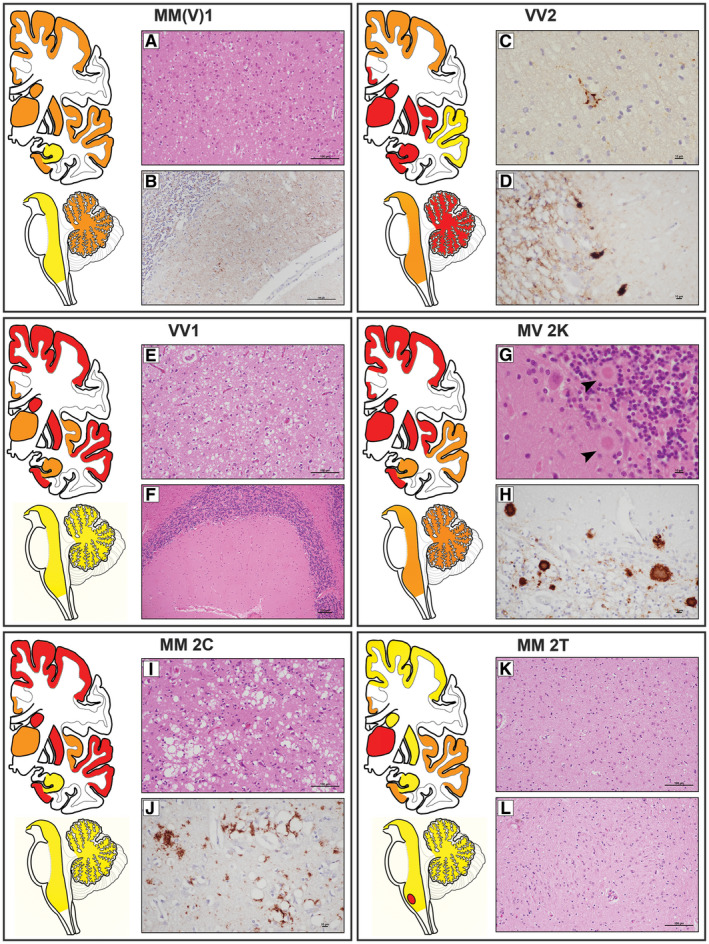
Lesion profiles and histopathological hallmarks of sporadic Creutzfeldt–Jakob subtypes. Analyzed brain regions have been highlighted in yellow, orange and red to indicate, respectively, “minimal to mild” (score ≤1), “mild to moderate” (score >1–2) and “moderate to severe” (score >2) neuropathological changes. Lesion profiles were obtained by averaging the pathological score of spongiosis (not detectable = 0, mild = 1, moderate = 2, severe = 3, status spongiosus = 4), astrogliosis and neuronal loss (not detectable = 0, mild = 1, moderate = 2, severe = 3). Lesion profile data are from “typical” cases (eg, with a disease duration representative of the subtype). Distinctive histopathological features of sCJD subtypes. A. Spongiform change characterized by small, fine, microvacuoles in the neuropil (H&E, ×200, temporal cortex), and (B) synaptic pattern of PrPSc deposition in the molecular and granular layers of cerebellum in sCJDMM(V)1 (IHC, ×400). PrP staining is punctate and diffuse in the molecular layer, while it stands out the cerebellar glomeruli in the granular cell layers. C. Perineuronal PrP staining in deep layers of frontal cortex (IHC, x600) and (D) plaque‐like PrPSc deposits at transition between the molecular and the granular cell layers of cerebellum in sCJDVV2 (IHC, ×400). E. Spongiform change characterized by non‐confluent vacuoles of “intermediate” size, relatively larger than those observed in sCJDMM(V)1, but overall smaller than those of sCJDMM2C, in the occipital cortex (H&E, ×200), and (F) minimal/mild pathological changes in the cerebellum of sCJDVV1 (H&E, ×100). G, H. Unicentric kuru amyloid plaques at transition between the molecular and the granular cell layers of cerebellum in sCJDMV2K (G, H&E, ×600; H, IHC, ×400). I. Spongiform change characterized by large, confluent vacuoles (H&E, ×200) and (J) coarse, dense PrPSc deposits with perivacuolar distribution in the temporal cortex of sCJDMM2C (IHC, 400×). K, L. Severe neuronal loss and reactive gliosis in the absence of significant spongiform change in medial‐dorsal thalamic nucleus (K) and inferior olive (L) of sCJDMM2T (H&E, ×200). Legend: H&E, hematoxylin and eosin stain; IHC, PrP immunochemistry with primary mAb 3F4.
Relatively few studies have contributed new knowledge about the phenotypic spectrum of sporadic CJD and FI in recent years. There has been a further characterization of an atypical sCJD variant showing kuru‐like PrP‐amyloid plaques limited to the white matter, but otherwise highly reminiscent of the MM(V)1 subtype, that was initially recognized in Japan 74, 146. In these cases, identified as p‐MM1, amyloid plaques show a widespread distribution involving the subcortical, deep nuclei and cerebellar white matter. Despite the relatively long disease duration (ie, mean 22 months) and the severe associated pathology, which notoriously favor the extent of plaque formation, evidence indicates that the amyloid plaques in this phenotype develop early in the course of the disease and independently from the severe white matter damage 74, 146. Moreover, in the cases with most numerous plaques, PrP immunostaining reveals focal coarse and tract‐like deposits in gray matter areas delimiting the white matter in the striatum and thalamus 146.
Two recent studies reported atypical neuropathological features, in association with an exceedingly long disease duration, in sCJD MM2C and MM2T. A patient with an overall disease duration of 44 months classified as sCJDMM2C+1 showed PAS‐positive, unicentric, amyloid plaques associated with a plaque‐like pattern of PrPSc deposition in the molecular layer of the cerebellum 12. This observation combined with the notion that the cerebellum is significantly spared in sCJDMM2C strongly suggests that, similarly to Aβ in AD, PrPSc spreads to the cerebellum only late in the disease course in sCJDMM2C; however, when present in a sufficient concentration, PrPSc would ultimately foster the formation of amyloid plaques at this site.
Similarly, in a recent series of European sCJDMM2T, the two subjects with the longest disease duration (54 and 96 months) showed severe spongiform change in the striatum, which is usually relatively spared in this subtype, and one of them also demonstrated an atypical pattern of PrP staining characterized by sparse, small plaque‐like deposits in the cerebral cortex 1. Thus, in long‐lasting sCJDMM2T cases, spongiform change may not be limited to the cerebral cortex and PrPSc aggregates may form small plaques in addition to the synaptic/fine granular pattern.
Neuropathologists involved in prion disease surveillance tend to agree on the existence of rare sCJD cases, which do not fit any of the subtypes established by the current consensus classification 125. Such cases are generally referred to as “atypical” sCJD. Despite the scientific interest, no comprehensive study has been carried out to date on atypical sCJD. Over the years, we have encountered a few cases with atypical histopathology associated with a distinctive immunoblot PrPSc profile characterized by the lack of the diglycosylated band and a type 1‐like migration pattern (P. Parchi personal communication). Recently, the clinicopathological features and transmission properties of a patient with an analog form of PrPSc have been reported 44, 180. Given that PrPC showed all three glycoforms, the lack of diglycosylated PrPSc in this case likely represents a strain‐specific property. The results of a transmission study in mice and bank voles supported this interpretation 44. PrP immunohistochemistry was also distinctive, revealing intraneuronal PrPSc deposits.
Genetic form
Genetic CJD (gCJD) accounts for 10%–15% cases of CJD and is linked to point mutations or insertions of octapeptide repeats (OPRI) in PRNP. It is generally assumed that the clinicopathological variability of gCJD largely reproduces that of sCJD since PrPSc types 1 and 2 and the codon 129 genotype are the main determinants of the disease phenotype also in gCJD. Intriguingly, PRNP mutations seem to mainly affect the disease susceptibility rather than the resulting phenotype, although the disease penetrance may vary significantly according to the mutation 28, 101. There are, however, a few significant exceptions to the general rule of the strict parallelism between the phenotypic spectrum of sporadic and genetic CJD, given that a few mutations co‐distribute with a phenotype, which deviates from that expected from the PRNP genotype/PrPSc type combination. As the most significant example, the histotype associated with the combination T183A‐129M/PrPSc type 2, which is characterized by spongiform change lacking large confluent vacuoles and primarily affecting the neocortices and the striatum with sparing of the cerebellum, would be more consistent with sCJDVV1 than with sCJDMM2 (both 2C and 2T subtypes) 51, 109. Moreover, the synaptic and plaque‐like patterns of PrPSc deposition through the neocortices and cerebellum 51, 109 and the microplaques in the molecular layer of cerebellum 51 that are seen in these cases are also inconsistent with the sCJDMM2 subtypes.
Similarly, several neuropathological features that are associated with the V180I‐129M/PrPSc type 2 combination, including the florid spongiform change lacking confluent vacuoles, as well as the faint synaptic type pattern of PrP immunostaining, do not fully match those of the sCJDMM2 subtypes 58, 63, 64.
Finally, gCJD linked to 5‐ and 6‐octapeptide repeat insertional (5‐ and 6‐OPRI) mutations, at variance with sCJD, is often characterized by relatively moderate spongiform change despite the long disease duration (usually >5 years) or even by the lack of distinctive histopathology (ie, no spongiform change and no amyloid PrPSc plaques) 26, 96, 156, 177. Nevertheless, PrPSc type 1 or 2 is detected in all these cases as in sCJD 26, 33, 65, 96, 156.
A detailed description of all phenotypes associated with the known PRNP haplotypes (mutation plus codon 129 in cis with the mutation) is out of the scope of this review and can be found elsewhere 28, 48. Focusing on the most recent contributions, a novel point mutation at codon 200, the most affected codon in humans, resulting in a glutamate‐to‐glycine substitution (E200G) rather than the prevalent E200K, has been described in a patient carrying MV at codon 129 with the mutation in cis with 129V 72. Neuropathological investigations showed spongiform change, a fine granular/synaptic PrP staining with a perivacuolar distribution and rare plaque‐like deposits in the cerebral cortices. Punctate, linear and curvilinear PrPSc deposits were also abundant in the basal ganglia, limbic structures, and brainstem. Western blot analysis revealed PrPSc type 2 with a glycoform ratio characterized by a predominant diglycosylated form and markedly underrepresented unglycosylated species.
A second novel variant, the A224V, also in cis with 129V, was recently reported in a patient from the US 175 showing clinical, neuropathological and molecular findings reminiscent of the sCJDVV1 subtype. However, whether this variant is truly pathogenic or only a risk factor remains controversial given that the patient’s father carried the same mutation but never developed CJD. Transmission properties of the patient’s brain resembled those obtained with sCJDVV1 inocula. Furthermore, transmission studies in mice lines expressing A224V revealed that this variant modulates the kinetics of replication of various prion strains, which supports the idea of A224V being a risk factor influencing the susceptibility to prion disease.
Another novel PRNP haplotype, the V215I‐129M, has been reported in three Spanish patients from two families 108. However, the non‐specific neuropathological findings of AD‐related pathology in all cases and the negative PrP immunostaining in one case showing spongiform change raise significant doubts about the pathogenicity of this mutation.
Although fatal familial insomnia (FFI), the genetic form of FI, belongs, like CJD, to the human prion diseases linked to PrP27‐30 (ie, types 1 and 2), the disease is associated with a peculiar neuropathological phenotype characterized by atrophy of the anteroventral and dorsomedial thalamic nuclei and inferior olivary nuclei in the absence of significant spongiform change 11, 115, 121. Conversely, the cortical histopathological changes include spongiosis, and are usually patchy and of mild to moderate severity, although significantly influenced by the disease duration. Notably, the paucity of spongiform change in FFI correlates with the overall amount of PrPSc, which is significantly lower than in CJD 115, 119. Consistently, except for some small, polymorphic granular deposits in the cerebral cortex and a strip‐like pattern in the molecular layer of the cerebellum 4, PrP immunochemistry is usually negative in most affected regions.
Although the leading pathogenic mechanisms in FFI are still unclear, recent findings pointed to dysfunction of several subunits of mitochondrial respiratory chain and alteration of protein synthesis machinery, being the latter strongly correlated with neuronal loss in the most affected regions 43. These findings are in line with transcriptomic studies showing a severe downregulation of genes linked to protein biogenesis and mitochondrial associated‐processes 43, 164.
Iatrogenic form
Iatrogenic CJD (iCJD) results from human‐to‐human transmission of CJD prions through medical procedures 21. The principal sources of infectious prions are contaminated dura mater graft (d‐iCJD) and human‐derived pituitary growth hormone (hGH‐iCJD) extracted from tissues obtained post‐mortem. Others include neurosurgical instruments, gonadotropin infusion, corneal graft and, finally, blood transfusion from patients with variant CJD (vCJD).
Although, as in sCJD, the pathological phenotype of iCJD largely depends on the PRNP codon 129 genotype and the PrPSc type, the phenotypic spectrum of iCJD does not fully reproduce that of the sporadic form. Specifically, to date, the MM2C, MM2T and VV1 subtypes have never been found among proven iCJD cases. Moreover, a “plaque‐type” phenotype linked to MM at codon 129 has been consistently observed in iCJD but not in sCJD 61, 78, 86, 103, 155, 178. Specifically, iCJD patients carrying MM at codon 129 may show two distinct phenotypes. While the most prevalent “non‐plaque‐type” reproduces the typical sCJDMM(V)1 and is linked to PrPSc type 1, the “plaque‐type” shows a widespread occurrence of kuru plaques and plaque‐like deposits, and is associated with a PrPSc fragment with a molecular mass intermediate between types 1 and 2 [type “i” (intermediate), 20 kDa, hence the name iCJD MMiK] (Figure 3) 73. Other distinctive pathological characteristics of iCJD MMiK include the occurrence of florid plaques in some cases 24, 86 and the presence of “stellate” cells highlighted by the pericellular deposition of PrPSc 24. Iatrogenic CJD MMiK has been extensively investigated in Japan in cases secondary to dura mater graft, but also recognized among hGH‐iCJD cases from the US and Europe 24, 25, 143. The atypical phenotypic features of iCJD MMiK are thought to depend on an incomplete adaptation of the sCJDV2 strain to the codon 129 MM genotype [77, 78, 161, see also the paragraph below on transmission studies]. Notably, because of the distinctive pathological and biochemical characteristics of iCJD MMiK and the absence of a similar histotype in sCJD, the combination of 129 MM genotype, kuru plaques and PrPSc type “i,” currently represents a reliable criterion for the identification of cases with iCJD even in the absence of a proven history of prion contamination 81.
Figure 3.
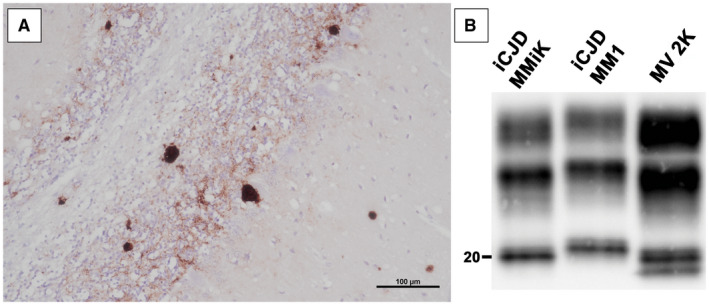
Histological features and western blot profile of iCJD MMiK. A. Unicentric kuru amyloid plaques in the molecular and granular cell layers, and in the white matter of cerebellum of iCJD MMiK (immunochemistry with primary Ab 3F4, ×200). B. Electrophoretic mobility of PK‐resistant PrPSc fragments in subtype iCJD MMiK compared with sCJDMM1 and sCJD MV2K. The molecular weight of the lower band (unglycosylated fragment) is ~20 kDa (PrPSc type “i”) in the iCJD MMiK case (courtesy from Prof. T. Kitamoto). Immunoblot was probed with the primary mAb 3F4.
Variant CJD
Variant CJD is an acquired, zoonotic prion disease caused by the oral exposure to food contaminated with material from bovine spongiform encephalopathy (BSE)‐affected cattle. Since 1995, the year of the first identification of the disease in the UK 176, vCJD has affected approximately 230 individuals, mostly in the UK. However, the disease has become exceedingly rare in recent years, following the decline of BSE in cattle 89. Interestingly, while for about two decades all definite vCJD cases occurred in subjects with MM at codon 129, the disease has also been recently found in a patient carrying 129 MV 105. Given the known influence of codon 129 on prion strain susceptibility, which ultimately determines the incubation time of disease in the acquired forms, the observation raised some concern about the possibility of a second wave of vCJD in subjects carrying a distinct PRNP genotype (ie, MV or even VV at codon 129) that would be associated with a longer incubation time. Pathological hallmarks of vCJD, which have also been confirmed in the case carrying MV at codon 129, include the presence of widespread and numerous florid plaques, characterized by a central eosinophilic amyloid core and a pale periphery of radiant fibrils surrounded by a corona of vacuoles, and PrPSc accumulation as small cluster plaques. PrPSc also deposits with a synaptic and perineuronal pattern in subcortical structures, mainly in the thalamus, striatum and brainstem. Spongiform change has a patchy distribution involving all cortical layers, assuming a confluent aspect in the molecular layer of the cerebellum and the striatum. The posterior thalamus shows predominant gliosis and severe neuronal loss rather than spongiform change. Hippocampus and brainstem are relatively spared. Given the “peripheral” pathogenesis and the lymphotropism of the BSE strain, PrPSc is also present in relatively high amount (ie, in comparison to classic CJD) outside the CNS, especially in the lymphoreticular tissues, allowing its detection by high sensitive western blot technique in tonsils, spleen and lymph nodes, and in low concentration in the adrenal gland, thymus and rectum 170.
PrPSc typing by western blotting showed, in both 129 MM and MV patients, a type 2B profile characterized by the predominance of the diglycosylated band of the protein 35, 117. The same biochemical signature characterized vCJD patients from different countries (UK, France) 19, which is consistent with the same strain of agent being involved in all cases.
Initial evidence, based on both PrPSc analyses 179 and transmission data 5, suggested a strain‐related biological heterogeneity of vCJD prions. However, the claim has not been confirmed in recent years. At variance, further analysis of PrPSc aggregates extracted from vCJD brains demonstrated homogeneous biochemical properties, consistent with a single strain and differing from those of the whole spectrum of sCJD prions 32, 112, 113, 152.
Secondary pathology in CJD and FI: tauopathy and microgliosis
Besides the primary tauopathies, in which the deposition of abnormal tau protein in the brain constitutes the dominant and often only pathology, several other neurodegenerative conditions with diverse etiologies demonstrate tau pathology. These conditions are known as secondary tauopathies, as other proteins play a central role in their pathogenesis. While GSS has been classified among secondary tauopathies for a long time 48, recently, a secondary tauopathy unrelated to AD pathology has also been documented in sCJD 85, 87, 140. Most frequently, tau deposits in CJD appear as either tiny rod‐ or stub‐like punctate inclusions in the neuropil or as thin neuritic threads 85, 87, 140. Notably, their morphology is also influenced by the pattern of PrPSc deposition, which ultimately depends on the disease subtype 87, 140. According to one study, the deposits of hyperphosphorylated tau (p‐tau) positively correlated with both the burden and the morphology of PrPSc aggregates 140. Intense PrPSc deposition and formation of small plaques elicited a stronger tau deposition, while perineuronal PrPSc aggregates were associated with a moderate density of tau particles. Furthermore, while synaptic PrPSc deposits correlated with tiny tau granules showing a homogeneous distribution, coarse granular PrPSc co‐localized with larger and well discernible tau granules 140. Notably, tau deposition in sCJD was not limited to the hippocampus and neocortical structures, but also involved the granular, molecular and Purkinje cell layers of the cerebellum 140. Although the prion‐specific secondary tauopathy may occur in all sCJD subtypes, it is especially seen in those linked to the V2 strain (ie, VV2 and MV2K subtypes) 87, in which PrPSc typically accumulates as kuru‐plaques and plaque‐like deposits. These findings and the relatively high prevalence of a secondary tauopathy in GSS and other PrP‐amyloidosis suggest a link between structural properties of PrPSc aggregates and tau hyperphosphorylation/deposition. In agreement with this conclusion, a large number of p‐tau immunoreactive neuritic profiles, often clustered around PrP‐amyloid deposits, have also been observed in vCJD in both the cerebral cortex and the cerebellum, in the absence of Aβ 49. In addition to sporadic and variant CJD, tau pathology has been observed in some phenotypes of gCJD, sometimes with atypical distribution and features [eg, neurofibrillary tangles occurring in brainstem nuclei of young patients carrying E200K 83, and granular p‐tau deposits in the medulla oblongata and pons in gCJD linked to the R208H substitution 93, 145]. Finally, tau deposition has also been described in the thalamus of a single case of FFI 67.
Inflammation is thought to be a major component of neurodegenerative diseases, although the precise role played by microglia, the key contributor to “neuroinflammation,” has yet to be defined. Experimental evidence supports the idea of a dual role of microglia in prion disease pathogenesis. On one hand, especially in the early disease stages, microglia would increase the phagocytosis of PrPSc aggregates, and therefore, have a protective role. On the other hand, during disease progression, both the accumulation of PrPSc and the neuronal damage would concur to switch microglia to a proinflammatory phenotype contributing to brain toxicity. This dual role of microglia appears to be regulated by multiple pathways, which polarize these cells into distinct phenotypes with characteristic functions 3. To date, studies investigating the microglial response in human prion disease support its role in disease pathogenesis. Microglial cell activation and proliferation occur early in CJD brains 42, 151, 157 where the microgliosis appears to be closely associated with PrPSc deposits 42, 52, 102, 107. Interestingly, besides the stage/severity of the disease, the molecular subtype significantly influences the degree of microglial reactivity 42, 154, 166. Other studies demonstrating that microglia markers, pro‐ and anti‐inflammatory cytokines, members of the complement system, regulatory proteins involved in microglial proliferation and key regulators of inflammation COX‐1/2 and PGE2, are all elevated in CJD brains 38, 50, 88 further speak for a crucial role of neuroinflammation in human prion disease.
Variably protease‐sensitive prionopathy
VPSPr is a recently recognized sporadic prion disease that owes its name to the peculiar properties of PrPSc aggregates 182. The estimated prevalence in the US, where most of ~40 cases reported to date worldwide have been recognized, is approximately 1%–2% of all prion diseases. Although VPSPr may affect subjects harboring each of the three PRNP codon 129 genotypes, it is most frequently linked to valine homozygosity 10, 45, 55, 56, 182. The clinical phenotype is more often consistent with an AD‐type or frontotemporal‐type of dementia, sometimes associated with atypical parkinsonism. However, atypical presentations may also occur as in two recently reported patients with autopsy‐confirmed VPSPr who received a clinical diagnosis of amyotrophic lateral sclerosis 167. Spongiform change is characterized by non‐confluent vacuoles of intermediate size [ie, larger than in sCJDMM(V)1, but smaller than in sCJDMM2C] and the lesion profile by a predominant involvement of cerebral cortices, striatum and thalamus, while the cerebellum is variably affected and the hippocampus relatively spared. PrP immunostaining reveals microplaques in the molecular layer of cerebellum and clusters of granular deposits often arranged in a target‐like pattern. Western blot analysis of PrPSc shows a ladder‐like profile of at least five mildly PK‐resistant fragments migrating in the 7–26 kDa range 182. The lack or minimal representation of diglycosylated PrPSc is another distinctive feature of VPSPr.
As per the disease name, both PrPSc amount and resistance to PK digestion are lower in VPSPr than in typical sCJDMM(V)1 152. However, PK‐resistance is also significantly influenced by the PRNP codon 129 genotype, being lowest in VPSPr‐129VV, highest in 129MM, and intermediate in 129MV. A recent study documented a regional variability of PrPSc properties in VPSPr involving both PK‐resistance and the presence of a CJD‐like fully glycosylated (ie, comprising the diglycosylated form) PrPSc form of 19 kDa 128. Further biochemical similarities with CJD were revealed by in vitro seeding assays, such as protein misfolding cyclic amplification (PMCA) and real time‐quaking induced conversion (RT‐QuIC), which showed striking similarities in the profile of newly converted PrPSc induced by both sCJDVV2 and VPSPr. Indeed, in VPSPr the converted PrPSc appears mainly determined by the 19 kDa CJD‐like fragment rather than by the GSS‐like ~7 kDa fragment 128.
Given the atypical clinical phenotype limiting the chance of a clinical diagnosis of suspected prion disease and the peculiar molecular and biochemical properties, requiring a modified western blot protocol for a consistent PrPSc detection, the prevalence of VPSPr is likely underestimated. Distinctive pathological features, which should direct to the correct diagnosis are the presence of cerebellar PrPSc microplaques and overall mild/moderate changes despite the relatively long disease duration (usually >20 months) (Figure 4).
Figure 4.
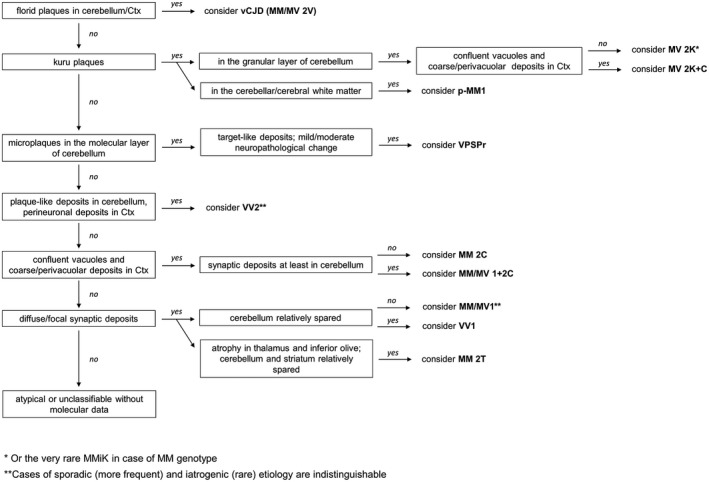
Updated diagnostic flowchart for diagnosis and classification of sporadic and acquired human prion disease histotypes. Adapted from Parchi et al 125. Abbreviations: ctx = cerebral cortex; V = variant.
Inherited PrP‐amyloidosis
Inherited PrP‐amyloidosis designates a group of genetic prion diseases characterized by the accumulation of an internal PrPSc fragment, truncated at both C‐ and N‐termini, mainly, although not exclusively, in the form of amyloid plaques or cerebral amyloid angiopathy. Depending on the causative mutation, the molecular mass of the abnormal PrPSc fragment may range from 6 to 11 kDa and can be occasionally associated with other PrP‐truncated species with higher molecular mass. The broad clinical and pathological heterogeneity observed in this group depends not only on the several disease‐associated PRNP mutations (ie, point and stop‐codon mutations, 7–12 extra octarepeat insertions, duplication in the hydrophobic domain), but also on the specific haplotype determined by the combination of PRNP codon 129 genotype/mutation, and other individual factors, exemplified by the significant intra‐familial phenotypic heterogeneity.
Gerstmann–Sträussler–Scheinker disease
GSS is the most prevalent inherited PrP‐amyloidosis and the first genetic prion disease that was linked to a PRNP mutation 62. GSS is often clinically characterized by slowly progressive ataxia, pyramidal signs and late dementia, while the accumulation of PrP‐amyloid plaques in the CNS, especially in the cerebral and cerebellar cortices, represents its pathological hallmark 46. Spongiform change is usually absent, sometimes limited to small foci and only occasionally widespread. The latter scenario occurs in a subgroup of cases carrying the P102L‐129M haplotype in which PrPSc aggregates include a CJD type 1‐like fragment in addition to the 8 kDa internal fragment, while those showing only the 8 kDa fragment manifest a typical GSS phenotype lacking spongiform change 118, 131. As stated previously, the GSS phenotype also comprises tau pathology in the form of neurofibrillary tangles and dystrophic neurites around the PrP‐amyloid plaques 48.
Some recently reported novel mutations highlighted once again the phenotypic heterogeneity of prion disease. The neuropathological characterization of GSS P105T (in cis with 129V) revealed severe spongiosis and neuronal loss throughout the cerebral cortices, while the cerebellum and basal ganglia were mildly affected. PrP immunochemistry showed a predominant synaptic staining, with some plaque‐like deposits and PrPSc plaques in the deeper cortical layers and in the molecular and granular cell layers of the cerebellum. Western blot analysis disclosed a 6 kDa PK‐resistant, unglycosylated fragment and two partially glycosylated fragments migrating at ~15 and ~25 kDa only after precipitation with phosphotungstic sodium acid suggesting a very low resistance to protease of these PrPSc species 136.
A typical GSS phenotype was also reported in association with the E211D mutation with a biochemical profile characterized by a prominent band migrating at 7 kDa 129. Interestingly an alternative missense mutation at the same codon (E211Q) was already reported in association with a CJD phenotype. The characterization of the two mutant proteins revealed that the relative stability of PrP underlies the generation of the distinct PK‐resistant fragments, which are in turn responsible for the phenotypic difference (ie, CJD vs GSS) 129.
Recently, a novel pathogenic pathway has been suggested for several GSS mutations. In an experimental murine model carrying an eight‐residue duplication (LGGLGGYV) in the hydrophobic region 60, 98, corresponding to a GSS‐causing mutation in humans, the pathogenic variant induced a conformational alteration in the unstructured N‐terminal portion of the protein leading to an early accumulation of a 16 kDa thermolysin‐resistant fragment. The latter showed similarities with the PrP species generated by stop‐codon mutations, and thought to be the precursor of the pathognomonic amyloidogenic 8 kDa fragment that is typically seen in GSS. Analysis of samples from other GSS patients carrying P102L, A117V, H187R and F198S revealed the same signature, suggesting a common molecular pathway.
PRNP truncating mutations and the widening spectrum of inherited prion amyloidoses
Premature stop‐codon mutations in PRNP uniquely result in an abnormal PrP isoform lacking the C‐terminal GPI anchor, which physiologically retains the prion protein at the cell surface. In most cases, the resulting anchorless PrP accumulates in the form of extracellular amyloid aggregates in both brain parenchyma and vessel walls. The discovery of this group of PRNP mutations has significantly expanded the phenotypic spectrum of PrP‐amyloidosis (Table 2). Indeed, besides GSS, PRNP stop‐codon mutations have been linked to cerebral amyloid angiopathy (PrP‐CAA) 47, 66, 70, 141 and, more recently, even found in patients with chronic diarrhea, progressive autonomic failure and peripheral polyneuropathy in association with widespread abnormal PrP deposition in both peripheral organs and CNS 29, 94, 97. To date, this form of systemic amyloidosis (PrP‐SA) has been linked to PRNP truncating mutations at codon 162 18, 163 29, 97, 169 29, and 203 94, 95 and is characterized by anchorless PrP deposition in the CNS and peripheral organs, including nerves, vessels, muscles, lung, kidney, liver, pancreas, gallbladder, bowel, uterus and skin. In most of these peripheral organs, staining for PrP showed granular and punctate deposits.
Table 2.
Pathology of inherited amyloidosis linked to PRNP truncating mutations.
| Haplotype | Reference | Onset (yrs) | Duration (yrs) | Pathological findings | PrPSc (kDa) |
|---|---|---|---|---|---|
| Y145X‐129M | 47 | 38 | 21 | No spongiform change; PrP amyloid deposits in the cerebral and cerebellar cortices; NFTs in the neocortex, hippocampus, subcortical nuclei; CAA. | 7 |
| Q160X‐129M | 70 | 20s‐50s | 5–20 | Diffuse cortical atrophy; PrP plaques in the cerebellum, and in the limbic and neocortical gray matter; NFTs and neuritic plaques in hippocampus, neocortex and midbrain. In one case co‐existence with neocortical Lewy body pathology. | 11 |
| Y162X‐129M | 18 | 45 | after 15 y still alive | Sural nerve biopsy: loss of small myelinated and unmyelinated fibers; PrP deposits surrounding vessel walls and nerve fibers. | n/a |
| Y163X‐129V | 97 | 30s‐50s | 18–33 | Mild spongiform change restricted to the cortical layer 1 and 2; widespread PrP plaques and tau‐related pathology (NFTs and neuropil threads); prominent PrP‐CAA in capillaries of subcortical regions. Granular PrP deposits in peripheral and autonomic nervous system. Fine, punctate PrP deposits in non‐extraneural tissues. | 10 |
| Y163X‐129V c.478_479insAAGTGTACT | 29 | 30s‐50s | 4–23 | Skin biopsy: fine, punctate deposits in the upper dermis at the transition to epidermis. | n/a |
| D178Efs25X (D203X) | 95 | 20s‐50s | 1–14 | Severe spongiform change and neuropil degeneration in the cerebral cortices, PrP coarse deposits in the cerebral cortex (often located alongside the small vessels), spinal cord, ganglions, femoral and sural peripheral nerves. In non‐CNS tissues, PrP mainly deposited in peripheral nerves, smooth muscles and blood vessels. | 9 |
| Y226X‐129V | 66 | 50s‐70s | 1–3 | Mild spongiform change and reactive astrocytic gliosis in all cortical and subcortical regions; PrP amyloid deposits and angiopathy involving small to medium‐sized arteries and arterioles of the cerebral and cerebellar cortices and leptomeninges; absence of tau neurofibrillary pathology. | n/a |
| Q227X‐129V | 66 | 37 | n/a | Frontal and caudate atrophy; multicentric, unicentric and diffuse PrP plaques (often located alongside capillaries) in all layers of the cerebral cortex and focally in the subcortical white matter associated with NFTs; no CAA. | 7 |
A secondary tauopathy characterized by widespread neurofibrillary tangles (NFTs) was observed in patients carrying the Y145X 46, Y160X 70, Y163X 97 and D203X 95 mutations. The pathology associated with the Y160X variant was also characterized by tau‐positive neurites in both limbic and neocortices 70, while a dot‐like pattern of tau deposition in the absence of NFTs featured the pathological phenotype in a patient carrying the Y226X mutation 66. Finally, there were abundant NFTs, neuropil threads, and dystrophic neurites in the neocortex of a patient carrying the Y227X variant, who showed a GSS phenotype without CAA 66.
Western blot analysis revealed the presence of small PK‐resistant fragments with molecular masses ranging from 7 to 11 kDa often associated with larger, smeared bands probably representing oligomers of abnormal PrP 46, 66, 70, 95, 97. Moreover, the lack of reactivity with antibodies to the C‐terminal indicates that both the oligomers and the smaller associated fragments are truncated and without the GPI anchor.
Co‐Pathology Related to Prion‐Like Protein Misfolding
Iatrogenic CJD
A number of recent studies have suggested that the age‐adjusted prevalence of Aβ co‐pathology in iCJD is unexpectedly high (up to 40% in patients <55 years) 25, 57, 69, 142. A similar high prevalence of Aβ co‐pathology has been reported in both d‐iCJD and hGH‐iCJD. Overall, the reported burden of Aβ deposits in iCJD is significantly higher than in age‐matched sCJD controls but lower than in AD 25. Aβ accumulation in iCJD frequently involves the vessels in the form of CAA 25, 142. Moreover, diffuse parenchymal deposits extending to the subpial spaces are more frequent than amyloid core plaques 25, 142.
Notably, evidence indicates that Aβ deposition in iCJD is an independent process rather than the result of a cross‐seeding mechanism with PrPSc. Indeed, in d‐iCJD, the regional distribution of Aβ aggregates is consistent with a spread from the graft (eg, subpial deposits) 54, 84. Most significantly, in a recent study, significant Aβ co‐pathology has also been observed in post‐mortem samples that received growth hormone treatment but did not develop CJD 142. Finally, another study suggested that neurosurgery with Aβ‐contaminated tools can transmit Aβ pathology and lead to intracerebral hemorrhage 57.
One study also documented Aβ contaminant in the hGH batches (old archived samples) and, after centrifugation, in the pellets, suggesting the presence of insoluble Aβ aggregates in the hormone preparation 39. Taken together these observations support the spreading of Aβ in a prion‐like manner through a peripheral route after subcutaneous hGH administration or centrally through use of dural mater grafts 54, 84, 142. This interpretation is in line with the results of transmission studies conducted in animal models showing that both intracerebral and intraperitoneal inoculation of Aβ induces CAA in APP‐transgenic mice 40, 99.
Given that AD pathology comprises a double proteinopathy, the above‐mentioned observations concerning Aβ raise the question about the role of tau. Two recent studies failed to demonstrate an increased prevalence of tau pathology in iCJD compared to sCJD 25, 142. Tau deposits, when present, were independent of Aβ deposits and compatible with primary age‐related tauopathy (PART). At variance, a very recent study in a French series of hGH‐iCJD brains 39 documented, in three cases, an atypical tau pathology characterized by positive staining of perikarya and neurites, and punctate deposits of a few microns in diameter in the hippocampus and temporal isocortex. Based on this finding and the detection of tau contaminant in hGH batches and pellets, a prion‐like transmission through the peripheral route was also suggested for tau.
Sporadic and genetic prion disease
Very few studies investigated the prevalence and type of neurodegenerative co‐pathologies in sporadic and genetic CJD. Moreover, they mainly focused on AD pathology while other reported associations were limited to case reports. Conflicting results also concern the frequency of the association between CJD and AD pathology, especially regarding the question of whether the co‐existence of the two diseases significantly exceeds that expected by chance based on their age‐related prevalence. A relatively old study that specifically addressed the prevalence of AD‐related pathology in sCJD 53 yielded a prevalence comparable to that observed in elderly non‐demented controls supporting the hypothesis of an age‐related unspecific co‐existence of the two pathologies. At variance, another study showed an inverse correlation between the total amount of accumulated Aβ and PrPSc in a subgroup of patients with the co‐occurrence of pathological changes typical for sCJD and AD, with patients with high Aβ load displaying significantly less PrPSc than those with low Aβ load. These results suggest that common pathways are involved in the generation of the two pathogenic proteins or their clearance or both 37.
Age‐related primary tauopathies, such as aging‐related tau astrogliopathy (ARTAG), argyrophilic grain disease (AGD) and PART have also been documented in sCJD, although in a minority of cases (5%–10%) 85, 87, 100. A predominantly gray matter type of ARTAG pathology characterized by granular/fuzzy astrocytes occurring in clusters, oligodendroglial coiled bodies, NFTs, pre‐tangles and neuropil threads with predominant localization in the limbic and frontotemporal cortices and, to a lesser extent, in thalamus and basal ganglia, has also been described in a patient carrying the V203I mutation 85.
Finally, in few case reports and small series, the CJD‐specific pathology was found to co‐exist with a synucleinopathy [sCJD+dementia with Lewy bodies (LBD) 41, 100, 168, 169, sCJD+preclinical multiple system atrophy 144] or a primary tauopathy [sCJD+progressive supranuclear palsy (PSP) 100]. Even more rarely, multiple proteinopathies have been documented in the same patient, namely sCJD+AD+LBD+AGD or CJD+AD+PSP+AGD 100. The very low prevalence of these cases suggests a by‐chance association rather than a common origin, although a possible cross‐seeding mechanism cannot be completely ruled out.
Cross‐seeding between PrP and other amyloidogenic proteins
Evidence supporting shared pathogenic mechanisms among neurodegenerative disorders caused by protein misfolding raised the question of the possible cross‐seeding between amyloidogenic proteins, an issue which is also relevant for the interpretation of the neuropathological studies described above on the prevalence of mixed neurodegenerative pathologies in autopsy series.
Current evidence suggesting a cross‐seeding role of PrP is limited to Aβ. However, the possibility of synergistic effect between prion and Aβ pathologies remains controversial. For example, published data indicated that PrPSc can exacerbate Aβ deposition at a time point where plaques are already present 106, with both pathologies working synergistically, but also that misfolded Aβ can interfere with prion pathogenesis 150 or do not affect prion pathology at all 139. Further work is needed to clarify this issue and also to understand the possible effect of the strain and the various routes of infection on PrPSc‐Aβ cross‐seeding.
PrPSc Properties and the molecular Basis of Prion Strains
The characterization of distinct isoforms of human PrPSc that strongly correlate with the disease phenotype has provided strong indirect support to the protein conformational theory of the origin of prion strains. It also led to the concept of molecular strain typing, in which the different PrPSc isoforms or types represent molecular markers for prion strains and provide a basis for the classification of the disease variants 116, 117, 118, 120, 131.
Studies on GSS‐affected brains originally showed that the PrPSc obtained by in vitro proteolysis of purified amyloid preparations mainly comprises unglycosylated 7–8 kDa PrP fragments with ragged N‐ and C‐termini, primarily composed of mutant PrP 118, 131, 132, 158, 159 (Figure 5).
Figure 5.
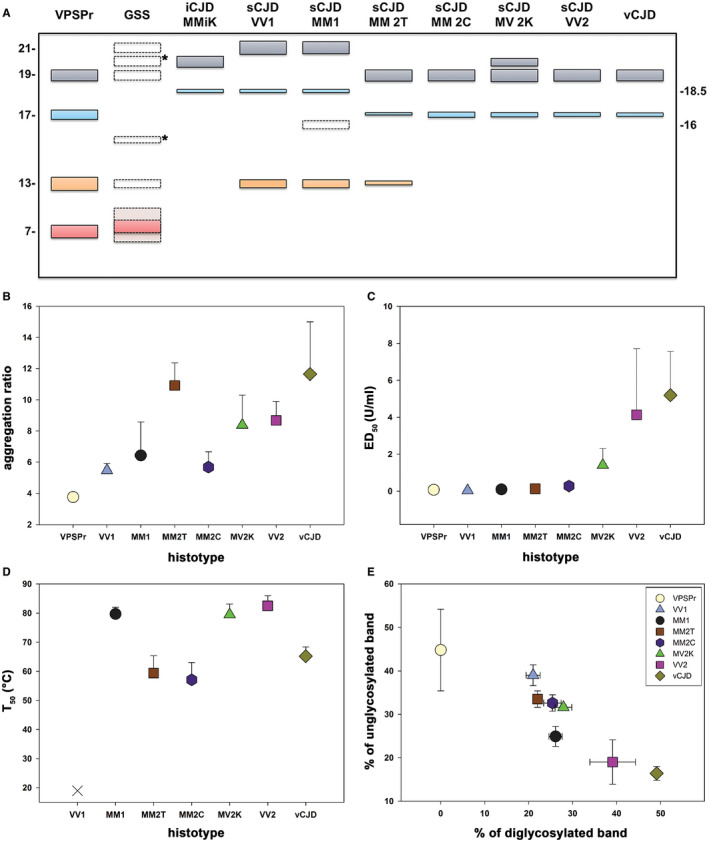
Spectrum of PrPSc physicochemical properties among phenotypic variants of human prion disease. A. Schematic representation of the spectrum of truncated PrPSc (unglycosylated) fragments observed in human prion disease. The classic N‐terminally truncated PrPSc fragments of ~21 (type 1), 20 (type “i”) and 19 (type 2) kDa, comprising the so‐called PrP27‐30, are shown in grey, while their anchorless counterparts of ~17 and 18.5 kDa are represented in light blue. C‐terminal associated fragments (CTF) of lower molecular weight (~13–15 kDa) are highlighted in orange, whereas the N‐ and C‐terminal ragged fragment of ~7–11 kDa linked to GSS and VPSPr are in light red. The bands with dotted lines symbolize PrPSc fragments that are only seen occasionally (eg, associated with specific GSS‐causing mutations) or in specific experimental conditions (eg, 16 kDa fragment seen in CJDMM1 in partially denaturing conditions). *Indicates PrPSc fragments (eg, 20 and 15 kDa in GSS) reported only once and lacking adequate comparison with control samples, leaving doubt about their precise molecular mass. B. Comparison of PrPSc “aggregation ratio” between CJD subtypes [data from Saverioni et al 152]. The ratio represents a semiquantitative index of the overall state of PrPSc aggregation, with values being proportional to the mean size of protein aggregates. C. Comparison of PrPSc PK‐resistance between CJD subtypes [data from Saverioni et. al. 152]. ED50 expresses the PK concentration needed to digest 50% of PrPSc. D. Comparison of PrPSc “thermostability” between CJD subtypes [data from Cescatti et al 32]. T50 expresses the temperature needed to solubilize 50% of PrPSc. The sCJDVV1 subtype is represented with a Χ since the calculated T50 is below 25°C. E. PrPSc glycoform ratio after PK digestion [data from Saverioni et al 152]. Data in B–E are based on densitometric analyses of immunoblots probed with mAb 3F4 or SAF60.
At variance, CJD and FI brains were invariably linked to either one or both of two protease‐resistant and N‐terminally truncated PrPSc fragments, which have been designated as types 1 and 2 116. PrPSc type 1 has the electrophoretic mobility of ~21 kDa and a primary cleavage site at residue 82, while PrPSc type 2 has a relative molecular mass of ~19 kDa and a primary cleavage site at residue 97 117, 122. Interestingly, PrPSc types 1 and 2 characterize all forms of CJD, that is, sporadic, inherited or acquired by infection 117, 122, indicating a common mechanism of PrPSc formation that is independent of the apparent etiology.
Later, it was also shown that GSS brains carrying different mutations might also occasionally show PrPSc fragments of higher molecular weight, including the CJD‐associated PrPSc type 1, in addition to the typical unglycosylated 7–8 kDa PrP fragments with ragged N‐ and C‐termini 118, 131 (Figure 5). Most significantly, GSS‐affected subjects carrying the most common mutation (P102L) manifesting a rapidly progressive CJD‐like phenotype with both spongiform change and amyloid plaques showed the co‐occurrence of PrPSc type 1‐like and the 8 kDa fragments, whereas those with a slowly progressive “pure” GSS phenotype correlated with the presence of amyloid plaques and the 8 kDa PrP fragment in the absence of the type 1‐like fragment 118, 131.
Despite the differences in the clinicopathological phenotype between VPSPr and GSS, the characterization of PrPSc physicochemical properties highlighted strong similarities that led to the hypothesis that the former may represent the sporadic variant of the latter 27, 134, 182. Indeed, PrPSc in VPSPr shows a striking, ladder‐like electrophoretic profile comprising at least four bands, including a prominent internal PrP fragment migrating at ~8 kDa (Figure 5).
The characterization of human strains by transmission studies (see also the dedicated section in this review) demonstrated an excess of biological variability with respect to PrPSc types 1 and 2 dichotomy (ie, five CJD strains vs. two protein types) 17, 76, 104, 124, which, according to the protein‐only hypothesis, would indicate that types 1 and 2 are structurally heterogeneous PrPSc species. Evidence from recent studies supports this contention. Indeed, a deeper analysis of western blot profiles revealed the presence of subtype‐specific PrPSc C‐terminal fragments of about 12 and 13 kDa (PrP‐CTF12/13) 181. Moreover, a further PrPSc fragment with an electrophoretic mobility of approximately 20 kDa, designated as PrPSc “i” (ie, with an intermediate molecular mass between types 1 and 2) has been characterized in both sporadic and iatrogenic CJD MV2K and in iCJDMMiK 78, 111, 120, 123, 124.
The glycoform ratio that is the ratio between the three differently glycosylated isoforms of PrPSc (ie, diglycosylated, monoglycosylated, and unglycosylated) also contributes to the heterogeneity of PrPSc between CJD subtypes, even within the type 1 and type 2 subgroups. Indeed, although the PrPSc glycotype in sCJD is most often characterized by an overrepresentation of the monoglycosylated form, there are differences in the relative abundance of the diglycosylated form among disease subtypes 120 that are strain‐specific 36, 120.
Interestingly, the relative amount of both diglycosylated PrPSc and CTF13 appears to inversely correlate with the degree of resistance of PrPSc to protease digestion, a property that was also shown to differentiate between certain animal strains and to be highly heterogeneous between sCJD isolates 152 (Figure 5). Indeed, PrPSc in sCJDVV2 shows the highest resistance to PK digestion, the most abundant diglycosylated band and no detectable CTF13, whereas, in contrast, PrPSc in sCJDVV1 is the most sensitive to proteases, has the lowest amount of diglycosylated form and the highest of PrP‐CTF13 152 (Figure 5).
The search for strain‐specific properties of PrPSc has also focused on biochemical features reflecting the tertiary and quaternary structures of PrPSc aggregates. Saverioni and coworkers 152 found a positive correlation between mean size and PK‐resistance of PrPSc aggregates in sporadic and variant CJD. However, PrPSc aggregates of the same size from sCJD MM(V)1 and VV2 maintained a significant difference in PK‐resistance suggesting that the latter property also stems from aggregate stability 152. In other words, the most PK‐resistant prions would comprise PrPSc aggregates of a larger size that are also kept together by stronger interactions.
In early 2000s, the description and characterization of a PK‐sensitive form of PrPSc (sPrPSc), which, like PrPC, is fully degraded at a relatively low PK activity seemed to open a potentially new perspective to the study of PrPSc properties and their relationship to prion strains 127, 165. Most significantly, Safar and colleagues 148 reported that sPrPSc represents an invariable and quantitatively significant component of human prions, contributing up to 90% of the whole PrPSc signal in sCJD. In apparent agreement with data obtained in experimental models of prion disease 147, suggesting a relationship between sPrPSc/rPrPSc ratio and strain‐specific properties such as the incubation time, these authors found also in sCJD an inverse correlation between the relative amount of sPrPSc and the clinical duration of the disease 71. More recently, however, Saverioni et al 152 and others 34 also looked for sPrPSc across the spectrum of sCJD prions and found a much lower proportion of sPrPSc with no significant differences between sCJD subtypes. In one study in particular, sPrPSc always (ie, in all CJD subtypes) represented a minor component of abnormal PrP not exceeding 10% of total detergent‐insoluble PrPSc.
To search for indirect evidence of structural PrPSc variation enciphering strain‐specific properties, the conformational stability of PrPSc aggregates has also been studied in sCJD, although using different experimental procedures. Guanidine hydrochloride (GdnHCl)‐induced unfolding, the most commonly used approach has been variably monitored measuring (i) the extent of exposure of a conformational epitope that is hidden in the native conformation (conformation‐dependent immunoassay or CDI), (ii) the progressive loss of PK‐resistance (conformational stability assay or CSA) or (iii) the increase in solubility of PrPSc (stability and solubility assay or CSSA) 130, 133, 147.
In the few studies performed to date in sCJD, PrPSc was shown to be more stable in sCJD MM(V)1 than in MM2C by both CSA and CSSA 23, 34, 133. Similarly, based on hydrogen/deuterium (H/D) exchange coupled with mass spectrometry, Safar et al 149 demonstrated that MM1 and MM2C PrPSc differ in their structural organization both at the level of the polypeptide backbone and the quaternary packing arrangements.
Finally, in a recent study 32, we obtained similar denaturation curves among the 6 sCJD subtypes analyzed by CSA, indicating that GdnHCl does not significantly perturb the strain‐specific structural organization of PrPSc. In the same study 32, we also investigated the effect of heating (in the presence of sodium dodecyl sulfate) on PrPSc aggregates. At variance with the negative results obtained with GdnHCl, the effect of temperature on PrPSc solubilization varied significantly according to the sCJD subtype. The calculated T50 was lowest for sCJD VV1, MM2T and MM2C (T50 < 55°C) and highest for sCJD MM1, VV2, and MV2K (T50 > 80°C). Interestingly, since the most thermostable prion protein types were those associated with the most prevalent phenotypes and most rapidly and efficiently transmitting strains (ie, M1 and V2) (Figure 6), the results suggest a direct correlation between strain replication efficiency and the thermostability of prion protein aggregates.
Figure 6.
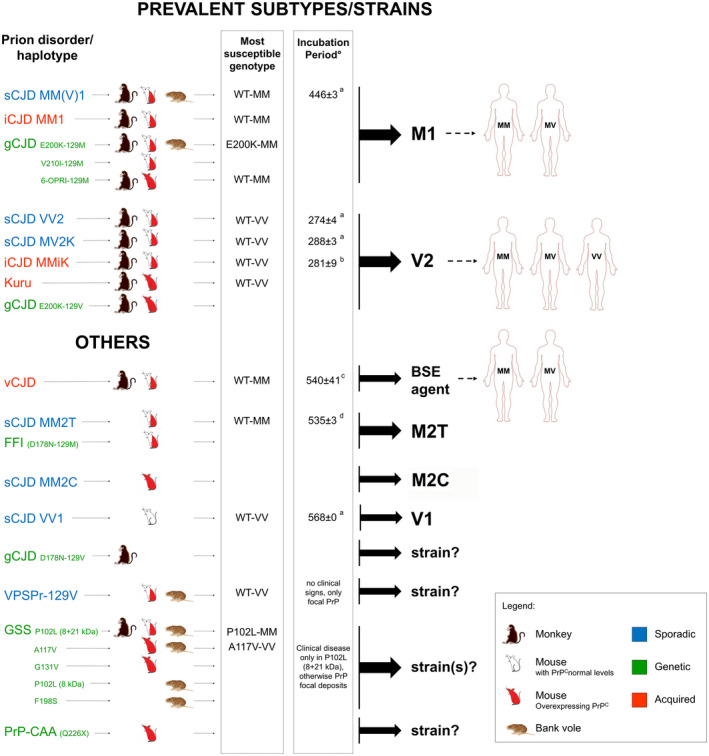
Successful transmissions of human prion disease variants: Most susceptible PRNP genotypes and resulting prion strain diversity . Uncolored mice refer to animals expressing physiological levels of PrPC, red mice to animals overexpressing PrPC. Half colored mice indicate successful transmissions in both types of mice. Susceptible genotypes are those associated with the shortest incubation time. °Only studies in mice expressing physiological levels of PrPC are listed. aBishop et al 2010 17; bKobayashi et al 2007 73; cTakeuchi et al 2013 160; dModa et al 2012 104.
Taken together, all these data indirectly support, but do not prove, the hypothesis that the strain‐specific properties of prions are enciphered within the structure (from secondary to quaternary) of PrPSc.
Prion Strain Isolation and Characterization: Transmission Studies
There have been relatively few transmission studies with human prions compared to those conducted with the scrapie agent. The transmission of human prions also started later than that of animal prions, mainly because the early pioneering studies by Gajdusek’s group which proved the transmissibility of human prion disease did not contribute any characterization of the isolated strains 20. Moreover, because of the difficulties in obtaining the transmissions in wild‐type rodents, the study of strain‐specific properties of human prion isolates has been mainly carried out in the era of transgenic mice. As the only exceptions, a few significant studies examined the transmission characteristics of human prions using bank voles or non‐human primates.
Sporadic prion disease
The results of the studies conducted to date with sCJD prions indicate that five of the sCJD subtypes defined by the current classification behave as different prion strains 17, 68, 76, 79, 82, 104, 110, 124, 174, 175 (Figure 6). As the only exception, prions associated with VV2 and MV2K shared transmission properties despite the different phenotypic features in the natural host, suggesting a host genotype effect related to PRNP codon 129. Specifically, sCJD MM1 and MV1 isolates showed identical transmission properties (M1 strain), which significantly differed from those of sCJD VV2 or MV2K (V2 strain) 17, 124. Furthermore, sCJD MM2C, MM2T and VV1 isolates generated distinctive transmission properties, suggesting they are caused by specific strains too 17, 68, 80, 82, 91, 104, 114, 174, 175.
In two recent studies, the transmission of brain inocula from either sCJD MM1+2C or atypical p‐MM1 gave results indistinguishable from those obtained with M1 prions 80, 146. Despite the analogy, the results might have different meanings. The most likely explanation of the former result is that the much higher susceptibility of PrP‐humanized knock‐in mice (as humans and non‐human primates) to M1 prions than to M2C prions prevented the appearance of histopathological or biochemical features related to M2C prions in the animal recipients. Serial passages of sCJDMM1+2C inocula in animals that are susceptible to M2C prions but not to M1 prions will be needed to further clarify the issue. At variance, the latter result [ie, sCJD p‐MM1 transmission] more likely suggests a host‐derived effect (ie, genetic predisposition to amyloid plaque formation in white matter). Indeed, to consider an alternative possibility, one would postulate the unlikely scenario of the co‐occurrence in sCJD p‐MM1 of a distinct prion strain besides sCJD M1, not inducing a distinctive cerebral grey matter pathology, not affecting PrPSc properties, and not transmissible to bank voles 146.
Genetic CJD and FFI
Genetic CJD has been linked to the same pool of strains isolated from sCJD, although, to date, transmission studies have examined only part of the phenotypic variants or PRNP haplotype combinations that characterize the disease. When propagated in transgenic mice, non‐human primates or bank voles, CJD inocula from carriers of E200K‐129M and V210I‐129M haplotypes and accumulating PrPSc type 1 in the brain showed similar transmission properties to M1 prions 92, 110, 124. Similar results were obtained after transmission of brain homogenates from a subject carrying a 6‐OPRI‐129M allele into transgenic mice expressing human PrPC and wild‐type FVB mice 96.
Interestingly, unlike with the E200K‐129M, a single E200K‐129V inoculum with PrPSc type 2 transmitted with prolonged incubation time and induced plaque‐like focal deposits in the affected mice 7, a result which parallels the findings obtained with sCJD MM1 and VV2 inocula in non‐human primates and transgenic mice 17, 124. On the same line, FFI and sFI inocula shared transmission properties when propagated into transgenic mice 91, 104.
Acquired CJD
Experimental evidence indicates that iCJD MMiK in Japan originated from an incomplete adaptation of the V2 strain to the PRNP codon 129 MM genotype 77, 78. Indeed, both transmission studies in mice 75, 76 and in vitro experiments using PMCA 143, 161 convincingly demonstrated that (i) the transmission of the V2 strain to recipients carrying 129MM results in an unusual PrPSc with altered conformational properties (PrPSc type “i”); (ii) the V2 strain rapidly re‐emerges when the PrPSc type “i” is transmitted to a host expressing 129VV. Notably, like iCJD MMiK, experimentally transmitted kuru also demonstrated the transmission properties of V2 prions, which led to the conclusion that it originated from an individual affected by sCJD VV2 or MV2K 124.
In contrast to prions from kuru and iCJD, vCJD prions showed transmission properties that were strikingly distinct from sCJD prions 22, 59. The vCJD prions transmitted the disease to wild‐type mice far more efficiently than any other form of human prion disease 22, 59, 172 and the faithful propagation of the vCJD phenotype in transgenic mice was dependent upon the homozygous expression of M at PRNP codon 129 6, 14, 16, 171. Transgenic mice homozygous for human PrP 129V showed a pronounced transmission barrier to vCJD prions and propagated a distinct clinicopathological phenotype 6, 16, 59, 160, 171. The latter results raised the possibility that the BSE‐vCJD strain may be associated with other human pathological phenotypes besides that observed in subjects carrying MM at codon 129.
Interestingly, given that at variance with sporadic and inherited prion disease, acquired prion diseases have all been linked to M1, V2 or BSE‐derived prions, it appears that the definition of “infectious” prions (ie, causing a disease that has propagated from human‐to‐human) may only apply to these three human strains. Indeed, strictly speaking, all other human disease variants would better fit the definition of prion‐like disorders (or prionoids), the terms currently used for transmissible but not‐infectious neurodegenerative disorders characterized by protein misfolding, aggregation and seeded polymerization (eg, synucleinopathies, tauopathies) 153.
GSS
With the significant exception of the GSS P102L associated with spongiform change and a PrPSc type 1‐like, which showed CJD‐like transmission properties in non‐human primates and wild‐type mice 90, 162, the other GSS variants have been more difficult to transmit to animals in comparison to CJD and FI. This led to the suggestion that these GSS phenotypes (ie, those lacking spongiform change) are not bona‐fide transmissible spongiform encephalopathies (TSE), and would be better designated as non‐transmissible prion disorders. However, more recently, the use of transgenic mice carrying GSS mutations, such as A117V or the mouse equivalent of P102L, led to successful transmissions, although with some significant differences between the two models 8, 9. More specifically, the inoculation of brain extracts from a GSS P102L patient with no spongiform change caused almost no clinical disease but induced striking PrP‐amyloid deposition in brains of several recipient mice; the extracts of those brains failed to transmit neurological disease on further passage but again induced PrP‐amyloid plaques in recipient mice 9. At variance, the transmission of A117V prions induced a typical TSE phenotype with spongiform change and tissue deposition of PrP27‐30 8.
In a recent study, brain homogenates from patients carrying the PRNP mutations G131V, Y226X and Q227X were injected intra‐cerebrally into transgenic mice overexpressing human PrP 138. Transmissions were successful with the former two patients but not with the one carrying Q227X. In detail, half of the animals injected with Y226X brain homogenate had PrPSc detectable in the brain by immunostaining, immunoblot, and PrP amyloid seeding activity assayed by RT‐QuIC, while G131V‐injected mice showed PrPSc deposition in the brain, although none were positive by immunoblot or RT‐QuIC assay. In contrast, none of the mice injected with Q227X brain had PrPSc or PrP‐amyloid seeding activity detectable by these methods 138. Notably, Y226X is the only known truncating PRNP mutation associated with a familial disease that has been shown to be transmissible.
Recently, homogenates from GSS brains expressing the F198S and A117V mutations and showing the typical 8 kDa fragment as the main form of PrPSc after PK digestion were successfully transmitted to bank voles, where they induced a classic spongiform encephalopathy 135.
Taken together the data gathered to date indicate that GSS is a truly transmissible disease. However, with the exception of the P102L‐129M phenotype linked to PrPSc type 1‐like and spongiform change, it seems that the disease mostly behaves in humans as a prion‐like disorder capable of prion protein amyloid seeding without generating infectious prions. The development of a spongiform encephalopathy rather than prion‐related amyloidosis in bank voles underlines the importance of the recipient host in determining the degree of transmissibility and the associated neurodegenerative changes induced by a given PrPSc isolate.
Transmissions to assess the possible zoonotic origin of human prions
Given the lack of a definite answer about the origin of sCJD, the notion that classic‐BSE prions are zoonotic agents causing vCJD in humans keeps alive the possibility that certain sCJD cases may also originate from animal prion disease cross‐transmission. Some concern in this regard came from the observation that a BSE strain variant, designated L‐BSE, responsible for an atypical phenotype of prion disease in aged cattle, propagated more easily than classical BSE in transgenic mice expressing human PrPC (tgHu) and shared biochemical PrPSc properties with sCJD M2C prions 13. However, a recent comparison of the transmission properties in tgHu mice between L‐BSE isolates and a series sCJD cases representative of the human prion strain diversity disclosed no etiological link between L‐BSE and sCJD M2C prions 68. Specifically, although the two isolates showed similar immunoblot profiles and conformational stabilities of PrPSc, a shared limited trophism for lymphoid tissue, and a comparable regional profile of PrPSc deposition, they significantly differed in the disease incubation time over serial passages and in the resistance to PK digestion of PrPSc aggregates.
Concern about the zoonotic potential of prion disease in ruminants was also kept alive by another transmission study in tgHu mice expressing human PrP codon 129 variants 30. In the latter, different scrapie isolates transmitted the disease to several tgHu mice models with an efficiency comparable to that of cattle BSE and led to the propagation of prions that were phenotypically identical to those causing sCJD in humans. If confirmed, these results will significantly strengthen the notion that scrapie prions have zoonotic potential and will generate new questions about the possible link between animal and human prions.
Conclusive remarks
There has been substantial progress on several aspects of human prion disease in recent years. The phenotypic spectrum of disease has been expanded with the addition or a better characterization of rare phenotypic variants such as sCJD p‐MM1, MM2T, VPSPr, iCJD MMiK, vCJD in codon 129 MV recipients, gCJD linked to E200G‐129V and A224V‐129V, GSS associated with P105T‐129V and, above all, PrP‐SA in patients carrying PRNP truncating mutations. Based on the finding of a specific form of tau deposition correlating with both amount and morphology of PrPSc aggregates, CJD has been added to the list of secondary tauopathies. Furthermore, recent studies on iCJD have raised concerns that Aβ may be transmissible from human‐to‐human through the injection of contaminated tissues, as it is the case for PrPSc. On a related issue, the increasing evidence supporting shared pathogenic mechanisms among neurodegenerative disorders caused by protein misfolding has also raised the question of the possible cross‐seeding between PrPSc and other amyloidogenic proteins such as Aβ and tau. However, despite the several studies, results remain controversial on this specific aspect and await further studies on both animal models and human brains.
Significant advances have also been made on the characterization of human prion strains and their molecular basis. The VPSPr, several GSS variants, and PrP‐CAA linked to the truncating mutation Y226X have been added to the list of prion diseases successfully transmitted to recipient animals. However, at variance with the CJD‐FI spectrum, in which current transmission studies indicate the existence of at least six well‐characterized strains, the above‐mentioned transmissions have not yet provided a conclusive picture regarding the extent of strain variation in GSS or VPSPr. Similarly, the critical question of whether or not the pathogenic mutations linked to genetic disease, which are thought to affect PrP conformation, are able to generate prion strains differing from those isolated from the sporadic disease remains unanswered. Finally, concern about the zoonotic potential of prion disease in ruminants remains alive based on the results of at least one study demonstrating the possible convergence of scrapie and sCJD prions into tgHu mice, and the occurrence of sCJD subtypes such as VV1 and MM2T in young adults with a mean age at onset incompatible with the hypothesis of a spontaneous etiology related to aging.
References
- 1. Abu‐Rumeileh S, Redaelli V, Baiardi S, Mackenzie G, Windl O, Ritchie DL et al (2018) Sporadic fatal insomnia in Europe: phenotypic features and diagnostic challenges. Ann Neurol 84:347–360. [DOI] [PubMed] [Google Scholar]
- 2. Aguzzi A, Heikenwalder M, Polymenidou M (2007) Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol 8:552–561. [DOI] [PubMed] [Google Scholar]
- 3. Aguzzi A, Zhu C (2017) Microglia in prion diseases. J Clin Invest 127:3230–3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Almer G, Hainfellner JA, Brücke T, Jellinger K, Kleinert R, Bayer G et al (1999) Fatal familial insomnia: a new Austrian family. Brain 122(Pt 1):5–16. [DOI] [PubMed] [Google Scholar]
- 5. Asante EA, Linehan JM, Desbruslais M, Joiner S, Gowland I, Wood AL et al (2002) BSE prions propagate as either variant CJD‐like or sporadic CJD‐like prion strains in transgenic mice expressing human prion protein. EMBO J 21:6358–6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Asante EA, Linehan JM, Gowland I, Joiner S, Fox K, Cooper S et al (2006) Dissociation of pathological and molecular phenotype of variant Creutzfeldt‐Jakob disease in transgenic human prion protein 129 heterozygous mice. Proc Natl Acad Sci USA 103:10759–10764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Asante EA, Gowland I, Grimshaw A, Linehan JM, Smidak M, Houghton R et al (2009) Absence of spontaneous disease and comparative prion susceptibility of transgenic mice expressing mutant human prion proteins. J Gen Virol 90:546–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Asante EA, Linehan JM, Smidak M, Tomlinson A, Grimshaw A, Jeelani A et al (2013) Inherited prion disease A117V is not simply a proteinopathy but produces prions transmissible to transgenic mice expressing homologous prion protein. PLoS Pathog 9:e1003643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Asante EA, Grimshaw A, Smidak M, Jakubcova T, Tomlinson A, Jeelani A et al (2015) Transmission properties of human PrP 102L prions challenge the relevance of mouse models of GSS. PLoS Pathog 11:e1004953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Assar H, Topakian R, Weis S, Rahimi J, Trenkler J, Höftberger R et al (2015) A case of variably protease‐sensitive prionopathy treated with doxycyclin. J Neurol Neurosurg Psychiatry 86:816–818. [DOI] [PubMed] [Google Scholar]
- 11. Ayuso Blanco T, Urriza Mena J, Caballero Martínez C, Iriarte Franco J, Munoz R, García‐Bragado F (2006) Fatal familiar insomnia: clinical, neurophysiological and histopathological study of two cases. Neurologia 21:414–420. [PubMed] [Google Scholar]
- 12. Baiardi S, Capellari S, Ladogana A, Strumia S, Santangelo M, Pocchiari M, Parchi P (2016) Revisiting the heidenhain variant of Creutzfeldt‐Jakob disease: evidence for prion type variability influencing clinical course and laboratory findings. J Alzheimers Dis 50:465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Béringue V, Andréoletti O, Le Dur A, Essalmani R, Vilotte J‐L, Lacroux C et al (2007) A bovine prion acquires an epidemic bovine spongiform encephalopathy strain‐like phenotype on interspecies transmission. J Neurosci 27:6965–6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Béringue V, Le Dur A, Tixador P, Reine F, Lepourry L, Perret‐Liaudet A et al (2008) Prominent and persistent extraneural infection in human PrP transgenic mice infected with variant CJD. PLoS One 3:e1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bessen RA, Marsh RF (1994) Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol 68:7859–7868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bishop MT, Hart P, Aitchison L, Baybutt HN, Plinston C, Thomson V et al (2006) Predicting susceptibility and incubation time of human‐to‐human transmission of vCJD. Lancet Neurol 5:393–398. [DOI] [PubMed] [Google Scholar]
- 17. Bishop MT, Will RG, Manson JC (2010) Defining sporadic Creutzfeldt‐Jakob disease strains and their transmission properties. Proc Natl Acad Sci USA 107:12005–12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bommarito G, Cellerino M, Prada V, Venturi C, Capellari S, Cortelli P et al (2018) A novel prion protein gene‐truncating mutation causing autonomic neuropathy and diarrhea. Eur J Neurol 25:e91–e92. [DOI] [PubMed] [Google Scholar]
- 19. Brandel J‐P, Heath CA, Head MW, Levavasseur E, Knight R, Laplanche J‐L et al (2009) Variant Creutzfeldt‐Jakob disease in France and the United Kingdom: evidence for the same agent strain. Ann Neurol 65:249–256. [DOI] [PubMed] [Google Scholar]
- 20. Brown P, Gibbs CJ, Rodgers‐Johnson P, Asher DM, Sulima MP, Bacote A et al (1994) Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol 35:513–529. [DOI] [PubMed] [Google Scholar]
- 21. Brown P, Brandel J‐P, Sato T, Nakamura Y, MacKenzie J, Will RG et al (2012) Iatrogenic Creutzfeldt‐Jakob disease, final assessment. Emerging Infect Dis 18:901–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A et al (1997) Transmissions to mice indicate that “new variant” CJD is caused by the BSE agent. Nature 389:498–501. [DOI] [PubMed] [Google Scholar]
- 23. Cali I, Castellani R, Yuan J, Al‐Shekhlee A, Cohen ML, Xiao X et al (2006) Classification of sporadic Creutzfeldt‐Jakob disease revisited. Brain 129:2266–2277. [DOI] [PubMed] [Google Scholar]
- 24. Cali I, Miller CJ, Parisi JE, Geschwind MD, Gambetti P, Schonberger LB (2015) Distinct pathological phenotypes of Creutzfeldt‐Jakob disease in recipients of prion‐contaminated growth hormone. Acta Neuropathol Commun 3:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cali I, Cohen ML, Haik S, Parchi P, Giaccone G, Collins SJ et al (2018) Iatrogenic Creutzfeldt‐Jakob disease with Amyloid‐β pathology: an international study. Acta Neuropathol Commun 6:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Capellari S, Vital C, Parchi P, Petersen RB, Ferrer X, Jarnier D et al (1997) Familial prion disease with a novel 144‐bp insertion in the prion protein gene in a Basque family. Neurology 49:133–141. [DOI] [PubMed] [Google Scholar]
- 27. Capellari S, Powers JM, Petersen RB, Gambetti P, Parchi P (1998) A novel molecular and clinico‐pathologic phenotype of prion disease in an American. Neurology 50:A235. [Google Scholar]
- 28. Capellari S, Strammiello R, Saverioni D, Kretzschmar H, Parchi P (2011) Genetic Creutzfeldt‐Jakob disease and fatal familial insomnia: insights into phenotypic variability and disease pathogenesis. Acta Neuropathol 121:21–37. [DOI] [PubMed] [Google Scholar]
- 29. Capellari S, Baiardi S, Rinaldi R, Bartoletti‐Stella A, Graziano C, Piras S et al (2018) Two novel PRNP truncating mutations broaden the spectrum of prion amyloidosis. Ann Clin Transl Neurol 5:777–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cassard H, Torres J‐M, Lacroux C, Douet J‐Y, Benestad SL, Lantier F et al (2014) Evidence for zoonotic potential of ovine scrapie prions. Nat Commun 5:5821. [DOI] [PubMed] [Google Scholar]
- 31. Caughey B, Baron GS, Chesebro B, Jeffrey M (2009) Getting a grip on prions: oligomers, amyloids, and pathological membrane interactions. Annu Rev Biochem 78:177–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cescatti M, Saverioni D, Capellari S, Tagliavini F, Kitamoto T, Ironside J et al (2016) Analysis of conformational stability of abnormal prion protein aggregates across the spectrum of Creutzfeldt‐Jakob disease prions. J Virol 90:6244–6254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen SG, Parchi P, Brown P, Capellari S, Zou W, Cochran EJ et al (1997) Allelic origin of the abnormal prion protein isoform in familial prion diseases. Nat Med 3:1009–1015. [DOI] [PubMed] [Google Scholar]
- 34. Choi YP, Gröner A, Ironside JW, Head MW (2011) Comparison of the level, distribution and form of disease‐associated prion protein in variant and sporadic Creutzfeldt‐Jakob diseased brain using conformation‐dependent immunoassay and Western blot. J Gen Virol 92:727–732. [DOI] [PubMed] [Google Scholar]
- 35. Collinge J, Sidle KC, Meads J, Ironside J, Hill AF (1996) Molecular analysis of prion strain variation and the aetiology of “new variant” CJD. Nature 383:685–690. [DOI] [PubMed] [Google Scholar]
- 36. Cracco L, Notari S, Cali I, Sy M‐S, Chen SG, Cohen ML et al (2017) Novel strain properties distinguishing sporadic prion diseases sharing prion protein genotype and prion type. Sci Rep 7:38280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Debatin L, Streffer J, Geissen M, Matschke J, Aguzzi A, Glatzel M (2008) Association between deposition of beta‐amyloid and pathological prion protein in sporadic Creutzfeldt‐Jakob disease. Neurodegener Dis 5:347–354. [DOI] [PubMed] [Google Scholar]
- 38. Deininger MH, Bekure‐Nemariam K, Trautmann K, Morgalla M, Meyermann R, Schluesener HJ (2003) Cyclooxygenase‐1 and ‐2 in brains of patients who died with sporadic Creutzfeldt‐Jakob disease. J Mol Neurosci 20:25–30. [DOI] [PubMed] [Google Scholar]
- 39. Duyckaerts C, Sazdovitch V, Ando K, Seilhean D, Privat N, Yilmaz Z et al (2018) Neuropathology of iatrogenic Creutzfeldt‐Jakob disease and immunoassay of French cadaver‐sourced growth hormone batches suggest possible transmission of tauopathy and long incubation periods for the transmission of Abeta pathology. Acta Neuropathol 135:201–212. [DOI] [PubMed] [Google Scholar]
- 40. Eisele YS, Obermüller U, Heilbronner G, Baumann F, Kaeser SA, Wolburg H et al (2010) Peripherally applied Abeta‐containing inoculates induce cerebral beta‐amyloidosis. Science 330:980–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fernández‐Vega I, Ruiz‐Ojeda J, Juste RA, Geijo M, Zarranz JJ, Sánchez Menoyo JL et al (2015) Coexistence of mixed phenotype Creutzfeldt‐Jakob disease, Lewy body disease and argyrophilic grain disease plus histological features of possible Alzheimer’s disease: a multi‐protein disorder in an autopsy case. Neuropathology 35:56–63. [DOI] [PubMed] [Google Scholar]
- 42. Franceschini A, Strammiello R, Capellari S, Giese A, Parchi P (2018) Regional pattern of microgliosis in sporadic Creutzfeldt‐Jakob disease in relation to phenotypic variants and disease progression. Neuropathol Appl Neurobiol 44:574–589. [DOI] [PubMed] [Google Scholar]
- 43. Frau‐Méndez MA, Fernández‐Vega I, Ansoleaga B, Blanco Tech R, Carmona Tech M, Antonio Del Rio J et al (2017) Fatal familial insomnia: mitochondrial and protein synthesis machinery decline in the mediodorsal thalamus. Brain Pathol 27:95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Galeno R, Di Bari MA, Nonno R, Cardone F, Sbriccoli M, Graziano S et al (2017) Prion strain characterization of a novel subtype of Creutzfeldt‐Jakob disease. J Virol 91:e02390–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gambetti P, Dong Z, Yuan J, Xiao X, Zheng M, Alshekhlee A et al (2008) A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol 63:697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ghetti B, Piccardo P, Frangione B, Bugiani O, Giaccone G, Young K et al (1996) Prion protein amyloidosis. Brain Pathol 6:127–145. [DOI] [PubMed] [Google Scholar]
- 47. Ghetti B, Piccardo P, Spillantini MG, Ichimiya Y, Porro M, Perini F et al (1996) Vascular variant of prion protein cerebral amyloidosis with tau‐positive neurofibrillary tangles: the phenotype of the stop codon 145 mutation in PRNP. Proc Natl Acad Sci U S A 93:744–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ghetti B, Piccardo P, Zanusso G (2018) Dominantly inherited prion protein cerebral amyloidoses – a modern view of Gerstmann‐Sträussler‐Scheinker. Handb Clin Neurol 153:243–269. [DOI] [PubMed] [Google Scholar]
- 49. Giaccone G, Mangieri M, Capobianco R, Limido L, Hauw JJ, Haïk S et al (2008) Tauopathy in human and experimental variant Creutzfeldt‐Jakob disease. Neurobiol Aging 29:1864–1873. [DOI] [PubMed] [Google Scholar]
- 50. Gómez‐Nicola D, Fransen NL, Suzzi S, Perry VH (2013) Regulation of microglial proliferation during chronic neurodegeneration. J Neurosci 33:2481–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Grasbon‐Frodl E, Lorenz H, Mann U, Nitsch RM, Windl O, Kretzschmar HA (2004) Loss of glycosylation associated with the T183A mutation in human prion disease. Acta Neuropathol 108:476–484. [DOI] [PubMed] [Google Scholar]
- 52. Guiroy DC, Wakayama I, Liberski PP, Gajdusek DC (1994) Relationship of microglia and scrapie amyloid‐immunoreactive plaques in kuru, Creutzfeldt‐Jakob disease and Gerstmann‐Sträussler syndrome. Acta Neuropathol 87:526–530. [DOI] [PubMed] [Google Scholar]
- 53. Hainfellner JA, Wanschitz J, Jellinger K, Liberski PP, Gullotta F, Budka H (1998) Coexistence of Alzheimer‐type neuropathology in Creutzfeldt‐Jakob disease. Acta Neuropathol 96:116–122. [DOI] [PubMed] [Google Scholar]
- 54. Hamaguchi T, Taniguchi Y, Sakai K, Kitamoto T, Takao M, Murayama S et al (2016) Significant association of cadaveric dura mater grafting with subpial Aβ deposition and meningeal amyloid angiopathy. Acta Neuropathol 132:313–315. [DOI] [PubMed] [Google Scholar]
- 55. Head MW, Knight R, Zeidler M, Yull H, Barlow A, Ironside JW (2009) A case of protease sensitive prionopathy in a patient in the UK. Neuropathol Appl Neurobiol 35:628–632. [DOI] [PubMed] [Google Scholar]
- 56. Head MW, Yull HM, Ritchie DL, Langeveld JP, Fletcher NA, Knight RS, Ironside JW (2013) Variably protease‐sensitive prionopathy in the UK: a retrospective review 1991–2008. Brain 136:1102–1115. [DOI] [PubMed] [Google Scholar]
- 57. Hervé D, Porché M, Cabrejo L, Guidoux C, Tournier‐Lasserve E, Nicolas G et al (2018) Fatal Aβ cerebral amyloid angiopathy 4 decades after a dural graft at the age of 2 years. Acta Neuropathol 135:801–803. [DOI] [PubMed] [Google Scholar]
- 58. Higuma M, Sanjo N, Satoh K, Shiga Y, Sakai K, Nozaki I et al (2013) Relationships between clinicopathological features and cerebrospinal fluid biomarkers in Japanese patients with genetic prion diseases. PLoS One 8:e60003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hill AF, Desbruslais M, Joiner S, Sidle KC, Gowland I, Collinge J et al (1997) The same prion strain causes vCJD and BSE. Nature 389:448 – 450, 526. [DOI] [PubMed] [Google Scholar]
- 60. Hinnell C, Coulthart MB, Jansen GH, Cashman NR, Lauzon J, Clark A et al (2011) Gerstmann‐Straussler‐Scheinker disease due to a novel prion protein gene mutation. Neurology 76:485–487. [DOI] [PubMed] [Google Scholar]
- 61. Hoshi K, Yoshino H, Urata J, Nakamura Y, Yanagawa H, Sato T (2000) Creutzfeldt‐Jakob disease associated with cadaveric dura mater grafts in Japan. Neurology 55:718–721. [DOI] [PubMed] [Google Scholar]
- 62. Hsiao K, Baker HF, Crow TJ, Poulter M, Owen F, Terwilliger JD et al (1989) Linkage of a prion protein missense variant to Gerstmann‐Sträussler syndrome. Nature 338:342–345. [DOI] [PubMed] [Google Scholar]
- 63. Iwasaki Y, Mori K, Ito M, Nagaoka M, Ieda T, Kitamoto T et al (2011) An autopsied case of V180I Creutzfeldt‐Jakob disease presenting with panencephalopathic‐type pathology and a characteristic prion protein type. Neuropathology 31:540–548. [DOI] [PubMed] [Google Scholar]
- 64. Iwasaki Y, Kato H, Ando T, Akagi A, Mimuro M, Miyahara H et al (2018) Autopsy case of V180I genetic Creutzfeldt‐Jakob disease presenting with early disease pathology. Neuropathology 38:638–645. [DOI] [PubMed] [Google Scholar]
- 65. Jansen C, van Swieten JC, Capellari S, Strammiello R, Parchi P, Rozemuller AJM (2009) Inherited Creutzfeldt‐Jakob disease in a Dutch patient with a novel five octapeptide repeat insertion and unusual cerebellar morphology. J Neurol Neurosurg Psychiatry 80:1386–1389. [DOI] [PubMed] [Google Scholar]
- 66. Jansen C, Parchi P, Capellari S, Vermeij AJ, Corrado P, Baas F et al (2010) Prion protein amyloidosis with divergent phenotype associated with two novel nonsense mutations in PRNP. Acta Neuropathol 119:189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jansen C, Parchi P, Jelles B, Gouw AA, Beunders G, van Spaendonk RML et al (2011) The first case of fatal familial insomnia (FFI) in the Netherlands: a patient from Egyptian descent with concurrent four repeat tau deposits. Neuropathol Appl Neurobiol 37:549–553. [DOI] [PubMed] [Google Scholar]
- 68. Jaumain E, Quadrio I, Herzog L, Reine F, Rezaei H, Andréoletti O et al (2016) Absence of evidence for a causal link between bovine spongiform encephalopathy strain variant L‐BSE and known forms of sporadic Creutzfeldt‐Jakob disease in human PrP transgenic mice. J Virol 90:10867–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jaunmuktane Z, Mead S, Ellis M, Wadsworth JDF, Nicoll AJ, Kenny J et al (2015) Evidence for human transmission of amyloid‐β pathology and cerebral amyloid angiopathy. Nature 525:247–250. [DOI] [PubMed] [Google Scholar]
- 70. Jayadev S, Nochlin D, Poorkaj P, Steinbart EJ, Mastrianni JA, Montine TJ et al (2011) Familial prion disease with Alzheimer disease‐like tau pathology and clinical phenotype. Ann Neurol 69:712–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kim C, Haldiman T, Cohen Y, Chen W, Blevins J, Sy M‐S et al (2011) Protease‐sensitive conformers in broad spectrum of distinct PrPSc structures in sporadic Creutzfeldt‐Jakob disease are indicator of progression rate. PLoS Pathog 7:e1002242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kim M‐O, Cali I, Oehler A, Fong JC, Wong K, See T et al (2013) Genetic CJD with a novel E200G mutation in the prion protein gene and comparison with E200K mutation cases. Acta Neuropathol Commun 1:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kobayashi A, Asano M, Mohri S, Kitamoto T (2007) Cross‐sequence transmission of sporadic Creutzfeldt‐Jakob disease creates a new prion strain. J Biol Chem 282:30022–30028. [DOI] [PubMed] [Google Scholar]
- 74. Kobayashi A, Arima K, Ogawa M, Murata M, Fukuda T, Kitamoto T (2008) Plaque‐type deposition of prion protein in the damaged white matter of sporadic Creutzfeldt‐Jakob disease MM1 patients. Acta Neuropathol 116:561–566. [DOI] [PubMed] [Google Scholar]
- 75. Kobayashi A, Asano M, Mohri S, Kitamoto T (2009) A traceback phenomenon can reveal the origin of prion infection. Neuropathology 29:619–624. [DOI] [PubMed] [Google Scholar]
- 76. Kobayashi A, Sakuma N, Matsuura Y, Mohri S, Aguzzi A, Kitamoto T (2010) Experimental verification of a traceback phenomenon in prion infection. J Virol 84:3230–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kobayashi A, Iwasaki Y, Otsuka H, Yamada M, Yoshida M, Matsuura Y et al (2013) Deciphering the pathogenesis of sporadic Creutzfeldt‐Jakob disease with codon 129 M/V and type 2 abnormal prion protein. Acta Neuropathol Commun 1:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kobayashi A, Matsuura Y, Mohri S, Kitamoto T (2014) Distinct origins of dura mater graft‐associated Creutzfeldt‐Jakob disease: past and future problems. Acta Neuropathol Commun 2:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kobayashi A, Teruya K, Matsuura Y, Shirai T, Nakamura Y, Yamada M et al (2015) The influence of PRNP polymorphisms on human prion disease susceptibility: an update. Acta Neuropathol 130:159–170. [DOI] [PubMed] [Google Scholar]
- 80. Kobayashi A, Matsuura Y, Iwaki T, Iwasaki Y, Yoshida M, Takahashi H et al (2016) Sporadic Creutzfeldt‐Jakob disease MM1+2C and MM1 are identical in transmission properties. Brain Pathol 26:95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kobayashi A, Parchi P, Yamada M, Mohri S, Kitamoto T (2016) Neuropathological and biochemical criteria to identify acquired Creutzfeldt‐Jakob disease among presumed sporadic cases. Neuropathology 36:305–310. [DOI] [PubMed] [Google Scholar]
- 82. Korth C, Kaneko K, Groth D, Heye N, Telling G, Mastrianni J et al (2003) Abbreviated incubation times for human prions in mice expressing a chimeric mouse‐human prion protein transgene. Proc Natl Acad Sci USA 100:4784–4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kovacs GG, Seguin J, Quadrio I, Höftberger R, Kapás I, Streichenberger N et al (2011) Genetic Creutzfeldt‐Jakob disease associated with the E200K mutation: characterization of a complex proteinopathy. Acta Neuropathol 121:39–57. [DOI] [PubMed] [Google Scholar]
- 84. Kovacs GG, Lutz MI, Ricken G, Ströbel T, Höftberger R, Preusser M et al (2016) Dura mater is a potential source of Aβ seeds. Acta Neuropathol 131:911–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kovacs GG, Rahimi J, Ströbel T, Lutz MI, Regelsberger G, Streichenberger N et al (2017) Tau pathology in Creutzfeldt‐Jakob disease revisited. Brain Pathol 27:332–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kretzschmar HA, Sethi S, Földvári Z, Windl O, Querner V, Zerr I, Poser S (2003) Iatrogenic Creutzfeldt‐Jakob disease with florid plaques. Brain Pathol 13:245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lattanzio F, Abu‐Rumeileh S, Franceschini A, Kai H, Amore G, Poggiolini I et al (2017) Prion‐specific and surrogate CSF biomarkers in Creutzfeldt‐Jakob disease: diagnostic accuracy in relation to molecular subtypes and analysis of neuropathological correlates of p‐tau and Aβ42 levels. Acta Neuropathol 133:559–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Llorens F, López‐González I, Thüne K, Carmona M, Zafar S, Andréoletti O et al (2014) Subtype and regional‐specific neuroinflammation in sporadic creutzfeldt‐jakob disease. Front Aging Neurosci 6:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Mackenzie G, Will R (2017) Creutzfeldt‐Jakob disease: recent developments. F1000Res 6:2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Masters CL, Gajdusek DC, Gibbs CJ (1981) Creutzfeldt‐Jakob disease virus isolations from the Gerstmann‐Sträussler syndrome with an analysis of the various forms of amyloid plaque deposition in the virus‐induced spongiform encephalopathies. Brain 104:559–588. [DOI] [PubMed] [Google Scholar]
- 91. Mastrianni JA, Nixon R, Layzer R, Telling GC, Han D, DeArmond SJ, Prusiner SB (1999) Prion protein conformation in a patient with sporadic fatal insomnia. N Engl J Med 340:1630–1638. [DOI] [PubMed] [Google Scholar]
- 92. Mastrianni JA, Capellari S, Telling GC, Han D, Bosque P, Prusiner SB, DeArmond SJ (2001) Inherited prion disease caused by the V210I mutation: transmission to transgenic mice. Neurology 57:2198–2205. [DOI] [PubMed] [Google Scholar]
- 93. Matěj R, Kovacs GG, Johanidesová S, Keller J, Matějčková M, Nováková J et al (2012) Genetic Creutzfeldt‐Jakob disease with R208H mutation presenting as progressive supranuclear palsy. Mov Disord 27:476–479. [DOI] [PubMed] [Google Scholar]
- 94. Matsuzono K, Ikeda Y, Liu W, Kurata T, Deguchi S, Deguchi K, Abe K (2013) A novel familial prion disease causing pan‐autonomic‐sensory neuropathy and cognitive impairment. Eur J Neurol 20:e67–69. [DOI] [PubMed] [Google Scholar]
- 95. Matsuzono K, Honda H, Sato K, Morihara R, Deguchi K, Hishikawa N et al (2016) “PrP systemic deposition disease”: clinical and pathological characteristics of novel familial prion disease with 2‐bp deletion in codon 178. Eur J Neurol 23:196–200. [DOI] [PubMed] [Google Scholar]
- 96. Mead S, Poulter M, Beck J, Webb TEF, Campbell TA, Linehan JM et al (2006) Inherited prion disease with six octapeptide repeat insertional mutation–molecular analysis of phenotypic heterogeneity. Brain 129:2297–2317. [DOI] [PubMed] [Google Scholar]
- 97. Mead S, Gandhi S, Beck J, Caine D, Gallujipali D, Carswell C et al (2013) A novel prion disease associated with diarrhea and autonomic neuropathy. N Engl J Med 369:1904–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Mercer RCC, Daude N, Dorosh L, Fu Z‐L, Mays CE, Gapeshina H et al (2018) A novel Gerstmann‐Sträussler‐Scheinker disease mutation defines a precursor for amyloidogenic 8 kDa PrP fragments and reveals N‐terminal structural changes shared by other GSS alleles. PLoS Pathog 14:e1006826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Meyer‐Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E et al (2006) Exogenous induction of cerebral beta‐amyloidogenesis is governed by agent and host. Science 313:1781–1784. [DOI] [PubMed] [Google Scholar]
- 100. Miguelez‐Rodriguez A, Santos‐Juanes J, Vicente‐Etxenausia I, Perez de Heredia‐Goñi K, Garcia B, Quiros LM et al (2018) Brains with sporadic Creutzfeldt‐Jakob disease and copathology showed a prolonged end‐stage of disease. J Clin Pathol 71:446–450. [DOI] [PubMed] [Google Scholar]
- 101. Minikel EV, Vallabh SM, Lek M, Estrada K, Samocha KE, Sathirapongsasuti JF et al (2016) Quantifying prion disease penetrance using large population control cohorts. Sci Transl Med 8:322ra9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Miyazono M, Iwaki T, Kitamoto T, Kaneko Y, Doh‐ura K, Tateishi J (1991) A comparative immunohistochemical study of Kuru and senile plaques with a special reference to glial reactions at various stages of amyloid plaque formation. Am J Pathol 139:589–598. [PMC free article] [PubMed] [Google Scholar]
- 103. Mochizuki Y, Mizutani T, Tajiri N, Oinuma T, Nemoto N, Kakimi S, Kitamoto T (2003) Creutzfeldt‐Jakob disease with florid plaques after cadaveric dura mater graft. Neuropathology 23:136–140. [DOI] [PubMed] [Google Scholar]
- 104. Moda F, Suardi S, Di Fede G, Indaco A, Limido L, Vimercati C et al (2012) MM2‐thalamic Creutzfeldt‐Jakob disease: neuropathological, biochemical and transmission studies identify a distinctive prion strain. Brain Pathol 22:662–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Mok T, Jaunmuktane Z, Joiner S, Campbell T, Morgan C, Wakerley B et al (2017) Variant Creutzfeldt‐Jakob disease in a patient with heterozygosity at PRNP codon 129. N Engl J Med 376:292–294. [DOI] [PubMed] [Google Scholar]
- 106. Morales R, Estrada LD, Diaz‐Espinoza R, Morales‐Scheihing D, Jara MC, Castilla J, Soto C (2010) Molecular cross talk between misfolded proteins in animal models of Alzheimer’s and prion diseases. J Neurosci 30:4528–4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Mühleisen H, Gehrmann J, Meyermann R (1995) Reactive microglia in Creutzfeldt‐Jakob disease. Neuropathol Appl Neurobiol 21:505–517. [DOI] [PubMed] [Google Scholar]
- 108. Muñoz‐Nieto M, Ramonet N, López‐Gastón JI, Cuadrado‐Corrales N, Calero O, Díaz‐Hurtado M et al (2013) A novel mutation I215V in the PRNP gene associated with Creutzfeldt‐Jakob and Alzheimer’s diseases in three patients with divergent clinical phenotypes. J Neurol 260:77–84. [DOI] [PubMed] [Google Scholar]
- 109. Nitrini R, Rosemberg S, Passos‐Bueno MR, da Silva LS, Iughetti P, Papadopoulos M et al (1997) Familial spongiform encephalopathy associated with a novel prion protein gene mutation. Ann Neurol 42:138–146. [DOI] [PubMed] [Google Scholar]
- 110. Nonno R, Di Bari MA, Cardone F, Vaccari G, Fazzi P, Dell’Omo G et al (2006) Efficient transmission and characterization of Creutzfeldt‐Jakob disease strains in bank voles. PLoS Pathog 2:e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Notari S, Capellari S, Giese A, Westner I, Baruzzi A, Ghetti B et al (2004) Effects of different experimental conditions on the PrPSc core generated by protease digestion: implications for strain typing and molecular classification of CJD. J Biol Chem 279:16797–16804. [DOI] [PubMed] [Google Scholar]
- 112. Notari S, Capellari S, Langeveld J, Giese A, Strammiello R, Gambetti P et al (2007) A refined method for molecular typing reveals that co‐occurrence of PrP(Sc) types in Creutzfeldt‐Jakob disease is not the rule. Lab Invest 87:1103–1112. [DOI] [PubMed] [Google Scholar]
- 113. Notari S, Strammiello R, Capellari S, Giese A, Cescatti M, Grassi J et al (2008) Characterization of truncated forms of abnormal prion protein in Creutzfeldt‐Jakob disease. J Biol Chem 283:30557–30565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Notari S, Xiao X, Espinosa JC, Cohen Y, Qing L, Aguilar‐Calvo P et al (2014) Transmission characteristics of variably protease‐sensitive prionopathy. Emerging Infect Dis 20:2006–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Parchi P, Castellani R, Cortelli P, Montagna P, Chen SG, Petersen RB et al (1995) Regional distribution of protease‐resistant prion protein in fatal familial insomnia. Ann Neurol 38:21–29. [DOI] [PubMed] [Google Scholar]
- 116. Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG et al (1996) Molecular basis of phenotypic variability in sporadic Creutzfeldt‐Jakob disease. Ann Neurol 39:767–778. [DOI] [PubMed] [Google Scholar]
- 117. Parchi P, Capellari S, Chen SG, Petersen RB, Gambetti P, Kopp N et al (1997) Typing prion isoforms. Nature 386:232–234. [DOI] [PubMed] [Google Scholar]
- 118. Parchi P, Chen SG, Brown P, Zou W, Capellari S, Budka H et al (1998) Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann‐Sträussler‐Scheinker disease. Proc Natl Acad Sci USA 95:8322–8327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Parchi P, Petersen RB, Chen SG, Autilio‐Gambetti L, Capellari S, Monari L et al (1998) Molecular pathology of fatal familial insomnia. Brain Pathol 8:539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Parchi P, Giese A, Capellari S, Brown P, Schulz‐Schaeffer W, Windl O et al (1999) Classification of sporadic Creutzfeldt‐Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 46:224–233. [PubMed] [Google Scholar]
- 121. Parchi P, Capellari S, Chin S, Schwarz HB, Schecter NP, Butts JD et al (1999) A subtype of sporadic prion disease mimicking fatal familial insomnia. Neurology 52:1757–1763. [DOI] [PubMed] [Google Scholar]
- 122. Parchi P, Zou W, Wang W, Brown P, Capellari S, Ghetti B et al (2000) Genetic influence on the structural variations of the abnormal prion protein. Proc Natl Acad Sci USA 97:10168–10172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Parchi P, Strammiello R, Notari S, Giese A, Langeveld JPM, Ladogana A et al (2009) Incidence and spectrum of sporadic Creutzfeldt‐Jakob disease variants with mixed phenotype and co‐occurrence of PrPSc types: an updated classification. Acta Neuropathol 118:659–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Parchi P, Cescatti M, Notari S, Schulz‐Schaeffer WJ, Capellari S, Giese A et al (2010) Agent strain variation in human prion disease: insights from a molecular and pathological review of the National Institutes of Health series of experimentally transmitted disease. Brain 133:3030–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Parchi P, de Boni L, Saverioni D, Cohen ML, Ferrer I, Gambetti P et al (2012) Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: an inter‐rater study among surveillance centres in Europe and USA. Acta Neuropathol 124:517–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Parchi P, Saverioni D (2012) Molecular pathology, classification, and diagnosis of sporadic human prion disease variants. Folia Neuropathol 50:20–45. [PubMed] [Google Scholar]
- 127. Pastrana MA, Sajnani G, Onisko B, Castilla J, Morales R, Soto C, Requena JR (2006) Isolation and characterization of a proteinase K‐sensitive PrPSc fraction. Biochemistry 45:15710–15717. [DOI] [PubMed] [Google Scholar]
- 128. Peden AH, Sarode DP, Mulholland CR, Barria MA, Ritchie DL, Ironside JW, Head MW (2014) The prion protein protease sensitivity, stability and seeding activity in variably protease sensitive prionopathy brain tissue suggests molecular overlaps with sporadic Creutzfeldt‐Jakob disease. Acta Neuropathol Commun 2:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Peoc’h K, Levavasseur E, Delmont E, De Simone A, Laffont‐Proust I, Privat N et al (2012) Substitutions at residue 211 in the prion protein drive a switch between CJD and GSS syndrome, a new mechanism governing inherited neurodegenerative disorders. Hum Mol Genet 21:5417–5428. [DOI] [PubMed] [Google Scholar]
- 130. Peretz D, Scott MR, Groth D, Williamson RA, Burton DR, Cohen FE, Prusiner SB (2001) Strain‐specified relative conformational stability of the scrapie prion protein. Protein Sci 10:854–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Piccardo P, Dlouhy SR, Lievens PM, Young K, Bird TD, Nochlin D et al (1998) Phenotypic variability of Gerstmann‐Sträussler‐Scheinker disease is associated with prion protein heterogeneity. J Neuropathol Exp Neurol 57:979–988. [DOI] [PubMed] [Google Scholar]
- 132. Piccardo P, Liepnieks JJ, William A, Dlouhy SR, Farlow MR, Young K et al (2001) Prion proteins with different conformations accumulate in Gerstmann‐Sträussler‐Scheinker disease caused by A117V and F198S mutations. Am J Pathol 158:2201–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Pirisinu L, Di Bari M, Marcon S, Vaccari G, D’Agostino C, Fazzi P et al (2010) A new method for the characterization of strain‐specific conformational stability of protease‐sensitive and protease‐resistant PrPSc. PLoS One 5:e12723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Pirisinu L, Nonno R, Esposito E, Benestad SL, Gambetti P, Agrimi U, Zou W‐Q (2013) Small ruminant nor98 prions share biochemical features with human gerstmann‐sträussler‐scheinker disease and variably protease‐sensitive prionopathy. PLoS One 8:e66405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Pirisinu L, Di Bari MA, D’Agostino C, Marcon S, Riccardi G, Poleggi A et al (2016) Gerstmann‐Sträussler‐Scheinker disease subtypes efficiently transmit in bank voles as genuine prion diseases. Sci Rep 6:20443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Polymenidou M, Prokop S, Jung HH, Hewer E, Peretz D, Moos R et al (2011) Atypical prion protein conformation in familial prion disease with PRNP P105T mutation. Brain Pathol 21:209–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Prusiner SB (1998) Prions. Proc Natl Acad Sci USA 95:13363–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Race B, Williams K, Hughson AG, Jansen C, Parchi P, Rozemuller AJM, Chesebro B (2018) Familial human prion diseases associated with prion protein mutations Y226X and G131V are transmissible to transgenic mice expressing human prion protein. Acta Neuropathol Commun 6:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Rasmussen J, Krasemann S, Altmeppen H, Schwarz P, Schelle J, Aguzzi A et al (2018) Infectious prions do not induce Aβ deposition in an in vivo seeding model. Acta Neuropathol 135:965–967. [DOI] [PubMed] [Google Scholar]
- 140. Reiniger L, Lukic A, Linehan J, Rudge P, Collinge J, Mead S, Brandner S (2011) Tau, prions and Aβ: the triad of neurodegeneration. Acta Neuropathol 121:5–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Revesz T, Holton JL, Lashley T, Plant G, Frangione B, Rostagno A, Ghiso J (2009) Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol 118:115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Ritchie DL, Adlard P, Peden AH, Lowrie S, Le Grice M, Burns K et al (2017) Amyloid‐β accumulation in the CNS in human growth hormone recipients in the UK. Acta Neuropathol 134:221–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Ritchie DL, Barria MA, Peden AH, Yull HM, Kirkpatrick J, Adlard P et al (2017) UK Iatrogenic Creutzfeldt‐Jakob disease: investigating human prion transmission across genotypic barriers using human tissue‐based and molecular approaches. Acta Neuropathol 133:579–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Rodriguez‐Diehl R, Rey MJ, Gironell A, Martinez‐Saez E, Ferrer I, Sánchez‐Valle R et al (2012) “Preclinical” MSA in definite Creutzfeldt‐Jakob disease. Neuropathology 32:158–163. [DOI] [PubMed] [Google Scholar]
- 145. Roeber S, Krebs B, Neumann M, Windl O, Zerr I, Grasbon‐Frodl E‐M, Kretzschmar HA (2005) Creutzfeldt‐Jakob disease in a patient with an R208H mutation of the prion protein gene (PRNP) and a 17‐kDa prion protein fragment. Acta Neuropathol 109:443–448. [DOI] [PubMed] [Google Scholar]
- 146. Rossi M, Saverioni D, Di Bari M, Baiardi S, Lemstra AW, Pirisinu L et al (2017) Atypical Creutzfeldt‐Jakob disease with PrP‐amyloid plaques in white matter: molecular characterization and transmission to bank voles show the M1 strain signature. Acta Neuropathol Commun 5:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Safar J, Prusiner SB (1998) Molecular studies of prion diseases. Prog Brain Res 117:421–434. [DOI] [PubMed] [Google Scholar]
- 148. Safar JG, Geschwind MD, Deering C, Didorenko S, Sattavat M, Sanchez H et al (2005) Diagnosis of human prion disease. Proc Natl Acad Sci USA 102:3501–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Safar JG, Xiao X, Kabir ME, Chen S, Kim C, Haldiman T et al (2015) Structural determinants of phenotypic diversity and replication rate of human prions. PLoS Pathog 11:e1004832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Sarell CJ, Quarterman E, Yip DC‐M, Terry C, Nicoll AJ, Wadsworth JDF et al (2017) Soluble Aβ aggregates can inhibit prion propagation. Open Biol 7:170158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Sasaki A, Hirato J, Nakazato Y (1993) Immunohistochemical study of microglia in the Creutzfeldt‐Jakob diseased brain. Acta Neuropathol 86:337–344. [DOI] [PubMed] [Google Scholar]
- 152. Saverioni D, Notari S, Capellari S, Poggiolini I, Giese A, Kretzschmar HA, Parchi P (2013) Analyses of protease resistance and aggregation state of abnormal prion protein across the spectrum of human prions. J Biol Chem 288:27972–27985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Scheckel C, Aguzzi A (2018) Prions, prionoids and protein misfolding disorders. Nat Rev Genet 19:405–418. [DOI] [PubMed] [Google Scholar]
- 154. Shi Q, Xie W‐L, Zhang B, Chen L‐N, Xu Y, Wang K et al (2013) Brain microglia were activated in sporadic CJD but almost unchanged in fatal familial insomnia and G114V genetic CJD. Virol J 10:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Shimizu S, Hoshi K, Muramoto T, Homma M, Ironside JW, Kuzuhara S et al (1999) Creutzfeldt‐Jakob disease with florid‐type plaques after cadaveric dura mater grafting. Arch Neurol 56:357–362. [DOI] [PubMed] [Google Scholar]
- 156. Skworc KH, Windl O, Schulz‐Schaeffer WJ, Giese A, Bergk J, Nagele A et al (1999) Familial Creutzfeldt‐Jakob disease with a novel 120‐bp insertion in the prion protein gene. Ann Neurol 46:693–700. [PubMed] [Google Scholar]
- 157. Szpak GM, Lewandowska E, Lechowicz W, Wierzba‐Bobrowicz T, Kulczycki J, Bertrand E et al (2006) The brain immune response in human prion diseases. Microglial activation and microglial disease. I. Sporadic Creutzfeldt‐Jakob disease. Folia Neuropathol 44:202–213. [PubMed] [Google Scholar]
- 158. Tagliavini F, Prelli F, Ghiso J, Bugiani O, Serban D, Prusiner SB et al (1991) Amyloid protein of Gerstmann‐Sträussler‐Scheinker disease (Indiana kindred) is an 11 kd fragment of prion protein with an N‐terminal glycine at codon 58. EMBO J 10:513–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Tagliavini F, Lievens PM, Tranchant C, Warter JM, Mohr M, Giaccone G et al (2001) A 7‐kDa prion protein (PrP) fragment, an integral component of the PrP region required for infectivity, is the major amyloid protein in Gerstmann‐Sträussler‐Scheinker disease A117V. J Biol Chem 276:6009–6015. [DOI] [PubMed] [Google Scholar]
- 160. Takeuchi A, Kobayashi A, Ironside JW, Mohri S, Kitamoto T (2013) Characterization of variant Creutzfeldt‐Jakob disease prions in prion protein‐humanized mice carrying distinct codon 129 genotypes. J Biol Chem 288:21659–21666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Takeuchi A, Kobayashi A, Parchi P, Yamada M, Morita M, Uno S, Kitamoto T (2016) Distinctive properties of plaque‐type dura mater graft‐associated Creutzfeldt‐Jakob disease in cell‐protein misfolding cyclic amplification. Lab Invest 96:581–587. [DOI] [PubMed] [Google Scholar]
- 162. Tateishi J, Kitamoto T, Hoque MZ, Furukawa H (1996) Experimental transmission of Creutzfeldt‐Jakob disease and related diseases to rodents. Neurology 46:532–537. [DOI] [PubMed] [Google Scholar]
- 163. Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R et al (1996) Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 274:2079–2082. [DOI] [PubMed] [Google Scholar]
- 164. Tian C, Liu D, Sun Q‐L, Chen C, Xu Y, Wang H et al (2013) Comparative analysis of gene expression profiles between cortex and thalamus in Chinese fatal familial insomnia patients. Mol Neurobiol 48:36–48. [DOI] [PubMed] [Google Scholar]
- 165. Tzaban S, Friedlander G, Schonberger O, Horonchik L, Yedidia Y, Shaked G et al (2002) Protease‐sensitive scrapie prion protein in aggregates of heterogeneous sizes. Biochemistry 41:12868–12875. [DOI] [PubMed] [Google Scholar]
- 166. Van Everbroeck B, Dobbeleir I, De Waele M, De Leenheir E, Lübke U, Martin J‐J, Cras P (2004) Extracellular protein deposition correlates with glial activation and oxidative stress in Creutzfeldt‐Jakob and Alzheimer’s disease. Acta Neuropathol 108:194–200. [DOI] [PubMed] [Google Scholar]
- 167. Vicente‐Pascual M, Rossi M, Gámez J, Lladó A, Valls J, Grau‐Rivera O et al (2018) Variably protease‐sensitive prionopathy presenting within ALS/FTD spectrum. Ann Clin Transl Neurol 5:1297–1302. 10.1002/acn3.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Vita MG, Tiple D, Bizzarro A, Ladogana A, Colaizzo E, Capellari S et al (2017) Patient with rapidly evolving neurological disease with neuropathological lesions of Creutzfeldt‐Jakob disease, Lewy body dementia, chronic subcortical vascular encephalopathy and meningothelial meningioma. Neuropathology 37:110–115. [DOI] [PubMed] [Google Scholar]
- 169. Vital A, Canron M‐H, Gil R, Hauw J‐J, Vital C (2007) A sporadic case of Creutzfeldt‐Jakob disease with beta‐amyloid deposits and alpha‐synuclein inclusions. Neuropathology 27:273–277. [DOI] [PubMed] [Google Scholar]
- 170. Wadsworth JD, Joiner S, Hill AF, Campbell TA, Desbruslais M, Luthert PJ, Collinge J (2001) Tissue distribution of protease resistant prion protein in variant Creutzfeldt‐Jakob disease using a highly sensitive immunoblotting assay. Lancet 358:171–180. [DOI] [PubMed] [Google Scholar]
- 171. Wadsworth JDF, Asante EA, Desbruslais M, Linehan JM, Joiner S, Gowland I et al (2004) Human prion protein with valine 129 prevents expression of variant CJD phenotype. Science 306:1793–1796. [DOI] [PubMed] [Google Scholar]
- 172. Wadsworth JDF, Joiner S, Linehan JM, Desbruslais M, Fox K, Cooper S et al (2008) Kuru prions and sporadic Creutzfeldt‐Jakob disease prions have equivalent transmission properties in transgenic and wild‐type mice. Proc Natl Acad Sci USA 105:3885–3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Walker LC, Jucker M (2015) Neurodegenerative diseases: expanding the prion concept. Annu Rev Neurosci 38:87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Watts JC, Giles K, Patel S, Oehler A, DeArmond SJ, Prusiner SB (2014) Evidence that bank vole PrP is a universal acceptor for prions. PLoS Pathog 10:e1003990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Watts JC, Giles K, Serban A, Patel S, Oehler A, Bhardwaj S et al (2015) Modulation of Creutzfeldt‐Jakob disease prion propagation by the A224V mutation. Ann Neurol 78:540–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A et al (1996) A new variant of Creutzfeldt‐Jakob disease in the UK. Lancet 347:921–925. [DOI] [PubMed] [Google Scholar]
- 177. Xiao X, Cali I, Dong Z, Puoti G, Yuan J, Qing L et al (2013) Protease‐sensitive prions with 144‐bp insertion mutations. Aging (Albany NY) 5:155–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Yamada M, Noguchi‐Shinohara M, Hamaguchi T, Nozaki I, Kitamoto T, Sato T et al (2009) Dura mater graft‐associated Creutzfeldt‐Jakob disease in Japan: clinicopathological and molecular characterization of the two distinct subtypes. Neuropathology 29:609–618. [DOI] [PubMed] [Google Scholar]
- 179. Yull HM, Ritchie DL, Langeveld JPM, van Zijderveld FG, Bruce ME, Ironside JW, Head MW (2006) Detection of type 1 prion protein in variant Creutzfeldt‐Jakob disease. Am J Pathol 168:151–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Zanusso G, Polo A, Farinazzo A, Nonno R, Cardone F, Di Bari M et al (2007) Novel prion protein conformation and glycotype in Creutzfeldt‐Jakob disease. Arch Neurol 64:595–599. [DOI] [PubMed] [Google Scholar]
- 181. Zou WQ, Capellari S, Parchi P, Sy MS, Gambetti P, Chen SG (2003) Identification of novel proteinase K‐resistant C‐terminal fragments of PrP in Creutzfeldt‐Jakob disease. J Biol Chem 278:40429–40436. [DOI] [PubMed] [Google Scholar]
- 182. Zou W‐Q, Puoti G, Xiao X, Yuan J, Qing L, Cali I et al (2010) Variably protease‐sensitive prionopathy: a new sporadic disease of the prion protein. Ann Neurol 68:162–172. [DOI] [PMC free article] [PubMed] [Google Scholar]