

Abstract
Graft inflammation impairs the induction of solid organ transplant tolerance and enhances acute and chronic rejection. Elucidating the mechanisms by which inflammation is induced after organ transplantation could lead to novel therapeutics to improve transplant outcomes. In this Review we describe endogenous substances — damage-associated molecular patterns (DAMPs) — that are released after allograft reperfusion and induce inflammation. We also describe innate immune signalling pathways that are activated after solid organ transplantation, with a focus on Toll-like receptors (TLRs) and their signal adaptor, MYD88. Experimental and clinical studies have yielded a large body of evidence that TLRs and MYD88 are instrumental in initiating allograft inflammation and promoting the development of acute and chronic rejection. Ongoing clinical studies are testing TLR inhibition strategies in solid organ transplantation, although avoiding compromising host defence to pathogens is a key challenge. Further elucidation of the mechanisms by which sterile inflammation is induced, maintained and amplified within the allograft has the potential to lead to novel anti-inflammatory treatments that could improve outcomes for solid organ transplant recipients.
Activation of the innate immune system after solid organ transplantation — a vital therapy for end-stage conditions — results in sterile inflammation, which enhances acute and chronic rejection and impairs transplant tolerance1–5. Our understanding of how the innate immune system contributes to the immune response after organ transplantation has increased over the past decade1,6,7; however, very little is known about the exact mechanisms by which inflammation is induced and maintained following organ transplantation.
In transplanted organs, sterile inflammation results from ischaemia–reperfusion injury (IRI), which occurs after the blood supply of an organ ceases and is then abruptly re-established. IRI is an obligatory component of the harvest and implantation of organs8 that has important implications for the fate of the transplant. Indeed, increasing ischaemic time, and thus IRI severity, enhances the risk of acute and chronic allograft rejection9. In human kidney transplantation, increasing cold ischaemic time from 6 h to 30 h augmented the risk of graft failure by 40%10. Ischaemic time also affects the fate of transplanted human hearts11–13. IRI of a graft is exacerbated by brain death in deceased donors14 and contributes to worse graft survival than with organs from living donors15. How brain death affects graft function requires further study, but experimental evidence shows that brain death activates a systemic immune response, with production of IFN-γ16. Mechanistically, the early induction of graft necrosis by IRI causes sterile inflammation by releasing ‘endogenous’ innate immune ligands known as damage-associated molecular patterns (DAMPs) (FIG. 1). DAMPs (BOX 1) are nuclear or cytosolic molecules that are typically hidden from the immune system inside cells. The signalling pathways triggered by DAMPs lead to migration of innate immune cells to kidney allografts, a process that precedes T-cell recruitment, which ultimately drives graft rejection17.
Figure 1 |. Inflammation initiation and maintenance after organ transplantation.
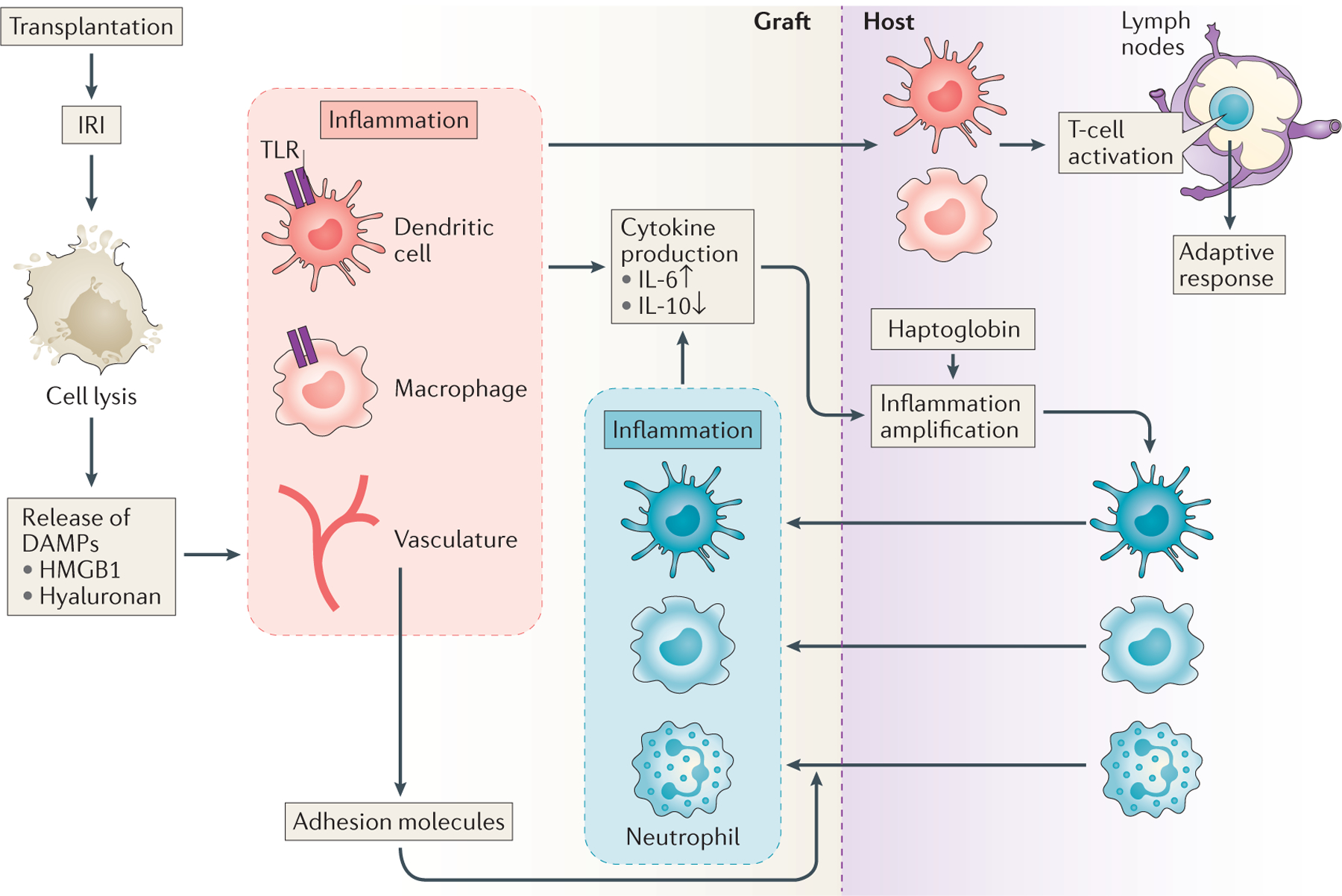
Ischaemia–reperfusion injury (IRI) induces a degree of graft cell necrosis and breakdown of the extracellular matrix. These processes lead to the release of damage-associated molecular patterns (DAMPs), which can heighten the intragraft inflammatory milieu by activating innate immune signalling pathways via the toll-like receptors (TLR). Graft cells (vascular endothelial cells, macrophages and dendritic cells) upregulate adhesion molecules (such as VCAM-1 and L-selectin) and co-stimulatory molecules (such as CD80). They also secrete inflammatory cytokines (such as IL-6 and TNF), which enhance immune cell recruitment into the graft and activate activate recipient cells to produce amplifiers of inflammation such as haptoglobin, which enter the circulation and amplify inflammation within the transplant. This increased inflammation further activates graft resident or infiltrated immune cells and induces their migration to the draining lymph nodes where they can activate naive anti-donor T cells to initiate adaptive anti-donor immune responses. The inflammatory reaction triggered by IRI can ultimately lead to graft rejection.
Box 1 |. Effectors of the sterile inflammation response.
PAMPs and DAMPs
Toll-like receptors (TLRs) and other innate immune receptors are collectively termed pattern recognition receptors (PRRs) as they recognize molecular patterns. When such patterns are expressed by pathogens, they are termed pathogen-associated molecular patterns (PAMPs). For example, lipopolysaccharide is a major component of the cell wall of Gram-negative bacteria and is the classic PAMP ligand for TLR4. When patterns are derived from endogenous sources following tissue damage, they are termed damage-associated molecular patterns (DAMPs). For example, HMGB1, a nuclear protein that binds DNA and modulates transcription in healthy cells, is released into the extracellular environment in response to cell damage and acts as a ligand for TLR4 and TLR9.
Toll-like receptors
TLRs are one of several classes of innate immune receptors that serve primarily as sentinels of the immune system and respond rapidly to the presence of non-self molecules, tissue damage or tissue stress125. They are highly conserved, germ-line encoded, transmembrane PRRs that are capable of recognizing PAMPs or DAMPs66. TLRs are characterized by a ligand-binding N-terminal leucine-rich repeats signalling domain, a transmembrane domain and an intracytoplasmic tail consisting of a Toll/IL-1R homology domain66. TLR1, TLR2, TLR4, TLR5, TLR6 and TLR11 span the plasma membrane and are ideally suited to respond to ligands present within the extracellular environment. TLR3 and TLRs 7–10 span endolysosomal membranes and are thereby well suited to respond to ligands composed of nucleic acids derived from invading organisms or damaged cells that have been processed within the endolysosome66.
Clinical and experimental evidence shows that inflammation has a critical effect on the fate of grafted organs. Thus, understanding how inflammation is initiated, amplified and resolved after solid organ transplantation could lead to new therapeutic paradigms and improve outcomes for recipients of solid organ transplants. In this Review, we discuss the contribution of sterile inflammation in solid organ transplantation, with a particular focus on DAMPs and their downstream signalling cascade triggered by Toll-like receptor (TLR) activation. Although pathogens induce inflammation and have a detrimental effect on transplant outcomes, we will not cover infectious inflammation as this subject has been reviewed elsewhere3. Likewise, the role of the complement system — another important component of the innate immune system — in organ transplantation has already been covered18. First, we discuss factors that are involved in activating and amplifying allograft inflammation. Then, we address specific innate immune signalling pathways that are activated after experimental organ transplantation with a focus on TLRs. We also consider clinical studies that have yielded evidence for the role of DAMPs and TLRs after solid organ transplantation. Lastly, we address future studies focused on developing novel therapeutics to dampen inflammation and improve outcomes for solid organ transplant recipients.
Inflammation activators and amplifiers
After an organ is transplanted, it undergoes IRI, which leads to a degree of graft necrosis and the subsequent release of DAMPs, which initiate sterile inflammation (FIG. 1). An increasing number of organs are perfused ex vivo to increase the donor organ pool19. With this new ex vivo approach, the therapeutic window of opportunity to treat organs (such as lungs or kidney) before implantation to reduce the deleterious effects of IRI may expand. Thus, identifying the key innate immune ligands that are released at the time of organ implantation could lead to novel therapies that, if applied locally to the organ before transplantation, could reduce graft inflammation, enable the dosages of systemic immunosuppressive medications to be decreased and improve patient outcomes.
Once released, DAMPs activate various graft cells such as endothelial cells and immune cells (macrophages and dendritic cells)2. Activation of graft-resident immune cells induces the production of chemokines, which recruit host immune cells such as neutrophils and macrophages into the graft. These host immune cells enhance inflammation and further promote the immune response to the graft (FIG. 1). Activation of the graft vasculature leads to the upregulation of adhesion molecules such as lymphocyte function-associated antigen 1 (LFA-1) and vascular cell adhesion protein 1 (VCAM-1) (REFS 20,21), which promote neutrophil attachment to the graft vasculature and facilitate neutrophil entry to the graft (FIG. 1). Importantly, host macrophages and dendritic cells that exit the graft after the initial inflammatory response following graft implantation traffic to the local draining lymph nodes where they initiate anti-donor adaptive immune responses22,23 (FIG. 1).
DAMPs released after organ transplantation
HMGB1.
High mobility group box 1 (HMGB1), a nuclear protein that regulates gene transcription, is one of the best-characterized of the intracellular DAMPs that induce inflammation in transplanted organs and affect the rate of development of acute allograft rejection. In non-transplanted murine models of hepatic and renal IRI, HMGB1 levels are increased in the damaged organs and pharmacological blockade of HMGB1 attenuates inflammation24–27. These results, together with findings from other studies which indicate that HMGB1 induces inflammation either by activating TLR4 (REF. 28) or the receptor for advanced glycation end-products (RAGE)29, have led researchers to examine the function of HMGB1 in experimental organ transplantation. A 2014 study found that inhibition of necroptosis, a form of regulated cell necrosis, reduced Hmgb1 expression in cardiac isografts and administration of recombinant HMGB1 increased the expression of the proinflammatory cytokine IL-17 in the grafts30. Consistent with these findings, other studies have shown that hypothermic preservation of cardiac allografts increases Hmgb1 expression after graft implantation, and IRI leads to release of HMGB1 from cardiac allografts31. Importantly, pharmacological blockade of HMGB1 activity extended cardiac allograft survival by 1 week, with reduced graft inflammation, in a full MHC-mismatched murine cardiac-transplant model32. In agreement with this latter study, another study of cardiac transplantation in mice found that inhibition of RAGE extended the median survival of cardiac allografts to almost 3 weeks, compared to just 1 week for allografts transplanted under control conditions33. Of note, RAGE is not critical for the induction of renal injury in non-transplant models of IRI in mice34 and a definitive role for RAGE in the inflammatory response to renal transplantation is yet to be determined. Additionally, in a rat-to-mouse cardiac xenograft model, expression of HMGB1 was upregulated in the xenograft and inhibition of HMGB1 function via an anti-HMGB1 monoclonal antibody increased xenograft survival by about 1 week, with evidence of reduced graft inflammation35.
The above findings indicate that HMGB1 participates in early allograft rejection and accelerates acute allograft rejection. Findings from a 2014 study suggest that HMGB1 also has a role in chronic allograft rejection, the leading cause of graft loss for which no effective new therapies are available. In an MHC class II mismatched murine model of chronic cardiac allograft rejection, levels of HMGB1 were elevated in the graft 2 months after transplantation36. Furthermore, administration of an anti-HMGB1 monoclonal antibody every 4 days during the first month after transplantation reduced the degree of graft vasculopathy by 50% and was accompanied with reduced graft levels of IL-17A, IFN-γ and inflammatory macrophages36.
Repetitive minor ischaemic insults, either to the organ of interest or to remote tissues, including muscle, might elicit a protective response within the organ through a process called ischaemic preconditioning37. In a mouse model of kidney IRI, prior administration of recombinant HMGB1 provided significant protection against kidney dysfunction, histological damage and inflammation38. Further mechanistic analyses revealed that HMGB1 preconditioning elicited upregulation of Siglec-G, a negative regulator of TLR4 signalling14. Findings from studies in experimental infection models suggest that innate immune cells can exhibit a form of memory — they can enhance immune responses after repeat stimulation through a phenomenon known as ‘trained immunity’ (REF. 39). Whether this phenomenon occurs in organ transplantation has not yet been determined but will need to be investigated if repeated administration of DAMPs is to be considered as a ischaemic-preconditioning strategy in patients undergoing organ transplantation.
Overall, these studies indicate that HMGB1 is released after organ implantation, contributes to graft inflammation and accelerates graft rejection. However, transplant survival in the various models was typically only extended from 1 week to 3 weeks after transplantation with HMGB1 inhibition32, indicating that HMGB1 is not the only contributor to graft inflammation.
Hyaluronan.
Hyaluronan (also called hyaluronic acid) is a glycosaminoglycan produced by mesenchymal cells that has been implicated in a variety of experimental models of sterile inflammation, most notably in bleomycin-induced lung injury40–42 and in murine non-transplant IRI models43,44. In quiescent, noninflammatory states, hyaluronan exists in a large molecular weight form, which undergoes degradation upon inflammation. Hyaluronan fragments can activate TLR2 and TLR4 to induce inflammation45, and the large form of hyaluronan is thought to be immunoregulatory46. However, a 2014 study found that overexpression of the enzyme that fragments hyaluronan in murine skin does not lead to skin inflammation, but rather promotes the migration of dendritic cells out of the skin, which reduces subsequent delayed-type hypersensitivity responses in the skin47. This study indicates that hyaluronan fragments could be important to control immune-cell emigration before immune activation. Whether this is also the case in internal organs requires further study.
As well as activating TLR2, TLR4 and their downstream cytoplasmic signal adaptor MYD8840, hyaluronan can activate CD44 in T cells in the lung41. Levels of hyaluronan are elevated in human kidney allografts undergoing acute rejection48, in minor mismatched skin grafts undergoing rejection and in human lung transplants with evidence of chronic rejection49, compared to levels in nonrejecting grafts. Fragmented, but not full length hyaluronan, acts via TLR2, TLR4 and MYD88 to activate dendritic cells to prime alloreactive T cells in vitro 49. These findings are compatible with the reported presence of hyaluronan in fibrotic lung tissue from human lung transplant recipients50. Interestingly, in an orthotopic lung allograft model in which transplant tolerance was induced by combined anti-CD154 and CTLA4 immunoglobulin treatment, fragmented, but not full length hyaluronan, abrogated transplant tolerance via a TLR2, TLR4 and MYD88-dependent pathway50. Fragmented hyaluronan is therefore likely to contribute to inflammation and acute graft rejection and might also impair the tolerance of some organ allografts. Investigation of the role of fragmented hyaluronan in chronic allograft rejection requires tools to efficiently deplete hyaluronan or hyaluronan fragments in murine models.
Other DAMPs.
Other factors that contribute to sterile inflammation, such as uric acid51 and mitochondrial DNA (which is involved in inflammation after trauma52), are not known to be causally involved in inflammation after solid organ transplantation. Some intracellular proteins, such as members of the heat shock protein-70 family, are thought to trigger inflammation; yet, when examined in experimental murine skin transplant models, Hsp1b deletion in the donor or Hsp1a deletion in both the donor and the recipient, did not affect the timing of allograft rejection53,54. Using proteomic approaches, the protein haptoglobin, which is known for its haeme binding and antioxidant properties55, was found to be upregulated in both syngeneic and allogeneic skin grafts in mice56. Purified haptoglobin activates dendritic cells via MYD88 to enhance the priming of alloreactive T cells56. In a minor histocompatibility mismatched skin transplant model in which MYD88 is required for graft rejection57, donor-derived haptoglobin accelerated acute graft rejection56. In a full MHC mismatched cardiac transplant model, recipient-derived haptoglobin, but not donor-derived haptoglobin, impaired the ability of CTLA4 immunoglobulin to induce indefinite cardiac allograft survival58. Haptoglobin amplified intragraft inflammation by stimulating the accumulation of dendritic cells in the graft, by increasing the expression of the proinflammatory cytokine IL-6 and of the neutrophil attracting chemokine CXCL2 (also known as MIP2), and by reducing the levels of the immunosuppressive cytokine IL-10 within the first 2 weeks of transplantation. Finally, haptoglobin was present in biopsy samples from human cardiac transplants that exhibited moderate cellular rejection, providing initial clinical translation of the experimental findings58. These studies indicate that discovery approaches such as proteomics can be powerful tools to identify novel DAMPs after organ transplantation.
Haptoglobin is probably not involved in the initiation of IRI-induced inflammation in the graft after transplantation. Therefore, we speculate that known (and possibly unknown) DAMPs, such as HMGB1, are released upon IRI and activate innate immune signalling pathways within the graft, which heighten the local inflammatory milieu and subsequently lead to the local release of proinflammatory cytokines such as IL-6 or IL-1. These inflammatory mediators then enter the circulation to induce the production of inflammatory amplifiers such as haptoglobin by the recipient (FIG. 1). These amplifiers tip the balance from graft acceptance to graft rejection by increasing inflammation, which leads to an increase in subsequent anti-donor T-cell responses.
Innate immunity in experimental grafts
When responding to an inflammatory insult, cellular innate immune recognition of pathogens occurs via cell surface receptors, endosomal receptors and cytosolic receptors59. Innate immune receptors can be categorized as inflammasome complexes60, TLRs and retinoic acid-inducible gene I-like receptors (RLRs)61,62. RLRs are yet to be examined in transplanted organs; data on the inflammasome and TLRs in transplantation are described below.
The inflammasome
The inflammasome is a group of intracellular multimeric protein complexes that are assembled in the cytoplasm in response to a priming signal, typically from DAMPs or PAMPs (BOX 1) via TLRs or other innate immune receptors60. The inflammasome complexes function as molecular scaffolds that recruit and activate caspase precursors, in particular procaspase-1 (REF. 60), via the adaptor protein PYCARD (also known as ASC). Inflammasome-mediated activation of caspase-1 leads to the production of IL-1β and IL-18.
Evidence for inflammasome activation after organ transplantation is beginning to emerge63. Specifically, in a murine cardiac allograft model, PYCARD and IL-1β were upregulated after transplantation in allografts, but not in syngeneic grafts63. A 2015 study identified PYCARD expression in rejecting human cardiac allograft specimens64. The mechanistic role of inflammasome activation in organ transplantation awaits further studies.
Toll-like receptor-mediated signalling
TLRs (BOX 1) are the best-characterized innate immune receptors in organ transplantation. Over 10 TLRs have been recognized across all mammalian species examined65. TLR expression and function has been documented in many cell types, in particular in innate immune cells such as monocytes, macrophages, dendritic cells, neutrophils and natural killer cells66. Lymphocytes, including the recently identified class of innate lymphoid cells, also express TLRs67. Expression of TLRs on endothelial and epithelial cells has also been well characterized and is of great relevance to tissue responses to transplantation44,68,69. Animal models and human studies have demonstrated that TLRs are upregulated in the grafted organ following IRI and during rejection44. This increase in TLR levels is attributable to increased TLR expression by bone-marrow-derived cells, but also by cells within the parenchyma of the transplanted organ, as shown by experiments using bone-marrow chimeras44,68. Other forms of organ injury that can occur following transplantation, including sterile injury caused by toxins and ischaemia, as well as non-sterile injury resulting from infection69, can also result in TLR upregulation and activation70.
As detailed in the previous section, numerous endogenous TLR ligands, such as HMGB1 and hyaluronan, are upregulated after organ transplantation44,57,68,71–74. Ligand binding to TLRs, often accompanied by hetero-dimerization of several TLR types, triggers a cascade of intracellular signalling events that culminate in the production of proinflammatory cytokines and chemokines66. Although TLRs signal through common downstream effectors, they also exhibit specificity as part of their downstream pathway is specific to individual TLRs and depends on the cell type involved. With the exception of TLR3, TLR signalling requires the recruitment of MYD88. TLR3 activation is mediated by the recruitment of TIR domain-containing adaptor molecule 1 (TICAM-1, also known as TRIF), whereas TLR4 signalling triggers both MYD88 and TICAM-1 pathways65. Signalling via the MYD88 pathway leads to the translocation of nuclear factor-κB from the cytosol to the nucleus, inducing the production of proinflammatory cytokines such as IL-6 and TNF, and chemokines, including CCL2 (also known as MCP-1) (REF. 66). Activation of the TLR/MYD88 pathway also initiates the transcription of two key inflammatory cytokine precursors, pro-IL-1β and pro-IL-18, and the assembly of the inflammasome. Signalling via the TICAM-1 pathway triggers production of type 1 interferons IFNα and IFNβ66.
In mice kidney IRI results in rapid upregulation of Tlr4 mRNA and TLR4, in tubular epithelial and endothelial cells in particular, and to a lesser extent in infiltrating leukocytes44. Deficiency of either Tlr4 or MYD88 affords substantial protection against kidney dysfunction, histological damage and inflammation following murine kidney IRI44. However, these results partly contrast with findings from another study showing that Tlr4 is critical for the induction of renal IRI in mice whereas MYD88 and Ticam-1 are not75. Differences in the degree of cross-clamping used in these studies could explain the discrepant findings with regard to the requirement of MYD88 for renal IRI44,75. Furthermore, the potential synergy between Ticam-1 and MYD88 has yet to be tested in double Ticam-1/MYD88-deficient mouse models of renal IRI.
The innate inflammatory response is necessary for the development of acute allograft rejection in a cardiac allograft model76 and, as mentioned above, TLRs are required for renal IRI. Thus, the impairment of TLR signalling could prevent acute rejection after transplantation. Deficiency of MYD88 in mice with minor HLA mismatches in a skin transplantation model was associated with reduced acute rejection and impaired dendritic cell maturation in draining lymph nodes52. In addition, loss of MYD88 promoted the induction of tolerance in skin allografts when combined with co-stimulatory blockade72,77, and downregulation of MYD88 and Ticam-1 through small interfering RNAs enhanced the survival of fully MHC-mismatched cardiac allografts to 40 days and indefinitely when combined with rapamycin78. Without co-stimulatory blockade, however, complete MYD88 deficiency only delayed acute rejection in a fully MHC-mismatched heart allograft model71 and did not prevent the rejection of fully HLA-mismatched skin grafts71. Finally, as mentioned above, MYD88 and Ticam-1 contribute to graft levels of IL-6, TNF and CCL2 3 h after reperfusion in syngeneic heart transplant models79. These results indicate that inhibiting TLR signalling synergizes with immune suppression to extend allograft survival.
In a life-sustaining, fully MHC-mismatched, murine kidney allograft model, MYD88 deficiency (in both donor and recipient strains) led to superior graft survival compared to wild-type allografts (100 days versus 40 days, respectively)73. At 14 days after transplantation, the time at which acute rejection usually occurs, MYD88-deficient allografts showed markedly less evidence of acute rejection and graft inflammation than did wild-type allografts. MYD88-deficient allografts exhibited normal kidney function and little evidence of chronic rejection 100 days after transplantation, whereas wild-type allografts developed marked albuminuria, elevated serum creatinine, prominent glomerulosclerosis, tubulointerstitial inflammation and fibrosis73. These findings support those of an earlier study performed in a murine kidney allograft model in which deficiency of Tlr2, Tlr4 or MYD88 in the recipient reduced chronic allograft damage and the number of macrophages and dendritic cells that infiltrated the allograft80. To investigate the possibility that MYD88 deficiency leads to donor-specific tolerance, third-party skin transplants were conducted on mice with long-term surviving skin allografts73. Wild-type survivors rejected skin grafts from matched donors and third-party donors, whereas MYD88-deficient survivors rejected third-party skin grafts but accepted skin grafts from donor-strain mice, confirming that these mice had achieved donor-specific tolerance73. Transplant tolerance was mediated at least in part by CD25+ regulatory T cells, as administration of a blocking anti-CD25 antibody abolished this tolerance73. Thus, the weight of experimental evidence clearly indicates that MYD88 is an important inducer of acute and chronic allograft rejection and prevents the induction of transplant tolerance.
Inflammation in clinical transplantation
Early inflammatory events
DAMP-mediated signalling.
HMGB1 is upregulated in human cadaveric kidney transplants74. Moreover, a follow-up analysis in human kidney transplant recipients showed that urinary HMGB1 levels were elevated as early as 3 h after transplantation81. In addition, the presence of HMGB1 in the peripheral blood of kidney transplant recipients was associated with an upregulation of TLR2 and TLR4 in peripheral blood leukocytes, which suggests that HMGB1 induces systemic TLR activation81. Similar observations have been made in liver transplant recipients82. Exposure of human tubular cells to HMGB1 in vitro induces the release of inflammatory cytokines (including IL-6, IL-1β, TNF and CCL2), suggesting that HMGB1 promotes inflammation in human organ transplantation74 (TABLE 1).
Table 1 |.
DAMPs and TLRs in clinical solid organ transplantation
| Allograft | Evidence of involvement of DAMPs and TLRs |
|---|---|
| Kidney |
|
| Lung |
|
| Liver | |
| Heart |
|
BAL, bronchoalveolar lavage; DAMP, damage-associated molecular pattern; TLR, Toll-like receptor.
Transcriptomic analysis of bronchoalveolar lavage fluid and lung tissue biopsy samples demonstrated that activation of innate immune pathways occurs in lung transplantation83,84. Specifically, gene expression analysis of bronchoalveolar lavage fluid before and after reperfusion in lung transplant recipients showed upregulation of the inflammasome (including NLRP3, IL-1β and caspase-1), inflammatory chemokines and cytokines (CXCL1, CXCL2, CCL7 and IL-6) and TLRs (TLR2, TLR4, TLR6 and TRL9) — key mediators of early graft dysfunction83. Of note, the gene expression analysis conducted in these studies does not detect post-translational modifications of proteins such as caspase-1 and pro-caspase-1, which are important inflammatory events. These studies also demonstrated that longer ventilation times were associated with a stronger inflammatory profile, including a higher expression of TLRs and inflammatory cytokines83,84, indicating that mechanical ventilation directly causes lung injury and inflammation, and impairs early graft function.
TLR-mediated signalling.
Interestingly, in lung transplant recipients expression of TLRs strongly correlated with that of inflammatory cytokines, providing evidence that TLRs are the main mediators of graft inflammation after human lung transplantation83 (TABLE 1). The importance of TLR4 signalling in IRI after organ transplantation has been further illustrated by genetic studies of specific single nucleotide polymorphisms (SNPs) in the Tlr4 allele. Presence of the Tlr4 loss-of-function alleles Asp299Gly or Thr399Ile was associated with lower rates of acute allograft rejection after kidney, lung and liver transplantation, and with lower intragraft levels of proinflammatory cytokines such as TNF and CCL2 (REFS 74,85,86). In addition, TLR gene-expression profiling of biopsy samples from kidney transplant recipients showed increased expression of TLR1, TLR2, TLR4, TLR7 and TLR8 in patients undergoing acute rejection, and TLR4 expression within the graft correlated with cellular infiltration and graft damage, according to the 2007 Banff classification87, indicating that TLRs, and TLR4 in particular, are important for graft inflammation after kidney transplantation74. However, in a 2015 study, TLR SNPs did not correlate with outcomes after kidney transplantation88. These discrepancies could stem from differences in cohort sizes, SNPs analysed and statistical methods. Nonetheless, given that loss-of-function of Tlr4 was affected by the expression of inflammatory cytokines in the graft74, we suggest that the weight of current evidence indicates a functional role of TLR4 in graft inflammation.
The mechanisms by which TLR4 is upregulated during IRI after organ transplantation remain unclear, although one can speculate that hypoxic and oxidative stress mediated by IRI or subsequent inflammation induced by DAMPs promote TLR4 upregulation in human epithelial and immune cells89–91. In support of a role for DAMPs in triggering TLR4 upregulation, release of RAGE after graft implantation is predictive of a poor short-term outcome after lung transplantation92. Specifically, plasma levels of RAGE 4 h after reperfusion correlate with a longer duration of mechanical ventilation and length of stay in the intensive care unit, independently of graft ischaemia time92.
Inflammation and long-term graft survival
TLRs.
Genetic analyses in 787 liver allografts identified three donor SNPs in the Tlr4 allele that were associated with graft failure93. However, in a separate study, when liver transplant recipients were stratified on the basis of their hepatitis C virus infection status, the association between Tlr4 polymorphisms and late graft failure was only found in patients with hepatitis C, suggesting that Tlr4 polymorphisms synergize with hepatitis C virus infection to enhance graft rejection86. In lung transplantation, recipient Tlr4 SNPs influence graft rejection. An analysis of 110 lung transplant recipients, including 20 patients with bronchiolitis obliterans syndrome (BOS), identified an association between polymorphisms in Tlr4, Tlr2 and Tlr9 and the development of BOS94, which indicates that these TLR polymorphisms enhance the development of chronic rejection after lung transplantation.
In kidney transplantation, loss-of-function polymorphisms in donor TLR4 are associated with a reduction in the risk of acute rejection95; however, these outcomes do not translate into improvements in long-term kidney allograft function. Indeed, allograft function 1–5 years after transplantation was reported as similar, regardless of donor polymorphism status96. Similarly, in a case-controlled study of kidney transplant recipients, graft SNPs in TLRs 1–8 were not associated with biopsy-proven graft rejection or mortality by 4 years after transplantation, although a comparison of kidney transplant recipients and donors identified an association between TLR SNPs and end-stage renal disease88.
Although genetic association studies have yielded inconsistent results, transcriptional analysis of tissue biopsy samples from starndard criteria living-donor kidney grafts showed an ongoing injury response and TLR-mediated inflammation after transplantation97,98. Specifically, development of inflammation during the first year after kidney transplantation is associated with upregulation of TLR4 and TLR2 (REFS 97,98). Higher TLR expression was only observed in patients with both graft interstitial fibrosis and cellular infiltrates and not in patients with normal graft histology or fibrosis alone97. TLR4 expression is also increased in human kidney grafts undergoing chronic rejection compared to stable controls99. Consistent with these findings, the expression pattern of MYD88 and TLR4 in the peripheral blood leukocytes of human kidney transplant recipients enables distinction between patients with chronic rejection and operationally tolerant recipients99. In heart transplant recipients, expression of TLR4 on circulating monocytes associates with endothelial dysfunction within the allograft up to 3 years after transplantation, although evidence of chronic rejection was not reported in this study100. Although upregulation of the TLR pathway has been reported in these studies, it is often associated (and sometimes masked) by the overexpression of genes involved in the adaptive immune response and other components of innate immunity, which illustrates the molecular complexity of the immune response during graft rejection97,101,102.
DAMPs.
DAMPs are present in chronically rejected human allografts50,103. Notably, lung transplant recipients with BOS have an accumulation of hyaluronan in the fibrous tissue of the airway lumen. These patients have a higher expression of hyaluronan synthase 1 in their lung tissue and higher levels of hyaluronan in their bronchoalveolar lavage fluid and plasma, as compared to grafted patients without BOS50. Extensive profiling of DAMPs in the bronchoalveolar lavage fluid of lung transplant recipients identified a specific biological pattern for BOS and for restrictive allograft syndrome. Specifically, levels of HMGB1 and S100 family proteins (including S100A8, S100A9, S100A8/A9, S100A12 and S100P), which can activate Tlr4 and induce innate immune-cell recruitment and endothelial cell activation104, were elevated in patients with restrictive allograft syndrome and BOS compared to control patients with stable graft function103. The production of these DAMPs was slightly increased in the restrictive allograft syndrome group compared to levels in the BOS group103. By contrast, expression of S100A8 in human kidney allografts was associated with a reduced incidence of chronic allograft vasculopathy105. This finding is consistent with experimental evidence showing that S100A8 contributes to resolution of inflammation after renal injury in non-transplant models of IRI101 and reduces inflammation after allotransplantation106,107. Differences between lung and kidney transplants (for example lung transplants are exposed to the environment whereas kidney grafts are not), might explain the divergent results in regard to S100 proteins.
Strategies to inhibit inflammation
Inhibition of TLR signalling.
Several antibodies and compounds that target TLR4 are in development or in clinical trials. Given that TLR4 is ubiquitously expressed and crucial for providing protection against infection, strategies that aim to nonspecifically block the TLR4 signalling pathway might incur a considerable risk of infectious complications.
Genetic deletion of pivotal molecules in the innate immune response (individual TLRs or molecules essential for their downstream signalling, such as MYD88) is achievable in mice, particularly when performed in specific pathogen-free environments, and has provided mechanistic insights into the pathogenic roles of these factors, as discussed above. Such major interruptions of innate immune signalling pathways are also theoretically achievable in humans through the use of blocking antibodies or small molecular antagonists, but are likely to carry high risks of infection and cancer. Inhibiting the ligands that activate TLRs directly in the allograft before implantation is a conceptually attractive strategy to avoid compromising host defence in transplant recipients. This strategy has yet to be tested experimentally or clinically. Despite the above-mentioned concerns, several clinical studies that inhibit TLRs are in progress.
A phase II clinical trial is currently testing the therapeutic efficacy of OPN-305, a monoclonal antibody that blocks TLR2, to dampen inflammation and prevent early dysfunction of extended-criteria-donor organs (donor age >60 years)108. Another clinical trial will assess the safety and tolerability of an anti-TLR4 antibody in healthy individuals109. The results of these studies will undoubtedly provide interesting insights as to the therapeutic potential of blocking specific TLRs to reduce inflammation after organ transplantation.
Stimulation of resolution pathways.
Tissues express a large array of evolutionary conserved cellular pathways that control tissue damage and counterbalance the deleterious effects of IRI110. Acute inflammation triggers inflammation resolution pathways that are required for tissue repair, including those regulated by haemoxygenase 1, heat shock proteins and nuclear factor erythroid 2-related factor 2 (NFE2-related factor 2, also known as NRF2)111,112. An experimental study found that heat shock preconditioning reduces damage after renal IRI through the expansion of regulatory T cells113. Hypoxia-inducible factor 1α (Hif-1α) is an oxygen-sensitive transcription factor that regulates tissue metabolism and angiogenesis114 and its induction reduces infarct size after myocardial infarction in mice115. Hif-1α is also critical for ischaemia preconditioning in murine models of myocardial IRI116. These results have given rise to strategies to precondition allografts using drugs that enhance the activity of Hif-1α or by inducing minor and remote ischaemic insults117. Such graft preconditioning should induce protective mechanisms that enable a greater degree of tolerance to tissue damage118. To date, the majority of the approaches being investigated in clinical trials to reduce inflammation have employed remote ischaemic preconditioning. In organ transplantation, this procedure consists of three 5-min cycles of left lower limb ischaemia, induced by an automated cuff inflator placed on the limb and inflated to 300 mm Hg, with an intervening 5 min of reperfusion during which the cuff is deflated119. However, this approach failed to demonstrate beneficial effects on early graft function or rates of acute rejection in human kidney transplantation119. Other experimental approaches that directly target stress responses in the damaged organ by enhancing Hif-1α activity via small molecules such as l-mimosine120, or promoting HO1 activity by exposing the graft to haeme arginate121 have shown beneficial effects and have provided an impetus for several ongoing clinical studies122–124. Patient safety data are already available for haeme arginate and this agent is now under investigation in a randomized controlled clinical trial in recipients of deceased donor kidneys122.
Future considerations and conclusions
Identification of the optimal strategy for blocking and managing TLR-mediated inflammation is bound to be a challenging process. The critical function of TLRs in innate immunity1, the dual role of TLR-mediated inflammation in providing host protection and inducing damage, the almost ubiquitous expression of TLRs, often underpinning cell-type-specific effects66, and our increasing, but vastly incomplete understanding of the roles of TLRs in health and disease, highlight some of the challenges involved in the control of TLR-regulated inflammation after transplantation. Several aspects of TLR inhibition will need to be clarified if therapeutic strategies are to be developed: cell specific versus general effects of TLR inhibition; the potential for off-target or adverse effects, especially infection; the optimal mode of delivery, which is particularly relevant for intracellular TLRs such as TLR9; the duration and stability of TLR blockade; and the potential for urgent reversibility in the event of an infection. Despite the challenges, the important roles of TLRs in inflammation after organ transplantation are undisputed and inhibiting inappropriate or excessive inflammatory responses by targeting TLRs, TLR ligands or the products of TLR activation, are attractive strategies that could reduce graft inflammation after solid organ transplantation. Additionally, therapeutic modulation of other inflammatory pathways, such as those regulated by the inflammasome, will require future experimental investigation.
Key points.
Sterile inflammation occurs in organs after their surgical removal and implantation into a recipient
Inflammation that occurs after solid organ transplantation can precipitate acute allograft rejection, impede transplant tolerance and enhance the development of chronic allograft rejection
Experimental and clinical studies have shown that several endogenous substances, also known as damage associated molecular patterns contribute to both acute and chronic allograft rejection
Toll-like receptors, which are among the best characterized innate immune receptors, induce inflammation and impair outcomes after solid organ transplantation
Clinical studies are investigating strategies to inhibit innate immune responses after organ transplantation; approaches to reduce inflammation without compromising host defence to pathogens would substantially improve outcomes for transplant recipients
Sterile inflammation .
Inflammation that occurs following the necrosis-mediated release of activators of inflammation in medical conditions such as ischaemia–reperfusion injury, crystalline-induced arthritis, acute lung injury and chronic inflammatory conditions, without an identifiable infectious precipitant.
Bronchiolitis obliterans syndrome.
A manifestation of chronic allograft rejection characterized by a decline in pulmonary function that affects more than half of lung recipients 5 years after transplantation and accounts for a considerable proportion of lung allograft losses and recipient deaths.
Operationally tolerant recipients.
Subgroup of patients who spontaneously tolerate their graft and maintain allograft function without the use of immunosuppressants for at least 1 year, in the absence of deleterious responses.
Restrictive allograft syndrome.
Newly described phenotype of chronic lung allograft dysfunction characterized by a persistent decline in vital and total lung capacities and allograft parenchymal fibrosis.
Acknowledgements
D.R.G.’s work is supported by NIH/NIA grant R01AG028082, S.J.C.’s work is supported by NHMRC Project Grant funding and S.B.’s work is supported by the CENTAURE foundation, the IHU-Cesti project, the French National Research Agency, and the Nantes Metropole and the Pays de la Loire Region.
Footnotes
Competing interests statement
The authors declare no competing interests.
References
- 1.Wu H & Chadban SJ Roles of Toll-like receptors in transplantation. Curr. Opin. Organ Transplant 19, 1–7 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Mori DN, Kreisel D, Fullerton JN, Gilroy DW & Goldstein DR Inflammatory triggers of acute rejection of organ allografts. Immunol. Rev 258, 132–144 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chong A & Alegre M The impact of infection and tissue damage in solid-organ transplantation. Nat. Rev. Immunol 12, 459–471 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McFarland-Mancini MM et al. Differences in wound healing in mice with deficiency of IL-6 versus IL-6 receptor. J. Immunol 184, 7219–7228 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Cameron GD et al. Haptoglobin phenotype correlates with development of cardiac transplant vasculopathy. J. Heart Lung Transplant 23, 43–49 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Shirali A & Goldstein D Tracking the toll of kidney disease. J. Am. Soc. Nephrol 19, 1444–1450 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Shirali A & Goldstein DR Activation of the innate immune system by the endogenous ligand hyaluronan. Curr. Opin. Organ Transplant 13, 20–25 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Chen GY & Nuñez G Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol 10, 826–837 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gjertson D Impact of delayed graft function and acute rejection on kidney graft survival. Clin. Transpl 467–480 (2000). [PubMed] [Google Scholar]
- 10.Debout A et al. Each additional hour of cold ischemia time significantly increases the risk of graft failure and mortality following renal transplantation. Kidney Int. 87, 343–349 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Stehlik J et al. The Registry of the International Society for Heart and Lung Transplantation: twenty-eighth adult heart transplant report — 2011. J. Heart Lung Transplant 30, 1078–1094 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Lund LH et al. The Registry of the International Society for Heart and Lung Transplantation: thirty-second official adult heart transplantation report — focus theme: early graft failure. J. Heart Lung Transplant 34, 1244–1254 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Chalasani G et al. The allograft defines the type of rejection (acute versus chronic) in the face of an established effector immune response. J. Immunol 172, 7813–7820 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Ponticelli C Ischaemia-reperfusion injury: a major protagonist in kidney transplantation. Nephrol. Dial. Transplant 29, 1134–1140 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Matas AJ et al. 2202 kidney transplant recipients with 10 years of graft function: what happens next? Am. J. Trans 8, 2410–2419 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Floerchinger B et al. Inflammatory immune responses in a reproducible mouse brain death model. Transpl. Immunol 27, 25–29 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Hoffmann SC et al. Molecular and immunohistochemical characterization of the onset and resolution of human renal allograft ischemia-reperfusion injury. Transplantation 74, 916–923 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Sheen JH & Heeger PS Effects of complement activation on allograft injury. Curr. Opin. Organ Transplant 20, 468–475 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ali F, Dua A & Cronin DC Changing paradigms in organ preservation and resuscitation. Curr. Opin. Organ Transplant 20, 152–158 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Li W et al. Intravital 2-photon imaging of leukocyte trafficking in beating heart. J. Clin. Invest 122, 2499–2508 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pittman K & Kubes P Damage-associated molecular patterns control neutrophil recruitment. J. Innate Immun 5, 315–323 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhuang Q & Lakkis FG Dendritic cells and innate immunity in kidney transplantation. Kidney Int. 5, 315–3 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lakkis FG Where is the alloimmune response initiated? Am. J. Transplant 3, 241–242 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Tsung A et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J. Exp. Med 201, 1135–1143 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu H et al. HMGB1 contributes to kidney ischemia reperfusion injury. J. Am. Soc. Nephrol 21, 1878–1890 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rabadi MM, Ghaly T, Goligorksy MS & Ratliff BB HMGB1 in renal ischemic injury. Am. J. Physiol. Renal Physiol 303, F873–F885 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kamo N et al. ASC/caspase-1/IL-1β signaling triggers inflammatory responses by promoting HMGB1 induction in liver ischemia/reperfusion injury. Hepatology 58, 351–362 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsung A et al. HMGB1 release induced by liver ischemia involves Toll-like receptor 4-dependent reactive oxygen species production and calcium-mediated signaling. J. Exp. Med 204, 2913–2923 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Helena Erlandsson Harris UA Mini-review: the nuclear protein HMGB1 as a proinflammatory mediator. Eur. J. Immunol 34, 1503–1512 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Zhang A et al. Necrostatin-1 inhibits Hmgb1-IL-23/IL-17 pathway and attenuates cardiac ischemia reperfusion injury. Transpl. Int 27, 1077–1085 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Syrjälä SO et al. Increased Th17 rather than Th1 alloimmune response is associated with cardiac allograft vasculopathy after hypothermic preservation in the rat. J. Heart Lung Transplant 29, 1047–1057 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Huang Y et al. Extracellular hmgb1 functions as an innate immune-mediator implicated in murine cardiac allograft acute rejection. Am. J. Transplant 7, 799–808 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Moser B et al. Blockade of RAGE suppresses alloimmune reactions in vitro and delays allograft rejection in murine heart transplantation. Am. J. Transplant 7, 293–302 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Dessing MC et al. RAGE does not contribute to renal injury and damage upon ischemia/reperfusion-induced injury. J. Innate Immun 4, 80–85 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li JH et al. Blockade of extracellular HMGB1 suppresses xenoreactive B cell responses and delays acute vascular xenogeneic rejection. Am. J. Transplant 15, 2062–2074 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Zou H et al. HMGB1 is involved in chronic rejection of cardiac allograft via promoting inflammatory-like mDCs. Am. J. Transplant 14, 1765–1777 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Hausenloy DJ & Yellon DM Ischaemic conditioning and reperfusion injury. Nat. Rev. Cardiol 10.1038/nrcardio.2016.5 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Wu H et al. Preconditioning with recombinant high-mobility group box 1 protein protects the kidney against ischemia-reperfusion injury in mice. Kidney Int. 85, 824–832 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Netea, Mihai G, Quintin J & van der Meer, Jos WM Trained immunity: a memory for innate host defense. Cell Host Microbe 9, 355–361 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Jiang D et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat. Med 11, 1173–1179 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Teder P et al. Resolution of lung inflammation by CD44. Science 296, 155–158 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Noble PW, McKee CM, Cowman M & Shin HS Hyaluronan fragments activate an NF-κB/I-κBα autoregulatory loop in murine macrophages. J. Exp. Med 183, 2373–2378 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rouschop KMA et al. Protection against renal ischemia reperfusion injury by CD44 disruption. J. Am. Soc. Nephrol 16, 2034–2043 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Wu H et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J. Clin. Invest 117, 2847–2859 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scheibner KA et al. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J. Immunol 177, 1272–1281 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Bollyky PL et al. Cutting edge: high molecular weight hyaluronan promotes the suppressive effects of CD4+CD25+ regulatory T cells. J. Immmunol 179, 744–747 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Muto J et al. Hyaluronan digestion controls DC migration from the skin. J. Clin. Invest 124, 1309–1319 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wells A et al. Increased hyaluronan in acutely rejecting human kidney grafts. Transplantation 55, 1346–1349 (1993). [DOI] [PubMed] [Google Scholar]
- 49.Tesar BM et al. The role of hyaluronan degradation products as innate alloimmune agonists. Am. J. Transplant 6, 2622–2635 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Todd JL et al. Hyaluronan contributes to bronchiolitis obliterans syndrome and stimulates lung allograft rejection through activation of innate immunity. Am. J. Respir. Crit. Care Med 189, 556–566 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kono H, Chen C-J, Ontiveros F & Rock KL Uric acid promotes an acute inflammatory response to sterile cell death in mice. J. Clin. Invest 120, 1939–1949 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shen H, Kreisel D & Goldstein DR Processes of sterile inflammation. J. Immunol 191, 2857–2863 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oh K-H et al. Targeted gene disruption of the heat shock protein 72 gene (hsp70.1) in the donor tissue is associated with a prolonged rejection-free survival in the murine skin allograft model. Transplant Immunol. 13, 273–281 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Tesar B & Goldstein DR Acute allograft rejections occurs independently of inducible HSP-70. Transplantation 11, 1513–1517 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Quaye IK Haptoglobin, inflammation and disease. Trans. R. Soc. Trop. Med. Hyg 102, 735–742 (2008). [DOI] [PubMed] [Google Scholar]
- 56.Shen H et al. Haptoglobin activates innate immunity to enhance acute transplant rejection in mice. J. Clin. Invest 122, 383–387 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goldstein DR, Tesar BM, Akira S & Lakkis FG Critical role of the Toll-like receptor signal adaptor protein MyD88 in acute allograft rejection. J. Clin. Invest 111, 1571–1578 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shen H et al. Haptoglobin enhances cardiac transplant rejection. Circ. Res 116, 1670–1679 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brubaker SW, Bonham KS, Zanoni I & Kagan JC Innate immune pattern recognition: a cell biological perspective. Annu. Rev. Immunol 33, 257–290 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guo H, Callaway JB & Ting JP Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med 21, 677–687 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoneyama M, Onomoto K, Jogi M, Akaboshi T & Fujita T Viral RNA detection by RIG-I-like receptors. Curr. Opin. Immunol 32, 48–53 (2015). [DOI] [PubMed] [Google Scholar]
- 62.Fitzgerald ME, Rawling DC, Vela A & Pyle AM An evolving arsenal: viral RNA detection by RIG-I-like receptors. Curr. Opin. Microbiol 20, 76–81 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seto T et al. Upregulation of the apoptosis-related inflammasome in cardiac allograft rejection. J. Heart Lung Transplant 29, 352–359 (2010). [DOI] [PubMed] [Google Scholar]
- 64.Shah KB, Mauro AG, Flattery M, Toldo S & Abbate A Formation of the inflammasome during cardiac allograft rejection. Int. J. Cardiol 201, 328–330 (2015). [DOI] [PubMed] [Google Scholar]
- 65.Pandey S, Kawai T & Akira S Microbial sensing by Toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harb. Perspect. Biol 7, a016246 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Takeuchi O & Akira S Pattern recognition receptors and inflammation. Cell 140, 805–820 (2010). [DOI] [PubMed] [Google Scholar]
- 67.Crellin NK et al. Regulation of cytokine secretion in human CD127+ LTi-like innate lymphoid cells by Toll-like receptor 2. Immunity 33, 752–764 (2010). [DOI] [PubMed] [Google Scholar]
- 68.Leemans JC et al. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J. Clin. Invest 115, 2894–2903 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patole PS et al. Toll-like receptor-4: renal cells and bone marrow cells signal for neutrophil recruitment during pyelonephritis. Kidney Int. 68, 2582–2587 (2005). [DOI] [PubMed] [Google Scholar]
- 70.Leventhal JS & Schroppel B Toll-like receptors in transplantation: sensing and reacting to injury. Kidney Int. 81, 826–832 (2012). [DOI] [PubMed] [Google Scholar]
- 71.Tesar BM, Zhang J, Li Q & Goldstein DR TH1 immune responses to fully MHC mismatched allografts are diminished in the absence of MyD88, a toll like receptor signal adaptor protein. Am. J. Transplant 4, 1429–1439 (2004). [DOI] [PubMed] [Google Scholar]
- 72.Walker WE et al. Absence of innate MyD88 signaling promotes inducible allograft acceptance. J. Immunol 177, 5307–5316 (2006). [DOI] [PubMed] [Google Scholar]
- 73.Wu H et al. Absence of MyD88 signaling induces donor-specific kidney allograft tolerance. J. Am. Soc. Nephrol 23, 1701–1716 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kruger B et al. Donor Toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proc. Natl Acad. Sci. USA 106, 3390–3395 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pulskens WP et al. Toll-like receptor-4 coordinates the innate immune response of the kidney to renal ischemia/reperfusion injury. PLoS ONE 3, e3596 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Oberbarnscheidt MH et al. Non-self recognition by monocytes initiates allograft rejection. J. Clin. Invest 124, 3579–3589 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen L et al. TLR engagement prevents transplantation tolerance. Am. J. Trans 6, 2282–2291 (2006). [DOI] [PubMed] [Google Scholar]
- 78.Zhang X et al. Induction of alloimmune tolerance in heart transplantation through gene silencing of TLR adaptors. Am. J. Trans 12, 2675–2688 (2012). [DOI] [PubMed] [Google Scholar]
- 79.Kaczorowski DJ et al. Mechanisms of Toll-like receptor 4 (TLR4)-mediated inflammation after cold ischemia/reperfusion in the heart. Transplantation 87, 1455–1463 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang S et al. Recipient Toll-like receptors contribute to chronic graft dysfunction by both MyD88- and TRIF-dependent signaling. Dis. Model. Mech 3, 92–103 (2010). [DOI] [PubMed] [Google Scholar]
- 81.Thierry A et al. The alarmin concept applied to human renal transplantation: evidence for a differential implication of HMGB1 and IL-33. PLoS ONE 9, e88742 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ilmakunnas M et al. High mobility group box 1 protein as a marker of hepatocellular injury in human liver transplantation. Liver Transplant. 14, 1517–1525 (2008). [DOI] [PubMed] [Google Scholar]
- 83.Cantu E et al. Gene set enrichment analysis identifies key innate immune pathways in primary graft dysfunction after lung transplantation. Am. J. Trans 13, 1898–1904 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Andrade CF et al. Toll-like receptor and cytokine gene expression in the early phase of human lung transplantation. J. Heart Lung Transplant 25, 1317–1323 (2006). [DOI] [PubMed] [Google Scholar]
- 85.Palmer SM et al. Donor polymorphisms in Toll-like receptor-4 influence the development of rejection after renal transplantation. Clin. Transplant 20, 30–36 (2006). [DOI] [PubMed] [Google Scholar]
- 86.Dhillon N et al. A single nucleotide polymorphism of Toll-like receptor 4 identifies the risk of developing graft failure after liver transplantation. J. Hepatol 53, 67–72 (2010). [DOI] [PubMed] [Google Scholar]
- 87.Dessing MC et al. Intragraft Toll-like receptor profiling in acute renal allograft rejection. Nephrol. Dial. Transplant 25, 4087–4092 (2010). [DOI] [PubMed] [Google Scholar]
- 88.Dessing MC et al. Toll-like receptor family polymorphisms are associated with primary renal diseases but not with renal outcomes following kidney transplantation. PLoS ONE 10, e0139769 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kuhlicke J, Frick JS, Morote-Garcia JC, Rosenberger P & Eltzschig HK Hypoxia inducible factor (HIF)-1 coordinates induction of Toll-like receptors TLR2 and TLR6 during hypoxia. PLoS ONE 2, e1364 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Powers KA et al. Oxidative stress generated by hemorrhagic shock recruits Toll-like receptor 4 to the plasma membrane in macrophages. J. Exp. Med 203, 1951–1961 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zarember KA & Godowski PJ Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J. Immunol 168, 554–561 (2002). [DOI] [PubMed] [Google Scholar]
- 92.Calfee CS et al. Plasma receptor for advanced glycation end-products predicts duration of ICU stay and mechanical ventilation in patients after lung transplantation. J. Heart Lung Transplant 26, 675–680 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Oetting WS et al. Donor polymorphisms of toll-like receptor 4 associated with graft failure in liver transplant recipients. Liver Transpl. 18, 1399–1405 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kastelijn EA et al. Polymorphisms in innate immunity genes associated with development of bronchiolitis obliterans after lung transplantation. J. Heart Lung Transplant 29, 665–671 (2010). [DOI] [PubMed] [Google Scholar]
- 95.Ducloux D et al. Relevance of Toll-like receptor-4 polymorphisms in renal transplantation. Kidney Int. 67, 2454–2461 (2005). [DOI] [PubMed] [Google Scholar]
- 96.Palmer C, Diehn M, Alizadeh AA & Brown PO Cell-type specific gene expression profiles of leukocytes in human peripheral blood. BMC Genomics 7, 115–115 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Park WD, Griffin MD, Cornell LD, Cosio FG & Stegall MD Fibrosis with inflammation at one year predicts transplant functional decline. J. Am. Soc. Nephrol 21, 1987–1997 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Park W, Griffin M, Grande JP, Cosio F & Stegall MD Molecular evidence of injury and inflammation in normal and fibrotic renal allografts one year posttransplant. Transplantation 83, 1466–1476 (2007). [DOI] [PubMed] [Google Scholar]
- 99.Braudeau C et al. Contrasted blood and intragraft toll-like receptor 4 mRNA profiles in operational tolerance versus chronic rejection in kidney transplant recipients. Transplantation 86, 130–136 (2008). [DOI] [PubMed] [Google Scholar]
- 100.Methe H, Zimmer E, Grimm C, Nabauer M & Koglin J Evidence for a role of toll-like receptor 4 in development of chronic allograft rejection after cardiac transplantation. Transplantation 78, 1324–1331 (2004). [DOI] [PubMed] [Google Scholar]
- 101.Naesens M et al. Progressive histological damage in renal allografts is associated with expression of innate and adaptive immunity genes. Kidney Int. 80, 1364–1376 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Khatri P et al. A common rejection module (CRM) for acute rejection across multiple organs identifies novel therapeutics for organ transplantation. J. Exp. Med 210, 2205–2221 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Saito T et al. Distinct expression patterns of alveolar “alarmins” in subtypes of chronic lung allograft dysfunction. Am. J. Trans 14, 1425–1432 (2014). [DOI] [PubMed] [Google Scholar]
- 104.Chan JK et al. Alarmins: awaiting a clinical response. J. Clin. Invest 122, 2711–2719 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Eikmans M et al. Expression of surfactant protein-C, S100A8, S100A9, and B cell markers in renal allografts: investigation of the prognostic value. J. Soc. Am. Neprol 16, 3771–3786 (2005). [DOI] [PubMed] [Google Scholar]
- 106.Dessing MC et al. The calcium-binding protein complex S100A8/A9 has a crucial role in controlling macrophage-mediated renal repair following ischemia/reperfusion. Kidney Int. 87, 85–94 (2015). [DOI] [PubMed] [Google Scholar]
- 107.Shimizu K et al. Loss of myeloid related protein-8/14 exacerbates cardiac allograft rejection. Circulation 124, 2920–2932 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.US National Library of Medicine. ClinicalTrials.gov [online], https://www.clinicaltrials.gov/ct2/show/NCT01794663 (2013).
- 109.US National Library of Medicine. ClinicalTrials.gov [online], https://www.clinicaltrials.gov/ct2/show/NCT01808469 (2013).
- 110.Soares MP, Gozzelino R & Weis S Tissue damage control in disease tolerance. Trends Immunol. 35, 483–494 (2014). [DOI] [PubMed] [Google Scholar]
- 111.Anders H-J Immune system modulation of kidney regeneration[mdash]mechanisms and implications. Nat. Rev. Nephrol 10, 347–358 (2014). [DOI] [PubMed] [Google Scholar]
- 112.Serhan CN, Chiang N & Van Dyke T Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol 8, 349–361 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kim M-G et al. The heat-shock protein-70-induced renoprotective effect is partially mediated by CD4+CD25+Foxp3+ regulatory T cells in ischemia/reperfusion-induced acute kidney injury. Kidney Int. 85, 62–71 (2014). [DOI] [PubMed] [Google Scholar]
- 114.Weidemann A & Johnson RS Biology of HIF-1α. Cell Death Differ. 15, 621–627 (2008). [DOI] [PubMed] [Google Scholar]
- 115.Kido M et al. Hypoxia-inducible factor 1-alpha reduces infarction and attenuates progression of cardiac dysfunction after myocardial infarction in the mouse. J. Am. College Cardiol 46, 2116–2124 (2005). [DOI] [PubMed] [Google Scholar]
- 116.Cai Z et al. Complete loss of ischaemic preconditioning-induced cardioprotection in mice with partial deficiency of HIF-1α. Cardiovasc. Res 77, 463–470 (2008). [DOI] [PubMed] [Google Scholar]
- 117.Eltzschig HK & Eckle T Ischemia and reperfusion — from mechanism to translation. Nat. Med 17, 1391–1401 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Medzhitov R, Schneider DS & Soares MP Disease tolerance as a defense strategy. Science 335, 936–941 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chen Y et al. Remote ischemic preconditioning fails to improve early renal function of patients undergoing living-donor renal transplantation: a randomized controlled trial. Transplantation 95, e4–6 (2013). [DOI] [PubMed] [Google Scholar]
- 120.Hill P et al. Inhibition of hypoxia inducible factor hydroxylases protects against renal ischemia-reperfusion injury. J. Am. Soc. Nephrol 19, 39–46 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ferenbach DA et al. The induction of macrophage hemeoxygenase-1 is protective during acute kidney injury in aging mice. Kidney Int. 79, 966–976 (2011). [DOI] [PubMed] [Google Scholar]
- 122.US National Library of Medicine. ClinicalTrials.gov [online], https://clinicaltrials.gov/ct2/show/NCT01430156 (2013).
- 123.US National Library of Medicine. ClinicalTrials.gov [online], https://clinicaltrials.gov/ct2/show/NCT01920594 (2013).
- 124.US National Library of Medicine. ClinicalTrials.gov [online], https://clinicaltrials.gov/ct2/show/NCT02299661 (2014).
- 125.Medzhitov R & Janeway C Jr. Innate immunity. N. Engl. J. Med 343, 338–344 (2000). [DOI] [PubMed] [Google Scholar]