

Abstract
Ubiquitin signaling is a sequence of events driving the fate of a protein based on the type of ubiquitin modifications attached. In the case of neurodegenerative diseases, ubiquitin signaling is mainly associated with degradation signals to process aberrant proteins, which form aggregates often fatal for the brain cells. This signaling is often perturbed by the aggregates themselves and leads to the accumulation of toxic aggregates and inclusion bodies that are deleterious due to a toxic gain of function. Decrease in quality control pathways is often seen with age and is a critical onset for the development of neurodegeneration. Many aggregates are now thought to propagate in a prion-like manner, where mutated proteins acting like seeds are transitioning from cell to cell, converting normal proteins to toxic aggregates. Modulation of ubiquitin signaling, by stimulating ubiquitin ligase activation, is a potential therapeutic strategy to treat patients with neurodegeneration diseases.
Subject terms: Biochemistry, Cell biology
Facts
Neurodegenerative diseases (NDs) affect millions of people worldwide
Protein misfolding and aggregation is inherently associated with neurodegenerative diseases
Misfolded proteins can be removed through various proteolytic systems
Mutations in E3 ubiquitin ligases can lead to neurodegenerative disorders
Open questions
Why does the ubiquitin machinery fail to eliminate aggregates?
How do some misfolded proteins self-propagate?
Are protein aggregates in proteinopathies protective or toxic?
Can ubiquitin ligases be targeted as therapeutic strategies in NDs?
Introduction
NDs are characterized by the aggregation of a vast population of proteins that would normally undergo degradation via the two main catabolism pathways, the ubiquitin-proteasome system (UPS) and the autophagy-lysosomal pathway (ALP). Ubiquitin signaling is a cascade reaction of tagging a ubiquitin residue on a substrate to modulate its function or promote its degradation. Building blocks of ubiquitin onto other ubiquitin molecules forms ubiquitin chains that will modulate the fate of the substrate by forming K48 or K63 ubiquitin chains for UPS or ALP degradation, respectively. Aggregated proteins often impair these two quality control pathways (QCPs) and promote a toxic gain of function that leads to neuronal death. NDs generally have a late onset, and the visible aggregates seen in the brains of postmortem patients are generally the end stage of the disease. Factors contributing to protein aggregates vary from ageing to genetic to environmental triggers. Although the vast majority of NDs are sporadic, a few early-onset and late-onset patients are linked to genome mutations notably Parkinson’s disease (PD) (PINK1, Parkin, DJ-1, LRRK2, α-synuclein), Alzheimer’s disease (AD) (APP, PSEN1, PSEN2,) or frontotemporal dementia (FTD) and Amyotrophic lateral sclerosis (ALS) (C9orf72, SOD1, TDP-43, FUS) [1]. However, toxic aggregates are not always observed in familial PD autopsy brains, and thus will not be developed further in this review. Other mutations can affect E3 ligases that have prominent functions in neurodevelopmental functions. UBE3A, ITCH, CHIP, PARKIN, TRAF-6, and many more have been implicated in neurodegeneration and neurodevelopmental diseases like Angelman syndrome or PD [2], or simply having neuroprotection effects. Furthermore, mitochondrial dysfunction has been implicated in NDs, notably in PD and AD, not as the initial cause but as an aggravating neurodegenerative process related to age [3]. However, the scope of this review is to focus on the cause of protein aggregation, as well as the signals and failure of ubiquitin-mediated degradation of aggregates.
Ubiquitin signaling in neurons
The ubiquitin machinery
One of the hallmarks of aging is diminished proteostasis in the brain. Protein homeostasis is tightly regulated by controlling protein synthesis, folding as well as degradation through its two main pathways, the UPS and ALP. These two QCPs are interconnected and rely on ubiquitin signaling to target proteins either to the proteasome or to the lysosome [4]. Ubiquitin is a small protein of 8.5 kDa that is covalently attached to proteins post-translationally in order to modulate substrate protein fate. This reversible process has been implicated in multiple functions, from substrate stability and activity to cellular localization and as a determinant for interaction with partners. Ubiquitination is tightly controlled by an enzymatic cascade reaction starting by activation by an E1 enzyme, conjugation to an E2 enzyme on its active-site cysteine and finally placement on its substrate via an E3 ligase enzyme that facilitates the covalent linkage of ubiquitin to amino groups on a lysine residue or an N-terminal methionine of the substrate. While only two E1 enzymes are found in human cells, about 40 E2 enzymes and over 600 E3 enzymes have been reported. Deubiquitinating enzymes (DUBs) ensure the reversibility of this process by specifically removing ubiquitin from substrates. The specificity of the signaling is driven by the E3 enzymes to synthesize various ubiquitin modifications on ubiquitin itself on any of its seven lysine residues creating chains of several molecules. Consequently, a substrate can be mono-ubiquitinated, multi-ubiquitinated (multiple mono-ubiquitination), or polyubiquitinated, where eight types of chains can be formed (see other reviews on this issue for further details). K48 and K63 chains are by far the best characterized ubiquitin chains and are the most relevant for the scope of this review. Linear ubiquitin linkages on N-terminal methionines also exist and function in innate immune signaling pathways [5].
The ubiquitin-proteasome system
The proteasome is a multicatalytic proteolytic system that degrades polyubiquitinated unfunctional or misfolded proteins into peptides. The 26S proteasome is comprised of two different subcomplexes arranged in a barrel-like structure, the 20S core particle (CP) and the 19S regulatory particle (RP). Proteasomal degradation of proteins can be divided into several steps; substrate recognition, deubiquitynation, and unfolding of the substrate that occurs at the RP before threading the substrate into the proteolytic channel and degradation inside the proteolytic chamber of the CP [6]. The proteasome can efficiently degrade soluble ubiquitinated proteins but is highly inefficient in degrading aggregated proteins due to the failure to unfold them before sliding the protein into the 20S CP. Under protein aggregation stress, or when the proteasome is otherwise overwhelmed, autophagy has been shown to compensate for the burden, illustrating the collaboration between the two QCPs [7, 8]. The peptides released after proteolysis are further degraded by amino and carboxypeptidases, while ubiquitin can be recycled for further rounds of ubiquitination. Although a decline in proteasomal degradation is associated with ageing [9], the downregulation of the proteasome is also a hallmark of many NDs [10]. Likewise, although challenging, the characterization of the molecular mechanisms underlying aberrant proteasomal degradation in a variety of NDs is opening new possible avenues to therapeutic strategies based on their specific modulations [11].
The autophagosome-lysosome pathway
Autophagy is a selective or nonselective cellular degradation pathway with high flexibility [12]. It can be triggered anywhere in the cell to sequester defective organelles or aggregated proteins within a double-membraned vesicle called an autophagosome. Autophagy can be divided into several steps, starting with the initiation of the phagophore, the elongation of a double membrane structure around the cargo, and the closure, forming an autophagosome encapsulating cytosolic substrates needed during starvation or to degrade defective or superfluous cytosolic structure(s) [13]. The autophagosome then fuses with lysosomes, forming an autophagolysosome that hydrolyzes its content [14]. The degraded content is then released in the form of amino acids, lipids, and glycosides that can be recycled. Autophagy in neurons is compartmentalized due to the spacial organization of neurons with autophagosome biogenesis observed in the somea as well as in distal axons. When formed in the axon, acidified vesicles are subsequently retrogradely transported toward the soma [15]. Evidence that autophagy is essential for the survival of neuronal cells was highlighted by the observation of neurodegeneration in mice with autophagy defects [16]. Although the proteasome mainly degrades soluble unfolded proteins, autophagy is able to engulf and degrade large protein aggregates in a selective process called aggrephagy [17]. Degradation of aggregated proteins is assisted by autophagy receptors (e.g., SQSTM1) that recognize the ubiquitin tag on aggregated proteins and mediate their association with the autophagosome via binding with the autophagosome-associated proteins mATG8 [18–22]. Chaperone mediated autophagy (CMA) is a noncanonical selective autophagy pathway targeting proteins that contain a specific motif in their sequence without any need for ubiquitination. The chaperone HSC70 binds this motif and delivers the substrate to the lysosome for degradation [23]. Although CMA cannot mediate the degradation of aggregated proteins, it acts as a first responder in case of protein misfolding. Similarly to observations with the UPS, autophagy declines with ageing [24] rendering it less efficient at clearing toxic protein aggregates found in the brain of patients suffering of neurodegeneration. Furthermore, the modulation of autophagy to clear aggregated proteins is a potential therapeutic target [25, 26].
Protein aggregates driving neurodegeneration diseases
Protein misfolding
Although perturbation of the ubiquitin-signaling pathways can lead to mis-localization of proteins, oxidative stress, defects in DNA repair, mitochondrial dysfunction, and many more deleterious events that have consequences for the development of neurodegenerative diseases, we focused here on protein aggregate turnover via the UPS and the ALP pathways.
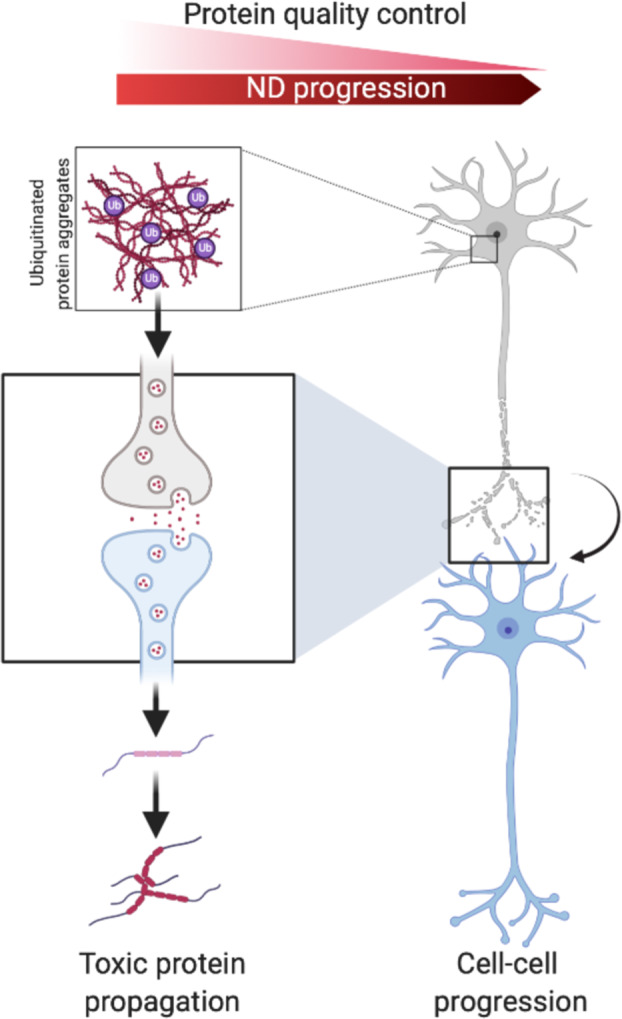
Aberrant protein folding is kept in constant check via chaperones which try to refold proteins post-translationally, or if unsuccessful will activate cellular programs that aim to eliminate the misfolded protein [27] (Fig. 1A). Misfolded proteins are particularly detrimental in the nervous system due to the postmitotic nature of neurons that cannot mitigate cytotoxic proteins by way of dilution during cell division. Progressive accumulation of protein aggregates in well-ordered structures is a hallmark of many NDs, and these protein aggregates are often ubiquitin-positive, highlighting the importance of understanding the involvement of UPS and ALP in NDs (Fig. 1B). One defining feature of most if not all characterized NDs is aggregation of one key protein, that can be used as a biomarker for the disease.
Fig. 1. Degradation of misfolded proteins by UPS and ALP.

A Newly synthesized proteins are folded by chaperones. Misfolded proteins that cannot be re-folded by chaperones can aggregate in ubiquitinated inclusions due to genetic and environmental factors. B Not-yet toxic aggregates can be targeted early by a functional UPS or ALP response, preventing neuron death and ND. On the other hand, due to factors like age or mutations in key genes, toxic aggregates develop, leading to deadly ND.
α-synuclein
Lewy bodies (LB) are large aggregates found in patients diagnosed with PD. These large aggregates are mainly comprised of α-synuclein (α-Syn) [28], a small (14 kDa) acidic protein normally lacking a defined secondary structure that has functions implicated in vesicle trafficking and neurotransmitter release [29, 30] (Fig. 2A). Current models hold that LB spread from cell to cell via secretion and uptake of α-Syn (called the Braak hypothesis), resulting in propagation throughout the nervous system [31]. Clinically, PD is characterized by motor dysfunction including movement disability, muscular rigidity, rest tremor, and postural instability [32]. The principal pathological feature is a loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc), which results in depletion in striatal dopamine. Studies in postmortem brains of PD patients showed ubiquitinated α-Syn accumulating in LBs with mainly mono-ubiquitinated and some partial polyubiquitinated residues [33]. The mode of clearance of α-Syn is still unclear and under heavy scrutiny as several reports revealed both proteasome and lysosome involvement, depending on localization and pathological state of α-Syn [34]. Recent evidence strongly implicates the UPS in PD pathogenesis [35, 36]. Interplay between aggregated α-Syn and the UPS has been reported, with selective interaction of α-Syn with the 19S RP, ultimately inhibiting proteasomal degradation [37]. Patients with PD dementia and LB disease were found to display a reduction of RPT6, a subunit of the 19S RP, further implicating dysfunction of the UPS in PD [38]. Another study, using a reporter consisting of a short degron fused to GFP showed that the ability of α-Syn to inhibit proteasome degradation was dependent on its ability to assemble into filaments [39]. In vivo work with rat dopaminergic neurons confirmed early-onset functional impairment of the UPS [40]. To understand the mechanisms involved in ubiquitin signaling, ubiquitylome analysis aimed at deciphering the conjugation of ubiquitin chains to α-Syn and its signaling consequences [41, 42]. Other efforts concentrated on the profiling of the ubiquitylome in SNpc in a pre-symptomatic PD mouse model in order to better understand the contribution of abnormally ubiquitinated proteins to dopaminergic neuron death [43]. As the site of ubiquitination on α-Syn seems to have different effects on proteasome turnover or aggregation and fibril formation [44–46], a number of E3 ubiquitin ligases were found to ubiquitinate α-Syn and prevent cell–cell propagation, hence counteracting LB formation (Table 1) [47–50]. However, it was shown that another PTM, SUMOylation, facilitated α-Syn aggregation by counteracting the pro-degradation role of ubiquitination, resulting in its accumulation [51]. Due to their polarized and terminally differentiated nature, neurons are likely to be especially sensitive to autophagy impairment. Furthermore, these postmitotic cells are unable to dilute damaged proteins by way of cell division. mATG8 homolog function was shown to be impaired by LBs sequestration of LC3 and SQSTM1 preventing a proper autophagy response [52]. Furthermore, α-Syn aggregates have been implicated in negatively regulating the retrograde transport of autophagosomes [53]. Another demonstration of the involvement of the autophagy machinery in NDs stems from the immunohistochemical examination of postmortem human brains showing the presence of the autophagy receptor NBR1 in LBs [54]. Exosomes are thought to facilitate transmission of α-Syn to other neuronal cells. Fibrillar α-Syn was shown to impair autophagy by upregulating the E3 ubiquitin ligase PELI1 in microglia, resulting in ubiquitin-mediated degradation of lysosome-associated membrane glycoprotein 2 through UPS and transmission of toxic α-Syn to healthy neurons [55]. Another study reported fibrillar α-Syn engulfment and subsequent damage of lysosomes in microglial cells resulting in recruitment of autophagy receptors in an attempt to control the damage done to the lysosome [56]. Exosome secretion was shown to be a compensatory mechanism when macroautophagy was impaired [57]. Microglia were reported to engulf and clear neuron-released non-aggregated α-Syn through a selective autophagy type called synucleinphagy mediated by Toll like receptor 4-NFk-B signaling, p62 upregulation, and ubiquitin [58]. Recent evidence thus tend to show that the proteasome is inhibited by α-Syn and that its ubiquitination by E3 ligases seems to drive this function. Furthermore, α-Syn prevents a proper autophagy response by sequestering important players or upregulating proteins with a negative impact on autophagy.
Fig. 2. Example of aggregate prone proteins.

A α-synuclein, involved in synapse function, can aggregate into LB, subsequently inhibiting UPS and ALP responses, progressing into PD. B Amyloid-β can degenerate into amyloid plaques as it inhibits the UPS response. C Tau can become phosphorylated and ubiquitinated, leading to neurofibrillary tangles and cross-talk with a-β, impairing ALP and UPS, leading to AD.
Table 1.
E3 ligases and DUBs modifiying aggregation-prone proteins.
| Protein | E3 ligase | DUB | Ub site | Branched form |
|---|---|---|---|---|
| α-synuclein | SCFFXBL5 [47] | |||
| CHIP [48] | ||||
| USP9X [49] | ||||
| UCH-L1 [163] | ||||
| USP8 [164] | ||||
| OTUB1 [165] | ||||
| Nedd4 [50] | ||||
| SIAH [166] | ||||
| K6, K10, K12, K21, K23, K32, K34, K43, K96 [166] | ||||
| Amyloid-β | UCHL1 [167] | |||
| USP25 [168] | ||||
| Tau | USP14 [169–171] | |||
| OTUB1 [172] | ||||
| USP10 [65] | ||||
| UCHL1 [173] | ||||
| CHIP [73] | ||||
| TRAF6 [74] | ||||
| MARCH7 [75] | ||||
| UBE3A [77] | ||||
| K264, K311, K353 [66] | ||||
| 28 sites [70] | ||||
| K6, K11, K48 [66] | ||||
| M1 [69] | ||||
| SOD1 | Ataxin-3 [174] | |||
| DORFIN [107] | ||||
| NEDL1 [108] | ||||
| MITOL [109] | ||||
| PARKIN [110] | ||||
| TDP-43 | UBPY [175] | |||
| USP14 [176] | ||||
| RNF112 [90] | ||||
| M1, K48, K63 [88] | ||||
| K102, 114,145,181 [177] | ||||
| AR | CHIP [131] | |||
| HTT | USP12 [178] | |||
| USP19 [179] | ||||
| K6, K27, K29 [117] | ||||
| K63 [119] | ||||
| TRAF6 [117] | ||||
| UBE3A [118, 122] | ||||
| WWP1 [119] | ||||
| Ube2W [121] | ||||
| UBR5 [180] | ||||
| CHIP [123] | ||||
| K6, K9, K15, K132, K337, K631, K804, K837, and K2097 [181] | ||||
| K444 [182] | ||||
| ATXN1 | CHIP [144] | |||
| K589 [143] | ||||
| ATXN3 | USP19 [179] | |||
| K8, K117, K190, K200, K291 [148] | ||||
| K48 [148] |
Amyloid-β and Tau
Intracellular neurofibrillary tangles (NFT) and amyloid plaques are pathological hallmarks of the brains of patients with AD. AD is a progressive ND characterized by the presence of insoluble aggregates as well as the loss of neurons and synapses. Amyloid plaques consist of aggregated amyloid-β (Aβ) peptide. Aβ is formed from sequential cleavage of the amyloid precursor protein (APP), generating peptides of various sizes ranging between 39 and 43 amino acid residues. The longest peptide tends to aggregate, leading to their accumulation in amyloid plaques (Fig. 2B).
NFTs are mainly composed of hyperphosphorylated Tau (p-TAU) [59]. Tau is a microtubule-associated protein that normally stabilizes the microtubule network within axons, facilitating transport of organelles and synaptic vesicles. Hyperphosphorylation of Tau attenuates its binding to microtubules, promoting its self-aggregation into NFTs, and resulting in impairment of neuron functions (Fig. 2C). Similar to observations made with α-Syn, Tau aggregates can be excreted and internalized into other cells, inducing fibrillization of normal Tau and transfer of NFTs [60, 61]. One aspect of the current research on AD is the investigation of which of these protein aggregation pathologies appears first. Evidence of a cross-talk between Aβ and Tau is exemplified by the finding that Aβ oligomers impair proteasome activity, contributing to the accumulation of Aβ and Tau [62]. UPS impairment is an important issue in AD pathogenesis, exemplified by the finding that a frameshift mutant of ubiquitin, UBB+1 is a dose dependent inhibitor of the UPS and has been found to accumulate in the brains of AD patients [63]. Stress granules (SG) have been suggested to promote Tau aggregation, and autophagy as well as the ubiquitin machinery were shown to have an important role in Tau aggregation through SG formation [64, 65]. A number of studies aimed at identifying ubiquitin linkages in aggregated proteins of AD brains identified not only K48-linked polyUb chains, but also K6, K11, and K63-linked forms in NFTs [66–69]. Mapping the brain ubiquitylome of postmortem AD patients contributed toward understanding the changes associated with AD neuropathology. Ubiquitination sites were found to be increased on Tau, notably around its phosphorylation sites suggesting a cross-talk between these two PTMs [70]. Furthermore, perturbation of the proteasome in AD brain is thought to prevent degradation of oxidized proteins, contributing to the pathology of AD [71]. Other studies investigated the influence of ubiquitin linkages on Tau aggregation and the influence it has on the fate of the aggregates [72]. Several E3 ligases including CHIP, TRAF6, and MARCH7 have been implicated in targeting Tau for degradation via the UPS (Table 1), and phosphorylation was shown to influence Tau ubiquitination [73–75]. This cross-talk between phosphorylation and ubiquitination PTMs is gaining traction and is thought to influence Tau filament structure [76]. The expression level of the E3 ligase Ube3A is decreased in neurons presenting with elevated Aβ oligomers, hinting at a role of this E3 ligase in AD pathogenesis [77]. An early immuno-electron microscopy study identified an accumulation of immature autophagosomes in AD brains, suggesting an impairment of the later steps of autophagy as a feature of AD pathobiology [78]. Consistent with this result, an accumulation of LC3 as well as the lysosomal protein LAMP1 was detected in postmortem AD brain patients, proposed to contribute to the development of tauopathies [79]. In accordance with the model of decreased autophagy in AD brains, Beclin-1 was found to be cleaved by caspase 3, with its cleavage product accumulating in plaque regions as well as within NFTs [80]. NUB1, a negative regulator of ubiquitin-like modifier 1, was shown to reduce the levels of phosphorylated and aggregated tau by modulating the autophagy lysosome pathway via SQSTM1 when degradation through the UPS was impaired [81]. In light of these recent studies, ubiquitination of Tau at specific sites seems to drive its pathological state. Furthermore, a decrease in autophagy is observed postmortem in AD patient brains.
Superoxide dismutase (SOD1), transactive response DNA-binding protein-43 (TDP-43) and fused in sarcoma (FUS)
FTD and ALS, also known as Lou Gehrig’s disease, are neurodegenerative disorders characterized by progressive degeneration of both upper and lower motor neurons leading to spasticity and paralysis, among other symptoms [82]. Pathological overlap between ALS and a subset of FTD suggests they represent two sides of the same ND. They are both linked to other genetic mutations of apparently unrelated proteins that show aggregation properties leading to intracellular inclusion bodies [83]. Under physiological conditions, transactive response DNA-binding protein-43 (TDP-43) is mainly localized in the nucleus, where it performs functions including gene repression or mRNA splicing. However, TDP-43 was found to be depleted from the nucleus and accumulated in cytoplasmic insoluble and ubiquitinated inclusions in postmortem brain of ALS patients and is considered a hallmark of ALS [84]. Cytoplasmic insoluble TDP-43 is found to be phosphorylated as well as ubiquitinated ultimately changing its structure, localization, and promoting its aggregation [85]. Furthermore, cross-talk between the two post-translational modifications seems to drive the neuropathology of aggregated TDP-43 [86]. The reasons behind the neurotoxicity of TDP-43 aggregation are still not clear. TDP-43’s solubility has been classified into six distinct pools, ranging from soluble monomer to undegradable macroaggregates. Clearance of TDP-43 via the UPS versus the ALP was found to be determined by its oligomerization status, with soluble TDP-43 cleared by the UPS and oligomeric TDP-43 cleared by the ALP. Macroaggregates, however, were found to be stable and difficult to clear by either pathway [87]. Immunohistochemical studies using linkage selective polyubiquitin chain antibodies identified linear polyubiquitin chains, K63-linked and K48-linked chains, with K63 and linear ubiquitin chains labeling the thicker TDP-43 aggregates, together with the autophagy receptor OPTN recruitment, hinting for a role of ALP in degradation of aggregates [88]. A study in a Drosophila model showed reduced UPS function upon TDP-43 accumulation, contributing to the progression of ALS-like dysfunctions [89]. Identifying the E3 ubiquitin ligases involved in ubiquitination of TDP-43 is still under scrutiny and only RNF112 has been identified to date [90]. Defects in autophagy have been associated with pathological mutations of several ALS-linked genes such as OPTN, TBK1, VCP, UBQLN2, and SQSTM1 [91–95]. Furthermore, SQSTM1 was shown to colocalize with aggregated TDP43 and to mediate its degradation [96].
Mutation in the RNA-binding proteins, fused in sarcoma (FUS) and TDP-43, are associated with ALS, but one main question is whether the neuron loss is due to the toxic gain of function of cytosolic aggregates or due to a loss of nuclear RNA-binding function. The mutated and disease-associated forms of TDP-43 and FUS become incorporated into SG. The formation of SG results from intrinsically disordered proteins coalescing together through liquid–liquid phase separation to form a membraneless organelle [97]. UBQLN2 was shown to colocalize with SGs and to undergo a ubiquitin-mediated modulation that disrupts UBQLN2 liquid–liquid phase separation to mediate trafficking of ubiquitinated substrate to the proteasome for degradation [98]. This suggests a possible link between ALS related proteins localized in SGs and their degradation through the UPS or ALP. Indeed, UBQLN2 maintains the solubility of FUS, resulting in the negative regulation of SGs. Furthermore, an ALS-linked mutation of UBQLN2 had a negative effect on its association with FUS and in regulating SGs [99]. The autophagy proteins ULK1/2, essential for initiation of canonical autophagy, were shown to regulate the disassembly of SGs by phosphorylating VCP. ULK1/2 disruption in mice promoted the formation of inclusions, thought to be composed of protein aggregates, positive for ubiquitin and TDP-43 [100]. Though FUS is less studied than TDP-43, a few studies revealed that pathological mutations in FUS promote its cytoplasmic localization, and its mis-localization to the cytoplasm was sufficient to induce ubiquitin and SQSTM1 positive aggregates [101]. Furthermore, mutated FUS promotes defects in autophagy in neuronal cells [102]. Although mutated FUS is mis-localized to the cytoplasm, mass spectrometry analysis showed it still retained its ability to bind to proteins involved in RNA metabolism, suggesting that the pathology of FUS-linked ALS does not rely only on cytosolic toxic gain of function but potentially on its inability to perform its nuclear functions [103].
The Cu, Zn-superoxide dismutase (SOD1) protein has been extensively characterized for its role in ALS, with a variety of mutations leading to its aggregation with ubiquitin deposits, ultimately causing motor neuron degeneration by a toxic gain of function [104]. ALP and UPS were shown to both participate comparably in the degradation of mutant and WT SOD1 [105]. Sequestration of ubiquitin into neurotoxic inclusions is thought to reduce the availability of free ubiquitin. SOD1 mutants were implicated in alteration of the homeostasis of ubiquitin, ultimately inhibiting the proteasome and perturbing cellular functions [106]. In contrast with TDP-43, several E3 ligases have been identified to localize in inclusion bodies with SOD1 (Table 1). Dorfin ubiquitinates mutant SOD1, promoting its degradation through the UPS and thus has a protective effect [107]. The HECT ubiquitin ligase NEDL1 binds and ubiquitinates mutant SOD1 and targets it for degradation [108]. Mutant SOD1 has been shown to localize to mitochondria and induce mitochondrial dysfunctions. Consequently MITOL, a mitochondrial outer membrane E3 ligase was found to interact with ubiquitinated SOD1, promoting its degradation [109]. Furthermore, another E3 ligase involved in mitochondrial homeostasis, Parkin, was reported to mediate K63-linked polyubiquitination of SOD1 mutants and promote pathology of SOD1 through the ALP [110]. An siRNA screen identified DCAF4, a substrate receptor of the E3 ligase CUL4-DDB1, to facilitate ubiquitination of OPTN leading to the autophagic degradation of mutant SOD1 [111]. ALS and FTD thus seem to originate from post-translational modification of key proteins, triggering their mis-localization, toxic gain of function, and ultimately reducing proteasome activity as well as inhibiting ALP.
Polyglutamine expansions in androgen receptor (AR), huntingtin (HTT), and ataxins (ATXN)
Polyglutamine (PolyQ) diseases encompass nine fatal NDs characterized by protein misfolding and aggregation, leading to neuronal dysfunction and death. They include Huntington’s Disease (HD), spinal and bulbar muscular atrophy (SBMA), dentatorubral-pallidoluysian atrophy (DRPLA), and six forms of spinocerebellar ataxia (SCA). Clinical features differ for each disorder due to a specific subset of neurons affected, but they are all hereditary neurodegenerative disorders caused by abnormal expansion of PolyQ tracts in the N-terminal region of widely differing mutant proteins. The threshold for the expansion to become aggregation prone and pathogenic is around 40 repeats. The polyQ expansion mutation is mainly seen as a toxic gain of function, but whether micro-aggregates and inclusions are toxic or protective was earlier seen as controversial [112].
The huntingtin gene (HTT) possesses a polyQ repeat of between 10 and 35 residues in a normal individual. However, repeats exceeding 35 residues cause HD. PolyQ expanded HTT, or mutant HTT (mHTT) confers to the protein its aggregation propensity resulting in cytotoxicity and molecular dysfunction in neuronal cells. This translates into motor disabilities, cognitive decline, and psychiatric problems [113]. mHTT is ubiquitinated in inclusion bodies suggesting a role in the ubiquitin system in its degradation [114]. Although mHTT was initially thought to impair proteasome activity, in vitro and in vivo studies did not show global impairment of the UPS [115, 116]. Inclusion bodies contain aggregated mHTT and ubiquitin ligases, hinting for a role of aggregates to inhibit ubiquitination that would normally target mHTT for proteasomal degradation. For example, TRAF6 was found to interact with and ubiquitinate WT and mHTT, promoting atypical K6, K27, and K29 ubiquitin chains, leading to increased aggregation [117]. Similarly, the HECT ligase Ube3A was shown to be strongly recruited to ubiquitinated mHTT aggregates, resulting in its loss of Ube3A function and inability to mediate mHTT degradation [118]. On the other hand, the E3 ligase WWP1 was reported to be upregulated in HD and to mediate K63-linked polyubiquitinated chains, which promoted mHTT protection from proteasomal degradation [119]. Furthermore, core components of the SCF complex (Skip1-Cul1-F-box protein) are downregulated in HD animal models, possibly participating in aggravating the pathology of HD [120]. The noncanonical E2 conjugating enzyme Ube2W is the only E2 known to initiate ubiquitination of the α-amino group of proteins containing disordered N-termini. The relatively disordered nature of the N-terminal domain of mHTT due to its polyQ extension, makes polyQ expanded forms of HTT targets of Ube2W and the HECT ligase Ube3A, promoting its oligomerization by ubiquitination and aggregation [121, 122]. Whereas the above described E2 and ubiquitin ligases seems to promote mHTT aggregation, another E3 ligase, CHIP were shown to be protective in HD by reducing polyQ aggregation [123].
Extensive studies have implicated autophagy in clearance of mHTT. HTT regulates the transport of various organelles, including autophagosomes. However, mHTT was found to prevent this transport, leading to accumulation of autophagosomes, ultimately resulting in autophagy dysfunction [124]. Furthermore, normal, non-pathologic HTT was identified as a scaffold protein for selective autophagy by interacting with p62 and facilitating its binding to K63-linked cargo [125]. The polyQ expansion is thought to impair HTT’s role in autophagy by preventing HTT to bind to autophagy receptors as well as release of ULK1 from the mTORC1 complex [126].
SBMA is a disease associated with a polyQ extension in the N-terminus of the androgen receptor (AR) protein, similar to HTT in HD [127]. SBMA is characterized by motor neuron loss in the bulbar region of the brain and in the anterior horns of the spinal cord. The toxicity of the mutant AR (mAR) is dependent on binding to its ligand testosterone. Under basal conditions, AR is located in the cytoplasm in a complex with heat shock proteins (HSPs). Binding to testosterone induces AR conformational changes, dissociation from HSPs, dimerization and translocation to the nucleus where AR participates in gene regulation. Misfolded and aggregated mAR when activated by testosterone translocates to the nucleus where it exerts a toxic gain of function [128]. Included in mAR aggregates are chaperons and components of the UPS that are heavily ubiquitinated, suggesting an impairment of the UPS [129]. However, a report suggested that testosterone-mediated aggregation of mAR is thought to be protective by sequestering toxic proteins into insoluble cytoplasmic aggregates and preserving the UPS from over-loading while neurotoxicity seems to be associated with nuclear aggregates [130]. The E3 protein CHIP was shown to interact with AR to promote its degradation [131] and its overexpression reduced nuclear mAR aggregates as well as improved motor symptoms in a mouse model [132]. Autophagy involvement in AR pathology was reported by work studying depletion and overexpression of SQSTM1 in a mouse model of SBMA. Depletion of p62 increased mAR aggregates while overexpression promoted cytoplasmic aggregates that were less toxic in the spine and muscle of mice, improving their overall condition [133]. Studies investigating the neurotoxicity of nuclear localized mAR discovered that cytoplasmic localized mAR does not display cytotoxicity and is cleared by autophagy. Furthermore, this group uncovered a critical role of androgens in promoting the pathology of SBMA in the nucleus, as targeted localization of mAR without associated testosterone in the nucleus is not sufficient for neurotoxicity [134]. The expression of the HSP HspB8 was shown to be induced in motoneurons of SBMA patients, where it seems to participate with the stress response. HspB8 was found to facilitate autophagic clearance of protein aggregates [135]. Later, investigation in muscle cells found BAG3 to be upregulated and work in concert with HspB8 and CHIP for SQSTM1 recognition of ubiquitinated substrates [136, 137]. Finally, AR promoted transcription factor EB (TFEB) transactivation, while mAR did not. Since TFEB activation leads to upregulation of autophagy-related genes, the failure of mAR to do so suggests a potential mechanism for SMBA pathology to spread [138].
SCAs are a group of heterogeneous NDs unified by being autosomal dominant and causing ataxia, due to cerebellar degeneration. They contain over 40 subtypes, including DRPLA. The pathogenesis of SCAs correlates with the aggregation of mutant ataxin proteins containing a polyQ, namely SCA1, SCA2, SCA3, SCA6, SCA7, and SCA17. SCA diseases are characterized by progressive disequilibrium of motor coordination and cerebellar atrophy [139]. SCA1 is caused by a polyQ expansion in the Ataxin1 (ATXN1) protein, leading to aberrant aggregated proteins and toxic gain of function. Phosphorylation at S776 by PKA promotes ATXN1 stabilization mediated by the chaperone 14–3–3, leading to its aggregation [140, 141]. Yeast two hybrid analysis identified the E2 conjugating enzyme UbcH6 to interact with and modulate ATXN1 stability [142]. A later study identified K589 of ATXN1 to be ubiquitynated [143]. The E3 ligase CHIP mediates non-extended and polyQ-extended ATXN1 ubiquitination for UPS degradation [144]. Surprisingly, CHIP promoted aggregation of ATXN1, and its action was impaired by mutation of S776. These observations contrast those seen with HTT and suggest that CHIP’s role in polyQ diseases varies greatly [145]. Intranuclear inclusions of mutant ATXN3 is a hallmark of Machado–Joseph disease (MJD), or SCA3. PolyQ stretches in ATXN3 promote its aggregation in nuclear inclusions that are positive for ubiquitin. ATXN3 is a DUB preferentially cleaving polyubiquitin chains of four or more ubiquitin units [146] and ubiquitination enhances its DUB activity [147]. Mass spectrometric analysis identified an altered ubiquitination site in mutant ATXN3 but no alteration in linkage pattern, potentially impacting MJD pathogenesis [148]. These alterations may compromise its DUB function, and thus promote UPS activity. For example, loss of ATXN3 DUB function affected important processes for neuronal development and function [149]. The ubiquitin-binding site 2 domain of ATXN3 can bind to the proteasome shuttle Rad23, to regulate proteasomal degradation of ATXN3. The binding between Rad23 and ATXN3 seems to be important to its autoprotective function as disturbing the interaction decreased the levels of mutant ATXN3 but, counterintuitively enhanced its toxicity [150]. Scaglione et al. showed the regulation of the neuroprotective E3 ligase CHIP by the E2 Ube2W and the DUB ATXN3, and how mutated ATXN3 contributes to the pathogenesis of SCA3 by increased affinity toward CHIP, resulting in its decreased neuroprotective effect [151]. Evidence of an alteration of the autophagy machinery in the brains of MJD patients was in the form of increased autophagy-related proteins, increased numbers of autophagosomes, and decreased beclin-1 levels [152, 153]. The Rubinsztein group later showed that ATXN3 interacts and deubiquitinates beclin-1 via its WT polyQ and protects it from UPS degradation, enabling autophagy to take place. However, a polyQ stretch was shown to compete and impair autophagy initiation, potentially contributing to late-onset polyQ disease [154]. Furthermore, two LIR domains were found in ATXN3 specific for LC3C and GABARAP. As ATXN3 was shown to localize to early autophagosomes but not with autolysosomes, the proposed role for ATXN3 is to regulate autophagy. It is noteworthy to point out that this regulation seems independent from the polyQ-dependent regulation of beclin-1 [155]. Contrary to other NDs, HD thus does not seem to inhibit the proteasome but displays a defect in ubiquitination by modulating E3 ligases. Similarly to events observed with other NDs, HD presents a defect in autophagy by modulating its initiation steps. Toxic gain of function also seems to be an important factor in PolyQ diseases.
Concluding remarks
In the light of recent developments, it seems that there is no all-encompassing paradigm for the neurotoxic mechanisms caused by protein aggregates. Some aggregates, like α-synuclein show toxicity by impairing UPS function as well as propagating through neuronal cells. Some other aggregates however, like AR seem to protect the UPS from overload by segregating inclusion bodies in the cytosol. These aggregates have nevertheless been reported to be toxic when in the nucleus, showing that protein aggregates are differentially toxic, depending on their subcellular localization. The specific brain region, as well as the type of cells affected by the toxic aggregates, seems to contribute in the efficiency of protein clearance pathways. Disruption of ubiquitin signaling was recently highlighted as a factor promoting ND. Indeed, loss of function mutations in CHIP were shown to be responsible for Gordon Holmes Syndrome and spinocerebellar autosomal recessive 16, resulting in its loss of ubiquitin ligase function [156, 157], demonstrating that disordered ubiquitination is a contributing factor leading to ND. There are still unanswered questions regarding the recent advances in protein aggregate spreading across the brain. This concept, while widely accepted in the prion disease field is now thought to occur in PD, AD, and many other in a transmissible misfolded protein paradigms [158]. However, the factors triggering protein misfolding, the mechanisms of spreading, the cellular pathways involved, and the specificity of the brain cells are still under investigation. Furthermore, the specificity of the ubiquitin branched chain of proteins associated with ND still warrants further investigation. Likewise, recent proteomics studies aimed at determining ubiquitination sites and the mechanisms of ubiquitination of aggregation-prone proteins, but many efforts are still needed in order to fully uncover the mechanisms of ubiquitin signaling involved in ND. In light of recent understanding of the mechanisms of protein aggregate modulation of ubiquitin signaling, many groups have proposed to modulate UPS function or autophagy to improve aggregate degradation (Box 1). Other groups proposed to overexpress E3 ligases to increase ubiquitin-mediated aggregate turn-over [159] (Fig. 3A). Caution will however be needed against modulating expression of an E3 ligase or a DUB for the purpose of therapy as some protein may have different functions in different cellular contexts. This is particularly the case of the E3 ligase CHIP, that is often cited as a candidate because of its reported neuroprotection effects in many NDs, except in SCA1 where it seems to act negatively. Similarly, while modulation of autophagy seems to be a great candidate to degrade aggregates, different strategies will be needed to avoid the side effects of autophagy modulation. Another interesting perspective is the possibility to disturb the native conformation of a protein by site-specific ubiquitination, leading to its proteasomal degradation [160]. Granting that some efforts would be necessary to apply this strategy for aggregated substrates, this would be an ideal way of specifically destabilizing and degrading toxic aggregates. Another aspect gaining traction recently is the development of PROTAC and AUTACs to selectively remove protein aggregates [161, 162] (Fig. 3B, C).
Fig. 3. Therapeutic strategies to mitigate aggregates: targeted protein degradation.

A Different strategies including modulating E3 ubiquitin ligases and DUBs, modulating the UPS and ALP using small molecules as well as PROTACs and AUTACs are currently explored to prevent toxic aggregate formation and development of NDs. B PROTAC is a bifunctional-hybrid compound that possess a ligand to the target protein, a ligand to an E3 ubiquitin ligase and a linker in between these two ligands. By spacially connecting an E3 ligase and the target protein, the E3 ligase will ubiquitinate the target protein and direct it to UPS degradation. PROTAC can then be recycled back for subsequent rounds of degradation. C AUTAC can come in two different flavors, the first one possessing one end consisting of a ligand to the target protein and the other end consisting of S-guanylation. The target protein will then be directed for selective autophagy degradation. S-guanylation is a standalone tag for ALP degradation. The second technology is based on small molecule screening that specifically recognize the polyQ stretch of mHTT and LC3, specifically targetting mHTT to the autophagosome.
Box 1 Therapeutic strategies to mitigate aggregated proteins.
Although most NDs are ultimately fatal for the patient and no cure has yet been found, different strategies have been adopted to clear deleterious aggregates. Focus on the UPS by refolding misfolded proteins or dissipation of an aggregate by overexpressing chaperones is an avenue currently prospected [183]. Likewise, given their importance in the aggregated state of ND-prone proteins, modulation of ubiquitin E3 ligases [184] and DUBs [185] to specifically target aggregates is under study. Specifically, the dual co-chaperone and E3 ubiquitin ligase CHIP could be modulated for ND treatment because of its specific degradation of toxic α-Syn, p-Tau, mHTT, and other polyQ aggregates [186]. Either overexpression of the E3 ubiquitin ligase via transfection or its modulation using small molecules agonists are potential therapeutic endeavors. Similarly, modulation of ubiquitin carboxyl-terminalhydrolase L1 (UCHL1), a DUB abundantly expressed in the neurons could be considered in order to treat PD and AD patients. However, contradicting reports may render its modulation challenging as overexpression was shown to regulate aβ production [167], while its inhibition was shown to prevent proteasomal impairment by Tau aggregates [173] as well as activating ALP [163]. Small molecules modulating UPS activity is also under current investigation. Another recently described strategy is the development of proteolysis-targeting chimeras (PROTAC), where E3 ligases specifically targets aggregates in order to direct them for UPS degradation. On the other hand, ALP, the other major degradation pathway besides UPS, can be modulated to specifically degrade aggregates. Small molecules are the major way to modulate autophagy, acting in various steps in the autophagy activation. Small molecules have a mechanism of action that is either m-TOR dependent or independent [187]. The autophagy counterparts of PROTACs are autophagy-targeting chimeras (AUTACs) and are currently under investigation [188, 189].
Acknowledgements
We thank Derek Narendra, Jose Vargas, and Achim Werner for their careful reading and insightful comments. This work was supported by the Intramural Program of NINDS. Figures were made with the help of BioRender.
Compliance with ethical standards
Conflict of interest
The authors declare no conflicts of interest.
Footnotes
Edited by F. Pentimalli
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Pihlstrøm L, Wiethoff S, Houlden H. Genetics of neurodegenerative diseases: an overview. Handb Clin Neurol. 2018;145:309–23. doi: 10.1016/B978-0-12-802395-2.00022-5. [DOI] [PubMed] [Google Scholar]
- 2.Upadhyay A, Amanullah A, Chhangani D, Mishra R, Mishra A. Selective multifaceted E3 ubiquitin ligases barricade extreme defense: Potential therapeutic targets for neurodegeneration and ageing. Ageing Res Rev. 2015;24:138–59. doi: 10.1016/j.arr.2015.07.009. [DOI] [PubMed] [Google Scholar]
- 3.Markaki M, Tavernarakis N. Mitochondrial turnover and homeostasis in ageing and neurodegeneration. FEBS Lett. 2020. 10.1002/1873-3468.13802. [DOI] [PubMed]
- 4.Dikic I. Proteasomal and autophagic degradation systems. Annu Rev Biochem. 2017;86:193–224. doi: 10.1146/annurev-biochem-061516-044908. [DOI] [PubMed] [Google Scholar]
- 5.Yau R, Rape M. The increasing complexity of the ubiquitin code. Nat Cell Biol. 2016;18:579–86. doi: 10.1038/ncb3358. [DOI] [PubMed] [Google Scholar]
- 6.Rousseau A, Bertolotti A. Regulation of proteasome assembly and activity in health and disease. Nat Rev Mol Cell Biol. 2018;19:697–712. doi: 10.1038/s41580-018-0040-z. [DOI] [PubMed] [Google Scholar]
- 7.Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010;584:1393–8. doi: 10.1016/j.febslet.2009.12.047. [DOI] [PubMed] [Google Scholar]
- 8.Limanaqi F, Biagioni F, Gambardella S, Familiari P, Frati A, Fornai F. Promiscuous roles of autophagy and proteasome in neurodegenerative proteinopathies. Int J Mol Sci. 2020;21:3028. doi: 10.3390/ijms21083028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keller J, Huang F, Markesbery W. Decreased levels of proteasome activity and proteasome expression in aging spinal cord. Neuroscience. 2000;98:149–56. doi: 10.1016/S0306-4522(00)00067-1. [DOI] [PubMed] [Google Scholar]
- 10.Saez I, Vilchez D. The mechanistic links between proteasome activity, aging and agerelated diseases. Curr Genomics. 2014;15:38–51. doi: 10.2174/138920291501140306113344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dantuma NP, Bott LC. The ubiquitin-proteasome system in neurodegenerative diseases: precipitating factor, yet part of the solution. Front Mol Neurosci. 2014;7:70. doi: 10.3389/fnmol.2014.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mizushima N. The ATG conjugation systems in autophagy. Curr Opin Cell Biol. 2020;63:1–10. doi: 10.1016/j.ceb.2019.12.001. [DOI] [PubMed] [Google Scholar]
- 13.Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. 2018;20:233–42. doi: 10.1038/s41556-018-0037-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yim WW-Y, Mizushima N. Lysosome biology in autophagy. Cell Discov. 2020;6:6. doi: 10.1038/s41421-020-0141-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stavoe AKH, Holzbaur ELF. Autophagy in neurons. Annu Rev Cell Dev Biol. 2019;35:477–500. doi: 10.1146/annurev-cellbio-100818-125242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 17.Yamamoto A, Simonsen A. The elimination of accumulated and aggregated proteins: a role for aggrephagy in neurodegeneration. Neurobiol Dis. 2011;43:17–28. doi: 10.1016/j.nbd.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zaffagnini G, Savova A, Danieli A, Romanov J, Tremel S, Ebner M, et al. p62 filaments capture and present ubiquitinated cargos for autophagy. EMBO J. 2018;37. 10.15252/embj.201798308. [DOI] [PMC free article] [PubMed]
- 19.Sarraf SA, Shah HV, Kanfer G, Ward ME, Youle RJ. Selective autophagic clearance of protein aggregates is mediated by the autophagy receptor, TAX1BP1. bioRxiv. 2019. 10.1101/558767.
- 20.Lu K, Psakhye I, Jentsch S. Autophagic clearance of PolyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell. 2014;158:549–63. doi: 10.1016/j.cell.2014.05.048. [DOI] [PubMed] [Google Scholar]
- 21.Isakson P, Holland P, Simonsen A. The role of ALFY in selective autophagy. Cell Death Differ. 2013;20:12–20. doi: 10.1038/cdd.2012.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kirkin V, Lamark T, Sou Y-S, Bjørkøy G, Nunn JL, Bruun J-A, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33:505–16. doi: 10.1016/j.molcel.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 23.Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. 2018;19:365–81. doi: 10.1038/s41580-018-0001-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stead ER, Castillo-Quan JI, Miguel VEM, Lujan C, Ketteler R, Kinghorn KJ, et al. Agephagy—adapting autophagy for health during aging. Front Cell Dev Biol. 2019;7:308. doi: 10.3389/fcell.2019.00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ravikumar B, Vacher C, Berger Z, Davies J, Luo S, Oroz L, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 26.Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, et al. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in α-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci. 2009;29:13578–88. doi: 10.1523/JNEUROSCI.4390-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jason G, Anthony C. Antonelli, Adil A, Sohrab V, et al. Protein misfolding and aggregation in neurodegenerative diseases: a review of pathogeneses, novel detection strategies, and potential therapeutics. Rev Neurosci. 2019;30:339–58. [DOI] [PubMed]
- 28.Spillantini MG, Schmidt ML, Lee VM-Y, Trojanowski JQ, Jakes R, Goedert M. α-Synuclein in Lewy bodies. Nature. 1997;388:839–40. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 29.Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, et al. Increased expression of α-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abeliovich A, Schmitz Y, Fariñas I, Choi-Lundberg D, Ho WH, Castillo PE, et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–52.. doi: 10.1016/S0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- 31.Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, et al. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–53.. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalia LV, Lang AE. Parkinson’s disease. Lancet. 2015;386:896–912. doi: 10.1016/S0140-6736(14)61393-3. [DOI] [PubMed] [Google Scholar]
- 33.Tofaris GK, Razzaq A, Ghetti B, Lilley KS, Spillantini MG. Ubiquitination of α-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J Biol Chem. 2003;278:44405–11. doi: 10.1074/jbc.M308041200. [DOI] [PubMed] [Google Scholar]
- 34.Stefanis L, Emmanouilidou E, Pantazopoulou M, Kirik D, Vekrellis K, Tofaris GK. How is alpha‐synuclein cleared from the cell? J Neurochem. 2019;150:577–90. doi: 10.1111/jnc.14704. [DOI] [PubMed] [Google Scholar]
- 35.Lim K-L, Tan JM. Role of the ubiquitin proteasome system in Parkinson’s disease. BMC Biochem. 2007;8:S13. doi: 10.1186/1471-2091-8-S1-S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zondler L, Kostka M, Garidel P, Heinzelmann U, Hengerer B, Mayer B, et al. Proteasome impairment by α-synuclein. PLoS One. 2017;12:e0184040. doi: 10.1371/journal.pone.0184040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Snyder H, Mensah K, Theisler C, Lee J, Matouschek A, Wolozin B. Aggregated and monomeric α-synuclein bind to the S6′ proteasomal protein and inhibit proteasomal function. J Biol Chem. 2003;278:11753–9. doi: 10.1074/jbc.M208641200. [DOI] [PubMed] [Google Scholar]
- 38.Alghamdi A, Vallortigara J, Howlett DR, Broadstock M, Hortobágyi T, Ballard C, et al. Reduction of RPT6/S8 (a proteasome component) and proteasome activity in the cortex is associated with cognitive impairment in Lewy body dementia. J Alzheimer’s Dis. 2017;57:373–86. doi: 10.3233/JAD-160946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nonaka T, Hasegawa M. A cellular model to monitor proteasome dysfunction by α-synuclein. Biochemistry. 2009;48:8014–22. doi: 10.1021/bi900619j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McKinnon C, De Snoo ML, Gondard E, Neudorfer C, Chau H, Ngana SG, et al. Early-onset impairment of the ubiquitin-proteasome system in dopaminergic neurons caused by α-synuclein. Acta Neuropathol Commun. 2020;8:17. doi: 10.1186/s40478-020-0894-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Na CH, Jones DR, Yang Y, Wang X, Xu Y, Peng J. Synaptic protein ubiquitination in rat brain revealed by antibody-based ubiquitome analysis. J Proteome Res. 2012;11:4722–32. doi: 10.1021/pr300536k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sugeno N, Hasegawa T, Tanaka N, Fukuda M, Wakabayashi K, Oshima R, et al. Lys-63-linked ubiquitination by E3 ubiquitin ligase Nedd4-1 facilitates endosomal sequestration of internalized α-synuclein. J Biol Chem. 2014;289:18137–51.. doi: 10.1074/jbc.M113.529461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He F, Zhang L, Qi G, Zhang Q, Cai H, Li T, et al. Global ubiquitome analysis of substantia nigra in doubly-mutant human alpha-synuclein transgenic mice. Behav Brain Res. 2020;380:112436. doi: 10.1016/j.bbr.2019.112436. [DOI] [PubMed] [Google Scholar]
- 44.Meier F, Abeywardana T, Dhall A, Marotta NP, Varkey J, Langen R, et al. Semisynthetic, site-specific ubiquitin modification of α-synuclein reveals differential effects on aggregation. J Am Chem Soc. 2012;134:5468–71. doi: 10.1021/ja300094r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abeywardana T, Lin YH, Rott R, Engelender S, Pratt MR. Site-specific differences in proteasome-dependent degradation of monoubiquitinated α-synuclein. Chem Biol. 2013;20:1207–13. doi: 10.1016/j.chembiol.2013.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mund T, Masuda-Suzukake M, Goedert M, Pelham HR. Ubiquitination of alpha-synuclein filaments by Nedd4 ligases. PLoS One. 2018;13:e0200763. doi: 10.1371/journal.pone.0200763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gerez JA, Prymaczok NC, Rockenstein E, Herrmann US, Schwarz P, Adame A, et al. A cullin-RING ubiquitin ligase targets exogenous α-synuclein and inhibits Lewy body-like pathology. Sci Transl Med. 2019;11. 10.1126/scitranslmed.aau6722. [DOI] [PMC free article] [PubMed]
- 48.Tetzlaff JE, Putcha P, Outeiro TF, Ivanov A, Berezovska O, Hyman BT, et al. CHIP targets toxic alpha-synuclein oligomers for degradation. J Biol Chem. 2008;283:17962–8. doi: 10.1074/jbc.M802283200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rott R, Szargel R, Haskin J, Bandopadhyay R, Lees AJ, Shani V, et al. α-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc Natl Acad Sci USA. 2011;108:18666–71.. doi: 10.1073/pnas.1105725108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tofaris GK, Kim HT, Hourez R, Jung J-W, Kim KP, Goldberg AL. Ubiquitin ligase Nedd4 promotes α-synuclein degradation by the endosomal–lysosomal pathway. Proc Natl Acad Sci. 2011;108:17004–9. doi: 10.1073/pnas.1109356108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rott R, Szargel R, Shani V, Hamza H, Savyon M, Abd Elghani F, et al. SUMOylation and ubiquitination reciprocally regulate α-synuclein degradation and pathological aggregation. Proc Natl Acad Sci USA. 2017;114:13176–81. doi: 10.1073/pnas.1704351114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tanji K, Mori F, Kakita A, Takahashi H, Wakabayashi K. Alteration of autophagosomal proteins (LC3, GABARAP and GATE-16) in Lewy body disease. Neurobiol Dis. 2011;43:690–7. doi: 10.1016/j.nbd.2011.05.022. [DOI] [PubMed] [Google Scholar]
- 53.Volpicelli-Daley LA, Gamble KL, Schultheiss CE, Riddle DM, West AB, Lee VM-Y. Formation of α-synuclein Lewy neurite–like aggregates in axons impedes the transport of distinct endosomes. Mol Biol Cell. 2014;25:4010–23. doi: 10.1091/mbc.e14-02-0741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Odagiri S, Tanji K, Mori F, Kakita A, Takahashi H, Wakabayashi K. Autophagic adapter protein NBR1 is localized in Lewy bodies and glial cytoplasmic inclusions and is involved in aggregate formation in α-synucleinopathy. Acta Neuropathol. 2012;124:173–86. doi: 10.1007/s00401-012-0975-7. [DOI] [PubMed] [Google Scholar]
- 55.Guo M, Wang J, Zhao Y, Feng Y, Han S, Dong Q, et al. Microglial exosomes facilitate α-synuclein transmission in Parkinson’s disease. Brain. 2020;143:1476–97. doi: 10.1093/brain/awaa090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bussi C, Peralta Ramos JM, Arroyo DS, Gallea JI, Ronchi P, Kolovou A, et al. Alpha-synuclein fibrils recruit TBK1 and OPTN to lysosomal damage sites and induce autophagy in microglial cells. J Cell Sci. 2018;131. 10.1242/jcs.226241. [DOI] [PMC free article] [PubMed]
- 57.Fussi N, Höllerhage M, Chakroun T, Nykänen N-P, Rösler TW, Koeglsperger T, et al. Exosomal secretion of α-synuclein as protective mechanism after upstream blockage of macroautophagy. Cell Death Dis. 2018;9:757. doi: 10.1038/s41419-018-0816-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choi I, Zhang Y, Seegobin SP, Pruvost M, Wang Q, Purtell K, et al. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat Commun. 2020;11:1386. doi: 10.1038/s41467-020-15119-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gallardo G, Holtzman DM. Amyloid-β and Tau at the crossroads of Alzheimer’s disease. Singapore: Springer; 2019. pp 187–203.
- 60.Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–13. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313:1781–4. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- 62.Tseng BP, Green KN, Chan JL, Blurton-Jones M, LaFerla FM. Aβ inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging. 2008;29:1607–18. doi: 10.1016/j.neurobiolaging.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gentier RJ, van Leeuwen FW. Misframed ubiquitin and impaired protein quality control: an early event in Alzheimer’s disease. Front Mol Neurosci. 2015;8:47. doi: 10.3389/fnmol.2015.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Silva JM, Rodrigues S, Sampaio-Marques B, Gomes P, Neves-Carvalho A, Dioli C, et al. Dysregulation of autophagy and stress granule-related proteins in stress-driven Tau pathology. Cell Death Differ. 2019;26:1411–27. doi: 10.1038/s41418-018-0217-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Piatnitskaia S, Takahashi M, Kitaura H, Katsuragi Y, Kakihana T, Zhang L, et al. USP10 is a critical factor for Tau-positive stress granule formation in neuronal cells. Sci Rep. 2019;9:10591. doi: 10.1038/s41598-019-47033-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cripps D, Thomas SN, Jeng Y, Yang F, Davies P, Yang AJ. Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-Tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation. J Biol Chem. 2006;281:10825–38. doi: 10.1074/jbc.M512786200. [DOI] [PubMed] [Google Scholar]
- 67.Paine S, Bedford L, Thorpe JR, Mayer RJ, Cavey JR, Bajaj N, et al. Immunoreactivity to Lys63-linked polyubiquitin is a feature of neurodegeneration. Neurosci Lett. 2009;460:205–8. doi: 10.1016/j.neulet.2009.05.074. [DOI] [PubMed] [Google Scholar]
- 68.Dammer EB, Na CH, Xu P, Seyfried NT, Duong DM, Cheng D, et al. Polyubiquitin linkage profiles in three models of proteolytic stress suggest the etiology of Alzheimer disease. J Biol Chem. 2011;286:10457–65.. doi: 10.1074/jbc.M110.149633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nakayama Y, Sakamoto S, Tsuji K, Ayaki T, Tokunaga F, Ito H. Identification of linear polyubiquitin chain immunoreactivity in tau pathology of Alzheimer’s disease. Neurosci Lett. 2019;703:53–57. doi: 10.1016/j.neulet.2019.03.017. [DOI] [PubMed] [Google Scholar]
- 70.Abreha MH, Dammer EB, Ping L, Zhang T, Duong DM, Gearing M, et al. Quantitative analysis of the brain ubiquitylome in Alzheimer’s disease. Proteomics. 2018;18:1800108. doi: 10.1002/pmic.201800108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tramutola A, Triani F, Di Domenico F, Barone E, Cai J, Klein JB, et al. Poly-ubiquitin profile in Alzheimer disease brain. Neurobiol Dis. 2018;118:129–41. doi: 10.1016/j.nbd.2018.07.006. [DOI] [PubMed] [Google Scholar]
- 72.Tan JMM, Wong ESP, Kirkpatrick DS, Pletnikova O, Ko HS, Tay S-P, et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet. 2008;17:431–9. doi: 10.1093/hmg/ddm320. [DOI] [PubMed] [Google Scholar]
- 73.Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, et al. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Investig. 2007;117:648–58.. doi: 10.1172/JCI29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Babu JR, Geetha T, Wooten MW. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J Neurochem. 2005;94:192–203. doi: 10.1111/j.1471-4159.2005.03181.x. [DOI] [PubMed] [Google Scholar]
- 75.Flach K, Ramminger E, Hilbrich I, Arsalan-Werner A, Albrecht F, Herrmann L, et al. Axotrophin/MARCH7 acts as an E3 ubiquitin ligase and ubiquitinates tau protein in vitro impairing microtubule binding. Biochim Biophys Acta. 2014;1842:1527–38. doi: 10.1016/j.bbadis.2014.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Arakhamia T, Lee CE, Carlomagno Y, Duong DM, Kundinger SR, Wang K, et al. Posttranslational modifications mediate the structural diversity of tauopathy strains. Cell. 2020;180:633–.e12. doi: 10.1016/j.cell.2020.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Olabarria M, Pasini S, Corona C, Robador P, Song C, Patel H, et al. Dysfunction of the ubiquitin ligase E3A Ube3A/E6-AP contributes to synaptic pathology in Alzheimer’s disease. Commun Biol. 2019;2:111. doi: 10.1038/s42003-019-0350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, et al. Extensive involvement of autophagy in Alzheimer Disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–22. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 79.Piras A, Collin L, Grüninger F, Graff C, Rönnbäck A. Autophagic and lysosomal defects in human tauopathies: analysis of post-mortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol Commun. 2016;4:22. doi: 10.1186/s40478-016-0292-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rohn TT, Wirawan E, Brown RJ, Harris JR, Masliah E, Vandenabeele P. Depletion of Beclin-1 due to proteolytic cleavage by caspases in the Alzheimer’s disease brain. Neurobiol Dis. 2011;43:68–78. doi: 10.1016/j.nbd.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Guarascio R, Salih D, Yasvoina M, Edwards FA, Cheetham ME, van der Spuy J. Negative regulator of ubiquitin-like protein 1 modulates the autophagy–lysosomal pathway via p62 to facilitate the extracellular release of tau following proteasome impairment. Hum Mol Genet. 2020;29:80–96. doi: 10.1093/hmg/ddz255. [DOI] [PubMed] [Google Scholar]
- 82.Chiò A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, et al. Prognostic factors in ALS: A critical review. Amyotroph Lateral Scler. 2009;10:310–23. doi: 10.3109/17482960802566824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ciryam P, Lambert-Smith IA, Bean DM, Freer R, Cid F, Tartaglia GG, et al. Spinal motor neuron protein supersaturation patterns are associated with inclusion body formation in ALS. Proc Natl Acad Sci USA. 2017;114:E3935–43.. doi: 10.1073/pnas.1613854114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–3. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 85.Kametani F, Obi T, Shishido T, Akatsu H, Murayama S, Saito Y, et al. Mass spectrometric analysis of accumulated TDP-43 in amyotrophic lateral sclerosis brains. Sci Rep. 2016;6:23281. doi: 10.1038/srep23281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hans F, Eckert M, von Zweydorf F, Gloeckner CJ, Kahle PJ. Identification and characterization of ubiquitinylation sites in TAR DNA-binding protein of 43 kDa (TDP-43) J Biol Chem. 2018;293:16083–99. doi: 10.1074/jbc.RA118.003440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cascella R, Fani G, Capitini C, Rusmini P, Poletti A, Cecchi C, et al. Quantitative assessment of the degradation of aggregated TDP‐43 mediated by the ubiquitin proteasome system and macroautophagy. FASEB J. 2017;31:5609–24. doi: 10.1096/fj.201700292RR. [DOI] [PubMed] [Google Scholar]
- 88.Nakayama Y, Tsuji K, Ayaki T, Mori M, Tokunaga F, Ito H. Linear polyubiquitin chain modification of TDP-43-positive neuronal cytoplasmic inclusions in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2020;79:256–65. doi: 10.1093/jnen/nlz135. [DOI] [PubMed] [Google Scholar]
- 89.Lee S, Jeon Y-M, Cha SJ, Kim S, Kwon Y, Jo M, et al. PTK2/FAK regulates UPS impairment via SQSTM1/p62 phosphorylation in TARDBP/TDP-43 proteinopathies. Autophagy. 2019;16:1396–412. [DOI] [PMC free article] [PubMed]
- 90.Lee Y-C, Huang W-C, Lin J-H, Kao T-J, Lin H-C, Lee K-H, et al. Znf179 E3 ligase-mediated TDP-43 polyubiquitination is involved in TDP-43- ubiquitinated inclusions (UBI) (+)-related neurodegenerative pathology. J Biomed Sci. 2018;25:76. doi: 10.1186/s12929-018-0479-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68:1440. doi: 10.1001/archneurol.2011.250. [DOI] [PubMed] [Google Scholar]
- 92.Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465:223–6. doi: 10.1038/nature08971. [DOI] [PubMed] [Google Scholar]
- 93.Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68:857–64. doi: 10.1016/j.neuron.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Deng H-X, Chen W, Hong S-T, Boycott KM, Gorrie GH, Siddique N, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477:211–5. doi: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347:1436–41. doi: 10.1126/science.aaa3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brady OA, Meng P, Zheng Y, Mao Y, Hu F. Regulation of TDP-43 aggregation by phosphorylation andp62/SQSTM1. J Neurochem. 2011;116:248–59. doi: 10.1111/j.1471-4159.2010.07098.x. [DOI] [PubMed] [Google Scholar]
- 97.Wolozin B, Ivanov P. Stress granules and neurodegeneration. Nat Rev Neurosci. 2019;20:649–66. doi: 10.1038/s41583-019-0222-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dao TP, Kolaitis R-M, Kim HJ, O’Donovan K, Martyniak B, Colicino E, et al. Ubiquitin modulates liquid-liquid phase separation of UBQLN2 via disruption of multivalent interactions. Mol Cell. 2018;69:965–.e6. doi: 10.1016/j.molcel.2018.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Alexander EJ, Ghanbari Niaki A, Zhang T, Sarkar J, Liu Y, Nirujogi RS, et al. Ubiquilin 2 modulates ALS/FTD-linked FUS-RNA complex dynamics and stress granule formation. Proc Natl Acad Sci USA. 2018;115:E11485–E11494.. doi: 10.1073/pnas.1811997115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang B, Maxwell BA, Joo JH, Gwon Y, Messing J, Mishra A, et al. ULK1 and ULK2 regulate stress granule disassembly through phosphorylation and activation of VCP/p97. Mol Cell. 2019;74:742–57.e8. doi: 10.1016/j.molcel.2019.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shiihashi G, Ito D, Yagi T, Nihei Y, Ebine T, Suzuki N. Mislocated FUS is sufficient for gain-of-toxic-function amyotrophic lateral sclerosis phenotypes in mice. Brain. 2016;139:2380–94. doi: 10.1093/brain/aww161. [DOI] [PubMed] [Google Scholar]
- 102.Soo KY, Sultana J, King A, Atkinson R, Warraich ST, Sundaramoorthy V, et al. ALS-associated mutant FUS inhibits macroautophagy which is restored by overexpression of Rab1. Cell Death Discov. 2015;1:15030. doi: 10.1038/cddiscovery.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang T, Jiang X, Chen G, Xu J. Interaction of amyotrophic lateral sclerosis/frontotemporal lobar degeneration–associated fused-in-sarcoma with proteins involved in metabolic and protein degradation pathways. Neurobiol Aging. 2015;36:527–35. doi: 10.1016/j.neurobiolaging.2014.07.044. [DOI] [PubMed] [Google Scholar]
- 104.Stieber A, Gonatas JO, Gonatas NK. Aggregation of ubiquitin and a mutant ALS-linked SOD1 protein correlate with disease progression and fragmentation of the Golgi apparatus. J Neurol Sci. 2000;173:53–62. doi: 10.1016/S0022-510X(99)00300-7. [DOI] [PubMed] [Google Scholar]
- 105.Kabuta T, Suzuki Y, Wada K. Degradation of amyotrophic lateral sclerosis-linked mutant Cu,Zn-superoxide dismutase proteins by macroautophagy and the proteasome. J Biol Chem. 2006;281:30524–33.. doi: 10.1074/jbc.M603337200. [DOI] [PubMed] [Google Scholar]
- 106.Farrawell NE, Lambert-Smith I, Mitchell K, McKenna J, McAlary L, Ciryam P et al. SOD1A4V aggregation alters ubiquitin homeostasis in a cell model of ALS. J Cell Sci. 2018;131. 10.1242/jcs.209122. [DOI] [PMC free article] [PubMed]
- 107.Niwa J-I, Ishigaki S, Hishikawa N, Yamamoto M, Doyu M, Murata S, et al. Dorfin ubiquitylates mutant SOD1 and prevents mutant SOD1-mediated neurotoxicity. J Biol Chem. 2002;277:36793–8. doi: 10.1074/jbc.M206559200. [DOI] [PubMed] [Google Scholar]
- 108.Miyazaki K, Fujita T, Ozaki T, Kato C, Kurose Y, Sakamoto M, et al. NEDL1, a novel ubiquitin-protein isopeptide ligase for dishevelled-1, targets mutant superoxide dismutase-1. J Biol Chem. 2004;279:11327–35.. doi: 10.1074/jbc.M312389200. [DOI] [PubMed] [Google Scholar]
- 109.Yonashiro R, Sugiura A, Miyachi M, Fukuda T, Matsushita N, Inatome R, et al. Mitochondrial ubiquitin ligase MITOL ubiquitinates mutant SOD1 and attenuates mutant SOD1-induced reactive oxygen species generation. Mol Biol Cell. 2009;20:4524–30. doi: 10.1091/mbc.e09-02-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yung C, Sha D, Li L, Chin L-S. Parkin protects against misfolded SOD1 toxicity by promoting its aggresome formation and autophagic clearance. Mol Neurobiol. 2016;53:6270–87. doi: 10.1007/s12035-015-9537-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Homma K, Takahashi H, Tsuburaya N, Naguro I, Fujisawa T, Ichijo H. Genome-wide siRNA screening reveals that DCAF4-mediated ubiquitination of optineurin stimulates autophagic degradation of Cu,Zn-superoxide dismutase. J Biol Chem. 2020;295:3148–58. doi: 10.1074/jbc.RA119.010239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–10. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 113.Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. 2014;10:204–16. doi: 10.1038/nrneurol.2014.24. [DOI] [PubMed] [Google Scholar]
- 114.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–3. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 115.Juenemann K, Schipper-Krom S, Wiemhoefer A, Kloss A, Sanz AS, Reits EAJ. Expanded polyglutamine-containing N-terminal huntingtin fragments are entirely degraded by mammalian proteasomes. J Biol Chem. 2013;288:27068–84. doi: 10.1074/jbc.M113.486076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schipper-Krom S, Juenemann K, Jansen AH, Wiemhoefer A, van den Nieuwendijk R, Smith DL, et al. Dynamic recruitment of active proteasomes into polyglutamine initiated inclusion bodies. FEBS Lett. 2014;588:151–9. doi: 10.1016/j.febslet.2013.11.023. [DOI] [PubMed] [Google Scholar]
- 117.Zucchelli S, Marcuzzi F, Codrich M, Agostoni E, Vilotti S, Biagioli M, et al. Tumor necrosis factor receptor-associated factor 6 (TRAF6) associates with huntingtin protein and promotes its atypical ubiquitination to enhance aggregate formation. J Biol Chem. 2011;286:25108–17. doi: 10.1074/jbc.M110.187591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Maheshwari M, Samanta A, Godavarthi SK, Mukherjee R, Jana NR. Dysfunction of the ubiquitin ligase Ube3a may be associated with synaptic pathophysiology in a mouse model of Huntington disease. J Biol Chem. 2012;287:29949–57.. doi: 10.1074/jbc.M112.371724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lin L, Jin Z, Tan H, Xu Q, Peng T, Li H. Atypical ubiquitination by E3 ligase WWP1 inhibits the proteasome-mediated degradation of mutant huntingtin. Brain Res. 2016;1643:103–12. doi: 10.1016/j.brainres.2016.03.027. [DOI] [PubMed] [Google Scholar]
- 120.Bhutani S, Das A, Maheshwari M, Lakhotia SC, Jana NR. Dysregulation of core components of SCF complex in poly-glutamine disorders. Cell Death Dis. 2012;3:e428–e428.. doi: 10.1038/cddis.2012.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang B, Zeng L, Merillat SA, Fischer S, Ochaba J, Thompson LM, et al. The ubiquitin conjugating enzyme Ube2W regulates solubility of the Huntington’s disease protein, huntingtin. Neurobiol Dis. 2018;109:127–36. doi: 10.1016/j.nbd.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bhat KP, Yan S, Wang C-E, Li S, Li X-J. Differential ubiquitination and degradation of huntingtin fragments modulated by ubiquitin-protein ligase E3A. Proc Natl Acad Sci USA. 2014;111:5706–11.. doi: 10.1073/pnas.1402215111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Miller VM, Nelson RF, Gouvion CM, Williams A, Rodriguez-Lebron E, Harper SQ et al. CHIP Suppresses polyglutamine aggregation and toxicity in vitro and in vivo. J Neurosci. 2005. 10.1523/JNEUROSCI.3001-05.2005. [DOI] [PMC free article] [PubMed]
- 124.Wong YC, Holzbaur ELF. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J Neurosci. 2014;34:1293–305. doi: 10.1523/JNEUROSCI.1870-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rui Y-N, Xu Z, Patel B, Chen Z, Chen D, Tito A, et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat Cell Biol. 2015;17:262–75. doi: 10.1038/ncb3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Franco-Iborra S, Plaza-Zabala A, Montpeyo M, Sebastian D, Vila M, Martinez-Vicente M. Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy 2020;1–18. [DOI] [PMC free article] [PubMed]
- 127.La SpadaAR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- 128.Breza M, Koutsis G. Kennedy’s disease (spinal and bulbar muscular atrophy): a clinically oriented review of a rare disease. J Neurol. 2019;266:565–73. doi: 10.1007/s00415-018-8968-7. [DOI] [PubMed] [Google Scholar]
- 129.Stenoien DL, Cummings CJ, Adams HP, Mancini MG, Patel K, DeMartino GN, et al. Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins, proteasome components and SRC-1, and are suppressed by the HDJ-2 chaperone. Hum Mol Genet. 1999;8:731–41. doi: 10.1093/hmg/8.5.731. [DOI] [PubMed] [Google Scholar]
- 130.Rusmini P, Sau D, Crippa V, Palazzolo I, Simonini F, Onesto E, et al. Aggregation and proteasome: the case of elongated polyglutamine aggregation in spinal and bulbar muscular atrophy. Neurobiol Aging. 2007;28:1099–111. doi: 10.1016/j.neurobiolaging.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 131.He B, Bai S, Hnat AT, Kalman RI, Minges JT, Patterson C, et al. An androgen receptor NH2-terminal conserved motif interacts with the COOH terminus of the Hsp70-interacting protein (CHIP) J Biol Chem. 2004;279:30643–53.. doi: 10.1074/jbc.M403117200. [DOI] [PubMed] [Google Scholar]
- 132.Adachi H, Waza M, Tokui K, Katsuno M, Minamiyama M, Tanaka F, et al. CHIP overexpression reduces mutant androgen receptor protein and ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model. J Neurosci. 2007. 10.1523/JNEUROSCI.1242-07.2007. [DOI] [PMC free article] [PubMed]
- 133.Doi H, Adachi H, Katsuno M, Minamiyama M, Matsumoto S, Kondo N, et al. p62/SQSTM1 differentially removes the toxic mutant androgen receptor via autophagy and inclusion formation in a spinal and bulbar muscular atrophy mouse model. J Neurosci. 2013;33:7710–27.. doi: 10.1523/JNEUROSCI.3021-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Montie HL, Cho MS, Holder L, Liu Y, Tsvetkov AS, Finkbeiner S, et al. Cytoplasmic retention of polyglutamine-expanded androgen receptor ameliorates disease via autophagy in a mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet. 2009;18:1937–50. doi: 10.1093/hmg/ddp115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rusmini P, Crippa V, Giorgetti E, Boncoraglio A, Cristofani R, Carra S, et al. Clearance of the mutant androgen receptor in motoneuronal models of spinal and bulbar muscular atrophy. Neurobiol Aging. 2013;34:2585–603. doi: 10.1016/j.neurobiolaging.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rusmini P, Polanco MJ, Cristofani R, Cicardi ME, Meroni M, Galbiati M, et al. Aberrant autophagic response in the muscle of a knock-in mouse model of spinal and bulbar muscular atrophy. Sci Rep. 2015;5:15174. doi: 10.1038/srep15174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cicardi ME, Cristofani R, Crippa V, Ferrari V, Tedesco B, Casarotto E, et al. Autophagic and proteasomal mediated removal of mutant androgen receptor in muscle models of spinal and bulbar muscular atrophy. Front Endocrinol. 2019;10:569. doi: 10.3389/fendo.2019.00569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cortes CJ, Miranda HC, Frankowski H, Batlevi Y, Young JE, Le A, et al. Polyglutamine-expanded androgen receptor interferes with TFEB to elicit autophagy defects in SBMA. Nat Neurosci. 2014;17:1180–9. doi: 10.1038/nn.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Diallo A, Jacobi H, Tezenas du Montcel S, Klockgether T. Natural history of most common spinocerebellar ataxia: a systematic review and meta-analysis. J Neurol. 2020;1–8. [DOI] [PubMed]
- 140.Pérez Ortiz JM, Mollema N, Toker N, Adamski CJ, O’Callaghan B, Duvick L, et al. Reduction of protein kinase A-mediated phosphorylation of ATXN1-S776 in Purkinje cells delays onset of ataxia in a SCA1 mouse model. Neurobiol Dis. 2018;116:93–105. doi: 10.1016/j.nbd.2018.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chen H-K, Fernandez-Funez P, Acevedo SF, Lam YC, Kaytor MD, Fernandez MH, et al. Interaction of Akt-phosphorylated ataxin-1 with 14-3-3 mediates neurodegeneration in spinocerebellar ataxia type 1. Cell. 2003;113:457–68. doi: 10.1016/S0092-8674(03)00349-0. [DOI] [PubMed] [Google Scholar]
- 142.Hong S, Lee S, Cho S-G, Kang S. UbcH6 interacts with and ubiquitinates the SCA1 gene product ataxin-1. Biochem Biophys Res Commun. 2008;371:256–60. doi: 10.1016/j.bbrc.2008.04.066. [DOI] [PubMed] [Google Scholar]
- 143.Kang A, Park SH, Lee S, Choi D-Y, Kim KP, Song HK, et al. A key lysine residue in the AXH domain of ataxin-1 is essential for its ubiquitylation. Biochim Biophys Acta. 2015;1854:356–64. doi: 10.1016/j.bbapap.2015.01.012. [DOI] [PubMed] [Google Scholar]
- 144.Al-Ramahi I, Lam YC, Chen H-K, de Gouyon B, Zhang M, Pérez AM, et al. CHIP protects from the neurotoxicity of expanded and wild-type ataxin-1 and promotes their ubiquitination and degradation. J Biol Chem. 2006;281:26714–24.. doi: 10.1074/jbc.M601603200. [DOI] [PubMed] [Google Scholar]
- 145.Choi JY, Ryu JH, Kim H-S, Park SG, Bae K-H, Kang S, et al. Co-chaperone CHIP promotes aggregation of ataxin-1. Mol Cell Neurosci. 2007;34:69–79. doi: 10.1016/j.mcn.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 146.Winborn BJ, Travis SM, Todi SV, Scaglione KM, Xu P, Williams AJ, et al. The deubiquitinating enzyme ataxin-3, a polyglutamine disease protein, edits Lys63 linkages in mixed linkage ubiquitin chains. J Biol Chem. 2008;283:26436–43.. doi: 10.1074/jbc.M803692200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Todi SV, Winborn BJ, Scaglione KM, Blount JR, Travis SM, Paulson HL. Ubiquitination directly enhances activity of the deubiquitinating enzyme ataxin-3. EMBO J. 2009;28:372–82. doi: 10.1038/emboj.2008.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kristensen LV, Oppermann FS, Rauen MJ, Hartmann-Petersen R, Thirstrup K. Polyglutamine expansion of ataxin-3 alters its degree of ubiquitination and phosphorylation at specific sites. Neurochem Int. 2017;105:42–50. doi: 10.1016/j.neuint.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 149.Neves-Carvalho A, Logarinho E, Freitas A, Duarte-Silva S, Costa M, do C, et al. Dominant negative effect of polyglutamine expansion perturbs normal function of ataxin-3 in neuronal cells. Hum Mol Genet. 2015;24:100–17. doi: 10.1093/hmg/ddu422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sutton JR, Blount JR, Libohova K, Tsou W-L, Joshi GS, Paulson HL, et al. Interaction of the polyglutamine protein ataxin-3 with Rad23 regulates toxicity in Drosophila models of spinocerebellar ataxia type 3. Hum Mol Genet. 2017;26:1419–31. doi: 10.1093/hmg/ddx039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Scaglione KM, Zavodszky E, Todi SV, Patury S, Xu P, Rodríguez-Lebrón E, et al. Ube2w and Ataxin-3 coordinately regulate the ubiquitin ligase CHIP. Mol Cell. 2011;43:599–612. doi: 10.1016/j.molcel.2011.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Nascimento-Ferreira I, Santos-Ferreira T, Sousa-Ferreira L, Auregan G, Onofre I, Alves S, et al. Overexpression of the autophagic beclin-1 protein clears mutant ataxin-3 and alleviates Machado–Joseph disease. Brain. 2011;134:1400–15. doi: 10.1093/brain/awr047. [DOI] [PubMed] [Google Scholar]
- 153.Sittler A, Muriel M-P, Marinello M, Brice A, den Dunnen W, Alves S. Deregulation of autophagy in postmortem brains of Machado-Joseph disease patients. Neuropathology. 2018;38:113–24. doi: 10.1111/neup.12433. [DOI] [PubMed] [Google Scholar]
- 154.Ashkenazi A, Bento CF, Ricketts T, Vicinanza M, Siddiqi F, Pavel M, et al. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature. 2017;545:108–11. doi: 10.1038/nature22078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Herzog LK, Kevei É, Marchante R, Böttcher C, Bindesbøll C, Lystad AH, et al. The Machado–Joseph disease deubiquitylase ataxin‐3 interacts with LC3C/GABARAP and promotes autophagy. Aging Cell. 2020;19. 10.1111/acel.13051. [DOI] [PMC free article] [PubMed]
- 156.Shi C-H, Schisler JC, Rubel CE, Tan S, Song B, McDonough H, et al. Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP. Hum Mol Genet. 2014;23:1013–24. doi: 10.1093/hmg/ddt497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Shi C, Rubel C, Soss SE, Sanchez-Hodge R, Zhang S, Madrigal SC, et al. Disrupted structure and aberrant function of CHIP mediates the loss of motor and cognitive function in preclinical models of SCAR16. PLOS Genet. 2018;14:e1007664. doi: 10.1371/journal.pgen.1007664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Soto C. Transmissible proteins: expanding the prion heresy. Cell. 2012;149:968–77. doi: 10.1016/j.cell.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chung E, Choi Y, Park J, Nah W, Park J, Jung Y, et al. Intracellular delivery of Parkin rescues neurons from accumulation of damaged mitochondria and pathological α-synuclein. Sci Adv. 2020;6:eaba1193. doi: 10.1126/sciadv.aba1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Carroll EC, Greene ER, Martin A, Marqusee S. Site-specific ubiquitination affects protein energetics and proteasomal degradation. Nat Chem Biol. 2020;16:866–75. [DOI] [PMC free article] [PubMed]
- 161.Burslem GM, Crews CM. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell. 2020;181:102–14. doi: 10.1016/j.cell.2019.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Verma R, Mohl D, Deshaies RJ. Harnessing the power of proteolysis for targeted protein inactivation. Mol Cell. 2020;77:446–60. doi: 10.1016/j.molcel.2020.01.010. [DOI] [PubMed] [Google Scholar]
- 163.Pukaß K, Richter-Landsberg C. Inhibition of UCH-L1 in oligodendroglial cells results in microtubule stabilization and prevents α-synuclein aggregate formation by activating the autophagic pathway: implications for multiple system atrophy. Front Cell Neurosci. 2015;9:163. doi: 10.3389/fncel.2015.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Alexopoulou Z, Lang J, Perrett RM, Elschami M, Hurry MED, Kim HT, et al. Deubiquitinase Usp8 regulates α-synuclein clearance and modifies its toxicity in Lewy body disease. Proc Natl Acad Sci USA. 2016;113:E4688–97. doi: 10.1073/pnas.1523597113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kumari R, Kumar R, Kumar S, Singh AK, Hanpude P, Jangir D, et al. Amyloid aggregates of the deubiquitinase OTUB1 are neurotoxic, suggesting that they contribute to the development of Parkinson’s disease. J Biol Chem. 2020;295:3466–84. doi: 10.1074/jbc.RA119.009546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Rott R, Szargel R, Haskin J, Shani V, Shainskaya A, Manov I, et al. Monoubiquitylation of alpha-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J Biol Chem. 2008;283:3316–28.. doi: 10.1074/jbc.M704809200. [DOI] [PubMed] [Google Scholar]
- 167.Zhang M, Cai F, Zhang S, Zhang S, Song W. Overexpression of ubiquitin carboxyl-terminal hydrolase L1 (UCHL1) delays Alzheimer’s progression in vivo. Sci Rep. 2015;4:7298. doi: 10.1038/srep07298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jung ES, Hong H, Kim C, Mook-Jung I. Acute ER stress regulates amyloid precursor protein processing through ubiquitin-dependent degradation. Sci Rep. 2015;5:8805. doi: 10.1038/srep08805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Boselli M, Lee B-H, Robert J, Prado MA, Min S-W, Cheng C, et al. An inhibitor of the proteasomal deubiquitinating enzyme USP14 induces tau elimination in cultured neurons. J Biol Chem. 2017;292:19209–25. doi: 10.1074/jbc.M117.815126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Jin YN, Chen P-C, Watson JA, Walters BJ, Phillips SE, Green K, et al. Usp14 deficiency increases Tau phosphorylation without altering Tau degradation or causing Tau-dependent deficits. PLoS ONE. 2012;7:e47884. doi: 10.1371/journal.pone.0047884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lee JH, Shin SK, Jiang Y, Choi WH, Hong C, Kim D-E, et al. Facilitated Tau degradation by USP14 aptamers via enhanced proteasome activity. Sci Rep. 2015;5:10757. doi: 10.1038/srep10757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wang P, Joberty G, Buist A, Vanoosthuyse A, Stancu I-C, Vasconcelos B, et al. Tau interactome mapping based identification of Otub1 as Tau deubiquitinase involved in accumulation of pathological Tau forms in vitro and in vivo. Acta Neuropathol. 2017;133:731–49. doi: 10.1007/s00401-016-1663-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Yu Q, Zhang H, Li Y, Liu C, Wang S, Liao X. UCH-L1 inhibition suppresses tau aggresome formation during proteasomal impairment. Mol Neurobiol. 2017;55:3812–21.. doi: 10.1007/s12035-017-0558-7. [DOI] [PubMed] [Google Scholar]
- 174.Wang H, Ying Z, Wang G. Ataxin-3 regulates aggresome formation of copper-zinc superoxide dismutase (SOD1) by editing K63-linked polyubiquitin chains. J Biol Chem. 2012;287:28576–85.. doi: 10.1074/jbc.M111.299990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hans F, Fiesel FC, Strong JC, Jäckel S, Rasse TM, Geisler S, et al. UBE2E ubiquitin-conjugating enzymes and ubiquitin isopeptidase Y regulate TDP-43 protein ubiquitination. J Biol Chem. 2014;289:19164–79.. doi: 10.1074/jbc.M114.561704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lee B-H, Lee MJ, Park S, Oh D-C, Elsasser S, Chen P-C, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467:179–84.. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Dammer EB, Fallini C, Gozal YM, Duong DM, Rossoll W, Xu P, et al. Coaggregation of RNA-binding proteins in a model of TDP-43 proteinopathy with selective RGG motif methylation and a role for RRM1 ubiquitination. PLoS ONE. 2012;7:e38658. doi: 10.1371/journal.pone.0038658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Aron R, Pellegrini P, Green EW, Maddison DC, Opoku-Nsiah K, Oliveira AO, et al. Deubiquitinase Usp12 functions noncatalytically to induce autophagy and confer neuroprotection in models of Huntington’s disease. Nat Commun. 2018;9:3191. doi: 10.1038/s41467-018-05653-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.He W-T, Zheng X-M, Zhang Y-H, Gao Y-G, Song A-X, van der Goot FG, et al. Cytoplasmic ubiquitin-specific protease 19 (USP19) modulates aggregation of polyglutamine-expanded ataxin-3 and huntingtin through the HSP90 chaperone. PLoS One. 2016;11:e0147515. doi: 10.1371/journal.pone.0147515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Koyuncu S, Saez I, Lee HJ, Gutierrez-Garcia R, Pokrzywa W, Fatima A, et al. The ubiquitin ligase UBR5 suppresses proteostasis collapse in pluripotent stem cells from Huntington’s disease patients. Nat Commun. 2018;9:2886. doi: 10.1038/s41467-018-05320-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sap KA, Guler AT, Bezstarosti K, Bury AE, Juenemann K, Demmers JAA, et al. Global proteome and ubiquitinome changes in the soluble and insoluble fractions of Q175 huntington mice brains. Mol Cell Proteom. 2019;18:1705–20. doi: 10.1074/mcp.RA119.001486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Dong G, Callegari E, Gloeckner CJ, Ueffing M, Wang H. Mass spectrometric identification of novel posttranslational modification sites in Huntingtin. Proteomics. 2012;12:2060–4. doi: 10.1002/pmic.201100380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ciechanover A, Kwon YT. Protein quality control by molecular chaperones in neurodegeneration. Front Neurosci. 2017;11:185. doi: 10.3389/fnins.2017.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Upadhyay A, Joshi V, Amanullah A, Mishra R, Arora N, Prasad A, et al. E3 ubiquitin ligases neurobiological mechanisms: development to degeneration. Front Mol Neurosci. 2017;10:151. doi: 10.3389/fnmol.2017.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Lim K-H, Joo J-Y, Baek K-H. The potential roles of deubiquitinating enzymes in brain diseases. Ageing Res Rev. 2020;61:101088. doi: 10.1016/j.arr.2020.101088. [DOI] [PubMed] [Google Scholar]
- 186.Zhang S, Hu Z, Mao C, Shi C, Xu Y. CHIP as a therapeutic target for neurological diseases. Cell Death Dis. 2020;11:727. doi: 10.1038/s41419-020-02953-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Djajadikerta A, Keshri S, Pavel M, Prestil R, Ryan L, Rubinsztein DC. Autophagy induction as a therapeutic strategy for neurodegenerative diseases. J Mol Biol. 2020;432:2799–821. doi: 10.1016/j.jmb.2019.12.035. [DOI] [PubMed] [Google Scholar]
- 188.Takahashi D, Moriyama J, Nakamura T, Miki E, Takahashi E, Sato A, et al. AUTACs: cargo-specific degraders using selective autophagy. Mol Cell. 2019. 10.1016/J.MOLCEL.2019.09.009. [DOI] [PubMed]
- 189.Li Z, Wang C, Wang Z, Zhu C, Li J, Sha T, et al. Allele-selective lowering of mutant HTT protein by HTT–LC3 linker compounds. Nature. 2019;575:203–9. doi: 10.1038/s41586-019-1722-1. [DOI] [PubMed] [Google Scholar]