

Abstract
Fluorochemicals are a widely distributed class of compounds and have been utilized across a wide range of industries for decades. Given the environmental toxicity and adverse health threats of some fluorochemicals, the development of new methods for their decomposition is significant to public health. However, the carbon-fluorine (C-F) bond is among the most chemically robust bonds; consequently, the degradation of fluorinated hydrocarbons is exceptionally difficult. Here, metalloenzymes that catalyze the cleavage of this chemically challenging bond are reviewed. These enzymes include histidine-ligated heme-dependent dehaloperoxidase and tyrosine hydroxylase, thiolate-ligated hemedependent cytochrome P450, and four nonheme oxygenases, namely, tetrahydrobiopterin-dependent aromatic amino acid hydroxylase, 2-oxoglutarate-dependent hydroxylase, Rieske dioxygenase, and thiol dioxygenase. While much of the literature regarding the aforementioned enzymes highlights their ability to catalyze C-H bond activation and functionalization, in many cases, the C-F bond cleavage has been shown to occur on fluorinated substrates. A copper-dependent laccase-mediated system representing an unnatural radical defluorination approach is also described. Detailed discussions on the structure-function relationships and catalytic mechanisms provide insights into biocatalytic defluorination, which may inspire drug design considerations and environmental remediation of halogenated contaminants.
Graphical Abstract
1. Introduction
Fluorine-containing hydrocarbons, also known as organofluorine compounds, are widely used in our daily lives. Fluorine substitution is known for its small steric effect, high electron-withdrawing properties, lipophilicity, and thermal stability.1–3 The unique chemical properties of fluorine have led to the applications of organofluorine compounds in diverse fields, including materials science, pharmaceutical industries, catalysis, energy, and agriculture.1–4 Human civilization has greatly benefited from the development and manufacture of fluorinated compounds. The fluorochemical market was valued at 24.6 billion USD with a global production volume of 4.2 million tonnes during 2018.5 It was also reported that about 30 percent of approved drugs are fluorine-containing compounds.6 Nevertheless, a significant characteristic of these fluorocarbon compounds is the strength of the carbon-fluorine (C-F) bond. For instance, the bond dissociation energy of the C-F bond in fluorobenzene is 526 J mol−1.7 To break such a chemically robust bond in a homolytic process is difficult under mild conditions. As a result, the prevalence and accumulation of fluorinated compounds, especially perfluorinated compounds, have become a severe, worldwide environmental threat.8–13 Series of per- and polyfluoroalkyl substances (PFAS) are manufactured in bulk and widely used; some are now globally banned due to ecological and human health concerns.14 These chemically inert carcinogens, once released, are challenging to decompose in the natural environment and thus result in significant contamination in soil and groundwater.15
Consequently, the degradation of fluorochemicals has attracted considerable attention and inspired research studies on C-F bond activation and cleavage. In recent years, various model complexes have been synthesized and shown to perform C-F bond cleavage by employing metals such as Fe, Al, Cu, and Mn and even some rare-earth metals.16–24 In most cases, it has been proposed that high-valent metal-oxo species play a critical role in the catalytic mechanism of these metal complexes.18,22,25 Currently, the primary methods for defluorination are the use of synthetic models and artificial catalysts, while the exploration of biocatalysts is still in its infancy. Although C-F bond cleavage requires a considerable input of energy, microorganisms that have adapted to polluted environments have been found to facilitate the biodegradation or biotransformation of fluorinated compounds.26–28 Studies on such microbial types of machinery have revealed several enzymes capable of C-F bond cleavage.27,29–31 In addition to the proteins identified in the catabolism of such microorganisms, there are other enzymes found to exhibit substrate promiscuity when confronted with noncanonical, fluorinated substrates.32–37 These findings provide a promising platform for engineering biocatalysts that can detoxify fluorochemicals. In general, these defluorinase proteins can be classified into two groups: metal-independent and metal-dependent enzymes. Examples of metal-independent systems are fluoroacetate dehalogenases,38–40 ATP-dependent reductases,41,42 certain hydroxylases and isomerases,43–46 and flavin-dependent monooxygenases.47–49 Metal-containing proteins with or intimately associated with C-F bond cleavage activities have been elucidated and have become increasingly popular. Metalloenzymes are proficient catalysts due to their oxygen activation characteristic, substrate selectivity, specificity, and structural stability. The formation of high-valent metal intermediates during catalysis empowers these proteins to mediate a broad spectrum of chemistries, including defluorination. In this review, metalloprotein-mediated defluorination, mostly on fluoroaromatics, is summarized. These metalloenzymes activate C-H bonds and are relevant to essential metabolism events, while C-F bond cleavage can take place on fluorinated substrates via distinguishing routes. Herein, the C-F bond cleavage reactions will be discussed in detail in terms of the molecular mechanism. It is expected that the study of enzymatic structure-function relationship coupled with the knowledge of the catalytic mechanism can shed light on the defluorination chemistry and pave the way for future applications.
2. C-F bond activation by histidine-ligated heme enzymes
2.1. Histidine-ligated heme-dependent dehaloperoxidase (DHP)
Metalloenzyme-mediated defluorination is a rare catalytic transformation found across the biosynthesis of natural metabolites. However, multifunctional globin-like DHP protein is a well-characterized example of an enzyme capable of promoting such chemistry. DHP was originally discovered in Amphitrite ornata, a marine polychaete worm of the family Terebellidae.50 In addition to binding and transporting molecular oxygen as hemoglobin, DHP has long been known to have multiple catalytic properties such as activity typical of an oxygenase, an oxidase, a peroxygenase, and even a peroxidase for dehalogenation.51–53 Herein, we will focus on discussing the peroxidase activity of this enzyme.
DHP oxidizes trihalophenol (TXP) into its corresponding quinone by utilizing hydrogen peroxide (H2O2) as the cosubstrate, resulting in the elimination of the para-substituted halogen (F, Cl, Br, or I) on TXP in the form of a halide anion, as shown in Fig. 1A. In the resting state, DHP possesses a histidine-ligated b-type ferric heme (Fig. 1B), as well as an overall fold that is typical of the globin family.54 The enzyme is predominantly dimeric in solution, and it crystallizes as a dimer in an asymmetric unit.30 The crystal structures of DHP in complex with the substrate analogs or inhibitors exhibit diverse binding modes in the active site, indicative of a spacious substrate cavity.55–58 The crystal structures of the enzyme-substrate (ES) complexes were determined by soaking them in a native substrate, namely, tribromophenol (TBP). However, the occupancy of TBP is extremely low in the determined ES complex structures (occupancy: ~10%; PDB entries: 4FH6 and 4FH7).56 Although the structures of the ES complex are contentious due to their low occupancies, TBP has been shown to bind at the active site, with its hydroxyl group pointing toward the heme center, while the para-substituent to be eliminated points away (Fig. 1B). This structural information excludes the possibility of direct activation at the para position by a hemebased oxidant during the catalytic reaction.
Fig. 1.
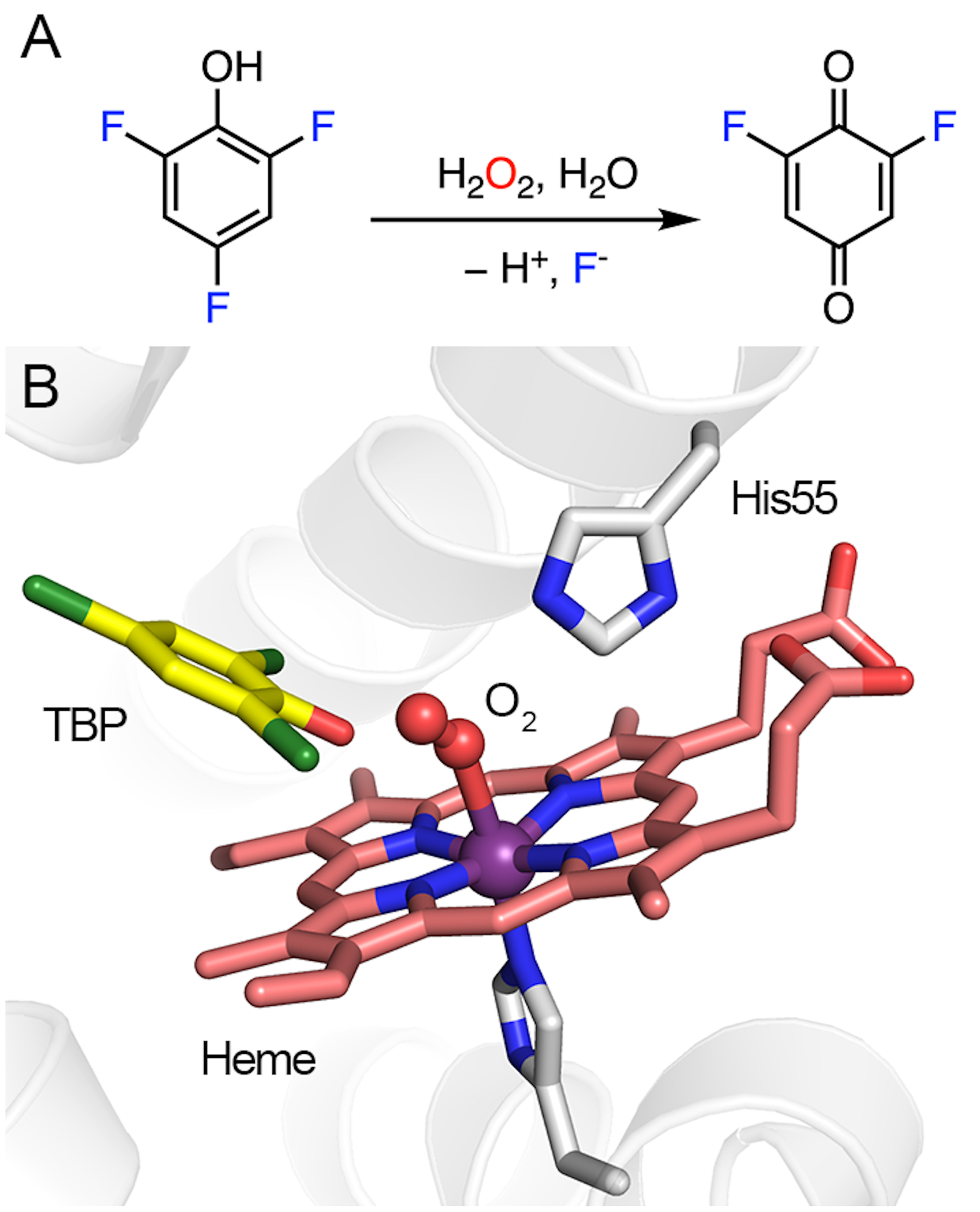
DHP-catalyzed reaction and its active-site architecture. (A) Oxidative C-F bond cleavage of trifluorophenol. (B) Crystal structure of DHP in a complex with TBP and oxygen. His55 interacts with oxygen via hydrogen bonding (PDB entry: 4FH6).
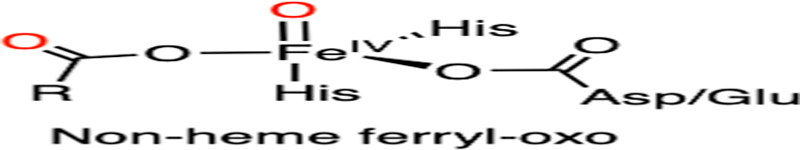
The dehalogenation mechanism of DHP is presumed to involve two one-electron oxidation processes mediated by the high-valent heme iron oxidants, i.e., Compound I (Cpd I)- and Compound II (Cpd II)-like species, respectively.59–62 Cpd I and Cpd II represent two reactive ferryl-oxo intermediates in the catalytic cycle of the thiolate-ligated cytochrome P450, where the former contains a porphyrin-based cation radical, which is absent in the latter. These high-valent species, or their equivalents with a histidine-ligated heme, are commonly proposed oxidants in the catalytic pathways of heme-containing enzymes.63,64 Fig. 2 shows the proposed defluorination mechanism of DHP. Even though the binding order of TXP and H2O2 is undetermined,55,65 it is believed that the conformational flexibility of a distal histidine, i.e., His55, plays a critical role in adopting an activated DHP.66,67 In the activated form, His55 is in close contact with the heme center, as shown in Fig. 1B. Then, the protein promotes the heterolytic cleavage of the O-O bond by providing a proton to the ferric hydroperoxo, resulting in a histidine-ligated Cpd I-like species.65,68 Such a species is chemically equivalent to the so-called Compound ES (Cpd ES), which is a Cpd I isoform with a ferryl heme and a nearby protein-based aromatic radical.69,70 Subsequently, Cpd I or Cpd ES proceeds with the first one-electron oxidation of the substrate, yielding a dissociable organic radical on the substrate and a Cpd II-like species.71,72 Then, the Cpd II-like species undergoes a second one-electron oxidation process on a resonance structure of the substrate radical, affording a phenoxy cation. The formation of the stable 2,6-difluoroquinone product proceeds through the elimination of fluoride, which occurs after the nucleophilic attack by a water molecule at the para-substituted carbon atom bearing the most cationic character.57,71 The18O-isotope-labeling studies confirmed that the oxygen atom incorporated into the final product is derived from water rather than the peroxide, which is consistent with the binding orientation of the substrate (as revealed by the crystal structures).57
Fig. 2.
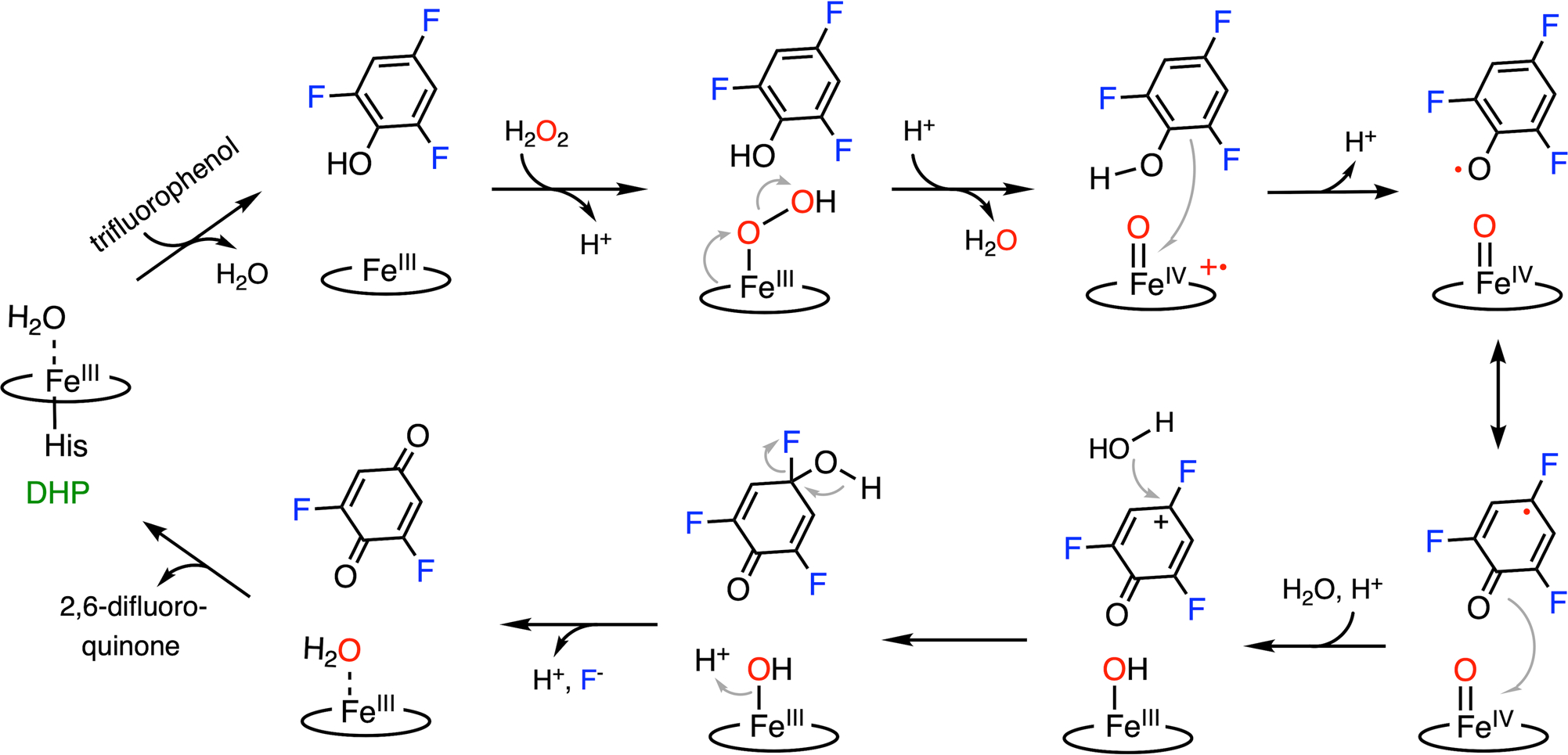
C-F bond cleavage pathway promoted by DHP.
Although DHP functions as a natural dehalogenase that oxidizes phenols with various halogen substituents, affinity and catalytic efficiency studies have shown that DHP favors brominated substrates under physiological conditions.50,73 Fluorinated phenols, on the other hand, have weak binding affinities and limited turnovers.50,58,73 However, the construction of an artificial DHP could be a promising method to develop competent biocatalysts that effectively cleave aromatic C-F bonds. Indeed, artificially created DHP activity has been achieved by mutating active-site residues in structurally related myoglobin.74 Although the defluorination activity of the native protein is yet to be reported, the engineered enzyme demonstrates a 1000-fold increase in efficiency on a trichlorophenol substrate. Consequently, engineered DHP is a prime candidate for defluorination activity, which confirms that residue-level modification is an effective strategy to achieve engineered enzymatic C-F bond cleavage of harmful fluorinated compounds.
2.2. Histidine-ligated heme-dependent tyrosine hydroxylase
A new class of biocatalysts hydroxylating L-tyrosine to L-dihydroxy-phenylalanine (DOPA) has been found in the biosynthetic pathways of antitumor and/or antibacterial antibiotics.75 While their protein structures remain to be determined, these tyrosine hydroxylases are heme-dependent proteins, hereafter referred to as heme TyrHs. A protein sequence analysis revealed that this type of enzymes comprised a b-type histidine-ligated heme.76,77 The first of this class of enzyme, namely, LmbB2, was identified from the biosynthetic gene cluster of lincomycin.78 In addition to LmbB2, other characterized heme TyrHs include the HrmE of hormaomycin,79 Orf13 of anthramycin,76 SibU of sibiromycin,80 TomI of tomaymycin,81 and Por14 of poranthramycin.82 The amino acid sequence alignment of these TyrHs shows no similarity with any known conserved domains or conserved motifs.76 Therefore, this group of proteins probably contains a novel structural fold, and determining the structural features of this enzyme family is imperative for mechanistic understandings. The heme-dependent TyrH is responsible for the first step in the construction of a pyrroline moiety in the biosynthetic pathways of the natural product by converting L-tyrosine into DOPA through hydrogen-peroxide-mediated oxidation. Unlike P450, nonheme TyrH, and other aromatic hydroxylases,83–87 the catalytic hemes in TyrHs are distinguished by an axial histidine ligand. Therefore, their mechanism of C-H bond activation and oxygen atom transfer cannot be simply extrapolated.32
LmbB2 has been shown to be capable of hydroxylating a range of L-tyrosine analogs with ring-deactivating meta-substituents such as 3-fluorotyrosine.32 Moreover, in addition to the expected C-H bond cleavage product, an additional product derived from the C-F bond cleavage has been observed (Fig. 3A). C-F bond cleavage requires two additional electrons for fluoride versus proton elimination; hence, the mechanism of C-F bond hydroxylation is likely to differ from native C-H bond hydroxylation. In the process of C-F bond cleavage, the consumption of additional peroxide and formation of oxygen were found, which suggests that the oxidation of peroxide into oxygen is presumably the source of two necessary electrons. Moreover, 18O-isotope-labeling experiments show that H2O2 is the oxygen source for hydroxylation in both C-H and C-F bond cleavage reactions. The detection of the double-scrambled product (with 18O atoms on both hydroxyl groups of DOPA) indicates a potential ketone or radical intermediate with broken aromaticity during catalysis. Collectively, a mechanism of C-F bond cleavage promoted by the heme-dependent TyrH was proposed (Fig. 4).
Fig. 3.
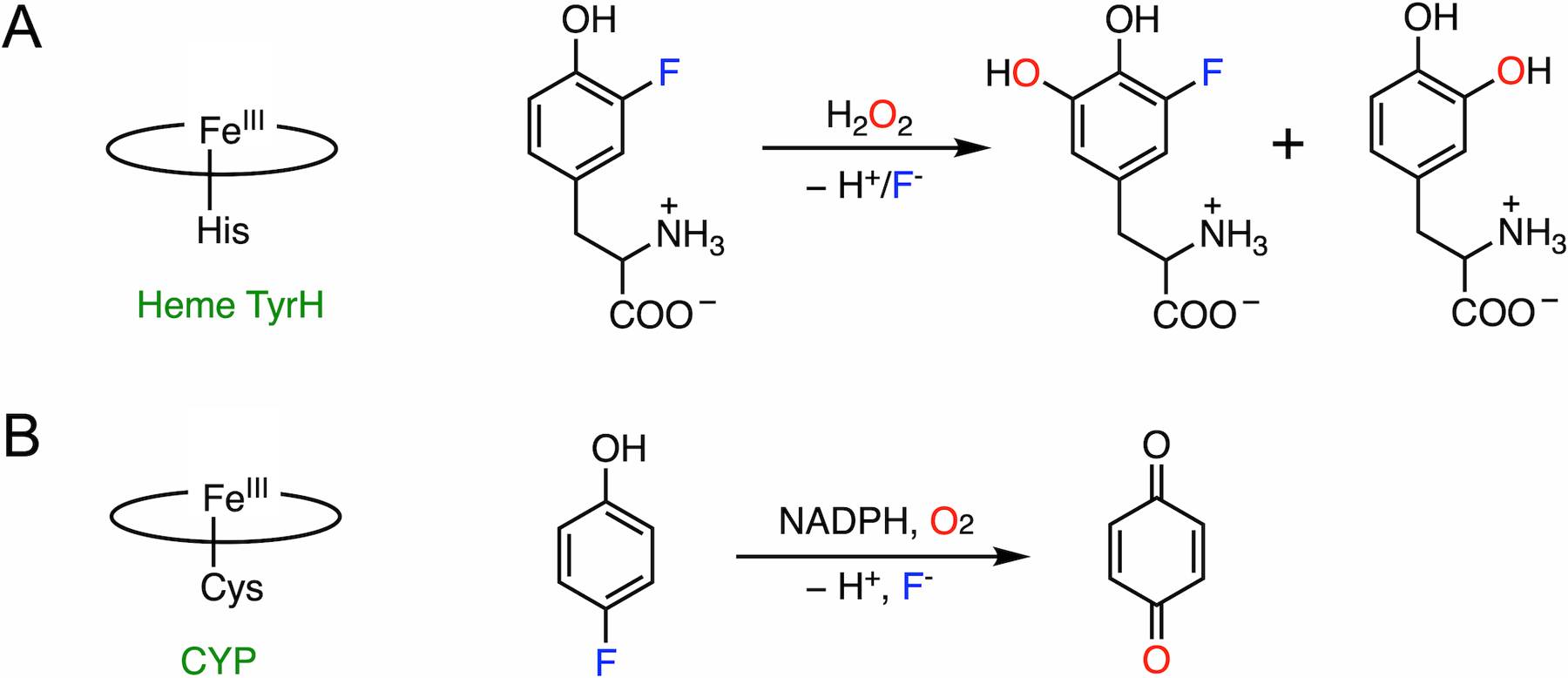
(A) Histidine-ligated heme TyrH catalyzes the defluorination of 3-fluorotyrosine. Two products are formed with C-H and C-F bond cleavages, respectively. (B) Thiolate-ligated CYP catalyzes the defluorination of 4-fluorophenol.
Fig. 4.
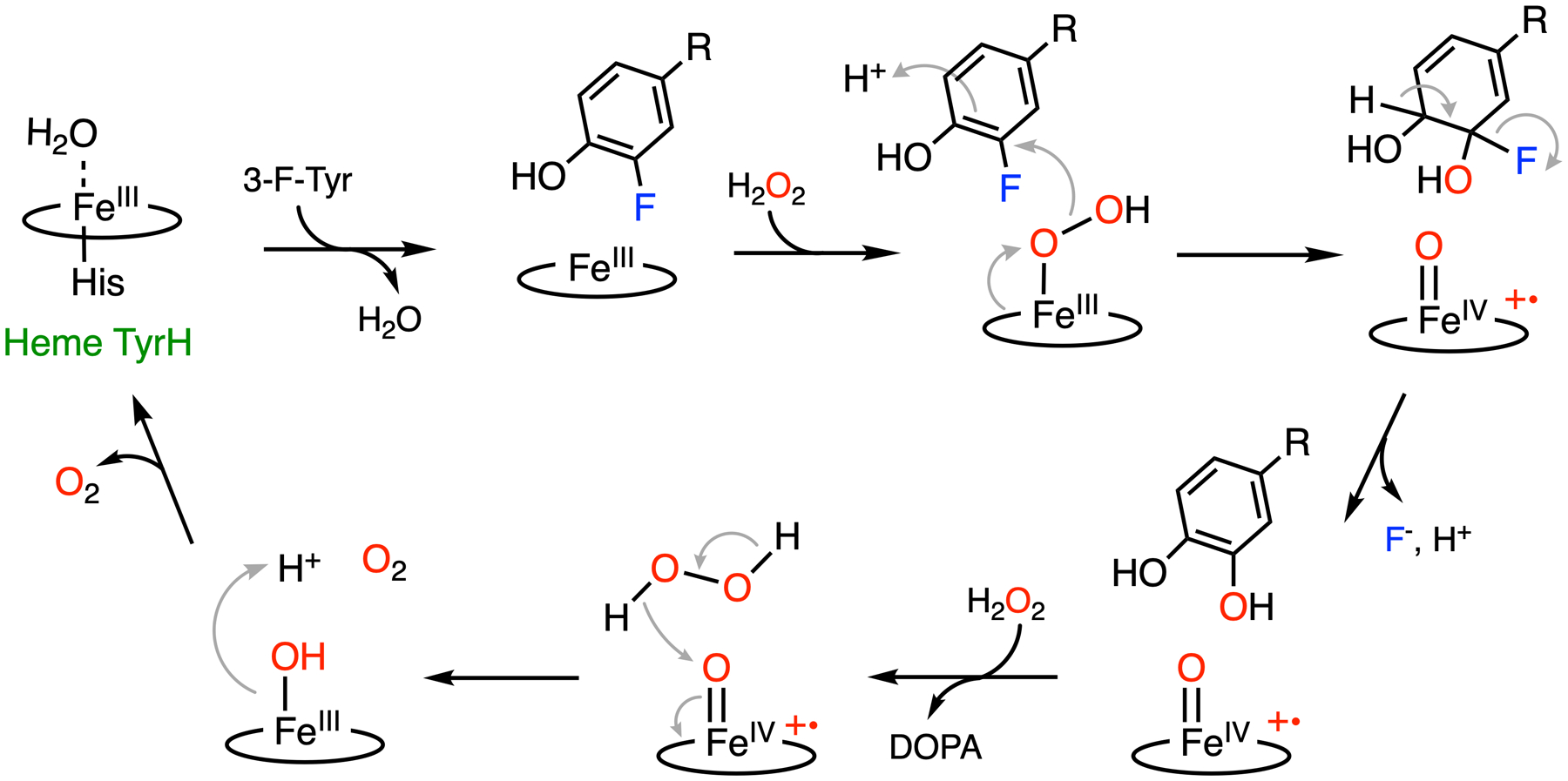
Proposed C-F bond cleavage mechanism promoted by heme TyrH.32 R: amino acid moiety.
Substrate binding to the active site allows for H2O2 activation by the ferric heme, affording a ferric-bound hydroperoxide. It has been speculated that 3-fluorotyrosine can bind in the active site with two distinct conformations, with the fluorine pointing toward or away from the heme center.32 Fluorine pointing away from the heme results in the native hydroxylated product, while fluorine oriented toward the heme results in the defluorinated product. In the latter case, due to the strong electron-withdrawing property of the fluorine substituent, the covalently bound carbon (C3) is partially electropositive. Therefore, a ferric heme-bound hydroperoxide can perform a nucleophilic attack at C3, resulting in a Cpd I-like species. In contrast to alkyl systems, fluoride is an excellent leaving group in a nucleophilic aromatic substitution reaction.88 It is eliminated from the ring to re-aromatize, yielding DOPA as the final product. The resulting Cpd I-like species can react with an additional equivalent of H2O2 to release oxygen and return to the resting state via catalase-like activity. Besides 3-fluorotyrosine, 3-chloro- and 3-iodotyrosine compounds have also been identified with dual reactivities. 3-Chloro-tyrosine has the best overall conversion (normal hydroxylation plus dehalogenation). At the same time, 3-fluorotyrosine exhibits the highest dehalogenation efficiency, despite the C-F bond being the most durable among all the carbon-halogen (C-X) bonds. The different distributions of hydroxylated products from two distinct reaction pathways are a cumulative outcome of the steric and inductive effects of these halogen substituents.
Cleaving an aromatic C-F bond is not a common feature among the histidine-ligated heme proteins because they typically function as globins or peroxidases to bind oxygen or transfer electrons. But a closer comparison between DHP and heme TyrH suggests that these two enzymes catalyze very different dehalogenation reactions (Table 1), even though they utilize the same biocatalytic cofactor and oxidant (H2O2). In the case of DHP, the para-substituted halogen is eliminated from the phenyl ring via a two-electron oxidation process, resulting in the elimination of the halide and formation of a quinone. The oxygen atom incorporated into the quinone is from a water molecule. In contrast, besides C-H bond cleavage, the heme TyrH promotes the elimination of the meta-substituted halogen via nonoxidative hydroxylation to generate catechol and halide. In addition, the order of dehalogenation reactivity of heme TyrH inversely reflects the size of the halogen atoms, whereas the opposite is true for DHP. Such contrary results indicate that the governing factor of catalysis is distinct between these two enzymes, which can be explained by their distinct key reactive species, i.e., Cpd I-like species for DHP and ferric-bound hydroperoxo for heme TyrH, respectively.
Table 1.
Comparison of the dehalogenation reactions catalyzed by two histidine-ligated heme enzymes, namely, DHP and heme-dependent TyrH
| DHP | Heme TyrH | |
|---|---|---|
| Cofactor | Histidine-ligated heme | Histidine-ligated heme |
| Reaction scheme | 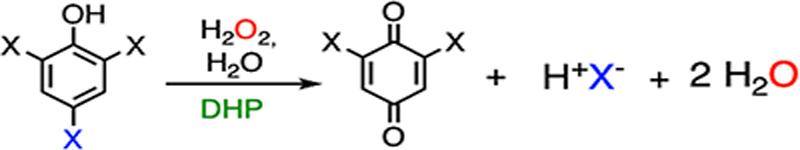 |
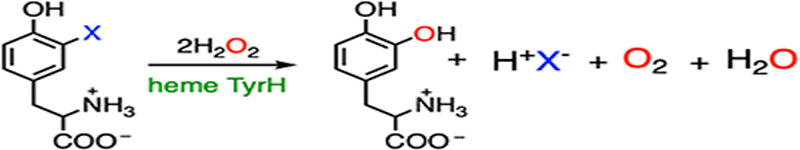 |
| Substrate | para-Substituted phenol | meta-Substituted phenol |
| Product | Oxidative C-X bond cleavage, quinone | Non-oxidative C-X cleavage, catechol |
| Source of oxygen | H2O | H2O2 |
| Dehalogenation reactivity | Br > Cl > F | F > Cl > 1 |
| Reactive species | Cpd l-like species | Ferric heme-bound hydroperoxo |
3. C-F bond cleavage by P450
The cytochrome P450 enzymes (CYPs) comprise a remarkable superfamily of monooxygenases that are ubiquitously present in all kingdoms of life as well as viruses.89 Notably, they are found in all mammalian tissues and are the most abundant in the liver and small intestine, which are participants in the metabolism of endogenous compounds, environmental pollutants, and carcinogens. For instance, at least 57 CYP genes have been found in humans until now, and 5 of the corresponding CYPs, namely, CYP 1A2, 2C9, 2C19, 2D6, and 3A4, are responsible for 90% of the oxidation of commercial drugs.90,91
With such prevalence and importance, the structures and functions of CYPs have been relatively well characterized. When compared with histidine-ligated heme enzymes, thiolate-ligated CYPs exhibit broad substrate specificity and promote a much broader spectrum of chemical reactions, e.g., hydroxylation, heteroatom oxygenation, oxidation, epoxidation, dealkylation, dehalogenation, desaturation, desulfurization, C-C bond formation, C-C bond cleavage, and O-demethylation.91–93 Although the mechanism varies in specific reactions, they share the same procedure to generate the common reactive intermediates, namely, Cpd I and II.63,64 As mentioned above, both Cpd I and II contain a ferryl-oxo intermediate but differ by one electron in the oxidation state of porphyrin. These ferryl species are almost exclusively responsible for a majority of the chemistry mediated by P450 enzymes. The active site of CYPs contains a proximal cysteine-ligated heme iron, usually deeply buried inside the protein with an access channel connecting it to the protein surface.91,94 Substrate binding induces a conformational change in the active site, triggering electron transfer from NADPH by NADPH-dependent P450 reductase or cytochrome b5. Subsequently, an oxygen molecule binds the ferrous heme and is activated to promote the heterolytic cleavage of the O-O bond, resulting in Cpd I (Fig. 5). Then, the subsequent substrate activation branches off to different reactions after the formation of the Cpd I oxidant. When compared with other metalloenzymes, CYPs are more frequently reported to perform defluorination reactions, typically on drugs or other small molecules: CYP 1A2 and 3A4 catalyzed the aromatic C-F bond hydroxylation on drugs such as sunitinib and famitinib to release reactive, potentially toxic metabolites.95,96 The CYP-mediated defluorination on imaging tracers have been reported from in vivo studies.97,98 Human liver CYPs defluorinate inhaled anesthetics such as halothane and sevoflurane to generate toxic metabolic intermediates.99–101 Multiple bacterial or mammalian liver CYPs have shown significant oxidative defluorination activity at the para position of fluorinated phenol or aniline, producing quinone or quinone imine and the corresponding halide anion.102–107
Fig. 5.
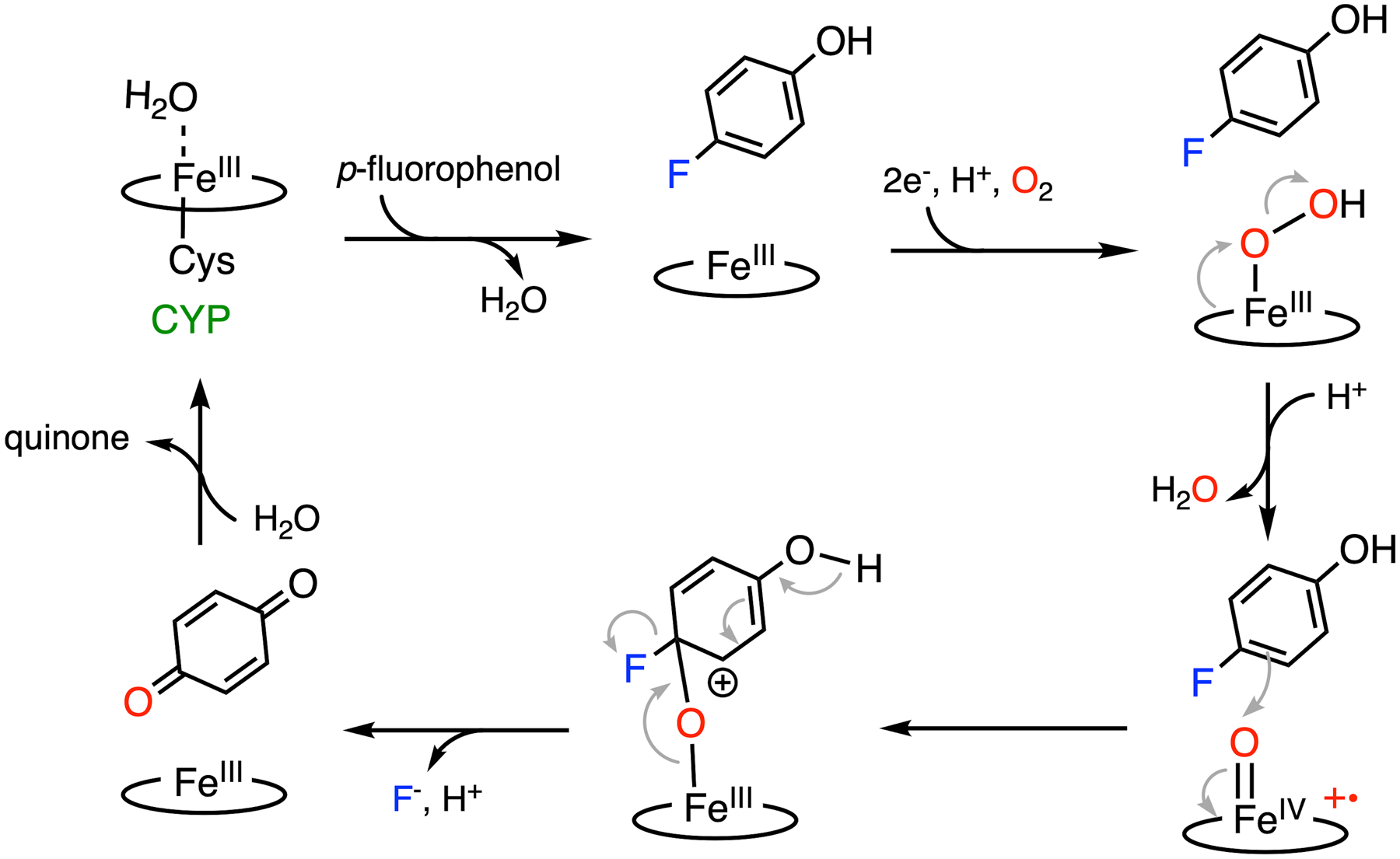
Mechanism of C-F bond cleavage promoted by CYP.
The mechanism of P450-mediated oxidative aromatic defluorination is illustrated using para-fluorinated phenol as an example (Fig. 3B). The most plausible defluorination proposal involves an electrophilic attack on the substrate by Cpd I, as shown in Fig. 5.63,103,105 Considering the electron-rich nature of the phenol, once Cpd I is formed at the active site, it readily performs an electrophilic attack on a nearby substrate producing a transient cationic intermediate, as proposed in the catalytic mechanism of nonheme TyrH discussed in the subsequent section. The rearrangement of the cationic adduct promotes heterolytic Fe-O bond cleavage to generate benzoquinone and fluoride, allowing heme to resume its resting state after releasing quinone. In the presence of NADPH or other reducing equivalents, hydroquinone can be observed due to the reduction of benzoquinone.63,102 Similarly, CYPs exhibit dehalogenation activity on the para-substituted chloro- and bromophenols as well, while a fluorinated substrate yielded the highest product concentration from the C-X bond cleavage.103,108,109 It is worth noting that fluoride is a better leaving group in nucleophilic aromatic substitution, but not necessarily in the case of electrophilic substitution (see section 7 for details). If the reaction indeed proceeds through Cpd I-directed electrophilic attack, the observed effect of the halogen substituent should be explained by other factors, such as steric hindrance and substrate-binding affinity.
In addition, some types of CYP-mediated defluorination reactions are found in metabolism to generate fluoride and resulting toxic metabolites, which can be triggered by O-demethylation, N-dealkylation, dealkylation, ortho-elimination of alcohol/amine, etc.110 It is not entirely surprising considering the exceptional chemistries of the superfamily of CYPs. Hence, drug defluorination promoted by CYPs and the toxicity of the resulting metabolites are indeed worthy of attention during the process of drug design, selection, optimization, and particularly toxicology studies in the drug development process.
4. C-F bond functionalization by nonheme iron hydroxylases
4.1. Tetrahydrobiopterin-dependent aromatic amino acid hydroxylase
Similar to heme-dependent enzymes, nonheme iron enzymes perform a multitude of oxidation chemistries. Like heme TyrH, a nonheme iron-dependent tyrosine hydroxylase (nonheme TyrH) also catalyzes the conversion of L-tyrosine to DOPA, and such a nonheme TyrH is biologically more significant than its heme counterpart. As a rate-limiting step in the biosynthesis of catecholamines, the reaction requires molecular oxygen and tetrahydrobiopterin (BH4) as the cosubstrate (Fig. 6B).87,111 One oxygen atom from molecular oxygen is inserted into the cosubstrate BH4, generating 4a-hydroxytetrahydropterin (4a-OH-BH4). The other oxygen atom is incorporated into the aromatic ring of tyrosine. The active enzyme is a tetramer, and each of the subunits contains a ferrous iron coordinated by two histidines and one glutamate as the cofactor, as shown in Fig. 6A.112,113 In the resting state, water molecules comprise the remaining ligands in a distorted octahedral complex. In some instances, the enzyme is activated by BH4 and oxygen, but without a bound L-tyrosine; this enzyme can self-hydroxylate its phenylalanine residue, i.e., Phe300, into 3-hydroxyl-phenylalanine (3-OH-Phe300).113 This alternative activity is likely to be an enzyme self-protection mechanism, as initially found in TfdA114 as well as other nonheme iron enzymes, to prolong the enzyme activity during undesired and uncoupled oxidation.115–118 The current understanding of the catalytic mechanism of nonheme TyrH is based on a variety of biochemical and spectroscopic studies.119–122 The hydroxylation reaction can be divided into two half-reactions. The first half-reaction involves BH4 hydroxylation to generate 4a-OH-BH4 and a ferryl-oxo intermediate. The second half-reaction starts with a high-valent iron species oxidizing tyrosine through an electrophilic addition, resulting in an Fe(ii)-bound cationic intermediate.123 Then, the proton is eliminated to liberate the final DOPA product.
Fig. 6.
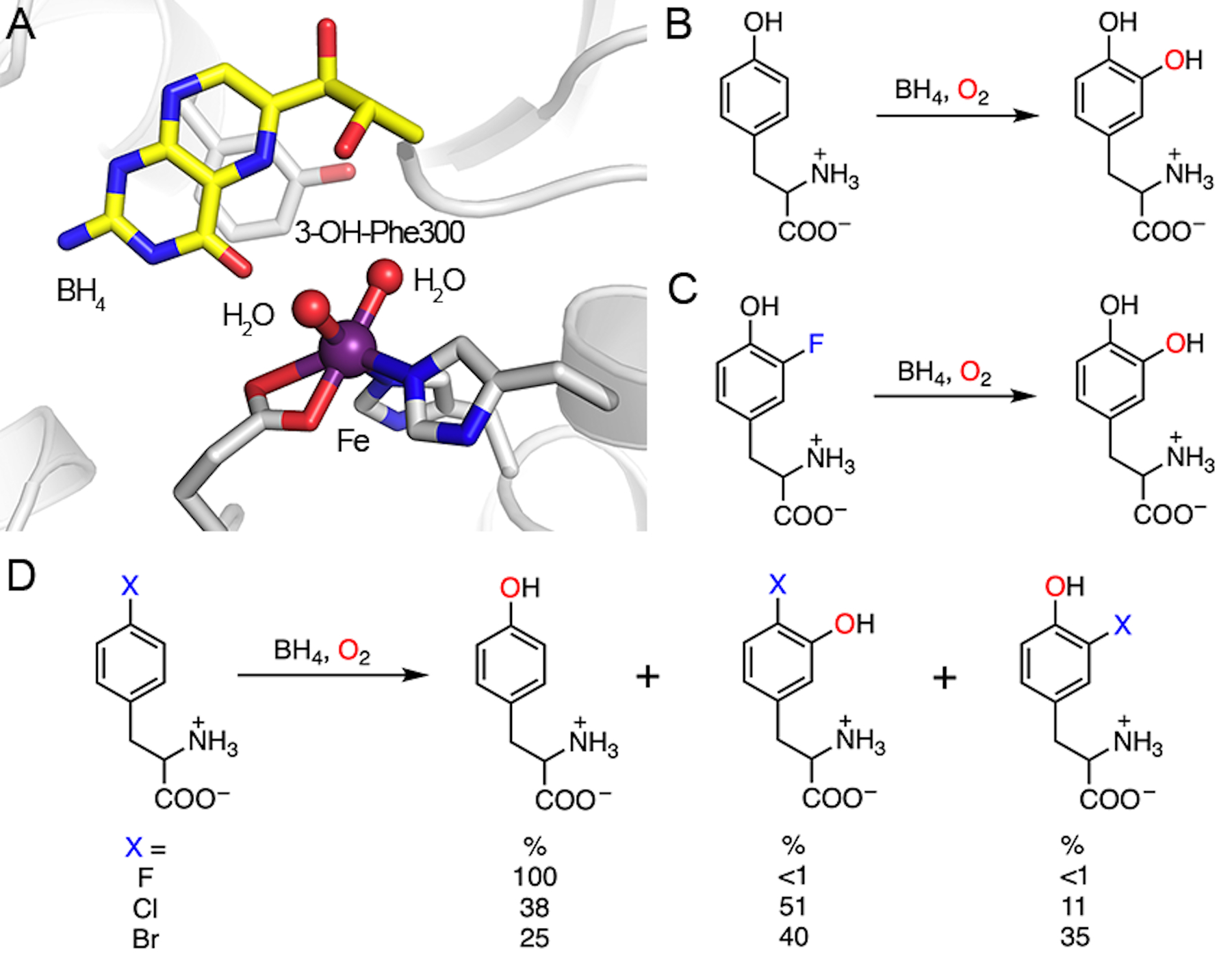
Active site of nonheme TyrH and the demonstrated reactions. (A) Crystal structure of nonheme TyrH in complex with BH4. Phe300 is self-hydroxylated to 3-OH-Phe300. PDB entry: 2TOH. (B) Natural hydroxylation of tyrosine. (C) Defluorination of 3-fluorotyrosine. (D) Dehalogenation of 4-halo-phenylalanine. 4-Fluorophenylalanine (X = F) only yields one product, namely, tyrosine. Other halogen substitutions (X = Cl, Br) afford multiple products.
When 3-fluorotyrosine is used as a substrate in place of L-tyrosine, the hydroxylation reaction takes place only on C3 due to the small size of fluorine and consequently produces a defluorinated product, i.e., DOPA (Fig. 6C). Notably, unlike heme TyrH, the reaction yields only the defluorinated product without hydroxylation at the C5 position. This selectivity may indicate that the active site recognizes and interacts specifically with the C3 substituent such that the monosubstituted tyrosine analog has a single binding mode to the active site that only allows defluorination. Other 3-halogenated tyrosine analogs have the same outcome with dehalogenation efficiencies of F > Cl > Br.33 However, the products derived from halogen atoms and the detailed dehalogenation mechanism remain unknown. The likelihood of the formation of a fluorine cation or a fluorine radical is extremely low in a biological system. If the fluorine substituent is eliminated as fluoride (as seen in most cases), the source of the additional electrons required relative to the C-H bond cleavage requires further investigation. The reaction stoichiometry of BH4 in the 3-fluorotyrosine reaction may reveal this mystery. It is also unclear if the Fe(iv)-oxo species involved in the hydroxylation of the native C-H bond is responsible for C-F hydroxylation.
Interestingly, 4-halogenated phenylalanine can react with nonheme TyrH, too. With the exception of 4-fluorophenylalanine, which produces only tyrosine, challenging nonheme TyrH with either 4-chloro- or 4-bromophenylalanine generates multiple products as the outcome of the NIH shift and dual hydroxylation sites (C3 and C4). In the case of nonheme TyrH, the NIH shift represents the 1,2-migration of the halogen functional group at the site of substitution in aromatic hydroxylation reactions. In addition to tyrosine, reactions on 4-chloro- and 4-bromophenylalanine also yield 3-halo-tyrosine and 3-hydroxy-4-halo-phenylalanine as the products (Fig. 6D). Nonheme TyrH can hydroxylate at both C3 and C4 positions of 4-halogenated phenylalanine depending on the size of the substituent at C4. It is proposed that hydroxylation at C3 results in 3-hydroxy-4-halo-tyrosine through the reaction route proposed for the hydroxylation of L-tyrosine. Hydroxylation at the C4 position yields an Fe(ii)-bound cationic intermediate, which can undergo either an NIH shift of the halogen substituent forming 3-halo-tyrosine or direct elimination of the halide yielding tyrosine.33,124 Here, 4-fluorophenylalanine is used to illustrate the defluorination catalyzed by nonheme TyrH (Fig. 7). After the first half-reaction forming 4a-OH-BH4 and ferryl-oxo intermediate, electrophilic substitution occurs only at C4 to generate an Fe(ii)-bound cationic intermediate. Without an NIH shift, fluoride atom is directly eliminated to create the re-aromatized product, i.e., tyrosine. For 3-chloro and 3-bromo-substituted phenylalanine analogs, the ratio of hydroxylation at C3 and C4 is nearly 1 : 1. In the case of C4 hydroxylation, a product with more NIH shift (3-halotyrosine) is observed as the size of the halogen atom increases (Br > Cl ≫ F).With halogen elimination, tyrosine is generated (F > Br, Cl). However, the fate of the halogen upon elimination is unresolved, although halide is the most plausible leaving agent of the reaction. There should be an electron source to provide the two necessary electrons and eventually generate halide as the final product. Unfortunately, neither such an electron donor nor a final halogen-containing product has been identified. Further investigation on this system is needed in order to gain a better mechanistic understanding.
Fig. 7.
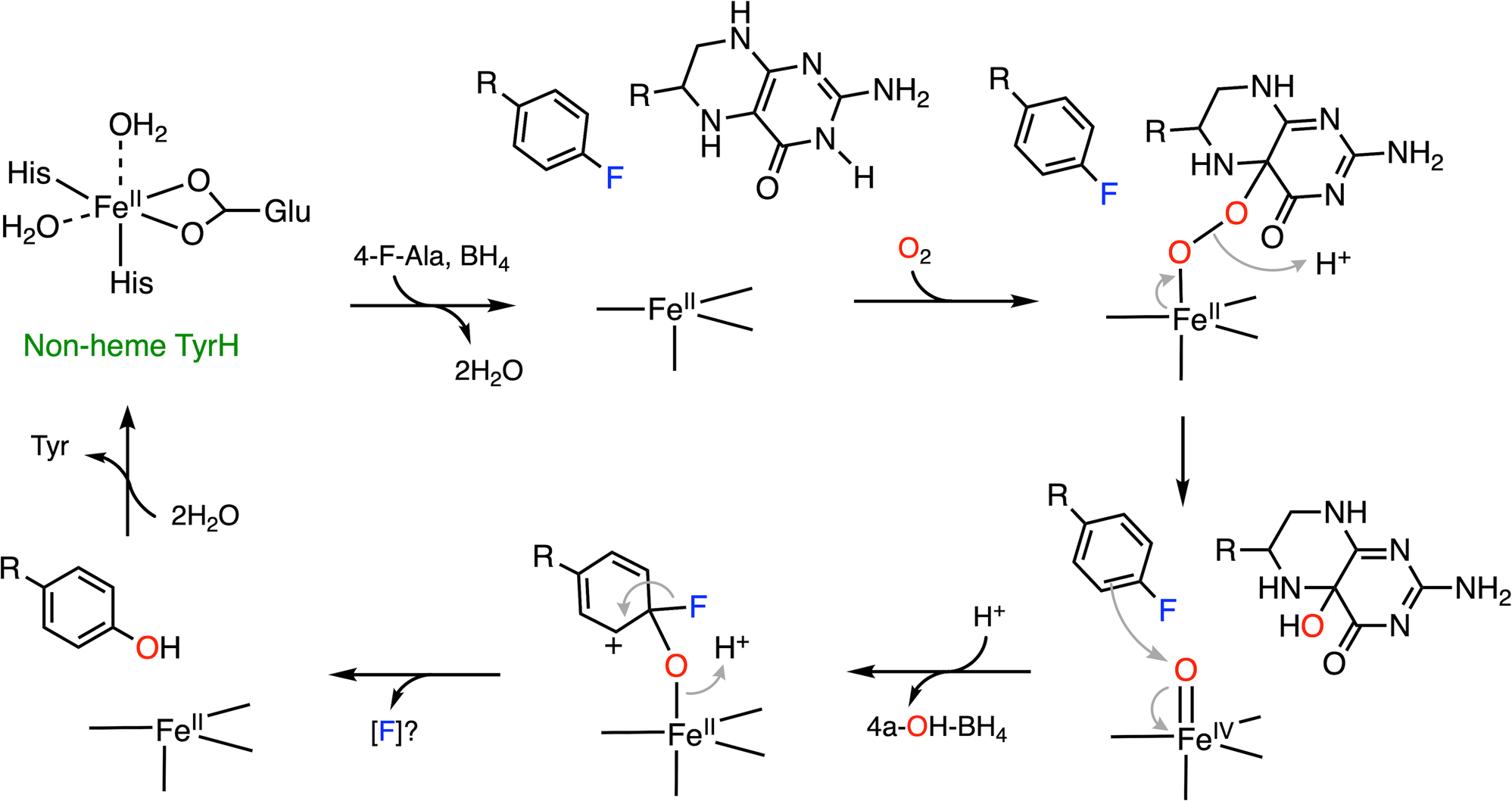
A plausible mechanism of C-F bond cleavage promoted by nonheme TyrH with which 4-fluorophenylalanine (4-F-Ala) is converted into tyrosine. BH4 represents tetrahydrobiopterin. R: amino acid moiety. (Note: The product derived from fluorine substitution is unclear and requires more experimental evidence for its production).
The nonheme TyrH has two other siblings. The mononuclear, nonheme, iron-dependent aromatic amino acid hydroxylases, namely, phenylalanine hydroxylase (PheH) and tryptophan hydroxylase (TrpH), have also been well studied. All the three enzymes are pterin-dependent hydroxylases, and they share a significant degree of homology in their catalytic domains. Spectroscopic data have revealed that their catalytic pathways may proceed through a similar Fe(iv)-oxo intermediate.125–127 The defluorination activity of PheH on 4-fluorophenylalanine to tyrosine has been reported in an earlier study.128 The formation of fluoride was detected, and the oxidation of BH4 was proposed to provide the required electrons, which is a feasible explanation for the questions raised in the TyrH system. Given the defluorination capacities of nonheme TyrH and PheH, it is reasonable to extrapolate that C-F bond cleavage is accessible by TrpH. Future studies regarding the activities and product distribution of fluorinated tryptophan analogs are anticipated.
Overall, the defluorination capacity of the nonheme iron enzyme TyrH is fairly interesting. This enzyme can cleave the C-F bond at different sites when facilitated by oxygen addition via electrophilic substitution. The cationic intermediate is highly destabilized due to fluorine substitution, which promotes re-aromatization to cleave the C-F bond. However, the nature of the fluorine elimination and potential electron source to balance the reaction require additional investigations.
4.2. Nonheme iron hydroxylase with 2-oxoglutarate as the cosubstrate
As discussed in nonheme TyrH that utilizes both BH4 and tyrosine as organic substrates, several nonheme iron, oxygen-dependent enzymes split dioxygen into two substrates via stepwise incorporation. Such an oxygen-activation method is also employed by a well-studied enzyme superfamily, i.e., 2-oxoglutarate-dependent hydroxylases. These enzymes are also nonheme iron dioxygenases that insert two oxygen atoms into a 2-oxoglutarate (2OG, also known as α-ketoglutarate) molecule and a primary substrate. The catalytic Fe(ii) center is coordinated by a 2-His-1-carboxylate facial triad and three water molecules, and the carboxylate group normally serves as a monodentate ligand.129,130 The cosubstrate 2OG ligates to the iron center in a bidentate fashion to replace two of the water molecules, which is followed by the binding of the primary substrate nearby to displace the remaining water.
Such a binding order triggers dioxygen ligation and activation, yielding ferric-ion-bound superoxide. The distal oxygen of the superoxide attacks the keto carbon of 2OG to form a bicyclic intermediate with a peroxide bridge. Heterolytic O-O bond cleavage affords an Fe(iv)-oxo species and promotes the oxidative decarboxylation of 2OG to release CO2. The Fe(iv)-oxo species has been observed by spectroscopic methods.131–134 After the oxygen transfer to the iron-bound 2OG, the succinate product remains bound to iron in a monodentate manner. The high redox power of Fe(iv)-oxo species allows hydrogen atom abstraction from the adjacent primary substrate, affording ferric hydroxo and a substrate radical.135 The second oxygen is delivered by hydroxyl radical rebound to the substrate radical, forming a hydroxylated product. The dissociation of the hydroxylated product and succinate from the active site completes the entire catalytic cycle. It is noteworthy that the process of hydrogen atom abstraction and oxygen rebound in 2OG-dependent hydroxylases is similar to CYP-mediated hydroxylation. Such a typical catalytic mechanism mediated by 2OG-dependent hydroxylases is well understood.136,137
If the site of hydroxylation has a fluorine substituent on the carbon, the hydroxylation can lead to defluorination after the oxidation of the substrate, as evident in prolyl hydroxylase.138–140 Prolyl hydroxylase is also a 2OG-dependent hydroxylase, which catalyzes 3- or 4-hydroxylation on proline residues in diverse proteins.137 Herein, our discussion will focus on prolyl-4-hydroxylase (P4H). As an important oxygen-sensing mechanism,141–143 the oxygen-dependent proline hydroxylation on a hypoxia-inducible factor is an irreversible and stereoselective process, which produces (2S,4R)-4-hydroxyproline (Hyp) in the target protein (Fig. 8A). With the multitude of substrate P4H reveals the wide distribution and various biological functions of the Hyp-presented proteins. Thus far, different P4Hs have been found to alter the protein conformation, tune enzymatic stability and activity, prepare for further modifications, as well as promote protein-protein interactions.144,145 Two biologically essential examples are hypoxia-inducible factor (HIF)- and collagen-related P4Hs; both are considered to be important therapeutic targets.146–148 Cellular adaptation to hypoxia involves the expression of multiple genes regulated by HIFs. Under hypoxic conditions, low oxygen availability limits the hydroxylation activity of P4H on the prolyl residues of HIFs and therefore prevents signaling for the proteasomal degradation of HIFs.148–150 Similar to all 2OG-dependent hydroxylases, P4H binds 2OG in a bidentate fashion, and the target proline residues are located nearby (Fig. 8B). In this particular structure, P4H is substituted with manganese and forms a complex with a fragment of HIF with Pro564 to be readily oxidized.150
Fig. 8.
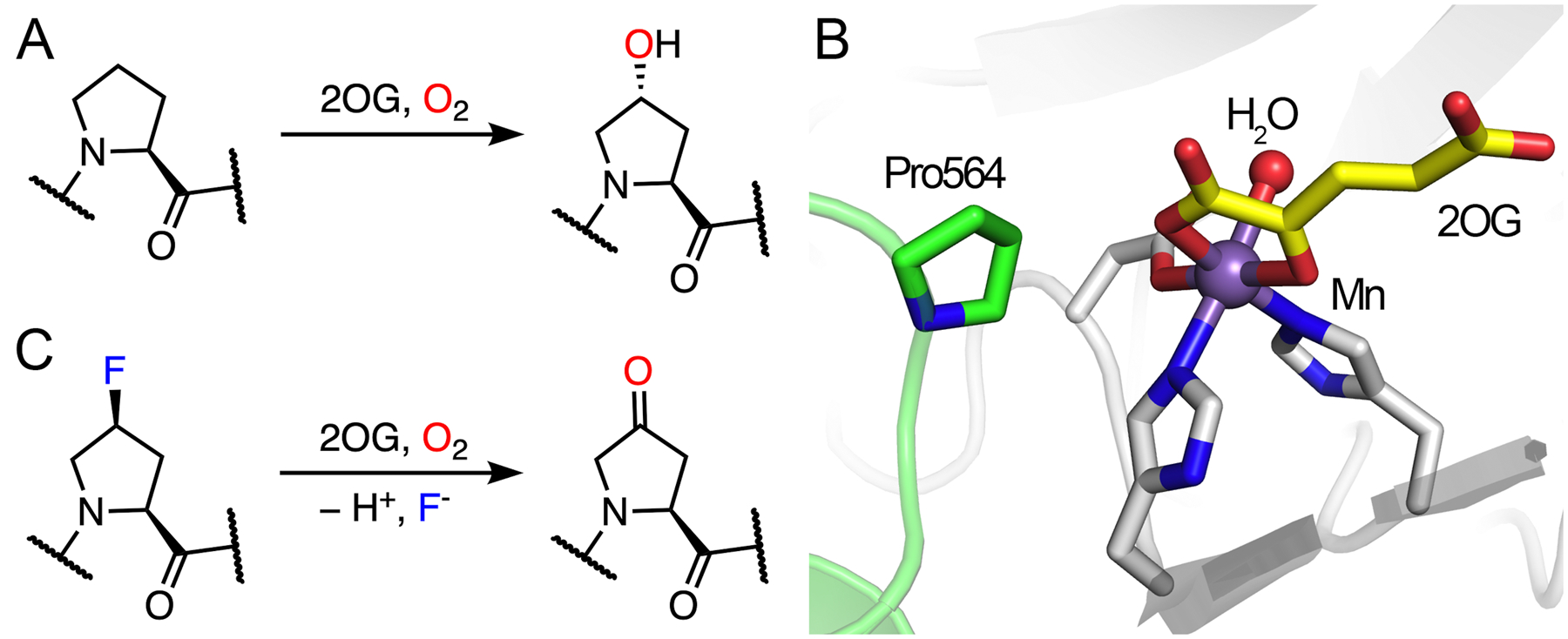
Demonstrated catalytic reactions and the crystal structure of P4H. (A) Native hydroxylation of P4H on a proline residue. (B) The catalytic active site of human P4H (white) in complex with 2OG (yellow) and a fragment of HIF peptide (green) (PDB entry: 5L9B). Catalytic iron was substituted with a manganese ion in this structure. Pro564 from HIF is the target for hydroxylation. (C) Defluorination of a fluorinated proline residue.
Collagen is a dominant protein of the extracellular matrix, which is composed of a three-stranded helix with high tensile strength.151 As a significant connective tissue in the human body, its stability is significantly increased by the formation of Hyp due to the establishment of water bridges and/or stereoelectronic effects.34,145,146,151 Studies have shown that the incorporation of (2S,4S)-4-fluoroproline (Flp) into collagen in addition to Hyp results in increased thermostability.34,152 However, the incorporation of Flp cannot be guaranteed in the presence of P4H due to its defluorination activity (Fig. 8C).138–140 After the formation of hydroxylated Flp through the typical 2OG-dependent hydroxylation mechanism, fluorine can be spontaneously eliminated from the hydroxylated product, affording (2S)-4-keto-proline (Kep) as the final product (Fig. 9). The elimination of fluoride rather than hydroxide occurs because fluoride is a much better leaving group. The steady-state kinetic parameters of an Flp-containing peptide are comparable to those of the natural substrate, which indicates that the rearrangement of the hydroxylated product to defluorination is not a rate-limiting step. The dehalogenation capacity of P4H has not been determined in halogens other than fluorine.
Fig. 9.
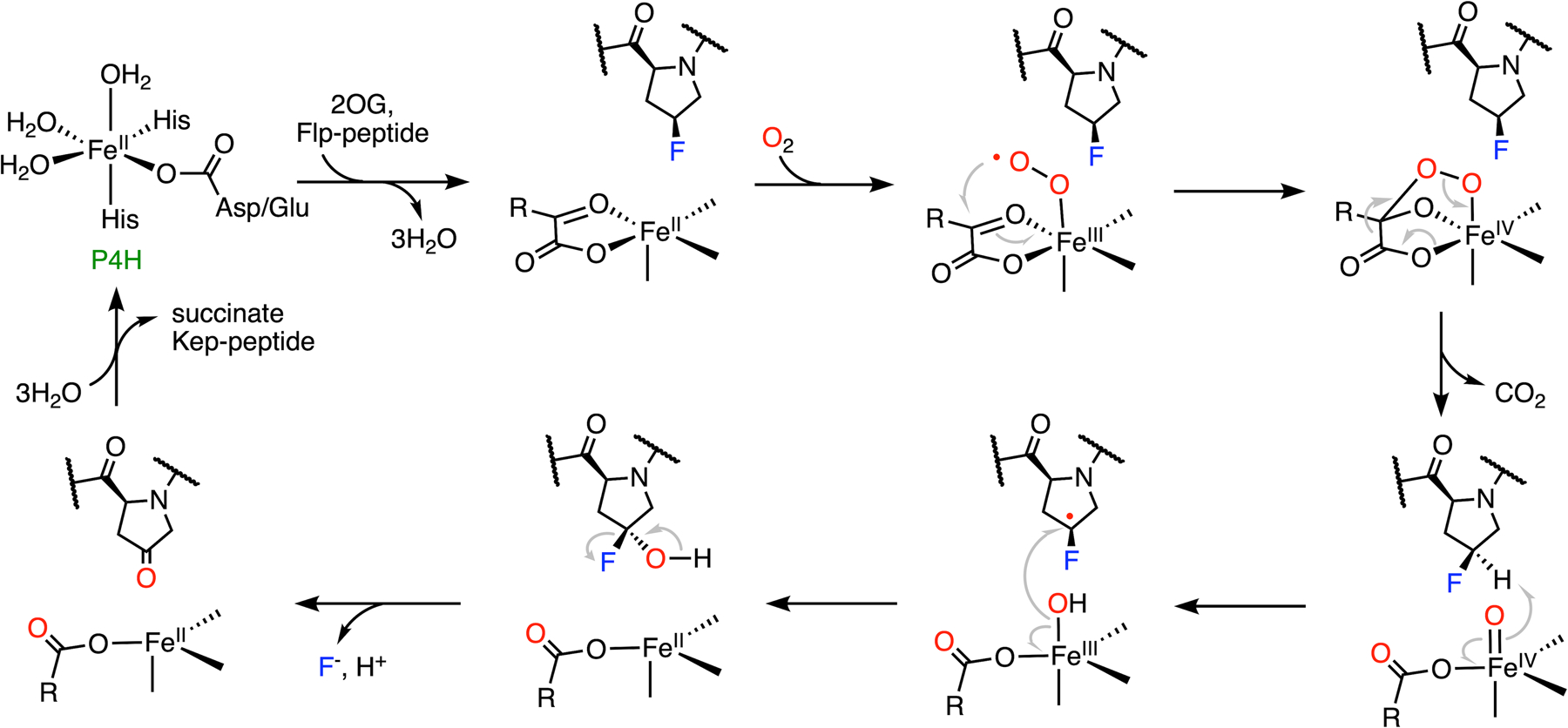
Mechanism of C-F bond cleavage mediated by prolyl-4-hydroxylase.
In addition to P4H, other 2OG-dependent hydroxylases that catalyze defluorination have also been reported, such as γ-butyrobetaine hydroxylase.153 The defluorination process is similar to the P4H-mediated reaction, which forms the keto product via fluoride elimination. The use of fluorinated substrate analogs to examine the activity of more enzymes in the 2OG-dependent superfamily seems to be a good strategy for further mechanistic exploration and identification of detoxification activity.
5. C-F bond functionalization by nonheme iron dioxygenases
5.1. Rieske nonheme iron dependent dioxygenase
Rieske oxygenases belong to another class of nonheme iron-containing enzyme family that catalyze a wide variety of biotransformations in diverse organisms by utilizing molecular oxygen.154 The most prominent reaction catalyzed by Rieske oxygenase discussed in the literature is the cis-dihydroxylation of aromatic compounds, which was first identified in the biodegradation pathway of Pseudomonas putida.155 Besides the same mononuclear iron-based catalytic center (2-His-1-carboxylate coordination) as tetrahydrobiopterin- and 2OG-dependent hydroxylases, Rieske oxygenases also bear an iron-sulfur cluster that is commonly found as a [2Fe-2S] cluster held by two cysteines and two histidines. These enzymes are typically trimeric in structure, and the iron-sulfur cluster mediates the transfer of electrons to the nonheme iron center across two different subunits, which are bridged by either a conserved aspartate or glutamate residue (Fig. 10A).
Fig. 10.
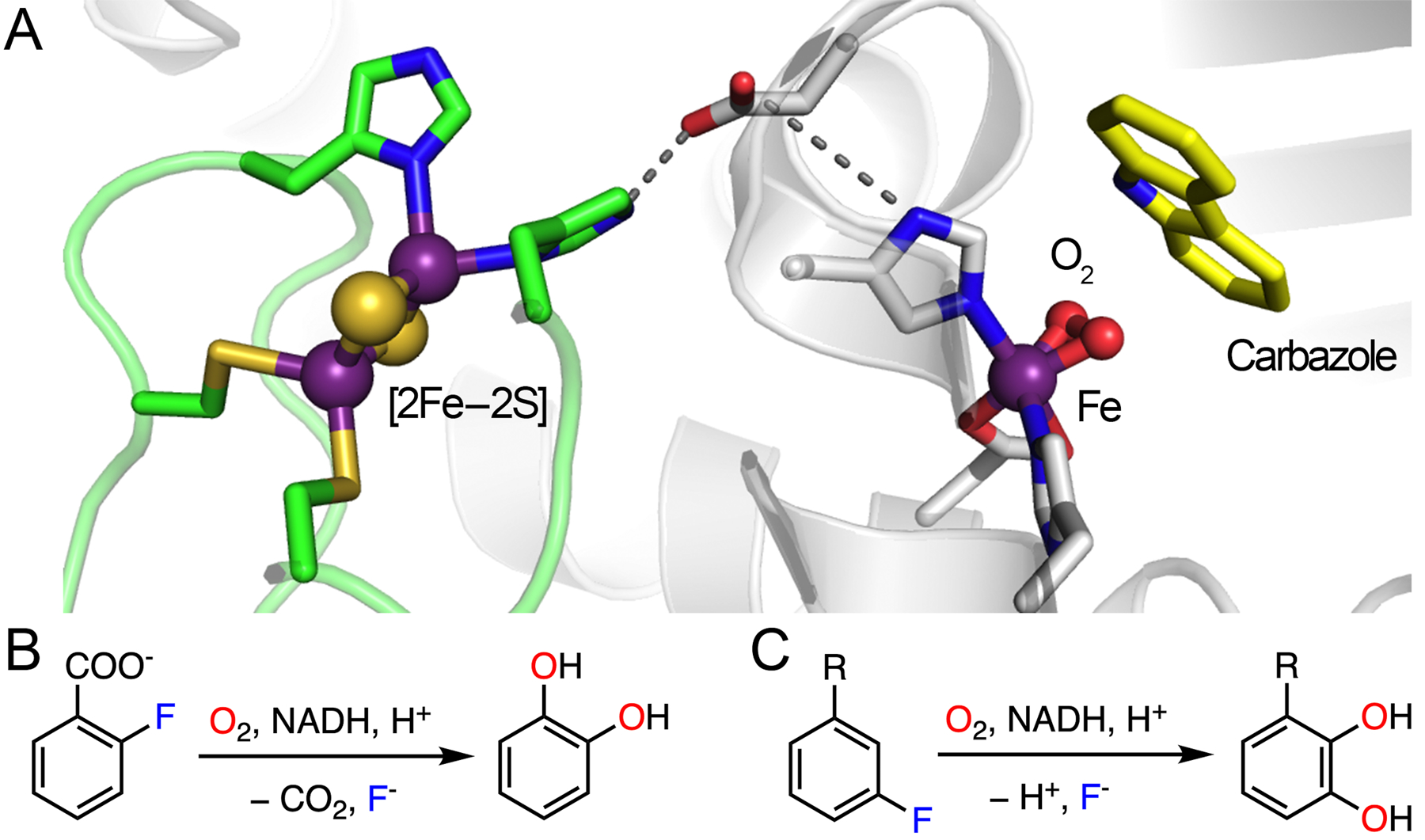
Architecture of Rieske oxygenase and the experimentally verified catalytic reactions. (A) Structure of enzyme in complex with carbazole (yellow) and dioxygen. The Rieske cluster and catalytic iron located in two subunits are represented by green loops and white ribbons, respectively, and they are connected by an Asp residue through H-bonding interactions. PDB entry: 3VMI. (B) Defluorination reaction catalyzed by 2HD. (C) Defluorination reaction catalyzed by toluene-1,2-dioxygenase. R: -CH3, −CN, −X, −OCH3, or −CF3.
A cis-dihydroxylation reaction mediated by a Rieske oxygenase enzyme involves a two-electron reduction in the presence of oxygen and NADPH. A reductase extracts two electrons from NADPH to initiate the electron transfer, and the Rieske center shuttles the electrons to the catalytic iron center potentially through the aspartate/glutamate residue.156 Rieske dioxygenases are capable of oxidizing four major types of aromatic compounds, namely, naphthalene, phenanthrene, benzoate, and toluene/biphenyl.157 Certain Rieske oxygenases have been reported to exhibit defluorination activity when exposed to fluorinated substrates, such as toluene-1,2-dioxygenase and 2-halobenzoate-1,2-dioxygenase.29,35 Herein, 2-halobenzoate-1,2-dioxygenase (2HD) is used as an example to illustrate the detailed mechanism of C-F bond cleavage promoted by Rieske dioxygenases. The 2HD enzyme controls the first step of biodegradation of 2-halobenzoate to generate a catechol by eliminating halide and CO2 (Fig. 10B), providing the substrate for subsequent oxidative ring cleavage by intradiol or extradiol dioxygenase enzymes.29,158,159
As shown in Fig. 11, the proposed catalytic cycle begins with the binding of an aromatic substrate to dispel a water molecule. The resulting five-coordinated ferrous center promotes oxygen ligation to generate a ferric-ion-bound superoxide intermediate, followed by reduction by the Rieske cluster and subsequent protonation to yield a ferric hydroperoxo species. The ferric hydroperoxo species has oxygen atoms chelated to the iron in a side-on manner, as revealed by the X-ray crystallographic data (Fig. 10A).160,161 The structural study revealed that a solvent molecule nearby may serve as the proton source.160 Homolytic O-O bond cleavage coupled with substrate oxidation yields a ferryl-oxo species and a substrate-based radical intermediate with a hydroxyl group added to the primary substrate. The high-valent iron complex further initiates the coupling of the second oxygen to the substrate radical, yielding a ferric alkoxy complex (Fig. 11).154,162 It is not clear which carbon becomes hydroxylated first, either the α-carbon or the fluorinated carbon, which may rely on their relative distances to the peroxide oxygen atom. The transfer of the second electron from the Rieske cluster and protonation results in the formation of a dihydroxylated product and regeneration of the resting state of the enzyme. Before the product is liberated from the active site, the energetically favorable re-aromatization induces the elimination of CO2 and fluoride from the ring (Fig. 11). The enzyme is promiscuous with very broad substrate selectivity. Catechol transformation has been observed in the reactions of 2-fluoro-, 2-chloro-, 2-bromo-, and 2-iodobenzoate with decreasing activity,29 which may result from the combined effect of steric hindrance and leaving group efficiency in the process of aromatic dehalogenation.
Fig. 11.
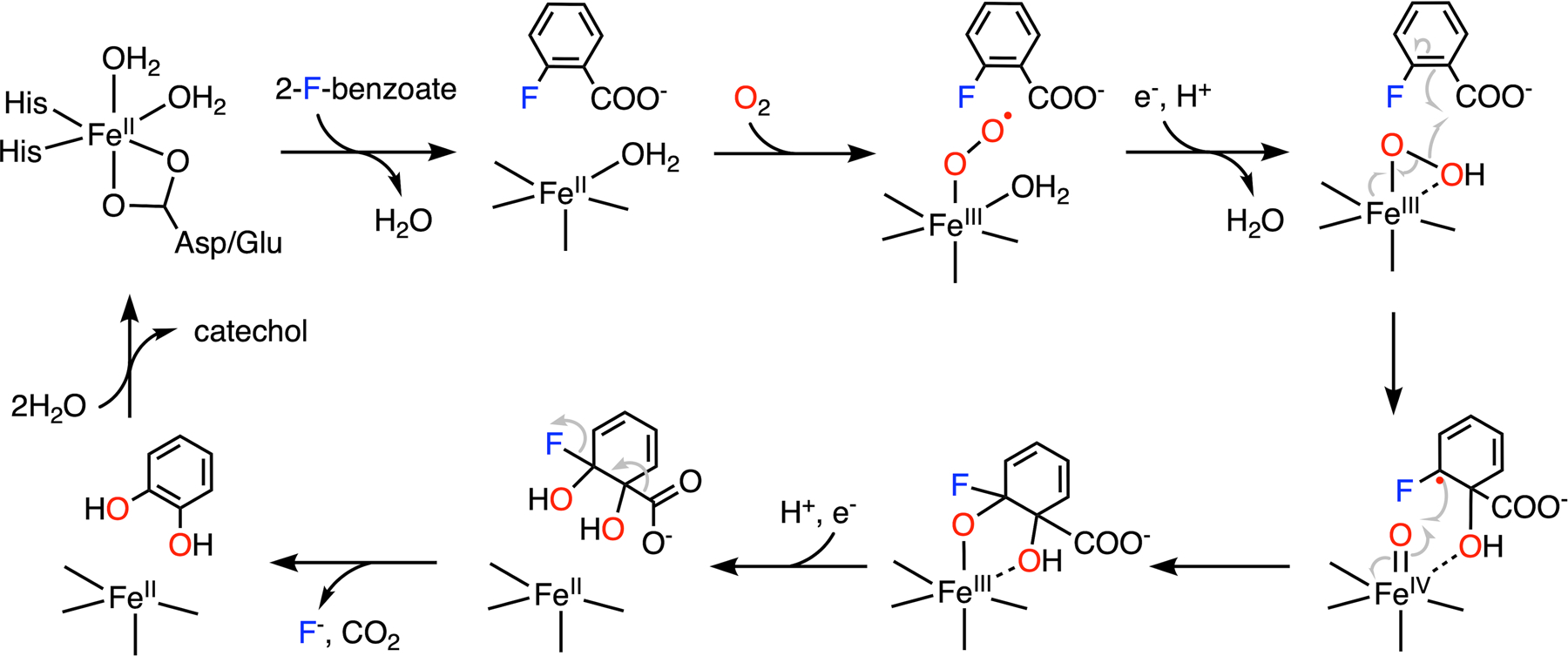
C-F bond cleavage of 2-fluorobenzoate promoted by 2-halobenzoate-1,2-dioxygenase.
Another Rieske enzyme capable of C-F cleavage is toluene-1,2-dioxygenase. It has been found that 3-fluoro-substituted benzene underwent defluorination when incubated with toluene-2,3-dioxygenase with the formation of fluoride (Fig. 10C).35 Collectively, Rieske nonheme iron oxygenase adds another member to the metalloenzymes that promote defluorination through hydroxylation.
5.2. Nonheme iron thiol dioxygenase
Cysteamine dioxygenase (ADO) and cysteine dioxygenase (CDO) are the only two known mammalian thiol dioxygenases.163 These enzymes share many common structural and functional features. They are nonheme iron-dependent enzymes that oxidize the thiol group of the substrate and insert both oxygen atoms of O2 into the same sulfur atom, affording sulfinic acids. The thiol dioxygenases function as thiol regulators and oxygen sensors in mammalian cells.164–166 Structurally, the active site of ADO/CDO is centered at a mononuclear, nonheme ferrous ion coordinated by three histidine residues along with a protein-derived, cysteine-tyrosine (Cys-Tyr) crosslinked cofactor located in the second coordination sphere.36,167,168 As shown in Fig. 12A, the Cys-Tyr cofactor is covalently crosslinked through a thioether bond between the side chains of a cysteine residue and a tyrosine residue (Cys93 and Tyr157 in human CDO, and Cys220 and Tyr222 in human ADO, respectively). The presence of the Cys-Tyr cofactor increases the catalytic efficiency by stabilizing substrate binding and potential intermediates.169–172 This post-translational modification has recently been studied through the genetic code expansion method to incorporate an unnatural amino acid, i.e., 3,5-difluorotyrosine (F2-Tyr), to replace the native tyrosine of the Cys-Tyr cofactor.36,37 This unnatural amino acid is introduced with the intention of disrupting the formation of a Cys-Tyr crosslink, since the C-F bond is more durable than the corresponding C-H bond, consequently increasing the difficulty of C-S bond formation. However, the enzyme-based oxidant is found to be sufficiently powerful, such that the added C-F bond cannot prevent the self-catalyzed formation of the Cys-Tyr cofactor. The C-F bond is cleaved in the engineered protein, resulting in a monofluorinated Cys-Tyr cofactor and a fluoride anion, as shown in Fig. 12B. This finding may provide a clue as to why some fluorinated compounds are toxic and carcinogenic, even though the C-F bond is thought to be inert. Iron-containing enzymes in humans could unexpectedly dismantle the fluorine-protected molecules, resulting in toxic metabolites before reaching their intended medical targets.
Fig. 12.
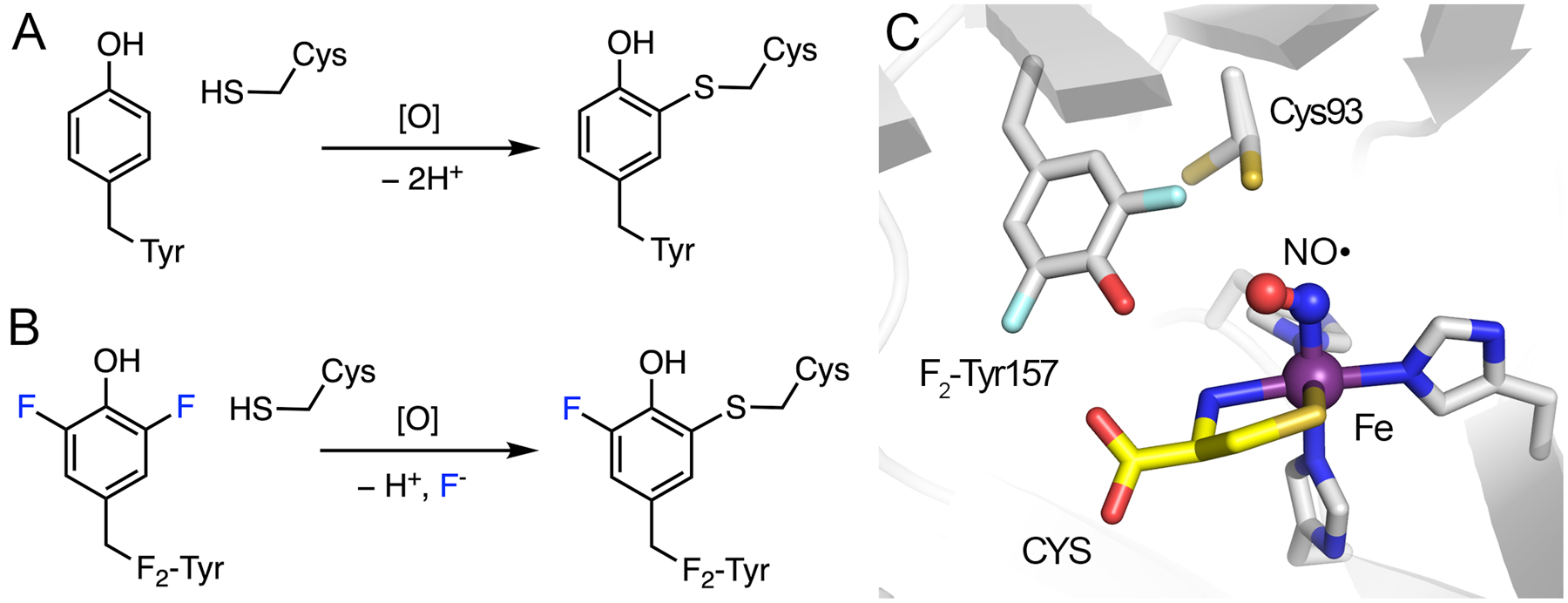
Cofactor biogenesis of CDO and its active site. (A) Crosslink formation in wild-type ADO/CDO with C-H bond cleavage. (B) Crosslink formation in F2-Tyr ADO/CDO with C-F bond cleavage. (C) Crystal structure of uncrosslinked CDO bound with L-cysteine (CYS) and nitric oxide (NO●). Cys93 exhibits two conformations. PDB entry: 6BGF.
The formation of the Cys-Tyr cofactor requires the presence of dioxygen and the primary substrate in both human thiol dioxygenases. An uncrosslinked ternary complex crystal structure of engineered human CDO, bound with the primary substrate and nitric oxide (Fig. 12C), has provided mechanistic insights into the cofactor biogenesis of these thiol dioxygenases and the C-F bond cleavage mechanism. It has been proposed that a cysteinyl radical generated by the nonheme iron-bound oxidant attacks the tyrosine aromatic ring to initiate the crosslink formation and promote C-F bond cleavage in the case of a fluorinated tyrosine.170
Fig. 13 shows the mechanism of C-F bond cleavage promoted by CDO. Once the enzyme-substrate complex is formed, oxygen is activated by the iron center to produce an iron-bound superoxide. Subsequent hydrogen atom abstraction from Cys93 by ferric superoxide leads to the formation of a cysteinyl radical and an iron-bound hydroperoxide. Then, F2-Tyr157 is oxidized by the thiyl radical to form a transient difluoro-tyrosyl radical that is covalently bound to Cys93 through a thioether bond. In order to develop the crosslink, however, the subsequent steps of wild-type and monofluorinated Cys-Tyr cofactor formation are distinct because the elimination of a proton or a fluoride differs by two electrons. In the creation of the native Cys-Tyr cofactor, the O2 bound to the nonheme iron ion is proposed to be reduced to H2O2.170 While in the creation of monofluorinated Cys-Tyr cofactor, two electrons leave the system with the fluoride anion, and superoxide functions only as a facilitator without being consumed. After the attack by the thiyl radical, the hydroxyl group of the difluoro-tyrosyl radical is deprotonated, forming a semiquinone-like intermediate upon fluorine elimination. One resonance form abstracts a hydrogen atom from the iron-bound hydroperoxide to generate the final, monofluorinated crosslinked cofactor. The iron center returns to the ferric superoxide form to proceed with oxygenation of the substrate, i.e., cysteine, forming cysteine sulfinic acid (CSA) as the product. Similarly, the incorporation of 3,5-dichlorotyrosine can also generate a monochlorinated cofactor with C-Cl bond cleavage; however, the efficiency of cofactor biogenesis is significantly decreased.37
Fig. 13.
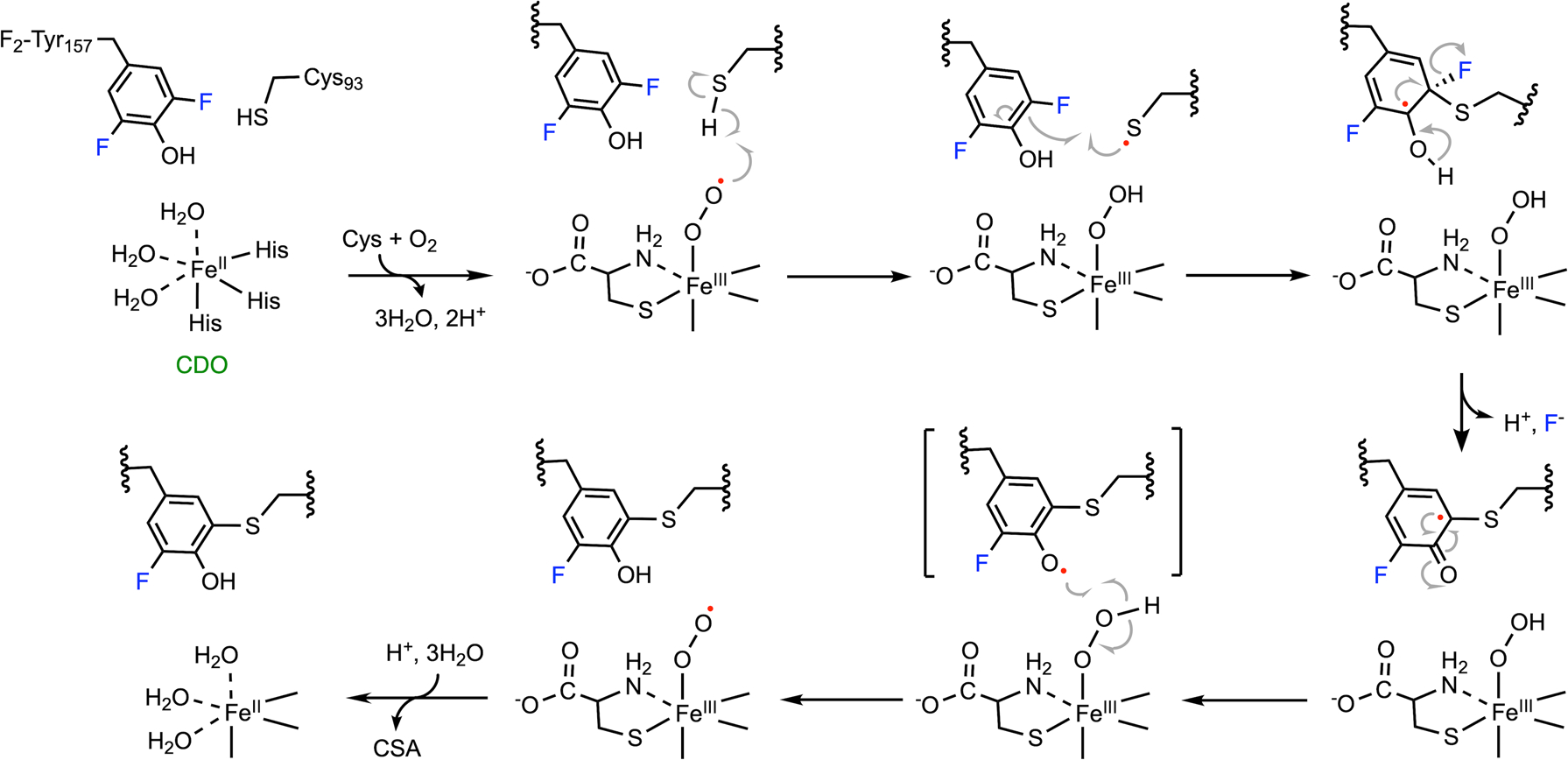
Mechanism of C-F bond cleavage promoted by CDO. Ferric superoxide is regenerated after cofactor biogenesis, and it proceeds with the dioxygenation of the ligated substrate, forming CSA as the product.
A computational study on the cofactor biogenesis of C-F bond cleavage in F2-Tyr157-incorporated CDO has been recently reported.173 The results, in general, support the mechanism proposed based on the experimental observations shown in Fig. 13.37,170 The computational work confirms the view that the cofactor biogenesis starts from a thiyl radical on Cys93 rather than Tyr157. The rate-limiting step is identified as the step of C-F bond dissociation with an energy barrier of 18.8 kcal mol−1. An intriguing deviation is proposed in the computational study, according to which an F atom transfers from C3 to C4 in F2-Tyr157 before the C-F bond cleavage takes place. The necessity of such F-atom migration remains unclear and requires experimental verification. This computational study also highlights the electrostatic influences of seven active-site residues on C-F bond cleavage. It is further suggested that the mutagenesis of Asp87 and Phe165 could improve the defluorination efficiency by lowering the energy barrier.173
In addition to CDO and ADO, there are other thiol dioxygenases, such as 3-mercaptopropionic acid dioxygenase (MDO) and 2-mercaptosuccinic acid dioxygenase, which are equipped with a similar protein scaffold and iron center, i.e., a mononuclear iron coordinated by three histidines.174–177 Although the Cys-Tyr cofactor is not always present in other thiol dioxygenases, such a crosslink could be generated by a specific mutation, e.g., G95C in MDO leads to the formation of an artificial Cys-Tyr cofactor.178 It would be interesting to investigate whether MDO G95C and other thiol dioxygenases can cleave the aromatic C-F bonds from exogenous ligands or their engineered Cys-Tyr cofactors.
6. Enzyme-/mediator-based radical approach for defluorination
The initiator of C-F bond cleavage in ADO/CDO is a thiyl radical generated by an iron-bound oxidant, which is distinct from other addressed iron-dependent enzymes that directly activate fluorinated substrates with their iron centers. Such a strategy correlates to a defluorination concept with a unique radical-mediated mechanism. The most representative case of such an approach is the defluorination catalyzed by the laccase-mediated system. Laccase is a multi-copper-containing oxidase that catalyzes the one-electron oxidation of four equivalents of a substrate while reducing molecular oxygen to water.179,180 Laccase from Trametes versicolor is crystallized as a monomer, which is organized in three sequentially arranged domains with similar β-barrel-type architecture (Fig. 14A).181 There are a total of four copper ions in each monomer, i.e., mononuclear (Cu1 of Type 1) and trinuclear (Cu2 and Cu3 of Type 3, and Cu4 of Type 2) centers. The trinuclear copper center is deeply buried within the protein matrix and located between domains 1 and 3, while Cu1 is embedded in domain 3 and closer to the protein surface. As shown in Fig. 14B, Cu2, Cu3, and Cu4 form an isosceles triangle with Cu4 at the vertex. The antiferromagnetic-spin-coupled Type 3 copper ions are coordinated by three histidines and shared by a hydroxyl ligand bridge. Type 2 copper is coordinated by two histidines and a water ligand in a trigonal planar configuration. Cu1 is held by a conserved His-Cys-His motif; methionine serves as the fourth ligand in some cases. The reduction of dioxygen to water takes place at the trinuclear copper site by converting a fully reduced protein-bound Cu+ to the oxidized form (Cu2+), passing through the formation of a peroxide intermediate.170,171 Next, the fully oxidized enzyme slowly catalyzes one-electron oxidation on four equivalents of the substrate at the mononuclear copper site.182
Fig. 14.
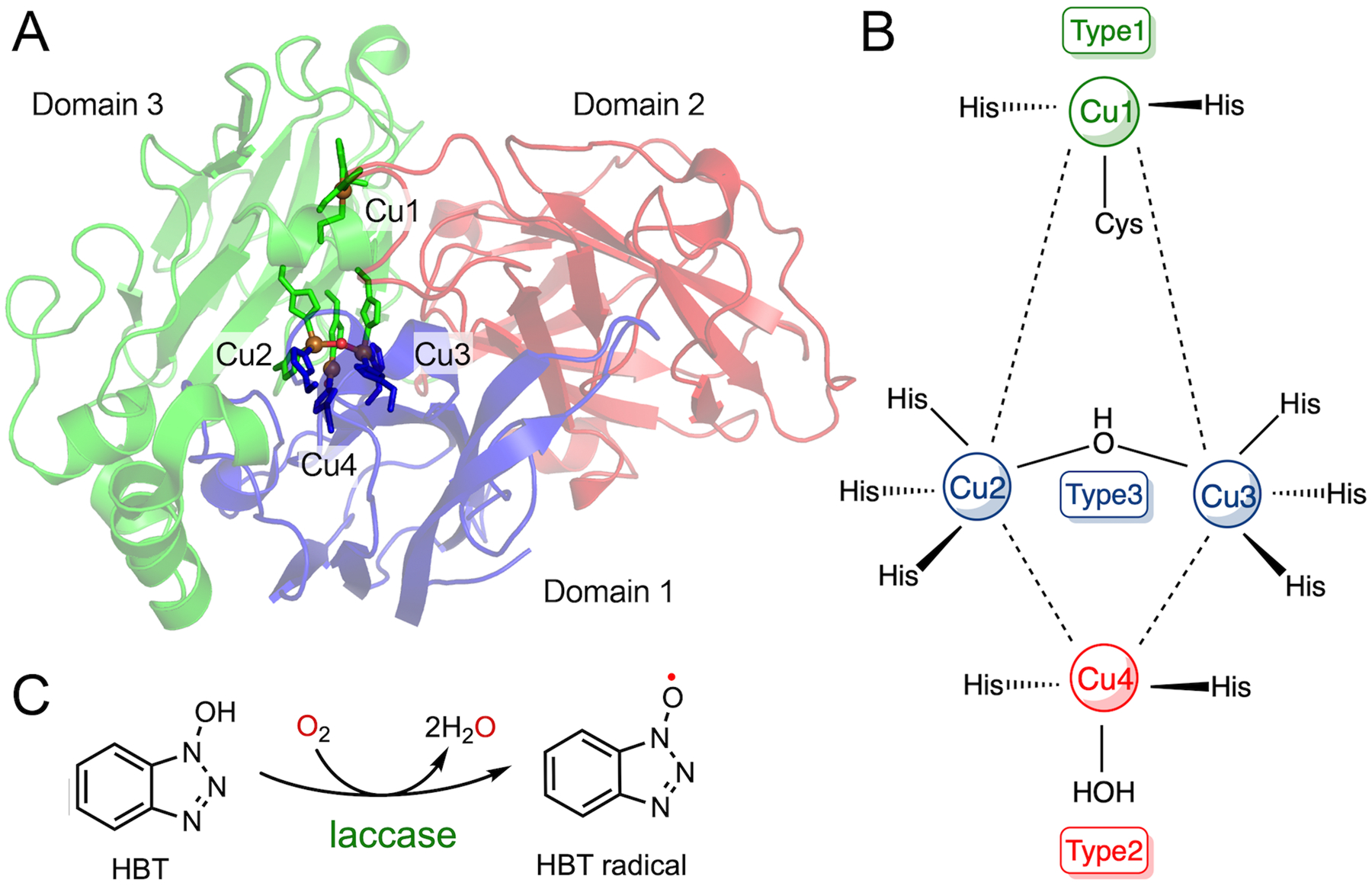
Laccase-/HBT-based radical approach for defluorination. (A) A global structure of laccase from Trametes versicolor. It is composed of three-domain polypeptide and four copper ions. PDB entry: 1GYC. (B) A simplified view of the laccase active site. (C) Reaction scheme of HBT radical formation.
In the presence of small-molecule redox mediators, the catalytic activity of laccase toward more recalcitrant compounds is largely expanded.183 First, a mediator is oxidized by laccase and is stabilized in the radical form. This acts as an electron shuttle by diffusing out from the active site to approach complex substrates. Substrates such as lignin polymers, with steric hindrance and high redox potential, are therefore oxidized.184 Ideally, four equivalents of mediator radicals can be generated in each turnover. Collectively, the high output of the reactive radicals enables laccase to become a capable candidate for degrading polymers, phenolic compounds, and other environmental contaminants. Biodegradation of peror polyfluorinated compounds by laccase-expressing microorganisms is of great interest, while the mechanism is yet poorly understood; therefore, it is in its infancy for general applications.185 However, past studies have attempted to exploit the advantages of a laccase-mediator system to decompose polyfluorinated chemicals. As studied in the degradation of perfluorooctanoic acid (PFOA) and perfluorooctanesulfonate (PFOS) contaminants, the synthetic mediator 1-hydroxybenzotriazole (HBT) can be used as a reducing substrate with laccase to initiate HBT radicals (Fig. 14C), which helps to decompose polyfluorinated hydrocarbons into short chains and fluoride through radical propagation and rearrangement.186,187 The detailed mechanism is not discussed here since it is a nonspecific radical process.
It is noteworthy that there are some fundamental challenges in a laccase-mediator system, which would need to be overcome before applications. A radical-based method will not be practically sound unless the problems of PFOA/PFOS enrichment, mediator toxicity, free-radical-based oxidation reaction specificity and selectivity, and enzyme instability are resolved. In essence, the enzyme-mediator system is genuinely an organic free-radical approach. When compared with a small-molecule mediator-based radical, a protein-based radical can target the substrate more specifically and effectively, as demonstrated by the example of ribonucleotide reductase.188–191 Similar long-range, remote catalysis via protein radical chemistry has also been established in other enzyme-based catalysis.192–196 We think that oxidizing covalently attached mediators to the enzyme can generate a well-behaved radical that can potentially be explored for radical-based defluorination.
7. Fluoroarene scavengers armed with iron
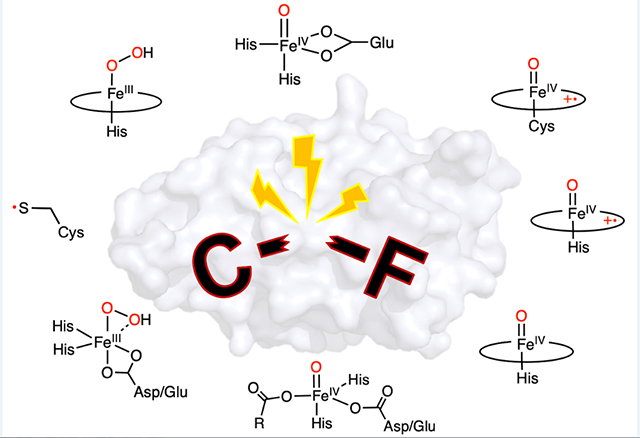
The defluorination reactions catalyzed by each of the iron-containing enzymes discussed above are summarized in Table 2, along with the proposed reactive intermediates and their respective dehalogenation activities. It is noteworthy that most substrates have fluorine substituted on aromatic moieties, except for 2OG-dependent hydroxylases; further, a protein-bound mononuclear iron is the only metal center involved in catalysis, regardless of whether it is nonheme or heme-dependent or in a ferrous or ferric state. As the reaction proceeds, iron oxidation to a higher-valent state by oxygen or H2O2 is necessary to complete the reaction. However, the high-valent iron species is not strictly required to achieve C-F bond cleavage as long as an oxidant with a high redox potential is generated.
Table 2.
Summary of the defluorination reactions catalyzed by iron-dependent enzymes: (A) DHP, (B) heme TyrH, (C) CYP, (D) nonheme TyrH, (E) P4H, (F) 2HD, and (G) ADO/CDO. The corresponding proposed reactive intermediates are also listed as well as their dehalogenation reactivities among halogenated substrates. R: amino acid moiety. Oxygen atoms from oxidants (H2O2 or O2) are highlighted in red
| A | 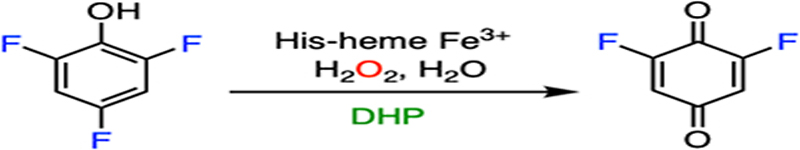 |
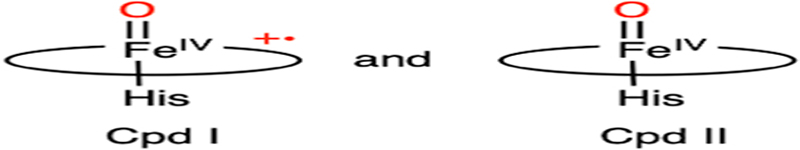 |
Br > Cl > F |
| B | 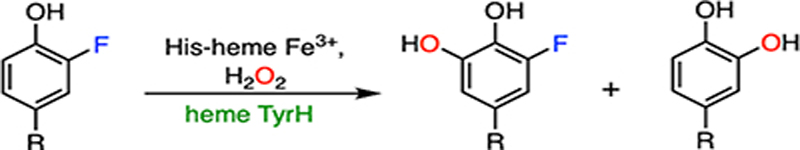 |
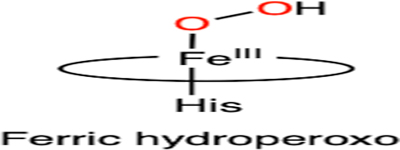 |
F > Cl > 1 |
| C |  |
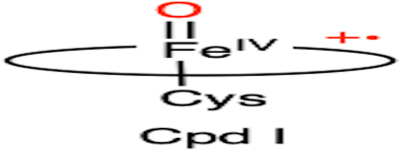 |
F > Cl > Br |
| D | 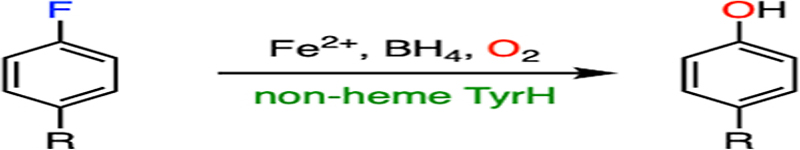 |
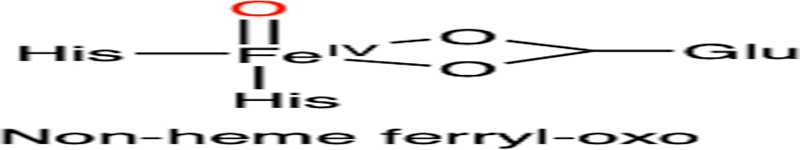 |
F > Cl > Br |
| E | 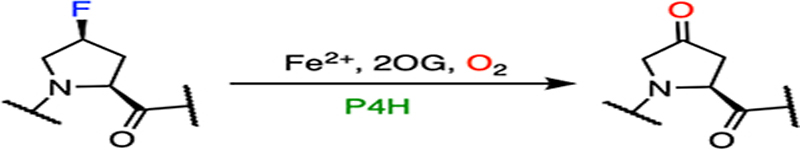 |
 |
Unknown |
| F | 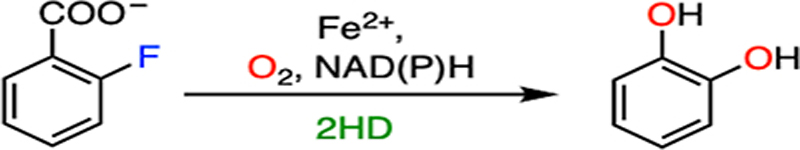 |
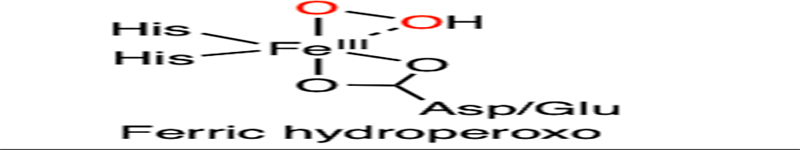 |
F > Cl > Br > 1 |
| G |  |
 |
F > Cl |
The fate of the fluorine atom and the identification of the final defluorinated organic products are essential for understanding the C-F bond chemistry. The cleaved fluorine atom has been exclusively detected as the fluoride anion,32,36,37 although the fluorine product in nonheme TyrH case has yet to be determined. Such consistency supports fluoride elimination directly from the aromatics and precludes the possibility of fluorine radical or cation formation during catalysis due to its instability. Therefore, although the bond dissociation energy of a C-F bond is extremely high, heterolytic bond cleavage yielding a fluoride is anticipated in a biological system. It is also evident that most defluorination reactions mediated by these metalloenzymes are the outcome of functionalization (mainly hydroxylation) of a C-H bond, which is the original chemistry carried out by these enzymes. Aside from the single-turnover cofactor biogenesis reaction shown in ADO and CDO for the formation of a thioether bond, all the defluorination reactions incorporate an oxygen atom into the fluoroarenes, producing either a quinone/keto or catechol/phenol with multiple turnovers. It is worth noting that these metalloenzymes can cleave the C-F bond via either an oxidative or a nonoxidative mechanism. On the one hand, the quinone/keto production in the cases of DHP, CYP, P4H, and potentially nonheme TyrH is considered to be oxidative C-F bond cleavage; therefore, it is promoted by the oxidized high-valent iron center, i.e., ferryl-oxo species. On the other hand, the catechol/phenol product formed by heme TyrH and 2HD is considered to be nonoxidative C-F bond cleavage, which requires additional electron supplement to balance fluoride formation, e.g., oxidation of H2O2 and formation of CO2. Overall, the C-F bond cleavage presented in these metalloenzymes produce environmentally benign products, which make them adequate to further develop into biocatalysts for fluorinated contaminants. While in drug discovery, metabolizable fluorine-containing compounds should undoubtedly sound the alarm on fluoride and resulting metabolites.
In addition, among the metalloenzymes reviewed, DHP is the only enzyme that performs defluorination as its natural chemistry, although fluorinated substrates are the least reactive among the halogenated substrates. The other enzymes discussed here mediate oxidative C-H bond functionalization as their natural reaction, while C-F bond cleavage is either an alternate or a side reaction occurring when a fluorinated substrate is positioned in place of the native substrate. However, the fluorine substitution of the substrate significantly alters the polarity and reactivity.2,3 In addition, there is a two-electron difference between the departure of fluoride versus proton. Therefore, the C-F bond cleavage requires a different mechanism than C-H bond cleavage. Although the mechanisms may share some similarities in the earlier stages of oxygen activation, they diverge into different pathways afterward. It is an exciting phenomenon that the C-F bond is predominantly the most readily cleaved, even though it is the most durable among all the C-X bonds in most enzymatic reactions (except DHP). The possible explanations for this include the notion that a fluorinated substrate has the highest binding affinity and the least steric effect. Further, fluorine could act as a better leaving group at certain intermediary stages, e.g., a substituted Meisenheimer complex in nucleophilic aromatic substitution (SNAr).197 Typically, the rate-determining step in SNAr is the initial nucleophilic attack resulting in the formation of a negatively charged Meisenheimer complex. Given the high energy barrier of the Meisenheimer complex, the elimination of halide is rapid once the intermediate is formed. Fluorine has the most substantial negative inductive effect to help stabilize the negatively charged intermediate,88 and therefore, it facilitates the defluorination process. Such an explanation can provide a rationale for the halogen reactivity of heme TyrH (F > Cl > I) but not DHP (Br > Cl > F) because the latter forms a positively charged intermediate even though both undergo SNAr. The concept that fluoride serves as a good leaving group can also be demonstrated by considering the example of glycosidases; its catalytic rate is monitored by the formation of fluoride using glycosyl fluorides as the substrates.198,199 In addition to the cases discussed in this study, the ability of fluoride to be an excellent leaving group has also been exploited in other enzymatic systems by the design of fluorinated substrate analogs as mechanism-based inhibitors or probes.200 CYP and nonheme TyrH are proposed to undergo electrophilic substitution, while P4H, 2HD, and ADO/CDO experience a radical rebound. In these mechanistic proposals, the steric effect may be the predominant determining factor for C-F bond functionalization by enzyme-based reactive intermediates.
Evidently, the determining factor for whether or not an enzymatic system can promote C-F bond cleavage is if it has the ability to generate the proposed reactive intermediates (as summarized in Table 2) or chemically equivalent species. Hence, apart from the specific metalloenzymes mentioned above, C-F bond cleavage is anticipated in enzymes that are capable of conducting similar oxidative chemistries. The enzyme families surveyed in this review include dioxygenase, hydroxylase, and peroxidase. We expect more defluorination activity to be reported among these enzyme families because of the mutual catalytic mechanisms and reactive intermediates. The beneficial aspects of developing biocatalysts that mediate C-F bond cleavage include unveiling the defluorination activity of other known enzymes and identifying putative novel enzymes capable of C-F bond cleavage through bioinformatics studies, which could significantly expand the defluorination chemistries. The next phase to develop the knowledge for applications may further benefit from enzyme engineering using the reported templates by direct evolution or cofactor manipulation.
8. Concluding remarks
Although the C-F bond exhibits extraordinary chemical and thermal stabilities, several distinct classes of metal-dependent enzymes have been shown to be able to cleave such a chemical bond either oxidatively or nonoxidatively. The biocatalytic defluorination reactions directly mediated by these metalloenzymes share many common features. The most apparent common feature is their utilization of a mononuclear iron as their catalytic center. Synthetic complexes containing mono- or binuclear 3d transition metals have been shown to catalyze C-F bond cleavage in a variety of systems,201–206 which echoes the potential defluorination reactivity of metalloenzyme with similar catalytic centers and ligand scaffolds. Future discovery of other 3d transition-metal-based or multimetal-dependent biocatalysts that mitigate the reliance on iron for C-F bond cleavage would be highly stimulating. In addition, a majority of the discussed metalloenzymes perform defluorination reactions through hydroxylation via a highly reactive iron-oxygen complex. Oxygen activation by a metal center to generate a powerful oxidant is the common trait to activate the inert C-F bond. With regard to the enzyme-/mediator-based radical approach, it can be a powerful defluorination method as long as the radical can be generated in a highly controlled manner. Other enzymatic or metal-complex systems capable of producing such radicals could potentially broaden such a strategy. Overall, a deeper understanding of these biocatalytic mechanisms will promote enzymatic applications as well as the development of catalysts in medicinal and environmental remediations.
Acknowledgements
This article was partially supported by the Lutcher Brown Distinguished Chair endowment fund. The authors’ research work in the nonheme iron-dependent thiol dioxygenases and heme iron-dependent tyrosine hydroxylase is supported by the National Science Foundation award CHE-1808637 and the National Institutes of Health grant R01GM108988, respectively.
Biographies

Yifan Wang
Yifan Wang is currently a PhD candidate at the University of Texas at San Antonio under the supervision of Dr Aimin Liu. She obtained her BSc from Zhejiang Sci-Tech University in 2015, with one semester at the University of Liverpool (UK) as a visiting student between 2014 and 2015. During her graduate study in San Antonio, she has developed a keen interest in mechanistic enzymology. Her research spans across a wide range of subjects, covering heme and nonheme iron-dependent enzymes related to amino acid metabolism, oxygen activation, C-H bond functionalization, natural product biosynthesis, and protein cofactor biogenesis.

Aimin Liu
Aimin Liu is a protein chemist with the scientific nickname Feradical. Originally from Mainland China with a bachelor’s degree from the University of Science and Technology of China, he received a doctorate from Stockholm University in Sweden. Aimin also received training at the University of Newcastle upon Tyne as a Royal Society Fellow and the University of Minnesota as a Postdoctoral Associate. He was a distinguished university professor at Georgia State University before taking the current Lutcher Brown Distinguished Professorship at the University of Texas at San Antonio. His research interest is centered around mechanistic enzymology and protein structure-function relationships.
Footnotes
Conflicts of interest
There are no conflicts to declare.
References
- 1.Berger R, Resnati G, Metrangolo P, Weber E and Hulliger J, Chem. Soc. Rev, 2011, 40, 3496–3508. [DOI] [PubMed] [Google Scholar]
- 2.Gillis EP, Eastman KJ, Hill MD, Donnelly DJ and Meanwell NA, J. Med. Chem, 2015, 58, 8315–8359. [DOI] [PubMed] [Google Scholar]
- 3.Shah P and Westwell AD, J. Enzyme Inhib. Med. Chem, 2007, 22, 527–540. [DOI] [PubMed] [Google Scholar]
- 4.Hernandes MZ, Cavalcanti SM, Moreira DR, de Azevedo Junior WF and Leite AC, Curr. Drug Targets, 2010, 11, 303–314. [DOI] [PubMed] [Google Scholar]
- 5.Analytics A, Global fluorochemical market: world market review by product type, by application, by end user industry (2019 Edition): opportunities and forecast (2019–2024), November, 2019.
- 6.Zhou Y, Wang J, Gu Z, Wang S, Zhu W, Acena JL, Soloshonok VA, Izawa K and Liu H, Chem. Rev, 2016, 116, 422–518. [DOI] [PubMed] [Google Scholar]
- 7.Cui B, Jia S, Tokunaga E and Shibata N, Nat. Commun, 2018, 9, 4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horner JM, Soc JR. Health, 1989, 109, 147–150. [DOI] [PubMed] [Google Scholar]
- 9.Camargo JA, Chemosphere, 2003, 50, 251–264. [DOI] [PubMed] [Google Scholar]
- 10.Steenland K, Fletcher T and Savitz DA, Environ. Health Perspect, 2010, 118, 1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jagtap S, Yenkie MK, Labhsetwar N and Rayalu S, Chem. Rev, 2012, 112, 2454–2466. [DOI] [PubMed] [Google Scholar]
- 12.Johnson PI, Sutton P, Atchley DS, Koustas E, Lam J, Sen S, Robinson KA, Axelrad DA and Woodruff TJ, Environ. Health Perspect, 2014, 122, 1028–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lehmann GM, LaKind JS, Davis MH, Hines EP, Marchitti SA, Alcala C and Lorber M, Environ. Health Perspect, 2018, 126, 096001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chemical & Engineering News, Governments endorse global PFOA ban, with some exemptions, https://cen.acs.org/environment/persistent-pollutants/Governments-endorse-global-PFOA-ban/97/web/2019/05.
- 15.Kuehn B, JAMA, 2019, 322, 1757. [DOI] [PubMed] [Google Scholar]
- 16.de Ruiter G, Carsch KM, Takase MK and Agapie T, Chemistry, 2017, 23, 10744–10748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jana A, Samuel PP, Tavcar G, Roesky HW and Schulzke C, J. Am. Chem. Soc, 2010, 132, 10164–10170. [DOI] [PubMed] [Google Scholar]
- 18.Sahu S, Quesne MG, Davies CG, Durr M, Ivanovic-Burmazovic I, Siegler MA, Jameson GN, de Visser SP and Goldberg DP, J. Am. Chem. Soc, 2014, 136, 13542–13545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suzuki N, Fujita T, Amsharov KY and Ichikawa J, Chem. Commun, 2016, 52, 12948–12951. [DOI] [PubMed] [Google Scholar]
- 20.Zamostna L, Sander S, Braun T, Laubenstein R, Braun B, Herrmann R and Klaring P, Dalton Trans, 2015, 44, 9450–9469. [DOI] [PubMed] [Google Scholar]
- 21.Garcia-Bosch I, Cowley RE, Diaz DE, Peterson RL, Solomon EI and Karlin KD, J. Am. Chem. Soc, 2017, 139, 3186–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Colomban C, Tobing A, Mukherjee G, Sastri C, Sorokin A and de Visser S, Chemistry, 2019, 25, 14320–14331. [DOI] [PubMed] [Google Scholar]
- 23.Lv H, Cai YB and Zhang JL, Angew. Chem., Int. Ed, 2013, 52, 3203–3207. [DOI] [PubMed] [Google Scholar]
- 24.Serrano-Plana J, Garcia-Bosch I, Miyake R, Costas M and Company A, Angew. Chem., Int. Ed, 2014, 53, 9608–9612. [DOI] [PubMed] [Google Scholar]
- 25.Sahu S, Zhang B, Pollock CJ, Durr M, Davies CG, Confer AM, Ivanovic-Burmazovic I, Siegler MA, Jameson GN, Krebs C and Goldberg DP, J. Am. Chem. Soc, 2016, 138, 12791–12802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang S and Jaffe PR, Environ. Sci. Technol, 2019, 53, 11410–11419. [DOI] [PubMed] [Google Scholar]
- 27.Murphy CD, Biotechnol. Lett, 2010, 32, 351–359. [DOI] [PubMed] [Google Scholar]
- 28.Liu J and Mejia Avendano S, Environ. Int, 2013, 61, 98–114. [DOI] [PubMed] [Google Scholar]
- 29.Fetzner S, Muller R and Lingens F, J. Bacteriol, 1992, 174, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.LaCount MW, Zhang E, Chen YP, Han K, Whitton MM, Lincoln DE, Woodin SA and Lebioda L, J. Biol. Chem, 2000, 275, 18712–18716. [DOI] [PubMed] [Google Scholar]
- 31.Kiel M and Engesser KH, Appl. Microbiol. Biotechnol, 2015, 99, 7433–7464. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y, Davis I, Shin I, Wherritt DJ, Griffith WP, Dornevil K, Colabroy KL and Liu A, ACS Catal, 2019, 9, 4764–4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hillas PJ and Fitzpatrick PF, Biochemistry, 1996, 35, 6969–6975. [DOI] [PubMed] [Google Scholar]
- 34.Bretscher LE, Jenkins CL, Taylor KM, DeRider ML and Raines RT, J. Am. Chem. Soc, 2001, 123, 777–778. [DOI] [PubMed] [Google Scholar]
- 35.Renganathan V, Appl. Environ. Microbiol, 1989, 55, 330–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Griffith WP, Li J, Koto T, Wherritt DJ, Fritz E and Liu A, Angew. Chem., Int. Ed, 2018, 57, 8149–8153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li J, Griffith WP, Davis I, Shin I, Wang J, Li F, Wang Y, Wherritt DJ and Liu A, Nat. Chem. Biol, 2018, 14, 853–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miranda-Rojas S and Toro-Labbe A, J. Chem. Phys, 2015, 142, 194301. [DOI] [PubMed] [Google Scholar]
- 39.Goldman P, J. Biol. Chem, 1965, 240, 3434–3438. [PubMed] [Google Scholar]
- 40.Chan PW, Yakunin AF, Edwards EA and Pai EF, J. Am. Chem. Soc, 2011, 133, 7461–7468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tiedt O, Mergelsberg M, Boll K, Muller M, Adrian L, Jehmlich N, von Bergen M and Boll M, mBio, 2016, 7(4), e00990–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tiedt O, Mergelsberg M, Eisenreich W and Boll M, Front. Microbiol, 2017, 8, 2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yevglevskis M, Lee GL, Sun J, Zhou S, Sun X, Kociok-Kohn G, James TD, Woodman TJ and Lloyd MD, Org. Biomol. Chem, 2016, 14, 612–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Solyanikova IP, Moiseeva OV, Boeren S, Boersma MG, Kolomytseva MP, Vervoort J, Rietjens IM, Golovleva LA and van Berkel WJ, Appl. Environ. Microbiol, 2003, 69, 5636–5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Williams L, Nguyen T, Li Y, Porter TN and Raushel FM, Biochemistry, 2006, 45, 7453–7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carvalho MF, Ferreira MI, Moreira IS, Castro PM and Janssen DB, Appl. Environ. Microbiol, 2006, 72, 7413–7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Husain M, Entsch B, Ballou DP, Massey V and Chapman PJ, J. Biol. Chem, 1980, 255, 4189–4197. [PubMed] [Google Scholar]
- 48.Driscoll JP, Aliagas I, Harris JJ, Halladay JS, Khatib-Shahidi S, Deese A, Segraves N and Khojasteh-Bakht SC, Chem. Res. Toxicol, 2010, 23, 861–863. [DOI] [PubMed] [Google Scholar]
- 49.Peelen S, Rietjens IM, Boersma MG and Vervoort J, Eur. J. Biochem, 1995, 227, 284–291. [DOI] [PubMed] [Google Scholar]
- 50.Chen YP, Woodin SA, Lincoln DE and Lovell CR, J. Biol. Chem, 1996, 271, 4609–4612. [PubMed] [Google Scholar]
- 51.Barrios DA, D’Antonio J, McCombs NL, Zhao J, Franzen S, Schmidt AC, Sombers LA and Ghiladi RA, J. Am. Chem. Soc, 2014, 136, 7914–7925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Malewschik T, de Serrano V, McGuire AH and Ghiladi RA, Arch. Biochem. Biophys, 2019, 673, 108079. [DOI] [PubMed] [Google Scholar]
- 53.Carey LM, Doctor of Philosophy Doctoral thesis, North Carolina State University, North Carolina, U. S. A., 2017. [Google Scholar]
- 54.Lebioda L, LaCount MW, Zhang E, Chen YP, Han K, Whitton MM, Lincoln DE and Woodin SA, Nature, 1999, 401, 445. [DOI] [PubMed] [Google Scholar]
- 55.Wang C, Lovelace LL, Sun S, Dawson JH and Lebioda L, Biochemistry, 2013, 52, 6203–6210. [DOI] [PubMed] [Google Scholar]
- 56.Zhao J, de Serrano V, Zhao J, Le P and Franzen S, Biochemistry, 2013, 52, 2427–2439. [DOI] [PubMed] [Google Scholar]
- 57.McGuire AH, Carey LM, de Serrano V, Dali S and Ghiladi RA, Biochemistry, 2018, 57, 4455–4468. [DOI] [PubMed] [Google Scholar]
- 58.Thompson MK, Davis MF, de Serrano V, Nicoletti FP, Howes BD, Smulevich G and Franzen S, Biophys. J, 2010, 99, 1586–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Coulter ED, Cheek J, Ledbetter AP, Chang CK and Dawson JH, Biochem. Biophys. Res. Commun, 2000, 279, 1011–1015. [DOI] [PubMed] [Google Scholar]
- 60.Davydov R, Osborne RL, Kim SH, Dawson JH and Hoffman BM, Biochemistry, 2008, 47, 5147–5155. [DOI] [PubMed] [Google Scholar]
- 61.D’Antonio J and Ghiladi RA, Biochemistry, 2011, 50, 5999–6011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Osborne RL, Raner GM, Hager LP and Dawson JH, J. Am. Chem. Soc, 2006, 128, 1036–1037. [DOI] [PubMed] [Google Scholar]
- 63.Guengerich FP, Chem. Res. Toxicol, 2001, 14, 611–650. [DOI] [PubMed] [Google Scholar]
- 64.Poulos TL, J. Biol. Inorg. Chem, 1996, 1(4), 356–359. [Google Scholar]
- 65.Belyea J, Gilvey LB, Davis MF, Godek M, Sit TL, Lommel SA and Franzen S, Biochemistry, 2005, 44, 15637–15644. [DOI] [PubMed] [Google Scholar]
- 66.Chen Z, de Serrano V, Betts L and Franzen S, Acta Crystallogr., Sect. D: Biol. Crystallogr, 2009, 65, 34–40. [DOI] [PubMed] [Google Scholar]
- 67.Thompson MK, Franzen S, Ghiladi RA, Reeder BJ and Svistunenko DA, J. Am. Chem. Soc, 2010, 132, 17501–17510. [DOI] [PubMed] [Google Scholar]
- 68.Hiner AN, Raven EL, Thorneley RN, Garcia-Canovas F and Rodriguez-Lopez JN, J. Inorg. Biochem, 2002, 91, 27–34. [DOI] [PubMed] [Google Scholar]
- 69.Coulson AFW, Erman JE and Yonetani T, J. Biol. Chem, 1971, 246(4), 917–924. [PubMed] [Google Scholar]
- 70.Gurbiel R, Huyett JE, Doan PE, Houseman AP, Sivaraja M, Goodin DB and Hoffman BM, J. Am. Chem. Soc, 1995, 117(35), 9033–9041. [Google Scholar]
- 71.Osborne RL, Coggins MK, Raner GM, Walla M and Dawson JH, Biochemistry, 2009, 48, 4231–4238. [DOI] [PubMed] [Google Scholar]
- 72.Franzen S, Gilvey LB and Belyea JL, Biochim. Biophys. Acta, 2007, 1774, 121–130. [DOI] [PubMed] [Google Scholar]
- 73.D’Antonio J, D’Antonio EL, Thompson MK, Bowden EF, Franzen S, Smirnova T and Ghiladi RA, Biochemistry, 2010, 49, 6600–6616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yin LL, Yuan H, Liu C, He B, Gao SQ, Wen GB, Tan XS and Lin YW, ACS Catal, 2018, 8, 9619–9624. [Google Scholar]
- 75.Colabroy KL, Biochim. Biophys. Acta, 2016, 1864, 724–737. [DOI] [PubMed] [Google Scholar]
- 76.Connor KL, Colabroy KL and Gerratana B, Biochemistry, 2011, 50, 8926–8936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Novotna J, Olsovska J, Novak P, Mojzes P, Chaloupkova R, Kamenik Z, Spizek J, Kutejova E, Mareckova M, Tichy P, Damborsky J and Janata J, PLoS One, 2013, 8, 483–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Peschke U, Schmidt H, Zhang HZ and Piepersberg W, Mol. Microbiol, 1995, 16, 1137–1156. [DOI] [PubMed] [Google Scholar]
- 79.Hofer I, Crusemann M, Radzom M, Geers B, Flachshaar D, Cai XF, Zeeck A and Piel J, Chem. Biol, 2011, 18, 381–391. [DOI] [PubMed] [Google Scholar]
- 80.Li W, Khullar A, Chou S, Sacramo A and Gerratana B, Appl. Environ. Microbiol, 2009, 75, 2869–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li W, Chou S, Khullar A and Gerratana B, Appl. Environ. Microbiol, 2009, 75, 2958–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Najmanova L, Ulanova D, Jelinkova M, Kamenik Z, Kettnerova E, Koberska M, Gazak R, Radojevic B and Janata J, Folia Microbiol, 2014, 59, 543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sariaslani FS, Crit. Rev. Biotechnol, 1989, 9, 171–257. [DOI] [PubMed] [Google Scholar]
- 84.Sono M, Roach MP, Coulter ED and Dawson JH, Chem. Rev, 1996, 96, 2841–2887. [DOI] [PubMed] [Google Scholar]
- 85.Leahy JG, Batchelor PJ and Morcomb SM, FEMS Microbiol. Rev, 2003, 27, 449–479. [DOI] [PubMed] [Google Scholar]
- 86.Rosenzweig AC and Sazinsky MH, Curr. Opin. Struct. Biol, 2006, 16, 729–735. [DOI] [PubMed] [Google Scholar]
- 87.Fitzpatrick PF, Biochemistry, 2003, 42, 14083–14091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fernandez I, Frenking G and Uggerud E, J. Org. Chem, 2010, 75, 2971–2980. [DOI] [PubMed] [Google Scholar]
- 89.Lamb DC, Lei L, Warrilow AG, Lepesheva GI, Mullins JG, Waterman MR and Kelly SL, J. Virol, 2009, 83, 8266–8269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rydberg P, Olsen L and Ryde U, Curr. Inorg. Chem, 2012, 2, 292–315(224). [Google Scholar]
- 91.Manikandan P and Nagini S, Curr. Drug Targets, 2018, 19, 38–54. [DOI] [PubMed] [Google Scholar]
- 92.Dornevil K, Davis I, Fielding AJ, Terrell JR and Liu A, J. Biol. Chem, 2017, 292, 13645–13657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nguyen RC, Yang Y, Wang Y, Davis I and Liu A, ACS Catal, 2020, 10, 1628–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Urban P, Lautier T, Pompon D and Truan G, Int. J. Mol. Sci, 2018, 19(6), 1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Amaya GM, Durandis R, Bourgeois DS, Perkins JA, Abouda AA, Wines KJ, Mohamud M, Starks SA, Daniels RN and Jackson KD, Chem. Res. Toxicol, 2018, 31, 570–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xie C, Zhou J, Guo Z, Diao X, Gao Z, Zhong D, Jiang H, Zhang L and Chen X, Br. J. Pharmacol, 2013, 168, 1687–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Huang YC, Chang YC, Yeh CN and Yu CS, Molecules, 2016, 21, 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sephton SM, Dennler P, Leutwiler DS, Mu L, Wanger-Baumann CA, Schibli R, Kramer SD and Ametamey SM, Am. J. Nucl. Med. Mol. Imaging, 2012, 2, 14–28. [PMC free article] [PubMed] [Google Scholar]
- 99.Ahr HJ, King LJ, Nastainczyk W and Ullrich V, Biochem. Pharmacol, 1982, 31, 383–390. [DOI] [PubMed] [Google Scholar]
- 100.Njoku D, Laster MJ, Gong DH, Eger EI 2nd, Reed GF and Martin JL, Anesth. Analg, 1997, 84, 173–178. [DOI] [PubMed] [Google Scholar]
- 101.Kharasch ED and Thummel KE, Anesthesiology, 1993, 79, 795–807. [DOI] [PubMed] [Google Scholar]
- 102.Harkey A, Kim HJ, Kandagatla S and Raner GM, Biotechnol. Lett, 2012, 34, 1725–1731. [DOI] [PubMed] [Google Scholar]
- 103.Ohe T, Mashino T and Hirobe M, Drug Metab. Dispos, 1997, 25, 116–122. [PubMed] [Google Scholar]
- 104.Cnubben NHP, Vervoort J, Boersma MG and Rietjens IMCM, Biochem. Pharmacol, 1995, 49, 1235–1248. [DOI] [PubMed] [Google Scholar]
- 105.den Besten C, van Bladeren PJ, Duizer E, Vervoort J and Rietjens IM, Chem. Res. Toxicol, 1993, 6, 674–680. [DOI] [PubMed] [Google Scholar]
- 106.Rietjens IM and Vervoort J, Chem. -Biol. Interact, 1991, 77, 263–281. [DOI] [PubMed] [Google Scholar]
- 107.Rietjens IM, Tyrakowska B, Veeger C and Vervoort J, Eur. J. Biochem, 1990, 194, 945–954. [DOI] [PubMed] [Google Scholar]
- 108.Vatsis KP and Coon MJ, Arch. Biochem. Biophys, 2002, 397, 119–129. [DOI] [PubMed] [Google Scholar]
- 109.Osman AM, Boeren S, Boersma MG, Veeger C and Rietjens IM, Proc. Natl. Acad. Sci. U. S. A, 1997, 94, 4295–4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pan Y, ACS Med. Chem. Lett, 2019, 10, 1016–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Daubner SC, Le T and Wang S, Arch. Biochem. Biophys, 2011, 508, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Goodwill KE, Sabatier C, Marks C, Raag R, Fitzpatrick PF and Stevens RC, Nat. Struct. Biol, 1997, 4, 578–585. [DOI] [PubMed] [Google Scholar]
- 113.Goodwill KE, Sabatier C and Stevens RC, Biochemistry, 1998, 37, 13437–13445. [DOI] [PubMed] [Google Scholar]
- 114.Liu A, Ho RYN, Que L, Ryle MJ, Phinney BS and Hausinger RP, J. Am. Chem. Soc, 2001, 123, 5126–5127. [DOI] [PubMed] [Google Scholar]
- 115.Ryle MJ, Liu A, Muthukumaran RB, Ho RYN, Koehntop KD, McCracken J, Que L and Hausinger RP, Biochemistry, 2003, 42, 1854–1862. [DOI] [PubMed] [Google Scholar]
- 116.Ryle MJ, Koehntop KD, Liu A, Que L and Hausinger RP, Proc. Natl. Acad. Sci. U. S. A, 2003, 100, 3790–3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mantri M, Zhang Z, McDonough MA and Schofield CJ, FEBS J, 2012, 279, 1563–1575. [DOI] [PubMed] [Google Scholar]
- 118.Yan W, Song H, Song F, Guo Y, Wu C-H, Sae Her A, Pu Y, Wang S, Naowarojna N, Weitz A, Hendrich MP, Costello CE, Zhang L, Liu P and Jessie Zhang Y, Nature, 2015, 527, 539–543. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 119.Fitzpatrick PF, Adv. Exp. Med. Biol, 1993, 338, 81–86. [DOI] [PubMed] [Google Scholar]
- 120.Frantom PA, Pongdee R, Sulikowski GA and Fitzpatrick PF, J. Am. Chem. Soc, 2002, 124, 4202–4203. [DOI] [PubMed] [Google Scholar]
- 121.Chow MS, Eser BE, Wilson SA, Hodgson KO, Hedman B, Fitzpatrick PF and Solomon EI, J. Am. Chem. Soc, 2009, 131, 7685–7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Krzyaniak MD, Eser BE, Ellis HR, Fitzpatrick PF and McCracken J, Biochemistry, 2013, 52, 8430–8441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Roberts KM and Fitzpatrick PF, IUBMB Life, 2013, 65, 350–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Guroff G, Daly JW, Jerina DM, Renson J, Witkop B and Udenfriend S, Science, 1967, 157, 1524–1530. [DOI] [PubMed] [Google Scholar]
- 125.Eser BE, Barr EW, Frantom PA, Saleh L, Bollinger JM Jr., Krebs C and Fitzpatrick PF, J. Am. Chem. Soc, 2007, 129, 11334–11335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Panay AJ, Lee M, Krebs C, Bollinger JM and Fitzpatrick PF, Biochemistry, 2011, 50, 1928–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bassan A, Blomberg MR and Siegbahn PE, Chemistry, 2003, 9, 4055–4067. [DOI] [PubMed] [Google Scholar]
- 128.Kaufman S, Biochim. Biophys. Acta, 1961, 51, 619–621. [DOI] [PubMed] [Google Scholar]
- 129.McDonough MA, Loenarz C, Chowdhury R, Clifton IJ and Schofield CJ, Curr. Opin. Struct. Biol, 2010, 20, 659–672. [DOI] [PubMed] [Google Scholar]
- 130.Clifton IJ, McDonough MA, Ehrismann D, Kershaw NJ, Granatino N and Schofield CJ, J. Inorg. Biochem, 2006, 100, 644–669. [DOI] [PubMed] [Google Scholar]
- 131.Price JC, Barr EW, Tirupati B, Bollinger JM Jr. and Krebs C, Biochemistry, 2003, 42, 7497–7508. [DOI] [PubMed] [Google Scholar]
- 132.Grzyska PK, Appelman EH, Hausinger RP and Proshlyakov DA, Proc. Natl. Acad. Sci. U. S. A, 2010, 107, 3982–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Riggs-Gelasco PJ, Price JC, Guyer RB, Brehm JH, Barr EW, Bollinger JM and Krebs C, J. Am. Chem. Soc, 2004, 126, 8108–8109. [DOI] [PubMed] [Google Scholar]
- 134.Wong SD, Srnec M, Matthews ML, Liu LV, Kwak Y, Park K, Bell CB, Alp EE, Zhao JY, Yoda Y, Kitao S, Seto M, Krebs C, Bollinger JM and Solomon EI, Nature, 2013, 499, 320–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Price JC, Barr EW, Glass TE, Krebs C and Bollinger JM Jr., J. Am. Chem. Soc, 2003, 125, 13008–13009. [DOI] [PubMed] [Google Scholar]
- 136.Martinez S and Hausinger RP, J. Biol. Chem, 2015, 290, 20702–20711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Islam MS, Leissing TM, Chowdhury R, Hopkinson RJ and Schofield CJ, Annu. Rev. Biochem, 2018, 87, 585–620. [DOI] [PubMed] [Google Scholar]
- 138.Gottlieb AA, Fujita Y, Udenfriend S and Witkop B, Biochemistry, 1965, 4, 2507–2513. [Google Scholar]
- 139.Gorres KL and Raines RT, Anal. Biochem, 2009, 386, 181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Smart TJ, Hamed RB, Claridge TDW and Schofield CJ, Bioorg. Chem, 2020, 94, 103386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Scotti JS, Leung IK, Ge W, Bentley MA, Paps J, Kramer HB, Lee J, Aik W, Choi H, Paulsen SM, Bowman LA, Loik ND, Horita S, Ho CH, Kershaw NJ, Tang CM, Claridge TD, Preston GM, McDonough MA and Schofield CJ, Proc. Natl. Acad. Sci. U. S. A, 2014, 111, 13331–13336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Taabazuing CY, Hangasky JA and Knapp MJ, J. Inorg. Biochem, 2014, 133, 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kaelin WG, Annu. Rev. Biochem, 2005, 74, 115–128. [DOI] [PubMed] [Google Scholar]
- 144.Gorres KL and Raines RT, Crit. Rev. Biochem. Mol. Biol, 2010, 45, 106–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Myllyharju J, Ann. Med, 2008, 40, 402–417. [DOI] [PubMed] [Google Scholar]
- 146.Vasta JD and Raines RT, J. Med. Chem, 2018, 61, 10403–10411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Smith TG and Talbot NP, Antioxid. Redox Signaling, 2010, 12, 431–433. [DOI] [PubMed] [Google Scholar]
- 148.Haase VH, Hemodial. Int, 2017, 21(Suppl 1), S110–S124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Fong GH and Takeda K, Cell Death Differ, 2008, 15, 635–641. [DOI] [PubMed] [Google Scholar]
- 150.Chowdhury R, Leung IK, Tian YM, Abboud MI, Ge W, Domene C, Cantrelle FX, Landrieu I, Hardy AP, Pugh CW, Ratcliffe PJ, Claridge TD and Schofield CJ, Nat. Commun, 2016, 7, 12673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Shoulders MD and Raines RT, Annu. Rev. Biochem, 2009, 78, 929–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Holmgren SK, Taylor KM, Bretscher LE and Raines RT, Nature, 1998, 392, 666–667. [DOI] [PubMed] [Google Scholar]
- 153.Rydzik AM, Leung IK, Kochan GT, Thalhammer A, Oppermann U, Claridge TD and Schofield CJ, ChemBioChem, 2012, 13, 1559–1563. [DOI] [PubMed] [Google Scholar]
- 154.Barry SM and Challis GL, ACS Catal, 2013, 3, 2362–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gibson DT, Koch JR and Kallio RE, Biochemistry, 1968, 7, 2653–2662. [DOI] [PubMed] [Google Scholar]
- 156.Parales RE, Parales JV and Gibson DT, J. Bacteriol, 1999, 181, 1831–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wackett LP, Enzyme Microb. Technol, 2002, 31, 577–587. [Google Scholar]
- 158.Francisco P Jr., Ogawa N, Suzuki K and Miyashita K, Microbiology, 2001, 147, 121–133. [DOI] [PubMed] [Google Scholar]
- 159.Wang Y, Li J and Liu A, J. Biol. Inorg. Chem, 2017, 22, 395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ashikawa Y, Fujimoto Z, Usami Y, Inoue K, Noguchi H, Yamane H and Nojiri H, BMC Struct. Biol, 2012, 12, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Karlsson A, Parales JV, Parales RE, Gibson DT, Eklund H and Ramaswamy S, Science, 2003, 299, 1039–1042. [DOI] [PubMed] [Google Scholar]
- 162.Kal S and Que L, J. Biol. Inorg. Chem, 2017, 22, 339–365. [DOI] [PubMed] [Google Scholar]
- 163.Stipanuk MH, Simmons CR, Karplus PA and Dominy JE Jr., Amino Acids, 2011, 41, 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Masson N, Keeley TP, Giuntoli B, White MD, Puerta ML, Perata P, Hopkinson RJ, Flashman E, Licausi F and Ratcliffe PJ, Science, 2019, 365, 65–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Dominy JE Jr., Simmons CR, Hirschberger LL, Hwang J, Coloso RM and Stipanuk MH, J. Biol. Chem, 2007, 282, 25189–25198. [DOI] [PubMed] [Google Scholar]
- 166.Stipanuk MH, Ueki I, Dominy JE Jr., Simmons CR and Hirschberger LL, Amino Acids, 2009, 37, 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Simmons CR, Liu Q, Huang QQ, Hao Q, Begley TP, Karplus PA and Stipanuk MH, J. Biol. Chem, 2006, 281, 18723–18733. [DOI] [PubMed] [Google Scholar]
- 168.Arjune S, Schwarz G and Belaidi AA, Amino Acids, 2015, 47, 55–63. [DOI] [PubMed] [Google Scholar]
- 169.Driggers CM, Kean KM, Hirschberger LL, Cooley RB, Stipanuk MH and Karplus PA, J. Mol. Biol, 2016, 428, 3999–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Li J, Koto T, Davis I and Liu A, Biochemistry, 2019, 58, 2218–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Davies CG, Fellner M, Tchesnokov EP, Wilbanks SM and Jameson GN, Biochemistry, 2014, 53, 7961–7968. [DOI] [PubMed] [Google Scholar]
- 172.Siakkou E, Rutledge MT, Wilbanks SM and Jameson GNL, Biochim. Biophys. Acta, 2011, 1814, 2003–2009. [DOI] [PubMed] [Google Scholar]
- 173.Song Z, Yue Y, Feng S, Sun H, Li Y, Xu F, Zhang Q and Wang W, Chem. Phys. Lett, 2020, 750, 137449. [Google Scholar]
- 174.Tchesnokov EP, Fellner M, Siakkou E, Kleffmann T, Martin LW, Aloi S, Lamont IL, Wilbanks SM and Jameson GN, J. Biol. Chem, 2015, 290, 24424–24437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Bruland N, Wubbeler JH and Steinbuchel A, J. Biol. Chem, 2009, 284, 660–672. [DOI] [PubMed] [Google Scholar]
- 176.Brandt U, Galant G, Meinert-Berning C and Steinbuchel A, Enzyme Microb. Technol, 2019, 120, 61–68. [DOI] [PubMed] [Google Scholar]
- 177.Brandt U, Schurmann M and Steinbuchel A, J. Biol. Chem, 2014, 289, 30800–30809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Fellner M, Aloi S, Tchesnokov EP, Wilbanks SM and Jameson GN, Biochemistry, 2016, 55, 1362–1371. [DOI] [PubMed] [Google Scholar]
- 179.Santhanam N, Vivanco JM, Decker SR and Reardon KF, Trends Biotechnol, 2011, 29, 480–489. [DOI] [PubMed] [Google Scholar]
- 180.Jones SM and Solomon EI, Cell Mol. Life. Sci, 2015, 72(5), 869–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Piontek K, Antorini M and Choinowski T, J. Biol. Chem, 2002, 277, 37663–37669. [DOI] [PubMed] [Google Scholar]
- 182.Solomon EI, Heppner DE, Johnston EM, Ginsbach JW, Cirera J, Qayyum M, Kieber-Emmons MT, Kjaergaard CH, Hadt RG and Tian L, Chem. Rev, 2014, 114, 3659–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Canas AI and Camarero S, Biotechnol. Adv, 2010, 28, 694–705. [DOI] [PubMed] [Google Scholar]
- 184.Christopher LP, Yao B and Ji Y, Front. Energy Res, 2014, 2, 12. [Google Scholar]
- 185.Parsons JR, Saez M, Dolfing J and de Voogt P, Rev. Environ. Contam. Toxicol, 2008, 196, 53–71. [DOI] [PubMed] [Google Scholar]
- 186.Luo Q, Lu JH, Zhang H, Wang ZY, Feng MB, Chiang SYD, Woodward D and Huang QG, Environ. Sci. Technol. Lett, 2015, 2, 198–203. [Google Scholar]
- 187.Luo Q, Yan X, Lu J and Huang Q, Environ. Sci. Technol, 2018, 52, 10617–10626. [DOI] [PubMed] [Google Scholar]
- 188.Stubbe J and van der Donk WA, Chem. Biol, 1995, 2, 793–801. [DOI] [PubMed] [Google Scholar]
- 189.Nordlund P and Reichard P, Annu. Rev. Biochem, 2006, 75, 681–706. [DOI] [PubMed] [Google Scholar]
- 190.Frey PA, Annu. Rev. Biochem, 2001, 70, 121–148. [DOI] [PubMed] [Google Scholar]
- 191.Kang G, Taguchi AT, Stubbe J and Drennan CL, Science, 2020, 368, 424–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Li X, Fu R, Lee SY, Krebs C, Davidson VL and Liu A, Proc. Natl. Acad. Sci. U. S. A, 2008, 105, 8597–8600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Yukl ET, Liu F, Krzystek J, Shin S, Jensen LMR, Davidson VL, Wilmot CM and Liu A, Proc. Natl. Acad. Sci. U. S. A, 2013, 110, 4569–4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Geng J, Dornevil K, Davidson VL and Liu A, Proc. Natl. Acad. Sci. U. S. A, 2013, 110, 9639–9644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Geng J, Davis I, Liu F and Liu A, J. Biol. Inorg. Chem, 2014, 19, 1057–1067. [DOI] [PubMed] [Google Scholar]
- 196.Rizzolo K, Cohen SE, Weitz AC, Lopez Munoz MM, Hendrich MP, Drennan CL and Elliott SJ, Nat. Commun, 2019, 10, 1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Senger NA, Bo B, Cheng Q, Keeffe JR, Gronert S and Wu W, J. Org. Chem, 2012, 77, 9535–9540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Lougheed B, Ly HD, Wakarchuk WW and Withers SG, J. Biol. Chem, 1999, 274, 37717–37722. [DOI] [PubMed] [Google Scholar]
- 199.Williams SJ and Withers SG, Carbohydr. Res, 2000, 327, 27–46. [DOI] [PubMed] [Google Scholar]
- 200.Pongdee R and Liu HW, Bioorg. Chem, 2004, 32, 393–437. [DOI] [PubMed] [Google Scholar]
- 201.Clot E, Eisenstein O, Jasim N, Macgregor SA, Mcgrady JE and Perutz RN, Acc. Chem. Res, 2011, 44, 333–348. [DOI] [PubMed] [Google Scholar]
- 202.Nova A, Mas-Balleste R and Lledos A, Organometallics, 2012, 31, 1245–1256. [Google Scholar]
- 203.Utsumi S, Katagiri T and Uneyama K, J. Fluorine Chem, 2013, 152, 84–89. [Google Scholar]
- 204.Ahrens T, Kohlmann J, Ahrens M and Braun T, Chem. Rev, 2015, 115, 931–972. [DOI] [PubMed] [Google Scholar]
- 205.Shen Q, Huang YG, Liu C, Xiao JC, Chen QY and Guo Y, J. Fluorine Chem, 2015, 179, 14–22. [Google Scholar]
- 206.Eisenstein O, Milani J and Perutz RN, Chem. Rev, 2017, 117, 8710–8753. [DOI] [PubMed] [Google Scholar]