

Abstract
During several months of 2002, severe acute respiratory syndrome (SARS) caused by SARS‐coronavirus (SARS‐CoV) spread rapidly from China throughout the world, causing more than 800 deaths due to the development of acute respiratory distress syndrome (ARDS), which is the severe form of acute lung injury (ALI). Interestingly, a novel homologue of angiotensin‐converting enzyme, termed angiotensin‐converting enzyme 2 (ACE2), has been identified as a receptor for SARS‐CoV. Angiotensin‐converting enzyme and ACE2 share homology in their catalytic domain and provide different key functions in the renin–angiotensin system (RAS). Angiotensin‐converting enzyme cleaves angiotensin I to generate angiotensin II, which is a key effector peptide of the system and exerts multiple biological functions, whereas ACE2 reduces angiotensin II levels. Importantly, our recent studies using ACE2 knockout mice have demonstrated that ACE2 protects murine lungs from ARDS. Furthermore, SARS‐CoV infections and the Spike protein of the SARS‐CoV reduce ACE2 expression. Notably, injection of SARS‐CoV Spike into mice worsens acute lung failure in vivo, which can be attenuated by blocking the renin–angiotensin pathway, suggesting that the activation of the pulmonary RAS influences the pathogenesis of ALI/ARDS and SARS.
The renin–angiotensin system (RAS) plays a central role in the control of cardiovascular and renal functions by maintaining the physiological homeostasis of blood pressure and electrolyte balance. Abnormal activation of the RAS has been associated with the pathogenesis of cardiovascular and renal diseases such as hypertension, myocardial infarction and heart failure (Ferrario, 1990; Nicholls et al. 1998; Fleming et al. 2006). The protease renin cleaves angiotensinogen to generate angiotensin (Ang) I. Angiotensin‐converting enzyme (ACE) cleaves Ang I to produce Ang II, which is a key regulator of the RAS and exerts biological functions through the specific receptors Ang II receptor type 1 (AT1R) and Ang II receptor type 2 (AT2R; Skeggs et al. 1980; Corvol et al. 1995). Thus, for many years, ACE has been known as the key enzyme in the regulation of the RAS (Skeggs et al. 1980; Turner & Hooper, 2002; Fig. 1).
Figure 1.
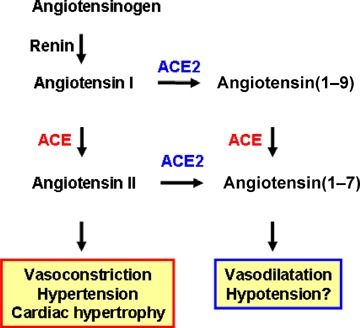
Current view of ACE and ACE2 functions Angiotensin I serves as a substrate for both ACE and ACE2. Angiotensin II is known to act as a vasoconstrictor in vivo. The function of angiotensin(1–9) is not well understood. Both ACE and ACE2 are involved in the production of the vasodilator peptide angiotensin(1–7).
In 2000, a homologue of ACE was cloned by two independent groups and termed angiotensin‐converting enzyme 2 (ACE2; Tipnis et al. 2000; Douglas et al. 2004). Despite the sequence similarity in their catalytic domains, ACE and ACE2 appear to act on different peptide substrates. While ACE cleaves Ang I into Ang II (Skeggs et al. 1980; Corvol et al. 1995), ACE2 removes a single residue from Ang I to yield Ang(1–9) (Tipnis et al. 2000; Douglas et al. 2004) and cleaves a single residue from Ang II to generate Ang(1–7) (Douglas et al. 2004). These biochemical differences are mirrored by the in vivo functions of these two key RAS enzymes. Gene targeting of ACE results in spontaneous hypotension, reduced sperm function and kidney malformations (Kakar et al. 1992). By contrast, when we created the first ACE2 mutant mouse using homologous recombination, disruption of the murine ace2 gene resulted in increased levels of Ang II and progressive worsening of cardiac contractility with age (Crackower et al. 2002). Moreover, independent studies showed that ACE2 mutant mice exhibit worsened heart failure following aortic banding (Yamamoto et al. 2006). Loss of ACE on an ACE2 background or pharmacological inhibition of the RAS can to a large extent reverse these cardiac phenotypes (Crackower et al. 2002). These data provided the first genetic evidence that ACE2 counterbalances the function of ACE and negatively regulates Ang II levels (Fig. 1).
Acute respiratory distress syndrome (ARDS) is the most severe form of acute lung injury (ALI). Many pathogenic conditions can trigger ARDS, including sepsis, gastric juice aspiration, pancreatitis, trauma, and pathogens such as Anthrax, Spanish flu virus, severe acute respiratory syndrome coronavirus (SARS‐CoV) and H5N1 avian influenza virus (Rubenfeld et al. 2005). To date, no pharmacological therapies have been developed to improve the clinical outcome of ARDS in large‐scale clinical trials, and the mortality associated with ARDS remains very high despite modern intensive care medicine (Ware, 2006). In 2003, a new disease termed ‘severe acute respiratory syndrome (SARS)’ caused by a novel coronavirus called SARS‐CoV spread quickly throughout the world, causing more than 800 deaths and severely disrupting socioeconomic life (http://www.who.int/csr/sars/en/WHOconsensus.pdf). The major cause of death in SARS was the development of ALI and ARDS. Interestingly, ACE2 was identified as a receptor for SARS infections in vitro (Li et al. 2003), and our own studies for the first time confirmed that ace2 knockout mice are protected from SARS infections in vivo (Kuba et al. 2005). These infection studies unequivocally showed that ACE2 is the key SARS‐CoV receptor in vivo. Importantly, SARS‐CoV infections and SARS Spike protein downregulate ACE2 expression, and ACE2 protects murine lungs from severe acute injury (Imai et al. 2005). Notably, ARDS in mice can be attenuated by blocking the renin–angiotensin pathway or by injection of catalytically active recombinant human ACE2 protein (Imai et al. 2005).
Angiotensin‐converting enzyme polymorphisms and ARDS
Factors predicting the onset or severity of ARDS are poorly defined. The relatively low incidence of ARDS in the relatively large group of patients at risk, however, suggests that genetic factors must contribute to the progression of ARDS. Moreover, large individual differences in plasma ACE concentrations have been reported among individuals, whereas the ACE plasma levels tend to be similar within families (Cambien et al. 1988). The human ACE gene (dcp1) is located on chromosome 17q23 and contains a restriction fragment length polymorphism within the coding sequence of intron 16 defined by the presence (insertion, I) or absence (deletion, D) of a 287 bp repeat. This polymorphism determines function, and the human ACE2 D allele confers increased ACE activity (Rigat et al. 1990). Importantly, recent cohort studies in ARDS patients showed a marked correlation between the ACE I/D polymorphism and the susceptibility and mortality of ARDS (Marshall et al. 2002). The D/D genotype was significantly increased in patients with ARDS compared with control patients with non‐ARDS respiratory failure ventilated in the intensive care unit (ICU), ICU patients undergoing coronary artery bypass grafting and individuals from a general population group. In addition, the ACE D/D allele correlated with mortality in the ARDS group. Another study showed that patients carrying the ACE I/I genotype have a significantly increased survival rate (Jerng et al. 2006). These data implicate genetic factors in susceptibility, progression and lethality of ARDS in human patients.
Angiotensin‐converting enzyme 2 protects from ARDS
Our group investigated the role of ACE2 in ALI/ARDS by using ace2 knockout mice. In three different ARDS models (acid aspiration‐induced ARDS, endotoxin‐induced ARDS and peritoneal sepsis‐induced ARDS), ace2 knockout mice show very severe disease compared with wild‐type mice (Imai et al. 2005). Loss of ACE2 expression in mutant mice resulted in enhanced vascular permeability, increased lung oedema, neutrophil accumulation and worsened lung function. Importantly, treatment with catalytically active recombinant ACE2 protein improved the symptoms of acute lung injury in wild‐type mice, as well as in ace2 knockout mice (Imai et al. 2005). Thus, ACE2 plays a protective role in acute lung injury. Mechanistically, the negative regulation of Ang II levels by ACE2 account, in part, for the protective function of ACE2 in ARDS. For example, AT1R inhibitor treatment or additional ace gene deficiency on an ace2 knockout background rescues the severe phenotype of ace2 single‐mutant mice in acute lung injury (Imai et al. 2005). In addition, ace knockout mice and at1a receptor knockout mice showed improved symptoms of acute lung injury (Imai et al. 2005). Therefore, in acute lung injury, ACE, Ang II, and AT1R function as lung‐injury‐promoting factors, while ACE2 protects from lung injury (Fig. 2).
Figure 2.
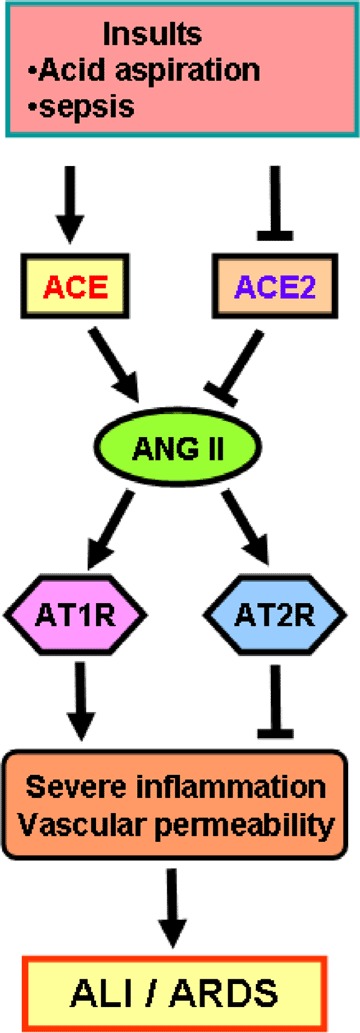
Schematic diagram of the proposed role of the renin–angiotensin system in development of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) Upon insults such as acid aspiration and sepsis, the generation of Ang II from Ang I is mediated by ACE. Angiotensin II contributes to acute lung failure through stimulation of the AT1R, while ACE2 and AT2R negatively regulate this pathway and protect from acute lung failure. However, additional ACE2‐regulated but Ang II‐independent pathways seem to contribute to ALI/ARDS.
Angiotensin‐converting enzyme 2 and SARS infections: a rational explanation for a killer virus?
Within months after the identification of the SARS‐CoV genome (Marra et al. 2003; Rota et al. 2003), ACE2 was identified as a potential receptor in in vitro cell line studies (Li et al. 2003). In these experiments, ACE2 protein was immunoprecipitated from cell lysates susceptible to SARS‐CoV infection using the recombinant Spike protein of SARS‐CoV (Li et al. 2003). Angiotensin‐converting enzyme 2 can bind to SARS‐CoV Spike, and this binding supports ‘syncytia formation’, the fusion of Spike protein‐expressing cells into large multinucleated cells that can also be observed in clinical SARS infections. Expression of ACE2 in cells that are usually not susceptible to SARS‐CoV infection allows entry of live virus into the cell (Li et al. 2003); however, it was unclear whether ACE2 is required for a natural in vivo SARS infection.
Using a SARS infection model in ace2 knockout mice, our group was able to show that ACE2 is indeed essential for SARS infections in vivo (Kuba et al. 2005). When ace2 knockout mice are infected with the SARS coronavirus, they were resistant to virus infection (Kuba et al. 2005). Virus titres from the lung tissues of infected ace2 knockout mice were 105‐fold lower than those from the lungs of wild‐type mice (Kuba et al. 2005). Moreover, none of lung histology from ace2 knockout mice challenged with SARS coronavirus showed signs of inflammation (Kuba et al. 2005), whereas some (but not all) SARS‐infected wild‐type mice displayed mild inflammation as defined by leukocyte infiltration (Hogan et al. 2004; Yang et al. 2004; Kuba et al. 2005). Thus, ACE2 is an essential receptor for SARS infections in vivo. One important question in SARS pathology is why infections with the SARS‐CoV trigger severe lung disease with such high mortality whereas infections with other coronaviruses result in rather mild disease. In addition, severe SARS infections are determined by the burden of viral replication as well as by the consequences of the host immune response. Our own studies have implicated the renin–angiotensin system in SARS pathogenesis: first, ACE2 is a critical SARS receptor in vivo; and second, ACE2 and other components of the RAS play a central role in controlling the severity of acute lung failure once the disease process has been initiated (Imai et al. 2005; Fig. 2).
Intriguingly, ACE2 expression in lungs is markedly downregulated in wild‐type mice infected with SARS‐CoV (Kuba et al. 2005). Similarly, treatment with recombinant SARS Spike protein, in the absence of any other virus components, downregulates ACE2 protein expression in cell lines in vitro and in lungs in vivo (Kuba et al. 2005). Thus, SARS‐CoV‐infected or Spike protein‐treated wild‐type mice resemble ace2 knockout animals. In addition, similar to ace2 mutant mice, Spike‐treated wild‐type mice show markedly more severe pathology in acute lung injury (Kuba et al. 2005). As a consequence, Ang II peptide levels were increased in Spike‐treated mice and these mice showed worsened ARDS symptoms that could be partly reversed by pharmacological inhibition of the AT1R. By contrast, SARS Spike challenge did not affect the ARDS symptoms in ace2 knockout mice (Kuba et al. 2005). Thus, the downregulation of ACE2 expression in SARS‐CoV infections might play a causal role in SARS pathogenesis, especially in disease progression to ARDS. These results also provide a rational explanation for the severe disease pathology in SARS patients (Fig. 3).
Figure 3.
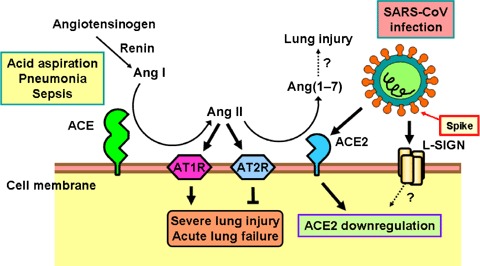
Schematic diagram of the role of the renin–angiotensin system in acute lung failure and proposed SARS‐CoV action In acute lung injury, such as acid aspiration, pneumonia or sepsis, the generation of Ang II from Ang I is enhanced by ACE, and Ang II induces acute lung failure through stimulation of the AT1R, while ACE2 and AT2R negatively regulate this pathway and protect from acute lung failure. In contrast, SARS‐CoV infection is mediated through binding of the SARS Spike protein to ACE2 or liver/lymph node‐specific intercellular adhesion molecule‐3‐grabbing nonintegrin (L‐SIGN) and downregulates the protective molecule ACE2, thus leading to severe lung injury and acute lung failure.
Pulmonary oedema
The pulmonary circulation is a potentially important target for the renin–angiotensin system in the lung. For instance, Ang II via AT1R induces pulmonary vasoconstriction in response to hypoxia, suggesting important roles of Ang II and AT1R in elevating pulmonary vascular tone that can result in pulmonary oedema (Kiely et al. 1997). Also, infusion of Ang I (Xu et al. 1998) or Ang II (Yamamoto et al. 1997) can produce pulmonary oedema independent of catecholamine release. In addition to increased vascular tone and a subsequent hydrostatic oedema formation, accumulating data suggest that Ang II also increases vascular permeability via AT1 receptors, whereas stimulation of AT2 receptors may exert opposite effects (Victorino et al. 2002). Several mediators have been implicated in Ang II‐regulated vascular permeability changes, including the eicosanoids leukotriene C4, prostaglandin E2 and prostaglandin I2, and vascular permeability factor (Suzuki et al. 2003). Few studies have addressed whether the RAS is involved in the increased vascular permeability, one of the hallmarks in the pathogenesis of ARDS. Our study demonstrated that loss of ACE2 expression results in increased vascular permeability using infusion of high‐molecular weight dextran or Evans Blue dye injections as an in vivo indicator of albumin leakage in mice (Imai et al. 2005). This vascular permeability was significantly attenuated in the lungs of AT1R knockout mice (Imai et al. 2005). These data indicate that loss of ACE2 expression and locally increased Ang II production trigger leakage of pulmonary blood vessels through AT1 receptor stimulation in ARDS (Imai et al. 2005; Fig. 2).
Additional ACE2 targets in ARDS
Both ACE and ACE2 are unspecific transmembrane metalloproteases and cleave additional substrates that, independent of Ang II levels and AT1R signalling, might also play important roles in ACE/ACE2‐regulated ARDS. One of the ACE targets is bradykinin, whereas ACE2 can catalyse the cleavage of bradykinin metabolites (Warner et al. 2004). Bradykinin is the key effecter in the kallikrein–kinin system and also functions as a major proinflammatory mediator. Bradykinin mediates its biological effects via B1 and B2 receptors and is degraded by two main kinases, which are ACE and neutral endopeptidase (Warner et al. 2004). The in vivo effects mediated via B1 or B2 receptors are still poorly characterized, but it appears that B2 receptors mediate most of the known effects of bradykinin, including its antiproliferative, anti‐oxidant and antithrombotic effects (Marceau & Regoli, 2004). Expression of B1 receptors can be also induced by inflammatory cytokines (Schanstra et al. 2000), albeit the role of bradykinin and B1 receptors in Ang II‐induced inflammation remains unclear.
In contrast to ACE, ACE2 does not metabolize bradykinin, but catalytically inactivates the bradykinin metabolites [des‐Arg9]‐bradykinin and lys[des‐Arg9]‐bradykinin (Donoghue et al. 2000; Vickers et al. 2002). Angiotensin‐converting enzyme 2 can also remove the C‐terminal residue from apelin and other vasoactive peptides, such as neurotensin and the neurotensin‐related peptide kinetensin. Moreover, the opioid peptides dynorphin A(1–13) and β‐casamorphin are ACE2 substrates in in vitro assays. Thus, although many ACE and ACE2 functions have been attributed to the regulation of Ang II levels, Ang II itself is probably only part of the RAS story, and other substrates might play a major role in mediating ACE2 functions.
Conclusions
When ACE2 was discovered, nobody had any idea of the amazing and exciting biological journey on which we would all embark. Angiotensin‐converting enzyme 2 has now been identified as a key factor for protection from ARDS/acute lung injury and it functions as a critical SARS receptor in vivo. Since SARS Spike protein‐mediated downregulation of ACE2 appears to contribute to the severity of lung failure, these findings may explain how the SARS coronavirus has turned into a lethal virus. In addition, in an acid aspiration ARDS mouse model, strong downregulation of ACE2 protein in the injured lung was observed, while ACE expression remained unchanged. Thus, recombinant ACE2 protein, which is under advanced development, could not only be a treatment to block the spread of SARS but could also protect SARS patients from developing lung failure. Furthermore, these findings could be applied to investigation of the therapeutic efficacy of ACE2 in the ARDS that develops in other emerging lung infectious diseases, such as avian influenza A (H5N1) or other yet unknown diseases that affect lung function. We look forward to the further elucidation of the pathophysiological role of the RAS in lung failure and to the use of recombinant ACE2 protein as a novel therapy for ARDS, which affects millions of people and for which no drug treatment yet exists.
Acknowledgements
J.M.P. is supported by grants from The National Bank of Austria, The Austrian Ministry of Science and Education, IMBA, and EUGeneHeart. Y.I. is supported by EUGeneHeart and the global COE program in Japan. K.K. is supported by the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan and Takeda Science Foundation.
References
- Cambien F, Alhenc‐Gelas F, Herbeth B, Andre JL, Rakotovao R, Gonzales MF et al (1988). Familial resemblance of plasma angiotensin‐converting enzyme level: the Nancy Study. Am J Hum Genet 43, 774–780. [PMC free article] [PubMed] [Google Scholar]
- Corvol P, Williams TA & Soubrier F (1995). Peptidyl dipeptidase A: angiotensin I‐converting enzyme. Methods Enzymol 248, 283–305. [DOI] [PubMed] [Google Scholar]
- Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE et al (2002). Angiotensin‐converting enzyme 2 is an essential regulator of heart function. Nature 417, 822–828. [DOI] [PubMed] [Google Scholar]
- Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N et al (2000). A novel angiotensin‐converting enzyme‐related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res 87, E1–E9. [DOI] [PubMed] [Google Scholar]
- Douglas GC, O'Bryan MK, Hedger MP, Lee DK, Yarski MA, Smith AI & Lew RA (2004). The novel angiotensin‐converting enzyme (ACE) homolog, ACE2, is selectively expressed by adult Leydig cells of the testis. Endocrinology 145, 4703–4711. [DOI] [PubMed] [Google Scholar]
- Ferrario CM (1990). The renin‐angiotensin system: importance in physiology and pathology. J Cardiovasc Pharmacol 15(Suppl. 3), S1–S5. [PubMed] [Google Scholar]
- Fleming I, Kohlstedt K & Busse R (2006). The tissue renin‐angiotensin system and intracellular signalling. Curr Opin Nephrol Hypertens 15, 8–13. [DOI] [PubMed] [Google Scholar]
- Hogan RJ, Gao G, Rowe T, Bell P, Flieder D, Paragas J et al (2004). Resolution of primary severe acute respiratory syndrome‐associated coronavirus infection requires Stat1. J Virol 78, 11416–11421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B et al (2005). Angiotensin‐converting enzyme 2 protects from severe acute lung failure. Nature 436, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerng JS, Yu CJ, Wang HC, Chen KY, Cheng SL & Yang PC (2006). Polymorphism of the angiotensin‐converting enzyme gene affects the outcome of acute respiratory distress syndrome. Crit Care Med 34, 1001–1006. [DOI] [PubMed] [Google Scholar]
- Kakar SS, Sellers JC, Devor DC, Musgrove LC & Neill JD (1992). Angiotensin II type‐1 receptor subtype cDNAs: differential tissue expression and hormonal regulation. Biochem Biophys Res Commun 183, 1090–1096. [DOI] [PubMed] [Google Scholar]
- Kiely DG, Cargill RI, Wheeldon NM, Coutie WJ & Lipworth BJ (1997). Haemodynamic and endocrine effects of type 1 angiotensin II receptor blockade in patients with hypoxaemic cor pulmonale. Cardiovasc Res 33, 201–208. [DOI] [PubMed] [Google Scholar]
- Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B et al (2005). A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus‐induced lung injury. Nat Med 11, 875–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA et al (2003). Angiotensin‐converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426, 450–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marceau F & Regoli D (2004). Bradykinin receptor ligands: therapeutic perspectives. Nat Rev Drug Discov 3, 845–852. [DOI] [PubMed] [Google Scholar]
- Marra MA, Jones SJ, Astell CR, Holt RA, Brooks‐Wilson A, Butterfield YS et al (2003). The Genome sequence of the SARS‐associated coronavirus. Science 300, 1399–1404. [DOI] [PubMed] [Google Scholar]
- Marshall RP, Webb S, Bellingan GJ, Montgomery HE, Chaudhari B, McAnulty RJ et al (2002). Angiotensin converting enzyme insertion/deletion polymorphism is associated with susceptibility and outcome in acute respiratory distress syndrome. Am J Respir Crit Care Med 166, 646–650. [DOI] [PubMed] [Google Scholar]
- Nicholls MG, Richards AM & Agarwal M (1998). The importance of the renin‐angiotensin system in cardiovascular disease. J Hum Hypertens 12, 295–299. [DOI] [PubMed] [Google Scholar]
- Rigat B, Hubert C, Alhenc‐Gelas F, Cambien F, Corvol P & Soubrier F (1990). An insertion/deletion polymorphism in the angiotensin I‐converting enzyme gene accounting for half the variance of serum enzyme levels. J Clin Invest 86, 1343–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP et al (2003). Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300, 1394–1399. [DOI] [PubMed] [Google Scholar]
- Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M et al (2005). Incidence and outcomes of acute lung injury. N Engl J Med 353, 1685–1693. [DOI] [PubMed] [Google Scholar]
- Schanstra JP, Marin‐Castano ME, Praddaude F, Tack I, Ader JL, Girolami JP et al (2000). Bradykinin B1 receptor‐mediated changes in renal hemodynamics during endotoxin‐induced inflammation. J Am Soc Nephrol 11, 1208–1215. [DOI] [PubMed] [Google Scholar]
- Skeggs LT, Dorer FE, Levine M, Lentz KE & Kahn JR (1980). The biochemistry of the renin‐angiotensin system. Adv Exp Med Biol 130, 1–27. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Ruiz‐Ortega M, Lorenzo O, Ruperez M, Esteban V & Egido J (2003). Inflammation and angiotensin II. Int J Biochem Cell Biol 35, 881–900. [DOI] [PubMed] [Google Scholar]
- Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G & Turner AJ (2000). A human homolog of angiotensin‐converting enzyme. Cloning and functional expression as a captopril‐insensitive carboxypeptidase. J Biol Chem 275, 33238–33243. [DOI] [PubMed] [Google Scholar]
- Turner AJ & Hooper NM (2002). The angiotensin‐converting enzyme gene family: genomics and pharmacology. Trends Pharmacol Sci 23, 177–183. [DOI] [PubMed] [Google Scholar]
- Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J et al (2002). Hydrolysis of biological peptides by human angiotensin‐converting enzyme‐related carboxypeptidase. J Biol Chem 277, 14838–14843. [DOI] [PubMed] [Google Scholar]
- Victorino GP, Newton CR & Curran B (2002). Effect of angiotensin II on microvascular permeability. J Surg Res 104, 77–81. [DOI] [PubMed] [Google Scholar]
- Ware LB (2006). Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Semin Respir Crit Care Med 27, 337–349. [DOI] [PubMed] [Google Scholar]
- Warner FJ, Smith AI, Hooper NM & Turner AJ (2004). Angiotensin‐converting enzyme‐2: a molecular and cellular perspective. Cell Mol Life Sci 61, 2704–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu ZH, Shimakura K, Yamamoto T, Wang LM & Mineshita S (1998). Pulmonary edema induced by angiotensin I in rats. Jpn J Pharmacol 76, 51–56. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Ohishi M, Katsuya T, Ito N, Ikushima M, Kaibe M et al (2006). Deletion of angiotensin‐converting enzyme 2 accelerates pressure overload‐induced cardiac dysfunction by increasing local angiotensin II. Hypertension 47, 718–726. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Wang L, Shimakura K, Sanaka M, Koike Y & Mineshita S (1997). Angiotensin II‐induced pulmonary edema in a rabbit model. Jpn J Pharmacol 73, 33–40. [DOI] [PubMed] [Google Scholar]
- Yang ZY, Kong WP, Huang Y, Roberts A, Murphy BR, Subbarao K & Nabel GJ (2004). A DNA vaccine induces SARS coronavirus neutralization and protective immunity in mice. Nature 428, 561–564. [DOI] [PMC free article] [PubMed] [Google Scholar]