

Abstract
Although radiation is widely used to treat cancers, resistance mechanisms often develop and involve activation of DNA repair and inhibition of apoptosis. Therefore, compounds that sensitize cancer cells to radiation via alternative cell death pathways are valuable. We report here that ferroptosis, a form of non-apoptotic cell death driven by lipid peroxidation, is partly responsible for radiation-induced cancer cell death. Moreover, we found that small molecules activating ferroptosis through system xc− inhibition or GPX4 inhibition synergize with radiation to induce ferroptosis in several cancer types by enhancing cytoplasmic lipid peroxidation, but not increasing DNA damage or caspase activation. Ferroptosis inducers synergized with cytoplasmic irradiation, but not nuclear irradiation. Finally, administration of ferroptosis inducers enhanced the anti-tumor effect of radiation in a murine xenograft model and in human patient-derived models of lung adenocarcinoma and glioma. These results suggest that ferroptosis inducers may be effective radiosensitizers that can expand the efficacy and range of indications for radiation therapy.
INTRODUCTION
Radiation therapy is one of the most important therapeutic modalities in the treatment of cancer, which provides both curative and palliative strategies for disease management1. DNA damage is thought to be the principal target of radiation, and its extent and repair are the most crucial factors determining intrinsic tumor cell death from radiation2. While radiation provides targeted local control of malignant lesions, the addition of systemic treatments is often required to provide radiosensitizing effects to tumors, as well as to manage undetected distant disease. To this effect, the combination of chemotherapy and radiation has become more common over the past 30 years3. However, tumor control still remains poor with combination chemoradiation therapy in many locally advanced cancers, such as sarcomas, gliomas and non-small cell lung cancers, which are historically considered radioresistant4, 5.
Radiation resistance mechanisms often involve activation of DNA repair pathways and inhibition of apoptosis6–8. At the same time, alternative radiation-induced cell death pathways, such as necroptosis and autophagy, have been suggested9, 10. If activated, these mechanisms might offer strategies for treating otherwise radioresistant tumors.
In addition to DNA damage, radiation also generates reactive oxygen species, which can result in oxidation of biomolecules, such as lipid oxidation11. While this effect has largely remained unexplored, a phospholipid-peroxidation-driven form of regulated cell death, ferroptosis, has recently been identified, and increasing evidence has been found to support its importance in a variety of biological and diseases processes12. Ferroptosis is induced when phospholipid-PUFA peroxidation overwhelms cellular defense systems, such as the capacity of the glutathione phospholipid peroxidase GPX4 and the CoQ10-regenerating enzyme FSP113. Ferroptosis inducers include system xc− inhibitors, which prevent cystine uptake into the cell, a building block of glutathione. By decreasing the biosynthesis of glutathione, system xc− inhibitors indirectly inhibit the lipid repair function of GPX4, which uses glutathione as a coenzyme. Direct inhibitors of GPX4 can also induce ferroptosis through this mechanism14. Numerous cancer cell lines, such as sarcomas, renal cell carcinoma, and diffuse large B-cell lymphomas, have been found to be particularly sensitive to ferroptosis14, 15; some of these cell lines are also sensitive in the context of xenograft tumor models15, 16. These data suggest the hypothesis that radiation’s anti-tumor efficacy may in some contexts be driven by triggering ferroptosis, and that ferroptosis inducers may be effective radiosensitizers.
RESULTS AND DISCUSSION
IKE and RSL3 synergize with radiation to promote clonogenic ferroptotic cell death in cell lines of multiple tumor types
We sought to determine first whether small molecule inducers of ferroptosis could synergize with radiation to promote cancer cell killing. Towards this end, we treated ferroptosis-sensitive HT-1080 fibrosarcoma cells with different doses of Cs-137 gamma radiation and either imidazole ketone erastin (IKE), a system xc− inhibitor, or Ras Synthetic Lethal 3 (RSL3), a GPX4 inhibitor, which are both small-molecule inducers of ferroptosis. We tested their ability to prevent clonogenic growth, along with DMSO-treated controls. The colony-forming ability of cells was measured, and the dose-responses to radiation of DMSO-treated, IKE-treated, and RSL3-treated groups were compared (Figure 1A). Both IKE and RSL3 significantly enhanced the effects of radiation in decreasing clonogenic survival. Given that radiation also induces apoptosis, necroptosis, and autophagy in different contexts, we also tested whether inducers of alternative cell death pathways could synergize with radiation under similar conditions. We found that the apoptosis inducers staurosporine and doxorubicin, the autophagy inducer rapamycin, and induction of necroptosis using a combination of TNFα, Z-VAD-FMK and birinapant17 were capable of only slightly enhancing radiation-induced cell death (Figure S1A), to a lesser degree than the enhancement observed using IKE and RSL3.
Figure 1. IKE and RSL3 increase radiation sensitivity in cancer cell lines through lipid peroxidation.
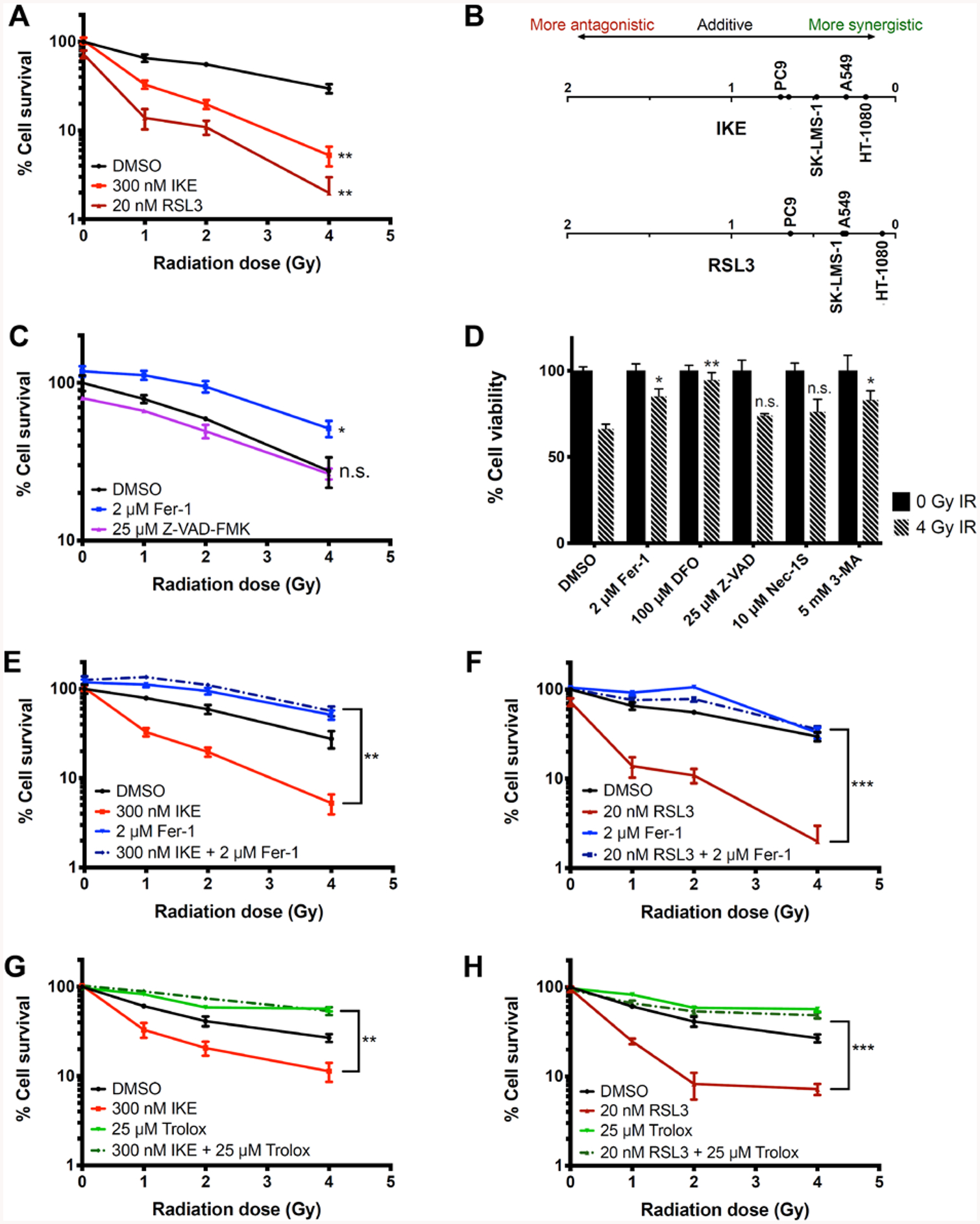
A) Dose response of HT-1080 cells treated with DMSO, IKE, or RSL3 to radiation measured by clonogenic assays. **p<0.01.
B) Coefficients of interaction between IKE (top) or RSL3 (bottom) and radiation observed for 5 tested cancer cell lines measured by clonogenic assays.
C) Dose response of HT-1080 cells treated with DMSO, ferrostatin-1, or Z-VAD-FMK to radiation measured by clonogenic assays. *p<0.05, n.s.: p>0.05.
D) Cell viability of HT-1080 cells treated with DMSO, ferrostatin-1, deferoxamine, Z-VAD-FMK, necrostatin-1S, or 3-methyladenine and co-treated with 0 or 4 Gy radiation for 24 hours. Data normalized to 0 Gy unirradiated controls for each treatment group. **p<0.01, *p<0.05, n.s.: p>0.05.
E) Dose response of HT-1080 cells treated with DMSO, IKE, ferrostatin-1, or IKE and ferrostatin-1 measured by clonogenic assays. **p<0.01. Significance is calculated between the group treated with ferroptosis inducer and the group co-treated with ferroptosis inducer and ferroptosis inhibitor in E), F), G) and H).
F) Dose response of HT-1080 cells treated with DMSO, RSL3, ferrostatin-1, or RSL3 and ferrostatin-1 measured by clonogenic assays. ***p<0.001.
G) Dose response of HT-1080 cells treated with DMSO, IKE, Trolox, or IKE and Trolox measured by clonogenic assays. **p<0.01.
H) Dose response of HT-1080 cells treated with DMSO, RSL3, Trolox, or RSL3 and Trolox measured by clonogenic assays. ***p<0.001.
Data are plotted as mean ± SEM; n = 3 side-by-side experiments for A), C), D), E), F), G), H). Three biologically independent experiments were performed with similar results.
The coefficient of drug interaction (CDI), used to compute interaction between two drugs, was used to quantify synergy between cell death inducers and radiation according to the formula CDI = AB / (A × B), where AB is the surviving fraction of the combination treatment, and A and B are the surviving fractions of the individual treatments. CDI < 1 indicates synergy, CDI = 1 indicates additivity, and CDI > 1 indicates antagonism18 (Table S1). The results indicate that ferroptosis inducers synergize with radiation to a greater degree than other compounds in HT-1080 cells, and suggest that, although a variety of mechanisms may participate in radiation-induced cell death in this model system, ferroptosis is the most pronounced.
We then tested whether cell death enhancement of radiation with ferroptosis inducers occurred across diverse tumor cell types. Using the same assay, we evaluated several cancer cell lines for synergistic cell killing with radiation and either IKE or RSL3 (Figure S1B–S1E). In addition to the initial ferroptosis-sensitive HT-1080 sarcoma cell line, glioma and lung cancer cells were evaluated, due to the clinical relevance of radiation therapy for its treatment. Enhanced cell killing was observed in all cell lines, SK-LMS-1 (uterine sarcoma), U87 (primary glioblastoma), and A549 and PC9 (lung carcinomas) when combining radiation with a ferroptosis inducer. The CDI values for each cell line were recorded at various doses of radiation and ferroptosis inducers, and the maximal CDI for each cell line was compared (Figure 1B and Table S2). The interactions between radiation and both ferroptosis inducers were synergistic for all the cell lines, ranging from CDI = 0.70 for IKE with radiation in PC9 cells to CDI = 0.09 for RSL3 with radiation in HT-1080 cells. Taken together, the results suggest that the cancer cell lines derived from radiation-sensitive tumors are synergistically killed by IKE and RSL3 and irradiation.
Radiation-induced cancer cell death is suppressed by ferroptosis inhibitors
It has been reported that radiation causes lipid peroxidation in cells11, in addition to its widely known ability to induce DNA damage. Thus, we hypothesized that cell death caused by radiation alone may partially be due to ferroptosis, particularly in contexts in which DNA damage does not induce apoptosis. To test this, we measured the effect of ferroptosis inhibitors ferrostatin-1 and deferoxamine, as well as the apoptosis inhibitor Z-VAD-FMK, on the colony-forming ability of HT-1080 cells treated with 1, 2 or 4 Gy radiation alone. In this experiment, the lipophilic radical-trapping agent and ferroptosis inhibitor ferrostatin-1 significantly rescued colony formation, whereas the apoptosis inhibitor Z-VAD-FMK did not (Figure 1C). Deferoxamine (DFO), a ferroptosis inhibitor and iron chelator, prevented cell proliferation and colony formation independent of radiation treatment, likely due to the requirement for iron for cell proliferation (data not shown). We then seeded cells more densely, and measured short term cell viability with an ATP-based luciferase assay to bypass this effect of DFO; cells treated with 4 Gy radiation for 24 h were rescued from death by co-treatment with either DFO or ferrostatin-1, but not by co-treatment with Z-VAD-FMK or with the necroptosis inhibitor necrostatin-1S (Figure 1D). The autophagy inhibitor 3-methyladenine also rescued cells in this format, suggesting that autophagy may also contribute to radiation-induced cell death in this model. Given that several autophagy-related genes are positive regulators of ferroptosis, one speculative explanation is that inhibiting autophagy also limits NCOA4-dependent ferritinophagy, therefore limiting intracellular redox-active iron availability and downregulating ferroptosis19.
We then evaluated whether the observed synergy in cell killing between radiation and ferroptosis inducers was due to enhanced ferroptosis. In this set of colony formation assays, we treated HT-1080 cells with the same doses of radiation and ferroptosis inducers, in the presence or absence of ferroptosis inhibitors ferrostatin-1 or trolox (Figure 1E–1H). Both of these lipophilic radical-trapping agents (which protect lipid membranes from oxidation) acted to suppress the synergy observed between either IKE or RSL3 and radiation. Consistent with the previous experiments, both inhibitors also partially rescued cell death induced by radiation alone, in the absence of ferroptosis inducers. These results suggest that ferroptosis and lipid peroxidation contribute to radiation-induced cell death in HT-1080 cells, and that this ferroptotic cell death can be enhanced by the addition of otherwise sublethal concentrations of IKE or RSL3.
Genetic and biochemical hallmarks of ferroptosis are observed in radiation-treated cancer cells
Based on the above results, we sought to evaluate further whether ferroptosis is a mechanism for radiation-induced cell death and IKE/RSL3-amplified death in these cells. To this end, we measured the mRNA expression level of prostaglandin-endoperoxide synthase 2 (PTGS2), a pharmacodynamic biomarker of ferroptosis15, using RT-qPCR in HT-1080 cells that were (1) radiated alone, (2) treated with 100 nM RSL3, (3) radiated and co-treated with RSL3, or (4) radiated and co-treated with 10 μM ferrostatin-1. We found that after 24 hours, PTGS2 mRNA was significantly induced in cells that were treated with 6 Gy radiation when compared to untreated cells (Figure 2A). Treating cells with ferrostatin-1 in combination with radiation reversed this induction of PTGS2. When radiation was combined with RSL3, the upregulation in PTGS2 mRNA was even further enhanced.
Figure 2. Markers of ferroptosis are elevated in HT-1080 cells treated with radiation.
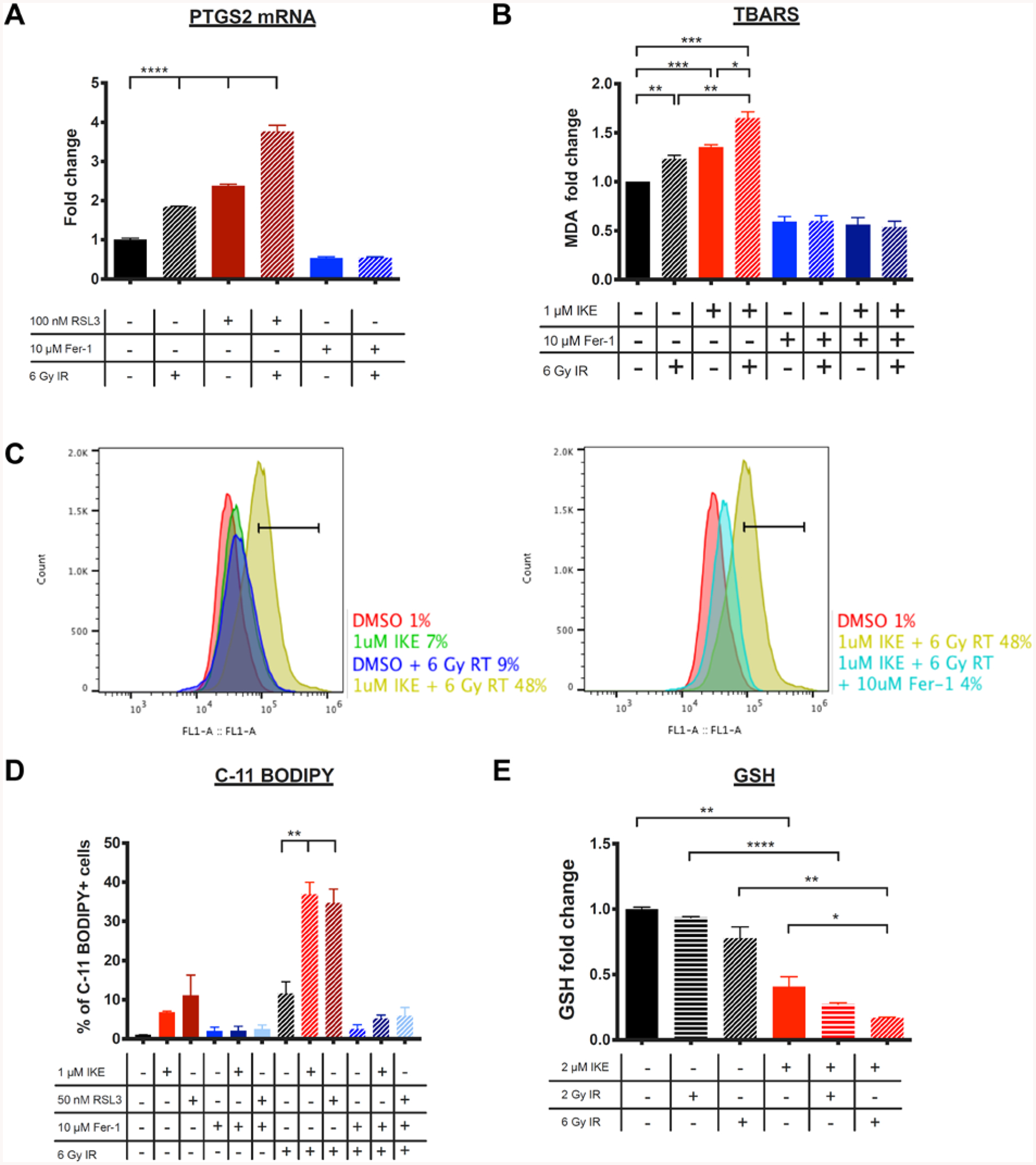
A) PTGS2 mRNA fold change measured by RT-qPCR in HT-1080 cells treated with DMSO, RSL3, or ferrostatin-1 and co-treated with 0 or 6 Gy radiation for 24 hours. ****p<0.0001.
B) MDA levels measured using the TBARS assay in HT-1080 cells treated with DMSO, IKE, or ferrostatin-1 and co-treated with 0 or 6 Gy radiation for 24 hours. ***p<0.001, **p<0.01, *p<0.05.
C) Representative histograms of HT-1080 cells treated with DMSO, IKE, or IKE + ferrostatin-1 and co-treated with 0 or 6 Gy radiation for 24 hours and stained with C-11 BODIPY measured by flow cytometry. Horizontal bars indicate C-11 BODIPY-positive cell populations.
D) C11-BODIPY staining of HT-1080 cells treated with ferroptosis modulators and co-treated with 0 or 6 Gy radiation measured by flow cytometry. **p<0.01.
E) Glutathione (GSH) level is detected in HT-1080 cells treated with DMSO or IKE and co-treated with radiation for 24 hours using a fluorometric assay. ****p<0.0001, **p<0.01, *p<0.05, n.s.: p>0.05.
Data are plotted as mean ± SEM; n = 3 technical replicates for A), B), D) and E). Three biologically independent experiments were performed with similar results.
Next, we sought to test the effects of radiation on cell membrane lipid peroxidation in a ferroptosis-sensitive cell context. We quantified levels of malondialdehyde (MDA), a biomarker for lipid peroxidation and ferroptosis, using an assay that measures thiobarbituric acid reactive substances (TBARS)20. In this assay, thiobarbituric acid (TBA) was added to cell lysates and heated under acidic conditions to form the MDA-TBA adduct, which was measured colorimetrically. MDA levels were found to be significantly elevated in cells treated for 24 h with 1 μM IKE, 6 Gy radiation, or a combination of the two, when compared to untreated cells (Figure 2B). Cells treated with 10 μM ferrostatin-1, either in the presence or absence of radiation, showed significantly lower levels of MDA compared to control cells.
To confirm that radiation causes lipid peroxidation in these cells, lipid peroxidation was also measured with C-11 BODIPY (581/591), a membrane-targeted lipid sensor dye. Flow cytometry analysis of HT-1080 cells treated with radiation, ferroptosis inducers, or a combination of both for 24 h and stained with C11-BODIPY showed that the combination treatment of either 1 μM IKE or 50 nM RSL3 with 6 Gy radiation significantly increased C-11 BODIPY fluorescence when compared to either radiation or ferroptosis inducer alone (Figure 2C, 2D). The resulting enhancement was reversed in both cases by also co-treating the cells with ferrostatin-1.
Ferroptosis inducers have been shown to alter the availability and consumption of intracellular glutathione (GSH). Class I ferroptosis inducers, such as IKE, inhibit system xc−, the cystine/glutamate antiporter on the plasma membrane that exchanges intracellular glutamate and extracellular cystine12, 14, 21. Cystine taken up by system xc− is reduced to cysteine, a building block in the biosynthesis of glutathione. The glutathione-depleting effect of IKE is thought to be its main mechanism of action that drives ferroptosis. Using a fluorometric GSH probe, we observed that treatment with 2 or 6 Gy radiation for 24 h depleted GSH in a dose-dependent manner in HT-1080 cells (Figure 2E). In addition, levels of glutathione further decreased when irradiated cells were co-treated with 2 μM IKE, suggesting that the two processes work in a cooperative fashion to deplete GSH. Indeed, the decrease in colony formation of HT-1080 cells following 2 or 4 Gy radiation was rescued by either glutathione methyl ester, or N-acetylcysteine, which is a biological precursor to glutathione (Figure S2). This finding provides a potential mechanism by which radiation and IKE act together to cause increased cell death.
IKE and RSL3 do not enhance radiation-induced DNA-damage signaling
Mechanisms of the cellular lethality from radiation are thought to be mainly derived from the downstream caspase-dependent apoptosis induced by DNA damage, including complex double-strand DNA breaks2, 22. Therefore, we wanted to determine if ferroptosis inducers and radiation induce DNA damage or affect DNA repair. We evaluated the extent of these effects of radiation by measuring DNA breaks and caspase activation in HT-1080 cells co-treated with radiation and ferroptosis inducers. Immunofluorescence staining of γH2AX, a marker for double-strand DNA (dsDNA) damage and repair, was performed in cells treated with 10 μM IKE, 1 μM RSL3, or 10 μM ferrostatin-1 along with DMSO control (Figures 3A, 3B). The cells were either treated with radiation (6 Gy) or not irradiated (0 Gy) as a control. After 30 minutes, numerous γH2AX foci were present in irradiated cells, but absent in control cells, suggesting that, as expected, radiation at this dose caused significant DNA damage.
Figure 3. IKE and RSL3 enhance radiation-induced cell death in HT-1080 cells through mechanisms independent of DNA damage or apoptosis.
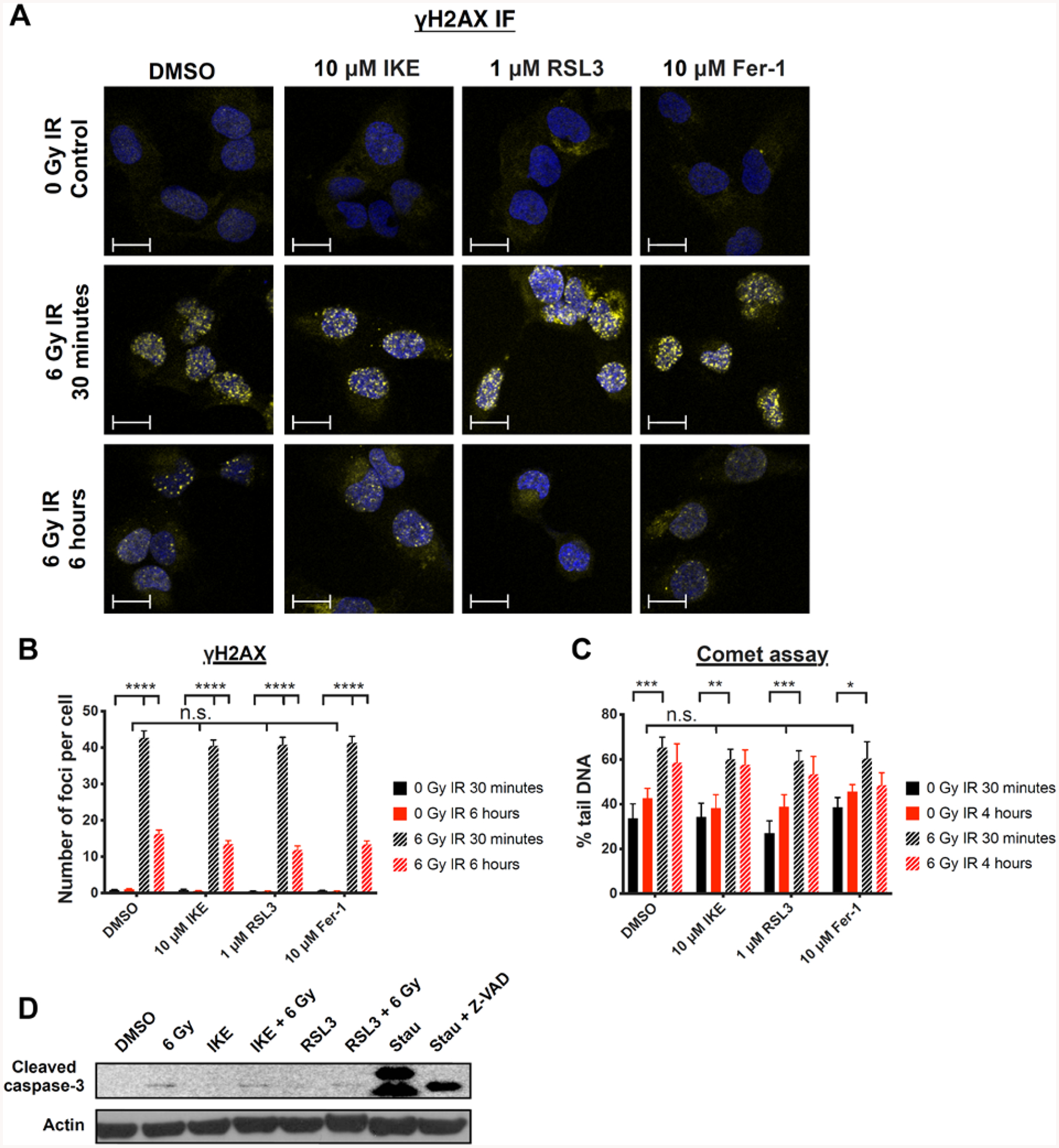
A) Representative images of γH2AX immunofluorescence staining in HT-1080 cells treated with DMSO, IKE, RSL3, or ferrostatin-1 and co-treated with 0 or 6 Gy radiation for 30 minutes or 6 hours. Blue: DAPI; Yellow: γH2AX-FITC. Scale bar, 10 μm.
B) Quantification of γH2AX immunofluorescence staining in HT-1080 cells treated with DMSO, IKE, RSL3, or ferrostatin-1 and co-treated with 0 or 6 Gy radiation for 30 minutes or 6 hours. ****p<0.0001, n.s.: p>0.05.
C) Quantification of percent tail DNA in HT-1080 cells using the comet assay. Cells were treated with DMSO, IKE, RSL3 or ferrostatin-1 and co-treated with 0 or 6 Gy radiation for 30 minutes or 4 hours. ***p<0.001, **p<0.01, *p<0.05, n.s.: p>0.05.
D) Western blot of cleaved caspase-3 in HT-1080 cells treated with DMSO, 1 μM IKE, 50 nM RSL3 and co-treated with 0 or 6 Gy radiation for 24 hours. Cells treated with 500 nM staurosporine and 500 nM staurosporine + 100 μM Z-VAD-FMK were included as positive and negative controls.
Data are plotted as mean ± SEM. Three biologically independent experiments were performed with similar results for B), C) and D).
However, pharmacological modulators of ferroptosis did not affect the number of observed γH2AX foci in any of the treatment groups, indicating that ferroptosis inducers alone do not cause double-strand DNA breaks, and that this type of DNA damage does not correlate with the radiation-sensitizing effects of IKE and RSL3, nor with the rescuing effect of ferrostatin-1, towards radiation-induced cell death. An experiment was then performed at 6 h post-treatment, at which point the majority of the γH2AX foci in cells treated with radiation alone disappeared, presumably due to DNA repair. Similarly, no differences were observed between the irradiated groups treated with the vehicle DMSO and those treated with IKE, RSL3, or ferrostatin-1, which suggested that DNA repair was not delayed by the co-treatment with ferroptosis inducers, nor enhanced by the co-treatment with ferrostatin-1. Treatment with IKE or RSL3 alone for 6 h did not result in γH2AX foci formation, with results similar to the 30-minute treatment. These results demonstrate that double-stranded DNA breaks do not correlate with the effects of ferroptosis inducers on cell viability in HT-1080 cells.
To test for other forms of DNA damage that cannot be detected by the γH2AX assay, we performed a comet assay, which detects single strand DNA damage in addition to double strand breaks, in HT-1080 cells treated under the same conditions. After 30 minutes, we observed a significant difference in percent of tail DNA between irradiated and unirradiated groups, demonstrating that DNA breaks had occurred following radiation treatment. We did not detect a significant increase in DNA single strand damage in cells treated with IKE or RSL3 alone, and no significant enhancement of DNA damage when IKE or RSL3 was combined with radiation, even when a proportion of cells had started to die at the 4-hour timepoint (Figure 3C). We also did not observe a significant protective effect of ferrostatin-1 towards radiation-induced DNA single strand damage. These results again reinforce the conclusion that although DNA damage occurs in HT-1080 cells exposed to radiation, it is not related to the synergistic cell death observed during co-treatment with ferroptosis inducers.
DNA damage is a potent inducer of apoptosis. Therefore, we also tested for the presence of radiation-induced apoptosis by measuring levels of cleaved caspase-3 in HT-1080 cells treated with 6 Gy radiation, or with IKE or RSL3, or with a combination of radiation plus ferroptosis inducer, for 24 h (Figure 3D). Levels of cleaved caspase-3 were minimally elevated in cells treated with radiation compared with those of non-irradiated cells, and the addition of ferroptosis inducers did not further increase the amount of cleaved caspase-3. Treatment with ferroptosis inducers alone did not induce detectable cleavage of caspase-3, as previously reported23. In contrast, the pro-apoptosis inducer staurosporine, used as a positive control at 500 nM for 6 h, induced cell death along with levels of caspase-3 cleavage, shown by bands at 17 and 19-kDa. When these cells were co-treated with staurosporine and 100 μM Z-VAD-FMK, a pan caspase inhibitor, the cells were rescued from staurosporine-induced cell death. Despite this, some quantity of 17-kDa cleaved caspase-3 was still detectable in the Z-VAD-FMK-treated sample. To further check for potential radiation-induced apoptosis in this model at a later time point, we attempted to rescue the effects of radiation using 100 μM Z-VAD-FMK after 48, 72, and 96 h (Figure S3). However, no significant rescue of cell viability was observed at any of these time points.
These findings suggest that ferroptosis driven by lipid peroxidation, not DNA damage or apoptosis, is the predominant radiosensitizing mechanism of ferroptosis inducers in HT-1080 cells.
Untargeted lipidomics reveals molecular features of ferroptosis in cells co-treated with IKE and radiation
To further probe the effects of radiation on cellular lipid composition and metabolism in cells that are ferroptosis-sensitive, we performed an ultra-performance liquid chromatography coupled to quadrupole- time-of-flight-mass-spectrometry-(UPLC-q-ToF MS)-based untargeted lipidomics analysis of HT-1080 cells treated with 0 or 6 Gy radiation for 24 h, and co-treated with either DMSO vehicle or 5 μM IKE for 12 h. Untargeted UPLC-MS analyses of the samples resulted in the detection of 1,304 and 561 features in the positive and negative electrospray ionization (ESI) modes, respectively. Unsupervised principal component analysis of the detected lipid features in both ESI modes showed clear clustering and separation among the groups (Figure 4A). Using two-way ANOVA (FDR corrected p value < 0.05 for both IKE-treated and IR-treated samples when compared to control samples), we found 18 lipid ions in the positive and 10 lipid ions the negative ESI modes whose abundances changed significantly among the groups (Table S3). By integrating the annotated lipid ions in both modes, we found 17 unique lipid species, including one free fatty acid (FA 16:1), 10 lysophospholipids (LysoPLs) and 6 diacylglycerols (DAGs), that increased significantly in cells treated with IKE or radiation, with even larger increases when IKE and radiation were combined (Figure 4B). Lysophospholipids, molecules generated following PUFA-containing phospholipid peroxidation by enzymatic cleavage of the oxidized PUFA tail, have been implicated in oxidative stress and accumulate during treatment with ferroptosis inducers16, 24, 25. Among these, lysophosphatidylinositol (LysoPI) 18:1 (interaction p value=0.01) and lysophosphatidylethanolamine (LysoPE) 18:1 (p=0.03) in particular had significantly interacting synergistic effects between IKE and radiation. Furthermore, the significantly elevated levels of diacylglycerols may have resulted from hydrolysis of triacylglycerols, which are enriched in ferroptosis-sensitive cell states of clear-cell carcinoma and have been shown to be accumulated by IKE in cell culture models of diffuse large B-cell lymphoma16, 26. Of these, DAG 16:0_16:1 also displayed significant interaction between IKE and radiation (p<0.05).
Figure 4. Untargeted lipidomic study reveals enhanced ferroptosis lipid signatures in cells co-treated with IKE and radiation in HT-1080 cells.
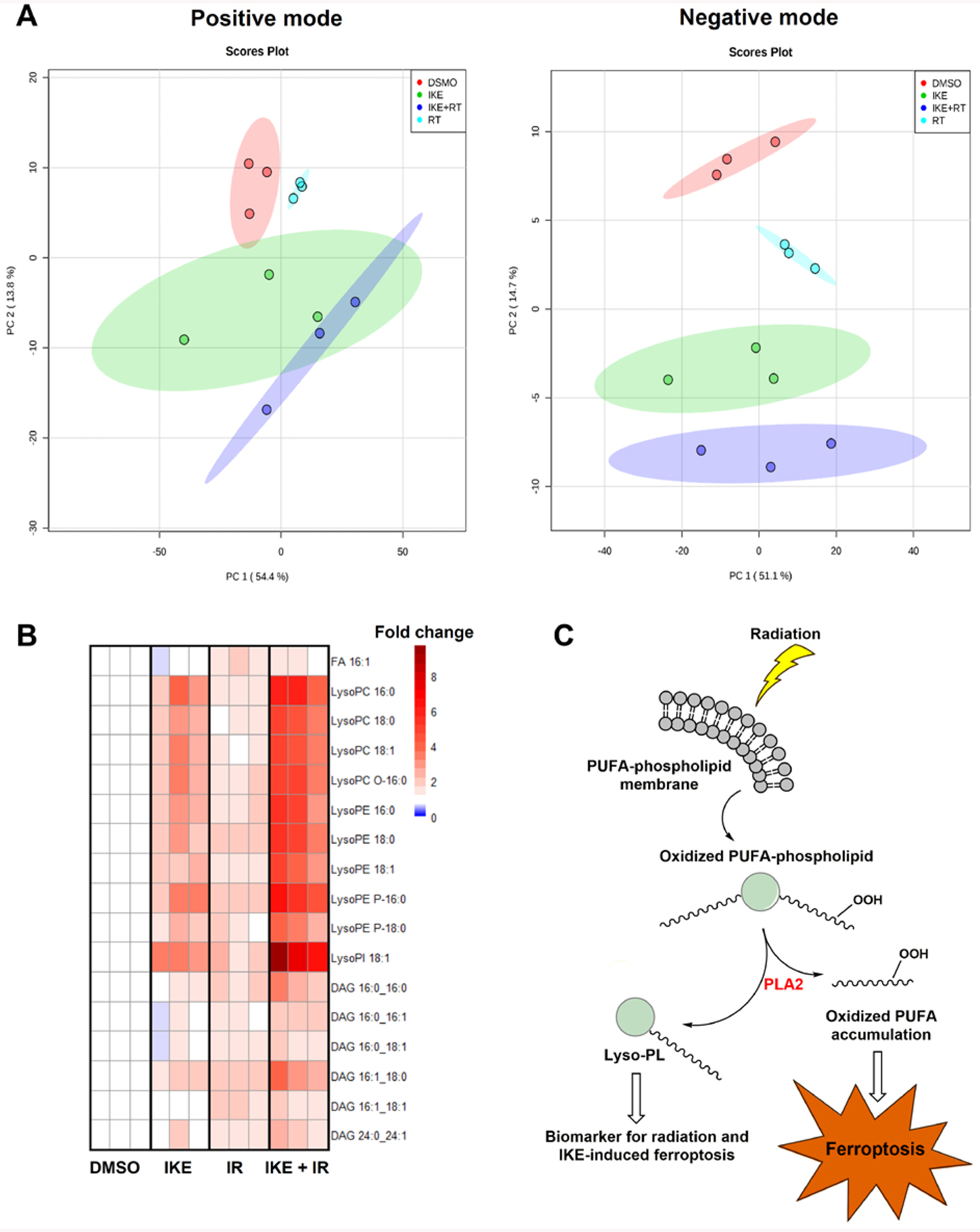
A) Principal component analysis of the extracted lipid features in samples treated with DMSO or 5 μM IKE for 12 hours, with or without 6 Gy radiation for 24 hours, in both positive and negative electrospray ionization modes.
B) Fold change heatmap of significantly changed lipid features from both IKE treatment and radiation treatment determined by two-way ANOVA (FDR corrected p value < 0.05) combined from both positive and negative ionization modes. Blue indicates decreased abundance compared to DMSO-treated controls (fold changes between 0.3 and 0.8); white indicates no change (fold changes between 0.8 and 1.2); red indicates increased abundance (fold changes between 1.2 and 10). n = 3 biologically independent samples. Abbreviations: FA, fatty acid; LysoPC, lysophosphatidylcholine; LysoPE, lysophosphatidylethanolamine; LysoPI, lysophosphatidylinositol; DAG, diacylglycerol.
C) Proposed model of how oxidation of membrane polyunsaturated fatty acids by IKE and radiation cause elevated lysophospholipids and cell death. Abbreviations: PUFA, polyunsaturated fatty acid; PLA2, phospholipase A2; Lyso-PL, lysophospholipid.
These results suggest that radiation-driven lipid peroxidation in ferroptosis-sensitive cells produces a downstream lipid signature similar to that produced by IKE alone, and consistent with the previous studies of cell lipidome changes during ferroptosis. In combination therapy, the oxidation of PUFA-phospholipids by radiation enhances the same effect driven by IKE, presumably potentiating ferroptosis through the accumulation of oxidized PUFAs, and producing lysophospholipids as a by-product that are as a ferroptosis biomarker (Figure 4C). However, the exact role of lysophospholipids in ferroptosis remains unexplored. We next utilized a precision charged particle microbeam to elucidate the consequence of lipid peroxidation with irradiation of sub-cellular compartments.
Targeted cytoplasmic, but not nuclear, microbeam radiation selectively synergizes with IKE and RSL3 to enhance clonogenic cell death
To further probe the mechanism by which radiation synergizes with IKE and RSL3 to cause cell death, we used a 5-MeV proton microbeam to deliver targeted radiation to either the nucleus or the cytoplasm of HT-1080 cells27. The microbeam consists of a single beam of proton radiation with a spot size of 4 micrometers that allows radiation damage to be precisely deposited at specific locations in a cell. This translates to delivery of a precise number of protons to either the cell nucleus or to the cytoplasm outside of the nucleus28, 29. The targetable nature of the microbeam allows us to distinguish the cytoplasmic effects of radiation from its nuclear effects, and test if the former is the predominant component that drives radiation-induced ferroptosis. To target the microbeam, cells were labeled with Hoechst stain and imaged. Nuclear radiation was delivered to the center of gravity of the cell nucleus, whereas cytoplasmic radiation was delivered to two sites 7 microns away from the nuclear edge along the nuclear long axis (Figure 5A). Using this method, we first established dose responses of these cells to nuclear and cytoplasmic radiation. The ED50 for clonogenic cell death was observed to be around 100 protons for nuclear radiation, and between 1,000 and 1,500 protons per site for cytoplasmic radiation (Figure S4). Compared to conventional photon radiation, these doses approximately correspond to 1 Gy to the nucleus and between 1–5 Gy to the cytoplasm, a therapeutically relevant dose range that is consistent with our previous experiments. Similar to previous reports, these results suggest that nuclear proton radiation was more lethal to cells than cytoplasmic proton radiation, presumably through direct radiation-induced damage to DNA and genome integrity30. This supports the view that the genotoxic effects of radiation are attributed mainly to direct damage to the nucleus.
Figure 5. Ferroptosis inducers synergize with cytoplasmic irradiation but not nuclear irradiation in HT-1080 cells.
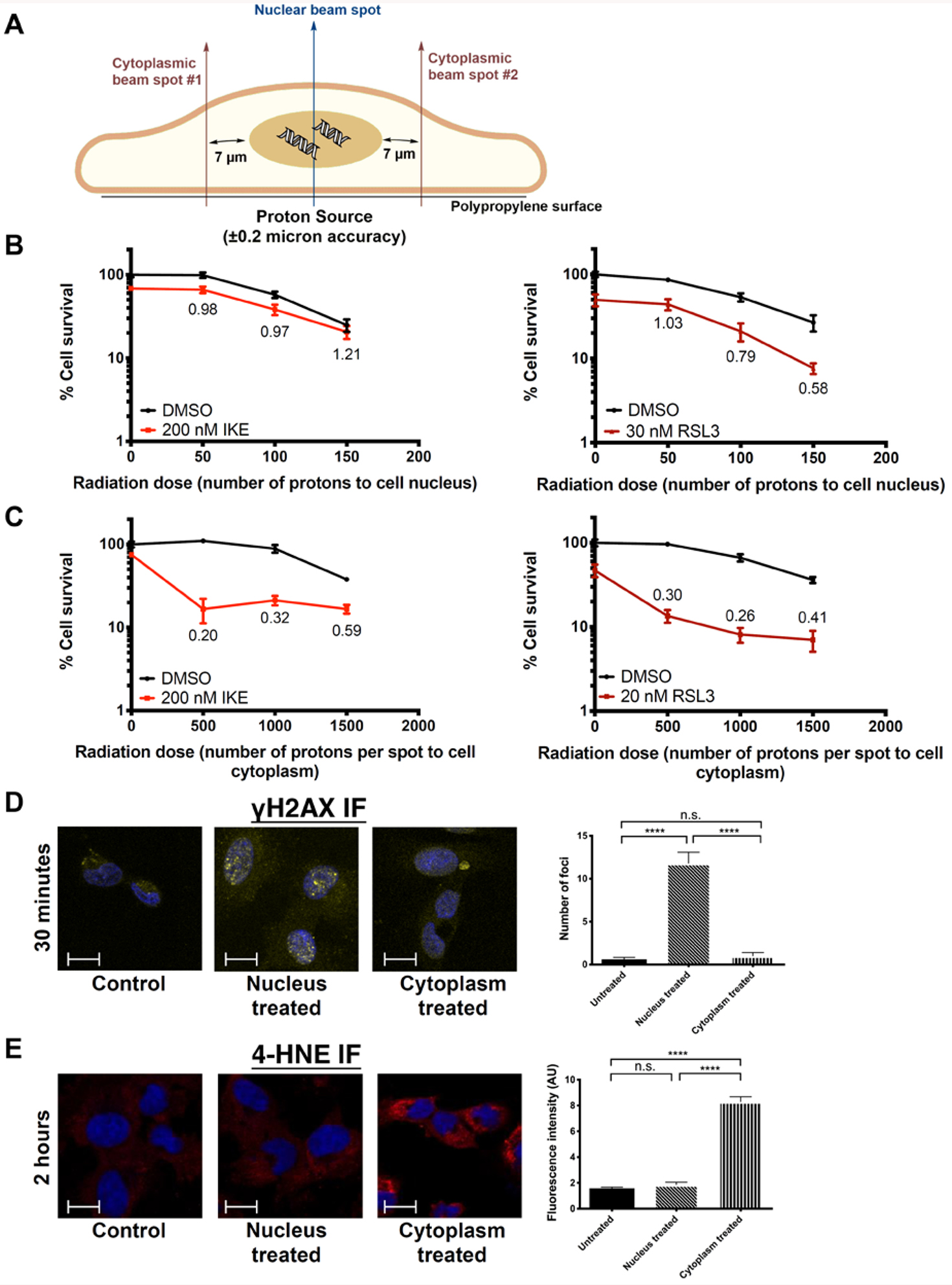
A) Diagram of microbeam setup showing locations of beam spots targeting either the nucleus or cytoplasm.
B) Clonogenic cell survival of HT-1080 cells treated with nuclear radiation and IKE or RSL3. CDI values are indicated.
C) Clonogenic cell survival of HT-1080 cells treated with cytoplasmic radiation and IKE or RSL3. CDI values are indicated.
D) Immunofluorescence staining of γH2AX in untreated cells, and cells treated with 100 protons to the nucleus or 2000 protons to the cytoplasm for 30 minutes. Blue: DAPI; Yellow: γH2AX-FITC. ****p<0.0001, n.s.: p>0.05. Scale bar, 10 μm.
E) Immunofluorescence staining of 4-HNE in untreated cells, and cells treated with 100 protons to the nucleus or 2000 protons to the cytoplasm for 2 hours. Blue: DAPI; Red: 4-HNE-Rhodamine Red. ****p<0.0001, n.s.: p>0.05. Scale bar, 10 μm.
Data are plotted as mean ± SEM; n = 3 side-by-side experiments. Three biologically independent experiments were performed with similar results.
However, when microbeam radiation was combined with inducers of ferroptosis, we observed that nuclear radiation had no synergy with IKE and RSL3, whereas cytoplasmic radiation synergized strongly with both compounds (Figure 5B, 5C). Notably, although no significant cell death was observed with 500 or 1,000 protons alone delivered per site to the cytoplasm, there was a large decrease in cell survival when irradiated cells were concurrently treated with sub-lethal doses of either of the two ferroptosis inducers, leading to synergistic CDI values between 0.2 and 0.4. By comparison, no such effect was observed when the cells were treated with nuclear irradiation. These results suggest that ferroptosis inducers sensitize cells to the effects of radiation primarily in the cytoplasm.
To further highlight the differences between nuclear and cytoplasmic microbeam radiation, and to examine if they represent two distinct forms of radiation-induced cell death, we sought to measure levels of DNA damage and lipid peroxidation in cells treated under the two conditions. To measure DNA damage, cells were treated either with 100 protons to the nucleus or 2,000 protons to each site in the cytoplasm. γH2AX immunofluorescence staining for dsDNA breaks was performed 30 minutes post-irradiation (Figure 5D). γH2AX foci were indeed present in cells treated with nuclear radiation, but absent from cells treated with cytoplasmic radiation. To examine the microbeam’s effects on lipid peroxidation, we performed immunofluorescence staining of 4-hydroxynonenal (4-HNE), a marker of lipid peroxidation, in cells with the same treatment conditions at 2 hours post-irradiation (Figure 5E). The 4-HNE signal was significantly increased in samples treated with cytoplasmic radiation relative to untreated cells, but not in those treated with nuclear radiation.
Taken together, these findings indicate that, although damage to the nucleus remains an important mechanism of radiation therapy in some contexts, inducers of ferroptosis serve to activate a distinct cell death mechanism based in the cytoplasm, which may become relevant in cancer cells that have acquired resistance to the traditional cell death and DNA damage pathways. Although it is not possible to differentially deliver cytoplasmic vs. nuclear radiation in clinical contexts, the microbeam is nevertheless a useful tool to separate the effects of radiation-induced lipid peroxidation and DNA damage and to examine how ferroptosis synergizes with the former but not the latter in different cells and tumors. Further mechanistic studies may reveal the targets contained within the cytoplasm required for this synergy, which could in principle be pharmacologically modulated for downstream clinical applications.
IKE and sorafenib enhance effects of radiation to inhibit tumor growth in a xenograft mouse model of sarcoma
We sought to evaluate the efficacy of the combined treatment regimen of radiation and ferroptosis inducer in an in vivo tumor model. Of the two ferroptosis inducers tested in cell culture, IKE was selected for in vivo studies due to its previously established stability and activity in xenograft mouse models of cancer16. Athymic nude mice were implanted with subcutaneous HT-1080 fibrosarcoma cells to form xenograft tumors. When the tumors reached an average volume of approximately 100 cubic millimeters, intraperitoneal (i.p.) injections of 40 mg/kg IKE or vehicle were delivered daily for 14 days, starting on day 0 and ending on day 13. On days two and four of IKE treatment, 0 or 6 Gy radiation was delivered to the tumor site using the Small Animal Radiation Research Platform (SARRP) system31. After two weeks, tumor volume was compared between mice treated with vehicle, IKE alone, radiation alone, or a combination of both (Figure 6A). Using two-sample t-tests, we observed a significant difference between the vehicle-treated control group and the groups treated with radiation alone; IKE alone was not strongly effective at this dose level in this model. We observed a significant further reduction in tumor volume between the single treatment groups when compared to the group treated with combination therapy, showing that IKE enhanced the effects of radiation in reducing tumor growth. Upon analysis of all groups with the two-way ANOVA test, we found statistical significance for treatment with IKE alone (p=0.03) and radiation alone (p=0.004). The two factors interacted positively with each other, although the interaction P value did not reach significance (p=0.34).
Figure 6. IKE and sorafenib, combined with stereotactic radiation therapy, suppress tumor growth in a mouse xenograft model of sarcoma and a patient-derived xenograft model of lung adenocarcinoma.
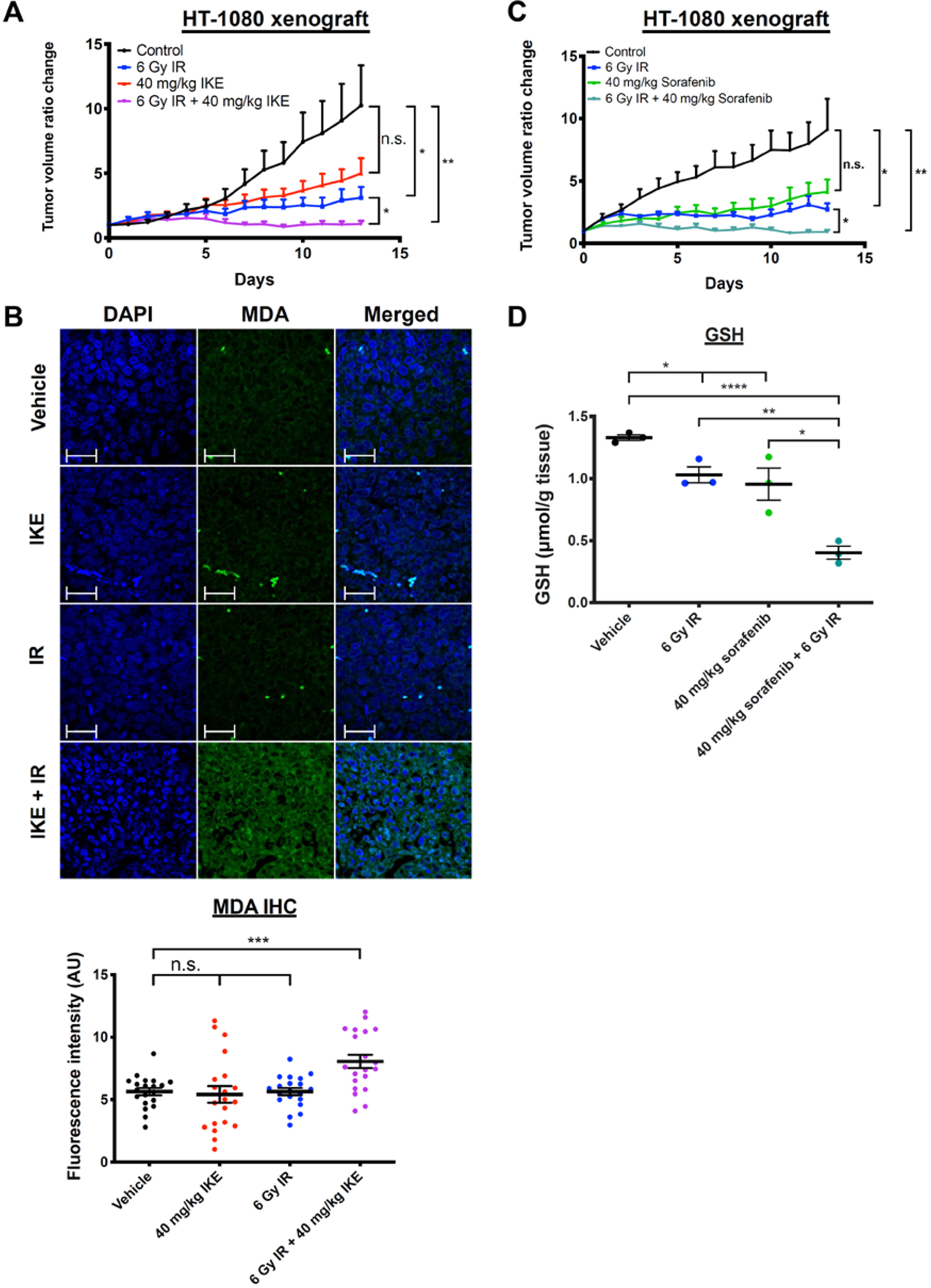
A) Tumor volume ratio change in HT-1080 xenograft tumors treated with vehicle or IKE for 14 days and co-treated with 0 or 6 Gy radiation on days 2 and 4. n = 7 or 8 mice per group. **p<0.01, *p<0.05, n.s.: p>0.05.
B) Immunofluorescence staining and quantification of MDA on paraffin-embedded tumor tissue sections measured by confocal microscopy. Blue: DAPI; Green: MDA-FITC. Scale bar, 50 μm. ***p<0.01, n.s.: p>0.05. n = 20 images with sections cut from four randomly chosen mice from each group, and five images captured from each section.
C) Tumor volume ratio change in HT-1080 xenograft tumors treated with vehicle or sorafenib for 14 days and co-treated with 0 or 6 Gy radiation on days 1 and 3. n = 4 or 5 mice per group. **p<0.01, *p<0.05, n.s.: p>0.05.
D) Glutathione (GSH) level is detected in HT-1080 xenograft tumors treated with vehicle or sorafenib for 14 days and co-treated with 0 or 6 Gy radiation on days 1 and 3, using a fluorometric assay. ****p<0.0001, **p<0.01, *p<0.05. n = 3 tumor samples from different animals per group.
Using weight loss as a measure of mice health, we did not observe any significant differences between any of the groups for the duration of the experiment (Figure S5A). This suggests that IKE and radiation and the combination were well tolerated at these dose levels for this length of exposure.
Next, we measured malondialdehyde (MDA) levels, as a biomarker for ferroptosis, in tumor tissue using immunohistochemistry on fixed and paraffin-embedded tumor samples resected at day 14 post-treatment (Figure 6B). We observed significantly elevated MDA signal in tumors treated with both IKE and radiation compared to that of tumors treated with vehicle, suggesting enhanced lipid peroxidation and ferroptosis in the co-treated tumors. No significant differences were observed between tumors treated with vehicle, IKE only, or radiation only. This again suggests synergistic pharmacodynamic effect of radiation and IKE in this tumor model.
We then sought to test the radiosensitizing effect of sorafenib, an FDA-approved chemotherapeutic drug, which also acts as an inhibitor of system xc− 21. First, the colony-forming ability of HT-1080 cells when treated with sorafenib, radiation or a combination was compared to that of untreated cells. We found that treatment with 5 μM sorafenib is synergistic with radiation at both 2 Gy (CDI=0.65) and 4 Gy (CDI=0.47), and that this effect is partially suppressible by co-treatment with ferrostatin-1 (Figure S6A). To confirm that the observed synergistic effect between sorafenib and radiation in HT-1080 cells is due to system xc− inhibition, we then measured levels of GSH in these cells treated with DMSO or sorafenib, and co-treated with 0 or 6 Gy radiation for 24 hours. Indeed, significant depletion of GSH was observed in the dual treated sample, when compared to samples treated with DMSO, sorafenib alone, or radiation alone (Figure S6B).
To test the radiosensitizing effects of sorafenib in vivo, we treated athymic nude mice implanted with HT-1080 xenograft tumors, as described above, with 40 mg/kg sorafenib tosylate or vehicle delivered i.p. daily for 14 days, with or without 6 Gy radiation delivered per mouse using SARRP on days 1 and 3. Using two-sample t-tests, we observed significant differences between the control, radiation-treated, and combination-treated groups (Figure 6C). No difference was observed between the control group and the group treated with sorafenib alone. Two-way ANOVA revealed statistical significance for treatment with sorafenib alone (p=0.03) and radiation alone (p=0.006). Similar to the experiment with IKE, the two factors interacted positively with each other but did not reach significance (p=0.18). No significant differences in weight were observed between any of the groups over the course of the experiment (Figure S5B).
We then sought to confirm that sorafenib inhibits system xc− in vivo, leading to a depletion of downstream intracellular glutathione. We measured GSH in tumor tissue taken from three randomly selected mice in each group and found significantly lower GSH in tumors treated with either radiation or sorafenib compared to those treated with vehicle. A further significant GSH depletion was observed in dual-treated tumors (Figure 6D).
Analysis of TCGA data suggests a role for SLC7A11 in radioresistance of gliomas
To determine if system xc− can potentially be an additional therapeutic target in tumors undergoing radiation therapy, we examined the association between SLC7A11 expression and methylation and clinical outcomes for all patients diagnosed with glioma in the Cancer Genome Atlas (TCGA) dataset32, 33. Comparing patient groups with low (quartile 1) or high (quartile 4) levels of SLC7A11 RNA expression, we found that high expression of SLC7A11 RNA was associated with decreased overall survival (OS) and disease-free survival (DFS, p < 0.001). Conversely, SLC7A11 DNA methylation was associated with improved OS and DFS (p < 0.001) (Figure 7A, S7). Given that our data suggests that inhibition of system xc− sensitizes glioma cells to radiation-induced ferroptosis, we would also expect that RNA expression and DNA methylation of SLC7A11 is preferentially important for patients treated with radiation over those who are not. In order to further determine whether survival outcomes were specific to radiation therapy, we conducted a subgroup analysis in the dataset, in which survival based on gene expression and methylation was stratified by receipt of radiation therapy. For patients who were not treated with radiotherapy, there was no association between survival and levels of SLC7A11 RNA expression or DNA methylation. However, in patients treated with radiation therapy, high SLC7A11 RNA expression was associated with decreased DFS (p < 0.001), while high DNA methylation was associated with improved DFS (p < 0.001). (Figure 7B). Taken together, this data supports a role for SLC7A11 in treatment resistance of gliomas towards radiation, and suggests a potential benefit for system xc− inhibition with IKE or sorafenib during radiation treatment.
Figure 7. SLC7A11 is a target for radiosensitization in human models of glioma and lung adenocarcinoma.
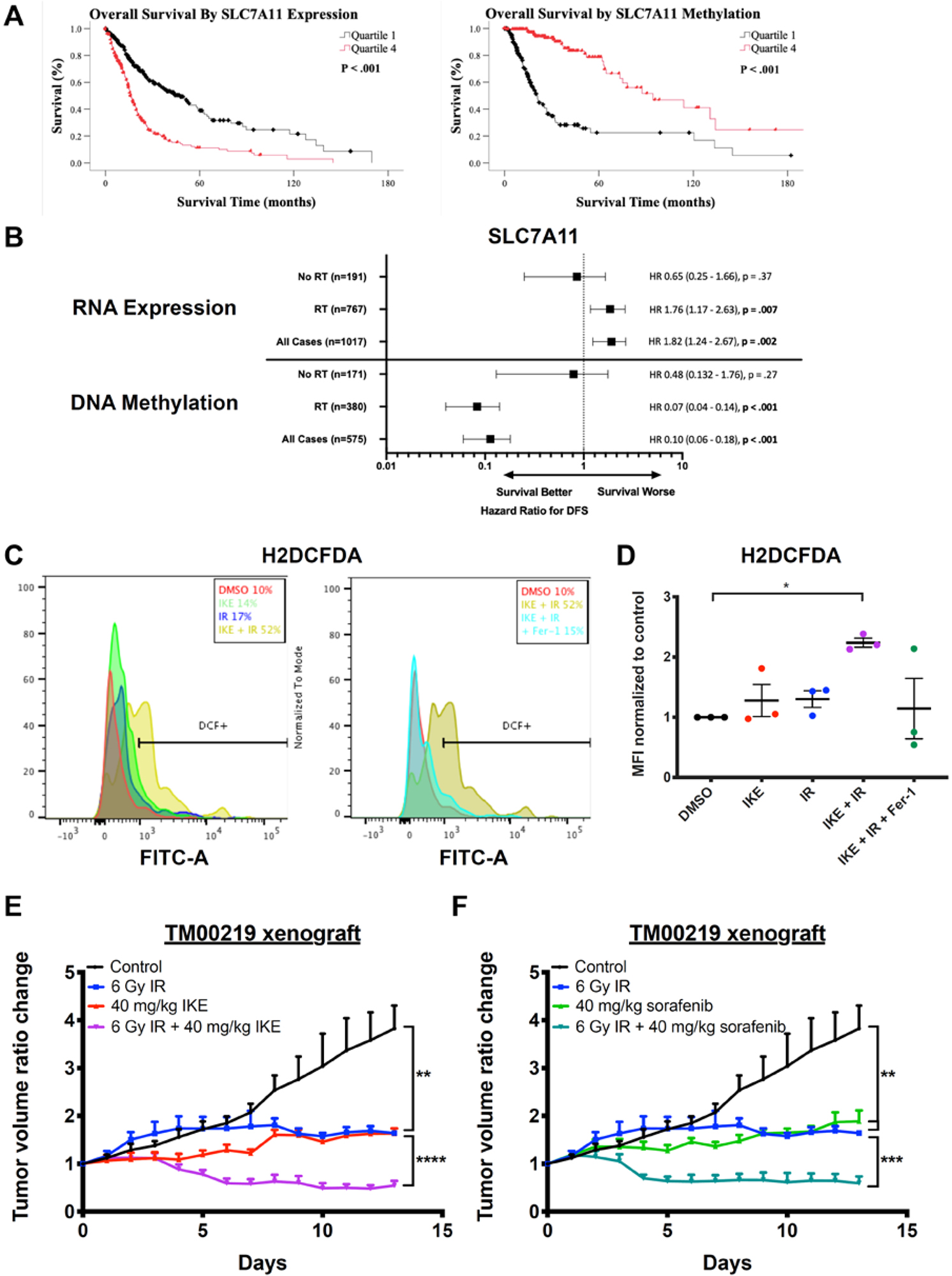
A) Kaplan-Meier survival analysis of overall survival of TCGA glioma patients in quartile 1 (low) and quartile 4 (high) of SLC7A11 RNA expression (left) or DNA methylation (right).
B) Hazard ratios for disease-free survival between patients in quartile 1 (low) and quartile 4 (high) of SLC7A11 RNA expression or DNA methylation either in the case of radiation treatment, no radiation treatment, or all cases.
C) Representative histograms of a human diffuse astrocytoma slice culture sample treated with DMSO, 10 μM IKE, or 10 μM IKE + 10 μM ferrostatin-1, co-treated with 0 or 2 Gy radiation for 24 hours, dissociated, stained with H2DCFDA, and measured by flow cytometry. Horizontal bars indicate H2DCFDA-positive cell populations.
D) H2DCFDA staining of three human glioma slice culture samples treated with DMSO, 10 μM IKE, or 10 μM IKE + 10 μM ferrostatin-1, co-treated with 0 or 2 Gy radiation for 24 hours, dissociated, stained with H2DCFDA, and measured by flow cytometry. *p<0.05. n = 3 samples from different human glioma patients.
E) Tumor volume ratio change in TM00219 patient-derived xenograft tumors treated with vehicle or IKE for 14 days and co-treated with 0 or 6 Gy radiation on day 1. n = 5 or 6 mice per group. ****p<0.0001, **p<0.01.
F) Tumor volume ratio change in TM00219 patient-derived xenograft tumors treated with vehicle or sorafenib for 14 days and co-treated with 0 or 6 Gy radiation on day 1. n = 5 or 6 mice per group. ***p<0.001, **p<0.01.
Data are plotted as mean ± SEM.
IKE combines with radiation to enhance lipid peroxidation in ex vivo slice cultures of mouse and human gliomas
To further study the potential of combining small-molecule ferroptosis inducers and radiation for human therapeutic use, we used ex vivo samples of human glioma, which have been immediately cut from freshly resected tumors and grown as organotypic slice cultures, as previously described34. De-identified patient information for these tumor samples are recorded in Table S4. The slices were then treated with 10 μM IKE, 2 Gy radiation, or a combination of both for 24 hours. Then, cells were dissociated and stained with H2DCFDA dye to measure formation of intracellular ROS using flow cytometry. A total of five human samples were tested. Of these, two glioblastomas did not demonstrate an increase in ROS generation following treatment with radiation, whereas three did respond to radiotherapy, including one high-grade oligodendroglioma and two astrocytomas. In the three responsive slice cultures, we also observed a significant enhancement of ROS generation with combination treatment when compared to control (Figure 7C,7D). The ROS accumulation was also partially suppressible by co-treating with 10 μM ferrostatin-1, indicating that part of the generated ROS originates from lipid membranes. Tumor cell viability within slices were not assessed, as the number of tumor cells embedded in each slice cannot be normalized between slices. Taken together, these experiments show that certain human gliomas may be susceptible to a combination therapy of a ferroptosis inducer and radiation. However, a mixed response to radiation among all tumors tested suggest that more experiments are needed to better understand which types of glioma might be most sensitive to the proposed treatment regimen.
IKE and sorafenib enhance effects of radiation to inhibit tumor growth in a patient-derived xenograft of lung adenocarcinoma
Finally, we tested the effect of ferroptosis-inducing system xc− inhibitors, in combination with radiation, on a patient-derived xenograft (PDX) model of lung adenocarcinoma, a type of cancer commonly treated with radiation therapy. NSG mice engrafted with a human lung adenocarcinoma tumor (TM00219) were evaluated. Intraperitoneal (i.p.) injections of 40 mg/kg IKE, 40 mg/kg sorafenib, or vehicle were delivered daily for 14 days, as described above in the HT-1080 xenograft study. On day one of treatment, 0 or 6 Gy radiation was delivered to the tumor site using the SARRP. At the conclusion of the study, we observed significant tumor growth inhibition in the combination-treated group compared to all other groups (Figure 7E, 7F). In addition, treatment with radiation alone, IKE alone, or sorafenib alone also showed significant tumor control compared to the group treated with vehicle only. This experiment demonstrates the potential applicability of this therapeutic strategy to patients whose cancers currently already receive radiation therapy as standard of care.
Traditionally, mechanisms of cellular lethality from radiation have focused on the role of clustered DNA damage, in particular double-strand DNA breaks, in the nucleus. This genotoxicity leads to downstream effects such as apoptosis and mitotic catastrophe, which are thought to be the predominant mechanisms of cancer cell death following irradiation35. Nevertheless, some prior reports have highlighted the capacity for radiation to produce hydroxyl radicals and even lipid peroxidation in cell membranes11, 36. With the framework of ferroptosis, a lipid-peroxidation-based form of regulated cell death that can be modulated by a wide arsenal of pharmacological agents and metabolic interventions, it may now be possible to enhance the radiation-induced lipid damage response to kill tumors. This alternative mechanism may be especially effective in tumors with either intrinsic or acquired resistance to the genotoxic effects of radiation, such as those with increased capacity for DNA repair or a defective apoptosis pathway.
Intriguingly, the ferroptosis-inducing molecules erastin and sulfasalazine have been shown previously to potentiate radiation in models of glioma, melanoma, and breast cancer37–39. Both of these molecules belong to class I ferroptosis inducers, which inhibit system xc− and decreases glutathione synthesis. While these studies noted lowered levels of glutathione to be the cause of the compounds’ radiosensitizing effect, the proposed mechanisms of synergy were proposed to be enhanced DNA damage and downstream apoptotic pathways. Our study is the first to link radiation to glutathione depletion resulting in lipid peroxidation and subsequent ferroptotic cell death. These studies together also suggest that altered glutathione metabolism may have the ability to activate distinct cell death mechanisms.
While its ability to deplete glutathione is one likely mechanism by which radiation synergizes with ferroptosis inducers, other possibilities may be considered. Gamma rays and X-rays, the most clinically relevant types of radiation therapy, do not damage biological molecules directly, but rather through hydroxyl radical intermediates. Polyunsaturated fatty acids (PUFAs), which are oxidized in ferroptosis, are particularly sensitive to this type of damage given their ability to stabilize a free radical in the bis-allylic position through conjugation, and are the most sensitive lipid species to destruction when exposed to high dose radiation40. Evidence to support this hypothesis includes reports from two groups that treatment of several cancer cell lines, including astrocytomas and colorectal cancers, with PUFAs resulting in enhanced cell killing by radiation41, 42. In addition, in a report identifying the role of ATM in iron metabolism and ferroptosis, the authors speculate that radiation-induced increase of intracellular iron may be induced through ATM expression, and that this increase may provide a further mechanism by which radiation can potentiate ferroptosis43. Furthermore, it was recently reported that radiotherapy-activated ATM suppresses SLC7A11, triggering ferroptosis through decreased cysteine uptake and lipid peroxidation44. Therefore, it is plausible that one of the ways in which glutathione depletion occurs in our models is due to ATM activation, although the authors agree with our view that many other mechanisms may be involved in this process. Additional studies are required to determine which of these mechanisms are the most relevant in diverse contexts.
While the upstream mechanism by which radiation oxidizes these lipids remains to be elucidated, our lipidomics data suggest that the effects of radiation on lipid species of treated cells as a whole overlap with those produced by a ferroptosis inducer such as IKE. In particular, the profound increase in lysophospholipids, a by-product of PUFA-phospholipid oxidation, has the potential to serve as a biomarker for ferroptosis induced by both radiation and IKE. For example, the levels of both LysoPI 18:1 and LysoPE 18:1 increased in both groups and showed strong interactions in the combination group, potentially acting as signatures of the synergistic effects between IKE and radiation.
The microbeam results we observed indicate that the interactions between radiation and the ferroptotic pathway occur primarily in the cytoplasm. At the same time, the lower threshold for cell killing by nuclear radiation compared to cytoplasmic radiation suggests that DNA damage is a major cell death modality of radiation. This is potentially because the charged particle radiation used for the microbeam experiments are known to have a higher Linear Energy Transfer (LET), causing more direct DNA damage when compared to gamma radiation used in cell culture experiments. Thus, the type of radiation may strongly impact the type of cell death activated, in addition to the tumor context. As ferroptosis inducers are optimized and developed as chemotherapeutic agents, these results suggest that they may be combined with radiation therapy in a variety of contexts, such as when cancers have evolved specific resistance mechanisms to DNA damage and downstream cell death modalities, such as enhanced DNA repair or deficient apoptotic machinery. In particular, the use of this combination therapy in cancers that have undergone EMT is potentially promising, as they have been identified as a resistant state susceptible to GPX4 inhibition and ferroptotic cell death45. In these cases, the synergistic effects of this therapeutic strategy may allow lower doses of radiation to be delivered, therefore reducing adverse effects of radiation in healthy tissues.
Finally, using a combination of radiation and IKE or sorafenib, we showed that synergistic tumor cell killing through ferroptosis can be extended to patient-derived models of glioma and lung cancer. Given that these two types of cancer are routinely treated with radiation therapy, our findings potentially pave the way for the first clinical trial focused on ferroptosis as an alternative cell death pathway for tumor control in a therapeutically relevant cancer type.
In summary, we report here that ferroptosis is a mechanism of radiation-induced cancer cell death, and that ferroptosis inducers act as radiosensitizers by potentiating the effects of radiation on cytoplasmic lipid peroxidation leading to cell death, in cell culture, xenograft mouse studies, and patient-derived xenografts and tumor slice cultures. These findings may open up new avenues of treatment for tumors that become resistant to conventional DNA damage and cell death pathways.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by an Emerson Collective grant to P.D.C. and B.R.S., National Cancer Institute grants R35CA209896 and P01CA087497-17 and National Institute for Neurological Disorders and Stroke grant R61NS109407 to B.R.S., an unrestricted research donation from B. Neustein to S.K.C., and a Louis V. Gerstner, Jr. Scholar Award to S.K.C. These studies used the resources of the Herbert Irving Comprehensive Cancer Center Shared Resources funded in part through Center Grant P30CA013696. The microbeam work was performed at the Radiological Research Accelerator Facility of Columbia University supported by the National Institute of Biomedical Imaging and Bioengineering (NIBIB) grant 5P41EB002033. The small animal radiation research platform was supported by the NIH Biomedical Research Support Shared Instrumentation Grant 1S10OD010631-01A1.
Footnotes
CONFLICT OF INTERESTS
B.R.S. holds equity in and serves as a consultant to Inzen Therapeutics.
Supporting Information Available: Supporting information, including methods and supplemental figures and tables, is available free of charge via the Internet.
REFERENCES
- [1].Delaney G, Jacob S, Featherstone C, and Barton M (2005) The role of radiotherapy in cancer treatment: estimating optimal utilization from a review of evidence-based clinical guidelines, Cancer 104, 1129–1137. [DOI] [PubMed] [Google Scholar]
- [2].Morgan MA, and Lawrence TS (2015) Molecular Pathways: Overcoming Radiation Resistance by Targeting DNA Damage Response Pathways, Clin Cancer Res 21, 2898–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Souhami RL, and Tobias JS (2003) Cancer and its management, 4th ed., Blackwell Science, Malden, Mass. [Google Scholar]
- [4].Tang Z, Zeng Q, Li Y, Zhang X, Ma J, Suto MJ, Xu B, and Yi N (2017) Development of a radiosensitivity gene signature for patients with soft tissue sarcoma, Oncotarget 8, 27428–27439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gerszten PC, Mendel E, and Yamada Y (2009) Radiotherapy and radiosurgery for metastatic spine disease: what are the options, indications, and outcomes?, Spine (Phila Pa 1976) 34, S78–92. [DOI] [PubMed] [Google Scholar]
- [6].Kim BM, Hong Y, Lee S, Liu P, Lim JH, Lee YH, Lee TH, Chang KT, and Hong Y (2015) Therapeutic Implications for Overcoming Radiation Resistance in Cancer Therapy, Int J Mol Sci 16, 26880–26913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Willers H, Azzoli CG, Santivasi WL, and Xia F (2013) Basic mechanisms of therapeutic resistance to radiation and chemotherapy in lung cancer, Cancer J 19, 200–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Goldstein M, and Kastan MB (2015) The DNA damage response: implications for tumor responses to radiation and chemotherapy, Annu Rev Med 66, 129–143. [DOI] [PubMed] [Google Scholar]
- [9].Nehs MA, Lin CI, Kozono DE, Whang EE, Cho NL, Zhu K, Moalem J, Moore FD Jr., and Ruan DT (2011) Necroptosis is a novel mechanism of radiation-induced cell death in anaplastic thyroid and adrenocortical cancers, Surgery 150, 1032–1039. [DOI] [PubMed] [Google Scholar]
- [10].Chaurasia M, Bhatt AN, Das A, Dwarakanath BS, and Sharma K (2016) Radiation-induced autophagy: mechanisms and consequences, Free Radic Res 50, 273–290. [DOI] [PubMed] [Google Scholar]
- [11].Walden TL, Hughes HN, and SpringerLink (Online service). (1988) Prostaglandin and Lipid Metabolism in Radiation Injury, Springer US,, Boston, MA. [Google Scholar]
- [12].Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, and Stockwell BR (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death, Cell 149, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE, Noel K, Jiang X, Linkermann A, Murphy ME, Overholtzer M, Oyagi A, Pagnussat GC, Park J, Ran Q, Rosenfeld CS, Salnikow K, Tang D, Torti FM, Torti SV, Toyokuni S, Woerpel KA, and Zhang DD (2017) Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease, Cell 171, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yang WS, and Stockwell BR (2016) Ferroptosis: Death by Lipid Peroxidation, Trends Cell Biol 26, 165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, Brown LM, Girotti AW, Cornish VW, Schreiber SL, and Stockwell BR (2014) Regulation of ferroptotic cancer cell death by GPX4, Cell 156, 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, Uchida K, O’Connor OA, and Stockwell BR (2019) Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model, Cell Chem Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liu Z, Silke J, and Hildebrand JM (2018) Methods for Studying TNF-Mediated Necroptosis in Cultured Cells, Methods Mol Biol 1857, 53–61. [DOI] [PubMed] [Google Scholar]
- [18].Pikman Y, Alexe G, Roti G, Conway AS, Furman A, Lee ES, Place AE, Kim S, Saran C, Modiste R, Weinstock DM, Harris M, Kung AL, Silverman LB, and Stegmaier K (2017) Synergistic Drug Combinations with a CDK4/6 Inhibitor in T-cell Acute Lymphoblastic Leukemia, Clin Cancer Res 23, 1012–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gao M, Monian P, Pan Q, Zhang W, Xiang J, and Jiang X (2016) Ferroptosis is an autophagic cell death process, Cell Res 26, 1021–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gaschler MM, Andia AA, Liu H, Csuka JM, Hurlocker B, Vaiana CA, Heindel DW, Zuckerman DS, Bos PH, Reznik E, Ye LF, Tyurina YY, Lin AJ, Shchepinov MS, Chan AY, Peguero-Pereira E, Fomich MA, Daniels JD, Bekish AV, Shmanai VV, Kagan VE, Mahal LK, Woerpel KA, and Stockwell BR (2018) FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation, Nat Chem Biol 14, 507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS, and Stockwell BR (2014) Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis, Elife 3, e02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lomax ME, Folkes LK, and O’Neill P (2013) Biological consequences of radiation-induced DNA damage: relevance to radiotherapy, Clin Oncol (R Coll Radiol) 25, 578–585. [DOI] [PubMed] [Google Scholar]
- [23].Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, Smith R, Lessnick SL, Sahasrabudhe S, and Stockwell BR (2007) RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels, Nature 447, 864–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Colles SM, and Chisolm GM (2000) Lysophosphatidylcholine-induced cellular injury in cultured fibroblasts involves oxidative events, J Lipid Res 41, 1188–1198. [PubMed] [Google Scholar]
- [25].Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, and Stockwell BR (2016) Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis, Proc Natl Acad Sci U S A 113, E4966–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, Tseng YY, Deasy R, Kost-Alimova M, Dancik V, Leshchiner ES, Viswanathan VS, Signoretti S, Choueiri TK, Boehm JS, Wagner BK, Doench JG, Clish CB, Clemons PA, and Schreiber SL (2019) A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis, Nat Commun 10, 1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Randers-Pehrson G, Johnson GW, Marino SA, Xu Y, Dymnikov AD, and Brenner DJ (2009) The Columbia University Sub-micron Charged Particle Beam, Nucl Instrum Methods Phys Res A 609, 294–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hei TK, Wu LJ, Liu SX, Vannais D, Waldren CA, and Randers-Pehrson G (1997) Mutagenic effects of a single and an exact number of alpha particles in mammalian cells, Proc Natl Acad Sci U S A 94, 3765–3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wu LJ, Randers-Pehrson G, Xu A, Waldren CA, Geard CR, Yu Z, and Hei TK (1999) Targeted cytoplasmic irradiation with alpha particles induces mutations in mammalian cells, Proc Natl Acad Sci U S A 96, 4959–4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhou H, Hong M, Chai Y, and Hei TK (2009) Consequences of cytoplasmic irradiation: studies from microbeam, J Radiat Res 50 Suppl A, A59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wu CC, Chaudhary KR, Na YH, Welch D, Black PJ, Sonabend AM, Canoll P, Saenger YM, Wang TJC, Wuu CS, Hei TK, and Cheng SK (2017) Quality Assessment of Stereotactic Radiosurgery of a Melanoma Brain Metastases Model Using a Mouselike Phantom and the Small Animal Radiation Research Platform, Int J Radiat Oncol Biol Phys 99, 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Cancer Genome Atlas Research, N., and Brat DJ, and Verhaak RG, and Aldape KD, and Yung WK, and Salama SR, and Cooper LA, and Rheinbay E, and Miller CR, and Vitucci M, and Morozsova O, and Robertson AG, and Noushmehr H, and Laird PW, and Cherniack AD, and Akbani R, and Huse JT, and Ciriello G, and Poisson LM, and Barnholtz-Sloan JS, and Berger MS, and Brennan C, and Colen RR, and Colman H, and Flanders AE, and Giannini C, and Grifford M, and Iavarone A, and Jain R, and Joseph I, and Kim J, and Kasaian K, and Mikkelsen T, and Murray BA, and O’Neill BP, and Pachter L, and Parsons DW, and Sougnez C, and Sulman EP, and Vandenberg SR, and Van Meir EG, and von Deimling A, and Zhang H, and Crain D, and Lau K, and Mallery D, and Morris S, and Paulauskis J, and Penny R, and Shelton T, and Sherman M, and Yena P, and Black A, and Bowen J, and Dicostanzo K, and Gastier-Foster J, and Leraas KM, and Lichtenberg TM, and Pierson CR, and Ramirez NC, and Taylor C, and Weaver S, and Wise L, and Zmuda E, and Davidsen T, and Demchok JA, and Eley G, and Ferguson ML, and Hutter CM, and Mills Shaw KR, and Ozenberger BA, and Sheth M, and Sofia HJ, and Tarnuzzer R, and Wang Z, and Yang L, and Zenklusen JC, and Ayala B, and Baboud J, and Chudamani S, and Jensen MA, and Liu J, and Pihl T, and Raman R, and Wan Y, and Wu Y, and Ally A, and Auman JT, and Balasundaram M, and Balu S, and Baylin SB, and Beroukhim R, and Bootwalla MS, and Bowlby R, and Bristow CA, and Brooks D, and Butterfield Y, and Carlsen R, and Carter S, and Chin L, and Chu A, and Chuah E, and Cibulskis K, and Clarke A, and Coetzee SG, and Dhalla N, and Fennell T, and Fisher S, and Gabriel S, and Getz G, and Gibbs R, and Guin R, and Hadjipanayis A, and Hayes DN, and Hinoue T, and Hoadley K, and Holt RA, and Hoyle AP, and Jefferys SR, and Jones S, and Jones CD, and Kucherlapati R, and Lai PH, and Lander E, and Lee S, and Lichtenstein L, and Ma Y, and Maglinte DT, and Mahadeshwar HS, and Marra MA, and Mayo M, and Meng S, and Meyerson ML, and Mieczkowski PA, and Moore RA, and Mose LE, and Mungall AJ, and Pantazi A, and Parfenov M, and Park PJ, and Parker JS, and Perou CM, and Protopopov A, and Ren X, and Roach J, and Sabedot TS, and Schein J, and Schumacher SE, and Seidman JG, and Seth S, and Shen H, and Simons JV, and Sipahimalani P, and Soloway MG, and Song X, and Sun H, and Tabak B, and Tam A, and Tan D, and Tang J, and Thiessen N, and Triche T Jr., and Van Den Berg DJ, and Veluvolu U, and Waring S, and Weisenberger DJ, and Wilkerson MD, and Wong T, and Wu J, and Xi L, and Xu AW, and Yang L, and Zack TI, and Zhang J, and Aksoy BA, and Arachchi H, and Benz C, and Bernard B, and Carlin D, and Cho J, and DiCara D, and Frazer S, and Fuller GN, and Gao J, and Gehlenborg N, and Haussler D, and Heiman DI, and Iype L, and Jacobsen A, and Ju Z, and Katzman S, and Kim H, and Knijnenburg T, and Kreisberg RB, and Lawrence MS, and Lee W, and Leinonen K, and Lin P, and Ling S, and Liu W, and Liu Y, and Liu Y, and Lu Y, and Mills G, and Ng S, and Noble MS, and Paull E, and Rao A, and Reynolds S, and Saksena G, and Sanborn Z, and Sander C, and Schultz N, and Senbabaoglu Y, and Shen R, and Shmulevich I, and Sinha R, and Stuart J, and Sumer SO, and Sun Y, and Tasman N, and Taylor BS, and Voet D, and Weinhold N, and Weinstein JN, and Yang D, and Yoshihara K, and Zheng S, and Zhang W, and Zou L, and Abel T, and Sadeghi S, and Cohen ML, and Eschbacher J, and Hattab EM, and Raghunathan A, and Schniederjan MJ, and Aziz D, and Barnett G, and Barrett W, and Bigner DD, and Boice L, and Brewer C, and Calatozzolo C, and Campos B, and Carlotti CG Jr., and Chan TA, and Cuppini L, and Curley E, and Cuzzubbo S, and Devine K, and DiMeco F, and Duell R, and Elder JB, and Fehrenbach A, and Finocchiaro G, and Friedman W, and Fulop J, and Gardner J, and Hermes B, and Herold-Mende C, and Jungk C, and Kendler A, and Lehman NL, and Lipp E, and Liu O, and Mandt R, and McGraw M, and McLendon R, and McPherson C, and Neder L, and Nguyen P, and Noss A, and Nunziata R, and Ostrom QT, and Palmer C, and Perin A, and Pollo B, and Potapov A, and Potapova O, and Rathmell WK, and Rotin D, and Scarpace L, and Schilero C, and Senecal K, and Shimmel K, and Shurkhay V, and Sifri S, and Singh R, and Sloan AE, and Smolenski K, and Staugaitis SM, and Steele R, and Thorne L, and Tirapelli DP, and Unterberg A, and Vallurupalli M, and Wang Y, and Warnick R, and Williams F, and Wolinsky Y, and Bell S, and Rosenberg M, and Stewart C, and Huang F, and Grimsby JL, and Radenbaugh AJ, and Zhang J (2015) Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas, N Engl J Med 372, 2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, Genovese G, Shmulevich I, Barnholtz-Sloan J, Zou L, Vegesna R, Shukla SA, Ciriello G, Yung WK, Zhang W, Sougnez C, Mikkelsen T, Aldape K, Bigner DD, Van Meir EG, Prados M, Sloan A, Black KL, Eschbacher J, Finocchiaro G, Friedman W, Andrews DW, Guha A, Iacocca M, O’Neill BP, Foltz G, Myers J, Weisenberger DJ, Penny R, Kucherlapati R, Perou CM, Hayes DN, Gibbs R, Marra M, Mills GB, Lander E, Spellman P, Wilson R, Sander C, Weinstein J, Meyerson M, Gabriel S, Laird PW, Haussler D, Getz G, Chin L, and Network TR (2013) The somatic genomic landscape of glioblastoma, Cell 155, 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Parker JJ, Lizarraga M, Waziri A, and Foshay KM (2017) A Human Glioblastoma Organotypic Slice Culture Model for Study of Tumor Cell Migration and Patient-specific Effects of Anti-Invasive Drugs, J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Eriksson D, and Stigbrand T (2010) Radiation-induced cell death mechanisms, Tumour Biol 31, 363–372. [DOI] [PubMed] [Google Scholar]
- [36].Shadyro OI, Yurkova IL, and Kisel MA (2002) Radiation-induced peroxidation and fragmentation of lipids in a model membrane, Int J Radiat Biol 78, 211–217. [DOI] [PubMed] [Google Scholar]
- [37].Sleire L, Skeie BS, Netland IA, Forde HE, Dodoo E, Selheim F, Leiss L, Heggdal JI, Pedersen PH, Wang J, and Enger PO (2015) Drug repurposing: sulfasalazine sensitizes gliomas to gamma knife radiosurgery by blocking cystine uptake through system Xc-, leading to glutathione depletion, Oncogene 34, 5951–5959. [DOI] [PubMed] [Google Scholar]
- [38].Nagane M, Kanai E, Shibata Y, Shimizu T, Yoshioka C, Maruo T, and Yamashita T (2018) Sulfasalazine, an inhibitor of the cystine-glutamate antiporter, reduces DNA damage repair and enhances radiosensitivity in murine B16F10 melanoma, PLoS One 13, e0195151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Cobler L, Zhang H, Suri P, Park C, and Timmerman LA (2018) xCT inhibition sensitizes tumors to gamma-radiation via glutathione reduction, Oncotarget 9, 32280–32297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hammer CT, and Wills ED (1979) The effect of ionizing radiation on the fatty acid composition of natural fats and on lipid peroxide formation, Int J Radiat Biol Relat Stud Phys Chem Med 35, 323–332. [DOI] [PubMed] [Google Scholar]
- [41].Vartak S, Robbins ME, and Spector AA (1997) Polyunsaturated fatty acids increase the sensitivity of 36B10 rat astrocytoma cells to radiation-induced cell kill, Lipids 32, 283–292. [DOI] [PubMed] [Google Scholar]
- [42].Cai F, Sorg O, Granci V, Lecumberri E, Miralbell R, Dupertuis YM, and Pichard C (2014) Interaction of omega-3 polyunsaturated fatty acids with radiation therapy in two different colorectal cancer cell lines, Clin Nutr 33, 164–170. [DOI] [PubMed] [Google Scholar]
- [43].Chen PH, Wu J, Ding CC, Lin CC, Pan S, Bossa N, Xu Y, Yang WH, Mathey-Prevot B, and Chi JT (2019) Kinome screen of ferroptosis reveals a novel role of ATM in regulating iron metabolism, Cell Death Differ. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lang X, Green MD, Wang W, Yu J, Choi JE, Jiang L, Liao P, Zhou J, Zhang Q, Dow A, Saripalli AL, Kryczek I, Wei S, Szeliga W, Vatan L, Stone EM, Georgiou G, Cieslik M, Wahl DR, Morgan MA, Chinnaiyan AM, Lawrence TS, and Zou W (2019) Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11, Cancer Discov. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, Viswanathan SR, Chattopadhyay S, Tamayo P, Yang WS, Rees MG, Chen S, Boskovic ZV, Javaid S, Huang C, Wu X, Tseng YY, Roider EM, Gao D, Cleary JM, Wolpin BM, Mesirov JP, Haber DA, Engelman JA, Boehm JS, Kotz JD, Hon CS, Chen Y, Hahn WC, Levesque MP, Doench JG, Berens ME, Shamji AF, Clemons PA, Stockwell BR, and Schreiber SL (2017) Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway, Nature 547, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.