

Abstract
The recent market approvals of recombinant adeno-associated virus (rAAV) gene therapies in Europe and the United States are landmark achievements in the history of modern science. These approvals are also anticipated to herald the emergence of a new class of therapies for monogenic disorders, which had hitherto been considered untreatable. These events can be viewed as stemming from the convergence of several important historical trends: the study of basic virology, the development of genomic technologies, the imperative for translational impact of National Institutes of Health–funded research, and the development of economic models for commercialization of rare disease therapies. In this review, these historical trends are described and the key developments that have enabled clinical rAAV gene therapies are discussed, along with an overview of the current state of the field and future directions.
Keywords: adeno-associated virus, AAV, gene therapy, genetic disease, genome editing, clinical trial
1. KEY MILESTONES IN THE HISTORY OF rAAV GENE THERAPY
1.1. The Discovery of AAV and AAV Latency
Adeno-associated virus (AAV) was discovered by Atchison et al. (1) by electron microscopy–based screening of adenovirus (Ad) preparations in the mid-1960s. Shortly thereafter, AAV was found in human isolates as a coinfecting agent during outbreaks of Ad-induced diarrhea, conjunctivitis, and other illnesses (220135).
The Berns laboratory (6) initially worked out the hairpin self-priming mechanism now established as the mechanism of replication of both wild-type and recombinant AAV (rAAV) (Figure 1). Subsequently, the Berns laboratory (7) discovered that AAV infection in the absence of Ad would establish a persistent or latent infection. With regard to latency, great interest developed around the fact that wild-type AAV2 demonstrated integration, with a strong preference for integration into a specific site on human chromosome 19 (the AAVS1 site) (8–10).
Figure 1.
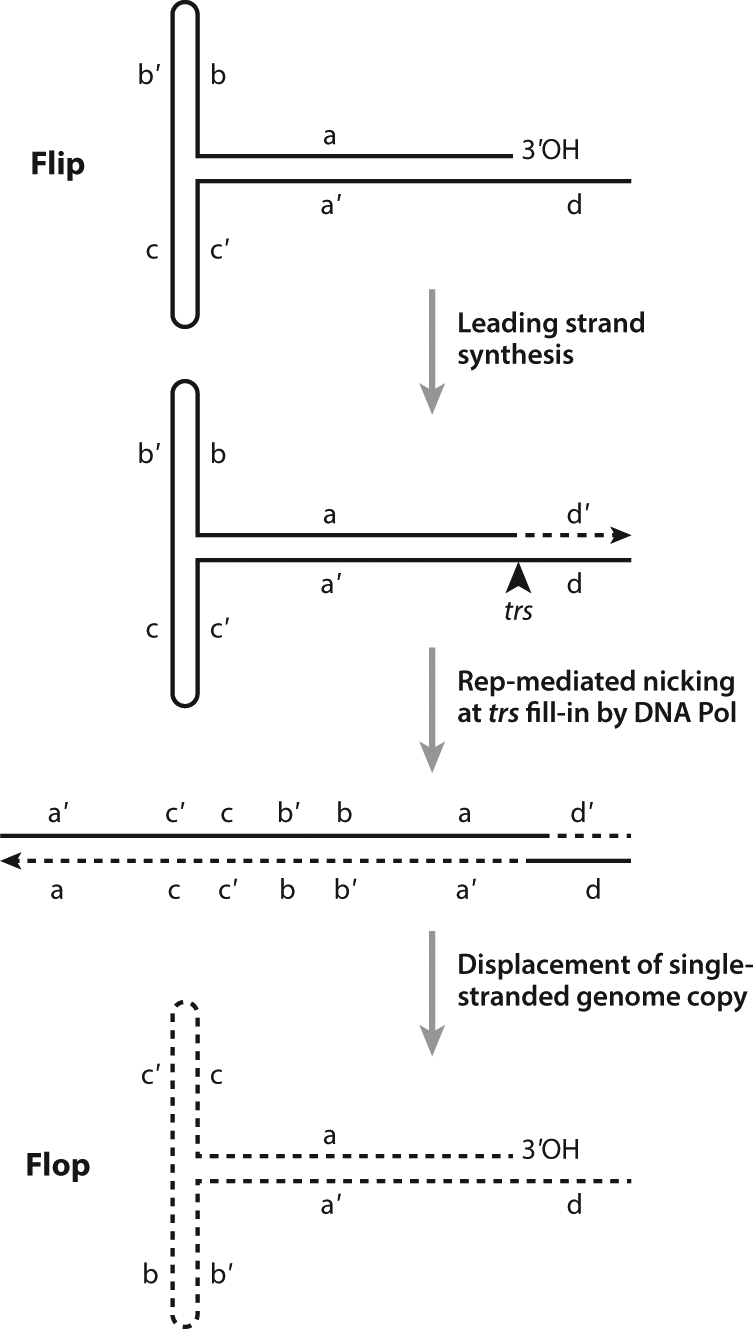
Adeno-associated virus (AAV) replication scheme. The steps in AAV replication and terminal resolution result in flip and flop configurations of inverted terminal repeats (ITR). The 3′ end of the hairpin configuration of the ITR acts as template for leading strand synthesis. Rep-mediated nicking at the trs allows for terminal resolution. Other abbreviations: trs, terminal resolution site, DNA Pol-DNA Polymerase. Figure adapted from Reference 148.
1.2. The Development of Recombinant Gene Transfer Vectors Based on AAV and Early Preclinical Studies
Initially, there was some technical difficulty with cloning the AAV2 genome into bacterial plasmids, most likely due to the secondary structure of the AAV inverted terminal repeats (ITRs), which are palindromes capable of hairpin formation. This technical hurdle was overcome by the Muzyczka laboratory at the University of Florida and the Carter laboratory at the National Institutes of Health. Shortly thereafter, Arun Srivastava and Ken Berns (11) published the sequence of the AAV2 genome. Then the Muzyczka and Carter labs (12–14) both succeeded in adapting recombinant AAV2 as a eukaryotic gene transfer vector.
A major breakthrough in the use of rAAV as a gene transfer vector came when Samulski, Chang, and Shenk (15, 16) developed a two-plasmid system in which AAV2-ITR-flanked payload was supplied on one plasmid and the AAV2 rep and cap on a second, ITR-deleted plasmid (Figure 2). Cotransfection of these two plasmids into Ad-infected HeLa cells enabled packaging of rAAV preparations that were relatively free of wild-type AAV (16). The system has been shown to be highly recombinogenic, and further modifications were required to eliminate all replication-competent AAV. However, the Samulski plasmids, pSub201(+) and pAAV/Ad, allowed for widespread use of rAAV for gene transfer experiments.
Figure 2.
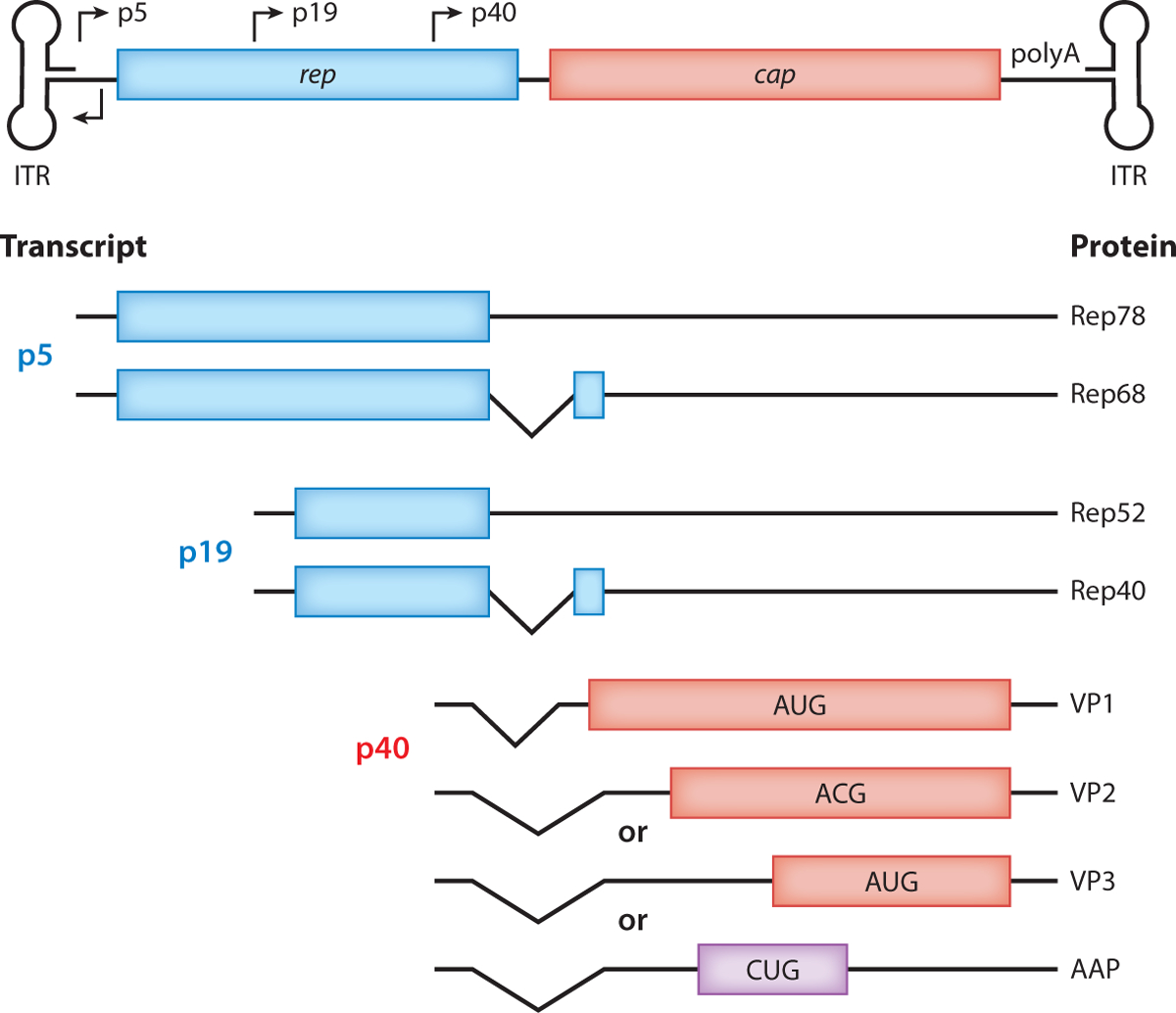
The wild-type AAV genome contains three parts: noncoding ITRs and two genes, rep and cap. From both alternative splicing and non-AUG start sites, eight different proteins are produced. Four Rep proteins are produced, two from the p5 promoter (Rep78 and Rep68) and two from the p19 promoter (Rep52 and Rep40). Three capsid and AAPs are produced from the p40 promoter. VP2, VP3, and AAP are produced from the same transcript but from different initiation start sites. Abbreviations: AAP, assembly-activating protein; AAV, adeno-associated virus; ITR, inverted terminal repeat.
The first concerted effort to develop rAAV-based therapy for a single-gene disorder was the effort by the Carter/Flotte laboratories and subsequently the Targeted Genetics Corporation to develop rAAV2-CFTR (cystic fibrosis transmembrane conductance regulator) vectors for in vivo use in the respiratory tract to treat patients with cystic fibrosis (CF). Initial efforts centered on the problem of packaging the CFTR coding sequence, which measures 4.4 kb, into rAAV with a suitable promoter and polyadenylation signal. These efforts resulted in the discovery of cryptic promoter elements within and near the AAV2-ITR (17). Packaging of rAAV2-CFTR enabled complementation of the CFTR chloride channel defect in CF bronchial epithelial cells in culture (18).
rAAV2-CFTR delivery to the airway in vivo was modeled using a fiberoptic bronchoscope to deliver rAAV2-CFTR to the airway epithelium of rabbits (19). These studies showed that the CFTR messenger RNA and protein could be expressed in vivo from a dose of up to 1 × 1010 physical particles of rAAV2-CFTR delivered to a single bronchopulmonary segment through the bronchoscope. This expression persisted at least 6 months, providing the pivotal proof of principle to justify moving forward to a clinical trial of rAAV2-CFTR to the nose and single bronchopulmonary segment of the lung in adult CF patients (20, 21).
A number of other early-stage in vivo rAAV2 gene therapy vector studies were published in the 1990s. Specific examples include studies using the lacZ reporter gene in skeletal muscle (22) and the central nervous system (CNS) (23, 24). Additional studies showed that rAAV2 delivery to muscle could mediate robust and sometimes stable expression of a secreted protein. This was initially tested with erythropoietin (25–27). More therapeutic examples of rAAV2 muscle injection for ectopic stable expression of secreted serum proteins included rAAV2-Factor IX (rAAV2-FIX) (28, 29) and rAAV2-alpha-1 antitrypsin (rAAV2-AAT) (30, 31).
The concept of using direct rAAV2 injection to transduce specific target cells and replace deficient gene products was also developed. This approach was used to create potential therapies to a number of disorders of the CNS and muscle. Specific examples included rAAV2-tyrosine hydroxylase vectors designed to increase dopamine formation in the basal ganglia in patients with Parkinson’s disease (PD) (32–34). Another example was direct intraparenchymal injections of rAAV2 vector encoding APSA (AAV2-ASPA) for Canavan disease (35, 36). The rAAV2-delta sarcoglycan vector was similarly developed for direct muscle injection in patients with limb girdle muscular dystrophy (37).
1.3. Description of AAV Capsid Variants and Tropisms
As more in vivo and potential clinical applications of rAAV emerged, it became clear that it could be advantageous to find AAV capsid variants that had higher efficiency for gene transfer into various target cell and tissue types or that were immunologically distinct. The logic was that capsid variants with a higher affinity for a specific cell type could be adapted to become therapeutic vectors with higher potency when delivered to those cell types.
While there were a few specific instances of using different serotype capsids prior (38), Xiao and colleagues (39) worked out a simple system in which the AAV2-ITRs would be used for all vector cargo and the AAV2 rep gene would be used as a complementing gene for all variants. However, these AAV2-ITR-flanked genomes would be cross-packaged into any of a variety of other AAV capsids by complementing the vector with the cap gene corresponding to that serotype or variant. Simply using the first five known AAV serotypes, Xiao and colleagues (40) demonstrated important differences in vector potency, specifically the very high potency of rAAV1 when injected into muscle.
1.3.1. Biodiversity of AAV.
The real breakthrough in vector potency came with Gao and Wilson and their colleagues discovering the tremendous biodiversity of AAV in nature (41, 42), with over 2,000 genetically distinct clades and hundreds of genomic variants (genomovars) found in human and nonhuman primate tissues. Subsequent studies have demonstrated that thousands of such variants exist naturally.
Early on in this work, it became clear that certain of the newer rAAV serotypes had dramatically improved efficacy in certain sites. rAAV8 was found to be very efficient for liver and muscle delivery (40). Meanwhile, rAAV9 and other members of its clade, AAVrh10 and AAVrh8, were found to be efficient for CNS and cardiac muscle gene transfer (43–45). The ability of rAAV9 vectors, when delivered systemically, to cross the blood-brain barrier was of particular interest (46).
1.3.2. Targeted capsid modifications.
While the identification of novel, natural AAV capsid variants was successful, the conceptual proof that changing the capsid could lead to great improvements in vector potency led others to conclude that designed modifications of the capsid could potentially be even more effective and confer a degree of specificity not generally observed with naturally occurring genomovars. The Muzyczka laboratory (47) performed alanine-scanning mutagenesis of the AAV2 capsid and identified a number of loci at which amino acid substitutions or insertions could be tolerated. Initially, only short polypeptide stretches were inserted, such as the short version of the serpin-enzyme complex receptor (47, 48). Subsequent work from Muzyczka and colleagues (49) has shown, however, that much larger domain insertions are tolerated when genetically engineered into the N terminus of VP2.
1.3.3. Synthetic capsid libraries and directed evolution and ancestral reconstruction.
A third approach to the generation of new capsids in which to deliver rAAV has explored a more empiric approach. Combinatorial AAV capsid libraries were generated with a number of methods, including both randomly generated variations in the variable cap regions (by synthesis or error-prone polymerase chain reaction) and so-called capsid shuffling where portions of the cap gene from multiple different serotypes are used to reconstitute intact chimeric capsids (50, 51). These combinatorial libraries of capsids were then passaged over cultured cells and/or in vivo, selecting for and thereby enriching for those capsids that enable efficient infection. The error-prone nature of AAV replication may enable a modest degree of additional genetic evolution during serial passage and selection. Serial passage under these selective conditions was used in several different applications, in many cases focusing on a specific cell or tissue type or animal host species (52–56). Almost opposite of directed evolution, reconstruction of ancestral AAVs has led to some interesting candidate capsids with unique properties and tropisms (57, 58).
1.3.4. Overview of tropisms.
With this new palette of AAV capsids, translational investigators have utilized multiple different serotypes for multiple different applications in order to optimize the serotype, dose, and delivery method that could potentially achieve a therapeutic goal. The tropism of each capsid variant is likely largely attributed to expression of viral receptors on target tissue cell types, which can vary greatly from species to species. As an interesting example, AAV3b shows almost no expression in mouse liver but robust expression in human xenograft models and nonhuman primates (59), presumably because AAV3 uses huHGFR as a coreceptor (60). Therefore, it is important to consider, and sometimes it is overlooked, that the tropisms that have been best described are those for mice, while newer data from larger animals and humans show some important distinctions in potency of different AAV capsid variants across species. With that caveat considered, the current pattern of preference of use by investigators, largely based on in vivo efficacy, is shown in Table 1.
Table 1.
Examples of adeno-associated virus (AAV) capsid specificities currently utilized for preclinical and clinical applications
| Organ | Mouse | Other large animal | Nonhuman primate | Human |
|---|---|---|---|---|
| Central nervous system | AAV9, AAVrh10, AAVrh8, AAV2, AAV5, ANC80L65, AAV-PHP.B | AAV9, AAVrh10, AAVrh8, AAV5 | AAV9, AAVrh10, AAVrh8, AAV5 | AAV2, AAV9, AAVrh10, AAVrh8 |
| Liver | AAV8, AAV9 | AAV8, AAV5 | AAV8, AAV9, AAV3B | AAV2, AAV5, AAV8, AAV9, AAV3B |
| Lung | AAV5, AAV6, AAV8, AAV1, AAV6.2, AAV6.2FF, AAVrh10, AAV2-ESGHGYF | AAV5, AAV2/HBoV1 | AAV1, AAV5, AAVrh10 | AAV2 |
| Muscle | AAV1, AAV8, AAVrh74, AAV9, AAV-B1 | AAV1, AAV8, AAVrh74, AAV9 | AAV1, AAV8, AAVrh74, AAV9 | AAV1, AAVrh74, AAV8, AAV9, AAV2.5 |
| Retina | AAV2, AAV5, AAV7, AAV8, AAV9, ANC80L65 | AAV2, AAV5 | AAV2, AAV5, AAV9, ANC80L65 | AAV2, AAV4, AAV5 |
Different AAV capsid variants have differing tropisms both for tissue targets and across different species. Many variants are initially tested preclinically in mouse models. However, the most promising capsids are tested in larger animal models and nonhuman primates often prior to initiating clinical trials in human patients.
1.4. Improvements in rAAV Manufacturing Technology
Two main technologies exist for manufacturing rAAV, transfection and helper virus systems (Table 2).
Table 2.
AAV packaging methods
| Packaging method | Host cell(s) | Source(s) of helper genes | Source of Rep and cap | Source of ITR construct | Reference(s) |
|---|---|---|---|---|---|
| Triple transfection | HEK-293 | Helper plasmid and host cell | Second helper plasmid | Proviral plasmid | 27 |
| Double transfection | HEK-293 | Dual helper plasmid and host cell | Dual helper plasmid | Proviral plasmid | 28, 29 |
| Baculovirus | Sf-9 | rBEV-Rep/cap | rBEV-Rep/cap | rBEV-rAAV | 18, 30 |
| HSV1 helper | Vero-27, BHK, HEK-293 | rHSV1 | rHSV1-Rep/cap | rHSV1-rAAV | 31–34 |
| Rescuable HeLa lines | HeLa | rcAd5 or rcAd2 | Integrated in HeLa | Integrated in HeLa | 35 |
Abbreviations: AAV, adeno-associated virus; HSV1, herpes simplex virus 1; ITR, inverted terminal repeat; rcAd, replication-competent Ad. Table adapted from Reference 153.
1.4.1. Transfection technology.
Initially, it was assumed that the maximum possible expression of all the Rep proteins in the rAAV producer cells would increase the efficiency packaging and the yields of vector. This was subsequently disproven. The Samulski laboratory (61) was the first to show that highly regulated rep gene expression (particularly moderating the expression of Rep78/69) could increase the efficiency of transfection-mediated rAAV manufacturing. This was initially done in the context of transfecting modified rep/cap plasmid helpers into Ad-infected cells. The approach was further improved by eliminating productive Ad infection from the producer cells and substituting transient transfection of three of the adenoviral helper genes (E2a, E4, and VA RNA) into cells that constitutively express adenoviral E1a and E1b genes, such as HEK-293 cells (62). The triple transfection of the ITR-flanked vector plasmid, the Rep/cap plasmid, and the adenoviral gene helper plasmid was thus established as the most commonly used packaging platform. This process has been further streamlined by combining the Ad-gene and rep/cap gene plasmids into a single helper plasmid, pDG (63, 64).
1.4.2. Helper virus-based technologies.
Several attempts to enhance the efficiency and scalability of manufacturing of rAAV have aimed to eliminate transient transfection from the upstream process. Ideally, this could be accomplished by creating stable producer cell lines, thus mimicking the most common upstream process for recombinant protein production. However, the toxic and cytostatic effects of the AAV-Rep proteins present a fundamental limitation to that approach. A more feasible alternative that has been successfully pursued is the use of infectious recombinant helper viruses. The three primary approaches of this type have been the use of replication-competent Ad (rcAd) in HeLa cells with inducible AAV-Rep, the use of infectious baculovirus recombinants in the Sf9 insect cell line, and the use of recombinant herpes simplex virus (HSV) constructs supplying helper virus function, Rep/cap complementation, and the ITR-flanked proviral rAAV construct.
1.4.2.1. Replication-competent adenovirus in Rep-HeLa cells.
As described above, the earliest transient transfection approaches to rAAV manufacturing all relied on infecting the transfected producer cells with rcAd, initially with wild-type Ad5 or Ad2. Early studies suggested that cell lines may be able to tolerate an integrated AAV2-rep gene, most likely because the AAV2-p5 promoter was active in HeLa cells only when lytic phase rcAd was supplied externally (65). Clark et al. (66) developed such cell lines as a sustainable method for manufacturing of rAAV vectors. In fact, this technology was used for later-phase studies of the first rAAV2-CFTR vectors (the initial phase 1 was performed with transfection-based material). The rcAd method has proven to be scalable and capable of generating high titers of rAAV, as well as being adaptable to multiple different serotypes.
1.4.2.2. Baculovirus-Sf9.
The baculovirus-Sf9 rAAV manufacturing system relies on the basics of the baculovirus expression system, namely the ability to use the baculovirus polyhedron protein promoter to mediate very high-level expression of proteins when induced. Interestingly, it was discovered that insect cells undergoing lytic infection with baculovirus were able to support helper virus functions as well as carry both the Rep/cap construct and the rAAV proviral plasmid (67). As with the rcAd system, the baculovirus system has proven to be very scalable and adaptable into suspension culture, resulting in net production of very high titer rAAV preparations (68).
1.4.2.3. Herpes simplex virus packaging.
Initial efforts combined HSV-1 amplicons capable of expression of AAV-rep and cap genes with replication-defective ICP27-deleted HSV-1 (69). Further optimization used a single rHSV1, capable of providing both helper virus functions and AAV-rep and cap gene functions (70), while separate rHSV-1 vectors can be used to provide the rAAV-proviral gene cassette. The rHSV-1 system has been used successfully to package rAAV vectors for both preclinical and clinical trials. Because all elements of the system are infectious, this system is also scalable into suspension cell bioreactor systems (71).
2. CLINICAL TRIALS AND CLINICAL PRODUCT DEVELOPMENT WITH rAAV
2.1. Early Cystic Fibrosis Transmembrane Conductance Regulator Work
As mentioned above, the rAAV2-CFTR program was the first clinical development program with rAAV vector. Following the rabbit proof-of-principle studies mentioned above, nonhuman primate studies for evaluating safety were performed (72, 73). Interestingly, the latter studies showed a clear demonstration that rAAV persistence in vivo was mediated by episomal persistence rather than integration, confirming a phenomenon that had been observed by the Flotte lab (74) two years prior. This episomal nature of rAAV persistence turned out to have major implications for the utility of rAAV, specifically that long-term persistence of rAAV was observed only in nondividing cells.
A number of human trials were then performed with the rAAV2-CFTR vector (20, 21, 75–80). In the case of the administration of rAAV2-CFTR both to the nasal and bronchial epithelium (21, 79) and in the maxillary sinus (75), rAAV2-CFTR delivery was associated with dose-related DNA transfer and expression of CFTR at low levels. Expression did not persist, as might have been predicted given the relatively rapid turnover of the airway epithelium in CF patients. However, these CF studies provided a strong set of evidence of the general safety of rAAV administration in vivo, setting the stage for later studies.
2.2. Hemophilia Trials
In a series of preclinical and clinical trials, a number of laboratories developed rAAV2-FIX vectors, initially delivered by intramuscular (IM) injection, for the continuous secretion of FIX in patients with hemophilia B (81–83). Subsequently, rAAV2-FIX, when delivered by intrahepatic artery, was shown to be capable of transient expression of FIX (84–87), which was the first trial that showed evidence of an effector T cell response to vector capsid being responsible for elimination of transduced cells. Nathwani et al. (88, 89) subsequently showed that rAAV8-FIX gene therapy was capable of sustained expression of a FIX from a self-complementary AAV construct. These studies also enabled the further elucidation of a common immune response profile with systemic rAAV vectors, the development of an effector T cell response against AAV capsid epitopes, occurring approximately 6 to 10 weeks after vector administration, apparently leading to a rise in serum transaminases and a suppression of transgene expression. These studies also demonstrated that this immune response could be controlled with oral anti-inflammatory corticosteroids. Similar studies have now been published by several other groups. More recently, a number of hemophilia B trials with rAAV8-FIX vectors have been developed by industry sponsors, including one with a gain-of-function variant, known as the Padua variant (R338L) (90).
Hemophilia A [Factor VIII deficiency (FVIII deficiency)] presented a more difficult challenge in the context of rAAV gene therapy, since the FVIII cDNA is substantially larger, exceeding the packaging capacity of most rAAV vector designs. The BioMarin group and Nathwani and colleagues have overcome this limitation with a B-domain deleted FVIII construct packaged in rAAV5 capsids (91).
2.3. Genetic Retinal Diseases
Another breakthrough in demonstrating the clinical efficacy of rAAV gene therapy came with a series of parallel programs aimed at using rAAV2-RPE65 vectors by subretinal injection as a means to treat inherited retinal dystrophy due to Leber congenital amaurosis RPE-65 deficiency (IRD-RPE65). In 2008, three similar trials of the first use of rAAV2-RPE65 were published (92–95). In light of these studies, the Spark Therapeutics program moved forward with carefully designing a multiluminance mobility test to measure the effect of the RPE-65 isomerase gene on retinoid recycling, specifically the restoration of dim light vision due to improvement in rod photoreceptor function (96). This multiluminance mobility test represents an important example of how gene therapies can diverge in design and endpoints based on their molecular mechanism of action. Because they often do not function in a manner equivalent to a small molecule drug, it is only logical that the most informative outcomes may also differ. These studies formed the basis for the market approval for the first Food and Drug Administration (FDA)-approved rAAV gene therapy product, voretigene neparvovec (Luxturna), in December 2017 and subsequent European Commission approval in November 2018, which has greatly impacted the field of AAV-based gene therapies moving forward. A number of other eye diseases are under investigation including retinitis pigmentosa, Leber hereditary optic neuropathy, choroideremia, and age-related macular degeneration.
2.4. Muscular Dystrophies
Several late-stage preclinical and clinical studies of rAAV delivery for treatment of muscular dystrophies have been performed ( NCT01519349, NNCT03375164, NCT03368742, NCT02354781, NCT00428935, NCT03362502) by direct IM injection, isolated limb perfusion, or systemic IV injection. Diseases that have been addressed in this manner include limb girdle muscular dystrophy and Duchenne muscular dystrophy. In general, the systemic delivery approach that has attracted the most attention has been the use of rAAV9 or rAAV9-like vectors, which are efficient for generalized gene transfer to skeletal and cardiac muscle after IV administration. As with hemophilia gene therapy, there has been some limitation on these studies because of immune responses. Surprisingly, the very high doses of rAAV9 required for these studies have also resulted in an early-phase toxicity occurring within the first week after administration, which had not previously been reported (97, 98).
2.5. Lipoprotein Lipase Deficiency and Alpha-1 Antitrypsin Deficiency
rAAV vectors have also been directed to muscle for the ectopic expression of proteins in single-gene disorders that are not associated with primary muscle disease. This has particularly been developed for AAT deficiency and lipoprotein lipase (LPL) deficiency. In the case of LPL deficiency, the deficiency state results in a form of hyperlipidemia that causes recurrent pancreatitis and premature atherosclerotic cardiovascular disease, including coronary artery disease. Clinical studies of rAAV1-LPL delivery to muscle resulted in actual clinical efficacy in patients with LPL deficiency (99–102). These results led to the European Medicines Agency (EMA) approval of Glybera for patients with LPL deficiency in 2012. The EMA approval of this therapy was, in fact, a milestone in rAAV therapeutic development, even though Glybera was subsequently withdrawn from the market in 2017 due to economic considerations.
In the case of AAT deficiency, the deficiency of the serum antiprotease AAT can result in emphysema due to degradation of interstitial elastin within the lungs. The use of muscle-based expression of AAT has been studied extensively in mice, rabbits, baboons, and rhesus macaques and more recently in clinical trials of both rAAV2-hAAT ( NCT00377416) and rAAV1-hAAT ( NCT00430768) delivery to the muscle of AAT-deficient patients. The use of rAAV2-hAAT for ectopic secretion of AAT was demonstrated in mice and baboons prior to the first human use (30, 31, 103, 104). Subsequently, the use of rAAV1 in muscle was shown to mediate a higher potency of gene transfer, and this platform was used to express wild-type (M) AAT from muscle in two human trials (105, 106). The more recent of these has been followed for five years with partial correction of biomarkers at a 2.5–3% level of serum protein reconstitution (107). In these studies, it was shown that rAAV1 delivery to muscle resulted in the generation of regulatory T cell responses to AAV1 capsid epitopes (108).
2.6. Spinal Cord Disease
A major therapeutic milestone was recorded by Mendell et al. (109), as a single IV injection of rAAV9-smn1 resulted in a dramatic restoration of lower motor neuron function in infants with spinal muscular atrophy 1 ( NCT02122952), a disorder that previously was uniformly fatal without mechanical ventilation. In May 2019, Zolegensma, a one-time treatment for spinal muscular atrophy in pediatric patients less than 2 years of age, became the second FDA approved AAV gene therapy. The dramatic benefit observed in this study has served as an important proof of concept for other gene therapies of lower motor neurons. Phase 1 studies of rAAV-rh10-mediated delivery of therapeutic anti-superoxide dismutase 1 microRNAs have demonstrated efficacy and safety in nonhuman primate studies for treatment of amyotrophic lateral sclerosis (110) and in early expanded access human studies (Brown, personal communication).
2.7. Monogenic Brain Disorders Including Lysosomal Storage Disorders
The use of rAAV in single-gene disorders affecting the brain has a more ambitious therapeutic target than for disorders affecting the spinal cord. Most clinical trials originally focused on using AAV2 for treatment of monogenic brain disorders such as Canavan disease (36) and late infantile neuronal ceriod lipofuscinosis (LINCF) ( NCT00151216) (111). New capsid variants, such as rAAVrh10 vectors, have been tested in patients with Batten disease (NCT0116576), metachromatic leukodystrophy ( NCT01801709), LINCF ( NCT01414985), and mucopolysaccharidosis IIIA ( NCT01474343) (112). rAAV9 vectors have been used in both late-state preclinical studies and patients with Batten disease ( NCT02725580), AADC deficiency (113, 114), and Canavan disease (115, 116). Meanwhile, late-stage preclinical studies with rAAVrh8 have been published in both GM1 gangliosidosis (117) and GM2 gangliosidosis, Tay-Sachs disease, and Sandhoff disease (118–122). Additionally, studies in patients with giant axonal neuropathy have added to the overall clinical experience with single-gene disorders affecting the brain ( NCT02362438).
2.8. Other Brain Disorders
The use of AADC as a therapeutic transgene has been undertaken for patients with acquired PD to restore l-dopa responsiveness. Muramatsu et al. (123) used rAAV2-hAADC in a phase I trial in which vector was injected directly into the putamen, resulting in measurable improvements in motor score and by positron emission tomography–computed tomography. Similar results were obtained by Christine et al. (124), with rAAV2-hAADC vector administered by intraputaminal injection in PD patients. As recently reviewed by Cartier and colleagues (125), gene therapy for Alzheimer disease is under development in a number of laboratories. Gene therapy for CNS diseases has been extensively reviewed elsewhere (126).
2.9. Inborn Errors of Metabolism, Glycogen Storage Disorders, and Other Liver Disease Trials
A number of other genetic disorders affecting the liver, including inborn errors of metabolism such as phenylketonuria and ornithine transcarbamylase deficiency ( NCT02991144), have been undertaken. Several clinical trials for Pompe disease ( NCT00976352, NCT02240407, NCT03533673) have been conducted, including a study looking at readministration of AAV; in the study, one leg is injected with immune modulation to ablate B cells followed by redosing of a contralateral leg four months later. Clinical trials have been initiated for other glycogen storage diseases such as addition glycogen storage disease type 1a ( NCT03517085).
3. LIMITATIONS AND RISKS OF rAAV
3.1. Packaging Capacity
The wild-type AAV2 genome, which is the backbone for most rAAV vectors, has a genome length of approximately 4,675 nucleotides, although it seems likely that there is natural variation of the wild-type genome length. One of the first systematic studies of the limitation on packaging capacity was published by Dong et al. (127), who showed a sharp decline in packaging efficiency between 4.8 and 5.0 kb. Subsequent studies indicated that certain oversized constructs might be packageable, especially in the context of certain serotypes or with elimination of VP2 expression (128, 129). Recently, it appears that the interpretation of many of these results was complicated by packaging of overlapping partial rAAV genomes, which were capable in some cases of recombining to regenerate full-length genomes in the target cell.
Given this, several strategies have been undertaken to bypass the packaging capacity limitation. The most consistently productive strategy has been to optimize the smallest possible designs for large transgenes. In the case of CFTR this has been done by using highly compact promoter, enhancer, and polyadenylation elements (130). In the case of larger coding sequences, such as dystrophin, the optimization of minigenes capable of restoring partial function has been an effective strategy (131, 132). Alternative approaches have included the use of overlapping constructs that may recombine by homologous recombination within the target cell or trans-splicing constructs. The typical trans-splicing approach includes a split intron, with the 5′-end of the transgene and the splice donor in one rAAV cassette and the 3′-end of the gene and the splice acceptor in a second cassette. The process of head-to-tail vector genome concatemerization allows such constructs to yield a functional expression unit within the target cell (133, 134).
3.2. Immune Response
Although AAV is generally considered a nonpathogenic virus, immune responses are still observed to both wild-type and recombinant viruses. Both capsid-specific and transgene-specific immune responses can hinder AAV gene therapy.
3.2.1. Humoral immune responses.
Preexisting immunity due to natural wild-type AAV infection continues to be a limitation for clinical rAAV applications. Neutralizing antibodies (NAbs) against different AAV capsid variants are readily found in the human population and are capable of blocking rAAV administration (84, 85). Several strategies are being tested to overcome NAb responses, but currently most clinical trials have seropositivity as an exclusion criterion.
It has been uniformly observed that the administration of rAAV vectors to any site outside the retina results in the generation of serum NAbs against the AAV capsid that was administered (135). Interestingly, cross NAbs against other AAV capsid variants have also been observed, at least in the case of rAAV1-IM administration (106). The prevalence of NAbs against different AAV capsid variants within healthy populations is a concern for gene therapies (136). Immune modulation of the blocking of the humoral immune responses may allow for readministration (137), and a number of clinical trials are now using immunosuppressive drugs that ablate B cells (138).
3.2.2. T cell–mediated responses.
The characterization of the immune responses observed in the early AAV2 hemophilia B trials has formed the basis for understanding of the primary T cell responses creating limitations to gene transfer in human applications. Similar findings have been demonstrated in subsequent trials with rAAV8-FIX (139), where investigators demonstrated that cytotoxic T cell responses were elicited against specific AAV capsid epitopes and lead to clearance of transduced cells (87). Many groups have begun using oral anti-inflammatory corticosteroids to control such responses, particularly with IV delivery. However, in two trials utilizing AAV1 by IM injection, a T-regulatory response to AAV capsid was associated with long-lived transgene expression (101, 108). T cell–mediated responses to AAV capsid are not easily modeled in animal models, making them difficult to study.
3.2.3. Innate immunity.
Initially, rAAV vectors were not commonly thought to activate early-phase innate immune response. However, some studies suggest that AAVs are sensed by Toll-like receptor 2 (TLR2) and TLR9 innate immune receptors in mice and humans (140–143). In addition, further questions about the innate immune response against AAV have been called into question with very high doses (2 × 1014 vg/kg) of rAAV9-like vectors (rAAVhu68 and rAAV-PHP.B) when given intravenously. In studies from the Wilson laboratory (97, 98), neonatal piglets and rhesus macaques were noted to have an early onset of liver toxicity with thrombocytopenia and toxicity of the dorsal root ganglia. Currently, it is unclear whether this represents a dose-limiting toxicity for rAAV generally or whether it is specific to certain variants.
3.2.4. Immune responses to transgene.
Transgenes can also elicit both B and T cell immune responses against transgenes, which is more frequently observed in patients who have null mutations. However, a T cell response against single amino acid common polymorphism was observed in a clinical trial for AAT deficiency (144). Additionally, delivery of therapeutic antibodies has been greatly limited by anti-antibody responses.
3.3. Risk of Tumorigenesis
There has been a long-standing controversy over whether wild-type AAV2 is occasionally associated with generation of tumors or whether in contrast it may actually be protective against tumorigenesis (145–147). While there is evidence on both sides of this question, it appears that the risk of carcinogenesis with this system is quite low, if it exists.
4. CURRENT TRENDS AND FUTURE DIRECTIONS OF rAAV GENE THERAPY
4.1. Further Technological Improvements in Capsids and Manufacturing
The widespread use of rAAV for gene therapy continues to be limited by the scalability of manufacturing methods. It is somewhat remarkable that there has not been a major change in the platforms in the past decade (148). It seems likely that the clinical development of rAAV will drive further investment in such innovations.
4.2. Use of rAAV to Deliver RNA Interference and Gene Editing Machinery
The ability of rAAV to deliver the molecular components required to silence genes by RNA inhibition and to engineer cells via gene editing is in its early stages of development (149). The Cas9 recombinase component of CRISPR/Cas9 systems will ideally be delivered transiently, which could be accomplished with rAAV in rapidly dividing cells, such as hematopoietic stem cells and epithelial surface cells. Meanwhile, ss-AAV genomes seem to be a particularly favorable template for homology directed repair–mediated editing. New Cas9s have considerable promise for in vivo genome editing using rAAVs with increased efficiencies, reduced size, and dinucleotide protospacer-adjacent motif sequences (150).
4.3. Future Clinical Applications
The future of rAAV gene therapy appears to be overwhelmingly positive, as clinical programs are advancing for a wide range of genetic diseases, including those affecting the retina, CNS, muscle, and liver. The most studied applications include hemophilia, Duchenne muscular dystrophy, and other retinal diseases, while the most remarkable improvements have been seen in patients with spinal muscular atrophy. These studies show promise of getting FDA approval, following in the footsteps of Luxturna and Zolgensma.
4.4. Economic Models of Rare Disease Treatment and Pricing of Approved Drugs
A considerable amount of resurgence in the field has come from biotechnology and pharmaceutical investment in gene therapy product development due to successes from Glybera and Luxturna but also economic models suggesting profitability from treatment of rare diseases. However, it is important to point out that of the two currently approved AAV-based therapies, Glybera has been removed from the market due to economic concerns. Glybera treatment costs were announced to be €1.11 million for an average adult (100, 151). However, the seemingly high price tag is not completely unreasonable when compared to yearly costs of enzyme replacement therapy, small molecule drugs, or monoclonal antibodies over a period of time, when AAV-based therapeutics would ideally be one-time treatments (152). Luxturna has also received criticism for a high price tag of $850,000 ($425,000 per eye), but it is difficult to put a price on regaining vision. Zolgensma treatment costs were announced at $2.1 million, or an annualized cost of $425,000 per year for 5 years, making it the world’s most expensive drug on the current market. However, when compared to Spinraza, the only other approved therapy for spinal muscular atrophy, which requires regular spinal infusions versus a one-time treatment, the 5-year costs for Zolgensma are lower considering Spinraza costs $750,000 the first year and $375,000 annually thereafter for life.
5. CONCLUSION
Our view is that rAAV will achieve its rightful place in the armamentarium of molecular therapies as the most safe and efficient means to correct single-gene defects in nondividing cells, such as neurons, retinal photoreceptors, and uninjured muscle and liver cells. It seems likely that the characteristics of AAV as a virus that exists in long-term symbiosis with its human host will suit it well as the vehicle for long-term gene transfer in humans. The limitations of this system related to immune responses seem likely to be overcome with specific immunosuppression, while those intrinsic to the packaging capacity of the virus will likely continue to exist. Overall, rAAV represents one of the most straightforward vehicles for direct translation of genomic revolution in medicine. In that context, rAAV-mediated gene therapy validates public investments in molecular genetics over the past half-century. The most valued outcome of such therapy will be to offer children and families with genetic diseases the opportunity to live longer and healthier lives.
ACKNOWLEDGEMENTS
T.R.F. would like to acknowledge funding from P01HL 131471-01 NIH/NHLBI.
Footnotes
DISCLOSURE STATEMENT
T.R.F. is a scientific cofounder of Applied Genetic Technologies, an unpaid advisor to Apic Bio, and a paid advisor to Beam Therapeutics, some of which may be developing technologies described here. The authors are not aware of any additional affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Atchison RW, Casto BC, Hammon WM. 1966. Electron microscopy of adenovirus-associated virus (AAV) in cell cultures. Virology 29:353–57 [DOI] [PubMed] [Google Scholar]
- 2.Blacklow NR, Hoggan MD, Kapikian AZ, Austin JB, Rowe WP. 1968. Epidemiology of adenovirus-associated virus infection in a nursery population. Am. J. Epidemiol 88:368–78 [DOI] [PubMed] [Google Scholar]
- 3.Blacklow NR, Hoggan MD, Rowe WP. 1967. Isolation of adenovirus-associated viruses from man. PNAS 58:1410–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blacklow NR, Hoggan MD, Sereno MS, Brandt CD, Kim HW, et al. 1971. A seroepidemiologic study of adenovirus-associated virus infection in infants and children. Am. J. Epidemiol 94:359–66 [DOI] [PubMed] [Google Scholar]
- 5.Schmidt OW, Cooney MK, Foy HM. 1975. Adeno-associated virus in adenovirus type 3 conjunctivitis. Infect. Immun 11:1362–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hauswirth WW, Berns KI. 1977. Origin and termination of adeno-associated virus DNA replication. Virology 78:488–99 [DOI] [PubMed] [Google Scholar]
- 7.Berns KI, Pinkerton TC, Thomas GF, Hoggan MD. 1975. Detection of adeno-associated virus (AAV)-specific nucleotide sequences in DNA isolated from latently infected Detroit 6 cells. Virology 68:556–60 [DOI] [PubMed] [Google Scholar]
- 8.Kotin RM, Linden RM, Berns KI. 1992. Characterization of a preferred site on human chromosome 19q for integration of adeno-associated virus DNA by non-homologous recombination. EMBO J 11:5071–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kotin RM, Menninger JC, Ward DC, Berns KI. 1991. Mapping and direct visualization of a region-specific viral DNA integration site on chromosome 19q13-qter. Genomics 10:831–34 [DOI] [PubMed] [Google Scholar]
- 10.Kotin RM, Siniscalco M, Samulski RJ, Zhu XD, Hunter L, et al. 1990. Site-specific integration by adeno-associated virus. PNAS 87:2211–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Srivastava A, Lusby EW, Berns KI. 1983. Nucleotide sequence and organization of the adeno-associated virus 2 genome. J. Virol 45:555–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laughlin CA, Tratschin JD, Coon H, Carter BJ. 1983. Cloning of infectious adeno-associated virus genomes in bacterial plasmids. Gene 23:65–73 [DOI] [PubMed] [Google Scholar]
- 13.Samulski RJ, Berns KI, Tan M, Muzyczka N. 1982. Cloning of adeno-associated virus into pBR322: rescue of intact virus from the recombinant plasmid in human cells. PNAS 79:2077–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Samulski RJ, Srivastava A, Berns KI, Muzyczka N. 1983. Rescue of adeno-associated virus from recombinant plasmids: gene correction within the terminal repeats of AAV. Cell 33:135–43 [DOI] [PubMed] [Google Scholar]
- 15.Samulski RJ, Chang LS, Shenk T. 1987. A recombinant plasmid from which an infectious adeno-associated virus genome can be excised in vitro and its use to study viral replication. J. Virol 61:3096–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samulski RJ, Chang LS, Shenk T. 1989. Helper-free stocks of recombinant adeno-associated viruses: Normal integration does not require viral gene expression. J. Virol 63:3822–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flotte TR, Afione SA, Solow R, Drumm ML, Markakis D, et al. 1993. Expression of the cystic fibrosis transmembrane conductance regulator from a novel adeno-associated virus promoter. J. Biol. Chem 268:3781–90 [PubMed] [Google Scholar]
- 18.Egan M, Flotte T, Afione S, Solow R, Zeitlin PL, et al. 1992. Defective regulation of outwardly rectifying Cl− channels by protein kinase A corrected by insertion of CFTR. Nature 358:581–84 [DOI] [PubMed] [Google Scholar]
- 19.Flotte TR, Afione SA, Conrad C, McGrath SA, Solow R, et al. 1993. Stable in vivo expression of the cystic fibrosis transmembrane conductance regulator with an adeno-associated virus vector. PNAS 90:10613–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flotte T, Carter B, Conrad C, Guggino W, Reynolds T, et al. 1996. A phase I study of an adeno-associated virus-CFTR gene vector in adult CF patients with mild lung disease. Hum. Gene Ther 7:1145–59 [DOI] [PubMed] [Google Scholar]
- 21.Flotte TR, Zeitlin PL, Reynolds TC, Heald AE, Pedersen P, et al. 2003. Phase I trial of intranasal and endobronchial administration of a recombinant adeno-associated virus serotype 2 (rAAV2)-CFTR vector in adult cystic fibrosis patients: a two-part clinical study. Hum. Gene Ther 14:1079–88 [DOI] [PubMed] [Google Scholar]
- 22.Fisher KJ, Kelley WM, Burda JF, Wilson JM. 1996. A novel adenovirus–adeno-associated virus hybrid vector that displays efficient rescue and delivery of the AAV genome. Hum. Gene Ther 7:2079–87 [DOI] [PubMed] [Google Scholar]
- 23.McCown TJ, Xiao X, Li J, Breese GR, Samulski RJ. 1996. Differential and persistent expression patterns of CNS gene transfer by an adeno-associated virus (AAV) vector. Brain Res. 713:99–107 [DOI] [PubMed] [Google Scholar]
- 24.Xiao X, Li J, McCown TJ, Samulski RJ. 1997. Gene transfer by adeno-associated virus vectors into the central nervous system. Exp. Neurol 144:113–24 [DOI] [PubMed] [Google Scholar]
- 25.Kessler PD, Podsakoff GM, Chen X, McQuiston SA, Colosi PC, et al. 1996. Gene delivery to skeletal muscle results in sustained expression and systemic delivery of a therapeutic protein. PNAS 93:14082–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao GP, Qu G, Faust LZ, Engdahl RK, Xiao W, et al. 1998. High-titer adeno-associated viral vectors from a Rep/Cap cell line and hybrid shuttle virus. Hum. Gene Ther 9:2353–62 [DOI] [PubMed] [Google Scholar]
- 27.Johnston J, Tazelaar J, Rivera VM, Clackson T, Gao GP, Wilson JM. 2003. Regulated expression of erythropoietin from an AAV vector safely improves the anemia of β-thalassemia in a mouse model. Mol. Ther 7:493–97 [DOI] [PubMed] [Google Scholar]
- 28.Herzog RW, Hagstrom JN, Kung SH, Tai SJ, Wilson JM, et al. 1997. Stable gene transfer and expression of human blood coagulation factor IX after intramuscular injection of recombinant adeno-associated virus. PNAS 94:5804–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Monahan PE, Samulski RJ, Tazelaar J, Xiao X, Nichols TC, et al. 1998. Direct intramuscular injection with recombinant AAV vectors results in sustained expression in a dog model of hemophilia. Gene Ther. 5:40–49 [DOI] [PubMed] [Google Scholar]
- 30.Flotte TR, Brantly ML, Spencer LT, Byrne BJ, Spencer CT, et al. 2004. Phase I trial of intramuscular injection of a recombinant adeno-associated virus alpha 1-antitrypsin (rAAV2-CB-hAAT) gene vector to AAT-deficient adults. Hum. Gene Ther 15:93–128 [DOI] [PubMed] [Google Scholar]
- 31.Brantly ML, Spencer LT, Humphries M, Conlon TJ, Spencer CT, et al. 2006. Phase I trial of intramuscular injection of a recombinant adeno-associated virus serotype 2 αl-antitrypsin (AAT) vector in AAT-deficient adults. Hum. Gene Ther 17:1177–86 [DOI] [PubMed] [Google Scholar]
- 32.Kaplitt MG, Leone P, Samulski RJ, Xiao X, Pfaff DW, et al. 1994. Long-term gene expression and phenotypic correction using adeno-associated virus vectors in the mammalian brain. Nat. Genet 8:148–54 [DOI] [PubMed] [Google Scholar]
- 33.During MJ, Samulski RJ, Elsworth JD, Kaplitt MG, Leone P, et al. 1998. In vivo expression of therapeutic human genes for dopamine production in the caudates of MPTP-treated monkeys using an AAV vector. Gene Ther. 5:820–27 [DOI] [PubMed] [Google Scholar]
- 34.Bankiewicz KS, Leff SE, Nagy D, Jungles S, Rokovich J, et al. 1997. Practical aspects of the development of ex vivo and in vivo gene therapy for Parkinson’s disease. Exp. Neurol 144:147–56 [DOI] [PubMed] [Google Scholar]
- 35.Janson C, McPhee S, Bilaniuk L, Haselgrove J, Testaiuti M, et al. 2002. Gene therapy of Canavan disease: AAV-2 vector for neurosurgical delivery of aspartoacylase gene (ASPA) to the human brain. Hum. Gene Ther 13:1391–412 [DOI] [PubMed] [Google Scholar]
- 36.Leone P, Shera D, McPhee SW, Francis JS, Kolodny EH, et al. 2012. Long-term follow-up after gene therapy for Canavan disease. Sci. Transl. Med 4:165ra3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li J, Wang D, Qian S, Chen Z, Zhu T, Xiao X. 2003. Efficient and long-term intracardiac gene transfer in δ-sarcoglycan-deficiency hamster by adeno-associated virus-2 vectors. Gene Ther. 10:1807–13 [DOI] [PubMed] [Google Scholar]
- 38.Beck SE, Jones LA, Chesnut K, Walsh SM, Reynolds TC, et al. 1999. Repeated delivery of adeno-associated virus vectors to the rabbit airway. J. Virol 73:9446–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rabinowitz JE, Rolling F, Li C, Conrath H, Xiao W, et al. 2002. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J. Virol 76:791–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Z, Zhu T, Qiao C, Zhou L, Wang B, et al. 2005. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat. Biotechnol 23:321–28 [DOI] [PubMed] [Google Scholar]
- 41.Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM. 2002. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. PNAS 99:11854–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gao G, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, et al. 2004. Clades of adeno-associated viruses are widely disseminated in human tissues. J. Virol 78:6381–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Inagaki K, Fuess S, Storm TA, Gibson GA, McTiernan CF, et al. 2006. Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol. Ther 14:45–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pacak CA, Mah CS, Thattaliyath BD, Conlon TJ, Lewis MA, et al. 2006. Recombinant adeno-associated virus serotype 9 leads to preferential cardiac transduction in vivo. Circ. Res 99:e3–9 [DOI] [PubMed] [Google Scholar]
- 45.Cearley CN, Wolfe JH. 2006. Transduction characteristics of adeno-associated virus vectors expressing cap serotypes 7, 8, 9, and Rh10 in the mouse brain. Mol. Ther 13:528–37 [DOI] [PubMed] [Google Scholar]
- 46.Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. 2009. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol 27:59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu P, Xiao W, Conlon T, Hughes J, Agbandje-McKenna M, et al. 2000. Mutational analysis of the adeno-associated virus type 2 (AAV2) capsid gene and construction of AAV2 vectors with altered tropism. J. Virol 74:8635–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang RL, McLaughlin T, Cossette T, Tang Q, Foust K, et al. 2009. Recombinant AAV serotype and capsid mutant comparison for pulmonary gene transfer of α−1-antitrypsin using invasive and noninvasive delivery. Mol. Ther 17:81–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Warrington KH Jr., Gorbatyuk OS, Harrison JK, Opie SR, Zolotukhin S, Muzyczka N. 2004. Adeno-associated virus type 2 VP2 capsid protein is nonessential and can tolerate large peptide insertions at its N terminus. J. Virol 78:6595–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li W, Asokan A, Wu Z, Van Dyke T, DiPrimio N, et al. 2008. Engineering and selection of shuffled AAV genomes: a new strategy for producing targeted biological nanoparticles. Mol. Ther 16:1252–60 [DOI] [PubMed] [Google Scholar]
- 51.Koerber JT, Jang JH, Schaffer DV. 2008. DNA shuffling of adeno-associated virus yields functionally diverse viral progeny. Mol. Ther 16:1703–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stiefelhagen M, Sellner L, Kleinschmidt JA, Jauch A, Laufs S, et al. 2008. Application of a haematopoetic progenitor cell-targeted adeno-associated viral (AAV) vector established by selection of an AAV random peptide library on a leukaemia cell line. Genet. Vaccines Ther 6:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang L, Jiang J, Drouin LM, Agbandje-McKenna M, Chen C, et al. 2009. A myocardium tropic adeno-associated virus (AAV) evolved by DNA shuffling and in vivo selection. PNAS 106:3946–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li W, Zhang L, Johnson JS, Zhijian W, Grieger JC, et al. 2009. Generation of novel AAV variants by directed evolution for improved CFTR delivery to human ciliated airway epithelium. Mol. Ther 17:2067–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maguire CA, Gianni D, Meijer DH, Shaket LA, Wakimoto H, et al. 2010. Directed evolution of adeno-associated virus for glioma cell transduction. J. Neurooncol 96:337–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lisowski L, Dane AP, Chu K, Zhang Y, Cunningham SC, et al. 2014. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature 506:382–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Santiago-Ortiz J, Ojala DS, Westesson O, Weinstein JR, Wong SY, et al. 2015. AAV ancestral reconstruction library enables selection of broadly infectious viral variants. Gene Ther. 22:934–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zinn E, Pacouret S, Khaychuk V, Turunen HT, Carvalho LS, et al. 2015. In silico reconstruction of the viral evolutionary lineage yields a potent gene therapy vector. Cell Rep. 12:1056–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li S, Ling C, Zhong L, Li M, Su Q, et al. 2015. Efficient and targeted transduction of nonhuman primate liver with systemically delivered optimized AAV3B vectors. Mol. Ther 23:1867–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ling C, Lu Y, Kalsi JK, Jayandharan GR, Li B, et al. 2010. Human hepatocyte growth factor receptor is a cellular coreceptor for adeno-associated virus serotype 3. Hum. Gene Ther 21:1741–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li J, Samulski RJ, Xiao X. 1997. Role for highly regulated rep gene expression in adeno-associated virus vector production. J. Virol 71:5236–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xiao X, Li J, Samulski RJ. 1998. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol 72:2224–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grimm D, Kern A, Rittner K, Kleinschmidt JA. 1998. Novel tools for production and purification of recombinant adenoassociated virus vectors. Hum. Gene Ther 9:2745–60 [DOI] [PubMed] [Google Scholar]
- 64.Grimm D, Kay MA, Kleinschmidt JA. 2003. Helper virus-free, optically controllable, and two-plasmid-based production of adeno-associated virus vectors of serotypes 1 to 6. Mol. Ther 7:839–50 [DOI] [PubMed] [Google Scholar]
- 65.Yang Q, Chen F, Trempe JP. 1994. Characterization of cell lines that inducibly express the adeno-associated virus Rep proteins. J. Virol 68:4847–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Clark KR, Voulgaropoulou F, Fraley DM, Johnson PR. 1995. Cell lines for the production of recombinant adeno-associated virus. Hum. Gene Ther 6:1329–41 [DOI] [PubMed] [Google Scholar]
- 67.Urabe M, Ding C, Kotin RM. 2002. Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum. Gene Ther 13:1935–43 [DOI] [PubMed] [Google Scholar]
- 68.Smith RH, Levy JR, Kotin RM. 2009. A simplified baculovirus-AAV expression vector system coupled with one-step affinity purification yields high-titer rAAV stocks from insect cells. Mol. Ther 17:1888–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Conway JE, Zolotukhin S, Muzyczka N, Hayward GS, Byrne BJ. 1997. Recombinant adeno-associated virus type 2 replication and packaging is entirely supported by a herpes simplex virus type 1 amplicon expressing Rep and Cap. J. Virol 71:8780–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Conway JE, Rhys CM, Zolotukhin I, Zolotukhin S, Muzyczka N, et al. 1999. High-titer recombinant adeno-associated virus production utilizing a recombinant herpes simplex virus type I vector expressing AAV-2 Rep and Cap. Gene Ther. 6:986–93 [DOI] [PubMed] [Google Scholar]
- 71.Thomas DL, Wang L, Niamke J, Liu J, Kang W, et al. 2009. Scalable recombinant adeno-associated virus production using recombinant herpes simplex virus type 1 coinfection of suspension-adapted mammalian cells. Hum. Gene Ther 20:861–70 [DOI] [PubMed] [Google Scholar]
- 72.Conrad CK, Allen SS, Afione SA, Reynolds TC, Beck SE, et al. 1996. Safety of single-dose administration of an adeno-associated virus (AAV)-CFTR vector in the primate lung. Gene Ther. 3:658–68 [PubMed] [Google Scholar]
- 73.Afione SA, Conrad CK, Kearns WG, Chunduru S, Adams R, et al. 1996. In vivo model of adeno-associated virus vector persistence and rescue. J. Virol 70:3235–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Flotte TR, Afione SA, Zeitlin PL. 1994. Adeno-associated virus vector gene expression occurs in nondividing cells in the absence of vector DNA integration. Am. J. Respir. Cell Mol. Biol 11:517–21 [DOI] [PubMed] [Google Scholar]
- 75.Wagner JA, Reynolds T, Moran ML, Moss RB, Wine JJ, et al. 1998. Efficient and persistent gene transfer of AAV-CFTR in maxillary sinus. Lancet 351:1702–3 [DOI] [PubMed] [Google Scholar]
- 76.Wagner JA, Moran ML, Messner AH, Daifuku R, Conrad CK, et al. 1998. A phase I/II study of tgAAV-CF for the treatment of chronic sinusitis in patients with cystic fibrosis. Hum. Gene Ther 9:889–909 [DOI] [PubMed] [Google Scholar]
- 77.Wagner JA, Messner AH, Moran ML, Daifuku R, Kouyama K, et al. 1999. Safety and biological efficacy of an adeno-associated virus vector–cystic fibrosis transmembrane regulator (AAV-CFTR) in the cystic fibrosis maxillary sinus. Laryngoscope 109:266–74 [DOI] [PubMed] [Google Scholar]
- 78.Wagner JA, Nepomuceno IB, Messner AH, Moran ML, Batson EP, et al. 2002. A phase II, double-blind, randomized, placebo-controlled clinical trial of tgAAVCF using maxillary sinus delivery in patients with cystic fibrosis with antrostomies. Hum. Gene Ther 13:1349–59 [DOI] [PubMed] [Google Scholar]
- 79.Flotte TR, Schwiebert EM, Zeitlin PL, Carter BJ, Guggino WB. 2005. Correlation between DNA transfer and cystic fibrosis airway epithelial cell correction after recombinant adeno-associated virus serotype 2 gene therapy. Hum. Gene Ther 16:921–28 [DOI] [PubMed] [Google Scholar]
- 80.Aitken ML, Moss RB, Waltz DA, Dovey ME, Tonelli MR, et al. 2001. A phase I study of aerosolized administration of tgAAVCF to cystic fibrosis subjects with mild lung disease. Hum. Gene Ther 12:1907–16 [DOI] [PubMed] [Google Scholar]
- 81.Kay MA, Manno CS, Ragni MV, Larson PJ, Couto LB, et al. 2000. Evidence for gene transfer and expression of factor IX in haemophilia B patients treated with an AAV vector. Nat. Genet 24:257–61 [DOI] [PubMed] [Google Scholar]
- 82.Manno CS, Chew AJ, Hutchison S, Larson PJ, Herzog RW, et al. 2003. AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood 101:2963–72 [DOI] [PubMed] [Google Scholar]
- 83.Buchlis G, Podsakoff GM, Radu A, Hawk SM, Flake AW, et al. 2012. Factor IX expression in skeletal muscle of a severe hemophilia B patient 10 years after AAV-mediated gene transfer. Blood 119:3038–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, et al. 2006. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med 12:342–47 [DOI] [PubMed] [Google Scholar]
- 85.Louis Jeune V, Joergensen JA, Hajjar RJ, Weber T. 2013. Pre-existing anti-adeno-associated virus antibodies as a challenge in AAV gene therapy. Hum. Gene Ther. Methods 24:59–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mingozzi F, Hasbrouck NC, Basner-Tschakarjan E, Edmonson SA, Hui DJ, et al. 2007. Modulation of tolerance to the transgene product in a nonhuman primate model of AAV-mediated gene transfer to liver. Blood 110:2334–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pien GC, Basner-Tschakarjan E, Hui DJ, Mentlik AN, Finn JD, et al. 2009. Capsid antigen presentation flags human hepatocytes for destruction after transduction by adeno-associated viral vectors. J. Clin. Invest 119:1688–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, et al. 2011. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med 365:2357–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nathwani AC, Reiss UM, Tuddenham EG, Rosales C, Chowdary P, et al. 2014. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med 371:1994–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Weber A, Engelmaier A, Voelkel D, Pachlinger R, Scheiflinger F, et al. 2018. Development of methods for the selective measurement of the single amino acid exchange variant coagulation factor IX Padua. Mol. Ther. Methods Clin. Dev 10:29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.McIntosh J, Lenting P, Rosales C, Lee D, Rabbanian, et al. 2013. Therapeutic levels of FVIII following single peripheral vein administration of rAAV vector encoding a novel human factor VIII variant. Blood 121(17):3335–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cideciyan AV, Aleman TS, Boye SL, Schwartz SB, Kaushal S, et al. 2008. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. PNAS 105:15112–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Maguire AM, Simonelli F, Pierce EA, Pugh EN Jr., Mingozzi F, et al. 2008. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N. Engl. J. Med 358:2240–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bainbridge JW, Smith AJ, Barker SS, Robbie S, Henderson R, et al. 2008. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N. Engl. J. Med 358:2231–39 [DOI] [PubMed] [Google Scholar]
- 95.Hauswirth WW, Aleman TS, Kaushal S, Cideciyan AV, Schwartz SB, et al. 2008. Treatment of Leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum. Gene Ther 19:979–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, et al. 2017. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet 390:849–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hinderer C, Katz N, Buza EL, Dyer C, Goode T, et al. 2018. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum. Gene Ther 29:285–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hordeaux J, Wang Q, Katz N, Buza EL, Bell P, Wilson JM. 2018. The neurotropic properties of AAV-PHP.B are limited to C57BL/6J mice. Mol. Ther 26:664–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Carpentier AC, Frisch F, Labbe SM, Gagnon R, de Wal J, et al. 2012. Effect of alipogene tiparvovec (AAV1-LPLS447X) on postprandial chylomicron metabolism in lipoprotein lipase-deficient patients. J. Clin. Endocrinol. Metab 97:1635–44 [DOI] [PubMed] [Google Scholar]
- 100.Gaudet D, Methot J, Dery S, Brisson D, Essiembre C, et al. 2013. Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPLS447X) gene therapy for lipoprotein lipase deficiency: an open-label trial. Gene Ther. 20:361–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ferreira V, Twisk J, Kwikkers K, Aronica E, Brisson D, et al. 2014. Immune responses to intramuscular administration of alipogene tiparvovec (AAV1-LPLS447X) in a phase II clinical trial of lipoprotein lipase deficiency gene therapy. Hum. Gene Ther 25:180–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gaudet D, Stroes ES, Methot J, Brisson D, Tremblay K, et al. 2016. Long-term retrospective analysis of gene therapy with alipogene tiparvovec and its effect on lipoprotein lipase deficiency-induced pancreatitis. Hum. Gene Ther 27:916–25 [DOI] [PubMed] [Google Scholar]
- 103.Song S, Morgan M, Ellis T, Poirier A, Chesnut K, et al. 1998. Sustained secretion of human alpha-1-antitrypsin from murine muscle transduced with adeno-associated virus vectors. PNAS 95:14384–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Song S, Scott-Jorgensen M, Wang J, Poirier A, Crawford J, et al. 2002. Intramuscular administration of recombinant adeno-associated virus 2 α−1 antitrypsin (rAAV-SERPINA1) vectors in a nonhuman primate model: safety and immunologic aspects. Mol. Ther 6:329–35 [DOI] [PubMed] [Google Scholar]
- 105.Brantly ML, Chulay JD, Wang L, Mueller C, Humphries M, et al. 2009. Sustained transgene expression despite T lymphocyte responses in a clinical trial of rAAV1-AAT gene therapy. PNAS 106:16363–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Flotte TR, Trapnell BC, Humphries M, Carey B, Calcedo R, et al. 2011. Phase 2 clinical trial of a recombinant adeno-associated viral vector expressing α1-antitrypsin: interim results. Hum. Gene Ther 22:1239–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mueller C, Gernoux G, Gruntman AM, Borel F, Reeves EP, et al. 2017. 5 year expression and neutrophil defect repair after gene therapy in alpha-1 antitrypsin deficiency. Mol. Ther 25:1387–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mueller C, Chulay JD, Trapnell BC, Humphries M, Carey B, et al. 2013. Human Treg responses allow sustained recombinant adeno-associated virus-mediated transgene expression. J. Clin. Invest 123:5310–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, et al. 2017. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med 377:1713–22 [DOI] [PubMed] [Google Scholar]
- 110.Borel F, Gernoux G, Sun H, Stock R, Blackwood M, et al. 2018. Safe and effective superoxide dismutase 1 silencing using artificial microRNA in macaques. Sci. Transl. Med 10:eaau6414. [DOI] [PubMed] [Google Scholar]
- 111.Worgall S, Sondhi D, Hackett NR, Kosofsky B, Kekatpure MV, et al. 2008. Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA. Hum. Gene Ther 19:463–74 [DOI] [PubMed] [Google Scholar]
- 112.Tardieu M, Zerah M, Husson B, de Bournonville S, Deiva K, et al. 2014. Intracerebral administration of adeno-associated viral vector serotype rh.10 carrying human SGSH and SUMF1 cDNAs in children with mucopolysaccharidosis type IIIA disease: results of a phase I/II trial. Hum. Gene Ther 25:506–16 [DOI] [PubMed] [Google Scholar]
- 113.Lee NC, Chien YH, Hu MH, Liu WS, Chen PW, et al. 2014. Treatment of congenital neurotransmitter deficiencies by intracerebral ventricular injection of an adeno-associated virus serotype 9 vector. Hum. Gene Ther 25:189–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lee NC, Muramatsu S, Chien YH, Liu WS, Wang WH, et al. 2015. Benefits of neuronal preferential systemic gene therapy for neurotransmitter deficiency. Mol. Ther 23:1572–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ahmed SS, Li H, Cao C, Sikoglu EM, Denninger AR, et al. 2013. A single intravenous rAAV injection as late as P20 achieves efficacious and sustained CNS gene therapy in Canavan mice. Mol. Ther 21:2136–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gessler DJ, Li D, Xu H, Su Q, Sanmiguel J, et al. 2017. Redirecting N-acetylaspartate metabolism in the central nervous system normalizes myelination and rescues Canavan disease. JCI Insight 2:e90807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gray-Edwards HL, Regier DS, Shirley JL, Randle AN, Salibi N, et al. 2017. Novel biomarkers of human GM1 gangliosidosis reflect the clinical efficacy of gene therapy in a feline model. Mol. Ther 25:892–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gray-Edwards HL, Brunson BL, Holland M, Hespel AM, Bradbury AM, et al. 2015. Mucopolysaccharidosis-like phenotype in feline Sandhoff disease and partial correction after AAV gene therapy. Mol. Genet. Metab 116:80–87 [DOI] [PubMed] [Google Scholar]
- 119.Rockwell HE, McCurdy VJ, Eaton SC, Wilson DU, Johnson AK, et al. 2015. AAV-mediated gene delivery in a feline model of Sandhoff disease corrects lysosomal storage in the central nervous system. ASN Neuro 7(2):1759091415569908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bradbury AM, Gray-Edwards HL, Shirley JL, McCurdy VJ, Colaco AN, et al. 2015. Biomarkers for disease progression and AAV therapeutic efficacy in feline Sandhoff disease. Exp. Neurol 263:102–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Golebiowski D, van der Bom IMJ, Kwon CS, Miller AD, Petrosky K, et al. 2017. Direct intracranial injection of AAVrh8 encoding monkey β-N-acetylhexosaminidase causes neurotoxicity in the primate brain. Hum. Gene Ther 28:510–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gray-Edwards HL, Randle AN, Maitland SA, Benatti HR, Hubbard SM, et al. 2018. Adeno-associated virus gene therapy in a sheep model of Tay-Sachs disease. Hum. Gene Ther 29:312–26 [DOI] [PubMed] [Google Scholar]
- 123.Muramatsu S, Fujimoto K, Kato S, Mizukami H, Asari S, et al. 2010. A phase I study of aromatic L-amino acid decarboxylase gene therapy for Parkinson’s disease. Mol. Ther 18:1731–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Christine CW, Starr PA, Larson PS, Eberling JL, Jagust WJ, et al. 2009. Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology 73:1662–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Alves S, Fol R, Cartier N. 2016. Gene therapy strategies for Alzheimer’s disease: an overview. Hum. Gene Ther 27:100–7 [DOI] [PubMed] [Google Scholar]
- 126.Hocquemiller M, Giersch L, Audrain M, Parker S, Cartier N. 2016. Adeno-associated virus-based gene therapy for CNS diseases. Hum. Gene Ther 27:478–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dong JY, Fan PD, Frizzell RA. 1996. Quantitative analysis of the packaging capacity of recombinant adeno-associated virus. Hum. Gene Ther 7:2101–12 [DOI] [PubMed] [Google Scholar]
- 128.Allocca M, Doria M, Petrillo M, Colella P, Garcia-Hoyos M, et al. 2008. Serotype-dependent packaging of large genes in adeno-associated viral vectors results in effective gene delivery in mice. J. Clin. Invest 118:1955–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Grieger JC, Samulski RJ. 2005. Packaging capacity of adeno-associated virus serotypes: impact of larger genomes on infectivity and postentry steps. J. Virol 79:9933–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yan Z, Sun X, Feng Z, Li G, Fisher JT, et al. 2015. Optimization of recombinant adeno-associated virus-mediated expression for large transgenes, using a synthetic promoter and tandem array enhancers. Hum. Gene Ther 26:334–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Watchko J, O’Day T, Wang B, Zhou L, Tang Y, et al. 2002. Adeno-associated virus vector-mediated minidystrophin gene therapy improves dystrophic muscle contractile function in mdx mice. Hum. Gene Ther 13:1451–60 [DOI] [PubMed] [Google Scholar]
- 132.Yue Y, Li Z, Harper SQ, Davisson RL, Chamberlain JS, Duan D. 2003. Microdystrophin gene therapy of cardiomyopathy restores dystrophin-glycoprotein complex and improves sarcolemma integrity in the mdx mouse heart. Circulation 108:1626–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Duan D, Yue Y, Engelhardt JF. 2001. Expanding AAV packaging capacity with trans-splicing or overlapping vectors: a quantitative comparison. Mol. Ther 4:383–91 [DOI] [PubMed] [Google Scholar]
- 134.Yan Z, Zhang Y, Duan D, Engelhardt JF. 2000. Trans-splicing vectors expand the utility of adeno-associated virus for gene therapy. PNAS 97:6716–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Halbert CL, Standaert TA, Aitken ML, Alexander IE, Russell DW, Miller AD. 1997. Transduction by adeno-associated virus vectors in the rabbit airway: efficiency, persistence, and readministration. J. Virol 71:5932–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Boutin S, Monteilhet V, Veron P, Leborgne C, Benveniste O, et al. 2010. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum. Gene Ther 21:704–12 [DOI] [PubMed] [Google Scholar]
- 137.Corti M, Cleaver B, Clement N, Conlon TJ, Faris KJ, et al. 2015. Evaluation of readministration of a recombinant adeno-associated virus vector expressing acid alpha-glucosidase in Pompe disease: preclinical to clinical planning. Hum. Gene Ther. Clin. Dev 26:185–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Corti M, Elder M, Falk D, Lawson L, Smith B, et al. 2014. B-cell depletion is protective against anti-AAV capsid immune response: a human subject case study. Mol. Ther. Methods Clin. Dev 1:14033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mingozzi F, Maus MV, Hui DJ, Sabatino DE, Murphy SL, et al. 2007. CD8+ T-cell responses to adeno-associated virus capsid in humans. Nat. Med 13:419–22 [DOI] [PubMed] [Google Scholar]
- 140.Hosel M, Broxtermann M, Janicki H, Esser K, Arzberger S, et al. 2012. Toll-like receptor 2-mediated innate immune response in human nonparenchymal liver cells toward adeno-associated viral vectors. Hepatology 55:287–97 [DOI] [PubMed] [Google Scholar]
- 141.Zaiss AK, Liu Q, Bowen GP, Wong NC, Bartlett JS, Muruve DA. 2002. Differential activation of innate immune responses by adenovirus and adeno-associated virus vectors. J. Virol 76: 4580–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhu J, Huang X, Yang Y. 2009. The TLR9-MyD88 pathway is critical for adaptive immune responses to adeno-associated virus gene therapy vectors in mice. J. Clin. Invest 119:2388–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Martino AT, Suzuki M, Markusic DM, Zolotukhin I, Ryals RC, et al. 2011. The genome of self-complementary adeno-associated viral vectors increases Toll-like receptor 9-dependent innate immune responses in the liver. Blood 117:6459–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Calcedo R, Somanathan S, Qin Q, Betts MR, Rech AJ, et al. 2017. Class I-restricted T-cell responses to a polymorphic peptide in a gene therapy clinical trial for α−1-antitrypsin deficiency. PNAS 114:1655–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nault JC, Datta S, Imbeaud S, Franconi A, Mallet M, et al. 2015. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat. Genet 47:1187–93 [DOI] [PubMed] [Google Scholar]
- 146.Berns KI, Byrne BJ, Flotte TR, Gao G, Hauswirth WW, et al. 2015. Adeno-associated virus type 2 and hepatocellular carcinoma? Hum. Gene Ther 26:779–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Srivastava A, Carter BJ. 2017. AAV infection: protection from cancer. Hum. Gene Ther 28:323–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Snyder RO, Flotte TR. 2002. Production of clinical-grade recombinant adeno-associated virus vectors. Curr. Opin. Biotechnol 13:418–23 [DOI] [PubMed] [Google Scholar]
- 149.Keeler AM, ElMallah MK, Flotte TR. 2017. Gene therapy 2017: progress and future directions. Clin. Transl. Sci 10:242–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Edraki A, Mir A, Ibraheim R, Gainetdinov I, Yoon Y, et al. 2019. A compact, high-accuracy Cas9 with a dinucleotide PAM for in vivo genome editing. Mol. Cell 73(4):714–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Narayanan G, Cossu G, Galli MC, Flory E, Ovelgonne H, et al. 2014. Clinical development of gene therapy needs a tailored approach: a regulatory perspective from the European Union. Hum. Gene Ther. Clin. Dev 25:1–6 [DOI] [PubMed] [Google Scholar]
- 152.Flotte TR. 2015. Ethical implications of the cost of molecularly targeted therapies. Hum. Gene Ther 26:573–74 [DOI] [PubMed] [Google Scholar]
- 153.Flotte TR, Keeler-Klunk AM. 2018. Regulatory pathway on the manufacture & quality control of recombinant adeno-associated virus vectors. Cell Gene Ther. Insights 4:89–99 [Google Scholar]