

Abstract
The angiotensin-converting enzyme 2 (ACE2) is an important regulator of the renin–angiotensin system and was very recently identified as a functional receptor for the SARS virus. The ACE2 sequence is similar (sequence identities 43% and 35%, and similarities 61% and 55%, respectively) to those of the testis-specific form of ACE (tACE) and the Drosophila homolog of ACE (AnCE). The high level of sequence similarity allowed us to build a robust homology model of the ACE2 structure with a root-mean-square deviation from the aligned crystal structures of tACE and AnCE less than 0.5 Å. A prominent feature of the model is a deep channel on the top of the molecule that contains the catalytic site. Negatively charged ridges surrounding the channel may provide a possible binding site for the positively charged receptor-binding domain (RBD) of the S-glycoprotein, which we recently identified [Biochem. Biophys. Res. Commun. 312 (2003) 1159]. Several distinct patches of hydrophobic residues at the ACE2 surface were noted at close proximity to the charged ridges that could contribute to binding. These results suggest a possible binding region for the SARS-CoV S-glycoprotein on ACE2 and could help in the design of experiments to further elucidate the structure and function of ACE2.
Keywords: ACE2, Structure prediction, SARS-CoV, S-Glycoprotein, Receptor, Binding
Enveloped viruses enter cells by binding their envelope glycoproteins to cell surface receptors followed by conformational changes leading to membrane fusion and delivery of the genome in the cytoplasm [1]. Very recently, the angiotensin-converting enzyme 2 (ACE2) was identified as a functional receptor for the SARS coronavirus (SARS-CoV) [2] and its binding site on the SARS-CoVS glycoprotein was localized between amino acid residues 303 and 537 [3]. ACE2 is a homolog of the metalloprotease angiotensin-converting enzyme ACE [4], [5] and was found to be an essential regulator of heart function [6]. ACE exists in two isoforms—somatic ACE, which has two homologous domains each containing an active catalytic site, and testis-specific ACE (tACE), which corresponds to the C domain of somatic ACE and has only one active site. ACE2 has a high level of similarity (sequence identities 43% and 35%, and similarities 61% and 55%, respectively) to tACE and the Drosophila homolog of ACE (AnCE). Recently, the crystal structures of tACE [7] and the Drosophila ACE homolog AnCE [8] have been determined at resolutions 2.0 and 2.4 Å, respectively.
These crystal structures were used as templates to build an accurate (root-mean-square deviation (rmsd) less than 0.5 Å) three-dimensional (3D) model of ACE2 by comparative (homology) modeling. Based on the ACE2 model, an analysis of the receptor-binding domain (RBD) of the SARS-CoV S-glycoprotein, and similarity with other interactions of viral envelope glycoproteins (Env) with receptors [9], we propose a possible mechanism of the ACE2 function as a receptor for the SARS virus. The analysis of the ACE2 model could also help in the design of experiments to further elucidate the structure and the dual function of ACE2.
Materials and methods
Homology modeling of the ACE2 structure. The sequences of ACE2, tACE, and AnCE were aligned by using the multiple sequence alignment program CLUSTALW [10]. The comparative modeling procedure COMPOSER [11], [12] implemented in SYBYL6.9 (Tripos, St. Louis, MO) was used to build a 3D model of the ACE2 structure. We used the tACE and AnCE structures to find out topologically equivalent residues based on structural alignment and the structurally conserved regions (SCRs) were modeled. The structurally variable regions (loops) were modeled by using loops either from the corresponding location of the homologous protein or from the general protein database. The 3D model of ACE2 was then subjected to energy minimization by using standard Tripos force fields and finally validated with the PROCHECK program [13]. The coordinates were deposited to the protein data bank (PDB) (code: 1RIX).
Modeling of a fragment containing the S-glycoprotein RBD. The threading was performed by the GeneFold module in SYBYL6.9 that uses three different scoring functions combining sequence and structure information [14]. The cytokine binding region of gp130 (PDB code 1BQU) was selected as a putative target structure with high scores from all the three scoring algorithms. The loops in the model were generated using protein loop search function of the GeneFold by scanning protein structural database for similar sequences. Among seven cysteine residues in the model, three pairs of disulfide bonds (S–S) were simulated and the resulting model was energy minimized. The model was validated with the PROCHECK program [13].
Modeling of glycosylation sites, electrostatic analysis, solvent accessibility, and surface hydrophobicity. The sequences of both ACE2 and the S RBD were scanned against the PROSITE [15] motifs in order to locate potential glycosylation sites. Six N-glycosylation sites with high probability of occurrences on ACE2 were predicted by PROSITE. Fully surface-exposed asparagine (N) residues were found at five of these sites, which were modeled by attaching N-acetylglucosamine moieties. Three N-glycosylation sites were found in the S RBD fragment and were modeled similarly. The areas of solvent accessibility (ASA) were calculated with a probe radius of 1.4 Å by using the Lee and Richards’ algorithm [16]. Electrostatistic potentials were calculated by using the program GRASP [17] with the following parameters: a protein dielectric constant of 2.0, a solvent dielectric constant of 80, an ion exclusion radius of 2.0 Å, a probe radius of 1.4 Å, and an ionic strength of 0.14 M. The calculated potentials were displayed at the solvent-accessible surface. The visualization of solvent accessibility, super-positioning of molecules, and calculation of surface hydrophobicity were performed by using InsightII. The hydrophobicity of the surface residues was calculated according to the Kyte–Doolittle method [18] with a window size of 5, and hydrophobic and hydrophilic levels of 0.7 and −2.4, respectively.
Results and discussion
The ACE2 model
To begin to understand the interactions between the SARS-CoV S-glycoprotein and its recently identified receptor, ACE2, we attempted to develop an accurate model of the ACE2 3D structure. Currently, the only computational methodology that allows prediction of protein 3D structures with high accuracy (an rms error lower than 2 Å) is comparative (homology) modeling. However, it requires sequence identity of the target protein with templates of known 3D structures higher than about 30% for accurate structure prediction. We found two proteins, tACE and AnCE, with available high resolution crystal structures, and ACE2 sequence identities of 43% and 35% (sequence similarities are 61% and 55%), respectively; the sequence alignment of ACE2 with tACE and AnCE2 is shown in Fig. 1 . Therefore, we have used homology modeling to build an accurate 3D model of ACE2 as described in Materials and methods.
Fig. 1.

Multiple sequence alignment of ACE2, tACE, and AnCE using CLUSTALW. The sequence numbering is the same as in the crystal structures. The N-glycosylation sites are underlined; putative binding residues in ACE2 are in boldface letters and boxed along with the corresponding aligned residues in tACE and AnCE. The identical and similar residues are shown in black and gray backgrounds, respectively.
The architecture of the ACE2 model is very similar to the crystal structure of tACE (Fig. 2A ). The superposition of the ACE2 model structure with the template structures of tACE and AnCE (Fig. 2B) shows very small deviation (rmsd less than 0.5 Å). A major feature of the ACE2 structure (and the template structures) is a deep channel on the top of the molecule that contains the catalytic site (Fig. 3A ). A comprehensive analysis of the structure and function of the catalytic site was very recently reported after our model was completed [19]; here we will not discuss the enzymatic function of ACE2 but rather use the enzymatic site location for reference purposes. The channel is surrounded by ridges containing loops, helices, and a portion of a β-sheet. The long loop between N210 and Q221 that is missing in tACE and AnCE (Fig. 1) is on the ACE2 surface (Fig. 2B); note that the orientation of ACE2 in Fig. 2B is different than in Fig. 2A in order to show this loop. Potential N-glycosylation sites were identified at six positions: 53, 90, 103, 322, 432, and 546, but only two of them (53 and 90) were aligned with the tACE structure (Fig. 1). They shared the pattern NXTX (except 103) and were modeled with a N-acetylglucosamine moiety (Fig. 3B). The direction of the main chain is illustrated in Fig. 3C.
Fig. 2.
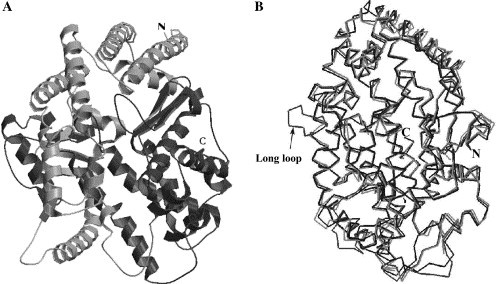
The model of the ACE2 structure. (A) A ribbon representation of the ACE2 model. The N- and C-termini are indicated. (B) Superposition of the ACE2 model structure with the crystal structures of tACE and AnCE based on the Cα-atoms of ACE2, tACE, and AnCE (ACE2, dark gray; tACE, light gray; and AnCE, black). The long loop inserted between N210 and Q221 that is unique for ACE2 is indicated.
Fig. 3.
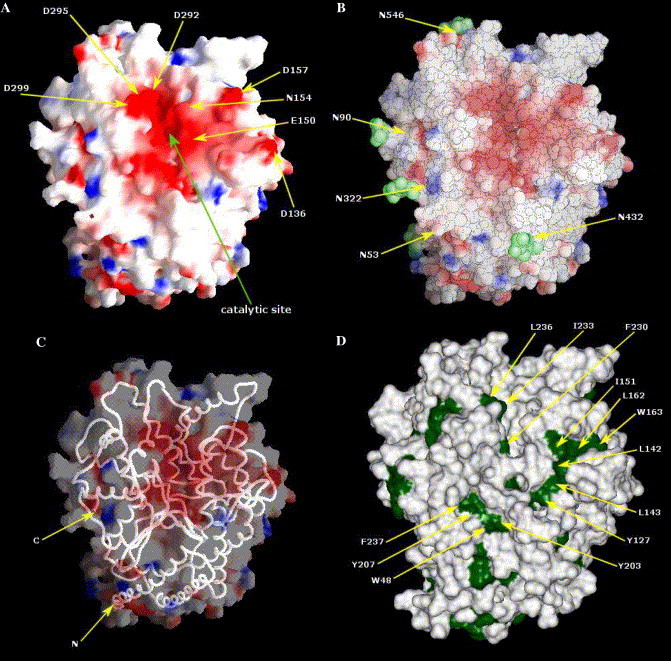
Analysis of the ACE2 model structure. (A) Representation of negative and positive molecular surface potentials in red and blue, respectively. The channel at the top of the molecule containing the catalytic site and the surrounding ridges containing negatively charged residues is indicated. (B) Distribution of glycosylation sites (green) on the ACE2 surface. (C) The backbone warm representing the orientation of the main chain. (D) Distribution of hydrophobic patches on the ACE2 surface.
ACE2 surface potential, solvent accessibility, hydrophobicity analysis, and carbohydrate distribution
Interactions of viral attachment proteins with protein receptor molecules are mostly determined by complementarity in surface charge distribution, hydrophobic interactions, and geometry; typically carbohydrates are excluded from the binding sites [9]. In an attempt to provide working hypothesis for possible regions involved in the interaction of the S-glycoprotein with its receptor we analyzed the ACE2 surface potential, solvent accessibility, hydrophobicity, and carbohydrate distribution. The surface of the deep channel at the top of ACE2 and the surrounding ridges is highly negatively charged (Fig. 3A). These ridges contain residues D136, E150, N154, D157, D292, D295, and D299 some of which have large ASA values, e.g., D136, N154, and D157 have 109, 108, and 80 Å2, respectively (Fig. 4 ). Comparison of these residues with the corresponding residues from ACE that do not support fusion mediated by the S-glycoprotein [2], and mouse ACE2 that binds to S but with somewhat lower affinity than human ACE2 (M. Farzan, personal communication) (Fig. 5 ) supports the possibility that some of these residues contribute to specific binding. The hydrophobicity analysis revealed distinct hydrophobic patches in close proximity to the negatively charged ridges (Fig. 3D). There are at least three hydrophobic regions comprising different residues including Phe, Trp, and Tyr which could contribute to binding in addition to the charged binding surface. All carbohydrate sites are topologically apart from the electronegative surface at the top of the molecule (Fig. 3B).
Fig. 4.
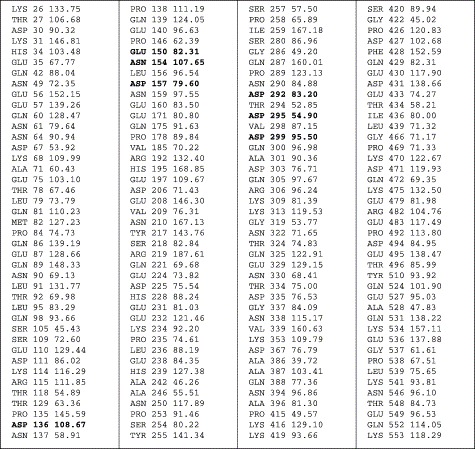
Solvent accessible surface areas (right column, in Å2) for ACE2 amino acid residues that are significantly exposed to solvent at the surface of the molecule. The cut-off for significant surface exposure here is assumed to be 45% ratio value defined as the ratio of side-chain surface area to a “random coil” value per residue in the tripeptide Gly–X–Gly. The middle column represents the amino acid residue number.
Fig. 5.

Conservation of amino acid residues in human ACE2, human ACE, and mouse ACE2 that could contribute to interactions with the S-glycoprotein. Identities are marked by a pipe (|), highly conservative replacements by a colon (:), and replacements with lower scores by a dot (·). The numbers denote the amino acid residue positions in the sequence. Note that the similarity of these ACE2 residues with the corresponding residues of mouse ACE2 is much higher than with the respective human ACE residues.
Proposition of receptor–S-glycoprotein binding interactions
The sequence similarity of the S-glycoprotein from the SARS virus with S-glycoproteins from other coronaviruses or other proteins whose structures are available in the PDB is about 20% or lower. The sequence similarity of the attachment glycoprotein (S1) from the SARS-CoV to other coronavirus S1 glycoproteins or other proteins with known 3D structures is even lower. Such low sequence similarity does not allow accurate homology modeling. Due to the absence of significant sequence similarity, we built a model by threading (data not shown) of a fragment (amino acid residues 300–537) containing the S RBD that we have recently identified [3]. The electrostatic analysis of the model revealed mostly positive charges on the surface and, particularly, an electronegative loop containing residues K439, R441, R444, H445, and K447. The hydrophobic analysis suggested several patches of hydrophobic residues around the positively charged loop region. One must note that although the size of the fragment is relatively small, the S RBD modeling has limitations in the absence of a template structure(s) with high sequence identity. Thus, the RBD model could significantly deviate or even be completely different from the real structure. In this aspect modeling of the much larger S1 and S2 units is even less reliable. For example, a recent model [20] of S1 and S2 proposed putative receptor (thought to be CD13) binding regions located in S2 instead of S1 where RBDs of coronaviruses should be. This is why we used our RBD model mostly as an illustration of possible complementary charged surfaces, hydrophobic patches, and β-sheets, and complemented it with an analysis of the secondary structure of the RBD fragment that also revealed predominance of β-sheets (data not shown). In progress are our experiments for the SARS-CoV S-glycoprotein RBD crystallization and determination of its 3D structure.
Typically virus receptors contain ridges that bind to cavities or to structures containing loops, cavities, and channels in the proteins mediating entry [9]. The model structure of ACE2 indicates that some or most of the ridges surrounding the cavity at the top of the molecule (Fig. 6 ) could serve as a likely binding region for the S-glycoprotein for the following reasons. First, the top of the molecule is far away of the membrane and is likely to be easier to reach than membrane proximal regions. Second, protruding structures are likely to be used for binding by viral proteins; it will also ensure geometric complementarity with concave surfaces as the S RBD domain could be based on our illustrative model (Fig. 6). Third, the negative charges of the ridges complement the positive charges of the RBD. Fourth, the hydrophobic patches around the charges could contribute to binding. Finally, the lack of carbohydrates at the top of the molecule could ensure high-affinity binding. Experiments currently in progress will determine the specific amino acid residues and their relative contribution to the interaction of ACE2 with the S-glycoprotein. The ACE2 model developed here, and this proposition of binding regions could help in the design and analysis of the experimental data, and the virus binding function of ACE2.
Fig. 6.
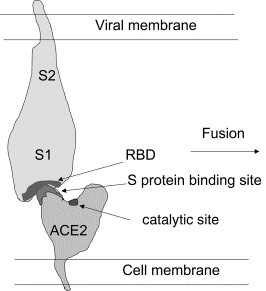
Schematic representation of the interaction between ACE2 and the SARS-CoV S-glycoprotein leading to binding and fusion. The RBD is depicted as a surface containing a cavity(s) that binds a ridge(s) close to the deep channel containing the catalytic site.
Acknowledgements
We thank M. Farzan for communicating unpublished data and interesting discussions, S. Ravichandran for helpful discussions, and the Advanced Biomedical Computing Center (ABCC), NCI-Frederick, for computing facilities.
References
- 1.Dimitrov D.S. Cell biology of virus entry. Cell. 2000;101:697–702. doi: 10.1016/s0092-8674(00)80882-x. [DOI] [PubMed] [Google Scholar]
- 2.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xiao X., Chakraborti S., Dimitrov A.S., Gramatikoff K., Dimitrov D.S. The SARS-CoV S-glycoprotein: expression and functional characterization. Biochem. Biophys. Res. Commun. 2003;312:1159–1164. doi: 10.1016/j.bbrc.2003.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Donoghue M., Hsieh F., Baronas E., Godbout K., Gosselin M., Stagliano N., Donovan M., Woolf B., Robison K., Jeyaseelan R., Breitbart R.E., Acton S. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 2000;87:E1–E9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 5.Tipnis S.R., Hooper N.M., Hyde R., Karran E., Christie G., Turner A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 6.Crackower M.A., Sarao R., Oudit G.Y., Yagil C., Kozieradzki I., Scanga S.E., Oliveira-dos-Santos A.J., da Costa J., Zhang L., Pei Y., Scholey J., Ferrario C.M., Manoukian A.S., Chappell M.C., Backx P.H., Yagil Y., Penninger J.M. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–828. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- 7.Natesh R., Schwager S.L., Sturrock E.D., Acharya K.R. Crystal structure of the human angiotensin-converting enzyme–lisinopril complex. Nature. 2003;421:551–554. doi: 10.1038/nature01370. [DOI] [PubMed] [Google Scholar]
- 8.Kim H.M., Shin D.R., Yoo O.J., Lee H., Lee J.O. Crystal structure of Drosophila angiotensin I-converting enzyme bound to captopril and lisinopril. FEBS Lett. 2003;538:65–70. doi: 10.1016/s0014-5793(03)00128-5. [DOI] [PubMed] [Google Scholar]
- 9.D.S. Dimitrov, Virus entry: molecular mechanisms and biomedical applications, Nat. Rev. Microbiol., 2004 (in press) [DOI] [PMC free article] [PubMed]
- 10.Thompson J.D., Higgins D.G., Gibson T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sutcliffe M.J., Hayes F.R., Blundell T.L. Knowledge based modelling of homologous proteins, Part II: Rules for the conformations of substituted sidechains. Protein Eng. 1987;1:385–392. doi: 10.1093/protein/1.5.385. [DOI] [PubMed] [Google Scholar]
- 12.Sutcliffe M.J., Haneef I., Carney D., Blundell T.L. Knowledge based modelling of homologous proteins, Part I: Three-dimensional frameworks derived from the simultaneous superposition of multiple structures. Protein Eng. 1987;1:377–384. doi: 10.1093/protein/1.5.377. [DOI] [PubMed] [Google Scholar]
- 13.Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:283–291. [Google Scholar]
- 14.Jaroszewski L., Rychlewski L., Zhang B., Godzik A. Fold prediction by a hierarchy of sequence, threading, and modeling methods. Protein Sci. 1998;7:1431–1440. doi: 10.1002/pro.5560070620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sigrist C.J., Cerutti L., Hulo N., Gattiker A., Falquet L., Pagni M., Bairoch A., Bucher P. PROSITE: a documented database using patterns and profiles as motif descriptors. Brief Bioinform. 2002;3:265–274. doi: 10.1093/bib/3.3.265. [DOI] [PubMed] [Google Scholar]
- 16.Lee B., Richards F.M. The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 17.Nicholls A., Sharp K.A., Honig B. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 18.Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 19.Guy J.L., Jackson R.M., Acharya K.R., Sturrock E.D., Hooper N.M., Turner A.J. Angiotensin-converting enzyme-2 (ACE2): comparative modeling of the active site, specificity requirements, and chloride dependence. Biochemistry. 2003;42:13185–13192. doi: 10.1021/bi035268s. [DOI] [PubMed] [Google Scholar]
- 20.Spiga O., Bernini A., Ciutti A., Chiellini S., Menciassi N., Finetti F., Causarono V., Anselmi F., Prischi F., Niccolai N. Molecular modelling of S1 and S2 subunits of SARS coronavirus spike glycoprotein. Biochem. Biophys. Res. Commun. 2003;310:78–83. doi: 10.1016/j.bbrc.2003.08.122. [DOI] [PMC free article] [PubMed] [Google Scholar]