

Abstract
Genome-scale screens for intraviral and virus–host protein interactions and the analysis of literature-curated datasets are able to provide a novel, comprehensive perspective of viruses, and virus-infected cells. Until now, large-scale interaction screens were predominantly performed with the yeast-two-hybrid (Y2H) system; however, alternative high-throughput technologies detecting binary protein interactions or protein complexes have been developed. Although many of the previous studies suffer from a rather poor validation of the results and few biological implications, these technologies potentially lead to a plethora of novel hypotheses. Here, we will give an overview of current approaches and their technical limitations, present recent examples and novel developments.
The Y2H system as the standard assay for the evaluation of interactomes
Essentially all high-throughput approaches to identify binary protein interactions on a genome-scale currently rely on the Gal4-based yeast-two-hybrid (Y2H) system (Figure 1 ) developed in 1989 [1]. In principle, two proteins are fused to separately expressed and nonfunctional domains of the Gal4 transcription factor, either the Gal4 DNA binding domain (bait) or the Gal4 activation domain (prey). Upon interaction of the two proteins of interest, the transcription factor activity is reconstituted in the yeast nucleus leading to the activation of one or several reporter genes. A major improvement was the introduction of a mating protocol in which pretransformed haploid yeast cells form diploids that carry both the bait and prey vector [2]. Compared to the previously used transformation protocol, this novel strategy is easier to perform and allows automated screening and the crosscombination of a large number of pretransformed bait and prey pairs. Another major improvement that further boosted genome-scale Y2H approaches was the combination with the highly efficient Gateway recombinational cloning system for the generation of larger clone collections [3]. Over the last 10 years, the interactomes of several previously sequenced organisms like H. pylori [4], C. jejuni [5], M. tuberculosis [6], P. falciparum [7], T. pallidum [8], S. cerevisiae [9, 10], C. elegans [11], D. melanogaster [12], and humans [13, 14] could be generated using the Y2H system. In most cases arrays were generated to test either individual or defined pools of open reading frames (ORFs) for interaction with each other (matrix screens). Alternatively, a number of individual baits were used to screen cDNA libraries. More recently, several viral interactomes with the herpesvirus family being by far the largest group analyzed have been generated (refs. [15••, 16••, 17•, 18••, 19••] and (E Fossum et al., in revision).
Figure 1.

High-throughput technologies to detect protein interactions. Graphic depiction of several approaches for the detection of binary protein interaction including the yeast-two-hybrid (Y2H) system, the nucleic acid programmable protein array (NAPPA), the LUMIER (luminescence-based mammalian interactome mapping), the protein fragment complementation assay (PCA), and the MAPPIT (mammalian protein–protein interaction trap). Multicomponent protein interactions can be analyzed by the TAP (tandem affinity purification) of ProtA-tagged bait proteins followed by mass spectrometry of associated proteins. Bait (x) and prey (y) proteins are indicated, fusion tags are shown in orange (AD: activation domain; DBD: DNA binding protein; ATG: start codon).
Limitations of the Y2H system
The reliability and the biological relevance of the Y2H system in general have been challenged repeatedly. Despite certain limitations the Y2H system is used by the majority of groups because of its enormous efficacy and the data discussed in this review are all based on Y2H screens as all currently published large-scale studies on intraviral or virus–host protein interactions are based on them. Clearly, since based on the nuclear localization of a transcriptional reporter system, it is limited in the analysis of transcriptional activators and proteins localized to membrane compartments. However, although the proteins to be tested are forced into the yeast nucleus for interaction, no bias between nuclear or non-nuclear proteins was observed [20••]. Interestingly, while the yeast system is not expected to provide translational modifications comparable to the mammalian cell, it is nonetheless able to introduce and report modification-dependent interactions up to a certain extent. Thus, the yeast cell offers an environment sufficiently natural for the analysis of protein interactions of other species [20••].
A major concern of the Y2H system particularly in its high-throughput application is the small overlap of identified interactions in comparative studies. This is exemplified by two S. cerevisiae proteome-wide Y2H approaches in which the screening of 6000 gene products identified 682 (Uetz-screen) and 843 (Ito-core) binary interactions [9, 10]. However, only 19% of the Uetz-screen and 15% of the Ito-core interactions were found in the respective other screen [9, 10]. A similarly small overlap was observed in several independently performed genome-wide herpesviral protein interaction screens [15••, 16••, 19••, 21, 22] (E Fossum et al., in revision). Recent large-scale efforts demonstrated by reinvestigating a set of random protein interactions of the S. cerevisiae and human interactomes that the low coverage is the result of low sensitivity rather than low specificity [20••, 23••, 24••]. Several validation protocols performed in parallel (see below) confirmed 25–30% of the Y2H interactions suggesting that the Y2H approach does not yield more false positives than other assays that detect binary interactions and is comparable in quality to literature-curated data [25••].
Several reasons are likely to account for the low sensitivity of Y2H approaches [23••]. First, only a fraction of all possible pairwise combinations are actually tested in a given screening situation. Second, depending on the assay or screening protocol applied different spaces are screened. This is demonstrated by studies on Hepatitis C virus (HCV) where the same set of proteins was screened by a yeast mating (IMAPI) as well as a transformation protocol (IMAPII) using two different human cDNA libraries [18••]. Although performed in the same laboratory, IMAPI and IMAPII shared only 22 interactions indicating that different screening protocols, vectors, and yeast strains as well as the quality and composition of cDNA libraries have a greater impact on the screening success than generally assumed. Third, high-throughput approaches are often hampered by technical limitations that can be improved for example by multiple screenings of the same set of proteins. Multiple sampling of the same search space allowed the identification of 80–90% while a single round revealed only 60% of all possible Y2H interactions [23••]. The ORFeomes of five evolutionarily related herpesviruses were used to systematically address the low coverage of Y2H screens by comparing the interactions identified in individual species followed by secondary methods [15••] (E Fossum et al., in revision). In the initial Y2H screens 283 interactions of the 41 core orthologous proteins were observed and 113 interactions were found in more than one species (E Fossum et al., in revision). On the basis of 55 Y2H interactions detected in KSHV, 59 of 92 interactions predicted for the corresponding orthologs in HSV-1, mCMV, and EBV could be identified by coprecipitation. In conclusion, the low coverage of the Y2H system — currently its major drawback — can be addressed either by technical improvements or the combination with other assays.
Alternative technologies
To address the biological significance of an interaction identified by Y2H, validation by one or more biochemical methods is required (Figure 1). Technologies similar to the Y2H, for example an assay based on closeness of two proteins if not their direct interaction likely leads to high confirmation rates, however be little complementary [20••]. Coexpression in mammalian cells of two proteins fused to distinct tags followed by immuno-coprecipitation or affinity-coprecipitation (CoP) was successfully applied to validate several Y2H studies [15••, 17•] (E Fossum et al., in revision). This method, however, is time consuming and thus not applicable to analyze large sets of interactions. Recently, a tool-box for high-throughput validation of Y2H interactions was developed (Figure 1; [20••]). Expression of proteins on array-printed template DNA using a coupled in vitro transcription–translation reaction (nucleic acid programmable protein array, NAPPA, [26]) follows a principle similar to the CoP. However, since performed in vitro under a defined but artificial environment it is technically challenging. A second method used on a rather large scale is the LUMIER (luminescence-based mammalian interactome mapping) pull-down assay where two proteins are expressed in mammalian cells. While the bait protein is expressed as a fusion to the protein A-tag or Flag-tag to immobilize the complex, the prey protein is expressed as a fusion with luciferase allowing the detection of the coisolated prey [27]. Protein fragment complementation assays (PCA), for example the split-YFP system in which interacting bait and prey proteins are fused to YFP domains, reconstitute an enzyme or a fluorescent protein and generally do not require an enrichment of the interactors. Thus, they might be more easily performed in an automated large-scale fashion [28]. Finally, the MAPPIT (mammalian protein–protein interaction trap) uses a bait protein fused to a hybrid erythropoietin–leptin receptor located to the plasmamembrane and a prey protein fused to gp130, which drives a signaling cascade resulting in the read-out of an endogenous transcriptional reporter [29].
Proteomic approaches that are able to detect indirect interactions are complementary to the Y2H system rather than confirmatory, and are therefore well suited to increase the coverage of a Y2H interactome. In the tandem affinity purification (TAP) approach individual proteins are fused to a cleavable tandem tag composed for example of protein A or G and a calmodulin binding peptide (CBP), and the isolated proteins present in the pulled-down complex are subsequently identified by mass spectrometry as done for yeast (Figure 1; [30, 31, 32]). Similarly, smaller subsets of human proteins have been analyzed in higher eukaryotic cells [33, 34, 35]. Since the systematic tagging of chromosomal genes is currently not feasible in these cells, tagged proteins have been introduced in addition to the endogenous proteins and thus compete with them in the pull-down analysis. Genetic systems are now available for many viral genomes (including large DNA viruses), and systematic functional TAP-tagging of viral proteins might be addressed in the near future. Proteomic analyses of purified virus particles have recently been performed for a few virus species and may provide further evidence for the interaction between viral proteins, particularly in conjunction with Y2H data (Figure 2 ) [36, 37].
Figure 2.
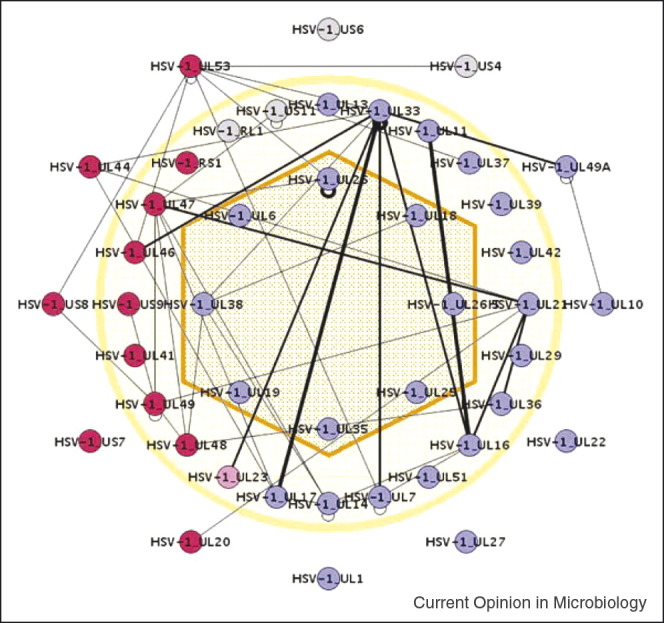
Scheme of HSV-1 virus particle with protein interactions detected in a genome-wide Y2H screen. Capsid, tegument, and glycoproteins are indicated depending on their localization in the virus particle. Proteins are colored according to their conservation, purple: conserved in all herpesviruses, red: in alpha herpesviruses, grey: in Herpes simplex virus, pink: in alpha and gamma herpesviruses.
Protein interaction networks in viruses
Initial ‘genome-wide’ studies concentrated on intraviral protein interactions of small RNA and DNA viruses. However, owing to the rather small number of proteins and protein interactions identified (Figure 3 ), a detailed network analysis could not be applied (reviewed by [38]). More recent approaches on intraviral interactomes include several members of the herpesvirus family [15••, 16••, 19••, 21, 22] (E Fossum et al., in revision) and the human SARS coronavirus (SARS-CoV, [17•]). The first combined virus–host interactomes were investigated in HCV [18••], EBV [16••], VZV and KSHV (Haas and collaborators). Virus–host protein interactions have been included into several public databases, or novel databases specific for virus–host interactions like VirHostNet have been set up [39].
Figure 3.

Global view of the interaction of two herpesviruses, VZV and KSHV, with the human proteome. Two experimental Y2H datasets were used to connect the two viral interactomes of VZV (red) and KSHV (pink) into a predicted high-confidence human interaction network consisting of 10 636 edges between 3169 nodes. Interactions between viral proteins are depicted in red or orange, interactions to cellular proteins in blue.
Hepatitis C virus (HCV)
In a systematic screen of 27 full-length proteins or domains of HCV against two human cDNA libraries, 314 virus–host interactions were identified [18••]. Taking published interactions into account, this is the first analysis on RNA viruses producing a large enough dataset to constitute a virus–human interaction network composed of 481 interactions involving 11 HCV and 421 human proteins. The most highly connected proteins were the NS3, NS5A, and core proteins with 214, 96, 76 interactors, respectively. Intriguingly, the insulin, Jak/STAT, and TGFβ signaling pathways were particularly enriched, which might be consistent with the metabolic disorders observed during chronic HCV infection. Focal adhesion complexes could represent a novel target of HCV suggesting a role of several HCV proteins in viral spreading, cell–cell interaction, and tissue reorganization. This analysis may thus help to identify potential new targets for HCV therapy.
Herpesviruses
The largest high-throughput approaches involving viruses have been performed with herpesviruses. Equipped with moderately large-sized genomes encoding a manageable but complex set of genes these viruses represent the ideal candidates for genome-wide Y2H approaches followed by bioinformatical analysis. Intraviral interactomes have been generated for several herpesviruses including the α-herpesviruses Herpes simplex virus type 1 (HSV-1, [21, 22], E Fossum et al., in revision) and Varicella zoster virus (VZV [15••]), the β-herpesvirus mouse Cytomegalovirus (mCMV), and the γ-herpesviruses Epstein–Barr virus (EBV [16••], E Fossum et al., in revision) and Kaposi's sarcoma herpesvirus (KSHV [15••, 19••]). In a comparison of the interactomes of five herpesviral species including 1007 intraviral interactions a core set of highly conserved protein interactions has been identified. Intriguingly, the interactions between the orthologous proteins were found to be conserved independent of their sequence homology. The topology of all herpesviral networks differed from cellular networks; however, it is difficult to judge whether this really reflects biological differences or artefacts caused by different setups used to evaluate them. In EBV [16••], HCV [18••], VZV and KSHV (Haas and collaborators), and KSHV (Haas and collaborators) viral proteins tend to interact with highly connected cellular proteins, which could be a general hallmark of many pathogen–host interactions [40••]. The datasets available to date are hampered by the low coverage of the Y2H screens performed, which makes it difficult to draw general biological conclusions. To reveal significant differences how different pathogens interact with the host proteome, protein interaction data with a considerably higher coverage of the screening space have to be generated.
To provide an example of the power of this approach, a comparison of the five herpesviral core networks identified the highly connected HSV-1 UL33 ortholog (VZV Orf25, mCMV M51, EBV BFRF4, and KSHV Orf67.5), which interacted with 14 tegument proteins (Figure 2, E Fossum et al., in revision). Interestingly, 11 out of 14 interactions were found in more than one species, a majority of which was confirmed by CoP. In addition, the interaction between UL33 orthologs and the HSV-1 UL31 orthologs (VZV Orf27, mCMV M53, EBV BFLF2, and KSHV Orf69) which in turn were found and previously published to interact with the HSV-1 UL34 orthologs (VZV Orf24, mCMV M50, EBV BFRF1, and KSHV Orf67) was highly conserved [41, 42, 43, 44, 45]. Likely these proteins form a large protein complex that mediates budding of capsids at the inner nuclear membrane of the host (Figure 4 ). The role of UL33 in DNA packaging and cleavage could point to a role in connecting capsid maturation with nuclear egress [46]. This is in line with these proteins being crucial for capsid formation and nuclear egress [41, 46, 47, 48, 49]. The identification of this protein complex thus demonstrates the potential of systems virology to reveal targets for alternative herpesviral therapies. This could be achieved by peptides or other small molecules introduced to interfere with targeting or assembly of the complex components.
Figure 4.

Nuclear egress of HSV-1 capsids. Herpesviral capsids are formed in the host nucleus and released to the cytoplasm by budding through the nuclear envelope. Primary envelopment at the inner nuclear membrane (INM) requires the membrane anchored UL34/UL31 family of proteins. The UL33 protein family interacts with this nuclear egress complex and may connect capsid packaging and nuclear egress (ER: endoplasmic reticulum; INM: inner nuclear membrane; ONM: outer nuclear membrane; NPC: nuclear pore complex).
SARS coronavirus (SARS-CoV)
Discovered in 2002, the SARS-CoV is thus far the largest RNA virus analyzed for genome-wide intraviral protein interactions [17•]. Its genome consists of approximately 29 700 nt and is predicted to encode 14 functional ORFs leading to approximately 30 structural and nonstructural protein products. Of the 900 pairwise interactions tested by a Y2H matrix analysis, 65 were positive and 35% of these could be confirmed by CoP. The intraviral interaction network revealed network parameters similar to herpesviruses [15••] and the combined published and experimental virus–host interactions suggest that SARS-CoV targets host functions like apoptosis, cell communication, and signaling.
Outlook
Large-scale protein interaction screens are able to provide large amounts of novel and unbiased data, but the extraction of biological implications from these screens is difficult, particularly if they are based on a technology with a rather low coverage as the Y2H system. In the near future, however, improved ‘deep’ screening technologies will lead to more comprehensive interaction maps of both intraviral and virus–host protein interactions, which, in combination with other genome-scale technologies like siRNA knock-down screens, transcriptional profiling, and spatial/temporal distribution studies may allow to setup improved models of virus-infected cells. The comparison of different viruses and, possibly, other pathogens may allow the identification of common strategies for infection and replication used by divergent pathogen groups, and the identification of targets for novel broad-spectrum antibiotics. On the other hand, it might reveal strategies that are specific for individual pathogens and help explain the characteristics of the infection with this particular pathogen. In combination with a genetic profiling of the infected host this approach will be even more powerful and might potentially lead to a step change in our understanding of viral infections.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
Acknowledgements
The work of the authors has been supported by grants provided by BayGene (Bayerisches Staatsministerium für Wissenschaft, Forschung und Kunst JH), DFG (SFB 576 JH, BA 1165/5-1 SB/JH), BMBF (NGFN+ JH), MRC (G050145, JH), and Scottish Funding Council (ICHAIR, JH).
Contributor Information
SM Bailer, Email: Bailer@mvp.uni-muenchen.de.
J Haas, Email: juergen.haas@ed.ac.uk.
References
- 1.Fields S., Song O. A novel genetic system to detect protein–protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 2.Fromont-Racine M., Rain J.C., Legrain P. Toward a functional analysis of the yeast genome through exhaustive two-hybrid screens. Nat Genet. 1997;16:277–282. doi: 10.1038/ng0797-277. [DOI] [PubMed] [Google Scholar]
- 3.Walhout A.J., Temple G.F., Brasch M.A., Hartley J.L., Lorson M.A., van den H.S., Vidal M. GATEWAY recombinational cloning: application to the cloning of large numbers of open reading frames or ORFeomes. Methods Enzymol. 2000;328:575–592. doi: 10.1016/s0076-6879(00)28419-x. [DOI] [PubMed] [Google Scholar]
- 4.Rain J.C., Selig L., De R.H., Battaglia V., Reverdy C., Simon S., Lenzen G., Petel F., Wojcik J., Schachter V. The protein–protein interaction map of Helicobacter pylori. Nature. 2001;409:211–215. doi: 10.1038/35051615. [DOI] [PubMed] [Google Scholar]
- 5.Parrish J.R., Yu J., Liu G., Hines J.A., Chan J.E., Mangiola B.A., Zhang H., Pacifico S., Fotouhi F., DiRita V.J. A proteome-wide protein interaction map for Campylobacter jejuni. Genome Biol. 2007;8:R130. doi: 10.1186/gb-2007-8-7-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raman K., Chandra N. Mycobacterium tuberculosis interactome analysis unravels potential pathways to drug resistance. BMC Microbiol. 2008;8:234. doi: 10.1186/1471-2180-8-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.LaCount D.J., Vignali M., Chettier R., Phansalkar A., Bell R., Hesselberth J.R., Schoenfeld L.W., Ota I., Sahasrabudhe S., Kurschner C. A protein interaction network of the malaria parasite Plasmodium falciparum. Nature. 2005;438:103–107. doi: 10.1038/nature04104. [DOI] [PubMed] [Google Scholar]
- 8.Titz B., Rajagopala S.V., Goll J., Hauser R., McKevitt M.T., Palzkill T., Uetz P. The binary protein interactome of Treponema pallidum — the syphilis spirochete. PLoS ONE. 2008;3:e2292. doi: 10.1371/journal.pone.0002292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uetz P., Giot L., Cagney G., Mansfield T.A., Judson R.S., Knight J.R., Lockshon D., Narayan V., Srinivasan M., Pochart P. A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- 10.Ito T., Chiba T., Ozawa R., Yoshida M., Hattori M., Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci U S A. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li S., Armstrong C.M., Bertin N., Ge H., Milstein S., Boxem M., Vidalain P.O., Han J.D., Chesneau A., Hao T. A map of the interactome network of the metazoan C. elegans. Science. 2004;303:540–543. doi: 10.1126/science.1091403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giot L., Bader J.S., Brouwer C., Chaudhuri A., Kuang B., Li Y., Hao Y.L., Ooi C.E., Godwin B., Vitols E. A protein interaction map of Drosophila melanogaster. Science. 2003;302:1727–1736. doi: 10.1126/science.1090289. [DOI] [PubMed] [Google Scholar]
- 13.Rual J.F., Venkatesan K., Hao T., Hirozane-Kishikawa T., Dricot A., Li N., Berriz G.F., Gibbons F.D., Dreze M., yivi-Guedehoussou N. Towards a proteome-scale map of the human protein–protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 14.Stelzl U., Worm U., Lalowski M., Haenig C., Brembeck F.H., Goehler H., Stroedicke M., Zenkner M., Schoenherr A., Koeppen S. A human protein–protein interaction network: a resource for annotating the proteome. Cell. 2005;122:957–968. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 15••.Uetz P., Dong Y.A., Zeretzke C., Atzler C., Baiker A., Berger B., Rajagopala S.V., Roupelieva M., Rose D., Fossum E., Haas J. Herpesviral protein networks and their interaction with the human proteome. Science. 2006;311:239–242. doi: 10.1126/science.1116804. [DOI] [PubMed] [Google Scholar]; See annotation to [16••].
- 16••.Calderwood M.A., Venkatesan K., Xing L., Chase M.R., Vazquez A., Holthaus A.M., Ewence A.E., Li N., Hirozane-Kishikawa T., Hill D.E. Epstein–Barr virus and virus human protein interaction maps. Proc Natl Acad Sci U S A. 2007;104:7606–7611. doi: 10.1073/pnas.0702332104. [DOI] [PMC free article] [PubMed] [Google Scholar]; Along with Refs. ([15••, 19••], Fossum et al., in revision) this study provides important insights into intraviral interactions of herpesviruses. Moreover, it presented the first virus–host interactome of a member of the herpesvirus family.
- 17•.von Brunn A., Teepe C., Simpson J.C., Pepperkok R., Friedel C.C., Zimmer R., Roberts R., Baric R., Haas J. Analysis of intraviral protein–protein interactions of the SARS coronavirus ORFeome. PLoS ONE. 2007;2:e459. doi: 10.1371/journal.pone.0000459. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study focused on the largest RNA virus thus far analyzed for genome-wide intraviral protein interactions. Identified virus–host interactions suggest that SARS-CoV targets host functions like apoptosis, cell communication, and signaling.
- 18••.de Chassey B., Navratil V., Tafforeau L., Hiet M.S., Aublin-Gex A., Agaugue S., Meiffren G., Pradezynski F., Faria B.F., Chantier T. Hepatitis C virus infection protein network. Mol Syst Biol. 2008;4:230. doi: 10.1038/msb.2008.66. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is the first analysis of virus–host interactions involving an RNA virus that produced a large enough dataset to constitute a virus–human interaction network. It represents an important step toward the identification of protein interactions relevant for HCV replication and spread.
- 19••.Rozen R., Sathish N., Li Y., Yuan Y. Virion-wide protein interactions of Kaposi's sarcoma-associated herpesvirus. J Virol. 2008;82:4742–4750. doi: 10.1128/JVI.02745-07. [DOI] [PMC free article] [PubMed] [Google Scholar]; The high-throughput protein interaction data of five different herpesviruses generated by Uetz et al. [15••], Calderwood et al. [16••], Rozen et al. [19••], as well as Fossum et al., in revision, represent the most comprehensive analysis of a family of viruses available to date. Identified interactions between evolutionarily conserved herpesvirus proteins enable the definition of a core orthologous network which may be useful to identify potential targets for alternative herpesviral therapies.
- 20••.Braun P., Tasan M., Dreze M., Barrios-Rodiles M., Lemmens I., Yu H., Sahalie J.M., Murray R.R., Roncari L., de Smet A.S. An experimentally derived confidence score for binary protein–protein interactions. Nat Methods. 2009;6:91–97. doi: 10.1038/nmeth.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]; A tool-box of high-throughput methods was tested for the validation of Y2H interactions including the mammalian protein–protein interaction trap (MAPPIT), the luminescence-based mammalian interactome (LUMIER), the yellow fluorescent protein (YFP) protein complementation assay (PCA), and the nucleic acid programmable protein array (NAPPA). This analysis aims at defining a confidence score for binary protein–protein interactions.
- 21.Vittone V., Diefenbach E., Triffett D., Douglas M.W., Cunningham A.L., Diefenbach R.J. Determination of interactions between tegument proteins of herpes simplex virus type 1. J Virol. 2005;79:9566–9571. doi: 10.1128/JVI.79.15.9566-9571.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee J.H., Vittone V., Diefenbach E., Cunningham A.L., Diefenbach R.J. Identification of structural protein–protein interactions of herpes simplex virus type 1. Virology. 2008;378:347–354. doi: 10.1016/j.virol.2008.05.035. [DOI] [PubMed] [Google Scholar]
- 23••.Yu H., Braun P., Yildirim M.A., Lemmens I., Venkatesan K., Sahalie J., Hirozane-Kishikawa T., Gebreab F., Li N., Simonis N. High-quality binary protein interaction map of the yeast interactome network. Science. 2008;322:104–110. doi: 10.1126/science.1158684. [DOI] [PMC free article] [PubMed] [Google Scholar]; These two sets of data define high-quality binary interaction maps of yeast and human protein networks. In particular, these datasets suggest that the Y2H data are more reliable than generally believed and that the Y2H screens currently performed suffer from low sensitivity rather than low specificity. Moreover, the distinct nature of binary versus cocomplex derived interactions is pointed out.
- 24••.Venkatesan K., Rual J.F., Vazquez A., Stelzl U., Lemmens I., Hirozane-Kishikawa T., Hao T., Zenkner M., Xin X., Goh K.I. An empirical framework for binary interactome mapping. Nat Methods. 2009;6:83–90. doi: 10.1038/nmeth.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]; These two sets of data define high-quality binary interaction maps of yeast and human protein networks. In particular, these datasets suggest that the Y2H data are more reliable than generally believed and that the Y2H screens currently performed suffer from low sensitivity rather than low specificity. Moreover, the distinct nature of binary versus cocomplex derived interactions is pointed out.
- 25••.Cusick M.E., Yu H., Smolyar A., Venkatesan K., Carvunis A.R., Simonis N., Rual J.F., Borick H., Braun P., Dreze M. Literature-curated protein interaction datasets. Nat Methods. 2009;6:39–46. doi: 10.1038/nmeth.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]; Curation of protein interactions from literature is a method applied to obtain comprehensive protein networks. However this analysis critically evaluates the current literature curation and shows that it is more error-prone and possibly of lower quality than generally assumed.
- 26.Ramachandran N., Hainsworth E., Bhullar B., Eisenstein S., Rosen B., Lau A.Y., Walter J.C., LaBaer J. Self-assembling protein microarrays. Science. 2004;305:86–90. doi: 10.1126/science.1097639. [DOI] [PubMed] [Google Scholar]
- 27.Barrios-Rodiles M., Brown K.R., Ozdamar B., Bose R., Liu Z., Donovan R.S., Shinjo F., Liu Y., Dembowy J., Taylor I.W. High-throughput mapping of a dynamic signaling network in mammalian cells. Science. 2005;307:1621–1625. doi: 10.1126/science.1105776. [DOI] [PubMed] [Google Scholar]
- 28.Nyfeler B., Michnick S.W., Hauri H.P. Capturing protein interactions in the secretory pathway of living cells. Proc Natl Acad Sci U S A. 2005;102:6350–6355. doi: 10.1073/pnas.0501976102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eyckerman S., Verhee A., der Heyden J.V., Lemmens I., Ostade X.V., Vandekerckhove J., Tavernier J. Design and application of a cytokine-receptor-based interaction trap. Nat Cell Biol. 2001;3:1114–1119. doi: 10.1038/ncb1201-1114. [DOI] [PubMed] [Google Scholar]
- 30.Gavin A.C., Bosche M., Krause R., Grandi P., Marzioch M., Bauer A., Schultz J., Rick J.M., Michon A.M., Cruciat C.M. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- 31.Ho Y., Gruhler A., Heilbut A., Bader G.D., Moore L., Adams S.L., Millar A., Taylor P., Bennett K., Boutilier K. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- 32.Krogan N.J., Cagney G., Yu H., Zhong G., Guo X., Ignatchenko A., Li J., Pu S., Datta N., Tikuisis A.P. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440:637–643. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- 33.Bouwmeester T., Bauch A., Ruffner H., Angrand P.O., Bergamini G., Croughton K., Cruciat C., Eberhard D., Gagneur J., Ghidelli S. A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol. 2004;6:97–105. doi: 10.1038/ncb1086. [DOI] [PubMed] [Google Scholar]
- 34.Burckstummer T., Bennett K.L., Preradovic A., Schutze G., Hantschel O., Superti-Furga G., Bauch A. An efficient tandem affinity purification procedure for interaction proteomics in mammalian cells. Nat Methods. 2006;3:1013–1019. doi: 10.1038/nmeth968. [DOI] [PubMed] [Google Scholar]
- 35.Ewing R.M., Chu P., Elisma F., Li H., Taylor P., Climie S., Broom-Cerajewski L., Robinson M.D., O’Connor L., Li M. Large-scale mapping of human protein–protein interactions by mass spectrometry. Mol Syst Biol. 2007;3:89. doi: 10.1038/msb4100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Viswanathan K., Fruh K. Viral proteomics: global evaluation of viruses and their interaction with the host. Expert Rev Proteomics. 2007;4:815–829. doi: 10.1586/14789450.4.6.815. [DOI] [PubMed] [Google Scholar]
- 37.Loret S., Guay G., Lippe R. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J Virol. 2008;82:8605–8618. doi: 10.1128/JVI.00904-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uetz P., Rajagopala S.V., Dong Y.A., Haas J. From ORFeomes to protein interaction maps in viruses. Genome Res. 2004;14:2029–2033. doi: 10.1101/gr.2583304. [DOI] [PubMed] [Google Scholar]
- 39.Navratil V., de C.B., Meyniel L., Delmotte S., Gautier C., Andre P., Lotteau V., Rabourdin-Combe C. VirHostNet: a knowledge base for the management and the analysis of proteome-wide virus–host interaction networks. Nucleic Acids Res. 2009;37:D661–D668. doi: 10.1093/nar/gkn794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40••.Dyer M.D., Murali T.M., Sobral B.W. The landscape of human proteins interacting with viruses and other pathogens. PLoS Pathog. 2008;4:e32. doi: 10.1371/journal.ppat.0040032. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper integrates protein interaction networks of several pathogens and identifies certain cellular processes that participate in interactions with different pathogen groups. This global view on infectious diseases may be a first step toward the identification of multiviral or multibacterial treatments.
- 41.Mettenleiter T.C. Budding events in herpesvirus morphogenesis. Virus Res. 2004;106:167–180. doi: 10.1016/j.virusres.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 42.Lake C.M., Hutt-Fletcher L.M. The Epstein–Barr virus BFRF1 and BFLF2 proteins interact and coexpression alters their cellular localization. Virology. 2004;320:99–106. doi: 10.1016/j.virol.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 43.Gonnella R., Farina A., Santarelli R., Raffa S., Feederle R., Bei R., Granato M., Modesti A., Frati L., Delecluse H.J. Characterization and intracellular localization of the Epstein–Barr virus protein BFLF2: interactions with BFRF1 and with the nuclear lamina. J Virol. 2005;79:3713–3727. doi: 10.1128/JVI.79.6.3713-3727.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Granato M., Feederle R., Farina A., Gonnella R., Santarelli R., Hub B., Faggioni A., Delecluse H.J. Deletion of Epstein–Barr virus BFLF2 leads to impaired viral DNA packaging and primary egress as well as to the production of defective viral particles. J Virol. 2008;82:4042–4051. doi: 10.1128/JVI.02436-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Santarelli R., Farina A., Granato M., Gonnella R., Raffa S., Leone L., Bei R., Modesti A., Frati L., Torrisi M.R., Faggioni A. Identification and characterization of the product encoded by ORF69 of Kaposi's sarcoma-associated herpesvirus. J Virol. 2008;82:4562–4572. doi: 10.1128/JVI.02400-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang K., Poon A.P., Roizman B., Baines J.D. Temperature-sensitive mutations in the putative herpes simplex virus type 1 terminase subunits pUL15 and pUL33 preclude viral DNA cleavage/packaging and interaction with pUL28 at the nonpermissive temperature. J Virol. 2008;82:487–494. doi: 10.1128/JVI.01875-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bubeck A., Wagner M., Ruzsics Z., Lotzerich M., Iglesias M., Singh I.R., Koszinowski U.H. Comprehensive mutational analysis of a herpesvirus gene in the viral genome context reveals a region essential for virus replication. J Virol. 2004;78:8026–8035. doi: 10.1128/JVI.78.15.8026-8035.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Farina A., Feederle R., Raffa S., Gonnella R., Santarelli R., Frati L., Angeloni A., Torrisi M.R., Faggioni A., Delecluse H.J. BFRF1 of Epstein–Barr virus is essential for efficient primary viral envelopment and egress. J Virol. 2005;79:3703–3712. doi: 10.1128/JVI.79.6.3703-3712.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lotzerich M., Ruzsics Z., Koszinowski U.H. Functional domains of murine cytomegalovirus nuclear egress protein M53/p38. J Virol. 2006;80:73–84. doi: 10.1128/JVI.80.1.73-84.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]