

Abstract
As one of the most abundant and highly conserved molecular chaperones, the 70‐kDa heat shock proteins (Hsp70s) play a key role in maintaining cellular protein homeostasis (proteostasis), one of the most fundamental tasks for every living organism. In this role, Hsp70s are inextricably linked to many human diseases, most notably cancers and neurodegenerative diseases, and are increasingly recognized as important drug targets for developing novel therapeutics for these diseases. Hsp40s are a class of essential and universal partners for Hsp70s in almost all aspects of proteostasis. Thus, Hsp70s and Hsp40s together constitute one of the most important chaperone systems across all kingdoms of life. In recent years, we have witnessed significant progress in understanding the molecular mechanism of this chaperone system through structural and functional analysis. This review will focus on this recent progress, mainly from a structural perspective.
Keywords: Hsp40, Hsp70, molecular chaperone, neurodegenerative diseases, protein folding, proteostasis
1. INTRODUCTION
As one of the most ubiquitous molecular chaperones, 70‐kDa heat shock proteins (Hsp70s) have been found in all species examined except for a number of archaea, and in all the cellular compartments of eukaryotes. Hsp70s regulate every aspect of cellular proteostasis including folding of both nascent and misfolded proteins, protein assembly, transportation into organelles, degradation, and preventing and dismantling protein aggregates.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 These fundamental housekeeping functions make Hsp70s essential especially for stressful conditions. It is not surprising that Hsp70s are one of the major molecular chaperones induced by wide ranges of stressful and pathological conditions such as elevated temperature, during which they provide essential protection for the cells and organisms to survive these harmful conditions.
It was proposed that the chaperone activity of Hsp70s is achieved through ATP‐regulated cycles of binding and releasing of polypeptide substrates. Hsp70s bind ATP, and possess a weak ATPase activity. At the same time, Hsp70s recognize and bind short stretches of exposed polypeptide segments rich in hydrophobic residues in extended conformations that are normally found in unfolded or misfolded proteins. Thus, Hsp70s have two essential intrinsic activities: first, as an ATPase and, second, in binding polypeptide substrates. These two activities are tightly coupled, and the chaperone activity is strictly dependent on this allosteric coupling.
In all the known proteostasis processes, the chaperone activities of Hsp70s are assisted by two classes of well‐characterized and essential cochaperones: Hsp40s and the nucleotide‐exchange factors (NEFs).2, 5, 6, 16, 17 The essential and functional conserved Hsp40s are characterized by the presence of the J‐domain and thus are also called J‐domain containing proteins. The well‐characterized role of Hsp40s is to directly speed up the rate of ATP hydrolysis by Hsp70s.18, 19, 20, 21, 22 At the same time, many Hsp40s have been shown to bind unfolded or partially folded polypeptides directly and thus have been proposed to bring polypeptide substrates to Hsp70s.20, 21, 22, 23, 24, 25 NEFs speed up the chaperone cycle of Hsp70s through accelerating the exchange of ADP for ATP after ATP hydrolysis.1, 26, 27 In summary, Hsp70s, Hsp40s, and NEFs constitute the most essential chaperone machinery for protein folding and proteostasis across all kingdoms of life. More recently, an important role of Hsp90s in assisting the Hsp70 folding machinery28, 29 and an emerging role of nucleotide‐exchange inhibitor in regulating Hsp70 in protein homeostasis30, 31 have been proposed.
2. THE Hsp70 MOLECULAR CHAPERONE
2.1. Domain organization and essential allosteric coupling
The sequences and structures of Hsp70s are highly conserved across all species examined.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 All Hsp70s contain two functional domains: a nucleotide‐binding domain (NBD) at the N‐terminus and a substrate‐binding domain (SBD) at the C‐terminus (Figure 1a), corresponding to the two essential intrinsic activities. Connecting these two functional domains is a short liner, the interdomain linker. The NBD is about 44 kDa, and it is the energy component. It is this domain that binds and hydrolyzes ATP to ADP to provide energy to power the chaperone activity. The SBD is the site to which polypeptide substrates bind so that Hsp70s can help them fold, assemble, transport across membranes, or degrade. In general, Hsp70s recognize short stretches of polypeptides of 5–7 residues in length with no sequence specificity but with a strong preference for hydrophobic residues, especially aliphatic amino acids, and residues with positive charges.32, 33, 34, 35, 36, 37, 38
Figure 1.
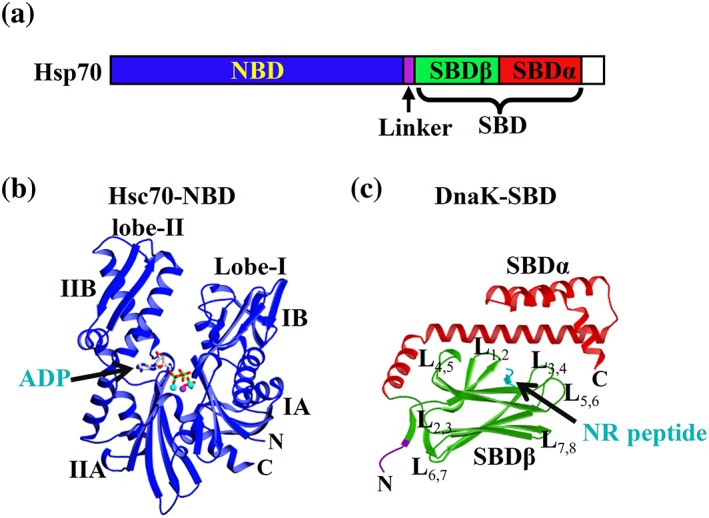
Domain organization and isolated domain structures of Hsp70s. (a) The domain organization of Hsp70s consisting of the nucleotide binding domain (NBD) and the substrate binding domain (SBD). (b) The isolated NBD structure in complex with ADP from bovine Hsc70 (PDB code: 1HPM). The bound ADP is shown as sticks. (c) The isolated SBD structure from DnaK (PDB code: 1DKX). Coloring is NBD (blue), interdomain linker (purple), SBDβ (green), SBDα (red), and the bound NR peptide (cyan). L stands for loop
Although each domain can bind their respective substrates tightly, the chaperone activity absolutely requires the functional coupling between these two domains upon ATP binding, the ATP‐induced allosteric coupling.1, 8, 39, 40, 41, 42, 43, 44, 45, 46 The nucleotide‐bound states of Hsp70s regulate the polypeptide‐substrate binding affinity and kinetics. In the absence of ATP, Hsp70s are in either the ADP‐bound form or the nucleotide‐free form (apo form). In these forms, there is little contact between the two domains.37, 39, 40, 43, 47, 48, 49, 50 The substrate‐binding properties behave as if the domains are isolated, characterized by relatively high affinity and slow kinetics.39, 40, 47, 49 In sharp contrast, upon ATP binding, extensive contacts form between the domains and these intimate contacts modulate both domains' activities. ATP binding to NBD reduces the peptide‐substrate binding affinity of the SBD by 2–3 orders of magnitudes through drastically accelerating the binding and release rates, especially for release.32, 51 At the same time, peptide‐substrate binding to SBD significantly speeds up the ATP hydrolysis rate of the NBD.5, 32 This ATP‐induced allosteric coupling ensures that the energy from ATP hydrolysis is efficiently used to power the chaperone activity. Recent advances in structural and biophysical studies have revolutionized our understanding of this allosteric coupling and the molecular mechanism of Hsp70 chaperone activity.8, 44, 52, 53, 54, 55, 56, 57 We will discuss these advances in detail below.
2.2. NBD structures
The first Hsp70 structure to be determined was the crystal structure of the isolated NBD from bovine Hsc70 in complex with ADP, published in 1990.58 Now, more than a dozen structures of the isolated NBD from a number of Hsp70s have been published or deposited to the Protein Data Bank (PDB).59, 60, 61, 62, 63, 64, 65, 66 Although bound with various nucleotides including ATP, all these structures are virtually identical in conformation. They all represent the ADP‐bound state regardless of what nucleotide is bound, or what ATP‐hydrolysis deficiency mutation is present. This suggests that the ADP‐bound conformation is much more stable for the isolated NBD than the active ATP‐bound conformation, and the NBD‐SBD contacts are required to maintain the active ATP‐bound conformation. As shown in Figure 1b, the NBD is composed of two large lobes: I and II. Between the two lobes is a deep cleft where the nucleotide binds. Each lobe is further divided into two subdomains: A and B. The bound nucleotide forms extensive contacts with all four subdomains, thus stabilizing this unique ADP‐bound conformation for the NBD.
NEFs are a class of well‐established cochaperones for Hsp70s. They were proposed to facilitate the chaperone activity by speeding up the exchange of ADP for ATP after ATP hydrolysis. A number of structural studies on the complexes of NEFs with isolated NBDs suggested that NEFs stabilize the nucleotide‐free state of NBD by prying open the nucleotide‐binding pocket.26, 27, 67, 68, 69, 70 The conformation of the nucleotide‐free state and mechanism of nucleotide exchange have been reviewed previously.8, 26, 27, 71
2.3. SBD structures
The SBD structure is represented by the crystal structure of the isolated SBD from DnaK, a major Hsp70 in E.coli.36 Since its identification as an Hsp70, DnaK has been widely used as a model Hsp70. This isolated DnaK SBD structure is the first SBD structure from an Hsp70. As shown in Figure 1c, the SBD is composed of two subdomains: SBDβ and SBDα. In this structure, a model peptide‐substrate NR (sequence NRLLLTG) was cocrystalized and bound to a narrow pocket formed in SBDβ. The SBDβ consists of eight β strands organized into two β sheets. The peptide‐binding pocket is formed between two loops, L1,2 and L3,4, and two β strands, β3 and β4. The SBDα covers the peptide‐binding pocket as a lid. All the following NMR and crystal structures on the isolated SBD revealed essentially the same conformation regardless of the presence or absence of peptide substrates.30, 36, 49, 52, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81 Except for one NMR study, all these structures have peptide substrates bound in the peptide‐binding pocket. Even the NMR study on SBD without peptide substrate reported virtually identical conformation but with more conformational dynamics.75 The bound peptide substrates were either added peptides, or the interdomain linker mimicking the peptide substrate. The bound peptide substrates could be the key determining factor for these crystallographic and NMR studies. Like the full‐length Hsp70s, the isolated SBDs tend to form poorly ordered oligomers.82, 83, 84, 85, 86, 87 Different from the functional dimers for Hsp70s,88, 89, 90 it is generally believed that these oligomers are formed through the binding of the interdomain linker to the peptide‐binding pocket of another molecule.17, 30, 49 This oligomerization may have some functional importance,91 but has presented challenges for structural studies.
2.4. Structures of full‐length Hsp70s in the ADP‐bound and nucleotide‐free states
After the publication of the isolated domain structures, the structures of full‐length Hsp70s, especially in the ATP‐bound state, the allosteric active state, were highly sought to understand the molecular mechanism of Hsp70 chaperone activity. However, this turned out to be challenging. Various biochemical and NMR studies have suggested that in the ADP‐bound and nucleotide‐free state, there is little contact between the two domains and each domain behaves as if isolated.37, 39, 40, 43, 47, 48, 49, 50 A NMR study on DnaK generated a positioning of the two functional domains in the ADP‐bound state (Figure 2a), providing a general picture of the overall conformation of this state.47 This positioning supports the idea that the two domains are relatively independent and there is no single fixed overall conformation for the ADP‐bound state. Two crystal structures of Hsp70s in complex with ADP have provided further support for this conformation.30, 49 The nucleotide‐free state structure of the bovine Hsc70 is the first crystal structure of a close‐ to full‐length Hsp70.73 In this structure, some small contacts between NBD and SBDα are revealed (Figure 2e); however, these contacts have been considered artifactual. One important feature for all these full‐length or close‐ to full‐length structures is that the SBD conformation is essentially identical to that of the isolated SBD. Although peptide substrates were not included in the crystallization conditions, either the interdomain linker or the truncated and unfolded C‐terminal of SBDα is bound to the peptide‐binding pocket, mimicking peptide‐substrate binding. In summary, these structural analyses are consistent with the biochemical studies and revealed a single conformation for the peptide‐binding pocket: closed on the bound peptide substrate (Figures 1c and 3a).
Figure 2.
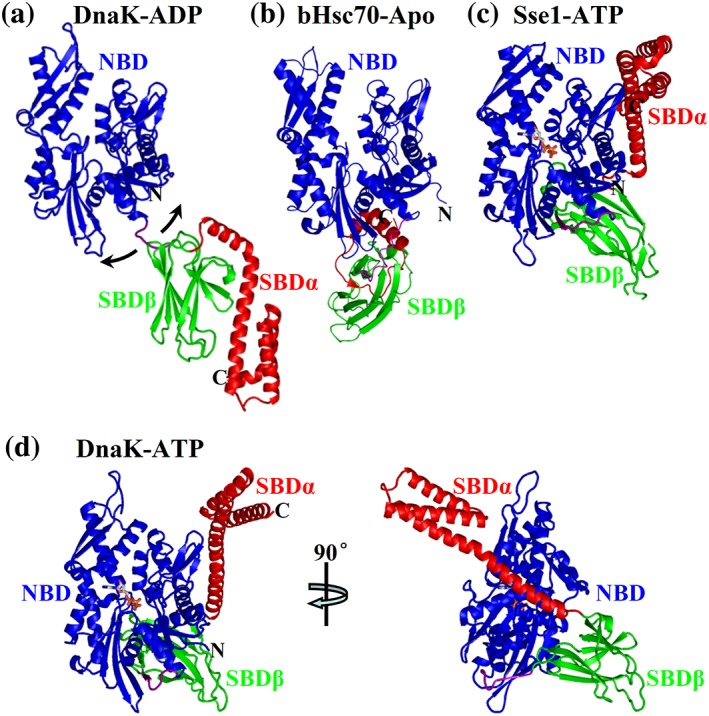
Representatives of full‐length structures of Hsp70s. (a) NMR positioning of DnaK NBD and SBD in a ADP‐bound state (PDB code: 2KHO). (b) The bHsc70 structure in a nucleotide‐free state (PDB code: 1YUW). (c) The structure of Sse1 in complex with ATP (PDB code: 2QXL). (d) The structure of DnaK in complex with ATP solved by our group (PDB code: 4JNE). Coloring is NBD (blue), interdomain linker (purple), SBDβ (green), and SBDα (red). ATP molecules are shown as sticks
Figure 3.
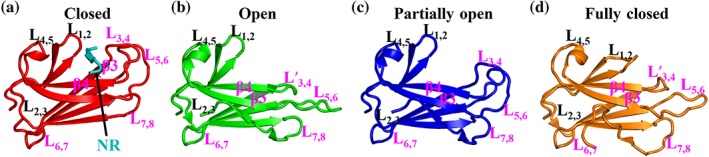
Conformations of the polypeptide‐binding pocket. The SBDβ structures from the isolated SBD of DnaK (a, PDB code: 1DKX), DnaK‐ATP solved by our group (b, PDB code: 4JNE), DnaK‐ATP solved by Mayer's group (c, PDB code: 4B9Q), BiP‐ATP2 (d, PDB code: 8ASY). For a, b, and c, the structures are aligned based on the Cα atoms of the L1,2 and L3,4. The BiP‐ATP2 structure (d) is aligned with the DnaK‐ATP structure (b) based on the Cα atoms of the β3‐β4 region
2.5. Structures of full‐length Hsp70s in the ATP‐bound state and the mechanism of allosteric coupling
In combination with recent NMR and FRET analysis, a number of structures of full‐length Hsp70s in the ATP‐bound state have provided unprecedented insights into allosteric coupling and have revolutionized our understanding of the molecular mechanism of Hsp70 chaperone activity.
Obtaining a crystal structure of a full‐length Hsp70 in the ATP‐bound state was challenging. There were three main hurdles to overcome: (a) the ATP‐bound state is short lived due to constant ATP hydrolysis; (b) the interaction between the two domains are too transient to be captured by structural analysis; and (c) purified Hsp70 proteins tend to form low‐ordered, flexible oligomers, which is incompatible with structural analysis. Moreover, all nonhydrolyzable ATP analogs tested so far have failed to induce allosteric coupling in Hsp70s in a way that ATP does. Stabilizing a functional ATP‐bound state that is compatible with structural analysis is the key.
The first full‐length Hsp70‐ATP structure determined was for Sse1 (Figure 2c).56 Sse1 is an Hsp110 chaperone from yeast. Hsp110s are distant homologs of Hsp70s.92, 93, 94 Sse1 has the advantage for crystallographic study that it does not have the aforementioned hurdles. This Sse1‐ATP structure revealed extensive domain contacts. More importantly, the domain contacts and relative orientation of the domains agree with many biochemical and NMR results on classic Hsp70s. One striking feature of this structure is the position of SBDα: it peels away from interacting with SBDβ in the isolated SBD structure and now docks on the side of NBD (compare Figure 2a,c). Without the SBDα lid covering, the peptide‐binding pocket is more accessible than the isolated SBD structure. This conformation is consistent with the fast kinetics and low affinity in the ATP‐bound state. However, this SBDα conformation alone is not sufficient to explain the drastically accelerated kinetics and reduced affinity in the ATP‐bound state since deletion of the SBDα only results in moderate decrease in affinity and increase in kinetics.75 Conformational changes in SBDβ, especially around the peptide‐binding pocket, must be essential for the allosteric coupling. However, the relatively low sequence conservation, especially in the SBDβ region, has limited the applicability of this Sse1‐ATP structure to Hsp70s.
Inspired by this Sse1‐ATP structure, two groups have published the first two intact Hsp70‐ATP structure using E. coli Hsp70 DnaK.53, 54 These two structures both took advantage of an ATPase‐deficient mutant T199A in the NBD.95 This mutation significantly reduced the ATPase activity but maintained allosteric coupling.40, 95 However, the T199A mutation alone is not sufficient for crystallization. The two groups used different strategies. The DnaK‐ATP structure solved by our group took advantage of a SBD mutation, L3,4′, which mimics the Sse1 sequence.96 The DnaK‐ATP structure solved by Mayer's group introduced a disulfide bond between NBD and SBDα based on the Sse1‐ATP structure, thus stabilizing the ATP‐bound state. Despite this difference, these two structures are almost identical. The overall conformations of these two DnaK‐ATP structures including the NBD conformation and location of the interdomain linker are highly similar to that of the Sse1‐ATP structure (Figure 2d), supporting the homology between Hsp70s and Hsp110s. Compared to the isolated SBD structures, which represent the ADP‐bound and nucleotide‐free states, there are two major conformational changes, which are dramatic. The first one is the position of SBDα, which was previously revealed by the Sse1‐ATP structure. The second major change is with the peptide‐binding pocket. In the isolated SBD structure, the two peptide‐binding loops close onto the bound peptide with favorable and extensive contacts (closed conformation, Figure 3a). In contrast, the L3,4 side of the peptide‐binding pocket, including L3,4 and its supporting loop L5,6, opens up in both the DnaK‐ATP structures although differing in the extend of opening. In the structure solved by our group, the L3,4 side is flipped wide open (open conformation, Figure 3b); whereas in Mayer's structure, the L3,4 side is only partially open (partially open confirmation, Figure 3c). Besides these loops, the bottom of the peptide‐binding pocket, formed by β3 and β4, is also flipped open at the L3,4 side. This occurs for both structures. Moreover, the L3,4 side in the DnaK‐ATP structure solved by Mayer's group has high B‐factors (constantly above 100 Å2, and some atoms are even over 200 Å2), and L5,6 in two of the four protomers are missing, suggesting high flexibility in these regions. Thus, the peptide‐binding pocket of the ATP‐bound state is dynamic and the two open conformations are most likely representatives of the conformational ensembles of the ATP‐bound state.
Based on the DnaK‐ATP structure solved by Mayer's group, structures of two specialized Hsp70s, Ssb1 and Ssz1, have been solved.57, 97 Our group solved a structure of human BiP, the major Hsp70 in ER, using an analogous construct as in the DnaK‐ATP structure.52 Almost identical conformation was observed for the peptide‐binding pocket for all these structures except for Ssz1 which has a nonclassical peptide‐binding pocket.
The open conformations of the peptide‐binding pocket are consistent with the fast kinetics and reduced affinity in the ATP‐bound state. However, with only these open conformations, it is hard to explain how Hsp70s decide when to bind and when to release substrates for productive chaperone cycles. Is binding and releasing substrate random or tightly controlled? Moreover, both the DnaK‐ATP structures depend on the ATPase‐deficient T199A mutation. The T199A mutation not only knocks out chaperone activity but also completely abolishes Hsp40 interaction.95, 98 It is still unclear which step of the ATP‐bound state that this T199A mutation represents. Interestingly, a new structure of human BiP, BiP‐ATP2, with a different conformation was solved in which the peptide‐binding pocket is completely closed (Figure 3d).55 Different from all the other published Hsp70‐ATP structures, this BiP‐ATP2 structure has a completely wild‐type (WT) NBD, that is, it does not carry the T199A mutation, although it contains the L3,4′ mutation. Thus, it most likely represents a different step in the ATP‐bound state. Solution studies further suggested that this fully closed conformation is the major conformation in solution for the ATP‐bound state. This conformation is incompatible with binding substrate. As the dominant conformation in solution for the ATP‐bound state, this conformation supports an active release of bound substrate upon ATP binding.
In summary, up to now, no WT Hsp70 structure in complex with ATP has been solved except the Sse1‐ATP structure. These structural studies revealed three conformations of the peptide‐binding pocket for the ATP‐bound state: open, partially open, and fully closed (Figure 3). A transition state from the ATP‐bound to the ADP‐bound has been proposed by an NMR study44 and is waiting for high‐resolution structural analysis.
2.6. Implications in chaperone activity and chaperone cycles
The classic paradigm for the Hsp70 chaperone cycle had evolved mainly based on essential allosteric coupling. In a simplified view, allosteric coupling is about how different nucleotide‐bound states control peptide‐substrate binding. Due to the fast kinetics in the ATP‐bound state, peptide substrate was proposed to bind in this state to start the chaperone cycle. Once the substrate binds, ATP hydrolysis is stimulated. With the high affinity and slow kinetics for peptide substrates, the ADP‐bound state holds the bound peptide for a while to prevent misfolding. Upon nucleotide exchange, Hsp70 reverts back to the ATP‐bound state. Due to the fast release rate, the bound peptide is released to fold on its own. The Hsp70‐ATP is available to bind other peptide substrates to restart a new chaperone cycle.
In combination with recent NMR and FRET studies, these new full‐length Hsp70‐ATP structures and their associated biochemical analysis have provided support for a more active chaperone cycle with tightly controlled substrate binding and release (Figure 4). The open conformations are for binding substrates to start chaperone cycles while the fully closed conformation is responsible for releasing bound substrates at the end of chaperone cycles after ATP rebinding. Moreover, Hsp40s were revealed to be the key to convert the fully closed conformation to the open conformations. In the ATP‐bound state, Hsp70 is mainly in the full‐closed conformation, and incompatible with substrate binding. Hsp40s not only bring substrates to Hsp70s but also convert Hsp70s to the open conformations to initial substrate binding. Once substrate binds, both substrate and Hsp40s stimulate the ATPase activity drastically. Now, Hsp70s are in ADP‐bound state, and Hsp40s are dissociated. Once ATP rebinds, Hsp70s revert back to the fully closed conformation, and the substrate is squeezed out of Hsp70s. This active release of bound substrate and Hsp40 regulation gives the chaperone cycle direction instead of random oscillation between the two nucleotide‐bound states.
Figure 4.
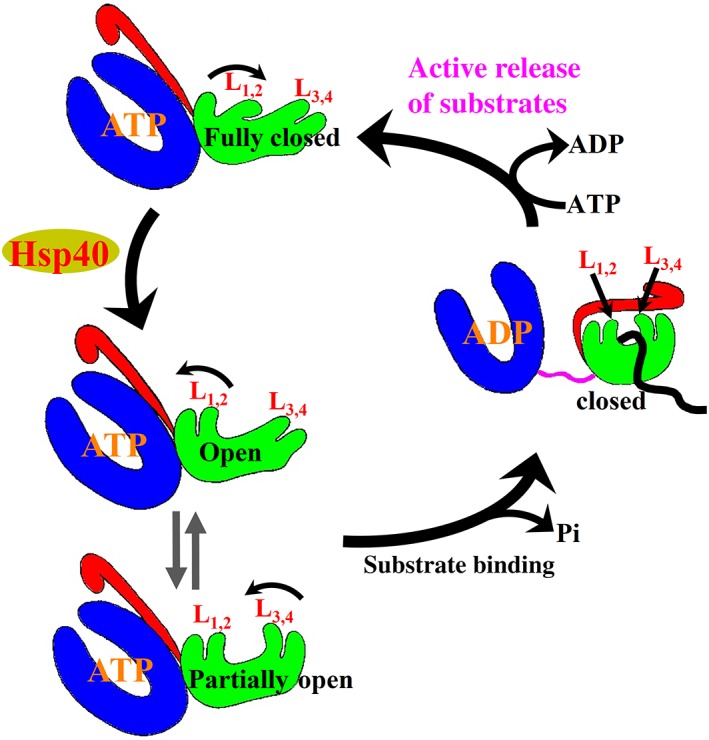
Schematic of the proposed Hsp70 chaperone cycle. Hsp70 domain color assignments are as in Figure 1a. Polypeptide substrate is highlighted in black
3. THE Hsp40 MOLECULAR CHAPERONES
3.1. Classification and functional diversity of Hsp40s
As a key class of cochaperones for Hsp70s, Hsp40s are more diverse than Hsp70s in both sequence and function. The conserved biochemical activity for all Hsp40s is to specifically interact with Hsp70‐ATP and stimulate ATP hydrolysis by the Hsp70.16, 19, 20, 23, 24, 99 Some Hsp40s can bind polypeptide substrates directly and show chaperone activity in preventing aggregation.17, 100, 101 Since the substrate binding preference of Hsp40s is different from that of Hsp70s, it was proposed that Hsp40s also serve as a substrate scanning factor for Hsp70s and bring polypeptide substrates to Hsp70s.102 Although Hsp40s are less conserved than Hsp70s, all share a conserved J‐domain named after the well‐studied E.coli Hsp40, DnaJ. The signature J‐domain is about 70 amino acids in length and has been shown to be critical for stimulating the ATPase activity of the Hsp70s.103, 104
Based on domain organizations, Hsp40s are classified into three classes: I, II, and III (Figure 5a). The class I and class II Hsp40s are relatively more conserved than the class III Hsp40 in both structure and function. This review mainly focuses on the first two classes. DnaJ, the founding member of class I, represents the domain organization of class I. Class I has five domains: J‐domain, glycine/phenylalanine rich (G/F) domain, carboxyl‐terminal domain 1 (CTD1) which contains a zinc finger‐like region (ZFLR), carboxyl‐terminal domain 2 (CTD2), and dimerization (D) domain. 16, 19, 20, 22 Class II has the same domain organization as DnaJ but with a CTD1 that does not contain ZFLR. CTD1 and CTD2 in both class I and class II Hsp40s are conserved in structure and are involved in binding polypeptide substrates. The role of ZFLR is still unclear although it seems essential for chaperone activity.105, 106 Class III is much more divergent with only the J‐domain conserved. Unlike the N‐terminal location in class I and class II Hsp40s, the J‐domain in class III Hsp40s can be in any location.
Figure 5.
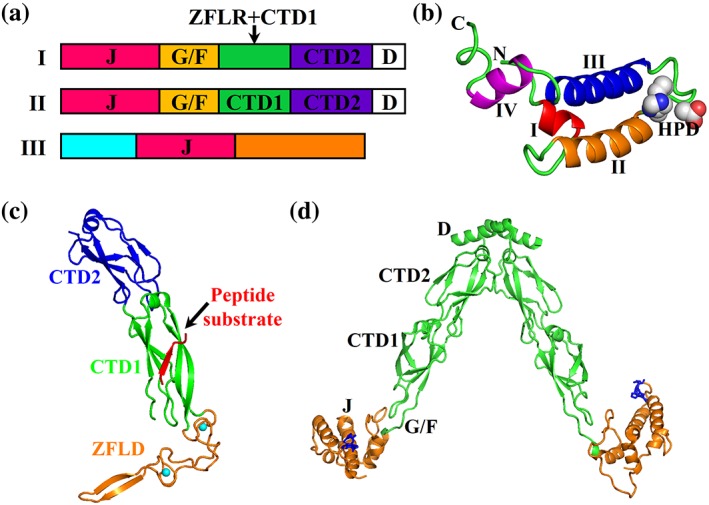
Hsp40 domain organization and structures. (a) Domain organization of Hsp40s (JDP). J, J‐domain; G/F, G/F rich domain; CTD, C‐terminal domain; ZFLR, zinc finger‐like region; D, dimerization domain. (b) Ribbon diagram of the J‐domain structure from the E. coli Hsp40 DnaJ (PDB code: 1XBL). (c) The CTD structure of a yeast Hsp40 Ydj1 (PDB code: 1NLT). The domains are labeled. The bound peptide substrate is highlighted in red, and the two associated Zn ions are colored in cyan. (d) The structural model of full‐length DnaJ from Thermus thermophiles (PDB code: 4 J80). The J‐domain is highlighted in orange, and the HPD motif is shown as blue sticks
The sequence and function diversity of Hsp40s was proposed to dictate the chaperone activity of Hsp70s and enable the diverse functions of Hsp70s.16, 19, 20, 23, 24, 99 Usually, there are more Hsp40 members than those of Hsp70s in any organism or a given cellular compartment. Various studies have suggested that multiple Hsp40s work with a single Hsp70 to specify the chaperone activity of the Hsp70. One of the well‐established roles of class I Hsp40s is assisting Hsp70s in general protein folding. Consistent with the sequence and domain diversity, many class III Hsp40s have specialized roles in assisting Hsp70s in proteostasis. A beautiful example is BiP, the single classic Hsp70 in ER, and its seven known Hsp40 cochaperones, ERdjs.25, 99, 107, 108 These ERdjs enable BiP to perform diverse functions in the ER: ERdj1 and ERdj2 are for protein import into ER,109, 110, 111 ERdj3 and ERdj6 are for protein folding,112, 113, 114, 115 and ERdj4 and ERdj5 are for protein degradation116 whereas the function of ERdj7 is unclear.99, 107 Moreover, a recent study suggested that class I and class II Hsp40s form complexes to assist Hsp70s in efficiently dismantling protein disaggregation.117 It is possible that different Hsp40s regulate the chaperone cycle of Hsp70s differently in order to achieve their diverse functions besides recognizing different substrates.
3.2. Isolated domain structures of Hsp40s
The NMR structures of the isolated J‐domains from two Hsp40s (E. coli Hsp40 DnaJ and human Hsp40 Hdj1) provided the first structural information for Hsp40s.118, 119, 120 These two Hsp40s belong to class I and class II Hsp40s, respectively. Following these structures, a number of crystal and NMR structures from all three classes were solved, and all revealed essentially the same fold for the J‐domain.121, 122, 123, 124, 125, 126, 127, 128, 129, 130, 131 The J‐domain is composed of four α‐helices: I to IV (Figure 5b). Between the helices II and III is the highly conserved HPD (histidine–proline–aspartate) motif. The HPD motif is essential for interacting with Hsp70s and stimulating the ATPase activity of Hsp70s.
The CTD including CTD1 and CTD2 is relatively conserved in class I and class II Hsp40s except for the presence of ZFLD in CTD1 of class I Hsp40s. The structure of CTD is represented by the crystal structure of the isolated CTD from Ydj1, a class I Hsp40 from yeast (Figure 5c).132 Except for the ZFLD, CTD1 and CTD2 have a similar fold. Each is made of two layers of β sheets. The CTD1 and CTD2 from class II hsp40 share a similar fold.128, 133, 134, 135 Interestingly, a peptide substrate was cocrystallized with the CTD of Ydj1 and revealed the structural basis of peptide binding. The peptide substrate (sequence: GWLYEIS) is bound to CTD1 by adding an antiparallel β‐strand to one of the two layers of the β‐sheets. The central Leu fits in a hydrophobic pocket formed on a conserved surface of CTD1. This Leu forms the most extensive and significant contacts with Ydj1, providing an explanation for the substrate preference of Hsp40s for hydrophobic residues. However, the shallow binding site for the peptide substrate raises concerns for the functional meaning. Thus, the precise mechanism of how class I and class II Hsp40s bind substrates is still under debate.
3.3. Full‐length structures of Hsp40s
At the present time, there is no high‐resolution full‐length structure known for either class I or class II Hsp40s. This could be due to the high conformational dynamics of these multidomain Hsp40s.22, 128 Both class I and class II Hsp40s are known to form dimers16, 20, 21, 23, 24, 136 although a recent study suggested that ERdj3, a class I Hsp40 in ER, functions as a tetramer.137 A structural model of full‐length DnaJ in dimeric form from T. thermophilus was built based on EPR and crystallographic results (Figure 5d).128 This DnaJ belongs to class II Hsp40s. The overall structure is a V‐shaped dimer with a large cleft formed between the two monomers. The two monomers dimerize through the extreme C‐terminal D domains. The J‐domains are at the end of each stalk formed by the two CTDs. Full‐length structures are available for a number of class III Hsp40s that facilitate the understanding of their unique chaperone activities.121, 133
4. THE Hsp70‐Hsp40 INTERACTIONS
The essential Hsp70‐Hsp40 interaction is one of the most important components for Hsp70 chaperone activity. Although Hsp70s are at the heart of this chaperone machinery, the chaperone activity in protein folding depends on the functional Hsp70‐Hsp40 interaction while Hsp70s by themselves have little activity in protein folding. As described above, Hsp40s assist Hsp70s in protein folding mainly through two mechanisms: stimulating ATP hydrolysis and bringing substrates to Hsp70s through directly binding to substrates. Recent advances in structural and biochemical analysis have made it possible to start to understand the molecular mechanism of the ATPase stimulation although the exact structural basis and molecular mechanism of the functional Hsp70‐Hsp40 interaction are still largely unknown.
Consistent with the well‐established role of Hsp40s in stimulating the ATP hydrolysis rate of Hsp70s, Hsp40s interact robustly with Hsp70s only in the ATP state.1, 5, 18, 19 However, this interaction is so transient that up to now, it has not been possible to capture any stable Hsp70‐Hsp40 complexes without chemical crosslinking or using protein fusion,138, 139 which raises concerns about associated artifacts. Moreover, as described above, the ATP‐bound state of Hsp70s itself is short lived, especially when interacting with Hsp40s. This could in part account for the transient nature of the Hsp70‐Hsp40 interaction. Hopefully, the recent advances in understanding the ATP‐bound state of Hsp70s will provide a foundation for studying the Hsp70‐Hsp40 interaction.140, 141, 142
Various biochemical analyses have shown that the well‐characterized J‐domain is critical for interacting with Hsp70s and stimulating the ATPase activity of Hsp70s. However, the J‐domain alone is not sufficient for either stimulating the ATPase activity of Hsp70s or forming a stable interaction with Hsp70s.143 Moreover, Hsp40s normally interact with both domains of Hsp70s106, 142, 144 with the J‐domain mainly interacting with the NBD.145, 146 A first breakthrough in understanding this interaction came from a genetic suppressor screen using E. coli Hsp70 DnaK and its Hsp40 DnaJ, which revealed a highly conserved R167 in DnaK as a contact site for DnaJ D35 in the conserved HPD motif of the J‐domain.144 Subsequent biochemical and structural analyses further suggested that the bottom cleft on NBD (where R167 resides) and the interdomain linker are also involved in the Hsp40 interaction.138, 142, 144, 145, 147, 148, 149 Inspired by these studies and recent advances in solving structures of full‐length Hsp70s in complex with ATP, two recent crystal structures and one molecular simulation analysis have shed light on the interaction between Hsp70s and the J‐domain.138, 140, 150
The first crystal structure of an Hsp70‐J‐domain complex was for the bovine Hsc70 NBD crosslinked with the J‐domain of Auxillin, a class III Hsp40, through the aforementioned R167‐D35 pair identified in the DnaK‐DnaJ interaction (Figure 6a).138 However, a very limited interface was revealed by this structure and the functional meaning was limited due to a number of issues including (a) the NBD of Hsc70 is in ADP‐bound conformation and (b) the presence of crosslinking. The second crystal structure was for a complex between the E. coli Hsp70 DnaK and the J‐domain of DnaJ (Figure 6b).139 To stabilize the complex, the J‐domain of DnaJ was fused to the N‐terminus of DnaK. The DnaK is the same construct used for crystallizing one of the first full‐length Hsp70s in the ATP‐bound state, carrying the ATPase deficient mutation T199A and an engineered disulfide bond. The T199A mutation has been shown to completely abolish the functional interaction between DnaK and DnaJ.98 Despite the concerns in using the T199A mutation and the fusion of J‐domain to DnaK, the interfaces revealed by this structure are consistent with a recent computational prediction using molecular simulations as well as many previous biochemical results,139, 150 suggesting the functional meaning of these interfaces. In this structure, the J‐domain binds on top of the interdomain linker, and at the same time, contacts both NBD and SBDβ. On the J‐domain side, these interactions mainly involve the helices II and III and the loop connecting these two helices that carries the highly conserved and functionally important HPD motif. Through the NBD interaction, the J‐domain accesses networks of interactions that connect to the catalytic site for ATP hydrolysis. At the same time, through SBDβ contacts, the J‐domain is connected to the pathway for transducing the peptide‐substrate binding signal to NBD. Thus, it was proposed that Hsp40s stimulate the ATPase activity of Hsp70s through tweaking these networks of interactions and holding the interdomain linker in place. Although this hypothesis sounds appealing, this structure failed to reveal how Hsp40s do so since the DnaK structure in this complex is essentially identical to the previously solved DnaK‐ATP structure by itself. Two fundamental factors could account for the lack of difference: (a) the T199A mutation used in forming the complex has been shown to completely abolish the functional Hsp70‐Hsp40 interaction and (b) the J‐domain alone is not able to stimulate the ATPase activity of Hsp70s. Interactions beyond the J‐domain must be important for the functional Hsp70‐Hsp40 interaction.
Figure 6.
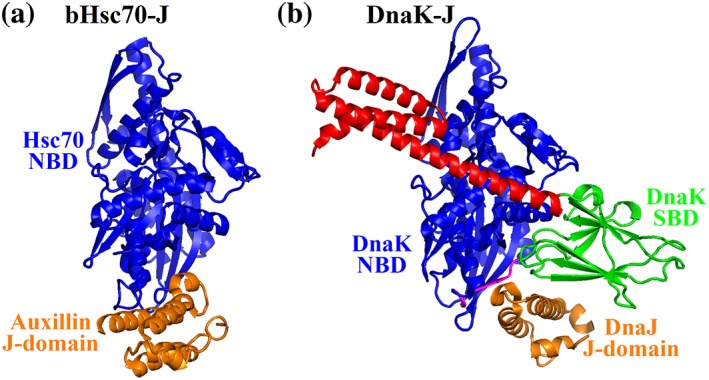
Structures of the Hsp70‐Hsp40 complexes. (a) The structure of bHsc70 NBD (blue) crosslinked with the J‐domain of Auxillin (orange) (PDB code: 2QWO). (b) The structure of DnaK fused to the DnaJ's J‐domain (PDB code: 5NRO). The domain coloring of DnaK is NBD (blue), interdomain linker (purple), SBDβ (green), and SBDα (red). The J‐domain of DnaJ is in orange
5. REMAINING QUESTIONS
Biochemical studies have shown that Hsp70s bind not only unfolded polypeptides but also larger substrates such as native proteins and protein aggregates.7, 8, 12, 80, 151, 152, 153 All available structural information on Hsp70s is on the interaction with model peptide substrates. How Hsp70s recognize and bind these large substrates is still a mystery. Another key question is how Hsp70s bind polypeptide substrate in the ATP‐bound state. All the available structures of Hsp70‐ATP have no peptide substrates. On the essential Hsp70‐Hsp40 interaction, two key questions still remain: (a) how do Hsp40s stimulate the ATP hydrolysis and (b) how do Hsp40s deliver substrate to Hsp70s? A structure of an Hsp70‐Hsp40 complex containing both full‐length Hsp70 and Hsp40 with and without substrates would fundamentally advance our understanding of the molecular mechanism. The experience in obtaining structures of full‐length Hsp70s in the ATP‐bound state will guide us to explore the even more challenging Hsp70‐Hsp40 structural analysis. Moreover, we now know Hsp90 is crucial for facilitating the Hsp70‐Hsp40 chaperone machinery in protein folding. How Hsp90 achieves this role is waiting for exploration. The recent structural and functional advances in studying the Hsp70‐Hsp40 chaperone machinery have paved a solid foundation for identifying inhibitor and chemical probes for this chaperone machinery for both basic research and medical purposes.71, 154, 155
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
ACKNOWLEDGMENTS
The authors tried to include as many relevant references as possible and apologize if important citations were inadvertently omitted. The authors were supported by NIH (R01GM098592 and R21AI140006 to Q.L.), Blick Scholar Award from the Virginia Commonwealth University (to Q.L.), and American Heart Association (17GRNT33660506 to Q.L.). L.Z. is partially supported by 1R01GM109193 from NIH.
Liu Q, Liang C, Zhou L. Structural and functional analysis of the Hsp70/Hsp40 chaperone system. Protein Science. 2020;29:378–390. 10.1002/pro.3725
Funding information American Heart Association, Grant/Award Number: 17GRNT33660506; Division of Intramural Research, National Institute of Allergy and Infectious Diseases, Grant/Award Number: R21AI140006; National Institute of General Medical Sciences, Grant/Award Numbers: 1R01GM109193, R01GM098592; Virginia Commonwealth University; NIH
REFERENCES
- 1. Mayer MP, Bukau B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hartl FU, Hayer‐Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16:574–581. [DOI] [PubMed] [Google Scholar]
- 3. Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. [DOI] [PubMed] [Google Scholar]
- 4. Bukau B, Deuerling E, Pfund C, Craig EA. Getting newly synthesized proteins into shape. Cell. 2000;101:119–122. [DOI] [PubMed] [Google Scholar]
- 5. Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell. 1998;92:351–366. [DOI] [PubMed] [Google Scholar]
- 6. Young JC. Mechanisms of the Hsp70 chaperone system. Biochem Cell Biol. 2010;88:291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu Q, Craig EA. Molecular biology: Mature proteins braced by a chaperone. Nature. 2016;539:361–362. [DOI] [PubMed] [Google Scholar]
- 8. Mayer MP, Gierasch LM. Recent advances in the structural and mechanistic aspects of Hsp70 molecular chaperones. J Biol Chem. 2019;294:2085–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hendershot LM. The ER function BiP is a master regulator of ER function. Mt Sinai J Med. 2004;71:289–297. [PubMed] [Google Scholar]
- 10. Fernandez‐Fernandez MR, Gragera M, Ochoa‐Ibarrola L, Quintana‐Gallardo L, Valpuesta JM. Hsp70‐a master regulator in protein degradation. FEBS Lett. 2017;591:2648–2660. [DOI] [PubMed] [Google Scholar]
- 11. Fernandez‐Fernandez MR, Valpuesta JM. Hsp70 chaperone: A master player in protein homeostasis. F1000Res. 2018;7:1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rosenzweig R, Nillegoda NB, Mayer MP, Bukau B. The Hsp70 chaperone network. Nat Rev Mol Cell Biol. 2019; https://www.nature.com/articles/s41580-019-0133-3. [DOI] [PubMed] [Google Scholar]
- 13. Boorstein WR, Ziegelhoffer T, Craig EA. Molecular evolution of the HSP70 multigene family. J Mol Evol. 1994;38:1–17. [DOI] [PubMed] [Google Scholar]
- 14. Zuiderweg ER, Hightower LE, Gestwicki JE. The remarkable multivalency of the Hsp70 chaperones. Cell Stress Chaperones. 2017;22:173–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Balchin D, Hayer‐Hartl M, Hartl FU. In vivo aspects of protein folding and quality control. Science. 2016;353:aac4354. [DOI] [PubMed] [Google Scholar]
- 16. Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11:579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schroder H, Langer T, Hartl FU, Bukau B. DnaK, DnaJ and GrpE form a cellular chaperone machinery capable of repairing heat‐induced protein damage. EMBO J. 1993;12:4137–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Craig EA, Huang P, Aron R, Andrew A. The diverse roles of J‐proteins, the obligate Hsp70 co‐chaperone. Rev Physiol Biochem Pharmacol. 2006;156:1–21. [DOI] [PubMed] [Google Scholar]
- 19. Fan CY, Lee S, Cyr DM. Mechanisms for regulation of Hsp70 function by Hsp40. Cell Stress Chaperones. 2003;8:309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li J, Qian X, Sha B. Heat shock protein 40: Structural studies and their functional implications. Protein Pept Lett. 2009;16:606–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kampinga HH, Andreasson C, Barducci A, et al. Function, evolution, and structure of J‐domain proteins. Cell Stress Chaperones. 2019;24:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alderson TR, Kim JH, Markley JL. Dynamical structures of Hsp70 and Hsp70‐Hsp40 complexes. Structure. 2016;24:1014–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Craig EA, Marszalek J. How do J‐proteins get Hsp70 to do so many different things? Trends Biochem Sci. 2017;42:355–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cyr DM, Ramos CH. Specification of Hsp70 function by type I and type II Hsp40. Subcell Biochem. 2015;78:91–102. [DOI] [PubMed] [Google Scholar]
- 25. Pobre KFR, Poet GJ, Hendershot LM. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J Biol Chem. 2019;294:2098–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hendrickson WA, Liu Q. Exchange we can believe in. Structure. 2008;16:1153–1155. [DOI] [PubMed] [Google Scholar]
- 27. Bracher A, Verghese J. The nucleotide exchange factors of Hsp70 molecular chaperones. Front Mol Biosci. 2015;2:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moran Luengo T, Kityk R, Mayer MP, Rudiger SGD. Hsp90 breaks the deadlock of the Hsp70 chaperone system. Mol Cell. 2018;70:545–552. [DOI] [PubMed] [Google Scholar]
- 29. Genest O, Hoskins JR, Camberg JL, Doyle SM, Wickner S. Heat shock protein 90 from Escherichia coli collaborates with the DnaK chaperone system in client protein remodeling. Proc Natl Acad Sci U S A. 2011;108:8206–8211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yan Y, Rato C, Rohland L, Preissler S, Ron D. MANF antagonizes nucleotide exchange by the endoplasmic reticulum chaperone BiP. Nat Commun. 2019;10:541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li Z, Hartl FU, Bracher A. Structure and function of Hip, an attenuator of the Hsp70 chaperone cycle. Nat Struct Mol Biol. 2013;20:929–935. [DOI] [PubMed] [Google Scholar]
- 32. Flynn GC, Chappell TG, Rothman JE. Peptide binding and release by proteins implicated as catalysts of protein assembly. Science. 1989;245:385–390. [DOI] [PubMed] [Google Scholar]
- 33. Flynn GC, Pohl J, Flocco MT, Rothman JE. Peptide‐binding specificity of the molecular chaperone BiP. Nature. 1991;353:726–730. [DOI] [PubMed] [Google Scholar]
- 34. Rudiger S, Buchberger A, Bukau B. Interaction of Hsp70 chaperones with substrates. Nat Struct Biol. 1997;4:342–349. [DOI] [PubMed] [Google Scholar]
- 35. Rudiger S, Germeroth L, Schneider‐Mergener J, Bukau B. Substrate specificity of the DnaK chaperone determined by screening cellulose‐bound peptide libraries. EMBO J. 1997;16:1501–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhu X, Zhao X, Burkholder WF, et al. Structural analysis of substrate binding by the molecular chaperone DnaK. Science. 1996;272:1606–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gragerov A, Zeng L, Zhao X, Burkholder W, Gottesman ME. Specificity of DnaK‐peptide binding. J Mol Biol. 1994;235:848–854. [DOI] [PubMed] [Google Scholar]
- 38. Blond‐Elguindi S, Cwirla SE, Dower WJ, et al. Affinity panning of a library of peptides displayed on bacteriophages reveals the binding specificity of BiP. Cell. 1993;75:717–728. [DOI] [PubMed] [Google Scholar]
- 39. Swain JF, Dinler G, Sivendran R, Montgomery DL, Stotz M, Gierasch LM. Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol Cell. 2007;26:27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Buchberger A, Theyssen H, Schroder H, et al. Nucleotide‐induced conformational changes in the ATPase and substrate binding domains of the DnaK chaperone provide evidence for interdomain communication. J Biol Chem. 1995;270:16903–16910. [DOI] [PubMed] [Google Scholar]
- 41. Mapa K, Sikor M, Kudryavtsev V, et al. The conformational dynamics of the mitochondrial Hsp70 chaperone. Mol Cell. 2010;38:89–100. [DOI] [PubMed] [Google Scholar]
- 42. Rist W, Graf C, Bukau B, Mayer MP. Amide hydrogen exchange reveals conformational changes in hsp70 chaperones important for allosteric regulation. J Biol Chem. 2006;281:16493–16501. [DOI] [PubMed] [Google Scholar]
- 43. Swain JF, Gierasch LM. The changing landscape of protein allostery. Curr Opin Struct Biol. 2006;16:102–108. [DOI] [PubMed] [Google Scholar]
- 44. Zhuravleva A, Clerico EM, Gierasch LM. An interdomain energetic tug‐of‐war creates the allosterically active state in Hsp70 molecular chaperones. Cell. 2012;151:1296–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schlecht R, Erbse AH, Bukau B, Mayer MP. Mechanics of Hsp70 chaperones enables differential interaction with client proteins. Nat Struct Mol Biol. 2011;18:345–351. [DOI] [PubMed] [Google Scholar]
- 46. Marcinowski M, Holler M, Feige MJ, Baerend D, Lamb DC, Buchner J. Substrate discrimination of the chaperone BiP by autonomous and cochaperone‐regulated conformational transitions. Nat Struct Mol Biol. 2011;18:150–158. [DOI] [PubMed] [Google Scholar]
- 47. Bertelsen EB, Chang L, Gestwicki JE, Zuiderweg ER. Solution conformation of wild‐type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc Natl Acad Sci U S A. 2009;106:8471–8476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mayer MP. Gymnastics of molecular chaperones. Mol Cell. 2010;39:321–331. [DOI] [PubMed] [Google Scholar]
- 49. Chang YW, Sun YJ, Wang C, Hsiao CD. Crystal structures of the 70‐kDa heat shock proteins in domain disjoining conformation. J Biol Chem. 2008;283:15502–15511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zuiderweg ER, Bertelsen EB, Rousaki A, Mayer MP, Gestwicki JE, Ahmad A. Allostery in the Hsp70 chaperone proteins. Top Curr Chem. 2012;328:99–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schmid D, Baici A, Gehring H, Christen P. Kinetics of molecular chaperone action. Science. 1994;263:971–973. [DOI] [PubMed] [Google Scholar]
- 52. Yang J, Nune M, Zong Y, Zhou L, Liu Q. Close and allosteric opening of the polypeptide‐binding site in a human Hsp70 chaperone BiP. Structure. 2015;23:2191–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Qi R, Sarbeng EB, Liu Q, et al. Allosteric opening of the polypeptide‐binding site when an Hsp70 binds ATP. Nat Struct Mol Biol. 2013;20:900–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kityk R, Kopp J, Sinning I, Mayer MP. Structure and dynamics of the ATP‐bound open conformation of Hsp70 chaperones. Mol Cell. 2012;48:863–874. [DOI] [PubMed] [Google Scholar]
- 55. Yang J, Zong Y, Su J, et al. Conformation transitions of the polypeptide‐binding pocket support an active substrate release from Hsp70s. Nat Commun. 2017;8:1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu Q, Hendrickson WA. Insights into Hsp70 chaperone activity from a crystal structure of the yeast Hsp110 Sse1. Cell. 2007;131:106–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gumiero A, Conz C, Gese GV, et al. Interaction of the cotranslational Hsp70 Ssb with ribosomal proteins and rRNA depends on its lid domain. Nat Commun. 2016;7:13563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Flaherty KM, DeLuca‐Flaherty C, McKay DB. Three‐dimensional structure of the ATPase fragment of a 70K heat‐shock cognate protein. Nature. 1990;346:623–628. [DOI] [PubMed] [Google Scholar]
- 59. Sriram M, Osipiuk J, Freeman B, Morimoto R, Joachimiak A. Human Hsp70 molecular chaperone binds two calcium ions within the ATPase domain. Structure. 1997;5:403–414. [DOI] [PubMed] [Google Scholar]
- 60. Wisniewska M, Karlberg T, Lehtio L, et al. Crystal structures of the ATPase domains of four human Hsp70 isoforms: HSPA1L/Hsp70‐hom, HSPA2/Hsp70‐2, HSPA6/Hsp70B', and HSPA5/BiP/GRP78. PloS One. 2010;5:e8625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Flaherty KM, Wilbanks SM, DeLuca‐Flaherty C, McKay DB. Structural basis of the 70‐kilodalton heat shock cognate protein ATP hydrolytic activity. II. Structure of the active site with ADP or ATP bound to wild type and mutant ATPase fragment. J Biol Chem. 1994;269:12899–12907. [PubMed] [Google Scholar]
- 62. Johnson ER, McKay DB. Mapping the role of active site residues for transducing an ATP‐induced conformational change in the bovine 70‐kDa heat shock cognate protein. Biochemistry. 1999;38:10823–10830. [DOI] [PubMed] [Google Scholar]
- 63. Wilbanks SM, McKay DB. How potassium affects the activity of the molecular chaperone Hsc70. II. Potassium binds specifically in the ATPase active site. J Biol Chem. 1995;270:2251–2257. [DOI] [PubMed] [Google Scholar]
- 64. Macias AT, Williamson DS, Allen N, et al. Adenosine‐derived inhibitors of 78 kDa glucose regulated protein (Grp78) ATPase: Insights into isoform selectivity. J Med Chem. 2011;54:4034–4041. [DOI] [PubMed] [Google Scholar]
- 65. Yan M, Li J, Sha B. Structural analysis of the Sil1‐Bip complex reveals the mechanism for Sil1 to function as a nucleotide‐exchange factor. Biochem J. 2011;438:447–455. [DOI] [PubMed] [Google Scholar]
- 66. Hughes SJ, Antoshchenko T, Chen Y, Lu H, Pizarro JC, Park HW. Probing the ATP site of GRP78 with nucleotide triphosphate analogs. PloS One. 2016;11:e0154862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Harrison CJ, Hayer‐Hartl M, Di Liberto M, Hartl F, Kuriyan J. Crystal structure of the nucleotide exchange factor GrpE bound to the ATPase domain of the molecular chaperone DnaK. Science. 1997;276:431–435. [DOI] [PubMed] [Google Scholar]
- 68. Polier S, Dragovic Z, Hartl FU, Bracher A. Structural basis for the cooperation of Hsp70 and Hsp110 chaperones in protein folding. Cell. 2008;133:1068–1079. [DOI] [PubMed] [Google Scholar]
- 69. Schuermann JP, Jiang J, Cuellar J, et al. Structure of the Hsp110: Hsc70 nucleotide exchange machine. Mol Cell. 2008;31:232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sondermann H, Scheufler C, Schneider C, Hohfeld J, Hartl FU, Moarefi I. Structure of a Bag/Hsc70 complex: Convergent functional evolution of Hsp70 nucleotide exchange factors. Science. 2001;291:1553–1557. [DOI] [PubMed] [Google Scholar]
- 71. Assimon VA, Gillies AT, Rauch JN, Gestwicki JE. Hsp70 protein complexes as drug targets. Curr Pharm Des. 2013;19:404–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cupp‐Vickery JR, Peterson JC, Ta DT, Vickery LE. Crystal structure of the molecular chaperone HscA substrate binding domain complexed with the IscU recognition peptide ELPPVKIHC. J Mol Biol. 2004;342:1265–1278. [DOI] [PubMed] [Google Scholar]
- 73. Jiang J, Prasad K, Lafer EM, Sousa R. Structural basis of interdomain communication in the Hsc70 chaperone. Mol Cell. 2005;20:513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Morshauser RC, Hu W, Wang H, Pang Y, Flynn GC, Zuiderweg ER. High‐resolution solution structure of the 18 kDa substrate‐binding domain of the mammalian chaperone protein Hsc70. J Mol Biol. 1999;289:1387–1403. [DOI] [PubMed] [Google Scholar]
- 75. Pellecchia M, Montgomery DL, Stevens SY, et al. Structural insights into substrate binding by the molecular chaperone DnaK. Nat Struct Biol. 2000;7:298–303. [DOI] [PubMed] [Google Scholar]
- 76. Stevens SY, Cai S, Pellecchia M, Zuiderweg ER. The solution structure of the bacterial HSP70 chaperone protein domain DnaK(393‐507) in complex with the peptide NRLLLTG. Protein Sci. 2003;12:2588–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wang H, Kurochkin AV, Pang Y, Hu W, Flynn GC, Zuiderweg ER. NMR solution structure of the 21 kDa chaperone protein DnaK substrate binding domain: A preview of chaperone‐protein interaction. Biochemistry. 1998;37:7929–7940. [DOI] [PubMed] [Google Scholar]
- 78. Liebscher M, Roujeinikova A. Allosteric coupling between the lid and interdomain linker in DnaK revealed by inhibitor binding studies. J Bacteriol. 2009;191:1456–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zhang P, Leu JI, Murphy ME, George DL, Marmorstein R. Crystal structure of the stress‐inducible human heat shock protein 70 substrate‐binding domain in complex with peptide substrate. PloS One. 2014;9:e103518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Clerico EM, Tilitsky JM, Meng W, Gierasch LM. How hsp70 molecular machines interact with their substrates to mediate diverse physiological functions. J Mol Biol. 2015;427:1575–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Otvos L Jr, Insug O, Rogers ME, et al. Interaction between heat shock proteins and antimicrobial peptides. Biochemistry. 2000;39:14150–14159. [DOI] [PubMed] [Google Scholar]
- 82. Benaroudj N, Batelier G, Triniolles F, Ladjimi MM. Self‐association of the molecular chaperone HSC70. Biochemistry. 1995;34:15282–15290. [DOI] [PubMed] [Google Scholar]
- 83. Osipiuk J, Georgopoulos C, Zylicz M. Initiation of lambda DNA replication. The Escherichia coli small heat shock proteins, DnaJ and GrpE, increase DnaK's affinity for the lambda P protein. J Biol Chem. 1993;268:4821–4827. [PubMed] [Google Scholar]
- 84. Liu Q, D'Silva P, Walter W, Marszalek J, Craig EA. Regulated cycling of mitochondrial Hsp70 at the protein import channel. Science. 2003;300:139–141. [DOI] [PubMed] [Google Scholar]
- 85. Schonfeld HJ, Schmidt D, Schroder H, Bukau B. The DnaK chaperone system of Escherichia coli: Quaternary structures and interactions of the DnaK and GrpE components. J Biol Chem. 1995;270:2183–2189. [DOI] [PubMed] [Google Scholar]
- 86. Carlino A, Toledo H, Skaleris D, DeLisio R, Weissbach H, Brot N. Interactions of liver Grp78 and Escherichia coli recombinant Grp78 with ATP: Multiple species and disaggregation. Proc Natl Acad Sci U S A. 1992;89:2081–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Burkholder WF, Zhao X, Zhu X, Hendrickson WA, Gragerov A, Gottesman ME. Mutations in the C‐terminal fragment of DnaK affecting peptide binding. Proc Natl Acad Sci U S A. 1996;93:10632–10637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sarbeng EB, Liu Q, Tian X, et al. A functional DnaK dimer is essential for the efficient interaction with Hsp40 heat shock protein. J Biol Chem. 2015;290:8849–8862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Morgner N, Schmidt C, Beilsten‐Edmands V, et al. Hsp70 forms antiparallel dimers stabilized by post‐translational modifications to position clients for transfer to Hsp90. Cell Rep. 2015;11:759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Liu Q, Li H, Yang Y, et al. A disulfide‐bonded DnaK dimer is maintained in an ATP‐bound state. Cell Stress Chaperones. 2016;22:201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Preissler S, Chambers JE, Crespillo‐Casado A, et al. Physiological modulation of BiP activity by trans‐protomer engagement of the interdomain linker. Elife. 2015;4:e08961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Shaner L, Morano KA. All in the family: Atypical Hsp70 chaperones are conserved modulators of Hsp70 activity. Cell Stress Chaperones. 2007;12:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wang XY, Subjeck JR. High molecular weight stress proteins: Identification, cloning and utilisation in cancer immunotherapy. Int J Hyperthermia. 2013;29:364–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Easton DP, Kaneko Y, Subjeck JR. The hsp110 and Grp1 70 stress proteins: Newly recognized relatives of the Hsp70s. Cell Stress Chaperones. 2000;5:276–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. McCarty JS, Walker GC. DnaK as a thermometer: Threonine‐199 is site of autophosphorylation and is critical for ATPase activity. Proc Natl Acad Sci U S A. 1991;88:9513–9517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Xu X, Sarbeng EB, Vorvis C, Kumar DP, Zhou L, Liu Q. Unique peptide substrate binding properties of 110‐kDa heat‐shock protein (Hsp110) determine its distinct chaperone activity. J Biol Chem. 2012;287:5661–5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Weyer FA, Gumiero A, Gese GV, Lapouge K, Sinning I. Structural insights into a unique Hsp70‐Hsp40 interaction in the eukaryotic ribosome‐associated complex. Nat Struct Mol Biol. 2017;24:144–151. [DOI] [PubMed] [Google Scholar]
- 98. Mayer MP, Laufen T, Paal K, McCarty JS, Bukau B. Investigation of the interaction between DnaK and DnaJ by surface plasmon resonance spectroscopy. J Mol Biol. 1999;289:1131–1144. [DOI] [PubMed] [Google Scholar]
- 99. Otero JH, Lizak B, Hendershot LM. Life and death of a BiP substrate. Semin Cell Dev Biol. 2010;21:472–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Gamer J, Multhaup G, Tomoyasu T, et al. A cycle of binding and release of the DnaK, DnaJ and GrpE chaperones regulates activity of the Escherichia coli heat shock transcription factor sigma32. EMBO J. 1996;15:607–617. [PMC free article] [PubMed] [Google Scholar]
- 101. Johnson JL, Craig EA. An essential role for the substrate‐binding region of Hsp40s in Saccharomyces cerevisiae . J Cell Biol. 2001;152:851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Rudiger S, Schneider‐Mergener J, Bukau B. Its substrate specificity characterizes the DnaJ co‐chaperone as a scanning factor for the DnaK chaperone. EMBO J. 2001;20:1042–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Liberek K, Marszalek J, Ang D, Georgopoulos C, Zylicz M. Escherichia coli DnaJ and GrpE heat shock proteins jointly stimulate ATPase activity of DnaK. Proc Natl Acad Sci U S A. 1991;88:2874–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Cyr DM, Lu X, Douglas MG. Regulation of Hsp70 function by a eukaryotic DnaJ homolog. J Biol Chem. 1992;267:20927–20931. [PubMed] [Google Scholar]
- 105. Fan CY, Ren HY, Lee P, Caplan AJ, Cyr DM. The type I Hsp40 zinc finger‐like region is required for Hsp70 to capture non‐native polypeptides from Ydj1. J Biol Chem. 2005;280:695–702. [DOI] [PubMed] [Google Scholar]
- 106. Linke K, Wolfram T, Bussemer J, Jakob U. The roles of the two zinc binding sites in DnaJ. J Biol Chem. 2003;278:44457–44466. [DOI] [PubMed] [Google Scholar]
- 107. Behnke J, Hendershot LM. The large Hsp70 Grp170 binds to unfolded protein substrates in vivo with a regulation distinct from conventional Hsp70s. J Biol Chem. 2014;289:2899–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Melnyk A, Rieger H, Zimmermann R. Co‐chaperones of the mammalian endoplasmic reticulum. Subcell Biochem. 2015;78:179–200. [DOI] [PubMed] [Google Scholar]
- 109. Dudek J, Greiner M, Muller A, et al. ERj1p has a basic role in protein biogenesis at the endoplasmic reticulum. Nat Struct Mol Biol. 2005;12:1008–1014. [DOI] [PubMed] [Google Scholar]
- 110. Brodsky JL, Goeckeler J, Schekman R. BiP and Sec63p are required for both co‐ and posttranslational protein translocation into the yeast endoplasmic reticulum. Proc Natl Acad Sci U S A. 1995;92:9643–9646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Zimmermann R, Muller L, Wullich B. Protein transport into the endoplasmic reticulum: Mechanisms and pathologies. Trends Mol Med. 2006;12:567–573. [DOI] [PubMed] [Google Scholar]
- 112. Shen Y, Hendershot LM. ERdj3, a stress‐inducible endoplasmic reticulum DnaJ homologue, serves as a cofactor for BiP's interactions with unfolded substrates. Mol Biol Cell. 2005;16:40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Petrova K, Oyadomari S, Hendershot LM, Ron D. Regulated association of misfolded endoplasmic reticulum lumenal proteins with P58/DNAJc3. EMBO J. 2008;27:2862–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Jin Y, Awad W, Petrova K, Hendershot LM. Regulated release of ERdj3 from unfolded proteins by BiP. EMBO J. 2008;27:2873–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Rutkowski DT, Kang SW, Goodman AG, et al. The role of p58IPK in protecting the stressed endoplasmic reticulum. Mol Biol Cell. 2007;18:3681–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Dong M, Bridges JP, Apsley K, Xu Y, Weaver TE. ERdj4 and ERdj5 are required for endoplasmic reticulum‐associated protein degradation of misfolded surfactant protein C. Mol Biol Cell. 2008;19:2620–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Nillegoda NB, Kirstein J, Szlachcic A, et al. Crucial HSP70 co‐chaperone complex unlocks metazoan protein disaggregation. Nature. 2015;524:247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Hill RB, Flanagan JM, Prestegard JH. 1H and 15N magnetic resonance assignments, secondary structure, and tertiary fold of Escherichia coli DnaJ(1‐78). Biochemistry. 1995;34:5587–5596. [DOI] [PubMed] [Google Scholar]
- 119. Qian YQ, Patel D, Hartl FU, McColl DJ. Nuclear magnetic resonance solution structure of the human Hsp40 (HDJ‐1) J‐domain. J Mol Biol. 1996;260:224–235. [DOI] [PubMed] [Google Scholar]
- 120. Pellecchia M, Szyperski T, Wall D, Georgopoulos C, Wuthrich K. NMR structure of the J‐domain and the Gly/Phe‐rich region of the Escherichia coli DnaJ chaperone. J Mol Biol. 1996;260:236–250. [DOI] [PubMed] [Google Scholar]
- 121. Cupp‐Vickery JR, Vickery LE. Crystal structure of Hsc20, a J‐type Co‐chaperone from Escherichia coli . J Mol Biol. 2000;304:835–845. [DOI] [PubMed] [Google Scholar]
- 122. Pinheiro GMS, Amorim GC, Iqbal A, Almeida FCL, Ramos CHI. Solution NMR investigation on the structure and function of the isolated J‐domain from Sis1: Evidence of transient inter‐domain interactions in the full‐length protein. Arch Biochem Biophys. 2019;669:71–79. [DOI] [PubMed] [Google Scholar]
- 123. Svard M, Biterova EI, Bourhis JM, Guy JE. The crystal structure of the human co‐chaperone P58(IPK). PloS One. 2011;6:e22337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Yu HY, Ziegelhoffer T, Osipiuk J, et al. Roles of intramolecular and intermolecular interactions in functional regulation of the Hsp70 J‐protein co‐chaperone Sis1. J Mol Biol. 2015;427:1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Schilke BA, Ciesielski SJ, Ziegelhoffer T, et al. Broadening the functionality of a J‐protein/Hsp70 molecular chaperone system. PLoS Genet. 2017;13:e1007084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Cheung J, Ginter C, Cassidy M, et al. Structural insights into mis‐regulation of protein kinase a in human tumors. Proc Natl Acad Sci U S A. 2015;112:1374–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Shrestha OK, Sharma R, Tomiczek B, et al. Structure and evolution of the 4‐helix bundle domain of Zuotin, a J‐domain protein co‐chaperone of Hsp70. PloS One. 2019;14:e0217098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Barends TR, Brosi RW, Steinmetz A, et al. Combining crystallography and EPR: Crystal and solution structures of the multidomain cochaperone DnaJ. Acta Crystallogr. 2013;D69:1540–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Huang K, Flanagan JM, Prestegard JH. The influence of C‐terminal extension on the structure of the “J‐domain” in E. coli DnaJ. Protein Sci. 1999;8:203–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Jiang J, Taylor AB, Prasad K, et al. Structure‐function analysis of the auxilin J‐domain reveals an extended Hsc70 interaction interface. Biochemistry. 2003;42:5748–5753. [DOI] [PubMed] [Google Scholar]
- 131. Mokranjac D, Bourenkov G, Hell K, Neupert W, Groll M. Structure and function of Tim14 and Tim16, the J and J‐like components of the mitochondrial protein import motor. EMBO J. 2006;25:4675–4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Li J, Qian X, Sha B. The crystal structure of the yeast Hsp40 Ydj1 complexed with its peptide substrate. Structure. 2003;11:1475–1483. [DOI] [PubMed] [Google Scholar]
- 133. Sha B, Lee S, Cyr DM. The crystal structure of the peptide‐binding fragment from the yeast Hsp40 protein Sis1. Structure. 2000;8:799–807. [DOI] [PubMed] [Google Scholar]
- 134. Suzuki H, Noguchi S, Arakawa H, Tokida T, Hashimoto M, Satow Y. Peptide‐binding sites as revealed by the crystal structures of the human Hsp40 Hdj1 C‐terminal domain in complex with the octapeptide from human Hsp70. Biochemistry. 2010;49:8577–8584. [DOI] [PubMed] [Google Scholar]
- 135. Hu J, Wu Y, Li J, Qian X, Fu Z, Sha B. The crystal structure of the putative peptide‐binding fragment from the human Hsp40 protein Hdj1. BMC Struct Biol. 2008;8:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Ramos CH, Oliveira CL, Fan CY, Torriani IL, Cyr DM. Conserved central domains control the quaternary structure of type I and type II Hsp40 molecular chaperones. J Mol Biol. 2008;383:155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Chen KC, Qu S, Chowdhury S, et al. The endoplasmic reticulum HSP40 co‐chaperone ERdj3/DNAJB11 assembles and functions as a tetramer. EMBO J. 2017;36:2296–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Jiang J, Maes EG, Taylor AB, et al. Structural basis of J cochaperone binding and regulation of Hsp70. Mol Cell. 2007;28:422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Kityk R, Kopp J, Mayer MP. Molecular mechanism of J‐domain‐triggered ATP hydrolysis by Hsp70 chaperones. Mol Cell. 2018;69:227–237. [DOI] [PubMed] [Google Scholar]
- 140. Suh WC, Lu CZ, Gross CA. Structural features required for the interaction of the Hsp70 molecular chaperone DnaK with its cochaperone DnaJ. J Biol Chem. 1999;274:30534–30539. [DOI] [PubMed] [Google Scholar]
- 141. Laufen T, Mayer MP, Beisel C, et al. Mechanism of regulation of hsp70 chaperones by DnaJ cochaperones. Proc Natl Acad Sci U S A. 1999;96:5452–5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Gassler CS, Buchberger A, Laufen T, et al. Mutations in the DnaK chaperone affecting interaction with the DnaJ cochaperone. Proc Natl Acad Sci U S A. 1998;95:15229–15234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Karzai AW, McMacken R. A bipartite signaling mechanism involved in DnaJ‐mediated activation of the Escherichia coli DnaK protein. J Biol Chem. 1996;271:11236–11246. [DOI] [PubMed] [Google Scholar]
- 144. Suh WC, Burkholder WF, Lu CZ, Zhao X, Gottesman ME, Gross CA. Interaction of the Hsp70 molecular chaperone, DnaK, with its cochaperone DnaJ. Proc Natl Acad Sci U S A. 1998;95:15223–15228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Greene MK, Maskos K, Landry SJ. Role of the J‐domain in the cooperation of Hsp40 with Hsp70. Proc Natl Acad Sci U S A. 1998;95:6108–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Landry SJ. Structure and energetics of an allele‐specific genetic interaction between dnaJ and dnaK: Correlation of nuclear magnetic resonance chemical shift perturbations in the J‐domain of Hsp40/DnaJ with binding affinity for the ATPase domain of Hsp70/DnaK. Biochemistry. 2003;42:4926–4936. [DOI] [PubMed] [Google Scholar]
- 147. Kumar DP, Vorvis C, Sarbeng EB, Cabra Ledesma VC, Willis JE, Liu Q. The four hydrophobic residues on the Hsp70 inter‐domain linker have two distinct roles. J Mol Biol. 2011;411:1099–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Davis JE, Voisine C, Craig EA. Intragenic suppressors of Hsp70 mutants: Interplay between the ATPase‐ and peptide‐binding domains. Proc Natl Acad Sci U S A. 1999;96:9269–9276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Wisen S, Bertelsen EB, Thompson AD, et al. Binding of a small molecule at a protein‐protein interface regulates the chaperone activity of hsp70‐hsp40. ACS Chem Biol. 2010;5:611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Malinverni D, Jost Lopez A, De Los Rios P, Hummer G, Barducci A. Modeling Hsp70/Hsp40 interaction by multi‐scale molecular simulations and coevolutionary sequence analysis. Elife. 2017;6:e23471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Gao X, Carroni M, Nussbaum‐Krammer C, et al. Human Hsp70 disaggregase reverses Parkinson's‐linked alpha‐synuclein amyloid fibrils. Mol Cell. 2015;59:781–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Mashaghi A, Bezrukavnikov S, Minde DP, et al. Alternative modes of client binding enable functional plasticity of Hsp70. Nature. 2016;539:448–451. [DOI] [PubMed] [Google Scholar]
- 153. Eisenberg E, Greene LE. Multiple roles of auxilin and hsc70 in clathrin‐mediated endocytosis. Traffic. 2007;8(6):640–646. [DOI] [PubMed] [Google Scholar]
- 154. Gestwicki JE, Shao H. Inhibitors and chemical probes for molecular chaperone networks. J Biol Chem. 2019;294:2151–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Brodsky JL, Chiosis G. Hsp70 molecular chaperones: Emerging roles in human disease and identification of small molecule modulators. Curr Top Med Chem. 2006;6:1215–1225. [DOI] [PubMed] [Google Scholar]