

Summary
During aging visceral adiposity is often associated with alterations in adipose tissue (AT) leukocytes, inflammation and metabolic dysfunction. However, the contribution of AT B cells in immunometabolism during aging is unexplored. Here, we show that aging is associated with an expansion of a unique population of resident non-senescent aged adipose B cells (AABs) found in fat-associated lymphoid clusters (FALCs). AABs are transcriptionally distinct from splenic age-associated B cells (ABCs) and show greater expansion in female mice, independently of ovarian steroids. Functionally, whole-body B cell depletion restores proper lipolysis and core body temperature maintenance during cold stress. Mechanistically, the age-induced FALC formation, AAB and splenic ABC expansion is dependent on the Nlrp3 inflammasome. Furthermore, AABs cells express IL-1R and inhibition of IL-1 signaling reduces their proliferation and increases lipolysis in aging. These data reveal that inhibiting Nlrp3-dependent B cell accumulation can be targeted to reverse metabolic impairment in aging AT.
Graphical Abstract
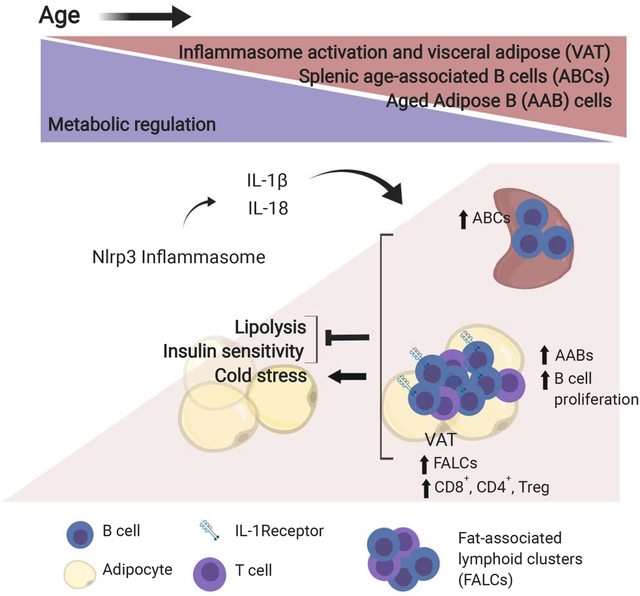
eTOC blurb
Camell et al. identify unique resident aged adipose B cells (AABs) that impair generation of fatty-acid substrates needed for ATP production during fasting and diminish the ability to maintain proper core body temperature upon cold stress in aging. AABs accumulation and age-induced defects in lipolysis depend on Nlrp3 inflammasome and IL-1 signaling.
Introduction
Increased visceral adiposity that is seen in aged individuals is accompanied by a decreased ability of adipose tissue (AT) to maintain homeostatic functions (Kennedy et al., 2014; Stenholm et al., 2008). These core functions of white AT include lipid storage and endocrine capability, both of which require intricate coordination between adipocytes and resident hematopoietic cells (Hotamisligil, 2006; Kanneganti and Dixit, 2012). White AT is highly heterogeneous containing defined microenvironment niches in which tissue-resident macrophages have distinct roles that facilitate tissue maintenance. Niches such as crown-like-structures (CLS), in which dying adipocytes are cleared by macrophages (Cinti et al., 2005; Martinez-Santibanez and Lumeng, 2014), and sympathetic nerve fibers, in which nerve-associated macrophages regulate local access to catecholamines that stimulate lipolysis (Bartness et al., 2014; Camell et al., 2017; Pirzgalska et al., 2017), have been implicated in metabolic pathogenesis during aging and obesity. Fat-associated lymphoid clusters (FALCs), predominantly composed of B1-innate B cells, serve as unique immunological sites that are acutely responsive to pathogens and increase with chronic inflammation (Benezech et al., 2015; Jackson-Jones et al., 2016; Lumeng et al., 2011; Morris et al., 2013). The contribution of FALCs and FALC-resident cells to age-related metabolic dysregulation remains unknown.
AT B cells have many functionally distinct roles, including antibody production, pro- and anti-inflammatory cytokine production and antigen presentation (Benezech et al., 2015; Frasca and Blomberg, 2017; Nishimura et al., 2013; Winer et al., 2011). In diet-induced-obesity, B cells produce IgG antibody and drive Th1 polarization contributing to insulin resistance and clearance of adipocytes in CLS (McDonnell et al., 2012; Winer et al., 2011). However, B1 and B regulatory subtypes favor insulin sensitization via IgM antibodies and anti-inflammatory cytokine production (Nishimura et al., 2013; Shen et al., 2015). Recent work suggests that B2 lymphocytes increase with age in AT of mice and ablation of the B cell-specific nuclear cofactor, Oct coactivator from B cells (OcaB), which controls B cell development, improves insulin-sensitivity in middle-aged mice (Carter et al., 2018), whereas peritoneal B1a cells are activated by commensal bacteria drive insulin resistance (Bodogai et al., 2018) and adipose B cells produce TNFα and IgG2c (Frasca and Blomberg, 2017); however, mechanisms that link impaired AT function to B cell homeostasis in aging are not well understood.
Recent studies demonstrate that the Nlrp3 inflammasome is among the major regulators of age-related inflammation and metabolic disturbance (Bauernfeind et al., 2016; Camell et al., 2017; Spadaro et al., 2016; Youm et al., 2013; Youm et al., 2012). The Nlrp3 inflammasome is an intracellular pattern recognition receptor in innate immune cells that is activated by a wide range of damage associated molecular patterns (DAMPs) (Kanneganti et al., 2007; Schroder and Tschopp, 2010). The identification of the Nlrp3 inflammasome in driving a number of age-related pathologies underscores the importance of innate immune cell-specific inflammation in aging (Bauernfeind et al., 2016; Goldberg and Dixit, 2015; Latz and Duewell, 2018; Youm et al., 2013; Youm et al., 2012).
We have previously shown that aging is associated with reduction in visceral AT (VAT) macrophage subsets which lack M1 or M2 polarization, but display senescent-like gene expression signatures that is in part dependent on the Nlrp3 inflammasome (Camell et al., 2017). Here we report that aging is associated with an expansion of non-senescent, aged adipose B cell (AABs) in FALCs of white VAT. To determine the nature and function of AABs, we performed flow cytometry phenotyping, whole mount confocal imaging and whole transcriptome expression analysis, which revealed the memory B cell profile. We found that ablation of the Nlrp3 inflammasome in aging protects against AAB expansion and FALC development, without altering their inflammatory transcriptional profile. Thus, we uncovered that age-related expansion of AABs and FALC development requires Nlrp3 inflammasome-dependent chronic inflammation. Further, we found that whole-body B cell depletion in aged mice restored the aberrant AT immune cell composition and improved the metabolic capacity of AT of aged mice.
Results
Aged white VAT shows a unique increase in memory-like B lymphocytes in FALCs
As there is an age-related decrease in VAT macrophages (Camell et al., 2017) along with an expansion of VAT CD3+ lymphocytes and Tregs (Cipolletta et al., 2015; Lumeng et al., 2011), we sought to investigate the impact of aging on AT B lymphocytes. To discriminate tissue resident cells from leukocytes in the AT vasculature, circulating cells were labelled with intravenous injection of a CD45.2 antibody (Figure S1A). Mice were sacrificed at 3 min post injection, which permitted labeling of all circulating cells, without CD45.2 antibody leaking into tissues or labelling cells extravasating into various tissues (Anderson et al., 2014). Quantification of tissue resident CD3+ and B220+ cells in mice revealed an increase in CD3+ lymphocytes and a 10-fold increase in the percentage of B220+ lymphocytes in the VAT from 24-month-old wild-type (WT) mice (Figure 1A & B). While only 20% of B220+ cells in AT from 3-month-old mice were tissue resident, nearly 100% of B220+ cells from 24-month-old mice were tissue resident in the AT (Figure 1C). Furthermore, age-related expansion in the frequency and cellularity of AT resident B cells was observed by 15-months of age and was markedly pronounced in female mice and was less profound in male animals (Figure 1D&E). There was no expansion of B cells in the spleen or VAT of ovariectomized female mice (Figure S1B) suggesting that ovarian-derived steroid and endocrine hormones do not regulate AT B cell expansion. A second cohort of male mice showed ~2-fold increase (Figure S1C), indicating that there is an age-related expansion in both genders, but it is of higher magnitude in female animals (~10 fold). Moreover, tissues from 2- or 24-month-old WT female mice housed at a separate animal facility (Pennington Biomedical, Baton Rouge) also showed a similar increase in VAT B cells (Figure S1D). These results demonstrate that the expansion of resident AT B cells initiates by 15-months-of age and is housing facility-independent, with greater cell expansion in female mice.
Figure 1.

Aged white VAT shows a unique increase in memory-like B lymphocytes in FALCs
(A) Contour plots gated through CD45+ iv antibody− live cells in stroma vascular fraction (SVF) of 3- and 24-month VAT. Values represent the mean percentage of B220+ cells. (B) Quantification of CD3+ and B220+ lymphocytes as a percentage of live CD45+ residents. (C) Quantification of resident and circulating cells as a percentage of total B220+ cells from 3- or 24-month mice. (A-C) n=3/group/cohort and pooled from two cohorts; WT female mice. Quantification of B220+ lymphocytes as (D) a percentage of live CD45+ iv− cells or (E) as cells per gram of AT in male or female mice at 5-, 10-, 15- or 20-months. (D-E) n= 5/group; WT male and female mice. (F) Quantification of resident (iv antibody−) B220+ cells as a percentage of live CD45+ iv− cells in brown (BAT), subcutaneous (SAT) and visceral AT (VAT). n=8 and 7/group; WT female mice. (G) Quantification of lymphocyte infiltration based on hematoxylin and eosin (H&E) staining in the VAT and SAT of 3- or 20-month-old mice. n=8/group; WT female. (H) Representative Masson’s trichrome staining showing a FALC in 22-month VAT. WT female. Representative H&E staining showing a (I) FALC in 22-month VAT or (J) inguinal lymph node in SAT of 22-month mouse. WT female. (K) Quantification of the percentage of Ki67+ cells, as a proportion of cell subsets, in spleen, VAT or mesenteric AT (MAT) cells of 24-month-old mice. n=3 to 9 samples/tissue; WT female mice. Representative whole mount confocal imaging in (L) 22-month-old VAT of Hec6ST mice to visualize HEVs (green) in B220+ FALCs (red). N=3; Female mice or (M) in 22-month-old VAT of ProxTom mice to visualize lymphatic vessels (tomato) in B220+ FALCs (green). N=3 female mice. Each symbol represents one mouse unless otherwise indicated. All error bars represent mean±SEM. *<0.05, **<0.01, ***<0.005. Statistical significance was determined by an ANOVA with posthoc test to adjust for multiple corrections. See also Figure S1.
Excess white VAT is associated with increased risk for metabolic impairments, whereas AT depots such as white subcutaneous or brown AT have been linked to insulin sensitivity and thermogenic capacity (Despres and Lemieux, 2006; Zamboni et al., 1997). Our quantification of resident B cells from various depots (Figure S1E), show that the age-related expansion in resident B cells was specific to white VAT (Figure 1F). Moreover, B220+ cells in the AT of 29-month-old WT mice express CD19 and do not express SiglecH (Figure S1F), a marker for B220+ plasmacytoid dendritic cells (pDCs). Histopathological scoring analysis revealed significant increases in the lymphocyte infiltration in the visceral, but not subcutaneous AT of 20-month-old mice (Figure 1G). These results suggest an association of AT B cell expansion with visceral adiposity seen in aging.
Previous reports describe immune cells within white VAT niches influence lipolysis, energy metabolism and tissue functionality (Camell et al., 2017; Xu et al., 2013). To understand the distribution of lymphocytes, whole mount imaging using an inverted microscope on fixed mesenteric and gonadal AT, revealed T and B cells in mesenteric lymph nodes and not throughout the AT of 7-month-old WT mice (Figure S1G). However, in 24-month-old WT mice, mesenteric and gonadal AT revealed FALCs throughout the tissue (identified previously: (Benezech et al., 2015; Lumeng et al., 2011; Moro et al., 2010). Lymphocytes accumulated within FALCs (Figure 1H&I) that were distinct from lymphocytes seen in a capsule-lined lymph node in subcutaneous inguinal AT (Figure 1J). B220+ B and CD3+ CD4+ T cells were found in the FALCs of the 22-month-old WT mice (Figure S1H). These data demonstrate that aging induces an increase in resident AT B cells localized to FALCs.
Adipose B cell express Ki67 and have access to the vasculature
Resident AT immune cells, such as macrophages in response to a high-fat diet, (Amano et al., 2014; Zamarron et al., 2017) and CD4+ Foxp3+ T regulatory cells during aging (Kolodin et al., 2015), are self-maintained through proliferation. Approximately 10% of B cells from spleen and AT of aged mice are positive for Ki67 (Figure 1K), a nuclear antigen expressed in actively dividing cells. These data indicate that a portion of age-expanded AT B cells are actively proliferating.
Lymphoid clusters in omentum contain high endothelial venules (HEVs), specialized post-capillary venules essential for naïve lymphocyte trafficking (Buscher et al., 2016; Rangel-Moreno et al., 2009). Whether HEVs and lymphatic vessels are present in the FALCs of aged VAT to transport B cells is unclear, therefore we imaged FALCs from aged reporter mice specific for HEV or lymphatic vessel markers (Ruddle, 2014; Truman et al., 2012). Hec6ST reporter mice at 22-months of age showed HEVs (GFP+) within FALCs and in close proximity to B220+ clusters (Figure 1L). Likewise, 22-month-old reporter mice (ProxTom mice), which express tdTomato under the control of a Prox1 promoter, a transcription factor required for lymphatic vessel formation, revealed the presence of lymphatic vessels (tomato+) within FALCs (Figure 1M). Lymphatic vessels in AT that were not within FALCs also contained B cells (Figure S1I). To confirm the presence of Prox1+ lymphatic vessels in WT aged mice, whole mount nuclear staining for Prox1 was performed on mesenteric lymph nodes (Figure S1J). Prox1 staining surrounded FALCs even at low magnification (Figure S1K) and confocal microscopy revealed clear Prox1+ nuclear staining in vessels near and surrounding FALCs (Figure S1L). Taken together these experiments identified a striking increase in the resident population of B lymphocytes in FALCs of white VAT and that B cell expansion may be supported by both active proliferation and local trafficking via lymphatic vessels and HEVs.
Age-expanded adipose B cells are a unique subset of tissue resident ABCs
During acute infection B1 cells, an innate-like B cell expressing CD11b and secreting natural IgM antibodies, in FALCs are activated and contribute to pathogen clearance (Jackson-Jones et al., 2016; Moro et al., 2010). CD11b+ AT B cells express higher levels of CD5, indicative of the B1-subset (Figure S2A). CD11b mean fluorescence intensity (MFI) and the proportions of CD11b+ (B1) or CD11b− (B2) resident B220+ cells were comparable in the resident VAT B cells from 3- or 24-month-old mice (Figure S2B, Figure 2A), suggesting that B cell subsets are equally expanded with age. To better understand the phenotype of age-expanded VAT B cells, we sorted B220+ cells from AT of 3- or 24-month-old WT mice for whole transcriptome analysis. Linear support vector regression analysis of those sorted cells revealed they express a memory-like B cell transcriptional profile (Figure 2B) (Newman et al., 2015).
Figure 2.

Age-expanded adipose B cells are a unique subset of tissue resident ABCs
(A) Quantification of the percentage of CD11b+ and CD11b− out of the total B220+ residents in VAT from 3- or 24-month-old male and female mice. n=3/group; WT mice. (B) Linear support vector analysis showing gene signature enrichment of AABs that overlap with plasma (light blue), memory (dark blue) or naïve (green) B cells (Newman et al., 2015). (C) Quantification of B cell subsets in spleen, MAT and VAT from 24-month female WT mice. n=4. (D) Contour plots (gated through CD45+ B220+ live cells) of CD21 and CD23 expression on B cells from 3-month-old mice after no stimulation (UNTX) or stimulation with AT media from 3- or 24-month-old mice. n=3–4; Repeated multiple times; WT female cells and media. (E) Quantification of Tbet+ cells as a percentage of live CD45+ B220+ cells in spleen and VAT. N=2 or 5/group; WT female mice. (F) PCA revealing separation of AT B cells and splenic ABCs. (G) Volcano plot showing log2 fold change and −log10 (padj) comparing AT B cells and splenic ABCs. Red dots are genes that are significantly over-represented, blue dots are under-represented and grey symbols signify no change. Each symbol represents one mouse unless otherwise indicated. Sequencing analysis is described in the methods. Otherwise, all error bars represent mean±SEM. *<0.05, **<0.01, ***<0.005. Statistical significance was determined by an ANOVA with posthoc test to adjust for multiple corrections. See methods for description on RNA seq. See also Figure S2.
During aging, the accumulation of a unique subset of B cells, termed age-associated B cells (ABCs), in the spleen, bone marrow and blood has been well described (Hao et al., 2011). ABCs are CD93− CD21− CD23− and evidence suggests that they are antigen-experienced, arising from their encounter with nucleic acid-containing antigens in the context of promoting cytokine production (Naradikian et al., 2016). Consistent with these reports, in 24-month-old WT mice 20–40% of splenic B cells were CD93−CD21−CD23−, but nearly 100% of resident AT B cells lacked CD93, CD21 or CD23 (Figure 2C). Notably, both young and aged AT-conditioned media was able to drive the phenotype of CD21− CD23− B cells from B cells from 3-month-old WT mice (Figure 2D). We next addressed how similar age-expanded AT B cells are to ABCs in spleen, which are reported to express the transcription factor Tbet and CD11c. AT B cells did not show Tbet expression (Figure 2E, S2D and E) or CD11c expression (Figure 2F and S2F). Furthermore, RNA sequencing analysis on sorted splenic ABCs and AT B cells (Figure S2G) revealed distinct clustering by tissue in principal component analysis (Figure 2F) and identified 2307 genes that were differentially regulated between the two populations (Figure 2G and S2H). Of interest is the differential regulation of pro-inflammatory IL-1β and chemokines in AT B cells which are not traditionally considered to be produced by splenic B cells (Figure 2G). These data are indicative of the inflammatory nature of AT B cells. Consistent with flow cytometry, Tbet (gene: Tbx21) was downregulated in the AT B cells (Figure S2I). This indicates that although AT B cells, now referred to as aged adipose B cells (AABs), share some similarities with splenic ABCs, they express a transcriptome that is based on their unique tissue-specific microenvironment.
Depletion of adipose B cells restores insulin sensitivity of aged mice
As the age-related expansion in AABs is localized to VAT and not to other AT depots (Figure 1F), we next determined whether a reduction in AABs would improve age-related insulin resistance. Intra-adipose injection of murine specific CD20 depleting antibody (CD20mAB) specifically depleted AABs (Figure S3A) and avoided systemic depletion of B cells. Body-weight and VAT weight in isotype (ISO)-treated and CD20mAB-treated mice were comparable (Figure S3B & C), but mice given CD20mAB showed a reduction in B cells in visceral, but not in subcutaneous AT, spleen, bone marrow or peritoneal cavity (Figure 3A, B, and S3D). When challenged with insulin, 20-month-old WT mice given the intra-adipose injection with CD20mAb showed improved insulin tolerance (Figure 3C). These results suggested that lipolytic capacity of aged AT may also be improved. We fasted the intra-adipose injected mice to induce lipolysis, a process that can be measured by release of glycerol from AT (Schweiger et al., 2014); however, there were no differences in glycerol release from the fed (Figure 3D) or fasted (Figure 3E) VAT in ISO-treated or CD20 mAb-treated mice. Collectively, these data indicate that specific depletion of the AABs was capable of improving age-related impaired insulin sensitivity, but not impaired fasting-induced lipolysis.
Figure 3.

Depletion of B cells restores AT metabolic capacity
(A) Fold change (% of B cell from CD20 mAb tissue/ average % of B cell from ISO tissue). (B) Frequency of B220+ cells gated through live CD45+ cells in VAT of 20-month-old WT mice given a single intra-AT injection of ISO or CD20 mAb. (C) Blood glucose following an insulin tolerance test (ITT) or (D) glycerol per gram of VAT in an ex vivo lipolysis assay from mice given treatment as described. (A-D) n=7/group WT female mice; 20m. (E) Glycerol per gram of tissue in VAT from mice that were fasted for 24 hours. n=9 and 8/group WT female mice; 20-months. (F) Histogram plots showing B220+ cells in tissues from 4- or 21-month-old mice given intraperitoneal injection of ISO or CD20 mAb. Values represent the mean percentage for that condition. n=6/4/5 at 4-/21-/21-month WT female mice. (G) Whole mount imaging showing representative FALCs in mesenteric AT from 22-month-old mice. (Top) FALCs from AT of mice treated with ISO control, (bottom) FALCs from mice treated with CD20 mAb. (H) Quantification of ABCs, FO B cells and MZ B cells in spleen of 5- or 21-month-old mice given ISO control or 21-month-old mice treated with CD20 mAb. n=3/4/4 at 5m/21m/21m WT female mice (I) Quantification of the percentage of CD4+ T cells out of the total CD45+ live cells in spleen or VAT. (right top) Quantification of CD4+ FoxP3+ CD25+ cells out of the total CD4+ T cells. (right bottom) Quantification PD1+ cells out of total CD4+ cells in the spleen and VAT tissue of conditions described. n=6/4/5 at 4m/21m/21m WT female mice. (J) Western blot showing phosphorylated HSL, ATGL, total HSL and ACTIN levels in VAT of 3-month, 22-month-old give isotype control or CD20 mAb as described. Representative of two individual experiments. n=2/3/4 at 3-/22-/22-month WT female mice. (K) Glycerol release (mM Glycerol per gram of tissue) from the VAT of 3-month-old WT mice that receive donor B cells via intraperitoneal injection. n=5/8/5 pooled from two separate experiments; WT female mice. (L) Rectal temperature at day 0 and day 3 in WT mice treated as described and exposed to four degrees. n=6/7/9 at 3–8-/23-/23-month-old WT female. Each symbol represents one mouse unless otherwise indicated. All error bars represent mean±SEM. *<0.05, **<0.01, ***<0.005. Statistical significance was determined by an ANOVA with posthoc test to adjust for multiple corrections. See also Figure S3.
Inhibition of systemic B cell expansion restores metabolic homeostasis in aged mice
To examine the consequence of both age-expanded ABCs in lymphoid tissues and AABs in metabolic dysregulation, we performed intraperitoneal (i.p.) injection of the ISO or CD20 mAb into 21-month-old mice to systemically deplete all B cells (Figure S3E). CD20 mAb reduced the percentage of B cells in the spleen and VAT in the 21-month-old mice (Figure 3F). FALCs tended to appear smaller and displayed reduced staining for B220 (Figure 3G) and percentages of ABCs in the spleen were significantly reduced (Figure 3H). To test whether B cell depletion can reverse age-related alterations in AT immune cell composition, we quantified the PD1+ effector T cells and FoxP3+ T regulatory (Treg) cells. The frequency of total CD4+ cells were increased (Figure 3I) in the B cell depleted mice, but there was a statistically significant VAT-specific reduction in the Treg and CD4+PD1+ sub-populations that typically increase in aged VAT (Figure 3I, S3H and S3I). These data indicate a single CD20 mAb injection to deplete B confers in part, a more young-like resident immune cell distribution in VAT from old mice.
Consistent with previous findings showing restored insulin tolerance in obese mice depleted of B cells and in aged mice depleted of Tregs (Bapat et al., 2015; Winer et al., 2011), we found that 20-month-old B cell-depleted mice had improved insulin sensitivity (Figure S3J and S3K). To address whether systemic depletion of B cells could restore lipolytic signaling and metabolic substrate availability in aged mice, 22-month-old control or B cell-depleted mice were fasted for 24 hours. The adipocyte lipases, adipose triglyceride lipase (ATGL) and hormone sensitive lipase (HSL), are required for triglyceride hydrolysis during fasting and are reduced in fasted aged VAT (Camell et al., 2017). B cell depletion resulted in increased levels of phosphorylated HSL and total ATGL during fasting, indicating that the age-related reduction in lipolytic signaling was restored with B cell depletion (Figure 3J). CD20mAB-treated mice also had increased isoproterenol-stimulated lipolysis (Figure S3L). Furthermore, i.p. transfer of ABCs cells from AT and spleen of 24-month-old mice impaired fasting induced-glycerol release in the 3-month-old hosts (Figure 3K).
Given that white AT lipolysis induced fatty acids are required for the generation of heat to induce thermogenesis and protect against cold stress (Schreiber et al., 2017; Shin et al., 2017) and that aged mice have impaired maintenance of proper core body temperature during cold stress (Figure S3M) (Berry et al., 2017), we tested whether B cell depletion improves defense against cold stress during aging. Consistent with the restored lipolysis providing additional metabolic substrates, 20-month-old B cell-depleted mice were able to maintain core body temperature when placed at four degrees Celsius (Figure 3L).
Age-induced adipose B cell expansion and FALC formation in adipose is regulated by the Nlrp3 inflammasome
In the context of acute inflammation, macrophage-derived TNFα promotes stromal cell activation and the accumulation of cells in FALCs (Benezech et al., 2015). We next studied the spatial association of macrophages with FALC resident B cells using aged mT/mG;LysM-cre reporter mice. FALCs contained dense capillary networks indicative of the vascular network (Figure 4A; S4A) (Benezech et al., 2015), and numerous B220+ cells, some of which could be found in close contact with macrophages (Figure 4A; S4A). Given this close association of cells within FALCs and our previous findings showing macrophage and Nlrp3 inflammasome regulation of AT homeostasis (Camell et al., 2017; Vandanmagsar et al., 2011), we asked whether the age-related expansion in AABs requires the Nlrp3 inflammasome. Compared to aged WT female mice, the aged Nlrp3-deficient mice showed a significant reduction in the percentage and the total numbers of B cells in VAT (two individual aging cohorts in Figure 4B and S4B). Although CD3+ T cell subsets, CD4+ and CD8+, are increased with age, the WT and Nlrp3-deficient mice showed comparable frequencies in aged AT (Figure S4C, D and E), indicating that the Nlrp3 inflammasome specifically promotes the age-related B cell expansion in VAT at 19-months, without affecting age-related T cell expansion.
Figure 4.

Age-induced adipose B cell expansion and FALC formation in adipose is regulated by the Nlrp3 inflammasome
(A) hole mount confocal imaging of FALC in VAT of 22-month-old mT/mG;LysMcre with B220 (yellow) and DAPI (blue) antibody staining. mTomato expressed on all cells and mGFP on myeloid cells. (B) Contour plots showing B220+ CD19+ AT B cells (gated through CD45+ live cells) from the VAT of 2- or 18–22-month-old WT and Nlrp3−/− mice. Percentage and cellularity (cells/g tissue) below. (C) Numbers of FALCs per AT in 24-month old WT or Nlrp3-deficient AT. n=4/5 female mice. (D) Whole mount confocal imaging to visualize Prox1+ (green) lymphatic endothelial cells in FALCs (B220+; red). (E) Quantification of Ki67+ cells (gated as a percentage of B220+ CD19+ cells) from the VAT of 19-month-old WT and Nlrp3−/− mice. n=3/group female mice. (F) Quantification of CD11b+ or CD11b− B cells (gated as a percentage of B220+ CD19+ cells). (B &F) n=(3) WT, (6) WT, or (6) Nlrp3−/− at 2-,19–22-, or 19–22-month-old female mice (G) Quantification of CD21−CD23− B cells (gated as a percentage of B220+ CD19+ cells) from the VAT of 19-month-old WT and Nlrp3−/− mice. N=3/3 female mice. (H) Quantification of CD11c MFI on B cells (gated as a percentage of B220+ MHCII+ cells) from the VAT of 24-month-old WT and Nlrp3−/− mice. N=4/5 female mice. (I) Differentially expressed genes in AABs from 19–22-month-old WT and Nlrp3−/− mice. Grey dots represent no significant change in gene expression, red dots represent significant changes in gene expression (FDR<0.1). (J) Volcano plot showing log2 fold change and −log10 (padj) comparing WT AABs and Nlrp3−/− AABs. Red dots are genes that are significantly over-represented, blue dots are under-represented and grey symbols signify no change. Each symbol represents one mouse unless otherwise indicated. All error bars represent mean±SEM. *<0.05, **<0.01, ***<0.005. Statistical significance was determined by a 2-sided, unpaired T-test or ANOVA with posthoc test to adjust for multiple corrections. RNA sequencing analysis is described in supplemental data. See also Figure S4.
To determine whether the Nlrp3 inflammasome is required for VAT T cell expansion at an earlier age, we quantified cell subsets in young WT mice and middle-aged WT or Nlrp3-deficient mice at a time (10-months) at which B cell expansion has not yet occurred. Consistent with previous findings (Camell et al., 2017), the 10-month old WT mice showed reduced CD11b+ myeloid cells, which was prevented by Nlrp3-deficiency (Figure S4F). Furthermore, AT CD4+ T cells were significantly elevated in the 10-month-old WT mice, and prevented by Nlrp3-deficiency (Figure S4G), with no age-related expansion in AABs at 10-month-of age (Figure S4H). These data support a model in which the Nlrp3 inflammasome initially controls age-related increases in total CD4+ T cells, and by 18-months of age, the Nlrp3 inflammasome is specifically required for resident AAB expansion.
We next asked whether the Nlrp3 inflammasome is also required for FALC development. Compared to 24-month-old WT mice, there was a significant reduction in the number of FALCs per AT in the aged Nlrp3-deficient mice (Figure 4C); however, we found that FALCs in the aged WT and Nlrp3/- contained Prox1+ lymphatic vessels (Figure 4D). These data suggested that although the Nlrp3 inflammasome is required for age-related B cell expansion, reduction in AABs is not because of reduced lymphatic vessel formation due to Nlrp3 inflammasome deficiency.
To address how Nlrp3-deficiency in aging protects against AAB expansion, we evaluated phenotype using flow cytometry. AABs from 19-month-old WT or Nlrp3-deficient mice had no difference in Ki67+ expression (Figure 4E), similar B1/B2 subset proportions (Figure 4F), ABC-phenotype (Figure 4G) and CD11c expression (Figure 4H). We then investigated the inflammatory potential of these age-expanded FALC resident B cells using unbiased RNA sequencing analyses. Surprisingly, this analysis revealed that compared to 19–22-month-old WT mice, the CD19+ B220+ CD93− CD21− CD23− AABs from age-matched Nlrp3-deficient mice displayed almost no differentially expressed genes (Figure 4I, J and S4I), with a total of 19 genes being upregulated and 17 genes being downregulated. Furthermore, the pro- or anti-inflammatory genes were not differentially regulated by Nlrp3-deficiency. Taken together with our previous findings (Camell et al., 2017), these data describe an Nlrp3 inflammasome-dependent role for age-related changes in AT resident macrophages and B cells. In contrast to the Nlrp3-dependent transcriptional regulation identified in macrophages (Camell et al., 2017), the transcriptional profile in B cells is primarily regulated in an Nlrp3-independent manner. Furthermore, Nlrp3 inflammasome-derived inflammation from macrophages acts upstream of VAAB expansion without altering their transcriptional phenotype. Thus, the Nlrp3 inflammasome-mediated reduction in AT inflammation during aging may result in part from numerical reduction in B cells.
Multiple receptor-ligand interactions among macrophages and B cells contribute to Nlrp3-mediated FALC development in aging
Given the role of the Nlrp3 inflammasome in resident B cell and FALC accumulation in aged AT, we asked whether aged AT macrophages directly control B cell recruitment. Using our previous AT macrophage whole genome sequencing analysis, (GSE93202) (Camell et al., 2017), we examined the mean chemokine gene expression in WT or Nlrp3-deficient AT macrophages from 3- or 24-month old mice. Hierarchical clustering analysis of chemokine expression did not reveal the group structure within the dataset suggesting that the Nlrp3 inflammasome is not a predominant regulator of B cell trafficking in aging AT (Figure S4J). To address the possibility that macrophage-B cell interaction contributes to the Nlrp3 inflammasome-regulated resident B cell expansion, we generated a macrophage-B cell interaction model by first curating a list of all possible receptors and ligands. To focus on the Nlrp3-dependent contribution, we identified macrophage ligands and receptors which are regulated in an Nlrp3 inflammasome-dependent manner and which have corresponding receptors expressed in the AABs (Figure S4K). All macrophage ligands and receptors identified showed potential to interact with B cells in one or more ways. Some interactions implied inflammatory signaling regulation (eg. Tlr4-S100a8/9 and Tnfrsf18-Tnfsf18 interaction) between macrophages and B cells, but no single interaction appears sufficient to explain the development of FALCs, suggesting that in context of low-grade chronic age-induced inflammation multiple interactions contribute to AAB expansion during aging.
Nlrp3-mediated inflammation and enhanced healthspan is associated with lower splenic ABC accumulation
During aging, the decrease in Nlrp3 inflammasome activation lowers systemic inflammation and protects against functional decline (Youm et al 2013). The increase in an antigen-experienced subset of ABCs in the spleen has been attributed to chronic levels of toll-like receptor activation and B cell receptor signaling (Hao et al., 2011; Rubtsova et al., 2015; Russell Knode et al., 2017). We wondered whether the age-related increase in splenic ABCs requires Nlrp3 inflammasome. There was significant elevation in the frequency of ABCs in the 24-month-old WT mice that was less pronounced in the 24-month-old Nlrp3-deficient mice (Figure 5A). We identified a corresponding age-regulated decrease in marginal zone and follicular B cells that was not restored with Nlrp3-deficiency (Figure 5B), suggesting that the Nlrp3 inflammasome is required for age-related ABC expansion, but not marginal or follicular B cell alterations. There were no differences in B cell subsets in bone marrow of 19-month-old WT and Nlrp3-deficient mice (Figure S5A), suggestive of tissue-specific protective mechanisms. Given that we had previously seen a lack of transcriptional control of aged AABs by Nlrp3-deficiency, we wondered whether splenic ABCs would reveal transcriptional profile unique to Nlrp3-deficiency. Consistent with AABs, the RNA sequencing analysis revealed limited transcriptional differences when comparing the WT and Nlrp3-deficient splenic ABCs (Figure 5C and D). Taken together, these data indicate a role for Nlrp3 inflammasome in driving age-related changes in B cell numbers in VAT and spleen, without altering the phenotype of those cells.
Figure 5.

B cell expansion is associated with reduced healthspan and lifespan
(A) Pseudocolor density plot gated through CD45+ B220+ MHCII+ splenocytes to show B cell subsets in 3- or 24-month-old WT and Nlrp3-deficient mice. (B) Quantification of splenic B cell subset frequency. MZ: marginal zone B cells; FO: follicular B cells. n= (10) 3-month WT, (6) 3-month Nlrp3−/−, (8) 24-month WT, or (5) 24-month Nlrp3−/− female mice. (C) Principal component analysis plot showing WT versus Nlrp3−/− splenic ABCs. (D) Volcano plot showing log2 fold change and mean of normalized counts comparing WT versus Nlrp3−/− splenic ABCs. Contour plots showing (E) CD21 and CD23 expression on B220+ CD19+ cells (gated through CD45+ live cells) from the spleen or (F) B220+ CD19+ B cells (gated through CD45+ live cells) from the VAT of 19-month-old WT and GHRKO female mice. Quantification of the percentage and cellularity (cells/g tissue) below each contour plot. (E-F) n=3 or 4/group female mice. Each symbol represents one mouse unless otherwise indicated. All error bars represent mean±SEM. *<0.05, **<0.01, ***<0.005. Statistical significance was determined by a 2-sided, unpaired T-test or ANOVA with posthoc test to adjust for multiple corrections. RNA sequencing analysis is described in supplemental data. See also Figure S5.
Reduction in adipose and splenic B cells is associated with increased longevity in mice
Reduction in GH-IGF1 somatotropic activity confers longevity in multiple animal models (Masternak and Bartke 2012). The long-lived mice with reduced growth hormone (GH) signaling have improved insulin sensitivity and reduced Nlrp3 inflammasome activation (Spadaro et al., 2016). Along with their extended lifespan and increased insulin sensitivity (Coschigano et al., 2003; Masternak and Bartke, 2012), male and female GHRKO mice have lower body weight, reduced VAT weight and decreased cellularity (Figure S5B and C). Quantification of ABCs in WT or GHRKO spleens revealed significant decreases in the percentage and total number of ABCs (Figure 5E, S5D and E). Furthermore, the VAT of GHRKO mice had significantly reduced percentage of B220+ cells (Figure 5F, S5F and G), but not T cell subsets (Figure S5H and I). These data highlight that reduction of aged B cells is associated with extended lifespan and may be related to improved metabolic outcomes associated with enhanced healthspan.
IL-1 signaling drives adipose B cell proliferation, inflammation and impaired lipolysis
Nlrp3 inflammasome activation results in release of the pro-inflammatory cytokines IL-1β and IL-18, which are implicated in age-related inflammation and pathologies (Youm et al 2013; Camell et al 2017). Our RNA sequencing analysis revealed significantly higher levels of IL1R on adipose B cells and this was the only interleukin receptor identified to be more highly expressed on the adipose B cells (Figure 6A). To address whether IL-1β signaling through the IL-1R drives B cell expansion and impaired lipolysis, 20-month-old female mice were depleted of B cells and co-treated with a vehicle control or IL-1R antagonist three times per week (Figure S6A). There were no differences in the body-weight in either group (Figure S6A and B); however, there was a reduction in the frequency of Ki67+ AABs upon IL-1R blockade (Figure 6B). Additionally, there was an inverse correlation between the frequency of Ki67+ and the frequency of B cells for the IL-1 receptor antagonist-treated group only (Figure 6C). Splenic ABCs showed comparable frequency of Ki67+ cells and no correlation for either treated group (Figure 6D& E). These results indicate that IL-1R signaling is required for B cell proliferation in the VAT, but not the spleen. There was a parallel reduction in pro-caspase-1 and active (p20) caspase-1 in the VAT of the mice treated with the IL-1R antagonist (Figure 6F and S6D). To address whether the reduced B cell proliferation and IL-1R inhibition is sufficient to increase metabolic capacity of aged AT, treated mice were fasted to evaluate lipolysis in the tissue. VAT of the mice treated with the IL-1R antagonist had elevated glycerol release (Figure 6G). These results show that IL-1 signaling drives B cell proliferation in the AT contributing to age-related inflammation and impaired lipolysis.
Figure 6.

IL-1 signaling drives adipose B cell proliferation, inflammation and impaired lipolysis
(A) Mean expression of chemokine receptors in sorted B cell sequencing analysis. (B) Quantification of Ki67+ cells, as a percentage of B220+ cells in the VAT of aged mice. (C) Correlation analysis in VAT, comparing % of Ki67+ and % of B cells. (D) Ki67+ cells, as a percentage of splenic ABCs of aged mice. (E) Correlation analysis in spleen ABCs, comparing % of Ki67+ and % of B cells. (F) Densitometric relative values of pro-caspase/actin and p20 (active caspase)/actin from VAT of mice treated as described. (G) Glycerol release (mM glycerol per gram of AT) in ex vivo assay from mice treated as described. Each symbol represents one mouse unless otherwise indicated. Each symbol represents a separate animal. n=8 or 9/group at 20-month-old female mice. All error bars represent mean±SEM. *<0.05, **<0.01, ***<0.005. Statistical significance was determined by a 2-sided, unpaired T-test. Pearson’s test was used to determine R2 and P value for correlation analysis. See also Figure S6.
Discussion
In this study we address the mechanism of age-related inflammation and AT dysfunction by investigating the resident AT B cell phenotype and function. Aging leads to expansion of resident AABs localized to FALCs in white visceral, but not subcutaneous or brown AT. Reminiscent of the splenic ABC expansion, AABs increase is primarily evident in female mice. Despite these broad similarities, RNA sequencing analysis identified inflammatory transcriptional differences between the splenic ABCs and the AABs. AABs exhibit a memory phenotype and their homeostatic expansion may require both proliferation and local trafficking. Functionally, treatment of aged mice with CD20 mAb caused a reduction in FALC-resident AABs and splenic ABCs, along with a restoration of CD4+ T cell subsets, restoration of lipolysis and improved capability to maintain cold-defense. The Nlrp3 inflammasome is required for AAB- and FALC expansion in aging, but surprisingly is not required for the inflammatory transcriptional phenotype of resident AABs. Furthermore, AABs express the IL-1R and treatment with an IL-1R antagonist to prevent signaling reduces AAB proliferative capacity and increases lipolysis in the VAT. Our data provide new information that the Nlrp3 inflammasome and IL-1R signaling drives resident AAB expansion and impaired AT metabolic capacity during aging.
That inflammation and metabolism are intimately linked, particularly in obesity, is well accepted (Ferrante, 2013; Hotamisligil, 2006). However, how aging disturbs immunometabolic inputs that impact maintenance of AT and metabolic homeostasis is less clear. Although we have uncovered that the Nlrp3 inflammasome-expansion of resident AABs contributes to impaired lipolysis and CD4+ T cell subpopulation expansion, how and why B cells localize to FALCs are likely regulated by additional mechanisms apart from the inflammasome.
In the young, the AT tertiary lymphoid structures are local sites of immune surveillance, contributing to the clearance of TLR agonists and pathogens (Benezech et al., 2015; Jackson-Jones et al., 2016; Moro et al., 2010); however, our data suggest that FALCs also control metabolic capacity of AT. Lipolysis is critical in infections and specific metabolic substrates help drive an efficient and effective immune response (Gazos-Lopes et al., 2017; Wang et al., 2018). Whether B cells regulate host energy homeostasis during various pathophysiological situations requiring metabolic substrates remains to be determined. Moreover, additional studies are needed to identify whether B cells secrete circulating factors or act directly to impair lipolytic capacity of adipocytes.
Our data offers novel insight into time-dependent and sex-dependent effects on AT hematopoietic cells. AAB and splenic ABC expansion with age is lower in male mice and the factors that drive B cell expansion predominantly in female mice is not clear. Aging is associated with loss of ovarian function, however ovariectomy in young mice that removes circulating ovarian steroids as well as granulosa cell derived hormones such as inhibin and activin did not mimic aging associated AAB expansion. Our experiments using male and female GHR−/− mice, both of which exhibit mild adiposity, insulin resistance and extended lifespan, revealed reduced age-related B cell expansion in both VAT and spleen. This suggests that the AAB expansion relies on the somatotropic signaling axis in a gender-independent manner. Interestingly, the B cell expansion is preceded by a reduction in macrophages as well as accumulation of T cell subsets suggesting potential role of B cells in determining immune landscape of aging AT. Furthermore, our B cell depletion experiments suggest a direct link between B cell expansion and T cell subset accumulation (Figure 3I).
Lymphatic vessels maintain fluid balance and regulate transport of antigen and immune cells (Ruddle, 2016); however they have also been implicated in driving adipogenesis, adiposity and insulin resistance (Harvey et al., 2005). Our study identified HEVs and lymphatic vessels within FALCs, suggesting regulated trafficking of lymphocytes. However, given previous findings describing the loss of functionality in lymphatic vessels and increased permeability in aging (Zolla et al., 2015), lymphatic vessels may also feed-forward into chronic age-related inflammation. FALC numbers are reduced in the aged Nlrp3-deficient mice, yet they still contain lymphatic vessels; whether improved functionality of lymphatic vessels contributes to improved metabolic capacity remains to be tested.
These data show a role for B cell depletion, which can rejuvenate B cell development in aged mice (Keren et al., 2011), and may also restore AT immune cell compartments, as well as tissue metabolic and immunologic capacity. Importantly, our data reveal previously unrecognized role of the Nlrp3 inflammasome in control of B cell expansion in aging and raise the possibility that FDA approved monoclonal antibodies to deplete B cells may be repurposed for reducing inflammation and reversing metabolic impairment in the elderly.
Limitations of the Study
This study provides evidence that the Nlrp3 inflammasome regulates B cell homeostasis in aging AT. AABs do not express Nlrp3 inflammasome, but do express the IL-1R, and are likely impacted in a direct manner by the inflammasome dependent cytokines IL-1β and IL-18 which increase with age. Contribution of IL-18 to AABs expansion is unknown. Furthermore, our RNA sequencing analysis indicates that AABs, themselves, are a source of IL-1β. Our experiment using the IL-1R antagonist reveal that inhibition of IL-1 signaling reduces AAB expression of the proliferation marker, Ki67, and increases lipolysis. Future studies requiring IL-1R deletion in AABs and evaluation of inflammation and metabolic capacity of VAT are needed to fully establish the contribution of IL-1 downstream of Nlrp3 inflammasome in AAB expansion in aging. However, our current results are consistent with a model of direct activity through the IL-1R that promotes AAB proliferation, which is in an interesting contrast with IL-1β suppressive activity on marrow B cells (Dorshkind, 1988; Kennedy and Knight, 2015; Pietras et al., 2016; Pioli et al., 2019). The reversal of lipolysis resistance in aged mice post B cell depletion and the impaired lipolysis following transfer of the ABCs, suggest that secreted factors from B cells either impact the lipolytic signaling in adipocytes or the sympathetic nervous system derived catecholamines or innervation itself. Despite comprehensive RNA sequencing analyses of B cells derived from VAT and spleen, no clear canonical regulators of lipolysis were revealed. Thus, the identity of AAB derived factors that control nerve-associated macrophages (Camell et al 2017) catecholamine degradation machinery or direct effectors remains to be identified. Moreover, unbiased interactome analyses from our RNA sequencing datasets of macrophage-B cell ligand receptor interactions did not reveal canonical TNF or lymphotoxin as mechanism of FALC development in aging. This may reflect unknown mechanisms specific to aging process in VAT that are reflective of chronic low-grade inflammation. These data also raise provocative question of whether therapeutic depletion of the pathogenic B cell in AT can promote longevity? Nonetheless, this study provides new evidence that the Nlrp3 inflammasome is a surprising new regulator of AAB expansion in aging and opens new avenues for future investigation of immunometabolic checkpoints to enhance healthy lifespan.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Vishwa Deep Dixit (vishwa.dixit@yale.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal care
All mice were housed in specific pathogen-free facilities in ventilated cage racks that deliver HEPA-filtered air to each cage with free access to sterile water through a Hydropac system at Yale School of Medicine. Sentinel mice in our animal rooms were negative for currently tested standard mouse pathogens (Ectromelia, EDIM, LCMV, Mycoplasma pulmonis, MHV, MNV, MPV, MVM, PVM, REO3, TMEV and Sendai virus) at various times while the studies were performed (Research Animal Diagnostic Laboratory). C57BL6/J (WT) mice were bred from our colony, purchased from Jackson Laboratories or received from the National Institute of Aging Rodent colony. Nlrp3−/− have been previously described and were bred in our facility, along with their WT controls (Mariathasan et al., 2006). LysM-cre mT/mG mice were bred in our facility. ProxTom and HEC6ST-GFP reporter mice were generated and bred by Nancy Ruddle (Bentley et al., 2011; Truman et al., 2012). GHRKO mice and their controls were generated and bred by Andrzej Bartke (Coschigano et al., 2003). All knockout mice were compared to WT controls raised in the same facility. Mice were fed a standard vivarium chow (Harlan 2018S) and housed under 12hr light/dark cycles. Mice were examined and only the mice that did not have lymphoma were used for experiments. All experiments and animal use were conducted in compliance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at Yale University. All mouse models used and the sex and number of animals are listed in the below sections.
Mouse models
For insulin tolerance test: Mice were fasted for 4 hours prior to ITT. Insulin was given by i.p. injection (0.8U/kg) and blood glucose levels were measured by glucometer.
For fasting: Mice were placed into a clean cage without food, but with water ad lib. Fast was complete after 16 hours (for B cell transfer experiment) or 24 hours (for B cell depletion experiments) when mice were euthanized for tissue harvest.
For cold challenge: Rectal temperature was taken using a rectal thermometer at 7am each day. Baseline body-temperature was taken at 7am at room temperature housing. Mice were then transferred to a cold room that is maintained at 4°C for three days. 12hour light/dark cycle and food and water were maintained throughout the cold challenge.
For B cell depletion: intra-adipose depletion was performed during survival surgery under anesthesia. A small ventral midline skin incision was made to access gonadal AT. 100ul of isotype control antibody (clone C1.18.4; bio X cell in vivo; catalogue#: BP0085) or CD20 monoclonal antibody (Genetech; clone 5D2) is injected into both left and right AT depots (50ug/depot). The tissue is replaced into the abdomen and the abdominal muscle and skin will be sutured with 4–0 Vicryl. Mice are monitored daily until incision heals. Systemic depletion was performed by i.p. injection of isotype control or CD20 mAB (100ug/mouse).
For IL-1R antagonist treatment: 300ug IL-1R antagonist (Cayman Chemical) in 10% DMSO or 10% DMSO alone as control was given by intraperitoneal injection to mice 3x per week.
Animal Experiments:
Experiment 1: Young vs old AT immune cell quantification (two individual cohorts compiled)
WT; N=3 at 3m; N=3 at 24m in each individual cohort; Female mice
Experiment 2: Time-course and sex comparison to quantify resident adipose B cells
WT; N= 5 at each time point for each sex.
Experiment 3: Young vs old adipose B cell comparison to test for increase in male mice
WT; N=3 at each age; Male mice
Experiment 4: Aging of mice in separate animal facility (Pennington Biomedical research center)
WT; N=3 at each age; female mice
Experiment 5: Quantification of pDCs in middle-aged and aged mice
WT; N=4 at 9m; N=5 at 29m; Female mice
Experiment 6: Adipose depot comparisons of resident adipose B cells in 7m and 24m old female mice
WT; N= 8 at 7m; N=7 at 24m; each pooled from multiple litters that were bred and housed at Yale University; Female mice
Experiment 7: Quantification of visceral and subcutaneous lymphocyte infiltration
WT; N= 8 at 3m and N =8 at 20m; Female mice
Experiment 8: Sorting of AABs and ABCs for RNA sequencing analysis
WT; N= 3 at 3m and N =2 at 24m pooled from 2 mice each*
Experiment 9: Ki67 expression in immune cells from aged animals (two individual cohorts compiled)
WT; N=3 and N=4 at 24m in two individual cohorts
Experiment 10: Aging of reporter mice and WT mice for confocal analysis
Hec6ST-GFP mice N=3; ProxTom mice N=3; WT N=3; all 20–22m; female mice
Experiment 11: Aging of WT and Nlrp3−/− mice for AT B and T cell quantification at young or elderly age
Part 1: N = 3 WT at 3m; N= 5 Nlrp3−/− at 3m; N=7 WT at 24m; N=5 Nlrp3−/− at 24m pooled from two separate experiments; Female mice
Part 2: N= 3 WT at 2m; N= 6 WT at 19–22m; N=6 Nlrp3−/− at 19–22m; Female mice
Experiment 12: Aging of WT and Nlrp3−/− mice for FALC quantification and Prox1+ staining
N= 5/5 WT/Nlrp3−/− at 24m for FALC quantification. N= 3/3 WT/Nlrp3−/− at 24m; female mice
Experiment 13: Aging of WT and Nlrp3−/− mice for AT B cell sorting and RNA seq analysis
Part 1: N=2 WT at 24m of age and N=3 Nlrp3−/− at 24m of age pooled from 2 mice each*
-
Part 2: N= 3 WT at 22m of age and N=3 Nlrp3−/− at 22m of age pooled from 2 female mice each*
N= 2 WT at 19m of age and N=1 Nlrp3−/− at 19m of age pooled from 2 female mice each; spleen samples harvested from these mice*
Experiment: 14: Aging of WT and Nlrp3−/− mice for quantification of AT B and T cells at 4m and 10m of age.
N= 4; female mice at 4m WT, 10m WT and 10m Nlrp3−/−
Experiment 15: Aging of WT and GHRKO mice for immune cell quantification
N = 7 WT and N=9 GHRKO at 20m, pooled from two separate experiments; Male mice
N= 3 WT and N= 4 GHRKO at 19m; Female mice
Experiment 16: Intra-adipose injection of CD20 mAB for visceral adipose-specific B cell depletion
N=7 ISO and N=7 CD20mAB; pooled from two separate experiments; female mice
Experiment 16: Intra-adipose injection of CD20 mAB for visceral adipose-specific B cell depletion and fasting challenge
N=9 ISO and N=8 CD20mAB; pooled from two separate experiments; female mice
Experiment 17: I.P. injection of CD20 mAB for systemic depletion of B cells for insulin tolerance test
N = 4 ISO and N= 5 CD20 mAB; female mice
Experiment 18: I.P. injection of CD20 mAB for systemic depletion of B cells with fasting challenge (two individual cohorts)
N= 3 3m ISO; N= 3 22m ISO; N= 4 22m CD20 mAB; repeated twice; representative data shown
Experiment 19: I.P. injection of 1.0×10^6 sorted donor cells into 3- month old mice. Hosts: N= 5 (no transfer); 8 (24m B cells); 5 (3m B cells). Pooled from experiments performed on two separate occasions. All female C57BL6 mice.
Experiment 20: 3 day cold challenge in 3–8m and 20–25m WT mice. N=9 (3–8m) and N=14 (20–25m) female mice
Experiment 21: I.P injection of CD20 mAB for systemic depletion of B cells with cold challenge (three individual cohorts compiled)
N= 6 3–8m ISO; N= 7 23m ISO; N=9 23m CD20m AB pooled together for rectal temperature; female mice
Experiment 22: I.P. injection of CD20mAB for systemic depletion of B cells with or without IL-1receptor antagonist treatment. N=8 mice given control treatment; N=9 mice given IL-1R antagonist; female mice at 20 months
All N’s represent a single animal (biological replicate), except for *RNA sequencing experiments which required pooling of AT from mice to collect the N’s indicated.
METHOD DETAILS
Adipose digestion and stromavascular staining
Visceral adipose was collected after euthanization and weighed. Tissue was enzymatically digested in 0.1% collagenase I or II (Worthington Biochemicals) in HBSS (Life Technologies) for 45 min at 37 °C. Tissue from control and experimental groups was digested and stained on the same day to eliminate minor procedure differences. The stromavascular fraction was pelleted by centrifugation at 500g for 10 min, then washed and filtered. Red blood cells are lysed using ACK lysis buffer. Cells were resuspended in 1 ml for counting before staining. For staining, the stromavascular fraction was incubated with FcBlock, surface antibodies for 30 min on ice, in the dark, then washed and stained with Fixable Viability Dye (eBioscience) and intracellular antibodies using cytofix (eBioscience/ThermoFisher). Analysis was performed on a BD LSRII and using FlowJo vX. For sorting: (1) B220+: CD3− B220+F4/80− CD11b− cells were sorted on a BD FACS Aria into RPMI with antibiotics/antimycotics (ThermoFisher) plus 20% FBS. Cells were pelleted and resuspended in trizol for RNA isolation. (2) Live Cd45+ B220+ Cd19+ CD3- CD11b- Cd93- CD21-CD23- were sorted on a BD FACS Aria into RLT with β-mercaptoethanol for RNA isolation. (3) Live Cd45+ B220+ Cd19+ CD3- CD11b- Cd93- CD21-CD23- or Live Cd45+ B220+ Cd19+ CD3- CD11b- Cd93- CD21+CD23+ from AT, lymph nodes and spleen were sorted into RPMI with antibiotics/antimycotics (ThermoFisher) plus 20% FBS for washing in sterile PBS and i.p. transfer into young hosts.
Lipolysis assay
For ex vivo lipolysis assay, AT was collected and 15 mg was cultured in 100 ul lipolysis buffer (Krebs buffer plus 0.1% glucose and 3.5% fatty acid free BSA; Sigma) in a 96-well plate for 2 h at 37 °C at 450 r.p.m. The glycerol assay (Sigma) was performed as per manufacturer’s instructions.
Western blot
Visceral adipose was snap frozen in liquid nitrogen immediately after harvest. Tissue was homogenized in RIPA buffer containing protease inhibitors. Protein concentration was quantified using the DC protein assay (Bio-Rad) and equal amounts of protein were run on an SDS-PAGE gel and transferred to nitrocellulose membrane. Blots were probed with primary antibodies and then incubated in secondary antibody of the appropriate species (ThermoFisher). Detection occurred using chemiluminescent visualization (Fisher, Bio-rad).
Whole-mount staining
Staining was performed similar to those previously published. In brief, AT was collected, fixed in 1% PFA and blocked with 5% BSA. Permeabilization was done using 0.1% Triton-X 100 in 5% BSA. The AT was then stained in primary antibodies over 1–2 days and secondary antibody, if needed, for 2.5 h in goat blocking serum. Confocal images were acquired using a laser scanning Leica SP8 or Leica SP5 and analyzed using Leica Application Suite AF.
For FALC counting, Images were acquired on a Zeiss Inverted Microscope (Axio) or Keyence (BZ-X710) using the stitching function within. FALCs from each tissue were counted using CD3+ staining.
Histology and pathological analysis:
Visceral and subcutaneous ATs were fixed in 10% formalin. Tissues were embedded, sectioned and stained with hematoxylin and eosin or masson’s trichrome by Mouse Research Pathology within Comparative Medicine at Yale University. Pathological analysis of lymphocyte infiltration was accomplished by a double-blind procedure Two pathologists made independent scores.
Antibodies
For tissue resident labeling: Intravascular labeling was performed by i.v. injection of 2.5ug CD45.2 diluted in 100ul PBS. Mice were euthanized exactly 3 minutes after injection for tissue collection.
For flow cytometry analysis, the following antibodies were used: Fixable Viability Dye Aqua; CD3-BV605, B220-PECy7, CD45.2-BV605, CD45.2-FITC, B220-APC, CD45-BV-711, CD4-BV605, FoxP3-APC, Ki67-PECy7, CD19-FITC, CD8-eF450, CD11b-FITC, MHCII-PECy7, B220-AF700, B220-eF450, CD25-PE, PD1-APC-e780, CD21/35-FITC, CD21-APC-e780, CD23-PE, CD23-eF450, F4/80-PE, CD11b-PerCP-Cy5.5, CD3-eF450, B220-FITC, B220-PerCP-Cy5.5. CD19-eF450, B220-APC, CD11c-APC-e780, Tbet-APC, SiglecH-PE
Intracellular staining: Intracellular antigens were detected using the eBioscience Fix/Perm nuclear staining kit as instructed by manufacture-supplied protocol.
For whole mount staining, the following antibodies were used: DAPI, B220-APC, B220-PE, CD3-FITC, Prox1 (Abcam; ab101851), Chicken anti-Rabbit-AF488 (Life Tech; A21441)
For western blot analysis, the following antibodies were used: pHSL (Ser563; 4139), HSL (4107), ATGL (2439), Beta-Actin (4967) (Cell signaling).
RNA extraction and gene expression analysis
RNA extraction and purification was performed using the trizol/chloroform method or by lysis in RLT buffer, and followed by use of the RNeasy kits (Qiagen) according to manufacturer’s instructions.
NA-sequence quality control
Total RNA quality is determined by estimating the 260 nm/A280 nm and A260 nm/A230 nm ratios by nanodrop. RNA integrity is determined by running an Agilent Bioanalyzer gel, which measures the ratio of the ribosomal peaks.
Library prep
mRNA is purified from approximately 500pg-1ng of total RNA using the Clontech SMARTer Ultra Low V4 RNA Kit. The kit utilizes an oligo(dT) primer, which primes the first-strand synthesis reaction. SPRI beads are used to selectively bind first-strand cDNA. ss cDNA is then amplified by LD PCR to make ds cDNA and is again purified using SPRI beads. During amplification the poly A sequence serves as the universal priming site for end-to-end cDNA amplification. Thus, cDNA without these sequences, such as prematurely terminated cDNAs, containing genomic DNA, or cDNA transcribed from Poly A minus RNA, are not exponentially amplified. The beads are then washed with 80% ethanol and eluted in purification buffer. The full-length cDNAs are then sheared using the Covaris, which results in 200–500bp DNA fragments. The fragmented cDNA library is then end-repaired, A-tailed, and adapters are ligated. Indexed libraries that meet appropriate cut-offs for both are quantified by qRT-PCR using a commercially available kit (KAPA Biosystems) and insert size distribution determined with the LabChip GX. Samples with a yield of ≥0.5 ng/ul are used for sequencing.
Flow cell preparation and sequencing
(1) Sample concentrations are normalized to 10 nM and loaded onto Illumina High-output flow cells at a concentration that yields 150–250 million passing filter clusters per lane. Samples are sequenced using 75 bp single-end sequencing on an Illumina HiSeq 2000 according to Illumina protocols. Each sample has a 6-bp index that is incorporated during the library prep. The index sequence is read using a different sequencing primer than the 75-bp sequencing read. The index read happens immediately after the 75-bp read. Data generated during sequencing runs are simultaneously transferred to the YCGA high-performance computing cluster. A positive control (prepared bacteriophage Phi X library) provided by Illumina is spiked into every lane at a concentration of 0.3% to monitor sequencing quality in real time.
(2) Sample concentrations are normalized to 10 nM and loaded onto an Illumina NovaSeq flow cell at a concentration that yields 25 million passing filter clusters per sample. Samples are sequenced using 100bp paired-end sequencing on an Illumina NovaSeq according to Illumina protocols. The 10bp dual index is read during additional sequencing reads that automatically follows the completion of read 1. Data generated during sequencing runs are simultaneously transferred to the YCGA high-performance computing cluster. A positive control (prepared bacteriophage Phi X library) provided by Illumina is spiked into every lane at a concentration of 0.3% to monitor sequencing quality in real time.
Data analysis and storage
Signal intensities are converted to individual base calls during a run using the system’s Real Time Analysis (RTA) software. Base calls are transferred from the machine’s dedicated personal computer to the Yale High Performance Computing cluster via a 1 Gigabit network mount for downstream analysis. Primary analysis - sample de-multiplexing and alignment to the human genome - is performed using Illumina’s CASAVA 1.8.2 software suite. The data are returned to the user if the sample error rate is less than 2% and the distribution of reads per sample in a lane is within reasonable tolerance. Data is retained on the cluster for at least 6 months, after which it is transferred to a tape backup system.
Primary data analysis of RNA-Seq data.
Raw fastq-files were checked by initial quality control using the FASTQC toolkit (http://www.bioinformatics.babraham.ac.uk/projects/fastqc, v0.11.7) in combination with MultiQC [PMID: 27312411]. Raw sequencing reads were pseudoaligned to the murine transcriptome model (mm10, GENCODE general assembly release vM16) using the Kallisto pipeline with default settings (Bray et al., 2016). Next, transcript levels were quantified and counts were imported into R using the tximport package (Soneson et al., 2015), while transcript information was summarized to gene-level count information. Further analysis was performed using the DESeq2 pipeline [v1.2; PMID: 25516281]. Tocheck for dendritic cell and macrophage enrichment we used cibersort analysis with standard parameters focusing on a subset of the provided signatures (macrophage subsets and DCs) [PMID: 25822800]. Following, normalization we determined differentially expressed genes using DESeq2 with standardsettings and IHW (Ignatiadis et al., 2016) filtering. Genes were considered as differentially expressed (DE) if the adjusted p value (IHW, weighted Benjamini and Hochberg correction) was less than 0.05. Thedifferences in gene expression were visualized as the dependency between the negative decadic logarithm of the p-value and the difference in gene expression as log2 fold change (volcano plot).
B-Cell macrophage interaction model.
To create a transcriptome-informed model of possible cell-to-cell ligand-receptor interactions we utilized the Fantom5 receptor-ligand database (Ramilowski et al., 2015). First, we downloaded the receptor-ligand interactions [http://fantom.gsc.riken.jp/data/, October/2016] and replaced the human receptor/ ligands with murine orthologs using ENSEMBL biomart (Ensembl Genes 92, GRCh38.p12). Genes that lacked a murine ortholog in this database were manually curated using the NCBI gene database. Interactions were filtered by DE genes of the respective comparison and visualized using the R package igraph 1.2.1 [https://cran.rproject.org/web/packages/igraph/index.html].
QUANTIFICATION AND STATISTICAL ANALYSIS
Experimental design
Blinding of investigators was not possible during experiments. Control and experimental groups were randomly assigned by cage. All experiments contained littermates and non-littermates, which were both randomly assigned to control and experimental group. Young control mice were raised or obtained from Jackson Laboratory (WT; C57BL6/J). Statistical significance was calculated by a two-tailed Student’s t-test or ANOVA using a post-hoc test to for multiple comparisons. *P < 0.05; **P < 0.005; ***P < 0.001; ****P < 0.0001. To determine exclusion criteria, GraphPad Prism was used to define statistical outliers, which were then excluded from data analysis. Aged mice with extreme frailty, tumor or other age-related pathology were excluded from the study. This amounted to less than 5% of all aged mice. A confidence interval of 95% was used for all statistical tests. All data were assumed to be normally distributed, unless the standard deviation was identified as significantly different between groups. Sample size for experiments were based upon previously published experiments. All statistical tests were performed using GraphPad Prism v7 for Windows (GraphPad Software). Data are expressed as mean ± s.e.m. Biological replicates and the number of independent experiment repetition are described in the figure legends.
DATA AND SOFTWARE AVAILABILITY
The RNA sequencing data has been uploaded to Gene Expression Omnibus (GSE137144). Previously published datasets that were used in the analysis are indicated by GEO number in the text.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD3-BV605 | Biolegend | cat: 100237; clone: 17A2 |
| CD45.2-FITC | ThermoFisher | cat: 11-0454; clone: 104 |
| CD45R (B220)-PE-Cy7 | ThermoFisher | cat: 25-0452; clone: RA3-6B2 |
| CD45.2-BV605 | Biolegend | cat: 109841; clone: 104 |
| CD45R (B220)-APC | ThermoFisher | cat: 17- 0452; clone RA3-6B2 |
| Cd45-BV711 | BDBioscience | cat: 563709; clone: 30-F11 |
| CD4-BV605 | Biolegend | cat: 100548; clone RM4-5 |
| FoxP3-APC | ThermoFisher | cat:17-5773; clone: FJK-16s |
| Ki67-PECy7 | BDBioscience | cat: 561283; clone: B56 |
| CD19-FITC | ThermoFisher | cat: 11-0191; clone: 1D3 |
| CD45R (B220)-AF700 | ThermoFisher | cat: 56-0452; clone RA3-6B2 |
| CD8-eF450 | ThermoFisher | cat:48-0081; clone 53-6.7 |
| CD11b-FITC | ThermoFisher | cat: 11-0112; clone: M1/70 |
| MHC ClassII (I-A/I-E) -PECy7 | ThermoFisher | cat: 25-5321; clone: M5/114.15.2 |
| CD45R (B220) -eF450 | ThermoFisher | cat: 48-0452; clone: RA3-6B2 |
| CD25 -PE | ThermoFisher | cat: 12-0251; clone: PC61.5 |
| CD279 (PD-1)- APC-e780 | ThermoFisher | cat: 47-9985; clone: J43 |
| CD21/CD35 -FITC | ThermoFisher | cat: 11-0212; clone: 8D9 |
| CD21/CD35 -APC-eFlour780 | ThermoFisher | cat: 47-0211; clone: 8D9 |
| CD23-PE | ThermoFisher | cat: 12-0232; clone: B3B4 |
| CD23-eF450 | ThermoFisher | cat: 48-0232; clone B3B4 |
| F4/80 -PE | ThermoFisher | cat: 12-4801; clone: BM8 |
| CD11b-PerCP-Cy5.5 | ThermoFisher | cat: 14-0112; clone: M1/70 |
| CD3e-eF450 | ThermoFisher | cat: 48-0031; clone: 145-2C11 |
| CD45R (B220)-PerCP-Cy5.5 | ThermoFisher | cat: 45-0452; clone: RA3-6B2 |
| CD19-eF450 | ThermoFisher | cat: 48-0193; clone: 1D3 |
| CD45R (B220)-APC | ThermoFisher | cat: 14-0452; clone RA3-6B2 |
| CD11c-APC-e780 | ThermoFisher | cat: 47-0114; clone N418 |
| Tbet-AF647 | Biolegend | cat: 644803; clone: 4B10 |
| SiglecH-PE | ThermoFisher | cat:12-0333; clone eBio440c |
| DAPI | Sigma | cat: D9542 |
| CD45R (B220)-PE | ThermoFisher | cat: 12-0452; clone: RA3-6B2 |
| CD3-FITC | ThermoFisher | cat: 11-0032; clone 17A2 |
| Anti-PROX1 antibody | Abcam | cat: ab101851 |
| Chicken anti-Rabbit-AF488 | LifeTech | Life Tech; A21441 |
| phospho-HSL (Ser563) Antibody | Cell Signaling | cat: 4139S |
| HSL Antibody | Cell Signaling | cat: 4107 |
| ATGL Antibody | Cell Signaling | cat: 2138 |
| Beta-Actin Antibody | Cell Signaling | cat: 4967 |
| CD93 (AA4.1)-PerCP-Cy5.5 | ThermoFisher | cat: 45-5892; clone: AA4.1 |
| CD3-PerCPCy5.5 | ThermoFisher | cat: 45-0031; clone: 145-2C11 |
| LIVE/DEAD Fixable Aqua Dead Cell Stain Kit | ThermoFisher | cat: L34966 |
| CD16/32 Monoclonal Antibody | ThermoFisher | cat: 14-0161; clone: 93 |
| CD20 monoclonal antibody | Genetech | clone: 5D2 |
| InVivoPlus mouse IgG2a isotype control | Bio X cell | cat: BP0085; clone: C1.18.4 |
| IL-1R antagonist | Cayman Chemical | cat: 21349 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| dimethyl-sulfoxide | Sigma-Aldrich | cat: 67-68-5 |
| Critical Commercial Assays | ||
| Glycerol Assay Kit | Sigma-Aldrich | cat: MAK117 |
| eBioscience Foxp3/Transcription Facotr Staining Buffer Set | ThermoFisher | cat: 00-5523-00 |
| Deposited Data | ||
| Expression profiling by high throughput sequencing | GEO DataSets | GSE93202 |
| Expression profiling by high throughput sequencing | GEO DataSets | GSE137144 |
| Experimental Models: Organisms/Strains | ||
| Mouse: wild-type: C57BL/6 | Jackson Lab | 000664 |
| Mouse: wild-type: C57BL/6 | National Institute of Aging Rodent Colony | C57BL/6 |
| Mouse: Nlrp3−/−: Nlrp3tm1Tsc | Genentech Inc | N/A |
| Mouse: LysM-cre: B6N.129P2(B6)-Lyz2tm1(cre)lfo/J | Jackson Lab | 004781 |
| Mouse: mT/mG: B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/Luo | Jackson Lab | 007676 |
| Mouse: ProxTom: B6;129S-Tg(Prox1-tdTomato)12Nrud/J | Dr. Nancy H. Ruddle, Yale University | 018128 |
| Mouse: HEC6ST-EGFP: B6.Cg-Tg(Chst4-EGFP)23Nrud/J | Dr. Nancy H. Ruddle, Yale University | 022787 |
| Mouse: GHRKO: Ghrtm1Jjk | Dr. Andrzej Bartke; Southern Illionois University | N/A |
| Software and Algorithms | ||
| Prism | GraphPad | N/A |
| FlowJo vX | TreeStar | N/A |
Highlights.
Adipose-resident aged B cells are increased in fat-associated lymphoid clusters (FALC)
Age-related FALC formation and B cell expansion are regulated by the Nlrp3 inflammasome
IL-1R signaling drives adipose B cell proliferation and impaired lipolysis
B cell depletion in aging restores lipolysis and improves cold tolerance
Context and Significance.
Age is associated with increased inflammation, visceral adiposity and metabolic diseases that are thought to be driven by a dysfunctional tissue-resident immune system. Here, Camell et al describe the identification of aged adipose B cells (AABs) that represent a unique resident population of B lymphocytes that accumulate in lymphoid clusters of visceral adipose tissue of older mice. AABs are proliferating, inflammatory cells that are able to impair metabolism during aging. The accumulation of AABs is dependent on innate sensor NLRP3 inflammasome and its production of the inflammatory cytokines IL-1β and IL-18. Approaches to target AABs and how they accumulate may provide potential therapeutic candidates for the treatment of visceral adiposity, inflammation and metabolic dysfunction in the elderly.
Acknowledgements:
We thank Genentech Inc for providing the Nlrp3-deficient mice and the mouse specific CD20mAb. We also thank The Yale Center on Genomic Analysis (YCGA) for RNAseq studies and the P. Cresswell laboratory for confocal microscopy support. J.L.S. was funded by the German Research Foundation (Deutsche Forschungsgemeinschaft) (SFB704, SFB645) and under Germany’s Excellence Strategy – EXC2151 – 390873048. C.D.C. was supported by AFAR (American Federation of Aging Research Postdoctoral transition fellowship) and NIA (K99AG058800). E.L.G was supported by AFAR (Postdoctoral fellowship) and NIA (K99AG058800). The Dixit laboratory is supported in part by NIH grants P01AG051459, AI105097, AG051459, AR070811, the Glenn Foundation on Aging Research and Cure Alzheimer’s Fund. A.B. was supported by R01AG019899. AB thanks J.J. Kopchick for providing GHRKO breeders.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no conflict of financial interest.
References
- Amano SU, Cohen JL, Vangala P, Tencerova M, Nicoloro SM, Yawe JC, Shen Y, Czech MP, and Aouadi M (2014). Local proliferation of macrophages contributes to obesity-associated AT inflammation. Cell metabolism 19, 162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KG, Mayer-Barber K, Sung H, Beura L, James BR, Taylor JJ, Qunaj L, Griffith TS, Vezys V, Barber DL, et al. (2014). Intravascular staining for discrimination of vascular and tissue leukocytes. Nature protocols 9, 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bapat SP, Myoung Suh J, Fang S, Liu S, Zhang Y, Cheng A, Zhou C, Liang Y, LeBlanc M, Liddle C, et al. (2015). Depletion of fat-resident Treg cells prevents age-associated insulin resistance. Nature 528, 137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartness TJ, Liu Y, Shrestha YB, and Ryu V (2014). Neural innervation of white AT and the control of lipolysis. Front Neuroendocrinol 35, 473–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauernfeind F, Niepmann S, Knolle PA, and Hornung V (2016). Aging-Associated TNF Production Primes Inflammasome Activation and NLRP3-Related Metabolic Disturbances. Journal of immunology 197, 2900–2908. [DOI] [PubMed] [Google Scholar]
- Benezech C, Luu NT, Walker JA, Kruglov AA, Loo Y, Nakamura K, Zhang Y, Nayar S, Jones LH, Flores-Langarica A, et al. (2015). Inflammation-induced formation of fat-associated lymphoid clusters. Nature immunology 16, 819–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley KL, Stranford S, Liao S, Mounzer RM, Ruddle FH, and Ruddle NH (2011). High endothelial venule reporter mice to probe regulation of lymph node vasculature. Advances in experimental medicine and biology 691, 35–44. [DOI] [PubMed] [Google Scholar]
- Berry DC, Jiang Y, Arpke RW, Close EL, Uchida A, Reading D, Berglund ED, Kyba M, and Graff JM (2017). Cellular Aging Contributes to Failure of Cold-Induced Beige Adipocyte Formation in Old Mice and Humans. Cell metabolism 25, 166–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodogai M, O’Connell J, Kim K, Kim Y, Moritoh K, Chen C, Gusev F, Vaughan K, Shulzhenko N, Mattison JA, et al. (2018). Commensal bacteria contribute to insulin resistance in aging by activating innate B1a cells. Science translational medicine 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscher K, Wang H, Zhang X, Striewski P, Wirth B, Saggu G, Lutke-Enking S, Mayadas TN, Ley K, Sorokin L, et al. (2016). Protection from septic peritonitis by rapid neutrophil recruitment through omental high endothelial venules. Nature communications 7, 10828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camell CD, Sander J, Spadaro O, Lee A, Nguyen KY, Wing A, Goldberg EL, Youm YH, Brown CW, Elsworth J, et al. (2017). Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature 550, 119–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter S, Miard S, Caron A, Salle-Lefort S, St-Pierre P, Anhe FF, Lavoie-Charland E, Blais-Lecours P, Drolet MC, Lefebvre JS, et al. (2018). Loss of OcaB Prevents Age-Induced Fat Accretion and Insulin Resistance by Altering B-Lymphocyte Transition and Promoting Energy Expenditure. Diabetes 67, 1285–1296. [DOI] [PubMed] [Google Scholar]
- Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, Wang S, Fortier M, Greenberg AS, and Obin MS (2005). Adipocyte death defines macrophage localization and function in AT of obese mice and humans. Journal of lipid research 46, 2347–2355. [DOI] [PubMed] [Google Scholar]
- Cipolletta D, Cohen P, Spiegelman BM, Benoist C, and Mathis D (2015). Appearance and disappearance of the mRNA signature characteristic of Treg cells in visceral AT: age, diet, and PPARgamma effects. Proceedings of the National Academy of Sciences of the United States of America 112, 482–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coschigano KT, Holland AN, Riders ME, List EO, Flyvbjerg A, and Kopchick JJ (2003). Deletion, but not antagonism, of the mouse growth hormone receptor results in severely decreased body weights, insulin, and insulin-like growth factor I levels and increased life span. Endocrinology 144, 3799–3810. [DOI] [PubMed] [Google Scholar]
- Despres JP, and Lemieux I (2006). Abdominal obesity and metabolic syndrome. Nature 444, 881–887. [DOI] [PubMed] [Google Scholar]
- Dorshkind K (1988). IL-1 inhibits B cell differentiation in long term bone marrow cultures. Journal of immunology 141, 531–538. [PubMed] [Google Scholar]
- Ferrante AW Jr. (2013). Macrophages, fat, and the emergence of immunometabolism. The Journal of clinical investigation 123, 4992–4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasca D, and Blomberg BB (2017). AT Inflammation Induces B Cell Inflammation and Decreases B Cell Function in Aging. Frontiers in immunology 8, 1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazos-Lopes F, Martin JL, Dumoulin PC, and Burleigh BA (2017). Host triacylglycerols shape the lipidome of intracellular trypanosomes and modulate their growth. PLoS pathogens 13, e1006800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg EL, and Dixit VD (2015). Drivers of age-related inflammation and strategies for healthspan extension. Immunological reviews 265, 63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, O’Neill P, Naradikian MS, Scholz JL, and Cancro MP (2011). A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood 118, 1294–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey NL, Srinivasan RS, Dillard ME, Johnson NC, Witte MH, Boyd K, Sleeman MW, and Oliver G (2005). Lymphatic vascular defects promoted by Prox1 haploinsufficiency cause adult-onset obesity. Nature genetics 37, 1072–1081. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS (2006). Inflammation and metabolic disorders. Nature 444, 860–867. [DOI] [PubMed] [Google Scholar]
- Ignatiadis N, Klaus B, Zaugg JB, and Huber W (2016). Data-driven hypothesis weighting increases detection power in genome-scale multiple testing. Nat Methods 13, 577–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson-Jones LH, Duncan SM, Magalhaes MS, Campbell SM, Maizels RM, McSorley HJ, Allen JE, and Benezech C (2016). Fat-associated lymphoid clusters control local IgM secretion during pleural infection and lung inflammation. Nature communications 7, 12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanneganti TD, and Dixit VD (2012). Immunological complications of obesity. Nature immunology 13, 707–712. [DOI] [PubMed] [Google Scholar]
- Kanneganti TD, Lamkanfi M, and Nunez G (2007). Intracellular NOD-like receptors in host defense and disease. Immunity 27, 549–559. [DOI] [PubMed] [Google Scholar]
- Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, Franceschi C, Lithgow GJ, Morimoto RI, Pessin JE, et al. (2014). Geroscience: linking aging to chronic disease. Cell 159, 709–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy DE, and Knight KL (2015). Inhibition of B Lymphopoiesis by Adipocytes and IL-1-Producing Myeloid-Derived Suppressor Cells. Journal of immunology 195, 2666–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren Z, Naor S, Nussbaum S, Golan K, Itkin T, Sasaki Y, Schmidt-Supprian M, Lapidot T, and Melamed D (2011). B-cell depletion reactivates B lymphopoiesis in the BM and rejuvenates the B lineage in aging. Blood 117, 3104–3112. [DOI] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodin D, van Panhuys N, Li C, Magnuson AM, Cipolletta D, Miller CM, Wagers A, Germain RN, Benoist C, and Mathis D (2015). Antigen- and cytokine-driven accumulation of regulatory T cells in visceral AT of lean mice. Cell metabolism 21, 543–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latz E, and Duewell P (2018). NLRP3 inflammasome activation in inflammaging. Seminars in immunology. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN, Liu J, Geletka L, Delaney C, Delproposto J, Desai A, Oatmen K, Martinez-Santibanez G, Julius A, Garg S, et al. (2011). Aging is associated with an increase in T cells and inflammatory macrophages in visceral AT. Journal of immunology 187, 6208–6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, and Dixit VM (2006). Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232. [DOI] [PubMed] [Google Scholar]
- Martinez-Santibanez G, and Lumeng CN (2014). Macrophages and the regulation of AT remodeling. Annual review of nutrition 34, 57–76. [DOI] [PubMed] [Google Scholar]
- Masternak MM, and Bartke A (2012). Growth hormone, inflammation and aging. Pathobiol Aging Age Relat Dis 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell ME, Ganley-Leal LM, Mehta A, Bigornia SJ, Mott M, Rehman Q, Farb MG, Hess DT, Joseph L, Gokce N, et al. (2012). B lymphocytes in human subcutaneous adipose crown-like structures. Obesity 20, 1372–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, Furusawa J, Ohtani M, Fujii H, and Koyasu S (2010). Innate production of T(H)2 cytokines by AT-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463, 540–544. [DOI] [PubMed] [Google Scholar]
- Morris DL, Cho KW, Delproposto JL, Oatmen KE, Geletka LM, Martinez-Santibanez G, Singer K, and Lumeng CN (2013). AT macrophages function as antigen-presenting cells and regulate AT CD4+ T cells in mice. Diabetes 62, 2762–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naradikian MS, Hao Y, and Cancro MP (2016). Age-associated B cells: key mediators of both protective and autoreactive humoral responses. Immunological reviews 269, 118–129. [DOI] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, and Alizadeh AA (2015). Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura S, Manabe I, Takaki S, Nagasaki M, Otsu M, Yamashita H, Sugita J, Yoshimura K, Eto K, Komuro I, et al. (2013). Adipose Natural Regulatory B Cells Negatively Control AT Inflammation. Cell metabolism. [DOI] [PubMed] [Google Scholar]
- Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, Lakshminarasimhan R, Chin CP, Techner JM, Will B, et al. (2016). Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol 18, 607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pioli PD, Casero D, Montecino-Rodriguez E, Morrison SL, and Dorshkind K (2019). Plasma Cells Are Obligate Effectors of Enhanced Myelopoiesis in Aging Bone Marrow. Immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirzgalska RM, Seixas E, Seidman JS, Link VM, Sanchez NM, Mahu I, Mendes R, Gres V, Kubasova N, Morris I, et al. (2017). Sympathetic neuron-associated macrophages contribute to obesity by importing and metabolizing norepinephrine. Nature medicine 23, 1309–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramilowski JA, Goldberg T, Harshbarger J, Kloppmann E, Lizio M, Satagopam VP, Itoh M, Kawaji H, Carninci P, Rost B, et al. (2015). A draft network of ligand-receptor-mediated multicellular signalling in human. Nature communications 6, 7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangel-Moreno J, Moyron-Quiroz JE, Carragher DM, Kusser K, Hartson L, Moquin A, and Randall TD (2009). Omental milky spots develop in the absence of lymphoid tissue-inducer cells and support B and T cell responses to peritoneal antigens. Immunity 30, 731–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsova K, Rubtsov AV, Cancro MP, and Marrack P (2015). Age-Associated B Cells: A T-bet-Dependent Effector with Roles in Protective and Pathogenic Immunity. Journal of immunology 195, 1933–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruddle NH (2014). Lymphatic vessels and tertiary lymphoid organs. The Journal of clinical investigation 124, 953–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruddle NH (2016). High Endothelial Venules and Lymphatic Vessels in Tertiary Lymphoid Organs: Characteristics, Functions, and Regulation. Frontiers in immunology 7, 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell Knode LM, Naradikian MS, Myles A, Scholz JL, Hao Y, Liu D, Ford ML, Tobias JW, Cancro MP, and Gearhart PJ (2017). Age-Associated B Cells Express a Diverse Repertoire of VH and Vkappa Genes with Somatic Hypermutation. Journal of immunology 198, 1921–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber R, Diwoky C, Schoiswohl G, Feiler U, Wongsiriroj N, Abdellatif M, Kolb D, Hoeks J, Kershaw EE, Sedej S, et al. (2017). Cold-Induced Thermogenesis Depends on ATGL-Mediated Lipolysis in Cardiac Muscle, but Not Brown AT. Cell metabolism 26, 753–763 e757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, and Tschopp J (2010). The inflammasomes. Cell 140, 821–832. [DOI] [PubMed] [Google Scholar]
- Schweiger M, Eichmann TO, Taschler U, Zimmermann R, Zechner R, and Lass A (2014). Measurement of lipolysis. Methods in enzymology 538, 171–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Chng MH, Alonso MN, Yuan R, Winer DA, and Engleman EG (2015). B-1a lymphocytes attenuate insulin resistance. Diabetes 64, 593–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H, Ma Y, Chanturiya T, Cao Q, Wang Y, Kadegowda AKG, Jackson R, Rumore D, Xue B, Shi H, et al. (2017). Lipolysis in Brown Adipocytes Is Not Essential for Cold-Induced Thermogenesis in Mice. Cell metabolism 26, 764–777 e765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spadaro O, Goldberg EL, Camell CD, Youm YH, Kopchick JJ, Nguyen KY, Bartke A, Sun LY, and Dixit VD (2016). Growth Hormone Receptor Deficiency Protects against Age-Related NLRP3 Inflammasome Activation and Immune Senescence. Cell reports 14, 1571–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenholm S, Harris TB, Rantanen T, Visser M, Kritchevsky SB, and Ferrucci L (2008). Sarcopenic obesity: definition, cause and consequences. Current opinion in clinical nutrition and metabolic care 11, 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truman LA, Bentley KL, Smith EC, Massaro SA, Gonzalez DG, Haberman AM, Hill M, Jones D, Min W, Krause DS, et al. (2012). ProxTom lymphatic vessel reporter mice reveal Prox1 expression in the adrenal medulla, megakaryocytes, and platelets. The American journal of pathology 180, 1715–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, and Dixit VD (2011). The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nature medicine 17, 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A, Huen SC, Luan HH, Baker K, Rinder H, Booth CJ, and Medzhitov R (2018). Glucose metabolism mediates disease tolerance in cerebral malaria. Proceedings of the National Academy of Sciences of the United States of America 115, 11042–11047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, Tsui H, Wu P, Davidson MG, Alonso MN, et al. (2011). B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nature medicine 17, 610–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Grijalva A, Skowronski A, van Eijk M, Serlie MJ, and Ferrante AW Jr. (2013). Obesity activates a program of lysosomal-dependent lipid metabolism in AT macrophages independently of classic activation. Cell metabolism 18, 816–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youm YH, Grant RW, McCabe LR, Albarado DC, Nguyen KY, Ravussin A, Pistell P, Newman S, Carter R, Laque A, et al. (2013). Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell metabolism 18, 519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youm YH, Kanneganti TD, Vandanmagsar B, Zhu X, Ravussin A, Adijiang A, Owen JS, Thomas MJ, Francis J, Parks JS, et al. (2012). The Nlrp3 inflammasome promotes age-related thymic demise and immunosenescence. Cell reports 1, 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamarron BF, Mergian TA, Cho KW, Martinez-Santibanez G, Luan D, Singer K, DelProposto JL, Geletka LM, Muir LA, and Lumeng CN (2017). Macrophage Proliferation Sustains AT Inflammation in Formerly Obese Mice. Diabetes 66, 392–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamboni M, Armellini F, Harris T, Turcato E, Micciolo R, Bergamo-Andreis IA, and Bosello O (1997). Effects of age on body fat distribution and cardiovascular risk factors in women. The American journal of clinical nutrition 66, 111–115. [DOI] [PubMed] [Google Scholar]
- Zolla V, Nizamutdinova IT, Scharf B, Clement CC, Maejima D, Akl T, Nagai T, Luciani P, Leroux JC, Halin C, et al. (2015). Aging-related anatomical and biochemical changes in lymphatic collectors impair lymph transport, fluid homeostasis, and pathogen clearance. Aging cell 14, 582–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequencing data has been uploaded to Gene Expression Omnibus (GSE137144). Previously published datasets that were used in the analysis are indicated by GEO number in the text.