

Abstract
Exocytosis is a widely observed cellular mechanism for delivering transmembrane proteins to the cell surface and releasing signaling molecules into the extracellular space. Calcium-evoked exocytosis, traditionally thought to be restricted to presynaptic specializations in neurons, has been described recently in many cells. Here, calcium-evoked dendritic exocytosis (CEDE) is visualized in living cultured hippocampal neurons. Organelles that undergo CEDE are in somata, dendrites, and perisynaptic regions, identified by using immunocytochemistry and correlative light and electron microscopy. CEDE is regulated developmentally: neurons <9 d in vitro do not show CEDE. In addition, CEDE is blocked by tetanus toxin, an inhibitor of regulated exocytosis, and nocodazole, an inhibitor of microtubule polymerization. Organelles that undergo CEDE often are found on the base of spines, putative sites of synaptic plasticity. CEDE therefore could be involved in structural and functional modification of spines and could play a role in synaptic plasticity, where it might involve changes in receptor/channel density, release of active compounds having effect on pre- and postsynaptic function, and/or growth of synaptic structures.
Keywords: exocytosis, trans-Golgi network, dendrite, pyramidal neurons, hippocampal culture, time-lapse imaging, FM1-43, immunocytochemistry, tetanus toxin, microtubules
Constitutive exocytosis is ubiquitous in eukaryotic cells. It is responsible for the recycling and renewal of plasma membrane components and for the secretion of molecules into the extracellular space (Alberts et al., 1989). In neurons the exocytosis of neurotransmitter molecules from presynaptic terminals represents the fundamental mechanism for the transfer of information. Although presynaptic vesicle exocytosis has been examined extensively, exocytosis has not yet been demonstrated directly in the dendritic regions of neurons, where it might provide a regulated means for effecting synaptic plasticity and/or homeostasis. Evidence exists that nonfunctional pools of postsynaptic membrane proteins are present (Hampson et al., 1992; Baude et al., 1995; Rubio and Wenthold, 1997) and that postsynaptic exocytosis is required for some forms of synaptic plasticity (Lledo et al., 1998). Calcium-evoked exocytosis has been demonstrated in neuronal somata (Huang and Neher, 1996) as well as in non-neuronal cells (Morimoto et al., 1995). In addition, lysosomes in fibroblasts, epithelial cells, and myoblasts undergo calcium-regulated exocytosis, which is temperature- and ATP-dependent (Rodriguez et al., 1997). In this study we demonstrate calcium-evoked exocytosis of dendritic compartments, consistent with the view that some forms of synaptic plasticity might occur via the regulated exocytosis of dendritic organelles.
In the past several years the mechanisms of presynaptic vesicle exocytosis have been examined in detail, including studies using fluorescent membrane probes. The styryl dye FM1-43 has been used effectively as a probe to monitor at the light microscope level the dynamics of presynaptic vesicles in living cells (Betz et al., 1992). A brief exposure to FM1-43, combined with the depolarization of the cell, induces selective internalization of the dye into the presynaptic terminals (Ryan et al., 1993). With triggered exocytosis the dye is released into the extracellular space, and the loss of fluorescence is used as a measure of exocytosis (Ryan et al., 1993).
FM1-43 nonspecifically attaches to the whole surface of a cell, and on endocytosis the dye becomes internalized. In fully polarized hippocampal neurons, endocytosis occurs over the entire somatodendritic surface (Parton and Dotti, 1993). Using these properties, we developed a method to label dendritic membranous compartments with FM1-43. This method allowed us to visualize the behavior of labeled compartments in living cultured hippocampal neurons and to determine the cellular mechanisms controlling their exocytosis. Furthermore, because there is evidence for mixing between endocytic and biosynthetic pathways (Trowbridge et al., 1993), studies monitoring the fate of endocytosed FM1-43 may shed light on the mechanisms by which newly synthesized membrane-bound proteins, like receptors, are transported and inserted into synapses.
MATERIALS AND METHODS
Cell culture
Cortical astrocytes were derived from 1-d-old rat pups and plated onto poly-l-lysine-coated coverslips. Hippocampal neurons were generated from 19-d-old rat embryos (Banker and Goslin, 1990) and plated onto a confluent monolayer of astrocytes. Astrocytes and neurons were plated at 50,000 and 30,000 cells per 18-mm-round glass coverslip (Fisher Scientific, Pittsburgh, PA), respectively. For relocation experiments the astrocytes and neurons were plated at 100,000 and 85,000 cells per 25-mm-square gridded glass coverslip (Bellco Glass, Vineland, NJ), respectively. Cultures were maintained in a serum-free medium (Banker and Goslin, 1990).
Labeling neurons with FM1-43
Acute FM1-43 loading. Cultures were exposed to 15 μm FM1-43 combined with 90 mm KCl for 1 min while on the microscope stage and washed for 10 min with constantly perfusing bath solution [containing (in mm) 119 NaCl, 2.5 KCl, 1.3 MgCl2, 2.5 CaCl2, 1 NaH2PO4, 26.2 NaHCO3, and 11 glucose].
Overnight FM1-43 loading. Cultures were exposed 16–36 hr to 1.5 μm FM1-43 at 35.5°C before transfer to the microscope stage. Cultures were rinsed with 0.1% DMSO for 1 min and maintained in constantly perfusing bath solution for 1 hr. The bath solution (see above) contained 1 μm tetrodotoxin to abolish action potentials.
Time-dependent loading. Mature cultures (>9 d in vitro, DIV) were loaded with 15 μm FM1-43 for 1–16 hr. After loading, they were transferred to a microscope stage and washed with the bath solution for 30 min. They were challenged with vehicle (0.1% DMSO) or 1 μm A23187 for 1 min. Calcium-evoked dendritic exocytosis (CEDE) was monitored for at least 15 min.
Calcium-evoked dendritic exocytosis
After overnight loading with 1.5 μm FM1-43, the cultures were transferred to the microscope stage and maintained in constantly perfusing bath solution for 1 hr. Calcium ionophore A23187 (1 μm) was applied directly onto the coverslip for 1 min. Fluorescence was monitored for 30 min before and 30 min after A23187 application, at 15 min intervals. An additional image was taken ∼5 min after ionophore application. All images in a given experiment were taken with the same exposure time (100–500 msec, chosen so that the fluorescent signal did not saturate the camera detection limit). A number of cells (>20) were tested for viability with 0.4% trypan blue. More than 90% of the neurons excluded the dye after A23187 challenge (data not shown), indicating that ionophore application did not have toxic effects. Viability also was confirmed in a number of experiments (>5) in which the subsequent application of A23187 combined with FM1-43 produced loading (data not shown).
EGTA treatment. Mature cultures (>9 DIV) were loaded with 1.5 μm FM1-43 overnight and washed as above. Cells were incubated in 5 mm EGTA, pH 7.0, in Ca2+-free artificial CSF for 10 min before A23187 challenge and for 15 min thereafter.
Tetanus toxin treatment. Mature cultures (>9 DIV) were loaded with 1.5 μm FM1-43 for 14 hr and then exposed for 5 hr to 10 nm tetanus toxin (generously provided by Dr. Giampetro Schiavo, Rockefeller University, New York, NY) and 1.5 μm FM1-43 at 35.5°C. Experiments were performed as above.
Nocodazole treatment. Mature cultures (>9 DIV), loaded overnight with 1.5 μm FM1-43, were exposed for 45 min to 2.5 μg/ml nocodazole and 1.5 μm FM1-43 at 35.5°C. Experiments were performed as above.
Field electrical stimulation. Mature cultures (>9 DIV), loaded overnight with 1.5 μm FM1-43, were stimulated with repetitive field stimuli. The current was delivered between the two silver wires placed 3 mm apart onto the coverslip with cultured neurons. Different frequencies (1, 10, 50, and 100 Hz) and numbers of stimuli were applied.
Measurement of released FM1-43. Neurons loaded overnight with 1.5 μm FM1-43 were washed in the same manner as for imaging experiments. They were incubated for 15 min in HEPES buffer, pH 7.2. An aliquot of this bathing solution (Apre) was recovered. Then the neurons were exposed to 1 μm A23187 for 1 min, followed by fresh HEPES buffer for 15 min. An aliquot from this bathing solution (Apost) was recovered.Apre and Apost solutions were pooled from 10 cultures. FM1-43 was extracted from these aqueous solutions with an equal volume of water-saturated butanol. The butanol fraction was recovered and evaporated. (3-[(3Cholamidopropyl)dimethylammonio]-1-propane-sulfonate) (CHAPS) (2%) was added (FM1-43 binds to CHAPS and increases fluorescence; Henkel et al., 1996). This solution was put in a quartz rectangular capillary, and FM1-43 fluorescence was measured (FITC filters; Zeiss Axioskop, Oberkochen, Germany). We measured the extracted fluorescence of HEPES buffer, Apre andApost. The amount of released FM1-43 was calculated as [Apost − buffer]/[Apre − buffer]. Calibration curves were generated from 1.5 nm to 1.5 μm FM1-43 before each experiment (n = 7).
Microscopy
Images were acquired with a computer-controlled cooled CCD camera (Photometrics, Tucson, AZ), using FITC filters (peak at 450 nm) on a Zeiss Axioskop epifluorescence microscope (50 W mercury lamp), and processed with Photometrics-supplied software (PMIS). Time-lapse images of mature neurons loaded with FM1-43 were acquired every 2 min over a 1 hr period, using FITC filters on a Zeiss Axiovert inverted microscope (50 W mercury lamp; 100× lens with additional 2.5× magnification).
Immunocytochemistry
Mature neurons (>12 DIV) were loaded with 1.5 μmFM1-43 overnight and then washed for 1 hr with the bathing solution. Cultures were fixed in 4% paraformaldehyde in 0.1 m PBS containing 0.12 m sucrose for 20 min at +4°C and immediately permeabilized with 0.3% Triton X-100 in PBS for 5 min at room temperature. FM1-43-labeled compartments were imaged after fixation (100× lens, FITC filter, Zeiss Axioskop, 75 W xenon lamp) to avoid confounding results if the labeled organelles moved before fixation. Imaging with Texas Red filters (Omega Optical, Brattleboro, VT) did not reveal any FM1-43 fluorescence. Cultures were incubated in blocking solution (10% horse serum, 1% goat-serum, and 0.1% Triton X-100 in PBS) for 1 hr at room temperature and then immunostained overnight with the primary antibody [polyclonal anti-synapsin I antibody: 1:10, polyclonal rabbit sera number 6246, generously provided by Dr. Pietro De Camilli (Yale University, New Haven, CT); polyclonal anti-transferrin receptor antibody raised in sheep: 215 μg/ml, Sigma (St. Louis, MO); polyclonal anti-mannose-6-phosphate receptor antibody: 1:50, generously provided by Dr. W. J. Brown (Cornell University, Ithaca, NY); polyclonal anti-cathepsin D antibody: 1:100, generously provided by Dr. W. J. Brown (Cornell University); polyclonal anti-inositol triphosphate (IP3) receptor antibody: 1:50, generously provided by Dr. T. Jayaraman (Mount Sinai School of Medicine, New York, NY); monoclonal anti-BiP antibody: 16 μg/ml, Stress Gen (Sidney, Canada)]. Primary antibody was washed out three times for 10 min with the blocking solution. Then the cells were incubated with the appropriate secondary biotinylated antibody (2.5 μg/ml; Cappel, West Chester, PA) for 1 hr at room temperature. After the washout of the biotinylated antibody, the cultures were incubated with Texas Red-conjugated avidin (25 μg/ml; Cappel) for 1 hr at room temperature. Immunoreactivity was analyzed immediately after the Texas Red–avidin was washed out with PBS (100× lens, Texas Red filter). Neurons were relocalized by using gridded coverslips (Bellco).
NBD C6-ceramide labeling. Mature neurons (>12 DIV) were loaded with 1.5 μm FM1-43 for 4 hr (see Fig. 6B) or overnight (see Fig. 3) and washed for 1 hr before fixation as described above. FM1-43-labeled compartments were imaged, and the area was exposed to UV light to bleach the FM1-43 fluorescence. When no FM1-43 fluorescence was detected, the neurons were incubated with 40 μm NBD C6-ceramide (Molecular Probes, Eugene, OR) for 1 hr at 37°C and washed with 2.5% fat-free bovine serum albumin three times for 20 min at room temperature. Fluorescence was analyzed with FITC filters.
Fig. 6.

FM1-43 reaches CEDE-competent compartments only after 8 hr of loading. A, Plot of mean ± SEM of CEDE observed 15 min after vehicle (0.1% DMSO; dotted line) or A23187 (solid line) application in mature neurons (>9 DIV) loaded for 1–16 hr with FM1-43 (FM1-43 at 1 hr, n = 13; all other time points,n = 5). B, Quantitative analysis of the correlation between FM1-43-labeled compartments and NBD C6-ceramide in neurons loaded with the FM1-43 for 4 or 16 hr. High correlation is observed only in neurons loaded for 16 hr with FM1-43 (n = 5), but not in neurons loaded for 4 hr (n = 2).
Fig. 3.
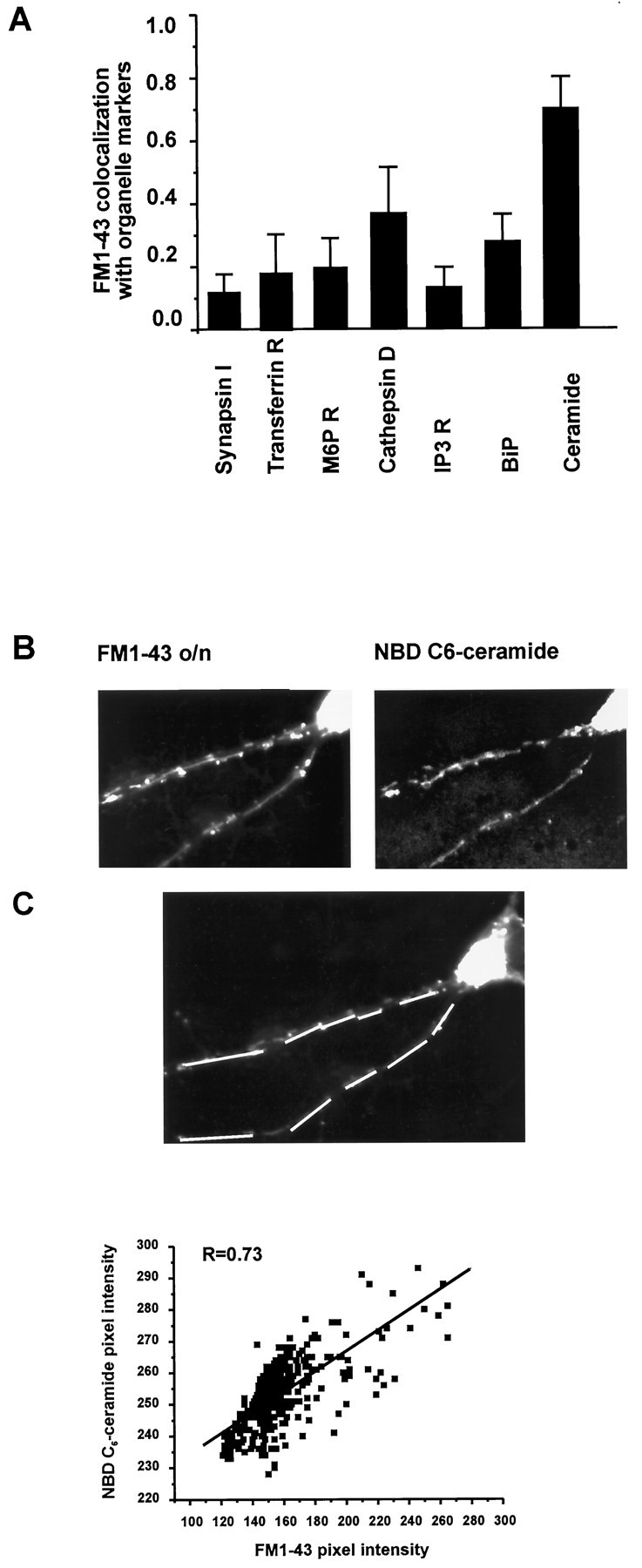
Organelles labeled by overnight exposure to FM1-43 colocalize with NBD C6-ceramide in dendrites, a label of SER/TGN-like structures. A, Quantitative analysis of the correlation between FM1-43-labeled compartments and specific organelles identified by immunocytochemistry. Bars show mean ± SEM of the correlation coefficient analyzed in two to eight regions per neuron. Significant correlation was observed only with a trans-Golgi network marker NBD C6-ceramide. B, Colocalization of FM1-43-labeled compartments and trans-Golgi network, identified by the fluorescent marker NBD C6-ceramide. Most FM1-43 label colocalizes with the trans-Golgi marker. Quantitative analysis of the correlation between FM1-43-labeled compartments and NBD C6-ceramide was performed along the indicatedlines. Scale bar, 10 μm. C, Quantitative analysis of the correlation between FM1-43-labeled compartments and NBD C6-ceramide for the neuron inB. For each pixel on the indicated lines, intensity in the channel measuring FM1-43 (ordinate) was plotted against intensity in the channel measuring NBD C6-ceramide (abscissa). The correlation coefficient, R, was calculated.
Electron microscopy
The 14 DIV hippocampal neurons, loaded with 1.5 μmFM1-43 overnight, were washed with PBS for 1 hr before fixation with 3% paraformaldehyde in 0.2 m cacodylate buffer, pH 7.30, for 45 min at room temperature. After being washed with 0.2m cacodylate buffer, secondary fixation was performed, using 1% OsO4 with 1.5% K4Fe(CN)6in cacodylate (90 min at room temperature), followed by dHOH washes and en bloc uranyl acetate staining. Cells were dehydrated through a graded series of ethanol and embedded in Epon–Araldite resin. Sections were collected on Formvar-coated cupri slot grids, counterstained with uranyl acetate and Reynold’s lead citrate, and examined in the Hitachi H-7000 at 75 kV.
Fluorescent and electron micrographs were superimposed in Adobe Photoshop 4.0. To align fluorescent and electron micrographs, we used fiduciary points chosen on the basis of regions clearly identified on both photographs (e.g., the intersection of dendrites and axons, and at the dendritic branching points). Electron micrographs were scanned (ScanJet Plus, Packard, Meriden, CT), and the image sizes of both micrographs were matched. Images were aligned with respect to fiduciary points. After alignment, the fluorescent image was pseudocolored for better visualization of the FM-labeled structures.
Data analysis
For quantitative measurements of CEDE, several regions of interest (2–5) were placed on dendrites (locations were chosen before post-treatment images were analyzed), and FM1-43 fluorescence was integrated. Background fluorescence, measured in regions next to the neurons, was subtracted. FM1-43 fluorescence values were normalized to the intensity observed immediately before A23187 application. Statistical analysis was done by one-way ANOVA.
The degree of colocalization between FM1-43 fluorescent sites and other fluorescent markers was quantified with PMIS software (Photometrics). Regions of interest from images of FM1-43 fluorescent sites were identified, and pixel intensity along lines traversing ∼50 μm was measured [IF(x)]. Pixel intensities along the same lines placed in the same regions from the image detecting another fluorescent marker were measured also [IM(x)]. We plotted IF(x) versusIM(x) and calculated a correlation coefficient,R (using Origin 4.0 software, Microcal, Amherst, MA). Each neuron that was examined had two to eight lines analyzed. Locations were chosen from FM1-43-loaded neurons before the corresponding marker-labeled image was analyzed.
RESULTS
FM1-43 labels dendritic compartments
Brief exposure of cultured neurons to FM1-43, combined with depolarizing stimuli, can produce a selective loading of presynaptic vesicles (Fig. 1A,C) (Ryan et al., 1993). In contrast, we found that cultured hippocampal neurons incubated with FM1-43 for 16–36 hr in the absence of stimulation loaded intracellular compartments in all processes and the soma (Figs. 1D, 2, 4). We focused our analysis on the characterization of FM1-43-labeled compartments in processes, anticipating that these may be more likely to be associated with synaptic plasticity.
Fig. 1.

FM1-43 fluorescent probe labels presynaptic or dendritic compartments in cultured hippocampal neurons, depending on the loading protocol. A, Location of FM1-43 dye after exposing 14 DIV neurons to FM1-43, combined with 90 mm KCl, for 1 min. Scale bar, 10 μm. B, Expression of synapsin I in characteristic punctate labeling pattern indicates presynaptic axon terminals that are closely apposed to the neuronal soma and major dendrite. The majority of FM1-43-labeled sites in Acorresponds to synapsin I immunoreactivity (arrow).C, Time-lapse images of a region after acute FM1-43 loading of 20 DIV cultured neuron. Little movement of spots is apparent. Spontaneous destaining (∼20%) over a 1 hr observation period was typical (display scale maximum was changed from 110 to 95 arbitrary units to facilitate the comparison of spot locations). The time is indicated in minutes. Scale bar, 2 μm. D, Overnight exposure of 14 DIV neurons to FM1-43 extensively labels compartments in soma and dendrites. Scale bar, 10 μm.E, Expression of synapsin I, again labeling presynaptic sites closely apposed to the major dendrite. FM1-43-labeled compartments and synapsin I do not overlap in the majority of sites, indicating dendritic (arrowheads) and presynaptic (arrows) localization of labeled sites, respectively.F, Time-lapse images of a region after overnight FM1-43 loading of 20 DIV cultured neuron. Note the location of FM1-43 spots at the base of small filopodial structures and coordinate movement of the filopodia and spots. The time is indicated in minutes. Scale bar, 2 μm.
Fig. 2.
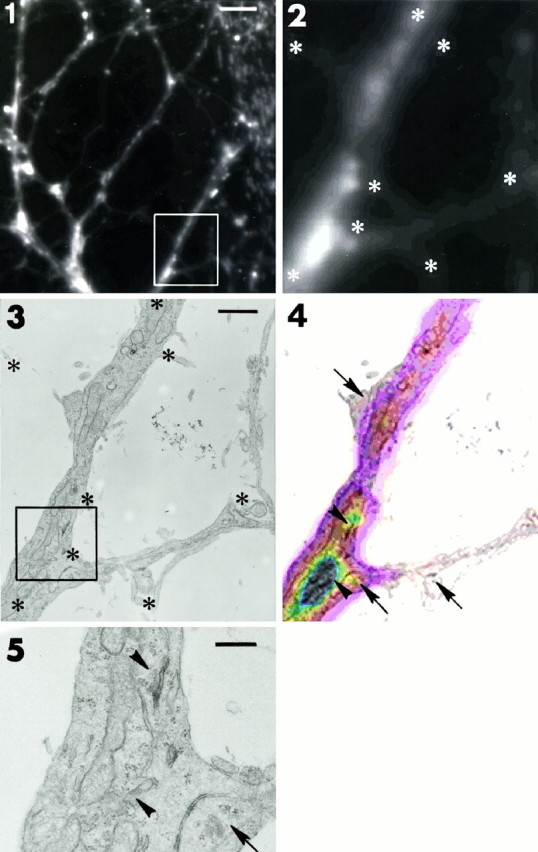
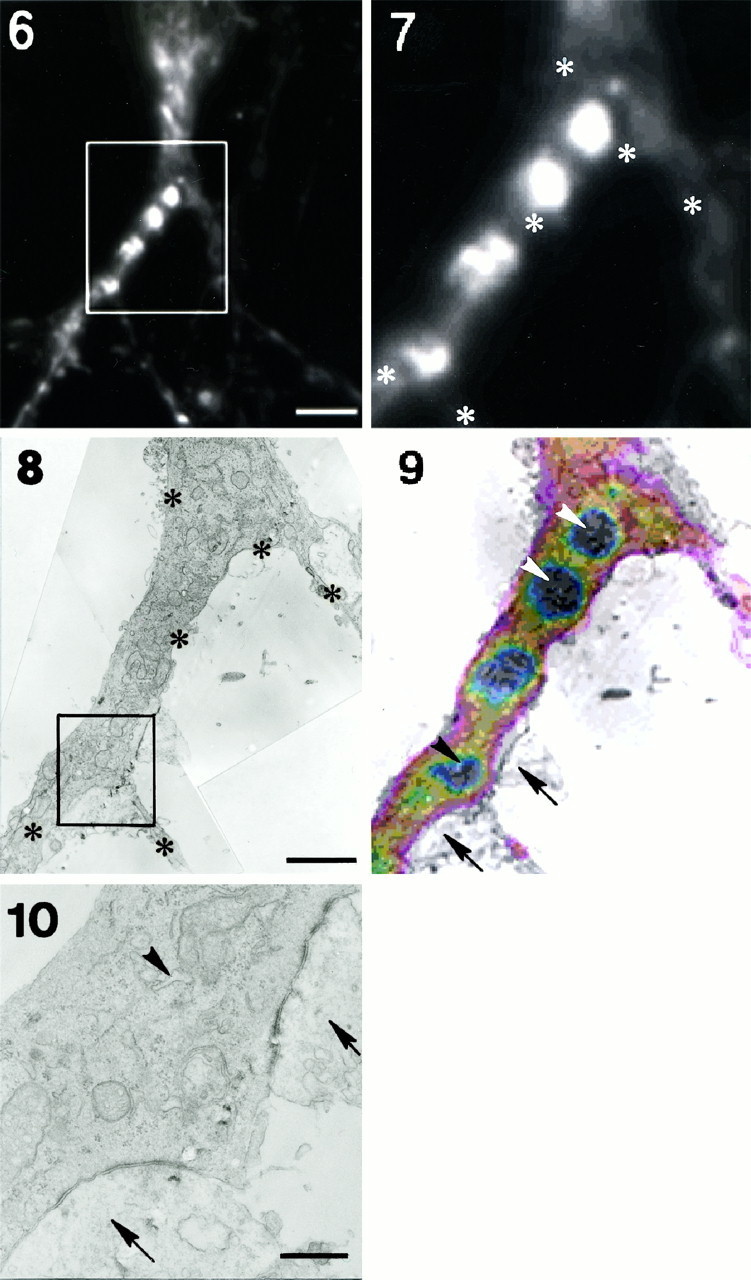
Identification of FM1-43-labeled compartments by electron microscopy (EM). Shown are two example sets (1-5, 6–10) of correlative fluorescent and electron microscopy of mature neurons exposed overnight to FM1-43. Fluorescent micrographs (1, 6) show FM1-43-labeled compartments. Scale bars, 10 μm. Outlined regions are enlarged (2,7) and are represented on the electron micrographs (3, 8). Scale bars: 1 μm in3; 1.8 μm in 8.Asterisks indicate fiduciary points used to superimpose fluorescent and electron micrographs. FM1-43-labeled compartments in dendrites (white arrowheads), opposite the presynaptic sites (arrows), and in areas containing SER/TGN organelles (black arrowheads) are evident on the superimposed pseudocolored fluorescent micrographs (4,9). Note the lack of FM1-43 in regions corresponding to presynaptic terminals. EM sections at other levels in the same region also failed to show presynaptic terminals in fluorescent regions.Outlined regions on the electron micrographs are enlarged in 5 and 10 for better identification of postsynaptic SER/TGN-like structures. Scale bars: 0.3 μm in 5; 0.5 μm in 10. Figure continues.
Fig. 4.

Calcium-evoked dendritic exocytosis (CEDE).A, Fluorescent images of a representative culture showing CEDE in response to ionophore application. Cultured hippocampal neurons (12 DIV) loaded overnight with 1.5 μm FM1-43 were challenged with 1 μm calcium ionophore A23187 (arrow) for 1 min after a prechallenge wash of 1 hr. The three images shown were taken at the times indicated, after the removal of FM1-43. Scale bar, 10 μm. B, Plot of the mean ± SEM of CEDE analyzed in two regions of interestoutlined in A, with respect to fluorescence observed immediately before A23187 application.
To determine the distribution of FM1-43 fluorescence in pre- and postsynaptic compartments after the two labeling protocols, we compared its labeling pattern with that of a presynaptic marker protein, synapsin I (Fig. 1B,E; n = 3). After standard acute exposure and depolarizing stimuli, FM1-43 and synapsin I showed significant colocalization, confirming the presynaptic localization of FM1-43 with this loading protocol (Fig.1A,B). However, with long exposure and no stimulation, FM1-43 and synapsin I showed little colocalization (Fig.1D,E). This indicates that the longer exposure selectively labels intracellular compartments that are not presynaptic terminals. Quantitated immunocytochemical analysis (see Materials and Methods) confirmed that, with overnight exposure to the dye, FM1-43 fluorescence in synapsin I-labeled compartments accounts for only 10% of the dye (Fig. 3A; R = 0.13 ± 0.06;n = 3).
Dendritic compartments labeled with FM1-43 often were seen at the base of small filopodial structures, proposed to be synaptic spine precursors (Fig. 1F) (Ziv and Smith, 1996). Time-lapse imaging over 1 hr revealed that FM1-43-loaded compartments maintained this association although the filopodial structures moved over time (∼2 μm/min; Fig. 1F). In contrast, presynaptic vesicles labeled with acute loading of FM1-43 did not move within 1 hr (n = 3; Fig. 1C), again supporting the conclusion that overnight FM1-43 exposure loads compartments that are not presynaptic terminals.
The dendritic localization of FM1-43-labeled compartments was confirmed further by correlative electron and fluorescent microscopy (Fig.2). Neurons loaded with FM1-43 overnight were imaged under high magnification (100× lens; Fig. 2-1,6) and then processed for electron microscopy (EM). The same regions were identified on the EM preparations (Fig. 2-3,8) and aligned with the light microscope images by using fiduciary points (Fig. 2-4,9). Under EM, FM1-43-fluorescent sites were dendritic and often close to synapses. The sites included different organelles, such as coated pits, endosomes, multivesicular bodies, endoplasmic reticulum, Golgi apparatus, and mitochondria (Fig. 2-5,10). No dense core granules were observed.
To identify which dendritic organelles are labeled by overnight exposure to FM1-43, we performed high-resolution immunocytochemical studies on FM1-43-loaded neurons (Fig.3A). After FM1-43 imaging, the cultures were fixed and labeled with different organelle-specific markers: transferrin receptor (Moos, 1996), cathepsin D (Nakanishi et al., 1994), and mannose-6-phosphate receptor (Couce et al., 1992), specific for endosome/lysosome organelles; IP3 receptor (Moschella et al., 1995) and BiP (Huovila et al., 1992), specific for endoplasmic reticulum; and NBD C6-ceramide (Pagano et al., 1989), a trans-Golgi fluorescent marker. Quantitative immunocytochemical analysis (see Materials and Methods) indicated little colocalization between FM1-43-labeled compartments and transferrin receptor (R = 0.2 ± 0.14;n = 3), cathepsin D (R = 0.4 ± 0.16; n = 3), mannose-6-phosphate receptor (R = 0.21 ± 0.1; n = 3), IP3 receptor (R = 0.14 ± 0.07;n = 3), and BiP (R = 0.3 ± 0.09;n = 3). However, we observed a consistently high correlation of FM1-43-labeled dendritic sites only with NBD C6-ceramide (R = 0.77 ± 0.11;n = 5; Fig. 3). This indicates that, after overnight exposure to FM1-43, dendritic organelles loaded with the FM1-43 are primarily dendritic Golgi-like or Golgi-derived structures.
Dendritic organelles undergo regulated exocytosis
To determine whether FM1-43-labeled compartments exhibit calcium-evoked exocytosis, we challenged neurons loaded with FM1-43 for 16–36 hr with the calcium ionophore A23187 (calcimycin) for 1 min. This produced a marked loss of FM1-43 fluorescence (Fig.4A) that was attributable to the exocytosis of internalized dye (see below). We measured this CEDE as the percentage of loss of background-subtracted FM1-43 fluorescence as compared with pre-ionophore background-subtracted fluorescence (Fig. 4B). Exposure to the carrier solution alone (0.1% DMSO) produced no loss of fluorescence (Fig. 5; n = 12). Similarly, calcimycin did not have any effect when it was applied in the presence of 5 mm EGTA (Fig. 5; n = 6). A23187 induced CEDE only in the presence of 2.5 mmcalcium. It could be recorded within 5 min after ionophore application and persisted for 15 min.
Fig. 5.

CEDE is age-dependent and blocked by tetanus toxin (TT) and nocodazole (noc). CEDE was analyzed in developing cultured neurons (5–21 DIV;EGTA, TT, and noc were tested at >9 DIV). Bar graph shows mean ± SEM of CEDE 15 min after A23187 application. Vehicle, 0.1% DMSO; *p< 0.05; **p < 0.01. Sample size inparentheses refers to the number of cultures examined.
Ionophore-induced exocytosis from neuronal cultures loaded overnight with FM1-43 was confirmed by recovering dye in the extracellular medium (Henkel et al., 1996). Extracellular medium aliquots from 10 pooled cultures were obtained before (Apre) and 15 min after (Apost) A23187 application. Dye from pooled samples was extracted with butanol and dissolved in 2% CHAPS. FM1-43 fluorescence intensity was measured in microcapillaries. The relative FM1-43 fluorescence intensity of solution after A23187 treatment was 3.32 ± 0.7 (n = 7;p < 0.05). This shows directly that a significant amount of FM1-43 was released by calcium ionophore treatment and could be recovered in the extracellular medium. Calibration curves generated from 1.5 nm to 1.5 μm FM1-43 indicate that the observed exocytosis corresponds to ∼35 nm FM1-43. This is ∼100 times more than the estimated FM1-43 release measured after similar experiments conducted on presynaptic exocytosis (0.3 nm; Henkel et al., 1996).
If the organelles undergoing CEDE are derived from the trans-Golgi network (TGN), then one may expect that endocytosed FM1-43 may require several hours before reaching such compartments. To examine this possibility, we measured the amount of CEDE as a function of the FM1-43 loading time (Fig. 6A). Mature cultures were exposed to FM1-43 for different periods of time (1–16 hr) and challenged with A23187. Only when neurons were exposed to FM1-43 for >8 hr was CEDE observed (Fig. 6A). Notably, FM1-43 showed little colocalization with NBD C6-ceramide fluorescence after 4 hr of loading (n = 2) and significant colocalization after 16 hr of FM1-43 loading (Fig. 6B). These results indicate that the passive loading of FM1-43 requires 8 hr before the dye reaches CEDE-competent organelles, at which time the dye colocalizes with NBD C6-ceramide. This result supports the view that organelles undergoing CEDE are TGN-like or TGN-derived structures.
We tested if CEDE uses exocytotic machinery with tetanus toxin, a specific inhibitor of exocytosis (Montecucco and Schiavo, 1994). CEDE was blocked completely by pretreatment with 10 nm tetanus toxin (see Fig. 5; n = 7), indicating regulated exocytosis rather than some nonspecific loss of the dye from intracellular compartments.
We found that CEDE is regulated developmentally. Although neurons of all ages that were examined (5–21 DIV) showed comparable loading after overnight exposure to FM1-43, only neurons that were 9 DIV or older were capable of producing CEDE (see Fig. 5; n = 11;p < 0.05). At this stage the cultured neurons have already established synaptic contacts and are fully differentiated. At earlier ages A23187 occasionally induced regrouping of labeled sites or intensified movement of fluorescently labeled organelles but caused no detectable CEDE.
Dendritic exocytosis of FM1-43 also could be elicited with repetitive field electrical stimulation (Fig. 7). We tested different numbers and frequencies of stimuli. Low-frequency stimuli (1 Hz) did not produce any detectable loss of fluorescence (n = 3). Medium-frequency stimuli (10 Hz) could produce loss of fluorescence, but only if sufficient stimuli were delivered. High-frequency stimuli (50 Hz) generally produced exocytosis (n = 10). Figure 7 shows a pattern of CEDE observed in a neuron stimulated with different parameters. This pattern generally was observed in the tested neurons. In addition, the application of 1 mm glutamate for 1 min could evoke the exocytosis of some dendritic structures (n = 2; data not shown). We characterized CEDE by using calcium ionophore as the stimulating agent, because it gave the most robust effect.
Fig. 7.

Field electrical stimulation elicits dendritic exocytosis. A, Fluorescent images of a neuron loaded overnight with FM1-43 before (left) and after (right) field electrical stimulation (50 Hz, 500 stimuli). Note the marked loss of fluorescence indicating dendritic and somatic exocytosis. B, Plot of dendritic exocytosis in the neuron (A) as a function of stimulation frequency. Note the requirement for higher frequencies and repeated stimuli, reminiscent of the requirements for LTP.
DISCUSSION
FM1-43 labels dendritic compartments
In this study we report on a novel biological process of regulated dendritic exocytosis. Cultured hippocampal neurons were exposed to FM1-43 for >16 hr with no stimulation. Because FM1-43 immerses into the outer leaflet of the plasma membrane, it is internalized inside the cells whenever there is endocytosis. On washout of the uninternalized dye from the outer surface, the remaining FM1-43 fluorescence marks the inner membrane of intracellular membranous compartments. Via the fusion of endosomes and endosome-derived compartments, over time, the dye has access to numerous intracellular compartments. Because the neurons are exposed continuously to FM1-43 for >16 hr, some FM1-43 can be found in intracellular compartments that are derived from endocytic organelles—like early endosomes (expressing transferrin receptor), late endosomes and lysosomes (expressing cathepsin D and mannose-6-phosphate receptor), and endoplasmic reticulum in soma (expressing IP3 receptor and BiP). However, quantitative analysis of the colocalization between FM1-43-labeled structures and intracellular markers of various organelles shows the greatest degree of colocalization with NBD C6-ceramide, indicating the labeling of TGN-like or TGN-derived structures (see Fig. 3).
The immunocytochemical analysis of labeled compartments is supported by correlative electron and light microscopy. Electron microscopy of FM1-43-labeled regions indicates that the majority of labeled sites clearly is in dendrites and not presynaptic terminals, has no dense core vesicles, has smooth endoplasmic reticulum (SER)/TGN organelles, and often is close to synapses. SER/TGN-like structures have been identified in spine apparatus and parent dendrites (Spacek and Harris, 1997). In addition, numerous organelles potentially involved in endocytosis and exocytosis have been visualized in spines under electron microscopic analysis (Harris and Kater, 1994; Spacek and Harris, 1997), although their function has only been hypothesized. Smooth vesicles fusing with the plasma membrane were seen in some spines, and exocytosis was suggested on the basis of these observations (Spacek and Harris, 1997).
In contrast to acute loading protocols, overnight exposure of neurons to FM1-43 appears to load little dye in presynaptic sites. This is indicated by several observations. First, organelles labeled with the overnight exposure to FM1-43 do not colocalize with synapsin I, a marker for presynaptic vesicles (see Fig. 1D,E). In addition, in associated electron micrographs the fluorescent regions are in dendrites and only occasionally in presynaptic terminals (see Fig. 2). Organelles loaded with overnight exposure to FM1-43 show considerable movement (∼2 μm/min) when observed during the 1 hr period, whereas organelles loaded with brief exposure to high potassium (which loads presynaptic terminals) do not move (see Fig.1F). After the neurons are exposed to FM1-43 in the absence of depolarization, 8 hr are required before the dye reaches exocytosis-competent organelles (see Fig. 6A). Presynaptic vesicles require only seconds after endocytosis to become exocytosis-competent (Ryan, 1996). Finally, compartments labeled with overnight exposure to FM1-43 colocalize with NBD C6-ceramide, which is found only in soma and dendrites and not in presynaptic terminals (see Figs. 3,6B). The significantly lower labeling of presynaptic sites during prolonged dye exposure may be attributable to their lower volume, mechanical perturbation (moving the coverslip to the recording chamber could induce presynaptic release), or extensive washing (generally >1 hr may preferentially induce presynaptic loss).
Calcium-evoked dendritic exocytosis
We find that a brief challenge of hippocampal neurons with calcium ionophore A23187 produces a robust dendritic exocytosis. CEDE of FM1-43 was demonstrated directly by measuring dye fluorescence in the extracellular medium. Calcium is required for CEDE, because ionophore did not produce any effect in the presence of the calcium chelator EGTA (see Fig. 5). CEDE was blocked by tetanus toxin, an agent known to block exocytosis (Montecucco and Schiavo, 1994). This indicates that the target of tetanus toxin, synaptobrevin, cellubrevin, or a homolog (Yamasaki et al., 1994; Chilcote et al., 1995), is necessary for CEDE.
CEDE can be detected within 5 min after ionophore application and reaches a plateau ∼15 min after stimulation. Individual sites do not disappear suddenly but destain continuously over this time. This is comparable to what has been described with presynaptic destaining, in which a single spot observed by light microscopy destains slowly rather than abruptly (Betz et al., 1992). The slow time course is explained by the presence of many small (50 nm) structures (labeled presynaptic vesicles) that cannot be viewed individually by light microscopy (Betz et al., 1992). The slow time course of CEDE may owe to a similar underlying process. For instance, CEDE destaining is consistent with the budding of small vesicles from dendritic SER/TGN-like structures and their fusion with the local dendritic plasma membrane. Such a process could occur over a time frame of minutes and would cause a relative decrease in the fluorescence of the SER/TGN-like compartments, rather than their complete destaining. Although small vesicles may be formed, they appear not to be transported for long distances along the dendrite, because all regions along a dendrite lose fluorescence with a similar time course. If vesicles were to move along a dendrite, then one would expect bright regions to lose fluorescence and neighboring dark regions (in the dendrite) to gain fluorescence. However, we see homogeneous loss of FM1-43 fluorescence over time, which indicates a delivery to the immediate surface. Therefore, organelles appear to be released locally.
Intact microtubules are required for CEDE, because nocodazole, an agent producing microtubule depolymerization (Goslin et al., 1989), prevents CEDE (see Fig. 5). An association between CEDE-competent compartments and microtubules is supported by the observation that these compartments undergo rapid movements that appear synchronized to local filopodial movements (see Fig. 1F). Thus, microtubules may provide a delivery system for TGN-like organelles to reach appropriate sites as well as a docking station for additional components that may be necessary to execute the numerous processes required for exocytosis (budding, docking, fusion, etc.).
Exocytosis of FM1-43-labeled sites also was observed in the soma, which is consistent with the recent publication demonstrating calcium-dependent exocytosis of compounds from the cell body of dorsal root ganglion cells (Huang and Neher, 1996).
Our experiments indicate that passive loading of FM1-43 requires 8 hr before the dye reaches structures that are competent to undergo CEDE. Studies of intracellular trafficking indicate that 8 hr is sufficient for endocytosed material to reach biosynthetic pathways like SER/TGN in cultured cells (Green and Kelly, 1992). Thus, these experiments further support the view that the organelles undergoing CEDE are TGN-derived structures and part of the biosynthetic pathway.
Under experimental conditions described in this study, in which calcium is raised everywhere in the cell, CEDE is widespread. However, the homogeneous loss of the dye in a nonhomogeneously labeled structure indicates that exocytosis is local. These observations suggest the existence of a general mechanism that is present throughout the cell. It is well known that physiological stimuli can produce a localized rise in postsynaptic Ca2+ concentration. Under such conditions one may expect localized CEDE, thereby ensuring spatial specificity to this process.
Possible Roles of CEDE
CEDE may underlie several previously described phenomena. Some of them include the release of dopamine from dendrites in substantia nigra neurons (Heeringa and Abercrombie, 1995) and somatic release of substance P observed after strong stimulation (Huang and Neher, 1996). Furthermore, there is evidence supporting a role for activity or calcium in the release of growth factors and β-amyloid (Querfurth and Selkoe, 1994; Blochl and Thoenen, 1995). It will be interesting to determine whether CEDE releases such compounds.
In addition, CEDE may be involved in the activity-dependent expression of transmembrane proteins. There are several reports on the activity-dependent expression of membrane-bound proteins, like neural cell adhesion molecules (NCAM) (Kiss et al., 1994). CEDE also might be involved in the redistribution of substance P receptors in spinal neurons after somatosensory stimulation (Mantyh et al., 1995) or the homeostatic control of transmembrane glucose transporter proteins (Cushman and Wardzala, 1980; Suzuki and Kono, 1980). Similarly, CEDE might play a role in the distribution of ion channels at the surface membrane, observed under different stimulus conditions (Harris et al., 1991; Turrigiano et al., 1994).
Could CEDE be involved in long-term potentiation (LTP)? Indeed, a recent report indicates that postsynaptic exocytosis is required to generate LTP (Lledo et al., 1998). Localized calcium entry during LTP induction might trigger the insertion of glutamate receptors (stored in intracellular compartments or perisynaptic regions) (Hampson et al., 1992; Baude et al., 1995) into synapses by a CEDE-like process. This would produce larger responses to a synaptically released transmitter. A similar process triggered by insulin appears to deliver GABA receptors to the cell surface (Wan et al., 1997). In addition to delivering receptors to synapses, substances concentrated inside the postsynaptic membranous structures could be released into the synaptic cleft, potentially affecting presynaptic function.
Finally, CEDE could provide new membrane at postsynaptic sites, allowing for the growth and formation of new synapses (Lisman and Harris, 1993). Indeed, SER and smooth vesicles have been localized at the base and inside dendritic spines (Spacek and Harris, 1997), but their function has not yet been clarified.
Conclusion
In summary, in this manuscript we have described a novel process of regulated exocytosis in cultured hippocampal neurons—CEDE. We have shown that CEDE has a number of important, physiologically relevant, properties: it is calcium-evoked and it requires mature neurons, exocytotic machinery, and intact microtubules. Compartments that undergo CEDE derive from SER/TGN-like organelles in dendrites and postsynaptic sites. This phenomenon thus could play a role in synaptic plasticity, where it might involve changes in glutamate sensitivity, release of active compounds having effect on pre- and postsynaptic function, and/or growth of the plasma membrane.
Footnotes
This study was supported by the Mathers Foundation and National Institutes of Health. We are grateful to Nancy Dawkins for preparing the cultures, Tamara Howard for assistance with the electron microscopy, Jason Kass for technical assistance with immunocytochemistry, and Irena Miloslavskaya for assistance in image processing.
Correspondence should be addressed to Dr. Roberto Malinow, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724.
REFERENCES
- 1.Alberts B, Bray D, Lewis J, Raff M, Roberts K, Watson JD. Molecular biology of the cell. Garland; Hamden, CT: 1989. [Google Scholar]
- 2.Banker GA, Goslin K. Culturing nerve cells. MIT; Cambridge, MA: 1990. [Google Scholar]
- 3.Baude A, Nusser Z, Molnar E, McIlhinney RA, Somogyi P. High-resolution immunogold localization of AMPA-type glutamate receptor subunits at synaptic and nonsynaptic sites in rat hippocampus. Neuroscience. 1995;69:1031–1055. doi: 10.1016/0306-4522(95)00350-r. [DOI] [PubMed] [Google Scholar]
- 4.Betz WJ, Mao F, Bewick GS. Activity-dependent fluorescent staining and destaining of living vertebrate motor nerve terminals. J Neurosci. 1992;12:363–375. doi: 10.1523/JNEUROSCI.12-02-00363.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blochl A, Thoenen H. Characterization of nerve growth factor (NGF) release from hippocampal neurons: evidence for a constitutive and an unconventional sodium-dependent regulated pathway. Eur J Neurosci. 1995;7:1220–1228. doi: 10.1111/j.1460-9568.1995.tb01112.x. [DOI] [PubMed] [Google Scholar]
- 6.Chilcote TJ, Galli T, Mundigl O, Edelmann L, McPherson PS, Takei K, De Camilli P. Cellubrevin and synaptobrevins: similar subcellular localization and biochemical properties in PC12 cells. J Cell Biol. 1995;129:219–231. doi: 10.1083/jcb.129.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Couce ME, Weatherington AJ, McGinty JF. Expression of insulin-like growth factor-II (IGF-II) and IGF-II/mannose-6-phosphate receptor in the rat hippocampus: an in situ hybridization and immunocytochemical study. Endocrinology. 1992;131:1636–1642. doi: 10.1210/endo.131.4.1396308. [DOI] [PubMed] [Google Scholar]
- 8.Cushman SW, Wardzala LJ. Potential mechanism of insulin action on glucose transport in the isolated rat adipose cell. Apparent translocation of intracellular transport systems to the plasma membrane. J Biol Chem. 1980;255:4758–4762. [PubMed] [Google Scholar]
- 9.Goslin K, Birgbauer E, Banker G, Solomon F. The role of cytoskeleton in organizing growth cones: a microfilament-associated growth cone component depends upon microtubules for its localization. J Cell Biol. 1989;109:1621–1631. doi: 10.1083/jcb.109.4.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green SA, Kelly RB. Low-density lipoprotein receptor and cation-independent mannose 6-phosphate receptor are transported from the cell surface to the Golgi apparatus at equal rates in PC12 cells. J Cell Biol. 1992;117:47–55. doi: 10.1083/jcb.117.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hampson DR, Huang XP, Oberdorfer MD, Goh JW, Auyeung A, Wenthold RJ. Localization of AMPA receptors in the hippocampus and cerebellum of the rat using an anti-receptor monoclonal antibody. Neuroscience. 1992;50:11–22. doi: 10.1016/0306-4522(92)90378-f. [DOI] [PubMed] [Google Scholar]
- 12.Harris HW, Jr, Strange K, Zeidel ML. Current understanding of the cellular biology and molecular structure of the antidiuretic hormone-stimulated water transport pathway. J Clin Invest. 1991;88:1–8. doi: 10.1172/JCI115263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris KM, Kater SB. Dendritic spines: cellular specializations imparting both stability and flexibility to synaptic function. Annu Rev Neurosci. 1994;17:341–371. doi: 10.1146/annurev.ne.17.030194.002013. [DOI] [PubMed] [Google Scholar]
- 14.Heeringa MJ, Abercrombie ED. Biochemistry of somatodendritic dopamine release in substantia nigra: an in vivo comparison with striatal dopamine release. J Neurochem. 1995;65:192–200. doi: 10.1046/j.1471-4159.1995.65010192.x. [DOI] [PubMed] [Google Scholar]
- 15.Henkel W, Lubke J, Betz WJ. FM1-43 dye ultrastructural localization in and release from frog motor nerve terminals. Proc Natl Acad Sci USA. 1996;93:1918–1923. doi: 10.1073/pnas.93.5.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang L-YM, Neher E. Ca2+-dependent exocytosis in the somata of dorsal root ganglion neurons. Neuron. 1996;17:135–145. doi: 10.1016/s0896-6273(00)80287-1. [DOI] [PubMed] [Google Scholar]
- 17.Huovila A-P, Eder AM, Fuller SD. Hepatitis B surface antigen assembles in a post-ER, pre-Golgi compartment. J Cell Biol. 1992;118:1305–1320. doi: 10.1083/jcb.118.6.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kiss JZ, Wang C, Olive S, Rougon G, Lang J, Baetens D, Harry D, Pralong WF. Activity-dependent mobilization of the adhesion molecule polysialic NCAM to the cell surface of neurons and endocrine cells. EMBO J. 1994;13:5284–5292. doi: 10.1002/j.1460-2075.1994.tb06862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lisman JE, Harris KM. Quantal analysis and synaptic anatomy—integrating two views of hippocampal plasticity. Trends Neurosci. 1993;16:141–147. doi: 10.1016/0166-2236(93)90122-3. [DOI] [PubMed] [Google Scholar]
- 20.Lledo PM, Zhang X, Sudhof TC, Malenka RC, Nicoll RA. Postsynaptic membrane fusion and long-term potentiation. Science. 1998;279:399–403. doi: 10.1126/science.279.5349.399. [DOI] [PubMed] [Google Scholar]
- 21.Mantyh PW, DeMaster E, Malhotra A, Ghilardi JR, Rogers SD, Mantyh CR, Liu H, Basbaum AI, Vigna SR, Maggio JE. Receptor endocytosis and dendrite reshaping in spinal neurons after somatosensory stimulation. Science. 1995;268:1629–1632. doi: 10.1126/science.7539937. [DOI] [PubMed] [Google Scholar]
- 22.Montecucco C, Schiavo G. Mechanism of action of tetanus and botulinum neurotoxins. Mol Microbiol. 1994;13:1–8. doi: 10.1111/j.1365-2958.1994.tb00396.x. [DOI] [PubMed] [Google Scholar]
- 23.Moos T. Immunohistochemical localization of intraneuronal transferrin receptor immunoreactivity in the adult mouse central nervous system. J Comp Neurol. 1996;375:675–692. doi: 10.1002/(SICI)1096-9861(19961125)375:4<675::AID-CNE8>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 24.Morimoto T, Popov S, Buckley KM, Poo MM. Calcium-dependent transmitter secretion from fibroblasts: modulation by synaptotagmin I. Neuron. 1995;15:689–696. doi: 10.1016/0896-6273(95)90156-6. [DOI] [PubMed] [Google Scholar]
- 25.Moschella MC, Watras J, Jayaraman T, Marks AR. Inositol 1,4,5-trisphosphate receptor in skeletal muscle: differential expression in myofibres. J Muscle Res Cell Motil. 1995;16:390–400. doi: 10.1007/BF00114504. [DOI] [PubMed] [Google Scholar]
- 26.Nakanishi H, Tominaga K, Amano T, Hirotsu I, Inoue T, Yamamoto K. Age-related changes in activities and localizations of cathepsins D, E, B, and L in the rat brain tissues. Exp Neurol. 1994;126:119–128. doi: 10.1006/exnr.1994.1048. [DOI] [PubMed] [Google Scholar]
- 27.Pagano E, Sepanski MA, Martin OC. Molecular trapping of a fluorescent ceramide analogue at the Golgi apparatus of fixed cells: interaction with endogenous lipids provides a trans-Golgi marker for both light and electron microscopy. J Cell Biol. 1989;109:2067–2079. doi: 10.1083/jcb.109.5.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parton RG, Dotti CG. Cell biology of neuronal endocytosis. J Neurosci Res. 1993;36:1–9. doi: 10.1002/jnr.490360102. [DOI] [PubMed] [Google Scholar]
- 29.Querfurth HW, Selkoe DJ. Calcium ionophore increases amyloid beta peptide production by cultured cells. Biochemistry. 1994;33:4550–4561. doi: 10.1021/bi00181a016. [DOI] [PubMed] [Google Scholar]
- 30.Rodriguez A, Webster P, Ortego J, Andrews NW. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J Cell Biol. 1997;137:93–104. doi: 10.1083/jcb.137.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rubio ME, Wenthold RJ. Glutamate receptors are selectively targeted to postsynaptic sites in neurons. Neuron. 1997;18:939–950. doi: 10.1016/s0896-6273(00)80333-5. [DOI] [PubMed] [Google Scholar]
- 32.Ryan TA. Endocytosis at nerve terminals: timing is everything. Neuron. 1996;17:1035–1037. doi: 10.1016/s0896-6273(00)80236-6. [DOI] [PubMed] [Google Scholar]
- 33.Ryan TA, Reuter H, Wendland B, Schweizer FE, Tsien RW, Smith SJ. The kinetics of synaptic vesicle recycling measured at single presynaptic boutons. Neuron. 1993;11:713–724. doi: 10.1016/0896-6273(93)90081-2. [DOI] [PubMed] [Google Scholar]
- 34.Spacek J, Harris KM. Three-dimensional organization of smooth endoplasmic reticulum in hippocampal CA1 dendrites and dendritic spines of the immature and mature rat. J Neurosci. 1997;17:190–203. doi: 10.1523/JNEUROSCI.17-01-00190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suzuki K, Kono T. Evidence that insulin causes translocation of glucose transport activity to the plasma membrane from an intracellular storage site. Proc Natl Acad Sci USA. 1980;77:2542–2545. doi: 10.1073/pnas.77.5.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trowbridge IS, Collawn JF, Hopkins CR. Signal-dependent membrane protein trafficking in the endocytic pathway. Annu Rev Cell Biol. 1993;9:129–161. doi: 10.1146/annurev.cb.09.110193.001021. [DOI] [PubMed] [Google Scholar]
- 37.Turrigiano G, Abbott LF, Marder E. Activity-dependent changes in the intrinsic properties of cultured neurons. Science. 1994;264:974–977. doi: 10.1126/science.8178157. [DOI] [PubMed] [Google Scholar]
- 38.Wan Q, Xiong ZG, Man HY, Ackerley CA, Braunton J, Lu WY, Becker LE, MacDonald JF, Wang YT. Recruitment of functional GABAA receptors to postsynaptic domains by insulin. Nature. 1997;388:686–690. doi: 10.1038/41792. [DOI] [PubMed] [Google Scholar]
- 39.Yamasaki S, Baumeister A, Binz T, Blasi J, Link E, Cornille F, Roques B, Fykse EM, Sudhof TC, Jahn R, Niemann H. Cleavage of members of the synaptobrevin/VAMP family by types D and F botulinal neurotoxins and tetanus toxin. J Biol Chem. 1994;269:12764–12772. [PubMed] [Google Scholar]
- 40.Ziv NE, Smith SJ. Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron. 1996;17:91–102. doi: 10.1016/s0896-6273(00)80283-4. [DOI] [PubMed] [Google Scholar]