

Abstract
Glutamate transporters clear glutamate from the extracellular space by high-affinity binding and uptake. Factors that regulate glutamate transporter expression and activity can thereby influence excitatory neurotransmission. Transporter function in GABAergic and other systems has been shown to be regulated by transporter substrates. Here, glutamate regulation of glutamate transport was studied using primary murine astrocyte cultures that express the GLAST (EAAT1) and GLT-1 (EAAT2) transporter subtypes. Glutamate was found to stimulate glutamate transport capacity (Vmax) in a dose- and time-dependent manner. The maximal increase was 100%, with an ED50 of 40 μm glutamate and with onset beginning ∼15 min after onset of glutamate exposure. The uptake stimulation was reproduced byd-aspartate, which is also a transporter substrate, but not by nontransported glutamate receptor agonists. Moreover, glutamate incubation did not stimulate transport when performed in a sodium-free medium, suggesting that the stimulatory effect of glutamate is triggered by increased transporter activity rather than receptor activation. Treatment with the actin-disrupting agents cytochalasin B or cytochalasin D prevented the glutamate-induced increase in glutamate uptake. Biotinylation labeling of membrane surface proteins showed that glutamate incubation produced an increase in GLAST expression at the astrocyte cell surface. These results suggest that cell-surface expression of GLAST can be rapidly regulated by glutamate through a process triggered by GLAST activity and involving the actin cytoskeleton. This feedback loop provides a mechanism by which changes in extracellular glutamate concentrations could rapidly modulate astrocyte glutamate transport capacity.
Keywords: EAAT1, EAAT2, GLT-1, GLAST, actin, cytochalasin, glutamate uptake, trafficking
Glutamate is the major excitatory neurotransmitter of the mammalian CNS (Fonnum, 1984). Glutamatergic transmission is ultimately terminated by binding of glutamate to its transporters and subsequent glutamate uptake (Robinson and Dowd, 1997). Glutamate uptake is accomplished primarily by a family of Na+-dependent, high-affinity glutamate transporters. Five subtypes of these transporters have been identified, EAAT1–EAAT5, and these have differing regional, cellular, and developmental distributions (Robinson and Dowd, 1997; Gegelashvili and Schousboe, 1998). Although glutamate transporters are present on both neurons and astrocytes, uptake by astrocytes appears to dominate in brain (McLennan, 1976; Rothstein et al., 1996). Astrocytes in mammalian forebrain express only the EAAT1 (GLAST) and EAAT2 (GLT-1) subtypes, with GLAST and GLT-1 denoting the rat homologs that were first cloned and described (Rothstein et al., 1994; Swanson et al., 1997; Gegelashvili and Schousboe, 1998).
Glutamate uptake is regulated at several levels. Expression of transporter protein is regulated by cAMP, neuronal factors, and in response to various brain injuries (Gegelashvili et al., 1996; Robinson and Dowd, 1997; Swanson et al., 1997; Gegelashvili and Schousboe, 1998;Schlag et al., 1998). The activity of expressed transporters can be regulated by phosphorylation (Casado et al., 1993), sulfhydryl oxidation (Trotti et al., 1997), Zn2+(Vandenberg et al., 1998), arachidonic acid (Zerangue et al., 1995), and other factors. In addition, at least some glutamate transporter subtypes can transit between the intracellular compartment and the membrane surface. EAAT3 has been shown to move to the membrane surface in C6 glioma cells after phorbol ester-mediated protein kinase C (PKC) activation (Davis et al., 1998), whereas astrocyte GLAST accumulation at the membrane may be inhibited by phorbol ester (Correale et al., 1998). In both instances, the changes in membrane localization of transporter correlate with changes in glutamate transport activity.
In the present study we show that in primary mouse astrocyte cultures expressing both GLT-1 and GLAST glutamate transporter subtypes, preincubation with glutamate produces a rapid and dose-dependent increase in glutamate uptake activity. This effect is triggered by glutamate transport itself, rather than by activation of glutamate receptors. The increase in activity is associated with an increase in cell-surface expression of GLAST and is blocked by inhibitors of actin polymerization. These results suggest a feedback loop whereby extracellular glutamate concentrations could rapidly influence astrocyte glutamate uptake capacity.
Part of this work has been published previously in abstract form (Duan et al., 1998).
MATERIALS AND METHODS
Immunopure immobilized monomeric avidin and sulfo-NHS-biotin were purchased from Pierce (Rockford, IL). Genistein, CF-109203X,trans-(±)-1-amino-1,3-cyclopentanedicarboxylic acid (t-ACPD),l(+)-2-amino-4-phosphonobutyric acid (l-AP4), (S)-4-carboxyphenylglycine (4-CPG), and 1-(5-isoquinolinesulfonyl)-2-methylpiperazine (H7) were purchased from RBI (Natick, MA); 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX),l-trans-pyrrolidine-2,4-dicarboxylate (t-PDC), and α-methyl-4-carboxyphenylglycine (MCPG) were purchased from Tocris (Ballwin, MO); and 1,2-bis(2-aminophenoxy)ethane-N,N,N,N-tetra-acetic acid acetoxymethyl ester (BAPTA AM) was purchased from Molecular Probes (Eugene, OR). All other reagents were obtained from Sigma (St. Louis, MO) except where noted.
Cell culture preparation. Primary murine cortical astrocyte cultures were prepared as described previously (Swanson and Seid, 1998). Tissue harvest was performed in accordance with National Institutes of Health guidelines in a manner that minimized animal suffering. In brief, cortices were harvested from 1-d-old mice (Simonsen, Gilroy, CA) under deep isofluorane anesthesia. The cortices were dissociated in papain/DNase, plated in 24-well tissue culture plates in Eagle's MEM containing 10% fetal bovine serum (FBS) (Hyclone, Ogden, UT), and 2 mm glutamine, and maintained at 37°C in a 5% CO2 incubator. At confluence (day 12–15), the cultures were treated for 48 hr with 10 μmcytosine arabinoside to prevent proliferation of other cell types, and the medium was replaced with MEM containing 2 mm glutamine, 3% FBS, and 0.15 mm dibutyryl cAMP to induce differentiation (Sensenbrenner et al., 1980; Swanson et al., 1997). The astrocyte cultures formed a confluent layer of process-bearing, glial fibrillary acidic protein (GFAP)-positive cells. Each study was repeated on cells from at least two different batches of astrocyte cultures at 28–35 d in vitro.
Experimental procedures. Incubations and uptake assays were performed in room air, 37°C, using a balanced salt solution (BSS) containing (in mm): NaCl 135, KCl 3.1, CaCl2 1.2, MgSO4 1.2, KH2PO4 0.5, PIPES 5, and glucose 2. pH was adjusted to 7.2 with NaOH. Osmolarity was measured with a vapor pressure osmometer (Wescor, Ogden, UT) and adjusted to 285–315 mOsm with water or NaCl if needed. Na+-free media was prepared by replacing NaCl with choline chloride and NaOH withN-methyl-d-glucamine. Ca2+-deficient medium was prepared by omitting CaCl2. All reagents were diluted into BSS from iso-osmolar stocks prepared in BSS or Na+-free BSS.
Glutamate uptake was determined after preincubation with glutamate or other agents. Preincubations were initiated 6 min after washing the cultures into 300 μl of BSS. The preincubations were terminated by double washes in BSS. The cultures remained in BSS at 37°C until initiation of a glutamate uptake assay (the “wash period”). The preincubation period was 60 min, and the wash period was 6 min except where otherwise noted.
Glutamate uptake assays were initiated by adding 0.167 μCi/ml14C-glutamate (American Radiochemicals, St. Louis, MO) plus 100 μm unlabeled glutamate to the culture wells in a final volume of 300 μl. Uptake was terminated after 7 min by two ice-cold washes with 500 μl BSS followed by immediate lysis in ice-cold 0.1N NaOH/0.01% lauryl sulfate. Aliquots of lysates were taken for scintillation counting and protein determinations. In separate experiments, uptake was confirmed to be linear through 10 min incubation (data not shown).
In some studies, glutamate uptake was assessed by HPLC measurements of medium glutamate concentrations. Glutamate (100 μm) was added to the wells in a final volume of 300 μl, and the medium was harvested 7 min later. Glutamate concentrations were determined by reversed-phase HPLC of the orthophthaldialdehyde derivatives using a fluorescence detector. Peak areas were measured with Nelson Analytical software (Norwalk, CT) and calibrated against BSS samples with known glutamate concentrations.
Biotinylation. Biotinylation of cell surface proteins was performed as described by Qian et al. (1997) and Davis et al. (1998)with some modifications. After a 1 hr preincubation period and a 6 min BSS wash period, the cultures were rinsed twice with PBS/Ca-Mg containing (in mm): 138 NaCl, 2.7 KCl, 1.5 KH2PO4, 9.6 Na2HPO4, 1 MgCl2, and 0.1 CaCl2, pH 7.3. Cultures were then incubated in sulfo-NHS-biotin solution (1 mg/ml in with PBS/Ca–Mg) for 20 min at 4°C. Biotinylation was terminated by washing twice in a quenching solution of PBS/Ca–Mg in which there was an equimolar substitution of 100 mm glycine for NaCl (to maintain 300 mOsm). This was followed by an additional 45 min incubation in the quenching solution at 4°C. Quenching solution was removed, and the cells were lysed with 100 μl/well of radioimmunoprecipitation assay (RIPA) buffer with protease inhibitors (100 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 1 mm iodoacetamide, 1 μg/ml leupeptin, 5 μg/ml aprotinin, and 250 μmphenylmethylsulfonyl fluoride) for 1 hr at 4°C with vigorous shaking. The lysates were centrifuged at 14,000 × g for 15 min at 4°C. Three hundred microliters of the supernatant were taken for Western analysis as the whole cell fraction. The rest of the supernatant (600 μl) was incubated with 300 μl avidin bead suspension for 1 hr at room temperature with gentle shaking. The avidin–lysate solution was then centrifuged for 15 min at 14,000 × g, and the supernatant was taken for Western analysis as the intracellular fraction. The pellet was washed four times with 1 ml RIPA buffer and resuspended in 300 μl Laemmli buffer (62.4 mm Tris-HCl, pH 7, 2% SDS, 20% glycerol, and 5% 2-mercaptethanol) for 1 hr with gentle shaking at room temperature. After centrifugation for 15 min at 14,000 × g, the supernatant was taken for Western analysis as the biotinylated (plasma membrane) fraction.
For Western analysis, the protein samples were loaded and run on a 10% SDS polyacrylamide gel. Samples were electrophoretically transferred onto polyvinylidene fluoride membranes (Boehringer Mannheim, Indianapolis, IN) and subsequently placed in blocking solution (5% skim milk/1% bovine serum albumin in 0.1 m phosphate buffer) for 1 hr at room temperature. The membranes were then transferred to a blocking solution containing polyclonal anti-actin antibody diluted 1:100 plus either polyclonal anti-GLAST antibody (0.2 ng/ml) or polyclonal anti-GLT-1 antibody (0.02 ng/ml) for 12 hr at 22°C. [The affinity-purified antibodies to GLAST and GLT-1 were kindly provided by Dr. Jeffrey Rothstein, Johns Hopkins University, and have been previously characterized (Rothstein et al., 1994; Swanson et al., 1997; Swanson and Seid, 1998)]. The membranes were washed three times in 0.1 m phosphate buffer containing 0.1% Tween 20 (PB-T) and placed in blocking solution containing a peroxidase-labeled anti-rabbit IgG antibody (Boehringer Mannheim), diluted 1:500, for 1 hr. After three additional washes in PB-T, the resulting peroxidase signal was detected using 3,3′-diaminobenzidene. The resulting bands were digitized, and densitometry was performed using the NIH Image program. The signal for each lane was calculated by summing the (area × (density-background)) measurements of the discrete monomer and multimer bands produced by GLAST and GLT-1.
Statistics. Statistical differences were determined by Student's t test for two-group comparisons, or by one-way ANOVA with the Tukey test for multiple comparisons among more than two groups, or by Dunnett's test when multiple groups were compared against a single control group.
RESULTS
Glutamate stimulates glutamate uptake in a dose- and time-dependent manner
Glutamate uptake under the standard assay condition of 100 μm glutamate was 7.5 ± 0.8 nmol per milligrams of protein per minute (n = 5). Preincubation of the cultures with glutamate produced a dose-dependent increase in glutamate uptake (Fig. 1A). The glutamate ED50 was ∼40 μm, and the maximal increase in uptake rate varied from 40 to 100% among 32 independent experiments using a 60 min glutamate preincubation and a 6 min interval (wash period) before the glutamate uptake assays.
Fig. 1.
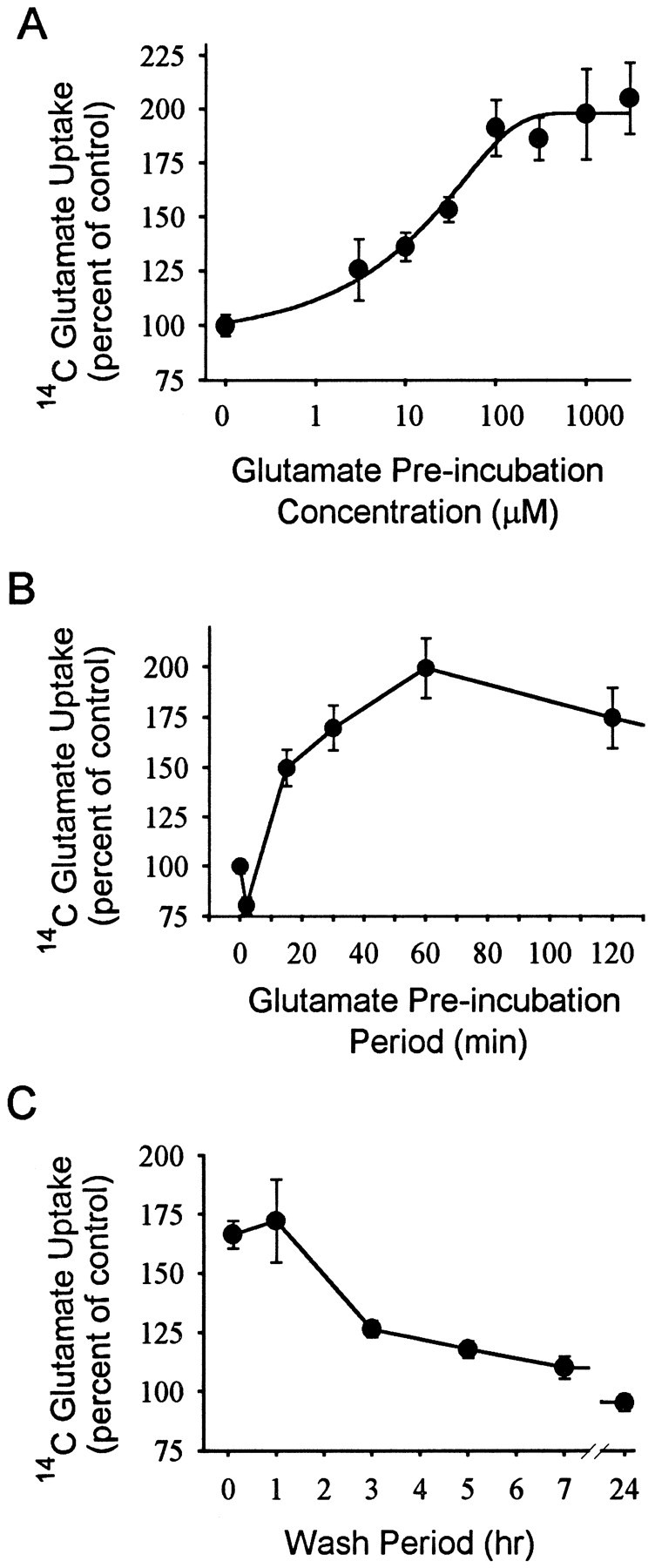
Glutamate-induced stimulation of glutamate uptake. Data are means ± SEM, n ≥ 6.A, Glutamate preincubation produced a concentration-dependent increase in subsequent glutamate uptake.B, Effect of varying glutamate preincubation periods. The glutamate preincubation concentration was 100 μm, and the wash period (interval before glutamate uptake assay) was 6 min.C, Effect of varying intervals between glutamate preincubation and beginning of glutamate uptake assay (wash periods). The glutamate preincubation concentration was 100 μm, and the preincubation period was 60 min.
The effects of differing preincubation and postincubation periods are shown in Figure 1. A significant increase in uptake was apparent with preincubation periods as short as 15 min but not 6 min. These intervals correspond to 21 and 12 min total elapsed time between the onset of glutamate preincubation and the initiation of glutamate uptake assays. A maximal effect was achieved within 60 min (Fig.1B). The increased uptake was maintained when the wash period was extended from 6 min to 1 hr, but slowly declined between 1 hr and 7 hr (Fig. 1C).
Eadie-Hofstee plots prepared using different glutamate concentrations during the glutamate uptake assay showed that glutamate preincubation increased the Vmax from 13.6 ± 0.3 to 22.4 ± 2.7 nmol per milligrams of protein per minute (p < 0.01). There was also a small increase in the apparent glutamateKm, from 72.9 + 0.3 to 91 + 17 μm, which was not statistically significant (Fig. 2). These studies and all subsequent studies were performed using a 60 min preincubation in 100 μm glutamate and a 6 min postincubation interval before beginning glutamate uptake assays. Uptake rates in sodium-free medium were <3% of normal medium at both high and low glutamate concentrations (data not shown), and consequently no correction was made for this small contribution of Na+-independent uptake.
Fig. 2.
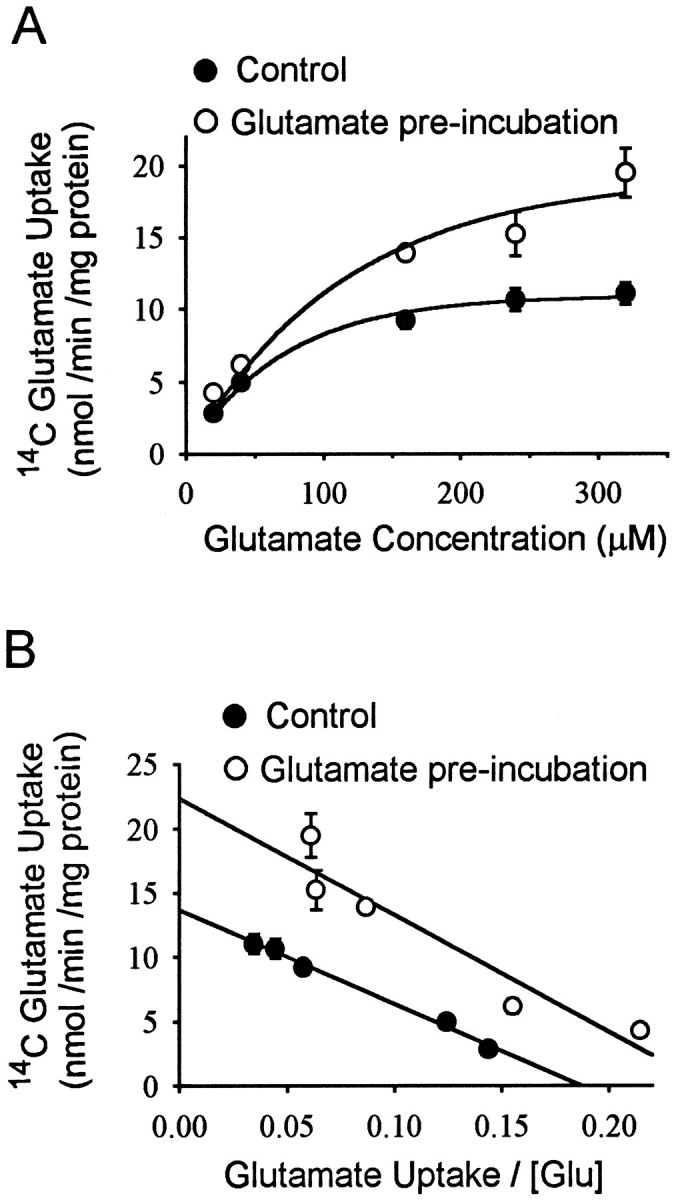
A, Glutamate uptake capacity was increased in glutamate-stimulated cultures. Data are means ± SEM,n = 6. B, Data shown as Eadie-Hofstee plot. Vmax was increased from 13.6 ± 0.3 to 22.4 ± 2.7 nmol per milligrams of protein per minute by glutamate preincubation (p < 0.01). The small increase in the apparent glutamateKm was not statistically significant.
HPLC measurements of glutamate in the astrocyte culture medium confirmed an increase in glutamate uptake rate after preincubation in glutamate (Fig. 3). HPLC was used because of the possibility that extracellular14C-glutamate could enter the astrocytes not only with net glutamate uptake but also by heteroexchange with unlabeled intracellular glutamate (Blitzblau et al., 1996), because intracellular glutamate concentrations are increased by glutamate preincubation. Bulk uptake of glutamate was measured by serial HPLC determinations of glutamate in the culture well media, whereas the standard 14C-glutamate uptake measurements were performed in sister culture wells at the same time. HPLC measurement of the nominally 100 μm glutamate BSS used for the uptake assays yielded a value of 104.8 ± 4.2 μm (n = 8). The glutamate remaining after 7 min incubation in culture wells preincubated with glutamate was 83.2 ± 2.0 μm, a value significantly lower than the 91.2 ± 2.0 μm glutamate measured in wells not preincubated with glutamate (n = 12, p < 0.01). As shown in Figure 3, the glutamate-induced increase in glutamate uptake measured by HPLC was quantitatively similar to that measured by the14C-glutamate method in sister culture wells.
Fig. 3.

The HPLC and 14C-glutamate methods of measuring glutamate uptake produced similar measures of glutamate-induced stimulation of glutamate uptake. Experiments were performed on sister culture wells on the same day. Data are means + SEM; data were pooled from two experiments. n = 12; **p < 0.01.
Glutamate stimulation of glutamate uptake is triggered by glutamate transport rather than by glutamate receptors
Glutamate receptor agonists and transporter substrates were tested for their ability to mimic the stimulating effect of glutamate on glutamate uptake. As shown in Figure4A, preincubation with the ionotropic glutamate receptor agonists NMDA and kainate failed to evoke glutamate uptake increase, whereas the transporter substrated-aspartate (d-ASP) did reproduce the glutamate effect. The apparentKm for astrocyted-ASP uptake is higher than that of glutamate uptake (Drejer et al., 1982; R. A. Swanson and K. Farrell, unpublished observations). Of note, the maximal effect produced byd-ASP was equivalent to the maximal effect of glutamate but required higher medium d-ASP concentrations. Preincubation with the metabotropic agonistsl-AP4 and t-ACPD also stimulated uptake, but interpretation of these results is complicated by the fact that both of these agents may themselves serve as substrates for glutamate transporters (Harris et al., 1987; Ye and Sontheimer, 1998). The metabotropic receptor antagonists MCPG and 4-CPG failed to block the stimulatory effect of glutamate when added with glutamate during the preincubation period (Fig. 4B and data not shown). A similar negative effect was seen with CNQX, a non-NMDA ionotropic glutamate receptor antagonist. In contrast, the glutamate transport inhibitor t-PDC did prevent the stimulatory effect of glutamate at a concentration that effectively inhibits glutamate uptake. Moreover, glutamate, d-ASP, andt-ACPD all failed to stimulate glutamate uptake activity when preincubated in Na+-free medium (Fig.4C). Taken together, these studies suggest that stimulation of glutamate uptake by glutamate preincubation is triggered by glutamate transporter activity rather than by glutamate receptor activation.
Fig. 4.
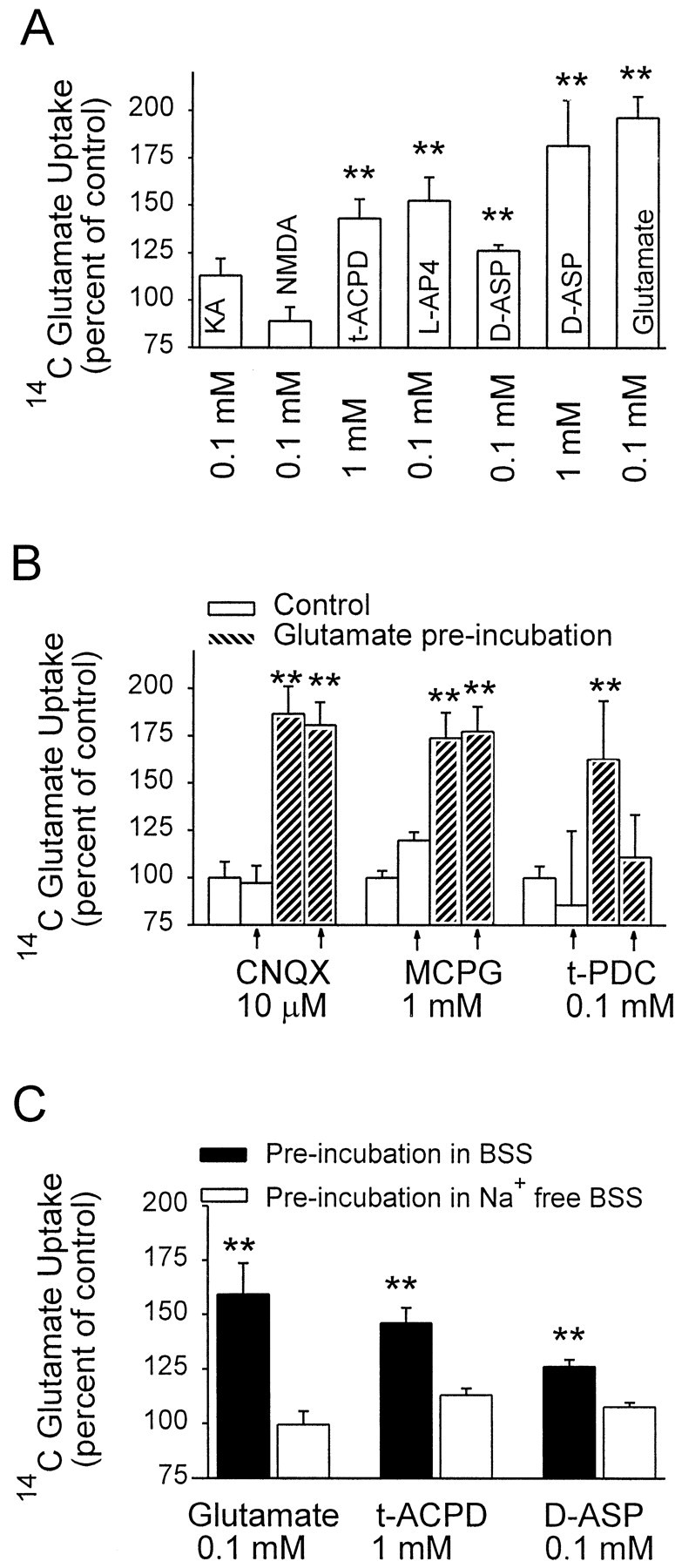
Glutamate stimulation of glutamate uptake is triggered by glutamate transport. Data are means + SEM,n ≥ 5; **p < 0.01.A, Preincubation with the nontransported glutamate receptor agonists kainate (KA) and NMDAdid not stimulate glutamate uptake, whereas the transportable agonistst-ACPD,d-ASP, and glutamate did stimulate uptake. B, The glutamate receptor antagonistsCNQX and MCPG failed to block the stimulatory effect of glutamate when added with glutamate during the preincubation period, but the glutamate transport inhibitort-PDC did block the glutamate effect.C, Preincubations performed in Na+-free medium failed to stimulate glutamate uptake.
Mammalian forebrain astrocytes express only the GLAST and GLT-1 transporter subtypes. The contribution of GLT-1-mediated uptake was assessed in glutamate-stimulated cultures by use of the GLT-1-specific inhibitor dihydrokainate (DHK) (Arriza et al., 1994; Robinson and Dowd, 1997). As shown in Figure 5A, 1 mm DHK had only a negligible effect on uptake under basal conditions, and a similar negligible effect was seen in glutamate-stimulated cells. DHK was also used to determine whether GLT-1 mediates the induction of uptake stimulation by glutamate. The uptake stimulation induced by 0.1 mm glutamate was not prevented by the presence of 1 mm DHK during the preincubation period (Fig. 5B). These results suggest that GLT-1 does not contribute to either the increase in uptake capacity induced by glutamate preincubation or the signaling mechanism that leads to this increase.
Fig. 5.
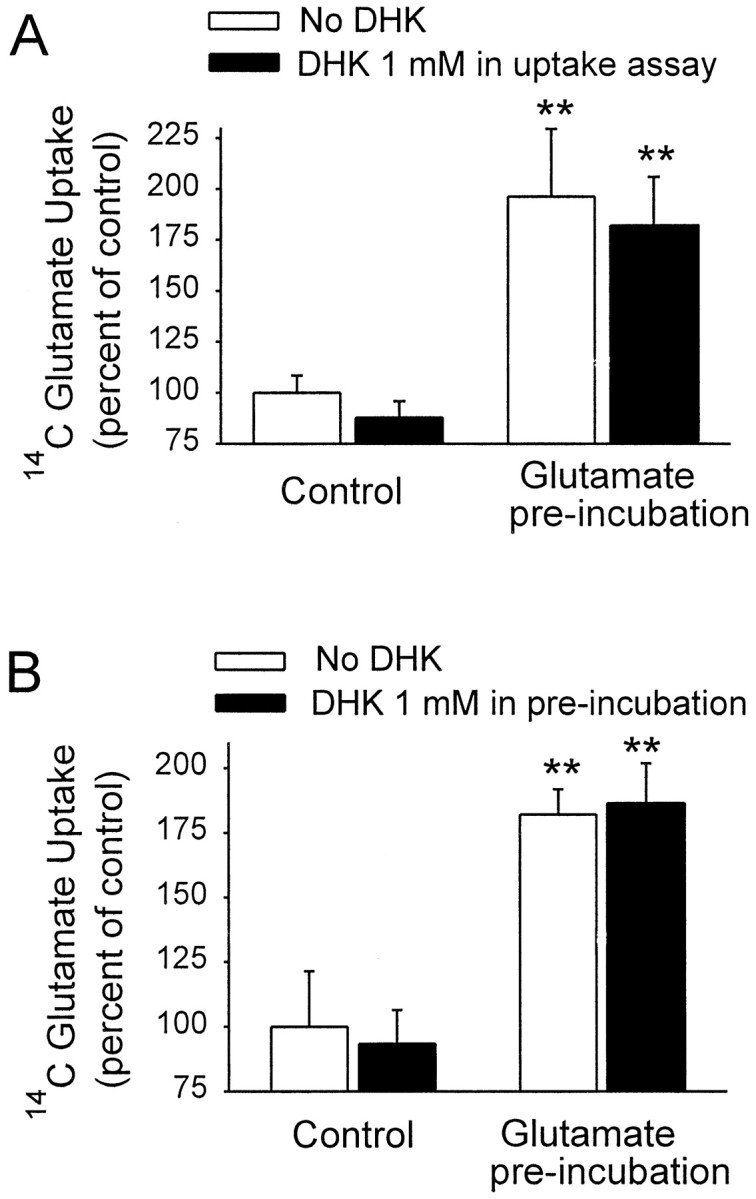
The GLT-1-specific uptake inhibitor dihydrokainate (DHK) had negligible effects on both basal and glutamate-stimulated glutamate uptake. Glutamate concentrations were 0.1 mm both in the preincubation period and during the glutamate uptake assay. Data are means + SEM, n = 6; **p < 0.01. A, Glutamate preincubation induced a large increase in subsequent glutamate uptake rate. DHK did not attenuate this increase and did not significantly reduce uptake under control conditions. B, The presence of saturating DHK concentrations during the preincubation period similarly had negligible effects on glutamate uptake under both control and glutamate-stimulated conditions.
Signal transduction mechanisms
Glutamate transport has several effects on astrocytes, including membrane depolarization, cell swelling, and increased intracellular Na+. Table 1shows results of studies designed to determine whether glutamate-induced stimulation of uptake might be signaled through these effects. Glutamate uptake was not stimulated by depolarizing K+ concentrations, by preincubation in 180 mOsm medium (to induce cell swelling), or by incubation in veratridine, a Na+ channel ionophore. In addition, preincubation with 1 mm of other Na+-dependent amino acid transporter substrates, cysteine, glycine, and GABA, also failed to affect the glutamate uptake activity.
Table 1.
Effects of preincubation conditions on subsequent glutamate uptake
| Preincubation condition | Glutamate uptake |
|---|---|
| Glutamate 0.1 mm | 180.1 ± 8.7* |
| K+ 60 mm | 109.1 ± 4.1 |
| Veratridine 12 μm | 106.4 ± 15 |
| 180 mOsm | 89.4 ± 7 |
| Cysteine 1 mm | 91.5 ± 3.6 |
| Glycine 1 mm | 85.7 ± 10.5 |
| GABA 1 mm | 92.9 ± 8.8 |
Preincubation conditions were maintained for 60 min, and glutamate uptake was measured after a wash period of 6 min. Glutamate uptake values are percentage of control; means ± SEM;n = 6–20.
*p < 0.01.
Glutamate transporters have multiple PKC and protein kinase A (PKA) phosphorylation sites, and PKC activation by phorbol ester is reported to modulate glutamate transporter activity in several cell systems (Casado et al., 1993; Dowd and Robinson, 1996; Conradt and Stoffel, 1997; Davis et al., 1998; Ganel and Crosson, 1998). However, pretreatment with the PKC inhibitor GF-109203X (Toullec et al., 1991) did not block the stimulatory effect of glutamate on uptake activity (Table 2), suggesting that PKC was not involved in the glutamate-induced uptake stimulation. The PKA and PKC inhibitor H7 (Lagord et al., 1993) also failed to block glutamate-induced uptake. Negative results were also obtained with pertussis toxin, an inhibitor of Gi/Go-protein; wortmannin, an inhibitor of phosphatidylinositol 3-kinase (PI3K) (Clarke et al., 1994); the tyrosine kinase inhibitor genistein; Ca2+-deficient BSS plus the calcium chelator BAPTA-AM; and calcium channel blockade with Cd2+ (Table 2).
Table 2.
Effects of signal transduction inhibitors on glutamate-stimulated glutamate uptake
| Inhibitor | Glutamate | Inhibitor + glutamate | |
|---|---|---|---|
| GF-109203X 5 μm | 119.7 ± 6.1 | 180.3 ± 21.6 | 175.4 ± 29.8 |
| H7 100 μm | 97.2 ± 7.4 | 181.4 ± 22 | 182 ± 31.2 |
| Genistein 100 μm | 118.5 ± 17.9 | 172.1 ± 34.1 | 171.1 ± 26.5 |
| Wortmannin 0.1 μm | 73.9 ± 4.1* | 162.5 ± 8.5 | 144.7 ± 12.4 |
| PTX 0.3 μg/ml | 96.7 ± 2.3 | 146.9 ± 4.4 | 141.6 ± 5.4 |
| Cd2+ 200 μm | 123.2 ± 16.9 | 198.6 ± 39.1 | 181.3 ± 25.7 |
| BAPTA AM 5 μm | 72.4 ± 6.7* | 201.2 ± 23.3 | 175.9 ± 3.7 |
Glutamate uptake values are percentage (means ± SEM) of uptake in control wells preincubated in the absence of glutamate or inhibitor. In all cases there was no significant difference between the groups preincubated with glutamate plus inhibitor and the groups preincubated with glutamate alone. Uptake in groups treated with inhibitor alone was in some cases lower than control (*p < 0.05), but uptake in all glutamate and all glutamate plus inhibitor preincubation groups was significantly greater than controls (p < 0.01; symbols omitted for clarity). BAPTA AM was added to cultures in Ca2+-deficient BSS for 30 min and removed before preincubation in Ca2+-deficient BSS. Cd2+ and GF-109203X were added 6 min before the studies and maintained during the preincubation period. Pertussis toxin (PTX) was added to the culture medium for 18 h before the preincubation period. All other inhibitors were added 30 min before and maintained during the preincubation period.
Recent studies indicate that cytoskeletal components are involved in several signal transduction pathways, as well as in transporter trafficking. As shown in Figure 6, pretreatment of astrocyte cultures with cytochalasin B, an inhibitor of actin polymerization (MacLean-Fletcher and Pollard, 1980; Ohmori et al., 1992), did not by itself affect glutamate uptake but did attenuate glutamate stimulation of glutamate uptake in a dose-dependent manner. Cytochalasin D, a similar but more specific agent (Rampal et al., 1980), had a similar inhibitory effect on glutamate-induced uptake increase. Pretreatment with nocodazole, an inhibitor of microtubule formation (Jordan and Wilson, 1998), did not inhibit glutamate-induced uptake.
Fig. 6.

Cytochalasin B (CytB) and cytochalasin D (CytD) attenuated the glutamate-induced stimulation of glutamate uptake, whereas nocodazole (Nocodz) had no effect. CytB,CytD, and Nocodz were added 1 hr before the studies and maintained during the preincubation period. Data are means + SEM, n = 6; *p < 0.05, **p < 0.01.
Glutamate preincubation increases cell surface expression of GLAST
Transporter trafficking to the cell membrane has been previously shown to regulate EAAC1 (EAAT3) glutamate transporter function in the C6 glioma cell line (Davis et al., 1998) and other transporters in several cell systems (Tsakiridis et al., 1994; Qian et al., 1997; Quick et al., 1997; Wang et al., 1998; Bernstein and Quick, 1999). We therefore used biotinylation labeling of cell surface proteins to determine whether the glutamate-stimulated increase in astrocyte glutamate uptake could be mediated by increased cell surface expression of transporters. Biotinylated proteins were separated from nonbiotinylated, intracellular proteins by using avidin-coated beads. Western blots were prepared from biotinylated and nonbiotinylated protein fractions, as well as from whole-cell fractions. The blots were probed with anti-actin antibody and with antibody to GLAST (Fig.7A) or to GLT-1 (Fig.7B). The actin band provides an index of intracellular proteins present in each preparation. GLAST and GLT-1 bands were present at molecular weights corresponding to both monomeric and multimeric forms, as reported previously (Haugeto et al., 1996; Davis et al., 1998). The effect of glutamate preincubation is apparent on the Western blots and in the pooled data from five independent studies shown in Figure 7C. Glutamate preincubation produced an increase in biotinylated (cell surface) GLAST protein and a decrease in nonbiotinylated (intracellular) GLAST, with no change in total cell GLAST protein. In contrast, glutamate preincubation produced no observable change in cell surface or total cell GLT-1 expression. Glutamate did appear to reduce the nonbiotinylated, intracellular pool in some studies, but it should be noted that interpretation of changes in the GLT-1 intracellular pool is complicated by the fact that it is small even under control conditions, and this small pool may be overestimated to varying degrees by small contaminations from the much larger cell-surface pool of GLT-1.
Fig. 7.
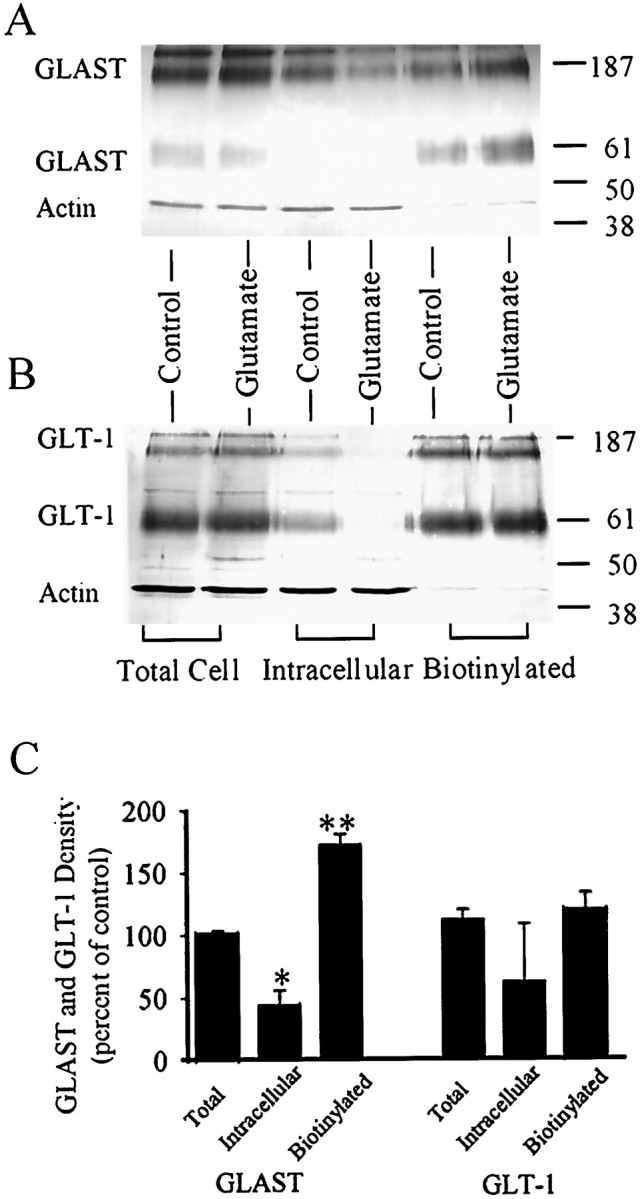
Glutamate preincubation increases cell-surface expression of GLAST. Western blots were probed with antibodies to GLAST and actin (A) or to GLT-1 and actin (B). GLAST and GLT-1 bands were present at molecular weights corresponding to both monomeric and multimeric forms. The actin bands provide an index of intracellular proteins present in each preparation. Actin immunoreactivity in the biotinylated fractions was <10% of that in the total cell fractions, and actin immunoreactivity was approximately equal in preparations from control and glutamate-treated cells. Densitometry measurements of the GLAST and GLT-1 bands were pooled from five independent experiments (C). GLAST, but not GLT-1, immunoreactivity was increased in the biotinylated (cell surface) fraction of astrocytes preincubated in 1 mm glutamate. GLAST immunoreactivity was significantly decreased in the nonbiotinylated, intracellular fraction in glutamate-treated cells. Glutamate preincubation did not affect total cell immunoreactivity of either GLAST or GLT-1. Data are means + SEM, n = 3; *p < 0.05, **p < 0.01.
DISCUSSION
The primary finding reported here is that glutamate can induce a rapid upregulation of astrocyte glutamate uptake. The studies further suggest that the glutamate-induced upregulation is signaled by increased activity of the GLAST (EAAT1) glutamate transporter subtype, and that the increase in uptake is mediated by increased cell-surface expression of GLAST. Additionally, a role for the actin cytoskeleton in this process is suggested by the finding that the increase in uptake activity is inhibited by cytochalasins.
Several factors support the conclusion that the glutamate-induced stimulation of glutamate uptake is triggered by increased activity of the glutamate transporters. The glutamate effect was mimicked byd-aspartate, which is also a transporter substrate, stimulation was blocked if the glutamate preincubation was performed in a Na+-free medium, and stimulation was not blocked by glutamate receptor antagonists that are not substrates for the transporters. In addition, maximal stimulation of uptake was observed only at glutamate concentrations that saturate glutamate transport, and these concentrations far exceed known receptor affinities for glutamate.
The membrane biotinylation studies provide evidence that the increase in uptake is mediated by transporter translocation to (or decreased removal from) the cell membrane. The observed increase in transportVmax with little or no change in glutamate Km is consistent with this mechanism. The time course of the glutamate stimulation also supports a trafficking mechanism, because the 12–21 min interval between onset of glutamate preincubation and the resultant increase in uptake activity shown in Figure 1B is too fast to involve de novo synthesis of new glutamate transporters but is typical of the time interval required for changes attributable to membrane trafficking (Davis et al., 1998; Bernstein and Quick, 1999; Lissin et al., 1999).
GLAST and GLT-1 are the only glutamate transporter subtypes known to be expressed by mammalian forebrain astrocytes, both in culture andin situ (Rothstein et al., 1994; Swanson et al., 1997;Gegelashvili and Schousboe, 1998). In the present study, glutamate-induced stimulation of glutamate uptake is attributed to GLAST because the GLT-1 selective inhibitor dihydrokainate had negligible effect, and because GLAST but not GLT-1 exhibited increased cell surface expression. It is possible that different, as yet unrecognized transporters contribute to the stimulated uptake, but this seems unlikely because the magnitude of the increase is large (∼100%).
Glutamate has previously been shown to regulate glutamate transport in other ways. Expression of GLAST protein in cultured astrocytes is stimulated by prolonged exposure (7 d) to glutamate or kainate (Gegelashvili et al., 1996), and this increase is accompanied by an increase in transport Vmax. Glutamate preincubation has also been reported to reduce extracellular glutamate concentrations in astrocyte cultures (Ye and Sontheimer, 1999). Interestingly, this effect displays a time course similar to that observed in the present studies, occurs in response to metabotropic glutamate receptor agonists as well as to glutamate itself, but is not blocked by metabotropic receptor antagonists. Because many of the metabotropic agonists can also serve as substrates for the transporters (Harris et al., 1987; Ye and Sontheimer, 1998), it is possible that this effect is also mediated by a substrate-stimulated increase in transporter activity.
Although the present studies suggest an upstream signaling mechanism by which increased extracellular glutamate concentrations may trigger the increase in glutamate uptake signal and a downstream effector mechanism by which the increase is accomplished, they do not establish an intervening signal transduction pathway. Previous studies have identified mechanisms controlling membrane trafficking of a predominately neuronal glutamate transporter, EAAC1 (EAAT3). In C6 glioma cells expressing EAAC1, PKC activation by phorbol ester leads to transporter movement from the intracellular compartment to the cell surface, whereas the PI3K inhibitor wortmannin decreased the membrane EAAC1 expression both in C6 glioma cells (Davis et al., 1998) and in neurons (Munir et al., 1998). Less is known about transporter trafficking in astrocytes, but it has been reported that membrane expression of GLAST, but not GLT-1, is inhibited by phorbol ester (Correale et al., 1998). In the present study neither the PKC inhibitor GF-109203X nor the PI3K inhibitor wortmannin affected glutamate-induced stimulation of uptake, and various other interventions also failed to affect stimulation of uptake (Tables 1, 2). However, translocation of membrane proteins does not necessarily require signal transduction pathways. G-protein-coupled receptors (Grady et al., 1997) and ionotropic glutamate receptors (Lissin et al., 1999) appear to involve the conformation-dependent interaction of the proteins with cytoplasmic regulatory proteins without intervening signal transduction, and this raises the possibility of a similar mechanism for glutamate transporters. Alternatively, the glutamate transporters themselves may serve as signal transducing units as proposed by Gegelashvili and Schousboe (1998), who noted that the third intracellular domains of GLAST, GLT-1, and EAAC1 each contain a motif similar to that of the IGF-II and α-adrenergic receptors that bind Gα-subunits of G-proteins.
Substrate-induced upregulation of transport activity mediated by membrane translocation of transporters, as described here, has been reported recently in other systems. The binding of substrates to the 5-HT transporter can rapidly increase 5-HT uptake by inhibiting PKC-dependent internalization of the 5-HT transporters (Qian et al., 1997; Ramamoorthy and Blakely, 1999). A substrate-induced rapid increase in GABA uptake was also observed in primary hippocampal cultures and in a cell line expressing the rat brain GABA transporter GAT1 (Bernstein and Quick, 1999), with the increase in GABA uptake correlating with increased membrane GAT1 expression. PKC is also involved in the regulation of GAT1 activity and trafficking (Quick et al., 1997), but in both the 5-HT and GABAergic systems the signals linking increased substrate exposure to PKC activity remain unknown.
The inhibitory effects of cytochalasin B and cytochalasin D on glutamate-stimulated glutamate uptake suggest that the actin cytoskeleton is important for either signal transduction or transporter translocation in this process. The cytochalasins impair actin polymerization and promote disruption of the actin cytoskeleton (MacLean-Fletcher and Pollard, 1980). Cytochalasin B has, in addition to effects on actin polymerization, a direct inhibitory effect on glucose transport (Rampal et al., 1980), but cytochalasin D does not have any known direct effect on glucose transporters or other transporter function.
The actin cytoskeleton has been linked to intracellular protein trafficking, transporter function, and signal transduction in several systems (Mills and Mandel 1994; Tsakiridis et al., 1994; Lamaze et al., 1997; Wang et al., 1998). Cytochalasin D inhibits insulin-stimulated glucose uptake and prevents insulin-induced trafficking of glucose transporters to the plasma membrane in L6 rat skeletal muscle cells (Tsakiridis et al., 1994) and 3T3-L1 adipocytes (Wang et al., 1998). In this system the actin filaments are thought to facilitate the insulin-induced relocation of PI3K to the intracellular glucose transporter compartment, rather than movement of the glucose transporters themselves (Wang et al., 1998). One potential link between actin and known signal transduction systems is that activation of the Rho family of the small G-protein can induce a reorganization of the actin cytoskeleton (Hall, 1998; Sasaki and Takai, 1998).
It is not known whether the two astrocyte glutamate transporters GLAST and GLT-1 have distinct physiological functions. Gene disruption studies suggest that GLAST, unlike GLT-1, may not be required for neuronal survival under normal conditions in brain (Tanaka et al., 1997; Watase et al., 1998). The rapid upregulation of GLAST transport capacity in response to elevated extracellular glutamate concentrations observed here suggests that GLAST may have a unique function in feedback regulation of extracellular glutamate concentrations and glutamatergic synaptic activity. Studies by Tong and Jahr (1994) and Diamond and Jahr (1997) show that transporter binding of glutamate contributes to an early, fast reduction in synaptic glutamate concentrations in addition to the slow, later phase attributable to actual glutamate uptake [but also see Otis et al. (1996)]. Translocation of GLAST to the astrocyte cell surface could therefore affect glutamatergic transmission in two ways: by increasing the number of glutamate binding sites and by accelerating subsequent glutamate uptake.
Footnotes
This work was supported by National Institutes of Health Grant RO1 NS31914 and the Veterans Affairs Merit Review program (R.A.S.). We thank Dr. Michael B. Robinson for helpful advice on the biotinylation studies.
Correspondence should be addressed to Dr. Raymond A. Swanson, (127) Neurology, V.A.M.C., 4150 Clement Street, San Francisco, CA 94121. E-mail: ray@itsa.ucsf.edu.
REFERENCES
- 1.Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci. 1994;14:5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bernstein EM, Quick MW. Regulation of gamma-aminobutyric acid (GABA) transporters by extracellular GABA. J Biol Chem. 1999;274:889–895. doi: 10.1074/jbc.274.2.889. [DOI] [PubMed] [Google Scholar]
- 3.Blitzblau R, Gupta S, Djali S, Robinson MB, Rosenberg PA. The glutamate transport inhibitor l-trans-pyrrolidine-2,4-dicarboxylate indirectly evokes NMDA receptor mediated neurotoxicity in rat cortical cultures. Eur J Neurosci. 1996;8:1840–1852. doi: 10.1111/j.1460-9568.1996.tb01328.x. [DOI] [PubMed] [Google Scholar]
- 4.Casado M, Bendahan A, Zafra F, Danbolt NC, Aragon C, Gimenez C, Kanner BI. Phosphorylation and modulation of brain glutamate transporters by protein kinase C. J Biol Chem. 1993;268:27313–27317. [PubMed] [Google Scholar]
- 5.Clarke JF, Young PW, Yonezawa K, Kasuga M, Holman GD. Inhibition of the translocation of GLUT1 and GLUT4 in 3T3–L1 cells by the phosphatidylinositol 3-kinase inhibitor, wortmannin. Biochem J. 1994;300:631–635. doi: 10.1042/bj3000631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conradt M, Stoffel W. Inhibition of the high-affinity brain glutamate transporter GLAST-1 via direct phosphorylation. J Neurochem. 1997;68:1244–1251. doi: 10.1046/j.1471-4159.1997.68031244.x. [DOI] [PubMed] [Google Scholar]
- 7.Correale DM, Kalandadze A, Zelenaia O, Robinson MB. Comparison of the regulation of cell surface expression of the GLT-1, GLAST, and EAAC1 subtypes of glutamate transporters. Soc Neurosci Abstr. 1998;24:2071. [Google Scholar]
- 8.Davis KE, Straff DJ, Weinstein EA, Bannerman PG, Correale DM, Rothstein JD, Robinson MB. Multiple signaling pathways regulate cell surface expression and activity of the excitatory amino acid carrier 1 subtype of Glu transporter in C6 glioma. J Neurosci. 1998;18:2475–2485. doi: 10.1523/JNEUROSCI.18-07-02475.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diamond JS, Jahr CE. Transporters buffer synaptically released glutamate on a submillisecond time scale. J Neurosci. 1997;17:4672–4687. doi: 10.1523/JNEUROSCI.17-12-04672.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dowd LA, Robinson MB. Rapid stimulation of EAAC1-mediated Na+-dependent l-glutamate transport activity in C6 glioma cells by phorbol ester. J Neurochem. 1996;67:508–516. doi: 10.1046/j.1471-4159.1996.67020508.x. [DOI] [PubMed] [Google Scholar]
- 11.Drejer J, Larsson OM, Schousboe A. Characterization of l-glutamate uptake into and release from astrocytes and neurons cultured from different brain regions. Exp Brain Res. 1982;47:259–269. doi: 10.1007/BF00239385. [DOI] [PubMed] [Google Scholar]
- 12.Duan S, Farrell K, Guenza JK, Stein BA, Swanson RA. Glutamate induces a rapid increase in glutamate uptake activity in mouse astrocyte cultures. Soc Neurosci Abstr. 1998;24:775. [Google Scholar]
- 13.Fonnum F. Glutamate: a neurotransmitter in mammalian brain. J Neurochem. 1984;42:1–11. doi: 10.1111/j.1471-4159.1984.tb09689.x. [DOI] [PubMed] [Google Scholar]
- 14.Ganel R, Crosson CE. Modulation of human glutamate transporter activity by phorbol ester. J Neurochem. 1998;70:993–1000. doi: 10.1046/j.1471-4159.1998.70030993.x. [DOI] [PubMed] [Google Scholar]
- 15.Gegelashvili G, Civenni G, Racagni G, Danbolt NC, Schousboe I, Schousboe A. Glutamate receptor agonists up-regulate glutamate transporter GLAST in astrocytes. NeuroReport. 1996;8:261–265. doi: 10.1097/00001756-199612200-00052. [DOI] [PubMed] [Google Scholar]
- 16.Gegelashvili G, Schousboe A. Cellular distribution and kinetic properties of high-affinity glutamate transporters. Brain Res Bull. 1998;45:233–238. doi: 10.1016/s0361-9230(97)00417-6. [DOI] [PubMed] [Google Scholar]
- 17.Grady EF, Bohm SK, Bunnett NW. Turning off the signal: mechanisms that attenuate signaling by G protein-coupled receptors. Am J Physiol. 1997;273:G586–G601. doi: 10.1152/ajpgi.1997.273.3.G586. [DOI] [PubMed] [Google Scholar]
- 18.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 19.Haugeto O, Ullensvang K, Levy LM, Chaudhry FA, Honore T, Nielsen M, Lehre KP, Danbolt NC. Brain glutamate transporter proteins form homomultimers. J Biol Chem. 1996;271:27715–27722. doi: 10.1074/jbc.271.44.27715. [DOI] [PubMed] [Google Scholar]
- 20.Harris EW, Stevens DR, Cotman CW. Hippocampal cells primed with quisqualate are depolarized by AP4 and AP6, ligands for a putative glutamate uptake site. Brain Res. 1987;418:361–365. doi: 10.1016/0006-8993(87)90104-1. [DOI] [PubMed] [Google Scholar]
- 21.Jordan MA, Wilson L. Use of drugs to study role of microtubule assembly dynamics in living cells. Methods Enzymol. 1998;298:252–276. doi: 10.1016/s0076-6879(98)98024-7. [DOI] [PubMed] [Google Scholar]
- 22.Lagord C, Carpentier G, Moraczewski J, Pons G, Climent F, Martelly I. Satellite cell myogenesis is highly stimulated by the kinase inhibitor iso-H7: comparison with HA1004 and staurosporine effects. Biochem Biophys Res Commun. 1993;191:928–936. doi: 10.1006/bbrc.1993.1306. [DOI] [PubMed] [Google Scholar]
- 23.Lamaze C, Fujimoto LM, Yin HL, Schmid SL. The actin cytoskeleton is required for receptor-mediated endocytosis in mammalian cells. J Biol Chem. 1997;272:20332–20335. doi: 10.1074/jbc.272.33.20332. [DOI] [PubMed] [Google Scholar]
- 24.Lissin DV, Carroll RC, Nicoll RA, Malenka RC, Zastrow M. Rapid, activation-induced redistribution of ionotropic glutamate receptors in cultured hippocampal neurons. J Neurosci. 1999;19:1263–1272. doi: 10.1523/JNEUROSCI.19-04-01263.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MacLean-Fletcher S, Pollard TD. Mechanism of action of cytochalasin B on actin. Cell. 1980;20:329–341. doi: 10.1016/0092-8674(80)90619-4. [DOI] [PubMed] [Google Scholar]
- 26.McLennan H. The autoradiographic localization of l-[3H]glutamate in rat brain tissue. Brain Res. 1976;115:139–144. doi: 10.1016/0006-8993(76)90828-3. [DOI] [PubMed] [Google Scholar]
- 27.Mills JW, Mandel LJ. Cytoskeletal regulation of membrane transport events. FASEB J. 1994;8:1161–1165. doi: 10.1096/fasebj.8.14.7958622. [DOI] [PubMed] [Google Scholar]
- 28.Munir M, Correale DM, Lu L, Robinson MB. Protein kinase C activation increases and N-methyl-d-aspartate (MNDA) receptor activation decreases cell surface expression of EAAC1 in neuron-enriched cultures. Soc Neurosci Abstr. 1998;24:2071. [Google Scholar]
- 29.Ohmori H, Toyama S, Toyama S. Direct proof that the primary site of action of cytochalasin on cell motility processes is actin. J Cell Biol. 1992;16:933–941. doi: 10.1083/jcb.116.4.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Otis TS, Wu YC, Trussell LO. Delayed clearance of transmitter and the role of glutamate transporters at synapses with multiple release sites. J Neurosci. 1996;16:1634–1644. doi: 10.1523/JNEUROSCI.16-05-01634.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qian Y, Galli A, Ramamoorthy S, Risso S, DeFelice LJ, Blakely RD. Protein kinase C activation regulates human serotonin transporters in HEK-293 cells via altered cell surface expression. J Neurosci. 1997;17:45–57. doi: 10.1523/JNEUROSCI.17-01-00045.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quick MW, Corey JL, Davidson N, Lester HA. Second messengers, trafficking-related proteins, and amino acid residues that contribute to the functional regulation of the rat brain GABA transporter GAT1. J Neurosci. 1997;17:2967–2979. doi: 10.1523/JNEUROSCI.17-09-02967.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramamoorthy S, Blakely RD. Phosphorylation and sequestration of serotonin transporters differentially modulated by psychostimulants. Science. 1999;285:763–766. doi: 10.1126/science.285.5428.763. [DOI] [PubMed] [Google Scholar]
- 34.Rampal AL, Pinkofsky HB, Jung CY. Structure of cytochalasins and cytochalasin B binding sites in human erythrocyte membranes. Biochemistry. 1980;19:679–683. doi: 10.1021/bi00545a011. [DOI] [PubMed] [Google Scholar]
- 35.Robinson MB, Dowd LA. Heterogeneity and functional properties of subtypes of sodium-dependent glutamate transporters in the mammalian central nervous system. Adv Pharmacol. 1997;37:69–115. doi: 10.1016/s1054-3589(08)60948-5. [DOI] [PubMed] [Google Scholar]
- 36.Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:1–20. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 37.Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 38.Sasaki T, Takai Y. The Rho small G protein family-Rho GDI system as a temporal and spatial determinant for cytoskeletal control. Biochem Biophys Res Commun. 1998;245:641–645. doi: 10.1006/bbrc.1998.8253. [DOI] [PubMed] [Google Scholar]
- 39.Schlag BD, Vondrasek JR, Munir M, Kalandadze A, Zelenaia OA, Rothstein JD, Robinson MB. Regulation of the glial Na+-dependent glutamate transporters by cyclic AMP analogs and neurons. Mol Pharmacol. 1998;53:355–369. doi: 10.1124/mol.53.3.355. [DOI] [PubMed] [Google Scholar]
- 40.Sensenbrenner M, Labourdette G, Delaunoy JP, Pettmann B, Devilliers G, Moonen G, Bock E. Morphological and biochemical differentiation of glial cells in primary culture. In: Giacobini E, editor. Tissue culture in neurobiology. Raven; New York: 1980. pp. 385–395. [Google Scholar]
- 41.Swanson RA, Seid LL. Barbiturates impair astrocyte glutamate uptake. Glia. 1998;24:365–371. doi: 10.1002/(sici)1098-1136(199812)24:4<365::aid-glia1>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 42.Swanson RA, Liu J, Miller JW, Rothstein JD, Farrell K, Stein BA, Longuemare MC. Neuronal regulation of glutamate transporter subtype expression in astrocytes. J Neurosci. 1997;17:932–940. doi: 10.1523/JNEUROSCI.17-03-00932.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- 44.Tong G, Jahr CE. Block of glutamate transporters potentiates postsynaptic excitation. Neuron. 1994;13:1195–1203. doi: 10.1016/0896-6273(94)90057-4. [DOI] [PubMed] [Google Scholar]
- 45.Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266:15771–15781. [PubMed] [Google Scholar]
- 46.Trotti D, Rizzini BL, Rossi D, Haugeto O, Racagni G, Danbolt NC, Volterra A. Neuronal and glial glutamate transporters possess an SH-based redox regulatory mechanism. Eur J Neurosci. 1997;9:1236–1243. doi: 10.1111/j.1460-9568.1997.tb01478.x. [DOI] [PubMed] [Google Scholar]
- 47.Tsakiridis T, Vranic M, Klip A. Disassembly of the actin network inhibits insulin-dependent stimulation of glucose transport and prevents recruitment of glucose transporters to the plasma membrane. J Biol Chem. 1994;269:29934–29942. [PubMed] [Google Scholar]
- 48.Vandenberg RJ, Mitrovic AD, Johnston GA. Molecular basis for differential inhibition of glutamate transporter subtypes by zinc ions. Mol Pharmacol. 1998;54:189–196. doi: 10.1124/mol.54.1.189. [DOI] [PubMed] [Google Scholar]
- 49.Wang Q, Bilan PJ, Tsakiridis T, Hinek A, Klip A. Actin filaments participate in the relocalization of phosphatidylinositol 3-kinase to glucose transporter-containing compartments and in the stimulation of glucose uptake in 3T3–L1 adipocytes. Biochem J. 1998;331:917–928. doi: 10.1042/bj3310917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watase K, Hashimoto K, Kano M, Yamada K, Watanabe M, Inoue Y, Okuyama S, Sakagawa T, Ogawa S, Kawashima N, Hori S, Takimoto M, Wada K, Tanaka K. Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur J Neurosci. 1998;10:976–988. doi: 10.1046/j.1460-9568.1998.00108.x. [DOI] [PubMed] [Google Scholar]
- 51.Ye ZC, Sontheimer H. Stereospecific transport of trans-ACPD and its impact on intracellular glutamate levels in astrocytes. Soc Neurosci Abstr. 1998;24:2070. [Google Scholar]
- 52.Ye ZC, Sontheimer H. Metabotropic glutamate receptor agonists reduce glutamate release from cultured astrocytes. Glia. 1999;25:270–281. [PubMed] [Google Scholar]
- 53.Zerangue N, Arriza JL, Amara SG, Kavanaugh MP. Differential modulation of human glutamate transporter subtypes by arachidonic acid. J Biol Chem. 1995;270:6433–6435. doi: 10.1074/jbc.270.12.6433. [DOI] [PubMed] [Google Scholar]