

Abstract
The retinoblastoma tumor suppressor protein (pRb) family is essential for cortical progenitors to exit the cell cycle and survive. In this report, we test the hypothesis that pRb collaborates with basic helix-loop-helix (bHLH) transcription factors to regulate cortical neurogenesis, taking advantage of the naturally occurring dominant-inhibitory HLH protein Id2. Overexpression of Id2 in cortical progenitors completely inhibited the induction of neuron-specific genes and led to apoptosis, presumably as a consequence of conflicting differentiation signals. Both of these phenotypes were rescued by coexpression of a constitutively activated pRb mutant. In contrast, Id2 overexpression in postmitotic cortical neurons affected neither neuronal gene expression nor survival. Thus, pRb collaborates with HLHs to ensure the coordinate induction of terminal mitosis and neuronal gene expression as cortical progenitors become neurons.
Keywords: neurogenesis, Id2, pRb, bHLH transcription factors, cortical development, neuronal gene expression, α-tubulin, neural progenitor cells, neurofilaments, apoptosis
During embryogenesis, cycling neural progenitor cells in the ventricular zones of the CNS commit to a neuronal fate, and as a consequence of that decision, coordinately undergo terminal mitosis and induce early, neuron-specific genes. One group of proteins known to be essential for this transition is the retinoblastoma tumor suppressor protein (pRb) family. In particular, inactivation of the Rb gene by homologous recombination led to ectopic mitoses and massive cell death in the developing nervous system (Clarke et al., 1992; Jacks et al., 1992; Lee et al., 1992, 1994), problems attributed to the essential role that this protein plays in cell cycle regulation (for review, see Slack and Miller, 1996). More recent studies defined a critical temporal requirement for the pRb family during cortical neurogenesis (Slack et al., 1998); these cell cycle regulators were essential for terminal mitosis and survival of progenitors but were dispensable for the induction of early neuronal genes. These findings thus implicated the pRb family as key regulators of terminal mitosis but left in question the intracellular mechanisms responsible for coordinately inducing neuronal gene expression.
One class of proteins known to play a key role in the induction of cell type-specific gene expression is the basic helix-loop-helix (bHLH) transcription factor family (for review, see Weintraub, 1993; Lee, 1998), whose role has been perhaps best defined during myogenesis and in Drosophila neurogenesis (Jan and Jan, 1993;Campos-Ortega, 1993). Interestingly, one of the key myogenic bHLHs, MyoD (Weintraub, 1993), is thought to interact with pRb to regulate muscle development (Gu et al., 1993). Although it is not known whether similar interactions between transcription factors and cell cycle proteins regulate neurogenesis, a significant body of work has defined a role for the positive bHLHs during mammalian neural development (Kageyama and Nakanishi, 1997; Lee, 1998). In particular, in the PNS, bHLHs such as Mash-1 (Johnson et al., 1990) and the neurogenins (Ma et al., 1996; Sommer et al., 1996) regulate the genesis of defined neuronal populations (Guillemot et al., 1993; Fode et al., 1998; Ma et al., 1998). Many positively acting bHLHs are also expressed in the developing CNS (for review, see Lee, 1998), and insights into their potential developmental roles are now starting to emerge. For example, in the absence of NeuroD1, NeuroD2, Math1, and/or Math2 (Ben-Arie et al., 1997; Miyata et al., 1999; Schwab et al., 2000), cerebellar and/or hippocampal granule cells fail to form appropriately, whereas progenitor cells are depleted in a defined region of the telencephalon in Mash1−/− mice (Casarosa et al., 1999; Torii et al., 1999). The rather limited nature of the CNS phenotypes in these mutant animals is likely attributable to compensation by other bHLH transcription factors, a phenomena that is well documented for myogenic bHLHs (Weintraub, 1993).
In this paper, we have chosen an alternative strategy to define the role of positively acting bHLH transcription factors during cortical neurogenesis. Specifically, we have chosen to inhibit all bHLH transcriptional activity by overexpressing Id2 (Christy et al., 1991;Sun et al., 1991; Biggs et al., 1992), a member of a family of naturally occurring dominant-inhibitory HLH proteins that lack a DNA-binding domain (Benezra et al., 1990). This family, which has four members (Id1–Id4) (for review, see Norton et al., 1998), binds to and inhibits the ubiquitously expressed E2A bHLHs (Jen et al., 1992), which are obligate partners for tissue-specific bHLHs such as MyoD and Mash-1 (Lassar et al., 1991; Norton et al., 1998). All four of the Id family members are expressed in the developing cortex in which some are expressed primarily in precursor cells and some (such as Id2) in both precursors and subpopulations of postmitotic neurons (Neuman et al., 1993; Riechmann and Sablitzky, 1995; Jen et al., 1997). Interestingly, unlike the other Id proteins, Id2 interacts with and regulates the activity of the pRb family (Iavarone et al., 1994; Lasorella et al., 1996), making endogenous Id2 a molecule that could regulate both cell cycle progression and neuronal gene expression in cortical progenitor cells.
Here, we report that overexpression of Id2 in cortical progenitor cells completely inhibited induction of neuron-specific genes and ultimately led to apoptosis. Both of these Id2-induced phenotypes could be rescued by coexpression of constitutively activated pRb. In contrast to progenitors, overexpression of Id2 in postmitotic cortical neurons affected neither survival nor neuronal gene expression. These findings support the hypothesis that positively acting bHLH transcription factors, in collaboration with the pRb family, are essential for cortical progenitors to become neurons and support the idea that interactions between endogenous pRb, Id2, and bHLH transcription factors play a key role in regulating the progenitor-to-neuron transition.
MATERIALS AND METHODS
Primary cultures of cortical progenitors and neurons.The preparation of cortical progenitors from embryonic day 12 (E12) to E13 mouse embryos has been described in detail previously (Slack et al., 1998; Gloster et al., 1999). Briefly, cortical tissue, obtained from E12–E13 mice, was dissected in ice-cold HBSS (Life Technologies, Burlington, CA) and then transferred into 37°C Neurobasal media (Life Technologies) containing 500 μm glutamine, 1% N2 supplement, 2% B27 supplement, and 1% penicillin–streptomycin (Life Technologies); this media was supplemented with 40 ng/ml basic fibroblast growth factor (bFGF) (Collaborative Research, Bedford, MA). The tissue was triturated with a fire-polished glass Pasteur pipette into small clusters of cells that were plated in multiwell tissue culture dishes (Nunc, Naperville, IL) or chamber slides precoated with laminin and poly-d-lysine (Collaborative Research). Cell density was 100,000 cells per well. Cultures were maintained at 37°C in a 5% CO2 incubator. Mature postmitotic neurons were prepared from E16 embryos, from which cortices were collected, triturated in culture media (Neurobasal with 0.5 mm glutamine, 1% penicillin–streptomycin, 1% N2, 2% B27 supplements, and 20 ng/ml bFGF), and plated at a density of 50–75,000 cells per well. For analysis of Tα1:nlacZ expression, cultures were prepared from the K6 line of transgenic mice, which has been characterized in detail previously (Gloster et al., 1994, 1999; Bamji and Miller, 1996). These animals are homozygous for the transgene, eliminating the need for genotyping.
Recombinant adenovirus vectors. The adenovirus vectors carrying the Escherichia coli lacZ expression cassette (Ad5CA17lacZ; gift of Dr. Frank Graham, McMaster University, Hamilton, Ontario, Canada) (Slack et al., 1998) and constitutively activated pRb (Chang et al., 1995) have been described previously. The adenoviruses expressing green fluorescent protein (GFP) and myc-tagged human Id2 were both constructed in the Ad5 backbone (Bett et al., 1994), which drives expression from the cytomegalovirus promoter. The GFP-expressing virus was obtained commercially from Quantum Biotechnologies (Montreal, Canada), and that expressing myc-tagged human Id2 was constructed using standard protocols, as we have described previously (Mazzoni et al., 1999).
All recombinant adenoviruses were purified on CsCl gradients, as we have described previously (Aloyz et al., 1998; Mazzoni et al., 1999), and extensively purified. Infectious titer was determined by plaque assay on HEK293 cells (Graham and Prevec, 1991).
Cortical progenitor cells were infected at the time of plating, and 24 hr after infection, half of the media was changed, and cells were fed every 2 d. The multiplicity of infection (MOI) indicates the number of plaque-forming units added per cell.
Immunocytochemistry of cultured cells. For immunocytochemical detection of nestin (1:1000; gift from Dr Ron McKay, National Institute of Neurological Disorders and Stroke, Bethesda, MD), neurofilament M (1:400; Chemicon, Temecula, CA), β-galactosidase (1:1000; ICN Biomedicals, Cleveland, OH), the myc-tag (1:200; PharMingen, San Diego, CA), and the hemagglutinin (HA)-tag [1:500; monoclonal from Boehringer Mannheim (Indianapolis, IN) and polyclonal from Babco (Richmond, CA)], cells were fixed for 20 min with 4% paraformaldehyde. Cultures were then washed with HEPES-buffered saline (HBS), pH 7.4, permeabilized for 5 min in 0.2% NP-40 in HBS, and then blocked for 45 min with buffer containing 6% goat serum and 0.5% bovine serum albumin. Cells were then incubated at 4°C overnight with primary antibodies in HBS containing 3% goat serum. After three washes with HBS, cells were incubated at room temperature for 60 min with indocarbocyanine (Cy3)-conjugated goat anti-mouse (1:400; Jackson ImmunoResearch, West Grove, PA), Cy3-conjugated anti-rabbit (1:400; Jackson ImmunoResearch), FITC-conjugated anti-mouse (1:100; Jackson ImmunoResearch), or FITC-conjugated anti-rabbit (1:100; Jackson ImmunoResearch) secondary antibodies (as necessary) prepared in HBS containing 3% goat serum. Samples were washed three times with HBS and then were counterstained for 2 min with Hoechst 33258 (Sigma, St. Louis, MO) before examination by fluorescence microscopy.
For quantitation, three to six random images of each treatment (per experiment) were captured and processed. Digital image acquisition and analysis was performed with the Northern Eclipse software (Empix Inc.) using a Sony (Tokyo, Japan) XC-75CE CCD video camera.
Cell survival assays and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling.MTT survival assays were performed as described previously (Slack et al., 1998; Vaillant et al., 1999). In brief, 20 μl of MTT reagent was added to the medium in each well of a 96-well plate containing the cultured progenitors or neurons. After a 2.5 hr incubation at 37°C, the medium–MTT mixture was removed, and the cells were lysed with 100 μl of isopropanol containing 2 μl/ml concentrated HCl. The absorbance of the lysate at 570 and 630 nm was determined using a Bio-Tek Instruments (Burlingame, CA) model Elx-800 UV plate reader (Mandel Scientific Inc.). All MTT assays were performed in triplicate.
For the terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) experiments, cells were briefly rinsed in HBS, pH 7.4, and fixed for 20 min in 4% paraformaldehyde (Sigma) in HBS. Cells were then permeabilized with 0.4% NP-40 in HBS for 5 min and washed three times with HBS. TUNEL reaction was performed for 1 hr at 37°C. Each 100 μl of TUNEL reaction mixture contained 20 μl of 5× terminal deoxynucleotidyl transferase (TdT) buffer, 1.5 μl of TdT enzyme (both from Promega, Madison, WI), and 1 μl of biotin-16-dUTP (Boehringer Mannheim). After the TUNEL reaction, cells were rinsed three times in HBS and incubated for 45 min at room temperature with Cy3-conjugated streptavidin (Jackson ImmunoResearch) diluted 1:2000 in HBS. In those experiments in which cells were double-labeled, cells were then incubated overnight in the primary antibody, as above, and immunocytochemical analysis was performed as described.
Western blot analysis and coimmunoprecipitations. For biochemistry, cortical progenitors or neurons were plated in 60 mm dishes and were lysed in TBS lysis buffer (Knusel et al., 1994) containing 137 mm NaCl, 20 mm Tris, pH 8.0, 1% (v/v) NP-40, 10% (v/v) glycerol, 1 mm PMSF, 10 μg/ml aprotinin, 0.2 μg/ml leupeptin, 1.5 mm sodium vanadate, and 0.1% SDS. Cells were collected in cold PBS by gentle scraping to detach them, were washed three times with the same buffer, and then were resuspended in 50–100 μl of lysis buffer, followed by rocking for 10 min at 4°C. After a 5 min centrifugation, the lysates were normalized for protein concentration using a BCA Protein Assay Reagent (Pierce, Rockford, IL). Equal amounts of protein (50–100 μg) were then boiled in sample buffer for 5 min and separated by 7.5–15% SDS-PAGE gradient gels. Alternatively, for Id2, samples were separated on tricine–SDS-PAGE gels, which are optimized for small proteins (Schagger and von Jagow, 1987). After electrophoresis, proteins were transferred to 0.2 μm nitrocellulose for 3 hr at 0.75 A, and the membrane was washed three times with TBS. For all antibodies, the membranes were blocked in 5% nonfat milk in TBS plus 0.2% Tween (TBST) (blotto) for 2 hr at room temperature. The membranes were then incubated overnight at 4°C with the primary antibodies in blotto:anti-myc (1:500), anti-HA (1:1000), anti-Id2 (1:200; Santa Cruz Biotechnology, Santa Cruz, CA) or anti-extracellular signal-regulated kinase (ERK) (1:500; Santa Cruz Biotechnology). After incubation with the primary antibodies, membranes were washed four times with TBST over 40 min and incubated with the secondary antibody for 1.5 hr at room temperature. The secondary antibodies (goat anti-mouse or goat anti-rabbit HRP from Boehringer Mannheim) were used at 1:10,000 dilution. After three washes with TBST, detection was performed using the ECL chemiluminescence reagent from Amersham Pharmacia Biotech (Arlington Heights, IL) and XAR x-ray film from Eastman Kodak (Rochester, NY).
For coimmunoprecipitations, HEK293 cells or sympathetic neurons of the superior cervical ganglion, cultured as described previously (Aloyz et al., 1998), were infected with recombinant adenoviruses expressing HA-pRb, myc-Id2, or β-galactosidase, cells were lysed in lysis buffer, and 200 μg of protein from the relevant lysates were mixed and incubated overnight at 4°C in the presence of anti-HA. Protein G-Sepharose (Amersham Pharmacia Biotech) was added to the lysates and incubated for 2 hr at 4°C, and the lysates were then centrifuged to collect the immunoprecipitated protein. Immunoprecipitates were washed twice with lysis buffer, boiled 5 min in 2× sample buffer, and loaded on 7.5–15% SDS-PAGE gradient gels for electrophoresis.
RESULTS
Expression of endogenous and exogenous Id2 in cortical progenitor cells
To address the role of HLH proteins in the progenitor-to-neuron transition, we used a culture system that we have characterized previously (Slack et al., 1998; Gloster et al., 1999).
Cultures of progenitor cells were derived from E12–E13 mouse cortex and were plated in the presence of bFGF. Upon plating, virtually all of the cells were dividing, nestin-positive progenitors (data not shown;Slack et al., 1998; Gloster et al., 1999). Over the ensuing 5 d, many of these cells exited the cell cycle to become postmitotic neurons that expressed the panneuronal markers βIII-tubulin, neuron-specific enolase, MAP-2, and neurofilament M (NFM), as well as a neuron-specific Tα1:nlacZ transgene composed of the Tα1 α-tubulin promoter linked to a nuclear β-galactosidase reporter gene (Gloster et al., 1994;Slack et al., 1998, 1999).
To determine whether positively acting bHLH transcription factors were essential for induction of the neuronal phenotype in cortical progenitors, we chose to inhibit their activity by overexpressing the dominant-inhibitory HLH, Id2. As a prelude to these studies, we characterized expression of endogenous Id2 in these cultures. Reverse transcription (RT)-PCR analysis demonstrated that Id2 mRNA was expressed in the E13 brain, the postnatal day 0 (P0) neocortex, and, at reduced levels, in the adult brain (Fig.1A). Id2 mRNA was also expressed in cultured cortical progenitors (Fig. 1A), as predicted by previous in situ hybridization studies localizing Id2 mRNA to the ventricular zone of the embryonic neocortex (Neuman et al., 1993). Western blot analysis confirmed this pattern of expression (Fig. 1A); Id2 was present at higher levels in the embryonic versus adult brain and was expressed by cortical progenitors.
Fig. 1.
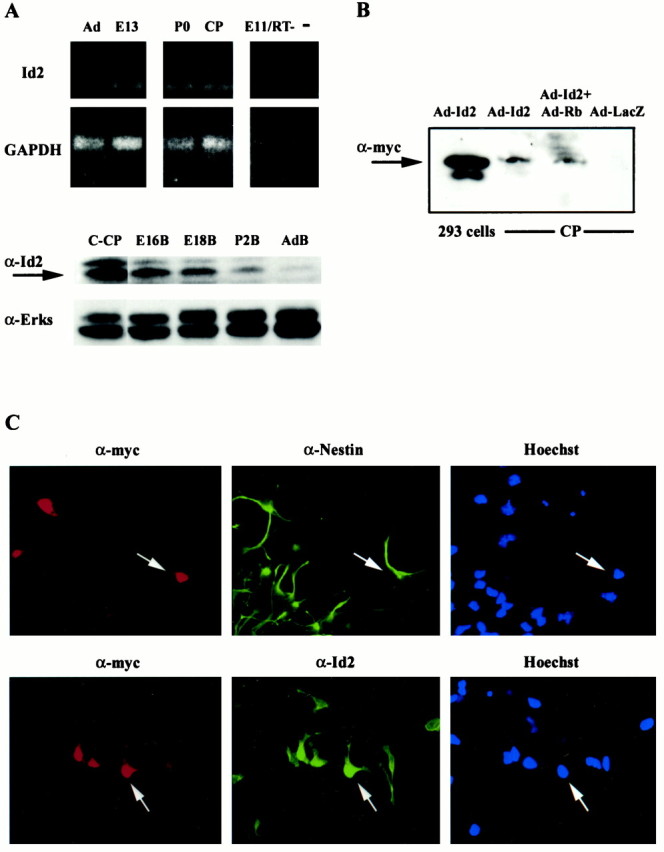
Expression of endogenous Id2 and a myc-tagged Id2 adenovirus in cortical progenitor cells. A,Top panels, RT-PCR analysis of Id2 and GAPDH mRNAs in cDNA isolated from adult cortex (Ad), E13 brain (E13), P0 brain (P0), or cortical progenitor cells 1 d after plating (CP). As controls, analysis was performed on embryonic brain samples in which the reverse transcriptase was eliminated from the cDNA reaction (E11/RT-) and on samples that contained no cDNA (−). Bottom panels, Western blot analysis for endogenous Id2 in cortical progenitors (C-CP), and E16 (E16B), E18 (E18B), P2 (P2B), and adult brain (AdB). The arrow indicates endogenous Id2. The same blot was reprobed with an antibody directed against ERKs 1 and 2 (α-Erks) to control for protein loading.B, Western blot analysis for myc-tagged Id2 in HEK293 cells and cortical progenitor cells (CP). Cells were infected with the Id2 adenovirus (Ad-Id2) and/or a virus expressing constitutively activated pRb (Ad-Rb), proteins were separated on tricine gels and transferred to nitrocellulose, and filters were probed with an anti-myc antibody. Control lysates of cells infected with a β-galactosidase adenovirus (Ad-LacZ) were analyzed on the same blot. A myc-tagged protein of ∼15 kDa was detected in both HEK293 cells and cortical progenitor cells infected with the Id2 adenovirus. C, Double-label immunocytochemical analysis of cortical progenitor cells infected with 50 MOI of an adenovirus expressing myc-tagged Id2 and analyzed for myc and for nestin (top panels) or for myc and Id2 (bottom panels). The right panelsare photomicrographs of the Hoechst staining for the same fields.Arrows indicate double-labeled cells.
To increase the level of expression of Id2 in cortical progenitors, we generated a recombinant adenovirus expressing myc-tagged Id2. We have previously used such an adenovirus approach to genetically manipulate cortical progenitor cells (Slack et al., 1998) and postmitotic neurons (Slack et al., 1996; Aloyz et al., 1998; Mazzoni et al., 1999; Vaillant et al., 1999). Western blot analysis of HEK293 cells infected with the Id2 adenovirus revealed that the recombinant Id2 protein was appropriately sized at 14–16 kDa (Fig. 1B). Progenitors infected with Id2, but not β-galactosidase, adenovirus also expressed a myc-positive protein of the appropriate size (Fig.1B). To confirm this result, we performed immunocytochemistry on progenitors infected with 50 MOI of Id2 adenovirus. Double-label analysis revealed that myc-tagged Id2 was expressed in nestin-positive progenitor cells (Fig. 1C). A similar analysis with antibodies to the myc-tag and to the endogenous Id2 protein demonstrated that endogenous Id2 was distributed in both the cytoplasm and nucleus as reported previously (Deed et al., 1996;Tzeng and de Vellis, 1998) and that the adenovirally driven Id2 was primarily targeted to the nucleus (Fig. 1C).
Id2 inhibits induction of neuronal gene expression in cortical progenitors but not in postmitotic cortical neurons
To determine whether positively acting bHLH transcription factors are essential for induction of neuron-specific gene expression, we infected progenitor cells with 50 MOI of the Id2 adenovirus upon plating and, 2.5 d later, monitored expression of two neuron-specific markers, the Tα1:nlacZ transgene and NFM. As controls, sister cultures were infected with 50 MOI of an adenovirus expressing GFP. Double-label immunocytochemistry revealed that Id2, but not GFP, completely inhibited induction of the Tα1:nlacZ transgene in cortical progenitors (Fig.2A). Quantitation demonstrated that Tα1:nlacZ was virtually never expressed in Id2-expressing cells, although it was expressed in the majority of control, GFP-expressing cells (Fig.2A,B). Id2 overexpression also inhibited NFM expression (Fig. 2B); Id2-expressing cells were virtually never NFM-positive, whereas control cells were (Fig. 2B). Similar results were obtained at MOIs of the Id2 virus ranging from 25 to 100 MOI (data not shown).
Fig. 2.
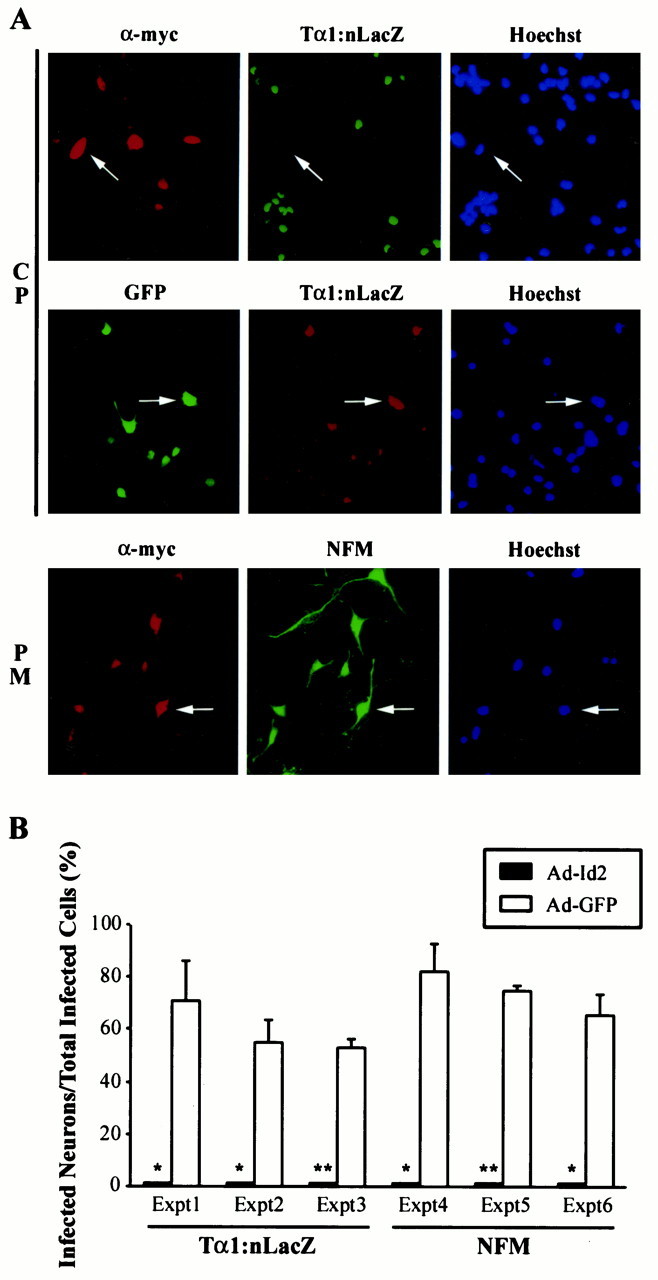
Id2 inhibits expression of neuron-specific genes in cortical progenitor cells as they become neurons but does not perturb neuronal gene expression in postmitotic cortical neurons.A, Double-label immunocytochemical analysis of cortical progenitor cells (CP) or postmitotic cortical neurons (PM) infected with adenoviruses expressing Id2 (α-myc) or GFP (left panels) and then analyzed for expression of the neuron-specific Tα1:nlacZ transgene (middle panels) or NFM 2.5 d later. The right panels are photomicrographs of the Hoechst staining for the same fields. Note that none of the myc-stained cortical progenitor cells express Tα1:nlacZ (arrows,top panels), whereas GFP and Tα1:nlacZ are colocalized in many cells (arrows, middle panels). In contrast to progenitors, expression of myc-tagged Id2 in cortical neurons does not inhibit neurofilament M expression (arrows, bottom panels).B, Quantitation of double-label immunocytochemical analysis similar to that in A, showing results for Tα1:nlacZ and NFM. For each treatment in each individual experiment, three to six random fields were analyzed. Cells expressing myc-tagged Id2 virtually never expressed either of these two neuronal marker genes. In contrast, many of the GFP-expressing cells coexpressed Tα1:nlacZ or NFM [range, 53 ± 3.3 (Expt 3) to 82 ± 10.5 (Expt 4)]. Results indicate the mean ± SE. *p < 0.05, **p < 0.005.
To determine whether the Id2-mediated inhibition of neuronal gene expression was specific to progenitors, we also examined postmitotic cortical neurons. Double-label immunocytochemical analysis revealed that cortical neurons expressing myc-tagged Id2 also expressed robust levels of NFM (Fig. 2A). Thus, Id2 inhibits the induction of neuronal gene expression in cortical progenitors cells, presumably by binding to and inhibiting positively acting bHLH transcription factors.
Id2 expression leads to apoptosis of cortical progenitors but not postmitotic neurons
We have demonstrated previously that inhibition of the pRb family in cortical progenitors led to apoptosis, although it had no effect on induction of neuronal gene expression (Slack et al., 1998). Because Id2 also interacts with members of the pRb family (Iavarone et al., 1994;Lasorella et al., 1996), we monitored survival of the Id2-infected progenitor cells. Specifically, cortical progenitor cells were infected with 10–100 MOI of Id2 or β-galactosidase adenovirus, and 4 d later, survival was determined using MTT assays, which measure mitochondrial function. This analysis revealed that expression of Id2 led to a dose-dependent decrease in cortical progenitor cell number, whereas similar MOIs of the control virus had no effect (Fig.3A). To confirm that this decrease in MTT reflected a decrease in survival, we performed TUNEL on cortical progenitor cells infected with 50 MOI of Id2 or β-galactosidase adenovirus. Double-label analysis revealed that, 4 d after infection, the majority of Id2-expressing progenitor cells were TUNEL-positive (Fig. 3C), whereas the vast majority of β-galactosidase-expressing cells were not (Fig.3C).
Fig. 3.
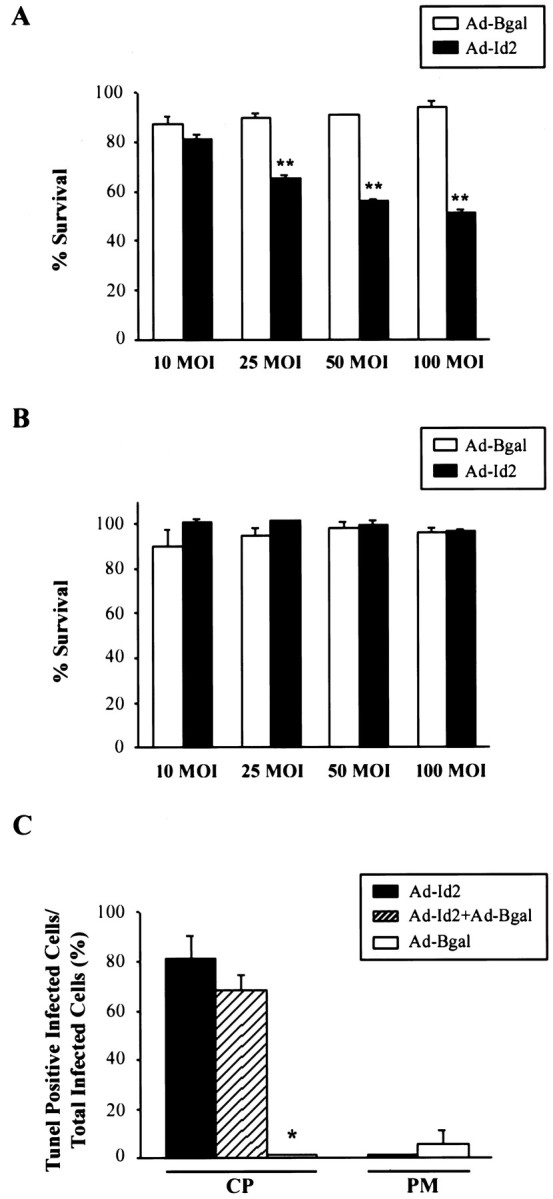
Overexpression of Id2 leads to apoptosis of cortical progenitors but not postmitotic cortical neurons.A, MTT assay to measure survival of cortical progenitor cells 4 d after infection with adenoviruses expressing Id2 (Ad-Id2) or β-galactosidase (Ad-Bgal) at MOIs ranging from 10 to 100 MOI. All points were performed in triplicate, and results represent the mean ± SE, with 100% survival being defined as the MTT value obtained for uninfected cells in the same experiment. **p < 0.005. B, MTT assay to measure survival of postmitotic cortical neurons after infection with adenoviruses expressing Id2 (Ad-Id2) or β-galactosidase (Ad-Bgal) at MOIs ranging from 10 to 100. All points were performed in triplicate, and results are as in A. C, Quantitation of the percentage of TUNEL-positive cortical progenitors (CP) and postmitotic cortical neurons (PM) infected with adenoviruses expressing myc-tagged Id2 (Ad-Id2) and/or β-galactosidase (Ad-Bgal) for 4 d. Results are the mean ± SE of data obtained from three to six randomly chosen fields. *p = 0.015 compared with cells infected with Ad-Id2. Note that the majority of Id2-expressing cortical progenitor cells are TUNEL-positive, whereas β-galactosidase-expressing progenitors are not.
Because inhibition of the pRb family led to apoptosis of cortical progenitors but not postmitotic neurons (Slack et al., 1998), we examined the effect of Id2 on survival of cortical neurons. Cortical neurons were plated at E16 and, 5 d later, were infected with various MOIs of adenovirus expressing either Id2 or β-galactosidase. MTT assays revealed that neither Id2 nor β-galactosidase had any effect on neuronal survival (Fig.3B). TUNEL analysis confirmed this conclusion; quantitative double-label analysis revealed that the vast majority of neurons expressing either Id2 or β-galactosidase were not TUNEL-positive 4 d after infection (Fig. 3C). Thus, overexpression of Id2 ultimately caused apoptosis of progenitors but not postmitotic neurons, an effect similar to that observed after inhibition of the pRb family (Slack et al., 1998).
Constitutively activated pRb rescues both the loss of neuronal gene expression and the progenitor cell apoptosis induced by Id2
Together, these data indicated that increased expression of Id2 in cortical progenitors (1) completely suppressed induction of neuronal gene expression, presumably by binding to and inhibiting positively acting bHLH transcription factors, and (2) led to progenitor cell apoptosis, potentially by interacting with pRb family members and disrupting terminal mitosis. To test this latter possibility, we asked whether pRb could rescue these Id2-induced phenotypes. To perform these experiments, we used a recombinant adenovirus expressing an HA-tagged, phosphorylation-deficient, constitutively activated form of pRb (Chang et al., 1995). Initially, we confirmed that this mutant form of pRb interacted with Id2, as does wild-type pRb (Iavarone et al., 1994;Lasorella et al., 1996). Lysates of HEK293 cells infected with Id2 or pRb adenoviruses were incubated together, and the mutant pRb was immunoprecipitated with anti-HA. These immunoprecipitates were then probed for the presence of myc-tagged Id2 using Western blots. This analysis revealed that the pRb adenovirus expressed an HA-tagged protein of ∼90–100 kb (the appropriate size) (Fig.4B) and that the mutant pRb and myc-tagged Id2 coimmunoprecipitated (Fig.4A), confirming an interaction between these two proteins. Confirmation that this interaction also occurred in neurons was obtained by performing similar coinfection experiments in sympathetic neurons. As seen in HEK293 cells, the mutant pRb and myc-tagged Id2 coimmunoprecipitated in this neuronal context (data not shown).
Fig. 4.
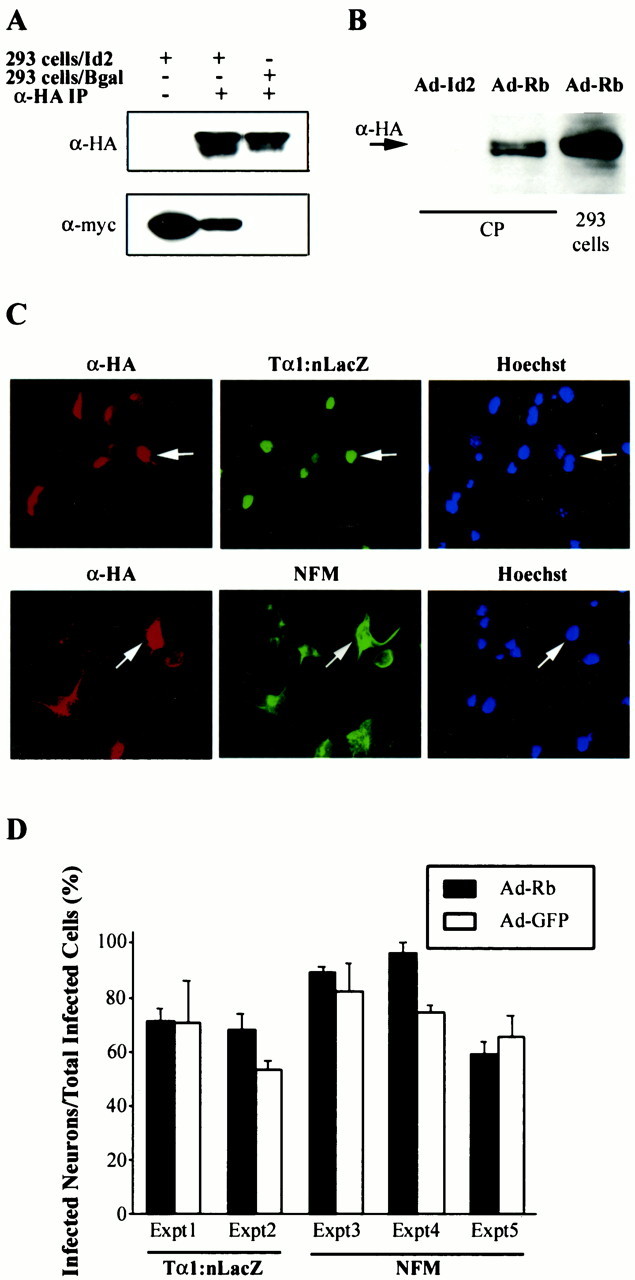
A, B, Constitutively activated pRb interacts with Id2. A, Lysates of HEK293 cells infected with adenoviruses expressing myc-tagged Id2 (293 cells/Id2), β-galactosidase (293 cells/Bgal), and/or HA-tagged activated pRb were mixed, the HA-tagged pRb was immunoprecipitated with anti-HA (α-HA IP), and then the immunoprecipitated proteins were probed with anti-HA to detect the immunoprecipitated pRb and anti-myc to detect coimmunoprecipitated Id2. B, Cortical progenitors (CP) or HEK293 cells were infected with adenoviruses expressing Id2 (Ad-Id2) or activated pRb (Ad-Rb), and lysates were analyzed with anti-HA on Western blots to detect expression of the pRb protein. Thearrow indicates a band of the appropriate size that is seen only in cells infected with Ad-Rb. C,D, Expression of constitutively activated pRb does not affect the induction of neuronal gene expression in cortical progenitor cells. C, Double-label immunocytochemical analysis of cortical progenitor cells infected with adenoviruses expressing HA-tagged constitutively activated pRb (α-HA;left panels) and then analyzed for expression of Tα1:nlacZ (middle panel, top) or NFM (middle panel, bottom). The right panels are photomicrographs of Hoechst staining for the same fields. Note that many of the HA-stained cells express either Tα1:nlacZ or neurofilament (arrows). D, Quantitation of double-label immunocytochemical analysis similar to that in C. For each treatment in each individual experiment, three to six random fields were analyzed. Many of the HA-tagged pRb-expressing cells coexpressed Tα1:nlacZ or NFM [range, 59 ± 4.9 (Expt 5) to 96 ± 3.7 (Expt 4)], a result similar to that obtained for cells expressing GFP [range, 53 ± 3.3 (Expt 2) to 82 ± 10.5 (Expt 3)]. Results indicate the mean ± SE. In all experiments, the percentage of pRb-positive cells coexpressing one of these two neuronal markers was statistically similar to the percentage of GFP-positive cells expressing the same marker (p > 0.05).
We next determined whether this mutant pRb had any effect on the progenitor-to-neuron transition. Progenitor cells were infected with 50 MOI of pRb adenovirus and analyzed for neuronal gene expression and cell survival. Western blot analysis demonstrated that pRb-infected cortical progenitors expressed an HA-tagged protein of the appropriate size (Fig. 4B), and immunocytochemistry demonstrated that progenitors infected with the mutant pRb adenovirus expressed detectable HA-immunoreactivity (Fig. 4C), whereas those infected with a control β-galactosidase adenovirus did not (data not shown). To analyze neuronal gene expression in these cultures, we performed double-label immunocytochemistry 2.5 d after infection. Most of the HA-positive, pRb-expressing progenitors also expressed NFM or the Tα1:nlacZ transgene (Fig. 4C, D), and there was no difference in neuronal marker gene expression between cells infected with the pRb versus control, GFP adenovirus (Fig.4D). Similarly, expression of mutant pRb had no effect on neuronal survival, as monitored by either MTT assays (Fig.5B) or quantitative TUNEL (Fig. 5C).
Fig. 5.
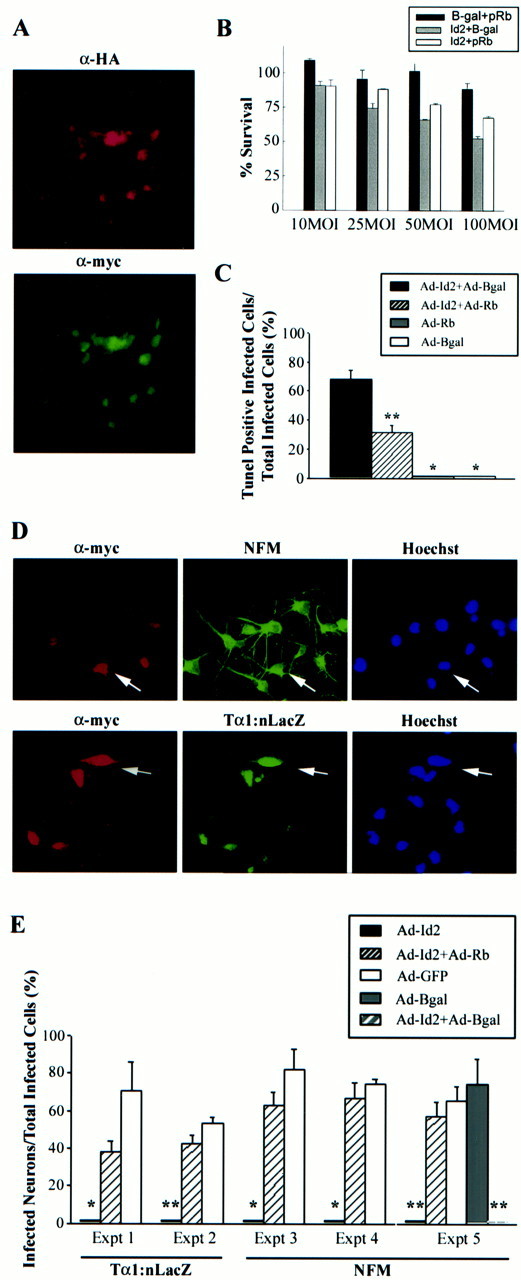
A, Coinfection of cortical progenitors with adenoviruses expressing HA-tagged pRb and myc-tagged Id2. Double-label immunocytochemical analysis for the HA-tag (α-HA, top panel) on pRb and the myc-tag (α-myc, bottom panel) on Id2 revealed that most of the cortical progenitors were infected with both adenoviruses and expressed both proteins. B,C, Constitutively activated pRb rescues the apoptosis induced by overexpression of Id2 in cortical progenitor cells.B, MTT assay to measure survival of cortical progenitor cells 4 d after infection with adenoviruses expressing Id2 and/or constitutively activated pRb or β-galactosidase (Bgal) at MOIs ranging from 10 to 100. All points were performed in triplicate, and results represent the mean ± SE, with 100% survival being defined as the MTT value obtained for uninfected progenitor cells in the same experiment. C, Quantitation of the percentage of TUNEL-positive cortical progenitors 4 d after infection with 50 MOI of adenoviruses expressing Id2 (Ad-Id2) and/or constitutively activated pRb (Ad-Rb) or β-galactosidase (Ad-Bgal). In the rescue experiments involving transduction with two adenovirally driven proteins, cells were double-labeled for TUNEL and anti-myc to detect those cells expressing myc-tagged Id2. Results indicate the mean ± SE. *p < 0.05, **p < 0.01 relative to cells infected with Ad-Id2 plus Ad-Bgal. D,E, Constitutively activated pRb rescues the inhibition of neuronal gene expression induced by overexpression of Id2 in cortical progenitor cells. D, Double-label immunocytochemical analysis of cortical progenitor cells infected with 50 MOI of adenoviruses expressing Id2 and constitutively activated pRb and then analyzed for expression of the myc-tagged Id2 protein (α-myc, left panel) and NFM or Tα1:nlacZ (middle panels). The right panels are photomicrographs of the Hoechst staining for the same field. Note that, when coinfected with constitutively activated pRb, many of the Id2-expressing cortical progenitors also express these two neuronal genes (arrows). E, Quantitation of double-label immunocytochemical analysis similar to that in D. For each treatment in each individual experiment, three to six random fields were analyzed. Cortical progenitor cells expressing myc-tagged Id2 virtually never coexpressed Tα1:nlacZ or NFM. In contrast, when coinfected with Ad-Rb, many of the Id2-positive cells were also positive for neuronal marker genes. Results indicate the mean ± SE. *p < 0.05, **p < 0.005.
Having ascertained that activated pRb had no effect on either neuronal gene induction or survival, we determined whether it could rescue the Id2-induced progenitor cell phenotype. Initially, we confirmed that coinfection of progenitor cells with the pRb virus did not alter expression of myc-tagged Id2 (Fig. 1B). We then used double-label immunocytochemistry to show that many of the progenitor cells expressed both myc-tagged Id2 and HA-tagged pRb (Fig.5A). Having performed these controls, we examined Id2-induced apoptosis. Cortical progenitor cells were coinfected with varying MOIs of adenoviruses expressing Id2 and mutant pRb, or Id2 and β-galactosidase, and survival was measured using MTT assays 4 d later. This analysis demonstrated that constitutively active pRb, but not β-galactosidase, partially rescued the decrease in neuronal survival induced by Id2 (Fig. 5B). This conclusion was confirmed by quantitative TUNEL; sister cultures were coinfected with 50 MOI each of Id2 and pRb or β-galactosidase adenoviruses, and the cultures were analyzed by double-labeling (Fig. 5C). When Id2-expressing cells were coinfected with the β-galactosidase virus, 68 ± 6.2% of the myc-tagged cells were TUNEL-positive, a result similar to that seen with the Id2 virus alone (Fig. 3C). In contrast, in cortical progenitors coinfected with the pRb virus, only 32 ± 4.9% of the myc-tagged cells were TUNEL-positive, a rescue of approximately half of the Id2-expressing progenitor cells. This rescue was statistically significant (p < 0.01) and was similar in magnitude to the rescue observed in the MTT assays (Fig. 5B). Thus, constitutively activated pRb substantially rescued the Id2-induced apoptosis, supporting the idea that Id2 interacts with endogenous pRb (and or pRb family members) to regulate terminal mitosis and ultimately, survival of cortical progenitors as they become postmitotic neurons.
We then determined whether activated pRb rescued the inhibition of neuronal gene expression seen with Id2. Progenitor cells were coinfected with 50 MOI each of adenoviruses expressing Id2 and pRb, or Id2 and GFP, and were analyzed for expression of the Tα1:nlacZ transgene or NFM. Double-labeling revealed that coinfection with activated pRb rescued the expression of the neuron-specific Tα1:nlacZ transgene in Id2-expressing progenitor cells (Fig.5D,E). Similarly, coinfection with activated pRb rescued the expression of NFM in Id2-expressing cells (Fig. 5D,E). In contrast, coinfection with a GFP- or β-galactosidase-expressing adenovirus was unable to rescue neurofilament gene expression in Id2-positive cells (Fig. 5E), indicating that activated pRb rescued not only Id2-induced apoptosis but also the Id2-induced inhibition of neuronal gene expression. Thus, inhibition of the pRb family does not on its own inhibit induction of neuronal genes in cortical progenitors, but activation of pRb is sufficient to rescue the Id2-induced loss of neuronal gene expression.
DISCUSSION
In this paper, we have tested the hypothesis that HLH proteins collaborate with pRb to regulate the progenitor-to-neuron transition. Data presented here support four major conclusions. First, these studies support the hypothesis that transcription by positively acting bHLHs is essential for induction of neuronal gene expression in cortical progenitor cells; when bHLH transcriptional activity is inhibited by overexpression of Id2, progenitor cells do not express neuronal genes. Second, these studies demonstrate that perturbation of the progenitor-to-neuron transition by overexpression of Id2 leads to apoptosis. We propose that this phenotype is attributable to inhibitory interactions between Id2 and endogenous pRb, because (1) this apoptotic phenotype is similar to that observed when the pRb family is ablated in these same progenitor cells (Slack et al., 1998), and (2) constitutively activated pRb can rescue the Id2-driven apoptosis. Third, our data indicate that once a progenitor cell has become a postmitotic cortical neuron, then Id2 overexpression does not inhibit neuronal gene expression or cause neuronal apoptosis, suggesting that the bHLH transcription factors and pRb are required for induction of the neuronal phenotype, but not necessarily for its maintenance. Fourth, our data demonstrate that constitutively activated pRb can rescue both the Id2-driven apoptosis and the inhibition of neuronal genes, suggesting that interactions between pRb and HLHs are likely to regulate both terminal mitosis and the induction of neuronal gene expression. Together, these studies indicate that collaborative interactions between the pRb family and HLHs play a key role in regulating cortical neurogenesis and suggest that endogenous Id2 may well be critical in enacting the progenitor-to-neuron decision.
The idea that interactions between the pRb family and HLH proteins may regulate cellular differentiation is a particularly attractive one when considering neurogenesis in the CNS. With a few exceptions, such as the precursors that migrate in the rostral migratory stream from the lateral ventricles to the olfactory bulb (Menezes et al., 1995), when CNS progenitor cells become postmitotic neurons, they coordinately undergo terminal mitosis and induce neuronal gene expression (Gloster et al., 1999). Moreover, when one aspect of this process is perturbed, such as occurs after functional ablation of the pRb family, the end result is cellular apoptosis (Slack et al., 1998). This integral relationship between terminal mitosis and neuronal gene expression suggests that the same molecules may well regulate both events. Data presented here suggest that Id2, which can interact with and inhibit pRb-driven terminal mitosis and bHLH-driven induction of neuronal gene expression, might be one such molecule.
It is likely that Id2 overexpression inhibits the induction of neuronal gene expression by directly binding to the ubiquitous E2A bHLH transcription factors (for review, see Norton et al., 1998), thereby titrating out the requisite binding partners for neurogenic bHLHs, such as Mash-1 (Johnson et al., 1990), neurogenins (Ma et al., 1996; Sommer et al., 1996), or NeuroD (Lee et al., 1995), although Id2 may also directly bind the neurogenic bHLHs themselves (Langlands et al., 1997). A similar mechanism is thought to underlie the ability of Id1 and Id3 to inhibit myogenesis (Jen et al., 1992; Atherton et al., 1996) and adipogenesis (Moldes et al., 1997) in cell lines and of Id1 (Sun, 1994) and Id2 (Morrow et al., 1999) to inhibit B cell and T cell developmentin vivo. Genetic evidence that endogenous Id proteins play a similar inhibitory role in the nervous system derives fromDrosophila (Ellis et al., 1990) (for review, seeCampos-Ortega, 1993; Jan and Jan, 1993) in which the extramachrochaete gene product (the Drosophilaequivalent of Id) antagonizes both Daughterless (DrosophilaE2A) and achaete-scute (Drosophila Mash) bHLH proteins, which are involved in sex determination and neurogenesis. Similarly, Id2 was also suggested to be involved in cell fate decisions in the chick neural crest (Martinsen and Bronner-Fraser, 1998). More recently, the targeted deletion of the Id1 and Id3 genes was shown to cause altered nervous system development accompanied by perturbed angiogenesis in the embryonic brain (Lyden et al., 1999). Deletion of either gene on its own produced no detectable phenotype, presumably because of compensation by different family members. An essential role for Id2 during development is also indicated by the recent report of Id2−/− mice, which display arrested development and lack lymph nodes and Peyer's patches (Yokota et al., 1999). Although no information is yet available regarding the nervous system of these animals and the underlying cellular deficit is unclear, these data do indicate that Id2 is essential for normal mammalian development.
Studies presented here also strongly support the idea that endogenous Id2 interacts with the pRb family to regulate neurogenesis and cell survival. As confirmed here, previous studies demonstrated that Id2 binds to the hypophosphorylated form of pRb and inhibits its ability to mediate growth arrest (Iavarone et al., 1994;Lasorella et al., 1996). Because pRb is required for cortical progenitors to exit the cell cycle and survive (Slack et al., 1998), then this finding predicts that increased Id2 would mimic the phenotype observed when the pRb family is ablated. In fact, this is what we observe; cortical progenitor cells, but not postmitotic neurons, undergo apoptosis when Id2 is overexpressed. Moreover, coexpression of a mutant pRb that is constitutively hypophosphorylated rescues this apoptotic phenotype, presumably by sequestering Id2 and allowing progenitor cells to undergo terminal mitosis. Thus, although our studies do not directly demonstrate that endogenous Id2 regulates the ability of pRb to mediate terminal mitosis in progenitor cells, they suggest that such may be the case.
Why does constitutively activated pRb rescue the deficit in neuronal gene expression induced by Id2 when (1) the hypophosphorylated pRb mutant does not, on its own, induce neuronal gene expression (shown here), and (2) functional ablation of the pRb family has no effect on induction of the same neuron-specific genes (Slack et al., 1998)? We propose that hypophosphorylated pRb, by binding to Id2, inhibits its ability to bind to and inhibit positively acting bHLH transcription factors. In this model, a cycling progenitor cell would have relatively high levels of Id2, and pRb would be primarily present as a hyperphosphorylated protein. At this point in time, Id2 levels would be sufficient to bind and inhibit any positively acting bHLH transcription factors and to bind and inhibit any hypophosphorylated pRb. In response to an as-yet undefined cue to become a postmitotic neuron, Id2 levels would decrease and/or Id2 activity would be altered by phosphorylation (Nagata et al., 1995; Hara et al., 1997), and the ratio of hypophosphorylated to hyperphosphorylated pRb would increase. Id2 would then no longer be able to sequester all of the hypophosphorylated pRb, thereby allowing pRb to “stop” the cell cycle. Moreover, the decrease in Id2 levels–activity, coupled with the increase in hypophosphorylated pRb, would ensure that little or no Id2 was available to bind and sequester E2A bHLHs, which could then form productive transcription complexes with neurogenic bHLHs. Thus, pRb, Id2, and neurogenic bHLHs would all collaborate to enact the neuronal commitment decision, once such a decision had been made. Such a mechanism would ensure (1) the coordinate induction of neuronal gene expression and terminal mitosis, and (2) the apoptosis of any progenitor cell that failed to properly enact this transition.
Footnotes
This work was supported by a grant from the Canadian Medical Research Council (MRC). H.E.-B. was supported by a Natural Sciences and Engineering Research Council of Canada (NSERC) fellowship during the course of these studies, and F.B.-H. was supported by an NSERC studentship. F.D.M. is an MRC Senior Scientist and a Killam Scholar. We thank Farid Arab-Said for assistance making the Id2 adenovirus, Xiuming Yang for performing some of the RT-PCR analysis, and all of the members of the Miller laboratory for helpful discussions.
J.G.T. and H.E.-B., and F.B.-H. contributed equally to this work.
Correspondence should be addressed to Freda Miller, Center for Neuronal Survival, Montreal Neurological Institute, 3801 rue University, Montreal, Quebec, Canada H3A 2B4. E-mail: mdfm@musica.mcgill.ca.
Dr. El-Bizri's present address: Novartis Pharmaceuticals, East Hanover, NJ 07936.
REFERENCES
- 1.Aloyz RS, Bamji SX, Pozniak CD, Toma JG, Atwal J, Kaplan DR, Miller FD. p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J Cell Biol. 1998;143:1691–1703. doi: 10.1083/jcb.143.6.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atherton GT, Travers H, Deed R, Norton JD. Regulation of cell differentiation in C2C12 myoblasts by the Id3 helix-loop-helix protein. Cell Growth Differ. 1996;7:1059–1066. [PubMed] [Google Scholar]
- 3.Bamji SX, Miller FD. Comparison of the expression of a Tα1 α-tubulin transgene and Tα1 α-tubulin mRNA in the mature central nervous system. J Comp Neurol. 1996;374:52–69. doi: 10.1002/(SICI)1096-9861(19961007)374:1<52::AID-CNE4>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 4.Ben-Arie N, Bellen HJ, Armstrong DL, McCall AE, Gordadze PR, Guo Q, Matzuk MM, Zoghbi HY. Math1 is essential for genesis of cerebellar granule neurons. Nature. 1997;390:169–172. doi: 10.1038/36579. [DOI] [PubMed] [Google Scholar]
- 5.Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H. The protein Id: a negative regulatory of helix-loop-helix binding proteins. Cell. 1990;61:49–59. doi: 10.1016/0092-8674(90)90214-y. [DOI] [PubMed] [Google Scholar]
- 6.Bett AJ, Haddara W, Prevec L, Graham FL. An efficient and flexible system for construction of adenovirus vectors with insertions or deletions in early regions 1 and 3. Proc Natl Acad Sci USA. 1994;91:8802–8806. doi: 10.1073/pnas.91.19.8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biggs J, Murphy E, Israel M. A human Id-like helix-loop-helix protein expressed during early development. Proc Natl Acad Sci USA. 1992;89:1512–1516. doi: 10.1073/pnas.89.4.1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campos-Ortega JA. Mechanisms of early neurogenesis in Drosophila melanogaster. J Neurobiol. 1993;24:1305–1327. doi: 10.1002/neu.480241005. [DOI] [PubMed] [Google Scholar]
- 9.Casarosa S, Fode C, Guillemot F. Mash1 regulates neurogenesis in the ventral telencephalon. Development. 1999;126:525–534. doi: 10.1242/dev.126.3.525. [DOI] [PubMed] [Google Scholar]
- 10.Chang MW, Barr E, Seltzer J, Jiang YQ, Nabel FJ, Nabel EG, Parmacek MS, Leiden JM. Cytostatic gene therapy for vascular proliferative disorders with a constitutively active form of the retinoblastoma gene product. Science. 1995;267:518–522. doi: 10.1126/science.7824950. [DOI] [PubMed] [Google Scholar]
- 11.Christy BA, Sanders LK, Lau LF, Copeland NG, Jenkins NA, Nathans D. An Id-related helix-loop-helix protein encoded by a growth factor-inducible gene. Proc Natl Acad Sci USA. 1991;88:1815–1819. doi: 10.1073/pnas.88.5.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clarke AR, Maandag ER, Van Roon M, Van der Lugt NMT, Van der Valk M, Hopper MI, Berns A, Te Riele H. Requirement for a functional Rb1 gene in murine development. Nature. 1992;359:328–330. doi: 10.1038/359328a0. [DOI] [PubMed] [Google Scholar]
- 13.Deed RW, Armitage S, Norton JD. Nuclear localization and regulation of Id protein through an E protein-mediated chaperone mechanism. J Biol Chem. 1996;271:23603–23606. doi: 10.1074/jbc.271.39.23603. [DOI] [PubMed] [Google Scholar]
- 14.Ellis HM, Spann DR, Posakony JW. Extramacrochaetae, a negative regulator of sensory organ development in Drosophila, defines a new class of helix-loop-helix proteins. Cell. 1990;61:27–38. doi: 10.1016/0092-8674(90)90212-w. [DOI] [PubMed] [Google Scholar]
- 15.Fode C, Gradwohl G, Morin X, Dierich A, LeMeur M, Goridis C, Guillemot F. The bHLH protein Neurogenin 2 is a determination factor for epibrachial placode-derived sensory neurons. Neuron. 1998;20:483–494. doi: 10.1016/s0896-6273(00)80989-7. [DOI] [PubMed] [Google Scholar]
- 16.Gloster A, Wu W, Speelman A, Weiss S, Causing C, Pozniak C, Reynolds B, Chang E, Toma JG, Miller FD. The Tα1 α-tubulin promoter specifies gene expression as a function of neuronal growth and regeneration in transgenic mice. J Neurosci. 1994;14:7319–7330. doi: 10.1523/JNEUROSCI.14-12-07319.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gloster A, El-Bizri H, Bamji SX, Rogers D, Miller FD. Early induction of Tα1 α-tubulin transcription in neurons of the developing nervous system. J Comp Neurol. 1999;405:45–60. doi: 10.1002/(sici)1096-9861(19990301)405:1<45::aid-cne4>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 18.Graham FL, Prevec L. Manipulation of adenovirus vectors. In: Murray EJ, editor. Methods in molecular biology. Humana; Totowa, NJ: 1991. pp. 109–128. [DOI] [PubMed] [Google Scholar]
- 19.Gu W, Schneider JW, Condorelli G, Kaushal S, Mahdavi V, Nadal-Ginard B. Interaction of myogenic factors and the retinoblastoma protein mediates muscle cell commitment and differentiation. Cell. 1993;72:309–324. doi: 10.1016/0092-8674(93)90110-c. [DOI] [PubMed] [Google Scholar]
- 20.Guillemot F, Lo LC, Johnson JE, Auerbach A, Anderson DJ, Joyner AL. Mammalian achaete-scute homolog-1 is required for the early development of olfactory and autonomic neurons. Cell. 1993;75:463–476. doi: 10.1016/0092-8674(93)90381-y. [DOI] [PubMed] [Google Scholar]
- 21.Hara E, Hall M, Peters G. Cdk2-dependent phosphorylation of Id2 modulates activity of E2A-related transcription factors. EMBO J. 1997;16:332–342. doi: 10.1093/emboj/16.2.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iavarone A, Garg P, Lasorella A, Hsu J, Israel MA. The helix-loop-helix protein Id-2 enhances cell proliferation and binds to the retinoblastoma protein. Genes Dev. 1994;8:1270–1284. doi: 10.1101/gad.8.11.1270. [DOI] [PubMed] [Google Scholar]
- 23.Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- 24.Jan YN, Jan LY. HLH proteins, fly neurogenesis, and vertebrate myogenesis. Cell. 1993;75:827–830. doi: 10.1016/0092-8674(93)90525-u. [DOI] [PubMed] [Google Scholar]
- 25.Jen Y, Weintraub H, Benezra R. Overexpression of Id protein inhibits the muscle differentiation program: in vivo association of Id with E2A proteins. Genes Dev. 1992;6:1466–1479. doi: 10.1101/gad.6.8.1466. [DOI] [PubMed] [Google Scholar]
- 26.Jen Y, Manova K, Benezra R. Each member of the Id gene family exhibits a unique expression pattern in mouse gastrulation and neurogenesis. Dev Dyn. 1997;208:92–106. doi: 10.1002/(SICI)1097-0177(199701)208:1<92::AID-AJA9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 27.Johnson JE, Birren SJ, Anderson DJ. Two rat homologues of Drosophila achaete-scute specifically expressed in neuronal precursors. Nature. 1990;346:858–861. doi: 10.1038/346858a0. [DOI] [PubMed] [Google Scholar]
- 28.Kageyama R, Nakanishi S. Helix-loop-helix factors in growth and differentiation of the vertebrate nervous system. Curr Opin Genes Dev. 1997;7:659–665. doi: 10.1016/s0959-437x(97)80014-7. [DOI] [PubMed] [Google Scholar]
- 29.Knusel B, Rabin SJ, Hefti F, Kaplan DR. Regulated neurotrophin receptor responsiveness during neuronal migration and early differentiation. J Neurosci. 1994;14:1542–1554. doi: 10.1523/JNEUROSCI.14-03-01542.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langlands K, Yin X, Anand G, Prochownik EV. Differential interactions of Id proteins with basic-helix-loop-helix transcription factors. J Biol Chem. 1997;272:19785–19793. doi: 10.1074/jbc.272.32.19785. [DOI] [PubMed] [Google Scholar]
- 31.Lasorella A, Iavarone A, Israel MA. Id2 specifically alters regulation of the cell cycle by tumor suppressor proteins. Mol Cell Biol. 1996;16:2570–2578. doi: 10.1128/mcb.16.6.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lassar AB, Davis RL, Wright WE, Kadesch T, Murre C, Voronova A, Baltimore D, Weintraub H. Functional activity of myogenic HLH proteins requires hetero-oligomerization with E12/E47-like proteins in vivo. Cell. 1991;66:305–315. doi: 10.1016/0092-8674(91)90620-e. [DOI] [PubMed] [Google Scholar]
- 33.Lee EHP, Chang CY, Hu N, Wang YCJ, Lai CC, Herrup K, Lee W, Bradley A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–294. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- 34.Lee EHP, Hu N, Yuan SSF, Cox LA, Bradley A, Lee W, Herrup K. Dual roles of the retinoblastoma protein in cell cycle regulation and neuron differentiation. Genes Dev. 1994;8:2008–2021. doi: 10.1101/gad.8.17.2008. [DOI] [PubMed] [Google Scholar]
- 35.Lee JE. Basic helix-loop-helix genes in neural development. Curr Opin Neurobiol. 1998;7:13–20. doi: 10.1016/s0959-4388(97)80115-8. [DOI] [PubMed] [Google Scholar]
- 36.Lee JE, Hollenberg SM, Snider L, Turner DL, Lipnick N, Weintraub H. Conversion of Xenopus ectoderm into neurons by NeuroD, a basic helix-loop-helix protein. Science. 1995;268:836–844. doi: 10.1126/science.7754368. [DOI] [PubMed] [Google Scholar]
- 37.Lyden D, Young AZ, Zagzag D, Yan W, Gerald W, O'Reilly R, Badfer BL, Hynes RO, Zhuang Y, Manova K, Benezra R. Id1 and Id3 are required for neurogenesis, angiogensis and vascularization of tumour xenografts. Nature. 1999;401:670–677. doi: 10.1038/44334. [DOI] [PubMed] [Google Scholar]
- 38.Ma Q, Kintner C, Anderson DJ. Identification of neurogenin, a vertebrate neuronal determination gene. Cell. 1996;87:43–52. doi: 10.1016/s0092-8674(00)81321-5. [DOI] [PubMed] [Google Scholar]
- 39.Ma Q, Chen Z, del Barco Barrantes I, de la Pompa JL, Anderson DJ. Neurogenin I is essential for the determination of neuronal precursors for proximal cranial sensory ganglia. Neuron. 1998;20:469–482. doi: 10.1016/s0896-6273(00)80988-5. [DOI] [PubMed] [Google Scholar]
- 40.Martinsen BJ, Bronner-Fraser M. Neural crest specification regulated by the helix-loop-helix repressor Id2. Science. 1998;281:988–991. doi: 10.1126/science.281.5379.988. [DOI] [PubMed] [Google Scholar]
- 41.Mazzoni IE, Said FA, Aloyz R, Miller FD, Kaplan D. Ras regulates sympathetic neuron survival by suppressing the p53-mediated cell death pathway. J Neurosci. 1999;19:9716–9727. doi: 10.1523/JNEUROSCI.19-22-09716.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Menezes JRL, Smith CM, Nelson KC, Luskin MB. The division of neuronal progenitor cells during migration in the neonatal mammalian forebrain. Mol Cell Neurosci. 1995;6:496–508. doi: 10.1006/mcne.1995.0002. [DOI] [PubMed] [Google Scholar]
- 43.Miyata T, Maeda T, Lee JE. NeuroD is required for differentiation of the granule cells in the cerebellum and hippocampus. Genes Dev. 1999;13:1647–1652. doi: 10.1101/gad.13.13.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moldes M, Lasnier F, Feve B, Pairault J, Djian P. Id3 prevents differentiation of preadipose cells. Mol Cell Biol. 1997;17:1796–1804. doi: 10.1128/mcb.17.4.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morrow MA, Mayer EW, Perez CA, Adelam M, Siu G. Overexpression of the helix-loop-helix protein Id2 blocks T cell development at multiple stages. Mol Immunol. 1999;36:491–503. doi: 10.1016/s0161-5890(99)00071-1. [DOI] [PubMed] [Google Scholar]
- 46.Nagata Y, Shoji W, Obinata M, Todokoro K. Phosphorylation of helix-loop-helix proteins Id1, Id2, and Id3. Biochem Biophys Res Commun. 1995;207:916–926. doi: 10.1006/bbrc.1995.1273. [DOI] [PubMed] [Google Scholar]
- 47.Neuman T, Keen A, Zuber MX, Kristjansson GI, Gruss P, Nornes HO. Neuronal expression of regulatory helix-loop-helix factor Id2 gene in mouse. Dev Biol. 1993;160:186–195. doi: 10.1006/dbio.1993.1297. [DOI] [PubMed] [Google Scholar]
- 48.Norton JD, Deed RW, Craggs G, Sablitzky F. Id helix-loop-helix proteins in cell growth and differentiation. Trends Cell Biol. 1998;8:58–65. [PubMed] [Google Scholar]
- 49.Riechmann V, Sablitzky F. Mutually exclusive expression of two dominant-negative helix-loop-helix genes, Id4 and Id3, in the developing brain of the mouse suggests distinct regulatory roles of these dnHLH protein during cellular proliferation and differentiation of the nervous system. Cell Growth Differ. 1995;6:837–843. [PubMed] [Google Scholar]
- 50.Schagger H, von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 51.Schwab MH, Bartholomae A, Heimrich B, Feldmeyer D, Druffel-Augustin S, Goebbels S, Naya FJ, Zhao S, Frotscher M, Tsai MJ, Navi KA. Neuronal basic helix-loop-helix proteins (NEX and BETA2/NeuroD) regulate terminal granule cell differentiation in the hippocampus. J Neurosci. 2000;20:3714–3724. doi: 10.1523/JNEUROSCI.20-10-03714.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Slack RS, Miller FD. Retinoblastoma in mouse neural development. Dev Genet. 1996;18:81–91. doi: 10.1002/(SICI)1520-6408(1996)18:1<81::AID-DVG9>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 53.Slack RS, Belliveau DJ, Rosenberg M, Atwal J, Aloyz R, Lochmuller H, Hagighi A, Lach B, Seth P, Cooper E, Miller FD. Adenovirus-mediated gene transfer of the tumor suppressor, p53 induces apoptosis in postmitotic neurons. J Cell Biol. 1996;135:1085–1096. doi: 10.1083/jcb.135.4.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Slack RS, El-Bizri H, Wong J, Belliveau DJ, Miller FD. A critical temporal requirement for the retinoblastoma protein family during neuronal determination. J Cell Biol. 1998;140:1497–1509. doi: 10.1083/jcb.140.6.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sommer L, Ma Q, Anderson DJ. Neurogenins, a novel family of atonal-related bHLH transcription factors, are putative mammalian neuronal determination genes that reveal progenitor cell heterogeneity in the developing CNS and PNS. Mol Cell Neurosci. 1996;8:221–241. doi: 10.1006/mcne.1996.0060. [DOI] [PubMed] [Google Scholar]
- 56.Sun XH. Constitutive expression of the Id1 gene impairs mouse B cell development. Cell. 1994;79:893–900. doi: 10.1016/0092-8674(94)90078-7. [DOI] [PubMed] [Google Scholar]
- 57.Sun XH, Copeland NG, Jenkins NA, Baltimore D. Id proteins Id1 and Id2 selectively inhibit DNA binding by one class of helix-loop-helix proteins. Mol Cell Biol. 1991;11:5603–5611. doi: 10.1128/mcb.11.11.5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Torii M, Matsuzaki F, Osumi N, Kaibuchi K, Nakamura S, Casarose S, Guillemot F, Nakafuku M. Transcription factors Mash-1 and Prox-1 delineate early steps in differentiation of neural stem cells in the developing central nervous system. Development. 1999;126:443–456. doi: 10.1242/dev.126.3.443. [DOI] [PubMed] [Google Scholar]
- 59.Tzeng S-F, de Vellis J. Id1, Id2, and Id3 gene expression in neural cells during development. Glia. 1998;24:372–381. doi: 10.1002/(sici)1098-1136(199812)24:4<372::aid-glia2>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 60.Vaillant AR, Mazzoni I, Tudan C, Boudreau M, Kaplan DR, Miller FD. Depolarization and neurotrophins converge on the phosphatidylinositol 3-kinase–Akt pathway to synergistically regulate neuronal survival. J Cell Biol. 1999;146:955–966. doi: 10.1083/jcb.146.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weintraub H. The MyoD family and myogenesis: redundancy, networks, and thresholds. Cell. 1993;75:1241–1244. doi: 10.1016/0092-8674(93)90610-3. [DOI] [PubMed] [Google Scholar]
- 62.Yokota Y, Mansouri A, Mori S, Sugawara S, Adachi S, Nishikawa S, Gruss P. Development of peripheral lymphoid organs and natural killer cells depends on the helix-loop-helix inhibitor Id2. Nature. 1999;37:702–706. doi: 10.1038/17812. [DOI] [PubMed] [Google Scholar]