

Significance
Human papillomaviruses (HPVs) cause 5% of all cancers, including cervical cancers and a growing number of oropharyngeal cancers, which are reaching epidemic proportion. While HPV oncogenes promote the acquisition of cancer hallmarks such as sustained proliferation and resistance to cell death by targeting the p53 and pRb pathways, the mechanism by which they induce genomic instability leading to cellular transformation remains largely unknown. This paper describes how the HPV oncoprotein E7 directly impedes the cellular response to DNA double-strand breaks by interacting with a previously uncharacterized domain of the E3 ubiquitin ligase RNF168. As the function of RNF168 is essential for proper DNA repair, our findings reveal a mechanism by which HPV induces genomic instability and fuels cancer progression.
Keywords: DNA double-strand break, high-risk human papillomavirus, 53BP1 nuclear bodies, E7 protein, RNF168
Abstract
High-risk human papillomaviruses (HR-HPVs) promote cervical cancer as well as a subset of anogenital and head and neck cancers. Due to their limited coding capacity, HPVs hijack the host cell’s DNA replication and repair machineries to replicate their own genomes. How this host–pathogen interaction contributes to genomic instability is unknown. Here, we report that HPV-infected cancer cells express high levels of RNF168, an E3 ubiquitin ligase that is critical for proper DNA repair following DNA double-strand breaks, and accumulate high numbers of 53BP1 nuclear bodies, a marker of genomic instability induced by replication stress. We describe a mechanism by which HPV E7 subverts the function of RNF168 at DNA double-strand breaks, providing a rationale for increased homology-directed recombination in E6/E7-expressing cervical cancer cells. By targeting a new regulatory domain of RNF168, E7 binds directly to the E3 ligase without affecting its enzymatic activity. As RNF168 knockdown impairs viral genome amplification in differentiated keratinocytes, we propose that E7 hijacks the E3 ligase to promote the viral replicative cycle. This study reveals a mechanism by which tumor viruses reshape the cellular response to DNA damage by manipulating RNF168-dependent ubiquitin signaling. Importantly, our findings reveal a pathway by which HPV may promote the genomic instability that drives oncogenesis.
Oncogenic viruses are estimated to contribute as much as 12% of all cancers worldwide (1, 2), and approximately 5% are specifically caused by a small group of human papillomaviruses (HPVs) classified as high-risk HPVs (HR-HPVs). These subtypes of HPV belong to the alpha genus and infect basal cells in the oral and genital mucosal epithelia (3). Accordingly, HR-HPVs promote the development of 99% of cervical cancers as well as a subset of anogenital and head and neck cancers, while low-risk HPVs of the same family induce the formation of benign genital warts (1, 4). In the past decade, the subset of oropharyngeal squamous cell carcinoma (a subtype of head and neck cancer) that are HPV-positive has drastically increased, such that the virus is now detected in 60 to 80% of cases (5–7). Although HPV is required for the development of these cancers, only a small fraction of infected patients will progress to cancer following persistent infection, indicating that additional mutations are required to promote carcinogenesis.
The genome of HPV is a circular double-stranded DNA of approximately 8 kb, which encodes 7 early proteins, E1, E2, E4, E5, E6, E7, and E8, as well as 2 structural proteins, L1 and L2, that are required for the formation of the capsid (8). In infected cells, the replication of the viral genome as an extra chromosomal element relies on the collaboration between the viral proteins E1 and E2 as well as the DNA replication machinery of the host cell. Intriguingly, an increasing amount of evidence now places the host cell DNA repair machinery as an essential player during viral replication (9, 10). For instance, proteins of the DNA double-strand break (DSB) signaling and repair pathways, such as ataxia telangiectasia mutated (ATM) protein and its substrate H2AX (γ-H2AX), the MRE11-RAD50-NBS1 (MRN) complex, p53-binding protein 1 (53BP1), the breast cancer susceptibility gene 1 (BRCA1), the replication protein A (RPA), and RAD51 (11–14), accumulate in nuclear structures termed “viral replication centers” (10). While the exact mechanism by which the DNA repair proteins contribute to viral replication is unknown, small molecules that inhibit the kinase activity of ATM, the exonuclease activity of MRN, or the interaction of RAD51 with DNA abolish viral DNA amplification upon the differentiation of infected keratinocytes, highlighting their role during viral life cycle (12, 15, 16). Consistent with this, an elegant study by Mehta and Laimins showed that the viral genome accumulates fewer DNA breaks than the host genome during productive replication (17).
DSB signaling and repair pathways are central to the maintenance of genome integrity and cell viability, as inappropriately repaired DSBs lead to genomic instability and mutations, a hallmark of cancer (18). Furthermore, genomic instability can lead to the acquisition of many of the other hallmarks of cancer, including resistance to programmed cell death, proliferation, and dissemination. Ultimately, if cells are unable to repair DSBs, apoptosis or senescence is triggered (19). Consequently, cells have evolved a highly orchestrated response that enables the detection, signaling, and repair of breaks. This process is regulated in a cell cycle-dependent manner and activates 2 main types of DNA repair, nonhomologous end-joining (NHEJ) and homology-directed recombination repair (HDR) (20). In the G1 phase of the cell cycle, cells rely on canonical NHEJ to religate DNA ends after limited end processing, while HDR is activated in the S/G2 phases to promote repair that is guided by the template created through newly replicated DNA. The latter process is driven by extensive 5′ to 3′ end resection at the break. Following DSBs, recruitment of DNA repair proteins is initiated locally by ATM-dependent phosphorylation and coordinated by ubiquitylation of chromatin surrounding the break (21, 22). The latter step is accomplished by the sequential recruitment of the E3 ligases RING finger protein 8 (RNF8) and RNF168, a process that culminates in the ubiquitylation of the N-terminal tail of H2A by RNF168 (23). This histone mark then amplifies the recruitment of RNF168 and also drives the recruitment of downstream effectors such as 53BP1 and RNF169, a paralog of RNF168 that counteracts the accumulation of 53BP1 at DSBs (24–28). By counteracting the recruitment of BRCA1 at DSBs, the recruitment of 53BP1 and its downstream effectors (RIF1 and the shieldin complex) promote DNA repair pathway by NHEJ by inhibiting resection of DNA ends (20, 29). RNF168 plays an essential role in the maintenance of genomic integrity, as knockout mice are predisposed to cancer development (30), and patients who lack functional RNF168 suffer from RIDDLE syndrome, a disease associated with immunodeficiency and radiosensitivity (31).
In HPV-positive (HPV+) primary tumors, RNF168 and 53BP1 accumulate in abnormally large foci, supporting the hypothesis that one or more HPV-gene products subverts the DSB signaling and repair pathway in these cells (32). Here, we show that the expression of RNF168 is specifically increased in HPV+ cancers and that HPV-transformed cell lines accumulate large foci of 53BP1 in the G1 phase of the cell cycle (53BP1 nuclear bodies; 53BP1-NBs), a marker of DNA damage that occurred in the previous S-phase. To gain insight into the molecular events that promote genomic instability in HPV+ cells, we sought to determine the impact of viral proteins on DSB signaling. Our results unexpectedly revealed that the E7 oncoprotein from HR-HPV directly targets a previously uncharacterized domain of RNF168 (herein termed the “E7-Binding Domain”; E7BD). We showed that the C-terminal domain of E7 mediates this interaction independently of DSB signaling, and that RNF168 is essential to promote viral genome amplification in differentiating keratinocytes. Importantly, we found that E7 hinders the function of the E3 ligase at DSBs, revealing a mechanism by which a viral oncoprotein induces genomic instability.
Results
RNF168 Is Expressed at High Levels in HPV+ Cancers.
As HPV+ tumors exhibit large nuclear foci of RNF168 and 53BP1, we first sought to determine if the levels of these proteins are specifically increased in these tumors. Strikingly, analysis of RNF168 mRNA expression in cancer samples from the cBioPortal database (33, 34) revealed that cervical and head and neck are among the few types of cancers in which the E3 ligase is overexpressed (SI Appendix, Fig. S1A). Next, we analyzed the Illumina HiSeq RNA-seq data from The Cancer Genome Atlas (TCGA) for 297 cervical carcinoma (CESC; 278 HPV+ and 19 HPV−) and 558 head and neck squamous cell carcinoma (HNSC; 73 HPV+, 442 HPV−, and 43 normal-adjacent tissues) cohorts for expression of RNF168 and 53BP1 (Fig. 1A). For comparison, the expression levels of 2 other ubiquitin ligases that also accumulate on chromatin following DSBs, RNF8 and RNF169, were analyzed. Strikingly, both types of HPV-related cancers were found to express high levels of RNF168 mRNA, as shown by the normalized RNA-seq absolute read count values. This increase is highly significant when comparing HPV+ and HPV− samples from the HNSC cohort (P ≤ 0.0001) and is observed independently of the HR-HPV type detected in these samples (Fig. 1A and SI Appendix, Fig. S1B). In samples from the HNSC cohort, expression levels of 53BP1 were also significantly increased, although this was not the case in the CESC cohort. In contrast to RNF168 and 53BP1, the expression levels for RNF8 and RNF169 were not increased in HPV+ samples. In fact, RNF8 expression was markedly reduced in HPV+ samples compared with HPV− tumors, indicating that the increased expression of RNF168 does not reflect a general up-regulation of DNA repair genes in these cancers. Consistent with these results, analysis of a panel of HPV+ cell lines isolated from cervical (SiHa, HeLa, CaSki) and head and neck carcinomas (UM-SCC-47; hereafter referred to as SCC47) revealed that the levels of RNF168 are markedly increased in HPV+ cell lines compared with HPV− CESC (C33-A) and HNSC (UM-SCC-1; referred to hereafter as SCC1) cell lines (Fig. 1B and SI Appendix, Fig. S1 C and D). The later observation suggested that viral proteins encoded by these integrated genomes lead to increased expression of RNF168 in HPV+ cells.
Fig. 1.

Expression and localization of RNF168 and 53BP1 in HPV+ carcinoma. (A) Expression of the indicated genes in cervical cancers (CESC) and head and neck cancers (HNSC) stratified by human papillomavirus (HPV) status. Normalized RNA-seq data extracted from The Cancer Genome Atlas (TCGA) database are presented. For HNSC, normal-adjacent control tissues were used as an extra control (black). Statistical significance was assessed by a Mann–Whitney U test. (B) Whole cell extracts (WCE) were analyzed by immunoblotting with the indicated antibodies. KAP1 and Tubulin were used as loading controls. (C) Representative images of 53BP1 and RNF168 foci observed in the indicated cell lines. Cells were fixed and processed for γ-H2AX, 53BP1, and RNF168 immunofluorescence. (Scale bar, 5 µm.) (D) Quantification of 53BP1 (Upper) and RNF168 (Lower) colocalizing with γ-H2AX. Data are presented as the mean ± SEM (n = 3; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001). ns, not significant. (E) Representative images of GFP-53BP1FFR and BRCA1 in CaSki cells treated or not with 1 Gy. Cyclin A staining was used to identify cells in S/G2 phase (positive) and G1 phase (negative).
When analyzed by immunochemistry, parafilm-embedded HPV+ tumor tissues were found to accumulate large foci of RNF168 and 53BP1 (32), a phenotype that is reminiscent of the formation of 53BP1-NBs, a marker of genomic instability induced by replication stress. These nuclear structures shield DNA ends from further processing until they reach the next late S phase, in which they will be repaired by RAD52-mediated repair (35–38). To determine whether the appearance of these foci is related to the HPV status of these cells, we analyzed the localization of RNF168 and 53BP1 in the panel of HPV+ and HPV− cells described here earlier by immunofluorescence microscopy. In these experiments, we found that the number of large 53BP1 bodies was significantly increased in HPV+ CESC and HNSC cell lines and directly correlated to the number of integrated viral genomes (Fig. 1 C and D and SI Appendix, Fig. S1E). These foci were present only during the G1 phase of the cell cycle (marked by the absence of cyclin A staining; Fig. 1E and SI Appendix, Fig. S1F) and were abolished by depletion of RNF168 (SI Appendix, Fig. S1 G–I), indicating that they correspond to 53BP1-NBs. Consistently, we found that BRCA1, but not the DNA repair factor RAD51, colocalizes with 53BP1 in these bodies (Fig. 1E and SI Appendix, Fig. S1J). Taken together, these results indicate that increased expression of RNF168 and accumulation of genomic instability are associated with the presence of HPV in human cancer cells.
HPV E7 Proteins Directly Interact with RNF168.
Many DSB signaling and repair proteins accumulate at viral replication centers during the viral replication cycle (10). Combined with the results described here earlier, this raises the possibility that RNF168-mediated signaling is directly subverted by one or more viral gene products. To explore this possibility, we used a LacO/LacR system that recapitulates the recruitment of DSB repair factors downstream of γ-H2AX in the absence of DNA damage (Fig. 2A) (24, 39). Specifically, we investigated if the viral replication proteins E1 and E2, E4, or the oncoproteins E6 and E7 from the prevalent HR-HPV31 could promote the recruitment of DSB-signaling proteins at the LacO-array, visible as a single nuclear focus in this cell-based assay. E5 and L1/L2 proteins were not tested, as they are, respectively, a transmembrane protein and capsid structural proteins only expressed in terminally differentiated keratinocytes (4). In this assay, mCherry-LacR fused to the C-terminal DNA cleavage domain of the endonuclease FOKI was used as positive control to monitor the recruitment of DSB-signaling factor at the LacO-array. Surprisingly, we found that mCherry-LacR-E6 and -E7 promoted a low but significant accumulation of 53BP1 at the LacO-array (Fig. 2 B and C and SI Appendix, Fig. S2 A and G). The absence of γ-H2AX at the array 6 h posttransfection shows that E6 and E7 recruit 53BP1 independently of DSB signaling in these conditions, unlike E2, which triggers the accumulation of the histone mark at the array, potentially through its ability to interact with TOPBP1, a factor that promotes ATR activation (40) (Fig. 2D and SI Appendix, Fig. S2B). Consistent with an interaction between the viral proteins and RNF168, we found that FLAG-RNF168, but not FLAG-RNF8, colocalized with the viral oncoproteins at the array in as many as 80% of the cells (Fig. 2 E and F and SI Appendix, Figs. S2 C, D, and H). Furthermore, while depletion of RNF8 drastically reduced the recruitment of 53BP1 to DSBs induced by targeting the nuclease FOKI at the LacO-array, it increased the amount of 53BP1 colocalizing with mCherry-LacR-E6 and -E7 (Fig. 2G and SI Appendix, Fig. S2 E and J). As RNF168 is a limiting factor during DSB signaling (32), it is possible that the depletion of RNF8 increases the amount of RNF168 available for recruitment to the array by E6 and E7.
Fig. 2.

E7 protein directly interacts with RNF168. (A) Schematic representation of the tethering LacO/LacR system inserted in U2OS 2-6-5. LacO, Lac operator; LacR, Lac repressor. (B) U2OS 2-6-5 cells transfected with plasmids expressing the indicated mCherry-LacR fusion protein or induced for the expression of ER-mCherry-LacR-FokI-DD were fixed and processed for 53BP1 immunofluorescence. (Scale bars, 5 µm.) (C–H) Quantification of the mCherry-LacR foci colocalizing with 53BP1 (C and G), γ-H2AX (D), and Flag (E and F) staining. In G, cells were depleted for RNF8 prior to transfection with mCherry-LacR constructs. In H, HPV31 E6 or E7 fused to a GFP tag were used. All of the quantifications are represented as the mean ± SD (n = 3; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001). (I) Pull-down of His-RNF168 with GST-HPV16 E7. GST alone and MBP-His were used as negative controls. Inputs and pull-down were analyzed by immunoblotting with the indicated antibodies.
HPV31 E7 and E6 interact with histone modifiers of the histone acetyl transferase (HAT) (41, 42) and deacetylase (HDAC) (43, 44) families. To exclude that E6 and E7 indirectly promote the recruitment of RNF168 to the array by inducing local epigenetic changes in chromatin, we investigated if mCherry-LacR-RNF168 also promoted the accumulation of the viral oncoproteins at the LacO-array. Under these conditions, only E7 was efficiently recruited to the array (Fig. 2H and SI Appendix, Fig. S2 F and I). Therefore, we decided to focus further analyses on the E7-mediated interaction. Using mCherry-LacR-E7, we also detected the accumulation of ubiquitin conjugates, as well as BRCA1, at the LacO-array (SI Appendix, Fig. S2 K–M), consistent with RNF168 being recruited to the array by HPV31 E7. However, although RNF169 is a paralog of RNF168, it is not recruited directly to the array by E7, as depletion of RNF168 prevents its recruitment (SI Appendix, Fig. S2N).
Finally, the interactions detected using the LacO-LacR tethering system could be indirect. To rule out this possibility, we showed that recombinant GST-HPV16 E7 protein efficiently pulled down recombinant His-RNF168 in vitro (Fig. 2I and SI Appendix, Fig. S2O), indicating that the interaction is direct and independent of posttranslational modifications. Taken together, these findings revealed that the HPV E7 protein specifically interacts with the E3 ligase RNF168 independently of the activation of the cellular response to DNA damage.
RNF168 Interacts with E7 from HR-HPVs and Is Required for Viral Replication.
Human papillomavirus E7 is essential for viral replication and HPV-induced carcinogenic transformation. E7 is a small protein of approximately 100 amino acids that is composed of 3 conserved regions (CR1–3; Fig. 3A). Mutations in any of these regions affect different biological activities of E7 (45). To gain insight into the role of the interaction between E7 and RNF168, we first investigated if this interaction is conserved among the E7s from different types of virus and which domain of E7 contributes to the interaction. According to their oncogenic potential, HPVs of the alpha genus are subdivided as low- and high-risk (4). HPV6 and 11 are classified as low-risk, as they are mainly associated with the emergence of benign genital warts. HPV16, 18, and 31 are part of a group of 12 HPV types classified as high-risk because of their association with cancer development (4). Interestingly, we observed that only the HPV E7 proteins from high-risk viruses promoted efficient recruitment of RNF168 to the LacO-array (Fig. 3B and SI Appendix, Fig. S3 A and B). These findings show that the interaction of E7 with RNF168 is conserved only among the alpha HR-HPV types. Surprisingly, we did not detect an interaction between RNF168 and E7 from HPV5 and HPV8, 2 types of virus that have been associated with the development of nonmelanoma skin cancers (46). In the LacO/LacR system, neither CR1 nor CR2, which comprises the pRb-binding motif (LXCXE), were sufficient to recapitulate the interaction with RNF168 (Fig. 3C and SI Appendix, Fig. S3 C and D). Accordingly, a point mutation or a small deletion that abolishes the interaction of E7 with the tumor suppressor pRb (C24G and DLYC) did not severely impact the recruitment of RNF168 by full-length E7 to the array (Fig. 3D and SI Appendix, Fig. S3 E and F). In contrast to CR1 and CR2, CR3 by itself was able to recruit RNF168 to the array, similarly to full-length E7 (Fig. 3C and SI Appendix, Fig. S3 C and D). This domain was also required for the interaction of E7 with RNF168 in immunoprecipitation experiments using cell extracts (Fig. 3E). CR3 is an atypical zinc finger-like domain that mediates the dimerization of E7 and is needed for its transforming activity (45). Using a panel of well described HPV16 E7 CR3 mutants (45, 47), we found that the recruitment of RNF168 to mCherry-LacR-E7 foci was only slightly impaired by a mutant that abolishes the interaction of E7 with HDACs (L67R; Fig. 3D and SI Appendix, Fig. S3 E and F). Interestingly, mutants of E7 that are transformation-defective and fail to interact with the cullin 2 ubiquitin ligase complex (CVQ, LEDLL) also significantly impair its interaction with RNF168.
Fig. 3.
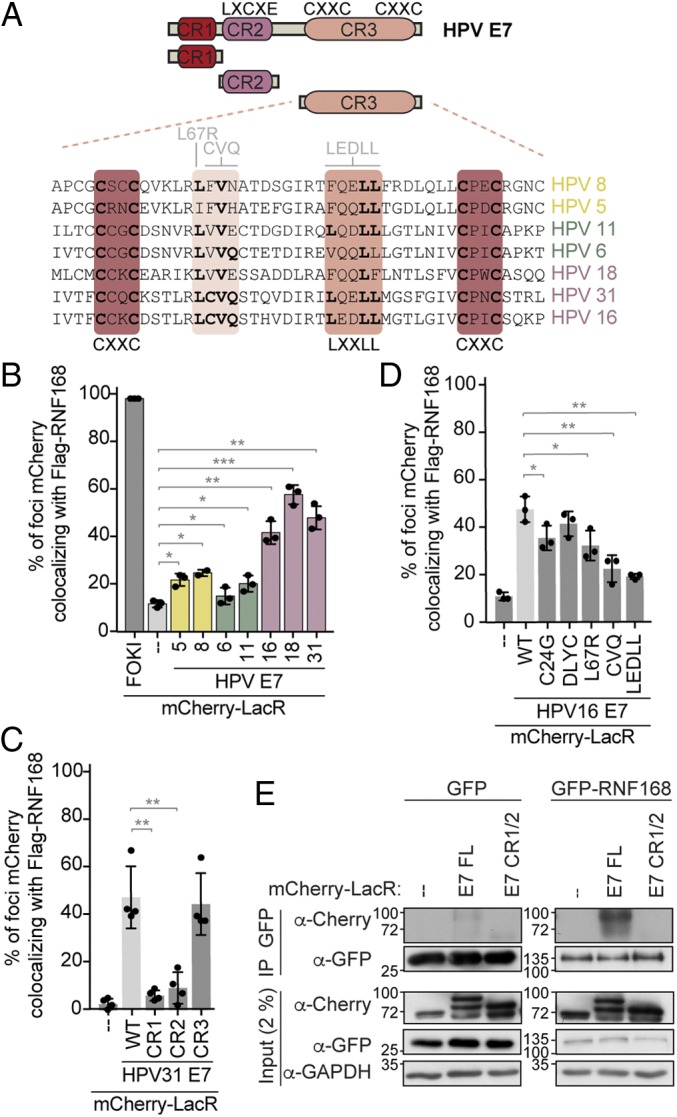
E7 from HR-HPVs binds to RNF168 through the CR3 domain. (A) Schematic representation of HPV E7 protein. CR, conserved region; CXXC, zinc-finger motif; LXCXE, pRb-binding sites. An alignment of the CR3 domain from different types of HPV (beta, yellow; alpha low-risk, green; alpha high-risk, pink) is also presented. Conserved residues are indicated in bold, the zinc-binding domains are indicated in dark red, and the residues mutated used in this study are shown in light gray. (B–D) Quantification of the mCherry-LacR-HPV E7 foci colocalizing with Flag-RNF168 were done as described in Fig. 2E. (*P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.) (E) Immunoprecipitation of GFP-RNF168 from U2OS 2-6-5 cell extracts. The indicated mCherry-LacR fusion proteins were transfected in GFP- or GFP-RNF168–expressing cells. GFP fusion proteins were purified over GFP-binding proteins beads (GFP-Trap) and analyzed by immunoblotting. GAPDH was used as a loading control.
As E7 is required for viral replication, we next investigated whether RNF168 is required for the replication of viral genomes. In CIN612 9E cells, which are derived from a biopsy of a low-grade cervical intraepithelial neoplasia (CIN1) and stably maintain multiple copies of the HPV31b genome in episomal form, depletion of RNF168 for 3 d had no impact on the amount of viral genome that is maintained in undifferentiated cells (shRNF168 vs. shScramble at T0; Fig. 4 A and B). Interestingly, Southern blot analysis of viral genome amplification, which occurs when CIN612 9E cells are induced to differentiate in high-calcium medium for 72 h, showed that RNF168 depletion substantially decreased the number of viral episomes compared with shScram (Fig. 4 A and B). In these experiments, the knockdown of RNF168 did not interfere with the ability of CIN612 9E cells to differentiate, as indicated by the increased levels of involucrin and keratin-10, 2 specific markers for differentiation. Consistent with the increased levels of RNF168 detected in HPV+ tumors, we also detected higher level of RNF168 in CIN612 9E compared with uninfected keratinocytes (Fig. 4C). Altogether, these results indicate that the expression of RNF168 E3 ligase is increased in HPV-infected cells and is required for productive viral replication upon differentiation. This phenotype is reminiscent of results obtained with knockdown or inhibition of ATM, MRE11, NBS1, RAD51, and BRCA1 (11, 12, 15, 16).
Fig. 4.

RNF168 is required for viral replication. (A) CIN612 9E cells were transiently transduced with lentiviral particles containing a control scrambled shRNA (shScram) or one of the 2 RNF168-specific shRNAs for 72 h. DNA and proteins were harvested at T0 (undifferentiated) or following differentiation in high-calcium medium for 72 h. DNA was digested with BamHI (does not cut the viral genome) and HindIII (linearizes the viral genome) and analyzed by Southern blotting using the HPV31 genome as a probe. WCEs were analyzed by immunoblotting with the indicated antibody. Involucrin and K-10 were used to demonstrate cellular differentiation. KAP1 and GAPDH were used as loading controls. (B) Quantification of the experiment present in A using densitometry of episomal bands with ImageJ software. Shown is the fold change normalized to T0 shScram, which is set to 1. Error bars represent means ± SD (n = 3; *P ≤ 0.05). (C) RNF168 expression in HFK and CIN612 9E cells was analyzed 0, 48, and 96 h after differentiation in high-calcium media by immunoblotting with the indicated antibodies. GAPDH and KAP1 were used as loading controls.
E7 Favors DNA Repair by HDR by Limiting the Function of RNF168 at DSBs.
Although the interaction of E7 with RNF168 can be easily detected at the LacO-array, the pan-nuclear localization of the viral protein observed in untreated cells remained unchanged in cells in which DNA lesions were induced by irradiation (IR), neocarzinostatin, or hydroxyurea, which induces lesions specifically in S-phase by blocking replication fork progression (SI Appendix, Fig. S4A). Similarly, E7 is not recruited to sites of intense DNA damage induced by targeting the FOKI nuclease to the LacO-array (Fig. 2H and SI Appendix, Fig. S2F). However, HPV16 E7 was recently reported to colocalize with 53BP1 using proximity-ligation assays, both in differentiated HPV+ keratinocytes and in CaSki (48), arguing that both proteins are near each other during viral replication and cellular transformation. Consistently, we found that E7 and the CR3 domain alone are recruited to foci of RNF168 that have been described as cellular replication factories in unperturbed conditions (Fig. 5A) (49). Thus, these results support a model in which E7 interacts with a subpopulation of RNF168 that localizes at replication forks.
Fig. 5.

E7 hinders the function of RNF168 at DSBs. (A) Representative images of U2OS cells transfected with the indicated mCherry-LacR protein and Flag-RNF168. (B and C) Quantification of γ-H2AX and 53BP1 foci in U2OS cells (B) and hTERT-HFK (C) treated with the indicated amount of Gy. In B, FK2 foci were also quantified. Expression of GFP-HPV31 E7 full-length and CR1/2 were induced 24 h before treatment. Results are represented as the mean ± SEM (n = 3; **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001). (D) Schematic representation of the HDR assay based on gene editing of the LMNA locus. (E) C33-A stable cell lines were transfected with Cas9, sgRNA against LMNA, and mClover donor vectors. Percentage of mClover-positive cells were analyzed by flow cytometry 72 h posttransfection and normalized over percentage of mClover-positive C33-A cells in each replicate. Data are represented as the mean ± SEM (n = 3).
RNF168 is a limiting factor during DSB signaling, as it quickly becomes saturated upon damage induction (32). This led us to investigate if E7 could reduce the amount of RNF168 at DSBs. Although the antibody against RNF168 detects high levels of DNA damage that accumulate in 53BP1-NBs (Fig. 1 C and D), it is less efficient at detecting the recruitment of RNF168 at irradiation-induced foci (SI Appendix, Fig. S1E; only 30% of γ-H2AX foci are RNF168-positive vs. 75% and 90% for conjugated-ubiquitin [FK2] and 53BP1 [Fig. 5B], respectively). Therefore, we quantified the formation of 53BP1 and FK2 foci as markers of RNF168 activity. To do so, U2OS cells stably expressing GFP-HPV31 E7 and E7 CR1-CR2 (referred to here as CR1/2) as a negative control were exposed to increasing doses of IR (Fig. 5B and SI Appendix, Fig. S4 B–F). As the accumulation of 53BP1 to DSBs is regulated in a cell cycle manner (20), we performed the experiment in U2OS cells in which the impact of E7 on DSB signaling can be uncoupled from its impact on cell cycle progression (pRb degradation). Indeed, pRB is always phosphorylated (inactive) in U2OS as a result of the absence of pRB dephosphorylation after mitosis (50). Interestingly, we observed that the total number of radiation-induced 53BP1 and FK2 foci were significantly reduced in cells expressing the viral oncoprotein compared with cells expressing a negative control only (Fig. 5B and SI Appendix, Fig. S4 B, E, and F). Importantly, the levels of γ-H2AX quantified in cells expressing HPV31 E7 were similar to those in the control cell lines (Fig. 5B and SI Appendix, Fig. S4 C, E and F). The same results were obtained when GFP-HPV31 E7 was expressed in human foreskin keratinocytes expressing the catalytic component of telomerase (hTERT-HFK; Fig. 5C and SI Appendix, Fig. S4 G and H) (51). Inhibition of RNF168 or 53BP1 leads to increased repair by HDR (20). Consistent with the reduced accumulation of RNF168 substrates at DSBs in E7-expressing cells, we also found that stable expression of E6/E7 in C33-A cell (HPV− cervical carcinoma) increases DNA repair by HDR (Fig. 5 D and E and SI Appendix, Fig. S4 I–M). These experiments were performed by measuring gene targeting efficiency of an mClover donor sequence at the LMNA locus in C33-A, C33-A stably expressing E6 and E7, and C33-A KO 53BP1 as positive control. Altogether, these observations support a model in which E7 directly hinders the function of RNF168 at DSBs.
E7 Interacts with RNF168 without Affecting Its E3 Ubiquitin Ligase Activity.
RNF168 is an E3 ubiquitin ligase that is composed of a RING domain (catalytic activity) and 2 ubiquitin recognition modules, which enable the recognition of RNF8’s substrate (UDM1) as well as its own substrate (UDM2; Fig. 6A). The latter activity is important for the autoamplification of RNF168 recruitment at DSBs. RNF168 also contains a PALB2-interacting domain (PID), which promotes its interaction with the complex PALB2/BRCA2 (52, 53). In vitro, RNF168 promotes ubiquitylation of histone H2A (23). As E7 reduces the accumulation of conjugated ubiquitin at DSBs (Fig. 5B), we investigated if E7 inhibits the E3 ligase activity of RNF168. Using in vitro ubiquitylation assay on mononucleosomes, we found that GST-HPV16 E7 does not interfere with H2A ubiquitylation by RNF168 (Fig. 6B). In this assay, along with a ubiquitylation assay that was performed without mononucleosomes, we found that GST-HPV16 E7 is not ubiquitylated (Fig. 6C and SI Appendix, Fig. S5A), suggesting that the viral protein is not a direct target of RNF168. We thus turned our attention to the interaction between RNF168 and E7 to gain insight into how the viral oncoprotein subverts the function of this E3 ubiquitin ligase. We assessed the ability of various mCherry-LacR-RNF168 fusion proteins to promote the recruitment of GFP-tagged E7 to the LacO-array. These experiments were done in RNF168-depleted cells to eliminate the endogenous protein as a potential confounding factor (24). Consistent with the results obtained in GST pull-down, we found that the catalytic activity of RNF168 was dispensable for its interaction with E7 (Fig. 6 D and E and SI Appendix, Fig. S5 B–D), but necessary for the recruitment of 53BP1 at the mCherry-LacR locus. Deletion of either one or both motifs of RNF168 that bind ubiquitin (MIUs; Fig. 6A) had little to no effect on the accumulation of GFP-HPV31 E7 at the LacO-array. Surprisingly, deletion of RNF168 amino acids 251 to 383 abolished its interaction with E7 both at the LacO-array and at RNF168-positive foci observed in unperturbed cells, but had no impact on its ability to recruit 53BP1 to the array (Fig. 6 D and E and SI Appendix, Fig. S5E). Furthermore, when residues 251 to 383 of RNF168 were fused to mCherry-LacR, this region was sufficient to recruit GFP-HPV31 E7 to the LacO-array. These results indicate that the ability of RNF168 to recruit 53BP1 and to interact with E7 resides in separate domains of the protein that can be uncoupled genetically. The necessity of the residues 251 to 383 in the interaction between RNF168 and HPV E7 was further confirmed by immunoprecipitation experiments in a U2OS RNF168 KO cell line (SI Appendix, Fig. S6 A–F). Altogether, our study has revealed a previously uncharacterized domain of RNF168, the E7-binding domain (E7BD), that is targeted by the oncoprotein E7 to hinder its signaling function at DSBs.
Fig. 6.

E7 interacts with a previously uncharacterized region of RNF168. (A) Schematic representation of RNF168 and its functional domains. LRM, LR-motif; PID, PALB2-interacting domain; R, Ring domain; UDM, Ub-dependent DSB recruitment module; UIM/MIU, motif-interacting with ubiquitin. (B) In vitro ubiquitylation assay (UbRx) on mononucleosomes (MNs) isolated from HeLa cells with or without a gradient of GST or GST-HPV16 E7. A reaction without ubiquitin was used as a negative control. (C) In vitro ubiquitylation assay on GST and GST-HPV16 E7 with MNs. A reaction without ubiquitin was used as a negative control. Asterisk indicates a nonspecific band. (D and E) RNF168-depleted U2OS 2-6-5 cells were transfected with plasmids expressing GFP-E7 and the indicated mCherry-LacR RNF168 construct and processed for 53BP1 immunofluorescence. Accumulation of GFP-HPV31 E7 (Left) and 53BP1 (Right) at the mCherry-LacR RNF168 foci are represented as the mean ± SD (n = 3) in D (*P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001). Representative images obtained with the indicated mutation/truncation of RNF168 are presented in E. (Scale bars, 5 µm.) (F) Proposed models for the inhibition of RNF168 function at DSBs by HR-HPV E7. P, phosphorylation; Ub, ubiquitin.
Discussion
In this study, we set out to understand how the HPV oncoproteins contribute to the overexpression and accumulation of RNF168 in large nuclear foci observed in HPV-associated tumors. Using a single-cell assay to determine the colocalization of DNA repair factors with mCherry-tagged viral proteins at an integrated LacO-array, we discovered a mechanism by which a host–pathogen protein interaction promotes genomic instability in the host cell. First, we discovered that a direct interaction between E7 from oncogenic HPV types and RNF168 inhibits DSB signaling, raising the possibility that this viral–host interaction underlies the acquisition of genomic instability characteristic of HPV+ lesions that progress to cancer. Second, we found that, in contrast to many other viruses, HPV relies on RNF168 to promote efficient viral replication (as detailed later). Third, our work identified a domain of RNF168 bound by E7, termed E7BD, which lies between the 2 highly conserved Ub-dependent DSB recruitment modules of RNF168, and, when targeted by oncogenic HPVs, disrupts host chromatin response to DNA breaks.
Hijacking host ubiquitin machineries is emerging as a common theme among viruses, and antiviral function of RNF168 has been described for other viruses (reviewed in refs. 10, 54). In the latter cases, viruses have evolved different strategies to inhibit the activity of RNF168. First, the viral protein ICP0 from herpes simplex virus type 1 (HSV1) targets both RNF8 and RNF168 for degradation and thus enables productive infection by inhibiting the intrinsic antiviral response triggered upon infection (55–57). Second, RNF168 was recently found to counteract porcine reproductive and respiratory syndrome virus (PRRSV) infection, although how the virus inhibits RNF168 is still unknown (58). Both the Epstein–Barr virus (EBV) and Kaposi’s sarcoma herpesvirus (KHSV), which cause Burkitt’s and Hodgkin’s lymphoma, nasopharyngeal carcinoma, stomach adenocarcinoma, and Kaposi’s sarcoma (2), inhibit the recruitment of RNF168 at DSBs. These effects are attributed to the EBV tegument protein BKRF4 and the KHSV LANA protein, which bind to chromatin and physically block the interaction of RNF168 with the nucleosome (59, 60). The consequences of RNF168 inhibition on viral replication and cancer progression induced by EBV and KHSV remain to be determined. In this study, we described a mechanism by which a viral oncoprotein hijacks the function of RNF168 through a direct interaction. Specifically, we showed that E7 accomplishes this task without affecting its enzymatic activity or inducing its degradation (SI Appendix, Fig. S6H). Importantly, and in contrast to previously described antiviral function, RNF168 is required for the replication of the HPV genome.
While our finding suggests that E7 titers RNF168 away from DSBs, 2 mechanisms by which E7 subverts the function of this E3 ubiquitin ligase could be envisioned from our findings. First, E7 may relocate RNF168 to safeguard the stability of viral replication forks within the viral replication centers (Fig. 6F, model 1), effectively sequestering this low-abundance protein. Second, the interaction of E7 with RNF168 may serve to rewire the ubiquitylome of RNF168 to modify alternative substrates (Fig. 6F, model 2). Both models are further discussed as follows.
RNF168 was reported to promote efficient DNA replication by stabilizing replication forks in S phase (49). Thus, E7 may redirect this E3 ligase to viral replication centers to promote the replication of its own genome (model 1). Consistent with this hypothesis, a lower number of DNA breaks are detected on the viral genome than on the host genome during productive viral replication, indicating that DNA breaks are either less frequent in the viral genome or repaired more rapidly (17). Furthermore, RAD51 and BRCA1 are bound to HPV chromatin and are essential for productive viral replication, emphasizing the possibility that these HDR factors are involved in viral replication either through DNA repair by HDR or by protecting stalled replication forks from degradation (15). In line with these findings, RNF168 was recently reported to interact with PALB2 and to promote HDR by facilitating the recruitment of RAD51 onto resected DNA-ends (52, 53). From this perspective, the interaction between E7 and RNF168 could facilitate the recruitment of RAD51 into viral replication centers to stabilize ssDNA generated at replication forks (Fig. 6F, model 1). Furthermore, 53BP1 has been shown to enable HDR repair of heterochromatin-DSBs (HC-DSBs) specifically in G2 phase (61). This is particularly intriguing given that 53BP1 localization to sites of HPV replication increases upon differentiation and that productive viral replication occurs in a G2-arrested environment (62). At HC-DSBs, 53BP1 promotes ATM-dependent phosphorylation of the heterochromatin building factor KAP1, leading to chromatin relaxation that facilitates the recruitment of HDR factors (63). RNF168 recruitment of 53BP1 may thus serve a similar role on viral chromatin during productive replication. Additional studies will be required to determine if E7/RNF168 can assemble in complexes with PALB2/BRCA2 and/or whether the E7–RNF168 interaction serves to preferentially recruit DNA repair factors to viral replication centers upon differentiation. Furthermore, characterization of the role of the newly identified domain in RNF168 on its ability to safeguard genomic stability will be instrumental for our understanding of why it is targeted by HPV E7.
It is likely that targeting of RNF168 by E7 plays additional roles in the HPV life cycle other than those described in this study. We can imagine several reasons why HPV would hijack the function of this E3 ubiquitin ligase. As suggested in model 2 (Fig. 6F), E7 may redirect the activity of the E3 ligase to promote ubiquitylation and subsequent degradation of proteins such as pRb, a tumor suppressor that is degraded by HR-HPV E7 (64). Consistent with this hypothesis, RNF168 induces the degradation of other proteins such as the histone demethylase KDM4A/JMJD2A (65). Furthermore, the interaction of E7 with RNF168 is reminiscent of its interaction with cullin 2, an E3 ubiquitin ligase usurped by E7 to promote pRB degradation and stabilize APOBEC3A (66–68). As E7 also uses distinct domains to interact with pRb and RNF168, it will be of interest to investigate if all 3 proteins can be part of the same complex.
HPV-positive cancer cells exhibit genomic instability and replication stress (69). In healthy cells, RNF168 expression is highly controlled. Its overexpression leads to a massive spreading of ubiquitin conjugates from the break point and concomitant recruitment of downstream DNA repair factors such as 53BP1 (32). When RNF168 is overexpressed, genomic instability emerges as a result of increased levels of mutagenic NHEJ, a phenotype that is observed in S-phase when HDR is inhibited by increased accumulation of 53BP1 at DSBs (70). While the exact mechanism underlying the overexpression of RNF168 in HPV+ tumors remains to be discovered, we propose that it results, at least in part, from a cellular adaptation to persistent hijacking and sequestration of RNF168 by E7. Consistent with this model, we show that HPV E7 reduces the numbers of 53BP1 and conjugate-ubiquitin foci in conditions in which RNF168 is rate-limiting (Fig. 5B). In these conditions, any process that reduces the pool of RNF168 available to promote DSB repair will have a direct impact on genomic stability by interfering with the recruitment of 53BP1 and thus DNA repair pathway choice. Consequently, we found that cervical cancer cells that express E6 and E7 exhibit higher levels of DNA repair by HDR (Fig. 5E).
In conclusion, our study has identified a mechanism by which HR-HPV E7, through its interaction with RNF168, likely promotes genomic instability that drives cancer progression. E7 appears to be the main viral driver of cancer progression (2, 45, 64), as it is expressed in virtually all HPV-associated cancers and rarely mutated in these cancers compared with other viral gene products (71). Therefore, the DNA repair deficiency induced by E7 in HPV-associated cancers may present new synthetic lethal opportunities to target the ∼5% of all human cancers caused by this important pathogen.
Materials and Methods
Cell Culture and Transfections.
Cell lines were maintained at 37 °C and 5% CO2. All culture media were supplemented with 10% fetal bovine serum (FBS). U-2-OS (U2OS) and U2OS 2-6-5, gift from Roger Greenberg, University of Pennsylvania, Philadelphia, PA (72), cell lines were cultured in McCoy’s medium (Life Technologies). C33-A, CaSki, SiHa, HeLa, SCC1, and SCC47 cells were purchased from ATCC and cultured in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies). The media of SCC1 and SCC47 was also supplemented with 1% minimum essential media (MEM) nonessential amino acid (Life Technologies). U2OS 2-6-5, U2OS, and U2OS RNF168 KO inducible cell lines were cultured in McCoy’s medium containing 40 mg/mL G418 and induced with 5 µg/mL doxycycline when indicated. U2OS Flp-in were cultured in McCoy’s medium containing 200 µg/mL hygromycin and 5 µg/mL blasticidin. hTERT-HFK cells were provided by Aloysius J. Klingelhutz (University of Iowa, Iowa City, IA) (51) and cultured in K-SFM media (Life Technologies) containing 0.16 ng/mL of epithelial growth factor and 25 µg/mL of bovine pituitary extract. Plasmid transfections were carried out using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. All cell lines were tested negative for mycoplasma contamination. CIN612 9E cells and HFK-31 cells were grown in E medium supplemented with 5 ng/mL mouse epidermal growth factor (BD Biosciences) in the presence of mitomycin C-treated J2 3T3 fibroblast feeder cells (73). When necessary, J2 feeders were removed from HPV+ cells by incubation with 1 mM EDTA in PBS. For differentiation, subconfluent cells were harvested at T0, and the remaining plates of cells were serum-starved overnight in basal keratinocyte growth medium (KGM; Lonza) with supplements for 16 h. Cells were then incubated in keratinocyte basal medium (KBM; Lonza) without supplements but with 1.8 mM CaCl2. Cells were allowed to differentiate for 48 to 96 h after addition of high-calcium medium. DNA and protein were harvested, and viral genome amplification was measured by Southern blotting as for each experiment to ensure activation of the productive phase of the viral life cycle.
Southern Blot Analysis.
DNA was isolated and Southern blot analysis was performed as described previously (74). Briefly, cells were harvested in buffer containing 400 mM NaCl, 10 mM Tris (pH 7.5), and 10 mM EDTA. Cells were lysed by the addition of 30 µL of 20% SDS and subsequently treated with 15 µL of 10 mg/mL proteinase K overnight at 37 °C. DNA was then extracted with phenol chloroform and precipitated using sodium acetate and ethanol. Five micrograms of resultant DNA were digested with BamHI (which does not cut the HPV31 genome) or HindIII (which linearizes the HPV31 genome), separated on 0.8% agarose gel for 15 h at 40 V, and subsequently transferred to a positively charged nylon membrane (Immobilon-Ny+; Millipore). The 32P-labeled linearized HPV31 genome was used as a probe.
RNA Interference.
Single siRNA duplexes targeting either RNF168 (D-007152-04) or a nontargeting sequence (D-001210-02-02) were purchased from Dharmacon. To knock down RNF8, a SMARTpools siRNA against RNF8 (D-006900-01-0020) was purchased from Dharmacon. Unless stated otherwise, all siRNAs were transfected in a forward transfection mode 24 h prior to cell processing using RNAimax (Invitrogen) according to the manufacturer’s protocol.
Generation of Lentiviruses.
Lentiviruses were produced as previously described (15). Briefly, plasmids expressing shRNAs or the indicated GFP-tagged proteins were cotransfected with vesicular stomatitis virus G (VSV-G) plasmid DNA and Gag-Pol-Tet-Rev plasmid DNA into HEK-293T cells using polyethyleneimine (PEI). At 48 to 72 h posttransfection, supernatants containing lentivirus were harvested, and CIN612 9E, U2OS, or hTERT-HFKs were transduced in the presence of 8 µg/mL hexadimethrine bromide (Polybrene; Sigma). Three days posttransduction, CIN612 9E were either harvested at T0 (undifferentiated sample) or differentiated in high-calcium medium for 72 h as described here earlier. Knockdown of RNF168 and the expression of the GFP constructs was confirmed for each experiment by Western blotting.
CRISPR-Cas9 Genome Editing of 53BP1 and RNF168.
C33-A and U2OS cells were transiently transfected with sgRNAs targeting 53BP1 or RNF168, respectively (SI Appendix, Table S1), and expressed from the pX459 vector containing Cas9 followed by the 2A-Puromycin cassette. The next day, cells were subcloned to form single colonies. Clones were screened by immunoblot and immunofluorescence to verify the loss of 53BP1 and RNF168. The genomic regions targeted by the CRISPR-Cas9 was subsequently characterized by PCR amplification, DNA sequencing, and TIDE analysis (75).
Immunofluorescence Microscopy.
Immunofluorescence was done as previously described (24) with the indicated antibodies (SI Appendix, Table S2). Micrograph images were taken using a Zeiss LSM700 laser-scanning microscope equipped with a 63× oil lens. In all micrographs, dashed lines indicated nucleus outlines. Insets represent a 10-fold magnification of the indicated region. Quantification was done on 3 biological replicates, and at least 50 cells were counted in each experiment. Unless stated otherwise, t test with Welch correction was realized to assess statistical significance.
Clover Assay.
C33-A stable cell lines were plated at 100,000 cells per well on 6-well plates 24 h post transfection with pX330-LMNAgRNA1 and pCR2.1-CloverLMNAdonor plasmids as previously described (76). At 72 h posttransfection, percentage of mClover-positive cells were analyzed by flow cytometry using an Accuri C6 (BD Biosciences).
Immunoprecipitation.
Cells were transfected with mCherry-LacR, mCherryLacR-HPV31 E7 full length, or mCherryLacR-HPV31 E7 CR1-CR2–expressing plasmids. At 15 h posttransfection, GFP, GFP-RNF168, GFP-RNF168 WT, and ∆E7BD expression were induced. Cells were lysed 9 h later in lysis buffer (50 mM Tris, pH 7.5, 300 mM NaCl, 1 mM EDTA, 1% Triton X-100) followed by Micrococcal nuclease (Sigma) treatment for 30 min. Cleared cell lysate (1 mg) was subjected to immunoprecipitation using 20 µL GFP Trap beads for 3 h at 4 °C. After 3 washing steps, beads were eluted in 2× Laemmli buffer for immunoblotting analyses.
Purification of Recombinant Protein.
GST-HPV16 E7, RNF168 WT, and ∆E7BD recombinant proteins were purified with technical help from Marie-Christine Caron from the laboratory of Jean-Yves Masson (Université Laval, Québec, QC, Canada) as previously described (53). For MBP-His recombinant protein purification, E. coli strain BL-21 was induced with 1 mM IPTG when they reached an OD600 of 0.6. Next, fusion proteins were purified on Ni-NTA agarose (Qiagen) according to the batch method described in the manufacturer’s manual. All recombinant proteins were stored in 20 mM Tris acetate, pH 8.0, 200 mM potassium acetate, 10% glycerol, 1 mM EDTA, 0.5 mM DTT.
Ubiquitylation Reaction.
Mononucleosomes were isolated from the HeLa cells and ubiquitylated as described previously (25). Briefly, 2.5 µg mononucleosomes were incubated with 30 nM Uba1, 1.5 μM UbcH5a, 4 μM HIS-RNF168, 22 μM ubiquitin (Boston Biochem), and 3.33 mM ATP in a buffer containing 50 mM Tris⋅HCl, pH 7.5, 100 mM NaCl, 10 mM MgCl2, 1 μM ZnCl2, and 1 mM DTT at 30 °C for 4 h. His-GST (4.44 µM) or GST-E7 (2.2 to 4.4 µM) proteins were added to the mixture. The reaction was quenched by adding 15 mM EDTA, pH 8.0. Denatured samples were separated on an SDS/PAGE and analyzed by Western blotting experiment.
GST Pull-Down.
For GST pull-down, 1 µg of purified GST-tagged protein was incubated with 4 µg of purified His-tagged protein in 1.5 mL of GSTB buffer (20 mM Hepes, pH 7.4, 150 mM KCl, 10% glycerol, 0.02% Triton X-100, 0.2 mM EDTA, 0.002 µg/mL aprotinin, 0.001 µg/mL pepstatin A, 0.002 µg/mL leupeptin, 1 mM AEBSF, 1 mM DTT, 1 mg/mL bovine serum albumin [BSA]) for 3 h at room temperature. Input was collected before the addition of glutathione Sepharose beads for 90 min at room temperature. Beads were then washed with GSTB buffer without BSA and directly eluted in 2× Laemmli buffer for immunoblotting analyses.
RNA-Seq Analyses.
Level 3 RNA-Seq by Expectation Maximization (RSEM) normalized Illumina HiSeq RNA expression data for the TCGA head and neck cancer (HNSC) and cervical carcinoma (CESC) cohorts were downloaded from the Broad Genome Data Analysis Center’s Firehose server (https://gdac.broadinstitute.org/). For all genes, the gene-level Firehose dataset was used. RSEM-normalized expression data were extracted, and the HPV status was manually curated based on published datasets as previously described (77). Primary patient samples with known HPV status were grouped as HPV-positive, HPV-negative, or normal-adjacent control tissue. For the boxplots, center lines show the medians, box limits indicate the 25th and 75th percentiles as determined by GraphPad Prism, and whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles. P values were assigned using a 2-tailed nonparametric Mann–Whitney U test.
Supplementary Material
Acknowledgments
We are grateful to Jacques Archambault, Jacques Côté, Agnel Sfeir, Alexandre Orthwein, Sabine Elowe, Kavi Mehta, and members of the A.F.-T. laboratory for critical reading of the manuscript; and to Daniel Durocher, Jacques Archambault, Jean-Yves Masson, Graham Dellaire, Roger Greenberg, and Karl Munger for essential reagents such as plasmids, purified proteins, and cell lines. We thank Vincent Tremblay for technical support to J.S., and Takashi Awaga for early work on establishing the LacO/LacR assay with HPV proteins. J.S. has received the doctoral fellowship “Luc Bélanger” from CHU de Québec–Université Laval Foundation.” S.A.B. and E.B. are supported by a master and a postdoctoral fellowship from the Fonds de Recherche du Québec - Santé (FRQS), respectively. This work was supported by Canadian Institutes of Health Research Project Grants 152948 (to A.F.-T.) and 142491 (to J.S.M.), and Scholarship for the Next Generation of Scientists 19590 from Cancer Research Society (to A.F.-T.). A.F.-T. is a tier 2 Canada Research Chair in Molecular Virology and Genomic Instability and is supported by the Foundation J.-Louis Lévesque. C.A.M. is supported by grants from the Health and Human Services (HHS) NIH National Cancer Institute (CA181581 and CA226523).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1906102116/-/DCSupplemental.
References
- 1.de Martel C., et al. , Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 13, 607–615 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Mesri E. A., Feitelson M. A., Munger K., Human viral oncogenesis: A cancer hallmarks analysis. Cell Host Microbe 15, 266–282 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Villiers E. M., Fauquet C., Broker T. R., Bernard H. U., zur Hausen H., Classification of papillomaviruses. Virology 324, 17–27 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Doorbar J., Egawa N., Griffin H., Kranjec C., Murakami I., Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 25 (suppl. 1), 2–23 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gillison M. L., Chaturvedi A. K., Anderson W. F., Fakhry C., Epidemiology of human papillomavirus-positive head and neck squamous cell carcinoma. J. Clin. Oncol. 33, 3235–3242 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Statistic Canada , Canadian cancer statistics 2016, special topic: HPV-associated cancers. http://www.cancer.ca/∼/media/cancer.ca/CW/cancer%20information/cancer%20101/Canadian%20cancer%20statistics/Canadian-Cancer-Statistics-2016-EN.pdf?la=en. Accessed 27 March 2019.
- 7.Nichols A. C., et al. , The epidemic of human papillomavirus and oropharyngeal cancer in a Canadian population. Curr. Oncol. 20, 212–219 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McBride A. A., Mechanisms and strategies of papillomavirus replication. Biol. Chem. 398, 919–927 (2017). [DOI] [PubMed] [Google Scholar]
- 9.McKinney C. C., Hussmann K. L., McBride A. A., The role of the DNA damage response throughout the papillomavirus life cycle. Viruses 7, 2450–2469 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weitzman M. D., Fradet-Turcotte A., Virus DNA replication and the host DNA damage response. Annu. Rev. Virol. 5, 141–164 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gillespie K. A., Mehta K. P., Laimins L. A., Moody C. A., Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J. Virol. 86, 9520–9526 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moody C. A., Laimins L. A., Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 5, e1000605 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakakibara N., et al. , Brd4 is displaced from HPV replication factories as they expand and amplify viral DNA. PLoS Pathog. 9, e1003777 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kadaja M., Isok-Paas H., Laos T., Ustav E., Ustav M., Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses. PLoS Pathog. 5, e1000397 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anacker D. C., Gautam D., Gillespie K. A., Chappell W. H., Moody C. A., Productive replication of human papillomavirus 31 requires DNA repair factor Nbs1. J. Virol. 88, 8528–8544 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chappell W. H., et al. , Homologous recombination repair factors, Rad51 and BRCA1, are necessary for productive replication of human papillomavirus 31. J Virol. 90, 2639–2652 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mehta K., Laimins L., Human papillomaviruses preferentially recruit DNA repair factors to viral genomes for rapid repair and amplification. MBio 9, e00064-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanahan D., Weinberg R. A., Hallmarks of cancer: The next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Sandell L. L., Zakian V. A., Loss of a yeast telomere: Arrest, recovery, and chromosome loss. Cell 75, 729–739 (1993). [DOI] [PubMed] [Google Scholar]
- 20.Hustedt N., Durocher D., The control of DNA repair by the cell cycle. Nat. Cell Biol. 19, 1–9 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Dantuma N. P., van Attikum H., Spatiotemporal regulation of posttranslational modifications in the DNA damage response. EMBO J. 35, 6–23 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson M. D., Durocher D., Reading chromatin signatures after DNA double-strand breaks. Philos. Trans. R. Soc. Lond. B Biol. Sci. 372, 20160280 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mattiroli F., et al. , RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell 150, 1182–1195 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Panier S., et al. , Tandem protein interaction modules organize the ubiquitin-dependent response to DNA double-strand breaks. Mol. Cell 47, 383–395 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Fradet-Turcotte A., et al. , 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 499, 50–54 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kitevski-LeBlanc J., et al. , The RNF168 paralog RNF169 defines a new class of ubiquitylated histone reader involved in the response to DNA damage. eLife 6, e23872 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu Q., Botuyan M. V., Cui G., Zhao D., Mer G., Mechanisms of ubiquitin-nucleosome recognition and regulation of 53BP1 chromatin recruitment by RNF168/169 and RAD18. Mol. cell 66, 473–487.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilson M. D., et al. , The structural basis of modified nucleosome recognition by 53BP1. Nature 536, 100–103 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Setiaputra D., Durocher D., Shieldin - the protector of DNA ends. EMBO Rep. 20, e47560 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bohgaki T., et al. , Genomic instability, defective spermatogenesis, immunodeficiency, and cancer in a mouse model of the RIDDLE syndrome. PLoS Genet. 7, e1001381 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stewart G. S., et al. , The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 136, 420–434 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Gudjonsson T., et al. , TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell 150, 697–709 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Cerami E., et al. , The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao J., et al. , Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pombo A., et al. , Regional and temporal specialization in the nucleus: A transcriptionally-active nuclear domain rich in PTF, Oct1 and PIKA antigens associates with specific chromosomes early in the cell cycle. EMBO J. 17, 1768–1778 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lukas C., et al. , 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 13, 243–253 (2011). [DOI] [PubMed] [Google Scholar]
- 37.Harrigan J. A., et al. , Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol. 193, 97–108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spies J., et al. , 53BP1 nuclear bodies enforce replication timing at under-replicated DNA to limit heritable DNA damage. Nat. Cell Biol. 21, 487–497 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Soutoglou E., Misteli T., Activation of the cellular DNA damage response in the absence of DNA lesions. Science 320, 1507–1510 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Donaldson M. M., et al. , An interaction between human papillomavirus 16 E2 and TopBP1 is required for optimum viral DNA replication and episomal genome establishment. J. Virol. 86, 12806–12815 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jha S., et al. , Destabilization of TIP60 by human papillomavirus E6 results in attenuation of TIP60-dependent transcriptional regulation and apoptotic pathway. Mol. Cell 38, 700–711 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Subbaiah V. K., et al. , E3 ligase EDD1/UBR5 is utilized by the HPV E6 oncogene to destabilize tumor suppressor TIP60. Oncogene 35, 2062–2074 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Longworth M. S., Laimins L. A., The binding of histone deacetylases and the integrity of zinc finger-like motifs of the E7 protein are essential for the life cycle of human papillomavirus type 31. J. Virol. 78, 3533–3541 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Longworth M. S., Wilson R., Laimins L. A., HPV31 E7 facilitates replication by activating E2F2 transcription through its interaction with HDACs. EMBO J. 24, 1821–1830 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roman A., Munger K., The papillomavirus E7 proteins. Virology 445, 138–168 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bouvard V., et al. ; WHO International Agency for Research on Cancer Monograph Working Group , A review of human carcinogens–Part B: Biological agents. Lancet Oncol. 10, 321–322 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Todorovic B., et al. , Systematic analysis of the amino acid residues of human papillomavirus type 16 E7 conserved region 3 involved in dimerization and transformation. J. Virol. 85, 10048–10057 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Squarzanti D. F., et al. , Human papillomavirus type 16 E6 and E7 oncoproteins interact with the nuclear p53-binding protein 1 in an in vitro reconstructed 3D epithelium: New insights for the virus-induced DNA damage response. Virol. J. 15, 176 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmid J. A., et al. , Histone ubiquitination by the DNA damage response is required for efficient DNA replication in unperturbed S phase. Mol. cell 71, 897–910.e8 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Broceño C., Wilkie S., Mittnacht S., RB activation defect in tumor cell lines. Proc. Natl. Acad. Sci. U.S.A. 99, 14200–14205 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kiyono T., et al. , Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature 396, 84–88 (1998). [DOI] [PubMed] [Google Scholar]
- 52.Zong D., et al. , BRCA1 haploinsufficiency is masked by RNF168-mediated chromatin ubiquitylation. Mol. Cell 73, 1267–1281.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luijsterburg M. S., et al. , A PALB2-interacting domain in RNF168 couples homologous recombination to DNA break-induced chromatin ubiquitylation. eLife 6, e20922 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dybas J. M., Herrmann C., Weitzman M. D., Ubiquitination at the interface of tumor viruses and DNA damage responses. Curr. Opin. Virol. 32, 40–47 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lilley C. E., et al. , A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J. 29, 943–955 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lilley C. E., Chaurushiya M. S., Boutell C., Everett R. D., Weitzman M. D., The intrinsic antiviral defense to incoming HSV-1 genomes includes specific DNA repair proteins and is counteracted by the viral protein ICP0. PLoS Pathog. 7, e1002084 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chaurushiya M. S., et al. , Viral E3 ubiquitin ligase-mediated degradation of a cellular E3: Viral mimicry of a cellular phosphorylation mark targets the RNF8 FHA domain. Mol. Cell 46, 79–90 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bai J., et al. , A high-throughput screen for genes essential for PRRSV infection using a piggyBac-based system. Virology 531, 19–30 (2019). [DOI] [PubMed] [Google Scholar]
- 59.Leung J. W., et al. , Nucleosome acidic patch promotes RNF168- and RING1B/BMI1-dependent H2AX and H2A ubiquitination and DNA damage signaling. PLoS Genet. 10, e1004178 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ho T. H., et al. , A screen for epstein-barr virus proteins that inhibit the DNA damage response reveals a novel histone binding protein. J. Virol. 92, e00262-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kakarougkas A., et al. , Opposing roles for 53BP1 during homologous recombination. Nucleic Acids Res. 41, 9719–9731 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang H. K., Duffy A. A., Broker T. R., Chow L. T., Robust production and passaging of infectious HPV in squamous epithelium of primary human keratinocytes. Genes Dev. 23, 181–194 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Noon A. T., et al. , 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat. Cell Biol. 12, 177–184 (2010). [DOI] [PubMed] [Google Scholar]
- 64.Moody C. A., Laimins L. A., Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 10, 550–560 (2010). [DOI] [PubMed] [Google Scholar]
- 65.Mallette F. A., et al. , RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 31, 1865–1878 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Westrich J. A., et al. , Human papillomavirus 16 E7 stabilizes APOBEC3A protein by inhibiting cullin 2-dependent protein degradation. J. Virol. 92, e01318-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huh K., et al. , Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J. Virol. 81, 9737–9747 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.White E. A., et al. , Systematic identification of interactions between host cell proteins and E7 oncoproteins from diverse human papillomaviruses. Proc. Natl. Acad. Sci. U.S.A. 109, E260–E267 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moody C. A., Impact of replication stress in human papillomavirus pathogenesis. J. Virol. 93, e01012-17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zong D., et al. , Ectopic expression of RNF168 and 53BP1 increases mutagenic but not physiological non-homologous end joining. Nucleic Acids Res. 43, 4950–4961 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mirabello L., et al. , HPV16 E7 genetic conservation is critical to carcinogenesis. Cell 170, 1164–1174.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tang J., et al. , Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat. Struct. Mol. Biol. 20, 317–325 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wilson R., Laimins L. A., Differentiation of HPV-containing cells using organotypic “raft” culture or methylcellulose. Methods Mol. Med. 119, 157–169 (2005). [DOI] [PubMed] [Google Scholar]
- 74.Fehrmann F., Klumpp D. J., Laimins L. A., Human papillomavirus type 31 E5 protein supports cell cycle progression and activates late viral functions upon epithelial differentiation. J. Virol. 77, 2819–2831 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brinkman E. K., Chen T., Amendola M., van Steensel B., Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pinder J., Salsman J., Dellaire G., Nuclear domain ‘knock-in’ screen for the evaluation and identification of small molecule enhancers of CRISPR-based genome editing. Nucleic Acids Res. 43, 9379–9392 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gameiro S. F., et al. , Analysis of class I major histocompatibility complex gene transcription in human tumors caused by human papillomavirus infection. Viruses 9, E252 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.