

Abstract
Potassium (K+) channels influence neurotransmitter release, burst firing rate activity, pacing, and critical dampening of neuronal circuits. Internal and external factors that further modify K+ channel function permit fine-tuning of neuronal circuits. Humanether-à-go-go-related gene (HERG) K+ channels are unusually sensitive to external calcium concentration ([Ca2+]o). Small changes in [Ca2+]o shift the voltage dependence of channel activation to more positive membrane potentials, an effect that cannot be explained by nonspecific surface charge screening or channel pore block. The HERG–calcium concentration–response relationship spans the physiological range for [Ca2+]o. The modulatory actions of calcium are attributable to differences in the Ca2+affinity between rested and activated channels. Adjacent extracellular, negatively charged amino acids (E518 and E519) near the S4 voltage sensor influence both channel gating and Ca2+dependence. Neutralization of these charges had distinct effects on channel gating and calcium sensitivity. A change in the degree of energetic coupling between these amino acids on transition from closed to activated channel states reveals movement in this region during channel gating and defines a molecular mechanism for protein state-dependent ligand interactions. The results suggest a novel extracellular [Ca2+]o sensing mechanism coupled to allosteric changes in channel gating and a mechanism for fine-tuning cell repolarization.
Keywords: human ether-à-go-go-related gene, potassium channel gating, HERG, calcium, allosteric, Monod–Wyman–Changeux
Plasticity and adaptive behavior in circuits of the nervous system depend on the dynamic properties of ion channels. Potassium channels modulate neuronal excitability and, in turn, are modulated by both intracellular and extracellular factors. The human ether-à-go-go-related gene (HERG) potassium channel has recently been discovered to play important roles in excitable tissues. Originally identified in hippocampus (Warmke and Ganetzky, 1994), HERG channels also play a role in myocardial repolarization (Sanguinetti et al., 1995). ERG channels have been implicated in neural crest cell development (Arcangeli et al., 1997), neoplastic cell survival (Bianchi et al., 1998), and neural spike frequency adaptation (Chiesa et al., 1997). Furthermore, theDrosophila erg is encoded at the seizure locus, originally defined by temperature-sensitive paralytic mutations in flies (Titus et al., 1997; X. J. Wang et al., 1997).
HERG channels (Warmke and Ganetzky, 1994; Trudeau et al., 1995) are six transmembrane (6 TM) family voltage-gated potassium channels and contain the typical motifs identified in these channels (S5-P-loop-S6, charged S4 voltage sensor, etc.). Unlike many other 6 TM K+ channels (Drosophila Shaker, Shal, Shab, Shaw, vertebrate Kv channels), HERG channels are highly sensitive to modest changes in extracellular divalent cation concentrations (Ho et al., 1998; Johnson et al., 1999; Po et al., 1999) around the physiological set point (1–3 mm). This sensitivity is much greater and qualitatively distinct from that in other voltage-gated delayed rectifier potassium channels (Frankenhaeuser and Hodgkin, 1957; Gilly and Armstrong, 1982; Johnson et al., 1999; Po et al., 1999) and cannot be explained by nonspecific surface charge screening or pore block. Changes in extracellular Ca2+ appear to discretely modify the voltage dependence of activation of HERG with little effect on channel inactivation (Johnson et al., 1999), suggesting that the voltage dependence of HERG K+ channel inactivation is distinct from the voltage dependence of activation gating as noted before (Spector et al., 1996; S. Wang et al., 1996, 1997; J. Wang et al., 1998; Zou et al., 1998). This specific interaction with one aspect of channel gating is intriguing and suggests interactions of Ca2+ with amino acids in regions of the channel involved in the voltage dependence of channel opening.
Interestingly, there are two nonconserved negatively charged amino acids (glutamic acids) located in the HERG extracellular loop between S3 and S4. These extracellular facing negative charges are immediately adjacent to the S4 voltage sensor and are good candidates for an interaction with Ca2+ or with some of the positive charges in the S4, leading to Ca2+ titratable changes in gating. The purpose of this investigation was to develop a better understanding of the molecular basis of modifications of HERG function by Ca2+ through analysis of the gating and [Ca2+]ointeractions in wild-type (WT) and HERG mutant channels in which these negative charges were neutralized.
MATERIALS AND METHODS
cDNA constructs and site-directed mutagenesis. The human ether-à-go-go-related gene (HERG) cDNA was obtained from Dr. Mark Keating (University of Utah) and ligated into the pSI mammalian expression plasmid (Promega, Madison, WI). Dr. Richard Horn (Jefferson Medical College) kindly provided the CD8 antigen gene in the EBO-pcD Leu2 vector. CD8, a human T lymphocyte surface antigen, was cotransfected with the channel construct to allow visual identification of transfected cells (Jurman et al., 1994). Mutagenesis was performed by the overlap-extension recombinant PCR technique using VENT® DNA polymerase (New England Biolabs, Beverly, MA). All constructs were assembled in the HERG pSI construct. Briefly, two separate PCR reactions were performed to generate overlapping products containing a desired point mutation in the overlap region. The first round PCR products were purified with QIAQuick purification columns (Qiagen, Hilden, Germany) to remove primers, and the two products were denatured and annealed to one another. Finally, the complete region was amplified in a second round of PCR with a nested set of primers. The PCR cassette was digested then and subcloned into the full-length cDNA using flanking restriction sites. Multiple clones were selected and analyzed by restriction digestion, and DNA sequencing of the entire region was amplified by PCR. For each mutant, two independent correctly assembled and fully sequenced clones were tested electrophysiologically.
Electrophysiology and solutions. HERG channel function was studied with the whole-cell patch-clamp technique by methods identical to those in Johnson et al. (1999). Cells were patch clamped 36–60 hr after transfection, and all experiments were performed at room temperature (23–25°C). The intracellular recording solution for all experiments contained (in mm): 110 KCl, 5 K2ATP, 5 K4BAPTA, 2 MgCl2, 10 HEPES, pH 7.2. The extracellular recording solution used for the experiments in Figure1A contained (in mm): 145 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose, pH 7.35. The base extracellular solution for all other figures contained (in mm): 145 NaCl, 4 KCl, 10 HEPES, 10 glucose. For solutions of Ca2+ concentrations from 100 μm to 10 mm, the appropriate amount of 1 mCaCl2 was added to a 2× concentrated form of the above base solution before addition of NaOH to bring the pH to 7.35 and the final dilution to 1× with deionized water. For Ca2+ concentrations from 3 to 30 μm, 1 mmH-ethylenediamine-N,N′,N′-triacetic acid was added to the base solution to buffer the Ca2+ concentration. The free Ca2+ concentrations were calculated assuming 25 μm contaminant Ca2+ using MaxChelator Sliders version 1.0 software (Chris Patton, Stanford University,www.stanford.edu/∼cpatton/maxc.html) using the Martell and Smith constants.
Fig. 1.
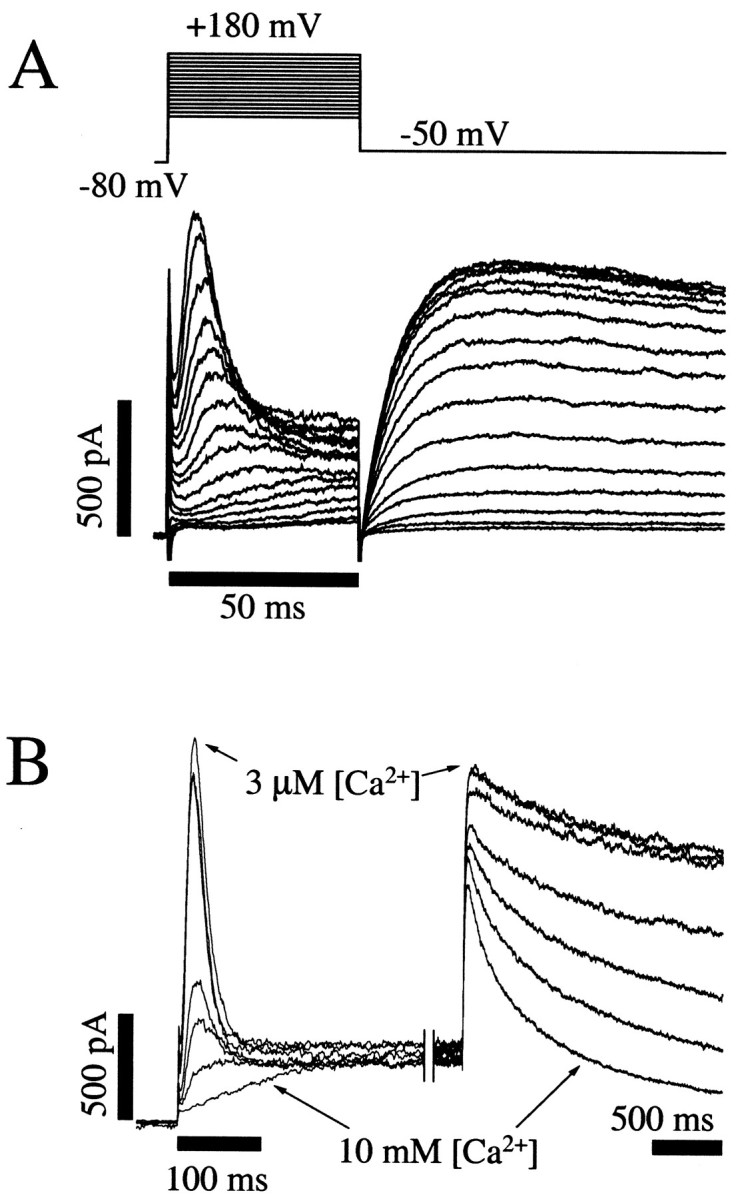
Direct observation of HERG channel inactivation and the effects of Ca2+ to slow the rate of channel activation. A, Activation and inactivation of HERG K+ currents measured during voltage-clamp steps to membrane potentials between +180 and +30 mV in 10 mV increments for 50 msec followed by a step to −50 mV for 100 msec. These recordings show rapid activation of HERG K+ currents followed by subsequent inactivation resulting in transient outward currents. On stepping to −50 mV, a large slow tail current is observed. The rate of activation (initial rising phase) is highly voltage dependent in this potential range, whereas the inactivation (decay) is not. The membrane potential was held at −80 mV before and after the test steps.B, Increases in the extracellular Ca2+ concentration caused a slowing of channel activation with no effect on inactivation, resulting in a truncation of the peak transient outward currents. The rate of tail current decay at −50 mV was also increased. Seven K+ currenttraces from the same cell, each in a different extracellular [Ca2+] (3 μm, 10 μm, 30 μm, 300 μm, 1 mm, 3 mm, and 10 mm), are superimposed. The membrane potential was held at −80 mV before stepping to +70 mV for 2 sec, then repolarized to −50 mV to measure tail currents. Note the two time scale barscorresponding to before and after the break in the current record. After ∼250 msec at +70 mV, the steady state current level was the same in all [Ca2+]; even the current in 10 mm Ca2+ had fully activated.
In figures with raw data, the zero current level is indicated by thebottom of the current calibration bar (vertical) in each figure. Leak correction was not used in any of the raw current traces shown. In a few cases, moderate linear leak correction was used offline using Clampfit. Cell capacitance and series resistance were compensated using the analog controls on the Axopatch 200.
Chinese hamster ovary K1 (CHO-K1) cells were obtained from the American Type Culture Collection (Rockville, MD) and maintained in HAMS F-12 media (Life Technologies, Grand Island, NY) supplemented with 1 mml-glutamine and 10% heat-inactivated fetal bovine serum (Life Technologies) in a humidified, 5% CO2 incubator at 37°C. CHO-K1 cells were cotransfected with the HERG and CD8 plasmids in a ratio of 4:1. Transfection was accomplished using the Lipofectamine transfection reagents and method (Life Technologies).
Data analysis. Data were analyzed and plotted using pClamp 6.03, Origin 5.0 (Microcal Software, Northhampton, MA), SigmaPlot 4.0 (SPSS Inc., Chicago, IL), and TableCurve 3D 3.0 (SPSS Inc.) software. The midpoints (V1/2) of channel activation were determined by fitting the data with a Boltzmann function of the form:
| Equation 1 |
where IMax is the limiting amplitude, V1/2 is the membrane potential whereI/IMax = 0.5, andkv is the slope factor. Concentration-effect data were fit with the Hill equation:
| Equation 2 |
where I is current,IMax is the maximal current,y0 is the current at a given voltage in the absence of Ca2+, A is an amplitude term, KD is the apparent Ca2+ dissociation constant, andb is the Hill coefficient.
A voltage-dependent Monod–Wyman–Changeux (MWC) model of allosteric protein function (Monod et al., 1965; Galzi et al., 1996; Cox et al., 1997; Changeux and Edelstein, 1998) was used to analyze the data. The model is described by Equation 3 below:
| Equation 3 |
F is the Faraday constant in coulombs per mole; T is temperature in degrees Kelvin;R is the gas constant in Joules per degree (Kelvin) per mole. V is membrane potential (volts). There are four free parameters in the equation: Q, Kc,Ka, and L0. The apparent gating charge, Q, that moves through the membrane electrical field during the transitions from closed to activated determines the voltage dependence, i.e., the steepness of the sigmoid curves in Figures 4 and 6. Kc is the Ca2+ dissociation constant for the closed channel. Ka is the Ca2+dissociation constant for the activated (open) channel. The degree of separation between the voltage-dependent activation curves in low and high Ca2+ is determined by the relative affinities of the closed and activated states (Kc andKa). A large difference between Kc andKa results in a larger separation between the voltage-dependent activation curves.L0 is the closed–open equilibrium constant in the absence of calcium, and it determines the lower limit of the midpoints of the voltage-dependent activation curves.L0 determines the relative position of the family of voltage-dependent activation curves on the voltage axis (see Fig. 4) or the baseline of theV1/2 versus Ca2+ concentration relationship (see Fig.5). Global fitting (Balser et al., 1990) of the allosteric MWC model to HERG currents simultaneously across multiple voltages and Ca2+ concentrations was performed with TableCurve 3D software using an iterative 64-bit Levenburg–Marquardt fitting algorithm. It should be noted that although the model is helpful for quantifying and interpreting the effect of the mutations and Ca2+, the primary conclusions are not dependent on the model.
Fig. 4.
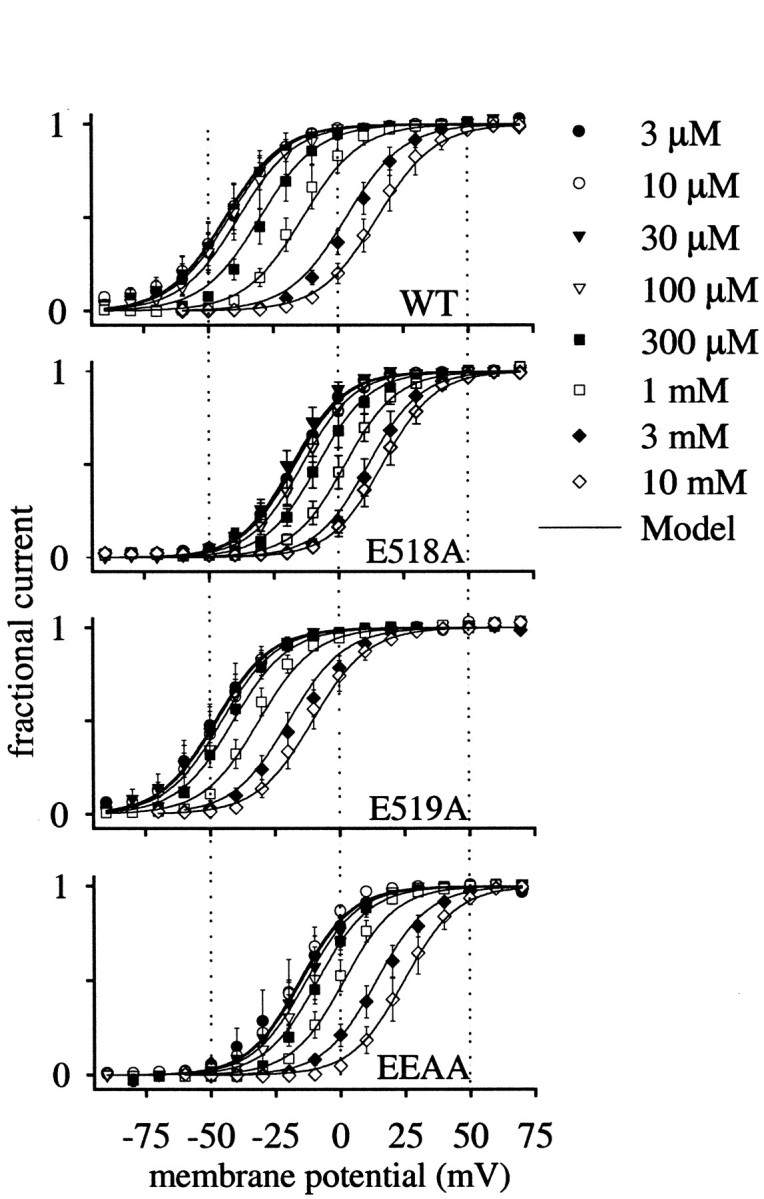
Effect of extracellular Ca2+ on the voltage dependence of activation of WT and mutant HERG channels. Voltage-dependent activation curves were determined from peak tail current amplitudes measured at −50 mV with a 2 sec step to the test potential (as in Fig. 2). Error bars indicate SEM (n = 3–15 for each concentration). Smooth curves correspond to the predicted values based on the three-dimensional fit of the voltage-dependent Monod–Wyman–Changeux model (see Fig. 6, Eq. 3).
Fig. 6.
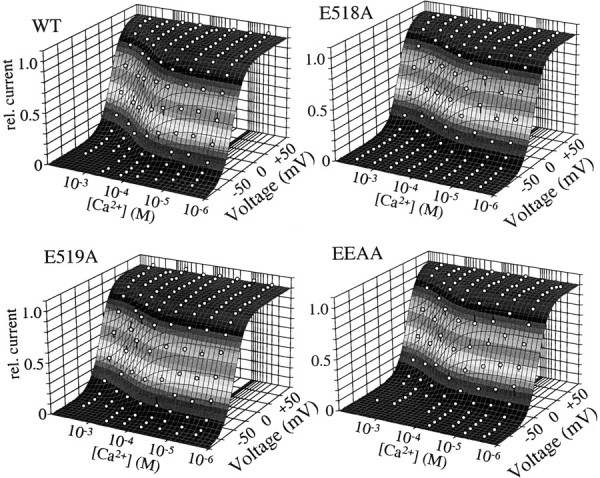
Ca2+-membrane voltage–response surface. Three-dimensional regression fits of the voltage-dependent Monod–Wyman–Changeaux model to relative K+currents for WT and mutant HERG measured in different Ca2+ concentrations and at different membrane potentials. The mean voltage– activation data for all Ca2+ concentrations are plotted as a function of membrane potential in a three-dimensional format. The vertical error bars on the symbols indicate the distance of the mean data point from the fitted plane.
Fig. 5.

Ca2+ dependence of the half maximal voltage (V1/2) of activation of WT and mutant HERG channels obtained from the curves in Figure 4. Error bars indicate SEM. Smooth curves correspond to the predicted values based on the three-dimensional fit of the voltage-dependent Monod–Wyman–Changeux model (see Fig. 6).
Thermodynamic cycle analysis. Channel alterations that energetically affect ligand binding or gating can be analyzed formally by evaluating the contributions of individual perturbations (Carter et al., 1984; Sali et al., 1991; Hidalgo and MacKinnon, 1995). For example, alterations in binding caused by a mutation can be defined in terms of perturbations in binding energies:
| Equation 4 |
where Kmutant andKWT are the equilibrium binding constants for mutant and wild-type channels, respectively. If different amino acids contribute to the binding but they contribute independently, then altering them should independently affect the outcome. The diagram below illustrates the method used for these calculations:
The bold lettering indicates a wild-type residue. The upper left construct is the fully WT channel, and the bottom right is the EEAA double mutant channel. X1 is the dissociation constant for the WT E518:E519 channel divided by that of the single mutant 518A:E519 and represents the change in free energy between the WT and E518A single mutant. Y1 is the dissociation constant for the single mutant 518A:E519divided by that of the double mutant 518A:519A and reflects the change in free energy between the E518A single mutant and the EEAA double mutant channel. These calculations were then extended to generateX2 and Y2. The null hypothesis is that, if there is no energetic coupling between the two residues, the change in free energy between the WT and EEAA channel will be independent of which residue was mutated first, generating an energetic coupling coefficient (Ω) of unity. Ω is described by the equation (Carter et al., 1984):
| Equation 5 |
In all cases, Ω values calculated byX1/X2 were identical to those calculated byY2/Y1.
RESULTS
Direct observation of HERG inactivation: effect of extracellular Ca2+ on channel gating
The HERG K+ channel was originally described as an inwardly rectifying channel based on the current–voltage relationship (Trudeau et al., 1995). Subsequent studies indicated that this apparent rectification results from inactivation gating (Schönherr and Heinemann, 1996; Smith et al., 1996; Spector et al., 1996). In this regard, HERG K+ channels seemed to be atypical, compared with other six transmembrane family K+ channels that are either delayed rectifiers or A-type channels with rapid channel activation followed by somewhat slower inactivation. Figure1A illustrates HERG K+ currents measured during brief (50 msec) steps between +30 and +180 mV. At these membrane potentials, the rate of channel activation became rapid relative to inactivation, and the inactivation process was directly observed. Although the rate of activation remained highly voltage-dependent, the rate of inactivation was nearly constant in this voltage range.
Changes in extracellular Ca2+ modify the voltage dependence of opening of this channel with little effect on channel inactivation (Johnson et al., 1999). Figure1B demonstrates this directly. Superimposed K+ current tracings from a single cell are shown in different extracellular Ca2+concentrations ranging from 3 μm to 10 mm. In the lowest concentrations of Ca2+, the voltage step to +70 mV elicited a rapidly activating current that quickly inactivated like an A-type K+ channel. As extracellular Ca2+ was increased, channel opening was delayed, but the rate of inactivation remained unchanged. The result was a truncation of the observed current peak. Note that marked modulation occurred between 0.3 and 3 mm. The specific interaction of Ca2+ with the voltage dependence of channel activation, but not inactivation, led us to investigate the role of amino acids in the vicinity of the channel voltage sensor in determining channel Ca2+sensitivity.
Extracellular negatively charged amino acids and the effect of [Ca2+]o
Ca2+ typically interacts with negatively charged amino acid side chains of proteins (Cowan, 1993;Eismann et al., 1994; Schreiber and Salkoff, 1997). This is attributable to the fact that ionized Ca2+has a full outer electron orbital, giving it a noble gas-like unreactive chemistry. As a result, Ca2+tends to interact with other molecules largely through electrostatic attraction and repulsion (Cowan, 1993). Likely candidates for Ca2+ interaction are extracellularly exposed negatively charged amino acids such as glutamate and aspartate. Because Ca2+ does not permeate HERG channels and the intracellular solution always contained a high affinity Ca2+ chelator (5 mmBAPTA), the hydrophilic extracellular loops of the channel are candidate regions for Ca2+ interaction. The S4 transmembrane segment has been identified as a voltage sensor of channels in the six transmembrane voltage-gated K+ channel family (Papazian et al., 1987;Tempel et al., 1987; Perozo et al., 1994; Mannuzzu et al., 1996). The extracellular S3–S4 linker of HERG contains only two acidic amino acids, adjacent glutamates at positions 518 and 519, and they are located just above the S4 transmembrane segment. These negatively charged amino acids were mutated individually and together to probe their role in the response to extracellular Ca2+. The phenotypes of the WT and mutant channels are shown in Figure 2. K+ current from cells expressing WT, E518A, E519A, or the double mutant (change of E518 and E519 to alanines: EEAA) are shown at numerous test potentials. Each mutation significantly altered the gating of HERG channels in control extracellular solutions. Interestingly, the E518A mutant appeared very similar to the WT channel in elevated [Ca2+]o, e.g., removal of the negative charge caused a decrease in outward current during depolarizations and enhanced the rates of tail current decay. Thus, removal of this negative charge seemed to resemble addition of positive charges (Ca2+) in the WT channel. In all three cases (E518A, E519A, and EEAA), outward currents during depolarizations were decreased, and the rates of decay of tail currents were increased. Further evaluation of these mutant channels compared with WT revealed unique differences (described below). First, we evaluated other channel properties to determine whether inactivation or permeation properties were altered.
Fig. 2.
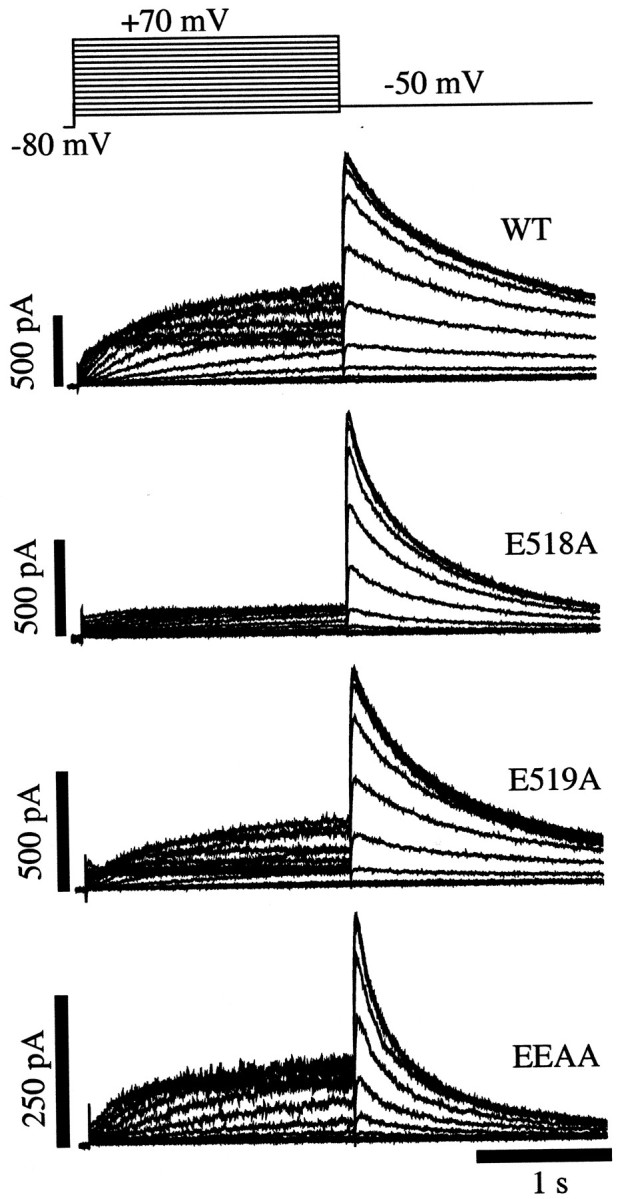
Comparison of HERG K+ currents in WT and S3–S4 charge neutralization mutants. Superimposed families of K+ current traces were recorded using the voltage-clamp protocol shown for each channel construct. Cells were held at −80 mV before stepping to test potentials between +70 and −60 mV for 2 sec. The membrane potential was then stepped to −50 mV for 2 sec to record tail currents. All records were made in the presence of 3 mm extracellular Ca2+.
Effect of negatively charged amino acids, mutations, or Ca2+ on inactivation
Our previous study demonstrated a significant effect of Ca2+ on HERG channel activation gating in the absence of an effect on inactivation (Johnson et al., 1999). Therefore, we wished to determine whether these amino acids that appeared to influence channel activation gating had an effect on channel inactivation. Despite the distinct current phenotypes of the mutant channels, their inactivation was not affected. Neutralization of E518, E519, or both together did not change the HERG channel inactivation time constants measured between 0 and +100 mV (p > 0.2; n = 4–8) (Fig.3). Thus, HERG inactivation gating is insensitive both to external Ca2+ and mutation of E518 and E519, in direct contrast to activation gating.
Fig. 3.

S3–S4 charge neutralization did not affect inactivation. The voltage-clamp protocol is shown at thetop. A representative family of currenttraces is shown in the middle panel. The step to +60 mV was 2 sec long; the scale bar to theright applies only to the current traceto the right of the break in the record. Time constants were obtained by fitting an exponential function to the decaying current during the third voltage step. Error bars indicate the SEM for 4–8 cells for each mutant and at each membrane potential.
Mutations did not change K+ selectivity
In contrast to changes in activation gating, we observed no effects on the ion permeation properties, suggesting that other channel properties were not affected by these changes. The ionic selectivity of all the mutant channels was normal, because there was no change in the K+ current reversal potential measured in 4 mm extracellular K+: WT, −83 ± 0.9 mV, n = 10; E518A, −81 ± 0.3 mV, n = 9; E519A, −82 ± 0.3 mV,n = 10; EEAA, −82 ± 0.6 mV, n = 10 (NS; p > 0.2).
Effect of negatively charged amino acids or Ca2+on channel activation
In Figure 4, voltage–activation curves are shown to compare the shifts in the voltage dependence caused by changes in [Ca2+]o in the WT and mutant HERG channels. Increasing Ca2+ caused progressive shifts of channel voltage-dependent activation relationships to more depolarized potentials in all channels, but the magnitude of the Ca2+ response and the relative position of the voltage dependence curves differed among the WT and charge-neutralized mutant channels. Note the differences in degree of separation of the curves in high and low Ca2+ and the relative shifts caused by change in Ca2+ among the different channels. There did not appear to be changes in the slope of the relationships, but the entire family of curves in a given channel (WT vs each mutant) was variously translated along the voltage axis. The degree to which elevation of Ca2+ was able to shift the curves was different among the channels as well. Initially it seemed paradoxical that channels with such similar and modest changes in amino acid sequence could behave so differently. A channel with the simple removal of a negative charge behaved very differently than when the adjacent charge was neutralized. These distinctions were very nicely accounted for by considering the allosteric nature of the channels. The solid curves through the data were generated by the allosteric gating model described below.
The Ca2+-independent voltage dependence of the channels (i.e., that measured in very low calcium: 3 μm Ca2+) differed among the channels as well. This can be seen more clearly in Figure5 in which the half maximal voltages of channel activation (V1/2) measured from data in Figure 4 are plotted as a function of the Ca2+ concentration. The double mutant (EEAA) and the E518A single mutant had a minimalV1/2 of approximately +30 mV more depolarized than that of the WT or E519A mutant channels (V1/2 in 3 μm[Ca2+]o: WT, −44 ± 6 mV; E518A, −17 ± 3 mV; E519A, −48 ± 5 mV; EEAA, −17 ± 8 mV). For all channels, theV1/2–[Ca2+] curve saturated before reaching the lowest tested [Ca2+] (3 μm). Therefore, the minimal V1/2 can be considered Ca2+ independent. Neutralization of glutamate 518, but not 519, resulted in channels with an altered Ca2+-independent voltage dependence. Channels in which E519 was neutralized had an altered Ca2+ response, but their Ca2+-independent voltage dependence was like that of WT channels.
The Ca2+ responses of the WT and mutant channels for currents measured at membrane potentials between −60 and +30 mV were fit with a Hill equation (Eq. 2; same data as in Fig. 4analyzed as a function of [Ca2+]o; data not shown). The fitted parameters (KD and Hill coefficient, b) were highly voltage dependent. The Hill coefficients from these fits varied from a minimum near one to a maximum greater than three depending on the membrane potential, suggesting multiple binding sites and cooperativity. Thus, a single Ca2+ binding site seems unlikely for several reasons. First, the Hill coefficient (b > 1, Eq. 2) values indicate increased complexity and suggest multiple Ca2+ binding sites. Secondly, HERG, like other six transmembrane domain voltage-gated K+ channels, is believed to form rotationally symmetric homotetrameric channels when expressed alone in heterologous systems. If all four subunits cooperated to create a single Ca2+ binding site on the extracellular face of the channel, this site would necessarily be centrally located and would therefore likely be in the channel pore. However, we know that Ca2+ does not block HERG channels as would be expected for a binding site in the pore lumen (Johnson et al., 1999). In the absence of a single centrally located Ca2+ binding site, a likely possibility for a homotetrameric channel is that each subunit has its own Ca2+ site(s). The similarities to hanatoxin modification of Kv2.1 and omega-Aga-IVA modification of calcium channels are also consistent with four Ca2+ sites and certainly more than one (Swartz and MacKinnon, 1995, 1997a,b; Li-Smerin and Swartz, 1998;Winterfield and Swartz, 2000).
The Hill fit-derived parameters must be interpreted cautiously, and the apparent voltage dependence of the Hill-derived KDs can be interpreted in several ways. First, this nonglobal analysis (i.e., fitting only a single independent variable at a time) neglects relevant information about the system, for example membrane potential. Although the Ca2+ response is obtained at several membrane potentials, each fit is independent and not constrained by data at other membrane potentials. The Hill fit analysis also lumps all interactions (possibly multiple KDs) into a single KD. In contrast, the global fit (discussed below) is constrained by information obtained across all membrane potentials and calcium concentrations. The voltage-dependent apparent KD values from the Hill fit analysis suggest several possible interpretations. First, the accessibility of the ligand to a single binding site of constant affinity could change as a function of membrane potential, because of the voltage-dependent channel conformational changes that must occur for channel function, i.e., the binding site is partially guarded. This would cause an apparent change in the affinity, but cannot account for the Hill coefficients that differ from unity. Another possible explanation for the voltage-dependent KD is that Ca2+ binding is influenced by the electric field through a direct effect on the positively charged ligand. In the Hill equation analysis, the fitted KDs ranged from ≤10−4m at −50 mV to 5 × 10−3m at +25 mV. These changes are consistent with a positively charged ligand binding with higher affinity at negative membrane potentials. This cannot readily explain the voltage-dependent changes in Hill slopes either.
In addition and not mutually exclusive, these observations could indicate that the affinity of the binding site for Ca2+ changes as a function of membrane potential because of a change in the protein conformation. For example, different channel conformations (closed or activated) may have distinct Ca2+ affinities. The data indicate that the resting closed channels bind Ca2+ with a higher affinity than the activated channel. Membrane potential determines the fraction of channels that are in the resting or activated states and the apparent KD increases at more depolarized potentials in which more channels are activated.
The voltage dependence of the Hill fits led us to consider an allosteric model for the Ca2+-channel interactions. Many multisubunit proteins including ion channels exhibit cooperativity, and conformational changes that confer differential ligand affinity have been described for many allosteric proteins (Monod et al., 1965; Hille, 1977; Hondeghem and Katzung, 1977; Marks and Jones, 1992; Galzi et al., 1996; Cox et al., 1997; Changeux and Edelstein, 1998; Horrigan and Aldrich, 1999; Rothberg and Magleby, 1999).
Allosteric modulation of gating by Ca2+
To refine the analysis of the Ca2+-HERG interaction, we considered voltage-dependent allosteric models of channel-gating and Ca2+ interactions. Because HERG is a voltage-gated channel (Sanguinetti et al., 1995; Trudeau et al., 1995), numerous states must be considered to account for channel voltage dependence (Patlak, 1991; Bezanilla et al., 1994; S. Wang et al., 1997). These channels are homotetramers when expressed heterologously. The four-fold symmetry of the channel naturally leads to at least five states to describe channels with 0–4 voltage sensors activated. Channel inactivation requires additional states. If only a single Ca2+ binds to each homotetrameric channel, then all four subunits must create a single Ca2+ binding site on the extracellular face of the channel. This site would necessarily be centrally located, as with tetraethylammonium (TEA) and certain toxins (Hurst et al., 1992), and would therefore likely be in the channel pore. However, Ca2+ does not block HERG channels (Johnson et al., 1999). The radial symmetry of the channel, the apparent cooperativity seen in the Ca2+ concentration–response curve Hill fits (data not shown), and the unlikelihood of a single central Ca2+ binding site suggest that each subunit has its own Ca2+ site(s). Minimally, to accommodate five voltage-dependent states and five calcium-liganded states requires a 25 state model. To include Ca2+ and voltage dependence and the allosteric changes from closed to activated channel conformations requires at least a two-tiered 50 state allosteric modelRothberg and Magleby, 1999). Nonindependent voltage sensors or Ca2+ binding would lead to even larger models (Horrigan et al., 1999).
As the complexity of these models increases, the number of free parameters increases, and confidence in the meaning of individual parameters decreases. Results shown below and in our previous work (Johnson et al., 1999) indicate that the inactivated states cannot be distinguished from open states with regard to Ca2+; hence, combining open and inactivated states that are occupied during depolarizations can be justified. Because the primary goal of this work was to understand the Ca2+ modulation of HERG, we can further simplify the analysis by combining the voltage sensing steps into a single concerted transition. There is reasonable evidence that in other voltage-gated K+ channels (Bezanilla, 2000), Ca2+-activated K+ channels (Cox et al., 1997; Horrigan et al., 1999; Rothberg and Magleby, 1999), and Ca2+ channels (Marks and Jones, 1992) a final cooperative step is indeed responsible for channels leaving the closed state.
Assuming four binding sites for the tetrameric channel, there are at least five types of states corresponding to the channel with 0–4 Ca2+ bound. The open to inactivated transitions of HERG are Ca2+ insensitive (Johnson et al., 1999), which permitted us to combine the open and inactivated states (referred to as activated). Making the assumption that only the voltage-dependent conformation of the channel (closed or activated) determines the affinity for Ca2+ and that each subunit binds Ca2+ independently from the others, the degree of Ca2+ binding then biases the fundamental voltage-dependent transition. The result is the simplified two-tiered, 10 state MWC model [see Cox et al. (1997) for in-depth discussion of this approach shown below in scheme 1 and described in Eq. 3] if Ca2+ only distinguishes closed and activated states (i.e., all closed state dissociation constants, Kc, are equivalent, as are all activated state dissociation constants, Ka). In this model, the receptor (here the HERG channel) protein exists in two possible conformations, tense or relaxed, corresponding in this case to the closed or activated channel (Monod et al., 1965; Changeux and Edelstein, 1998).
This voltage-dependent allosteric model (see Eq. 3) was fit to all of the data simultaneously as a function of the known membrane potentials and Ca2+ concentrations. Figure6 shows the voltage- and Ca2+-response surface that was fitted to these data sets. The shaded gray bands indicate the height along the vertical axis. Round symbols show the mean data points, and the vertical lines on the symbols show the distance of the data points from the fitted plane, illustrating the quality of the fit.
This simple model fit the observed data remarkably well. The smooth curves in Figures 4 and 5, as well as the shaded gray surface in Figure 6, are all from the model fit. The fitted parameters are shown in Figure 7. The sensitivity of the channels to changes in membrane potential was similar in all channels and at all Ca2+ concentrations, as reflected by the fitted Q (apparent gating charge), which was the same for all the channels (mean ± SE; WT = 2.3 ± 0.05 e−, E518A = 2.5 ± 0.04 e−, E519A = 2.4 ± 0.05e−, EEAA = 2.5 ± 0.002 e−). The constancy of Q among the different channels suggests that the E518 and E519 do not constitute part of the gating charge and are not directly responsible for voltage sensing. The change in free energy required to move the channels from closed to activated states in the absence of Ca2+ and voltage (L0) differed considerably among the channels. This is apparent not only from fittedL0 values (Fig. 7), but also by the increased voltage threshold for channel activation in the E518A and EEAA mutants visible in Figures 4 and 5. In contrast, the E519A mutant had an L0 similar to that of the wild-type channel. Removal of the negative charge at position 518 had little effect on the Ca2+ dissociation constant of the closed channel (Kc; Fig. 7), but channels bearing an E519A mutation had a Kc twice that of the WT or E518A mutant, indicating that neutralization of this negative charge decreased the affinity of the closed channel for Ca2+.
Fig. 7.

Fitted parameters of the voltage-dependent MWC model for WT and mutant HERGs. L0 is the equilibrium constant for the allosteric transition between the closed and activated states in the absence of Ca2+ at 0 mV.Q is the equivalent gating charge ine−. Kc is the Ca2+ dissociation constant for the closed channel.Ka is the Ca2+ dissociation constant for the activated channel. The parameter uncertainties (SDs) are shown as error bars and were determined from the covariance matrix.
We found that the E518A mutant had a reduced Ka, indicating an enhanced affinity of activated channels for Ca2+, despite the fact that Kawas not reduced in the EEAA double mutant. This observation suggests that the 518 and 519 amino acids are not entirely independent with respect to their influence on activated channel Ca2+ binding. To quantify the degree of energetic coupling between these amino acids, we used thermodynamic mutant cycle analysis (see Scheme FS1 and Eq. 4) (Carter et al., 1984;Sali et al., 1991; Hidalgo and MacKinnon, 1995; Schreiber and Fersht, 1995; Horovitz, 1996; Ranganathan et al., 1996; Frisch et al., 1997). The distinct Ca2+ dissociation constants for the closed (Kc) and activated (Ka) states and the equilibrium constant (L0) for the distribution among closed and activated states for each channel were used to estimate the energetic interactions between these charged amino acids in the different conformational states. Using these parameters, a coupling coefficient (Ω) was calculated (Eq. 4). AnΩ of unity indicates independence and a lack of energetic coupling. Deviations of Ω from unity indicate energetic coupling between the amino acids. The Ω values were:L0, 21.2; Kc, 1.1;Ka, 2.4. The large Ω for theL0 indicates that there is energetic coupling between these amino acids in determining the equilibrium change in free energy between the closed and activated states, although clearly E518 was more critical in determining theL0 value (Fig. 7). The coupling coefficient for the closed channel Ca2+dissociation constant was near unity, indicating a lack of coupling between these residues in the closed states. In contrast,Ω for activated channels was 2.4, indicating that an interaction between these amino acids influences Ca2+ binding on channel opening. ThisΩ value is relatively small, but it is noteworthy because these adjacent residues did not display coupling in the closed state. This implies a change in the microenvironment around E518 and E519 between the closed and activated channels. The negative charge at position 518 influences Ca2+ association when the channel is in the activated state but not when the channel is closed.
Fig. FS1.

DISCUSSION
Our data revealed a novel mechanism for extracellular calcium modulation of an important ion channel and indicated that the affinity of HERG channels for Ca2+ is dependent on its conformational state. Significant changes in gating occur with very modest changes in [Ca2+]obecause the modulation occurs in the middle of the normal extracellular range of calcium concentrations. This suggests that rather modest fluctuations in [Ca2+]o in clefts or restricted spaces can have marked changes on this repolarizing K+ current.
We find that negatively charged amino acids in the S3–S4 linker critically bias normal voltage and Ca2+dependence of the channel. Ca2+ is well known as an intracellular second messenger, but recently it has also been found to be an important first messenger extracellularly (Brown, 1999). Extracellular Ca2+ receptors are involved in regulation of serum Ca2+. These receptors, like HERG, have millimolar affinity for Ca2+. Even small changes in extracellular Ca2+ can have large effects on HERG current density under conditions resembling those in vivo(Johnson et al., 1999). A low-affinity interaction between Ca2+ and extracellular sites on proteins like HERG and the Ca2+ receptor is mandatory if physiological fluctuations in Ca2+ are to dynamically modify protein function. At normal mammalian serum Ca2+concentrations of ∼1.3 mm (Brown, 1999), a high-affinity site, like an EF hand motif, would be continuously occupied, preventing modulation.
Our results indicate that the glutamate at position 518 is critical in determining the normal voltage dependence of HERG channels. In contrast, the glutamate at position 519 has little effect on the intrinsic voltage dependence of the channel, but instead is involved in determining the response of the channel to extracellular Ca2+. What is the physical explanation for these effects? The diagram in Figure8 shows one possibility. Transmembrane domains of a single channel subunit emphasizing the positively charged S4 transmembrane voltage sensor domain are represented. In the diagram, activation of the voltage sensor is shown as a movement of the S4 as recent studies indicate (Larsson et al., 1996; Cha and Bezanilla, 1997,1998; Cha et al., 1999; Bezanilla, 2000; Horn, 2000). The negative charge of a glutamate (E518) in the extracellular loop connecting S3 and S4 electrostatically interacts with positive charge(s) of the S4 voltage sensor, encouraging (attracting) the activating motion of the S4. Removing the negative charge at position 518 (E518A mutation) increases the energy required for activation and thus shifts the voltage dependence of activation to more positive potentials. Removing the negative charge at position 518 (E518A mutation) also increases the affinity of the channel for Ca2+ during channel activation (opening) relative to the WT channel, suggesting that the Ca2+ binding motif is better able to associate stably with Ca2+. The negative charge of a glutamate (E519) participates in the association of Ca2+ with the channel protein, and thus the E519A mutation alters the Ca2+response. A conformational change in the S3–S4 loop during channel activation gating (opening) interferes with the association of Ca2+ with the channel protein, lowering the affinity.
Fig. 8.

Top, Amino acid alignment of S3–S4 segments of several voltage-gated potassium channels. Standard single letter amino acid abbreviations are used. Glutamate 518 (E518) in HERG is indicated by an arrow. Kv1.1,Kv1.5, Kv2.1, Kv3.1, andKv4.3 indicate the human channels corresponding to KCNA1, KCNA5, KCNB1, KCNC1, and KCND3, respectively.eag is Drosophilaether-à-go-go, and HERG corresponds to KCNH2.Bottom, Diagram of a plausible physical explanation for the interactions between negative charges in S3–S4, positive charges in S4, and extracellular Ca2+. Ca2+ interacts at the surface of the channel at a site near the S4 voltage sensor by attraction to the negative charge of glutamate 519. The data suggest that E519 may participate in the Ca2+ binding site. The negative charge of glutamate 518 (E518) attracts or stabilizes positive charges in S4 and promotes or facilitates channel opening. Thus, E518 affects the Ca2+-independent voltage-dependence of the channel by electrostatic interaction with the voltage sensor. Adapted fromPapazian and Bezanilla (1999) for HERG channels.
It should be noted that we have no direct proof that either of the two glutamic acid residues that we investigated directly interacts with Ca2+. They may merely influence its effect on the channel, perhaps through the surrounding electrostatic field projected from the protein surface or through other allosteric mechanisms. Our data do show that the actions are fairly specific and that these amino acids are involved. Calcium ions are typically coordinated by multiple negatively charged amino acids, and mutation of one of the coordinate sites is highly disruptive of high-affinity interactions. The interaction seen here is rather low affinity, unlike typical Ca2+ coordination sites, for example in calmodulin, so perhaps there is no such coordination, but simply an electrostatic interaction. This could explain both the lower affinity and the modest disruption caused by the mutations, but it requires other interaction sites.
Interestingly, many peptide toxins also modify voltage-gated ion channels by altering voltage dependence through an association with the S3–S4 extracellular domain. The voltage dependences of Na+, Ca2+, and K+ channel gating are modified by differences in this region or by binding of toxins to the S3–S4 linker domains (Rogers et al., 1996; Dib-Hajj et al., 1997; McDonough et al., 1997; Cestele et al., 1998). Splice variants of α1A (Bourinet et al., 1999) and α1B (Hans et al., 1999; Lin et al., 1999) Ca2+ channel subunits that differ in this region are distinguishable by their altered voltage dependence of gating.
Other K+ channels are also sensitive to changes in the extracellular residues near the voltage sensor (Elinder and Århem, 1999). Tang et al. (2000) characterized the effects of Mg2+ on Drosophila ether-à-go-go K+ channel (eag). They found that Mg2+ slowed activation eag-gating kinetics. Splice variants of bovine eag that differ in the length of the S3–S4 loop show distinct Mg2+ sensitivities (Frings et al., 1998).Drosophila eag contains a DRED (Asp-Arg-Glu-Asp) motif at amino acids 333–337 at the C-terminal end of S3. Tang et al. (2000)deleted these charged amino acids and found a depolarizing shift in the voltage–activation curves consistent with removal of a negative surface charge, but gating was still modified by Mg2+. They concluded that the DRED sequence does not constitute an Mg2+binding site. HERG channels do not possess DRED amino acids at analogous positions. Interestingly, mutation of leucine at position 342 to histidine (L342H) in S3–S4 eliminated the modulation by Mg2+. It is unlikely that leucine is directly involved in Mg2+ binding, although a mutation at this position may perturb Mg2+ binding or the coupling of the Mg2+ effect to the voltage sensor. Alternatively, addition of a charged histidine may prevent access to an Mg2+ site by introduction of a local positive charge. More recently, Silverman et al. (2000) provided evidence that aspartic acids at positions 278 and 327 in eag constitute an Mg2+ binding site. Mutations of these negatively charged amino acids to alanines greatly altered the response to Mg2+, although no concentration dependence was defined. As such it is not possible to compare the binding affinities or energetics. The composition of the S3–S4 linker affects the voltage sensitivity of Shaker K+ channel gating (Mathur et al., 1997), and natural toxins affect K+ channel gating by binding near this site (Swartz and MacKinnon, 1997a,b), all consistent with our observations on HERG channels that S3–S4 is a critical modulatory domain. Together, these observations of interactions between extracellular effectors and channel S3–S4 regions near the voltage sensor reveal the general importance of this key modulatory domain (Li-Smerin and Swartz, 1998; Winterfield and Swartz, 2000).
We conclude that extracellular Ca2+ is an allosteric modulator of the HERG K+ channel. Ca2+ associates with closed channels (occupied at negative membrane potentials) with higher affinity than with activated channels (occupied at less negative membrane potentials), thereby stabilizing the closed state. Two glutamates in the S3–S4 linker domain, E518 and E519, are critical for normal HERG function. Neutralization of E518 shifts the voltage dependence of channel opening to more depolarized membrane potentials promoting closed states, possibly by removing a negative charge that is normally attracting the S4 positive charge. Neutralization of E518 does not affect the affinity of the closed channel for Ca2+. Neutralization of E519 does not appear to affect the membrane potential sensed by the voltage sensor but does decrease the affinity of the closed channel for Ca2+. The E518 and E519 residues are energetically coupled in the activated channel but not in the closed channel. These results emphasize the importance of the S3–S4 linker in channel function and present a mechanism of Ca2+ action on this physiologically important ion channel.
Footnotes
This work was supported by National Institutes of Health Grants T32 HL07411, T32 GM07628, HL 51197, and HL 46681. We thank Drs. Louis J. DeFelice and Christoph Fahlke for their insightful discussions and Dr. Christoph Fahlke for critical reading of this manuscript.
Correspondence should be addressed to Dr. Paul B. Bennett, Senior Director, Ion Channel Research, WP42-209 (Pharmacology), Merck Research Laboratories, 770 Sumneytown Pike, West Point, PA 19486. E-mail:paul_bennett@merck.com.
REFERENCES
- 1.Arcangeli A, Rosati B, Cherubini A, Crociani O, Fontana L, Ziller C, Wanke E, Olivotto M. HERG- and IRK-like inward rectifier currents are sequentially expressed during neuronal development of neural crest cells and their derivatives. Eur J Neurosci. 1997;9:2596–2604. doi: 10.1111/j.1460-9568.1997.tb01689.x. [DOI] [PubMed] [Google Scholar]
- 2.Balser JR, Roden DM, Bennett PB. Global parameter optimization for cardiac potassium channel gating models. Biophys J. 1990;57:433–444. doi: 10.1016/S0006-3495(90)82560-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bezanilla F. The voltage sensor in voltage-dependent ion channels. Physiol Rev. 2000;80:555–592. doi: 10.1152/physrev.2000.80.2.555. [DOI] [PubMed] [Google Scholar]
- 4.Bezanilla F, Perozo E, Stefani E. Gating of Shaker K+ channels: II. The components of gating currents and a model of channel activation. Biophys J. 1994;66:1011–1021. doi: 10.1016/S0006-3495(94)80882-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bianchi L, Wible B, Arcangeli A, Taglialatela M, Morra F, Castaldo P, Crociani O, Rosati B, Faravelli L, Olivotto M, Wanke E. Herg encodes a K+ current highly conserved in tumors of different histogenesis: a selective advantage for cancer cells. Cancer Res. 1998;58:815–822. [PubMed] [Google Scholar]
- 6.Bourinet E, Soong TW, Sutton K, Slaymaker S, Mathews E, Monteil A, Zamponi GW, Nargeot J, Snutch TP. Splicing of alpha 1A subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nat Neurosci. 1999;2:407–415. doi: 10.1038/8070. [DOI] [PubMed] [Google Scholar]
- 7.Brown EM. Physiology and pathophysiology of the extracellular calcium-sensing receptor. Am J Med. 1999;106:238–253. [PubMed] [Google Scholar]
- 8.Carter PJ, Winter G, Wilkinson AJ, Fersht AR. The use of double mutants to detect structural changes in the active site of the tyrosyl-tRNA synthetase (Bacillus stearothermophilus). Cell. 1984;38:835–840. doi: 10.1016/0092-8674(84)90278-2. [DOI] [PubMed] [Google Scholar]
- 9.Cestele S, Qu Y, Rogers JC, Rochat H, Scheuer T, Catterall WA. Voltage sensor-trapping: enhanced activation of sodium channels by beta-scorpion toxin bound to the S3–S4 loop in domain II. Neuron. 1998;21:919–931. doi: 10.1016/s0896-6273(00)80606-6. [DOI] [PubMed] [Google Scholar]
- 10.Cha A, Bezanilla F. Characterizing voltage-dependent conformational changes in the Shaker K+ channel with fluorescence. Neuron. 1997;19:1127–1140. doi: 10.1016/s0896-6273(00)80403-1. [DOI] [PubMed] [Google Scholar]
- 11.Cha A, Bezanilla F. Structural implications of fluorescence quenching in the Shaker K+ channel. J Gen Physiol. 1998;112:391–408. doi: 10.1085/jgp.112.4.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cha A, Snyder GE, Selvin PR, Bezanilla F. Atomic scale movement of the voltage-sensing region in a potassium channel measured via spectroscopy. Nature. 1999;402:809–813. doi: 10.1038/45552. [DOI] [PubMed] [Google Scholar]
- 13.Changeux JP, Edelstein SJ. Allosteric receptors after 30 years. Neuron. 1998;21:959–980. doi: 10.1016/s0896-6273(00)80616-9. [DOI] [PubMed] [Google Scholar]
- 14. Chiesa N, Rosati B, Arcangeli A, Olivotto M, Wanke E. A novel role for HERG K+ channels: spike-frequency adaptation. J Physiol (Lond) 501 1997. 313 318 [Erratum (1997) 502:715] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cowan JA. Inorganic biochemistry: an introduction. VCH; New York: 1993. [Google Scholar]
- 16.Cox DH, Cui J, Aldrich RW. Allosteric gating of a large conductance Ca-activated K+ channel. J Gen Physiol. 1997;110:257–281. doi: 10.1085/jgp.110.3.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dib-Hajj SD, Ishikawa K, Cummins TR, Waxman SG. Insertion of a SNS-specific tetrapeptide in S3–S4 linker of D4 accelerates recovery from inactivation of skeletal muscle voltage-gated Na channel mu1 in HEK293 cells. FEBS Lett. 1997;416:11–14. doi: 10.1016/s0014-5793(97)01154-x. [DOI] [PubMed] [Google Scholar]
- 18.Eismann E, Muller F, Heinemann SH, Kaupp UB. A single negative charge within the pore region of a cGMP-gated channel controls rectification, Ca2+ blockage, and ionic selectivity. Proc Natl Acad Sci USA. 1994;91:1109–1113. doi: 10.1073/pnas.91.3.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elinder F, Århem P. Role of individual surface charges of voltage-gated K channels. Biophys J. 1999;77:1358–1362. doi: 10.1016/S0006-3495(99)76984-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frankenhaeuser B, Hodgkin AL. The action of calcium on the electrical properties of squid axons. J Physiol (Lond) 1957;137:218–244. doi: 10.1113/jphysiol.1957.sp005808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frings S, Brull N, Dzeja C, Angele A, Hagen V, Kaupp UB, Baumann A. Characterization of ether-a-go-go channels present in photoreceptors reveals similarity to IKx, a K+ current in rod inner segments. J Gen Physiol. 1998;111:583–599. doi: 10.1085/jgp.111.4.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frisch C, Schreiber G, Johnson CM, Fersht AR. Thermodynamics of the interaction of barnase and barstar: changes in free energy versus changes in enthalpy on mutation. J Mol Biol. 1997;267:696–706. doi: 10.1006/jmbi.1997.0892. [DOI] [PubMed] [Google Scholar]
- 23.Galzi JL, Edelstein SJ, Changeux JP. The multiple phenotypes of allosteric receptor mutants. Proc Natl Acad Sci USA. 1996;93:1853–1858. doi: 10.1073/pnas.93.5.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gilly WF, Armstrong CM. Divalent cations and the activation kinetics of potassium channels in squid giant axons. J Gen Physiol. 1982;79:965–996. doi: 10.1085/jgp.79.6.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hans M, Urrutia A, Deal C, Brust PF, Stauderman K, Ellis SB, Harpold MM, Johnson EC, Williams ME. Structural elements in domain IV that influence biophysical and pharmacological properties of human alpha1A-containing high-voltage-activated calcium channels. Biophys J. 1999;76:1384–1400. doi: 10.1016/S0006-3495(99)77300-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hidalgo P, MacKinnon R. Revealing the architecture of a K+ channel pore through mutant cycles with a peptide inhibitor. Science. 1995;268:307–310. doi: 10.1126/science.7716527. [DOI] [PubMed] [Google Scholar]
- 27.Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J Gen Physiol. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ho WK, Kim I, Lee CO, Earm YE. Voltage-dependent blockade of HERG channels expressed in Xenopus oocytes by external Ca2+ and Mg2+. J Physiol (Lond) 1998;507:631–638. doi: 10.1111/j.1469-7793.1998.631bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hondeghem LM, Katzung BG. Time- and voltage-dependent interactions of antiarrhythmic drugs with cardiac sodium channels. Biochim Biophys Acta. 1977;472:373–398. doi: 10.1016/0304-4157(77)90003-x. [DOI] [PubMed] [Google Scholar]
- 30.Horn R. A new twist in the saga of charge movement in voltage-dependent ion channels. Neuron. 2000;25:511–514. doi: 10.1016/s0896-6273(00)81055-7. [DOI] [PubMed] [Google Scholar]
- 31.Horovitz A. Double-mutant cycles: a powerful tool for analyzing protein structure and function. Fold Des. 1996;1:R121–R126. doi: 10.1016/S1359-0278(96)00056-9. [DOI] [PubMed] [Google Scholar]
- 32.Horrigan FT, Aldrich RW. Allosteric voltage gating of potassium channels II: Mslo channel gating charge movement in the absence of Ca2+. J Gen Physiol. 1999;114:305–336. doi: 10.1085/jgp.114.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hurst RS, Kavanaugh MP, Yakel J, Adelman JP, North RA. Cooperative interactions among subunits of a voltage-dependent potassium channel. Evidence from expression of concatenated cDNAs. J Biol Chem. 1992;267:23742–23745. [PubMed] [Google Scholar]
- 34.Johnson JPJ, Mullins FM, Bennett PB. Human ether-a-go-go-related gene K+ channel gating probed with extracellular Ca2+. Evidence for two distinct voltage sensors. J Gen Physiol. 1999;113:565–580. doi: 10.1085/jgp.113.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jurman ME, Boland LM, Liu Y, Yellen G. Visual identification of individual transfected cells for electrophysiology using antibody-coated beads. Biotechniques. 1994;17:876–881. [PubMed] [Google Scholar]
- 36.Larsson HP, Baker OS, Dhillon DS, Isacoff EY. Transmembrane movement of the shaker K+ channel S4. Neuron. 1996;16:387–397. doi: 10.1016/s0896-6273(00)80056-2. [DOI] [PubMed] [Google Scholar]
- 37.Lin Z, Lin Y, Schorge S, Pan JQ, Beierlein M, Lipscombe Alternative splicing of a short cassette exon in alpha1B generates functionally distinct N-type calcium channels in central and peripheral neurons. J Neurosci. 1999;19:5322–5331. doi: 10.1523/JNEUROSCI.19-13-05322.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li-Smerin Y, Swartz KJ. Gating modifier toxins reveal a conserved structural motif in voltage-gated Ca2+ and K+ channels. Proc Natl Acad Sci USA. 1998;95:8585–8589. doi: 10.1073/pnas.95.15.8585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mannuzzu LM, Moronne MM, Isacoff EY. Direct physical measure of conformational rearrangement underlying potassium channel gating. Science. 1996;271:213–216. doi: 10.1126/science.271.5246.213. [DOI] [PubMed] [Google Scholar]
- 40.Marks TN, Jones SW. Calcium currents in the A7r5 smooth muscle-derived cell line. An allosteric model for calcium channel activation and dihydropyridine agonist action. J Gen Physiol. 1992;99:367–390. doi: 10.1085/jgp.99.3.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mathur R, Zheng J, Yan Y, Sigworth FJ. Role of the S3–S4 linker in Shaker potassium channel activation. J Gen Physiol. 1997;109:191–199. doi: 10.1085/jgp.109.2.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McDonough SI, Lampe RA, Keith RA, Bean BP. Voltage-dependent inhibition of N- and P-type calcium channels by the peptide toxin omega-grammotoxin-SIA. Mol Pharmacol. 1997;52:1095–1104. doi: 10.1124/mol.52.6.1095. [DOI] [PubMed] [Google Scholar]
- 43.Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: a plausible model. J Mol Biol. 1965;12:88–1181. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 44.Papazian DM, Bezanilla F. Voltage-dependent activation of ion channels. Adv Neurol. 1999;79:481–491. [PubMed] [Google Scholar]
- 45.Papazian DM, Schwarz TL, Tempel BL, Jan YN, Jan LY. Cloning of genomic and complementary DNA from Shaker, a putative potassium channel gene from Drosophila. Science. 1987;237:749–753. doi: 10.1126/science.2441470. [DOI] [PubMed] [Google Scholar]
- 46.Patlak J. Molecular kinetics of voltage-dependent Na+ channels. Physiol Rev. 1991;71:1047–1080. doi: 10.1152/physrev.1991.71.4.1047. [DOI] [PubMed] [Google Scholar]
- 47.Perozo E, Santacruz-Toloza L, Stefani E, Bezanilla F, Papazian DM. S4 mutations alter gating currents of Shaker K channels. Biophys J. 1994;66:345–354. doi: 10.1016/s0006-3495(94)80783-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Po SS, Wang DW, Yang IC, Johnson JP, Jr, Nie L, Bennett PB. Modulation of HERG potassium channels by extracellular magnesium and quinidine. J Cardiovasc Pharmacol. 1999;33:181–185. doi: 10.1097/00005344-199902000-00002. [DOI] [PubMed] [Google Scholar]
- 49.Ranganathan R, Lewis JH, MacKinnon R. Spatial localization of the K+ channel selectivity filter by mutant cycle-based structure analysis. Neuron. 1996;16:131–139. doi: 10.1016/s0896-6273(00)80030-6. [DOI] [PubMed] [Google Scholar]
- 50.Rogers JC, Qu Y, Tanada TN, Scheuer T, Catterall WA. Molecular determinants of high affinity binding of alpha-scorpion toxin and sea anemone toxin in the S3–S4 extracellular loop in domain IV of the Na+ channel alpha subunit. J Biol Chem. 1996;271:15950–15962. doi: 10.1074/jbc.271.27.15950. [DOI] [PubMed] [Google Scholar]
- 51.Rothberg BS, Magleby KL. Gating kinetics of single large-conductance Ca2+-activated K+ channels in high Ca2+ suggest a two-tiered allosteric gating mechanism. J Gen Physiol. 1999;114:93–124. doi: 10.1085/jgp.114.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sali D, Bycroft M, Fersht AR. Surface electrostatic interactions contribute little of stability of barnase. J Mol Biol. 1991;220:779–788. doi: 10.1016/0022-2836(91)90117-o. [DOI] [PubMed] [Google Scholar]
- 53.Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- 54.Schönherr R, Heinemann SH. Molecular determinants for activation and inactivation of HERG, a human inward rectifier potassium channel. J Physiol (Lond) 1996;493:635–642. doi: 10.1113/jphysiol.1996.sp021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schreiber G, Fersht AR. Energetics of protein–protein interactions: analysis of the barnase-barstar interface by single mutations and double mutant cycles. J Mol Biol. 1995;248:478–486. doi: 10.1016/s0022-2836(95)80064-6. [DOI] [PubMed] [Google Scholar]
- 56.Schreiber M, Salkoff L. A novel calcium-sensing domain in the BK channel. Biophys J. 1997;73:1355–1363. doi: 10.1016/S0006-3495(97)78168-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Silverman WR, Tang CY, Mock AF, Huh KB, Papazian DM. Mg2+ modulates voltage-dependent activation in ether-a-go-go potassium channels by binding between transmembrane segments S2 and S3. J Gen Physiol. 2000;116:663–678. doi: 10.1085/jgp.116.5.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smith PL, Baukrowitz T, Yellen G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature. 1996;379:833–836. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- 59.Spector PS, Curran ME, Zou A, Keating MT, Sanguinetti MC. Fast inactivation causes rectification of the IKr channel. J Gen Physiol. 1996;107:611–619. doi: 10.1085/jgp.107.5.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Swartz KJ, MacKinnon R. An inhibitor of the Kv2.1 potassium channel isolated from the venom of a Chilean tarantula. Neuron. 1995;15:941–949. doi: 10.1016/0896-6273(95)90184-1. [DOI] [PubMed] [Google Scholar]
- 61.Swartz KJ, MacKinnon R. Hanatoxin modifies the gating of a voltage-dependent K+ channel through multiple binding sites. Neuron. 1997a;18:665–673. doi: 10.1016/s0896-6273(00)80306-2. [DOI] [PubMed] [Google Scholar]
- 62.Swartz KJ, MacKinnon R. Mapping the receptor site for hanatoxin, a gating modifier of voltage-dependent K+ channels. Neuron. 1997b;18:675–682. doi: 10.1016/s0896-6273(00)80307-4. [DOI] [PubMed] [Google Scholar]
- 63.Tang CY, Bezanilla F, Papazian DM. Extracellular Mg2+ modulates slow gating transitions and the opening of Drosophila ether-a-go-go potassium channels. J Gen Physiol. 2000;115:319–338. doi: 10.1085/jgp.115.3.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tempel BL, Papazian DM, Schwarz TL, Jan YN, Jan LY. Sequence of a probable potassium channel component encoded at Shaker locus of Drosophila. Science. 1987;237:770–775. doi: 10.1126/science.2441471. [DOI] [PubMed] [Google Scholar]
- 65.Titus SA, Warmke JW, Ganetzky B. The Drosophila erg K+ channel polypeptide is encoded by the seizure locus. J Neurosci. 1997;17:875–881. doi: 10.1523/JNEUROSCI.17-03-00875.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–95. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- 67.Wang J, Trudeau MC, Zappia AM, Robertson GA. Regulation of deactivation by an amino terminal domain in human ether-a-go-go-related gene potassium channels. J Gen Physiol. 1998;112:637–647. doi: 10.1085/jgp.112.5.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang S, Morales MJ, Liu S, Strauss HC, Rasmusson RL. Time, voltage and ionic concentration dependence of rectification of h-erg expressed in Xenopus oocytes. FEBS Lett. 1996;389:167–173. doi: 10.1016/0014-5793(96)00570-4. [DOI] [PubMed] [Google Scholar]
- 69.Wang S, Liu S, Morales MJ, Strauss HC, Rasmusson RL. A quantitative analysis of the activation and inactivation kinetics of HERG expressed in Xenopus oocytes. J Physiol (Lond) 1997;502:45–60. doi: 10.1111/j.1469-7793.1997.045bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang XJ, Reynolds ER, Deak P, Hall LM. The seizure locus encodes the Drosophila homolog of the HERG potassium channel. J Neurosci. 1997;17:882–890. doi: 10.1523/JNEUROSCI.17-03-00882.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Warmke JW, Ganetzky B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc Natl Acad Sci USA. 1994;91:3438–3442. doi: 10.1073/pnas.91.8.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Winterfield JR, Swartz KJ. A hot spot for the interaction of gating modifier toxins with voltage-dependent ion channels. J Gen Physiol. 2000;116:637–644. doi: 10.1085/jgp.116.5.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zou A, Xu QP, Sanguinetti MC. A mutation in the pore region of HERG K+ channels expressed in Xenopus oocytes reduces rectification by shifting the voltage dependence of inactivation. J Physiol (Lond) 1998;509:129–137. doi: 10.1111/j.1469-7793.1998.129bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]