

Abstract
An analysis of programmed cell death of several populations of developing postmitotic neurons after genetic deletion of two key members of the caspase family of pro-apoptotic proteases, caspase-3 and caspase-9, indicates that normal neuronal loss occurs. Although the amount of cell death is not altered, the death process may be delayed, and the cells appear to use a nonapoptotic pathway of degeneration. The neuronal populations examined include spinal interneurons and motor, sensory, and autonomic neurons. When examined at both the light and electron microscopic levels, the caspase-deficient neurons exhibit a nonapoptotic morphology in which nuclear changes such as chromatin condensation are absent or reduced; in addition, this morphology is characterized by extensive cytoplasmic vacuolization that is rarely observed in degenerating control neurons. There is also reduced terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling in dying caspase-deficient neurons. Despite the altered morphology and apparent temporal delay in cell death, the number of neurons that are ultimately lost is indistinguishable from that seen in control animals. In contrast to the striking perturbations in the morphology of the forebrain of caspase-deficient embryos, the spinal cord and brainstem appear normal. These results are consistent with the growing idea that the involvement of specific caspases and the occurrence of caspase-independent programmed cell death may be dependent on brain region, cell type, age, and species or may be the result of specific perturbations or pathology.
Keywords: cell death, neurons, apoptosis, caspases, mouse, embryo, spinal cord
Cysteine proteases comprising the caspase family are considered to be among the most highly conserved molecules involved in apoptosis and programmed cell death (PCD) during development, being expressed in animals as diverse as worms and humans (Cryus and Yuan, 1998; Li and Yuan, 1999; Kuan et al., 2000). They are one of the major classes of pro-apoptotic molecules that are thought to be essential for many of the degradative events that occur during the cell-death process. By cleaving specific substrates in the nucleus and cytoplasm, caspases are responsible for many of the biochemical and cytological changes that define apoptotic PCD.
Previous in vitro studies have shown that various kinds of stimuli that normally induce apoptosis with chromatin condensation and DNA degradation fail to do so in neuronal and non-neuronal cells that are caspase-3-deficient or in which caspase-3 or other caspases are inhibited (Jänicke et al., 1998a,b; Stefanis et al., 1998, 1999;Woo et al., 1998; Zheng et al., 1998; Bortner and Cidlowski, 1999;Cregan et al., 1999; Ferrer, 1999; Keramaris et al., 2000;Tanabe et al., 1999; Xue et al., 1999; D'Mello et al., 2000; Sperandio et al., 2000; Williams et al., 2000). Accordingly, caspase-3, and by extrapolation, caspase-9, which is thought to be upstream of and required for caspase-3 activation, both appear to be primarily required for the nuclear changes that occur during apoptosis. In contrast,in vivo studies of caspase-3- and caspase-9-deficient mice indicate that these proteases are in fact essential for both the nuclear changes and the normal PCD of many developing neurons (Kuida et al., 1996, 1998; Hakem et al., 1998; Roth et al., 1999; Kuan et al., 2000), whereas many types of PCD outside of the nervous system are delayed but nonetheless occur in caspase-3- and caspase-9-deleted animals (Cecconi et al., 1998; Hakem et al., 1998; Kuida et al., 1998;Yoshida et al., 1998; Chautan et al., 1999). However, previous studies of neuronal PCD in caspase-3- and caspase-9-deficient animals focused almost exclusively on mitotically active or immature neurons in the forebrain that undergo normal cell death during early developmental stages before the formation of synaptic connections. The regulation of neuronal PCD in vitro or in populations in vivothat undergo PCD before the formation of synaptic connections may differ from the well known target-dependent type of naturally occurring neuronal death that occurs when synaptic connections are being formed (Oppenheim, 1999). Therefore, these in vitro and in vivo studies on the role of caspase-3 and caspase-9 may not accurately reflect the in vivo role of these proteases in this type of PCD. To examine this, we have focused on several populations of developing neurons that undergo PCD as postmitotic cells while establishing synaptic connections.
MATERIALS AND METHODS
Animals and histology. Heterozygous caspase-3 and caspase-9 mutant mice were maintained in a mixed C57BL/6 and 129sv strain background and were interbred to generate homozygous mutants for this study. Sibling animals at indicated embryonic stages were collected and individually genotyped by PCR as described previously (Kuida et al., 1996, 1998). Given the variable severity of caspase-3 and caspase-9 mutant phenotypes, only mutant embryos showing forebrain abnormalities (exencephaly) were included in our analysis. Because most caspase-9-deficient embryos die in utero after embryonic day 16.5 (E16.5) (Kuida et al., 1998), only E16.5 or younger animals were examined here. Most of the embryos and postnatal animals were immersion-fixed in either Bouins' or Carnoy's solution; the spinal cord (with attached DRG) and brain were processed separately, embedded in paraffin, sectioned serially (10 μm), and stained with either thionine or hematoxylin eosin (Li et al., 1994). Some animals were also fixed in 4% paraformaldehyde and 2% glutaraldehyde, post-fixed in 2% osmium tetraoxide, dehydrated in graded alcohol, and embedded in plastic (Epon); semiserial sections were stained with toludine blue. A polyclonal antibody specific for the 17 kDa cleaved subunit of caspase-3 (BD 559565; PharMingen, San Diego, CA) was used for in situ detection of caspase-3 activation. FITC-conjugated anti-rabbit IgG secondary antibody (Jackson ImmunoResearch, West Grove, PA) was used for visualization of the staining. Spinal cord sections stained with bisbenzimide for cell nuclei and with FITC-labeled activated caspase-3 were imaged using aZeiss LSM510 microscope (Zeiss, Thornwood, NY). Simultaneous confocal–two-photon excitation using an argon laser (488 nm) for FITC, and an infrared pulse laser (760 nm) for bisbenzimide was used to localize cells expressing the active form of caspase-3. For terminal deoxynucleotidyl transferase (TdT)-mediated biotinylated UTP nick end labeling (TUNEL) staining, 10 μm paraffin-embedded tissue sections were first rehydrated and permeabilized with 0.2% Triton X-100 before nick end-labeling with 0.32 U/μl TdT and 2 mmbiotin-16-dUTP (Boehringer Mannheim, Indianapolis, IN). Texas Red-conjugated streptavidin (PerkinElmer Life Sciences, Emeryville, CA) was used for visualization of the TUNEL-positive signals. The number of TUNEL-positive neurons was counted in an equal number of sections spanning 470 μm in two wild-type (WT) and two caspase-3 knock-out (KO) lumbar spinal cords on E14.5.
Morphometric analysis. Neurons were identified and counted according to criteria described by Clarke and Oppenheim (1995), who indicated that reliable and accurate cell counts can be attained without the use of correction factors or stereological techniques. For each neuronal population examined, all cells that met these criteria were counted in every fifth or 10th section, and these totals were multiplied by 5 or 10 as an estimate of the total number of neurons in that population. Because neurons dying in the absence of caspase-3 or caspase-9 exhibit a modified degenerating morphology when examined using the light microscope (see Results), we have included these cells as well as typical pyknotic cells in our counts of degenerating neurons. Counts of surviving and dying neurons in the intermediate gray matter of the lumbar spinal cord excluded the dorsal and ventral horns, and cells were counted on only one side (hemisection) in every 10th section. The degeneration data from caspase-3 and caspase-9 embryos (control and mutant) were similar and thus have been pooled for further analysis. Finally, because we have never observed any differences in neuronal numbers between caspase-3 or caspase-9 homozygotes (+/+) and heterozygotes (+/−), these cell counts have been pooled and compared with caspase-3- and caspase-9-deficient (−/−) littermates.
RESULTS
Gross morphology and cell counts of surviving neurons
In contrast to the severe and obvious forebrain malformations observed in all of the caspase-3- and caspase-9-deficient animals (Kuida et al., 1996, 1998), the organization and morphology of the brainstem, spinal cord, and peripheral ganglia of these same animals appeared completely normal at both embryonic and postnatal stages (Figs.1–3).
Fig. 3.
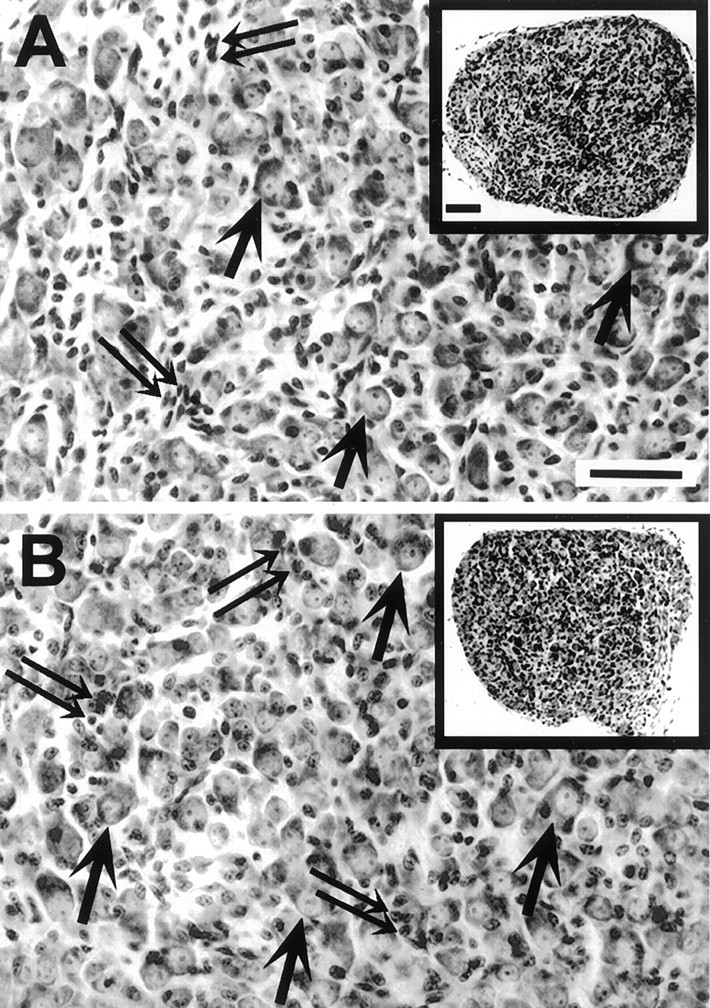
Transverse sections through the middle of the superior cervical ganglion (SCG) of homozygote (A) and caspase-3 KO (B) littermates at P8. The overall size and organization of the ganglia appear normal in both low-magnification (insets) and high-magnification photomicrographs from KO animals.Arrows indicate some of the SCG neurons; double arrows indicate non-neuronal cells. Scale bars: A, B at full size, 25 μm; insets, 40 μm.
Fig. 1.
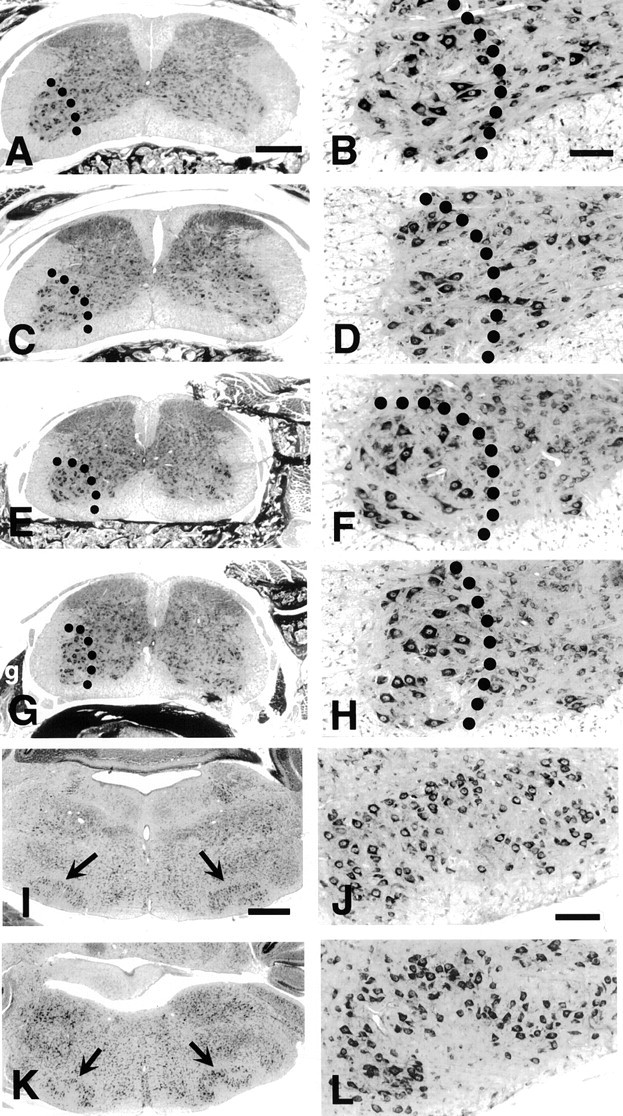
The spinal cord and brainstem of caspase-3-deficient mice appear morphologically normal. Nissl-stained transverse sections of postnatal day 10 (P10) mouse spinal cord (A–H) and brainstem–facial motor nucleus (I–L) of caspase-3-deficient (A, B, E, F, I, J) and control animals (C, D, G, H, K, L) are shown. A–D, Brachial;E–H, lumbar; I–L, brainstem–facial nucleus. Scale bars: A, C, E, G, 300 μm (shown inA); B, D, F, H, 50 μm (shown inB); I, K, 250 μm (shown inI); J, L, 120 μm (shown inJ). Dotted lines indicate ventral horns; an arrow indicates the facial motor nucleus.
A quantitative analysis of cell numbers in a variety of different populations representing spinal interneurons and motor, sensory, and autonomic neurons showed that cell numbers were comparable with control values at stages during and after the normal period of naturally occurring cell death (Tables 1,2). Because of embryonic lethality, neuronal numbers in caspase-9-deficient animals were only examined on E16.5, when the normal cell-death period for spinal motoneurons (MNs) and DRG cells is almost complete. Although the cell-death period for cranial MNs and spinal interneurons begins embryonically and extends into the early postnatal period (Wright et al., 1983; Lawson et al., 1997; Grieshammer et al., 1998), cell numbers in these populations in caspase-3-deficient animals were also comparable with control values when assessed after the cessation of their normal period of cell death (Table 1). Because the normal PCD of sympathetic neurons occurs postnatally, we were unable to assess this population in caspase-9 KO animals. Although not shown in Table 1, MN numbers in the lumbar spinal cord at the beginning of the cell-death period on E13.5 were similar in control and caspase-3-deficient embryos (controls, 3617 ± 360, n = 4 vs caspase-3-deficient, 4009 ± 479, n = 3).
Table 1.
Neuronal numbers (mean ± SD) in postnatal caspase-3-deficient mice
| P0-P2 | P8-P201-a | P42 | ||||
|---|---|---|---|---|---|---|
| +/+ | −/− | +/+ | −/− | +/+ | −/− | |
| Lumbar MNs | 1738 ± 179 | 1895 ± 220 | 1806 ± 173 | 1788 ± 205 | 1650 | 1687 |
| n = 8 | n = 8 | n = 7 | n = 6 | n = 2 | n = 2 | |
| Brachial MNs | — | — | 1710 ± 98 | 1770 ± 113 | — | — |
| n = 6 | n = 6 | |||||
| Facial MNs | 3145 ± 196 | 3220 ± 260 | 2914 ± 200 | 2980 ± 221 | — | — |
| n = 3 | n = 3 | n = 5 | n = 4 | |||
| Hypoglossal MNs | — | — | 1434 ± 81 | 1273 ± 113 | — | — |
| n = 5 | n = 5 | |||||
| Trigeminal MNs | — | — | 978 ± 61 | 936 ± 54 | — | — |
| n = 4 | n = 4 | |||||
| Abducens MNs | — | — | 174 ± 13 | 178 ± 18 | — | — |
| n = 5 | n = 5 | |||||
| Oculomotor MNs | — | — | 531 ± 40 | 542 ± 33 | — | — |
| n = 3 | n = 3 | |||||
| Interneurons (lumbar) | — | — | — | — | 20,280 | 22,317 |
| n = 2 | n = 2 | |||||
| DRG (L4) | — | — | 4455 ± 210 | 4185 ± 237 | 5691 | 5359 |
| n = 4 | n = 4 | n = 2 | n = 2 | |||
| SCG1-b | — | — | 11,853 ± 747 | 12,276 ± 1324 | — | — |
| n = 4 | n = 4 | |||||
Includes data from P8, P10, and P20.
Data from P8 only.
Table 2.
Neuronal numbers (mean ± SD) in caspase-3- and caspase-9-deficient mice on E16.5
| Lumbar MNs | Lumbar DRG (L4) | Facial MNs | Hypoglossal MNs | ||||
|---|---|---|---|---|---|---|---|
| Caspase-9 | Caspase-3 | Caspase-9 | Caspase-9 | Caspase-3 | Caspase-9 | Caspase-3 | |
| Control (+/+, +/−) | 2372 ± 169 | 2224 ± 200 | 4197 ± 311 | 3985 ± 671 | 4000 ± 480 | 1790 ± 465 | 1610 ± 360 |
| n = 6 | n = 5 | n = 3 | n = 4 | n = 5 | n = 4 | n = 5 | |
| Mutant (−/−) | 2421 ± 183 | 2430 | 3833 ± 269 | 3614 ± 510 | 3600 | 1715 ± 410 | 1550 |
| n = 5 | n = 2 | n = 3 | n = 3 | n = 2 | n = 4 | n = 2 | |
Assessment of dying neurons
Many neurons undergoing PCD in normal animals exhibit a morphology featuring a distinct kind of nuclear pyknosis characteristic of an apoptotic type of degeneration, and the dying cells fragment into cytoplasmic–nuclear-containing apoptotic bodies (Fig. 2) (Chu-Wang and Oppenheim, 1978; Oppenheim et al., 1986; Li et al., 1998). When examined using an electron microscope (Fig.4), these same cells exhibit the ultrastructural nuclear and cytoplasmic hallmarks of an apoptotic-like PCD (Chu-Wang and Oppenheim, 1978; Clarke, 1990; Yaginuma et al., 1996;Li et al., 1998; Shiraiwa et al., 2001). In contrast, apparently dying neurons in caspase-3- and caspase-9-deficient embryos exhibit a somewhat different morphology. When examined with the light microscope, these cells exhibit cell shrinkage, increased density of the cytoplasm and nucleus, little if any fragmentation of the cell into apoptotic bodies, and very little chromatin condensation (Fig. 2). Neurons exhibiting these characteristics were rarely observed in control embryos. These same morphological differences were seen even more clearly when the degenerating neurons were examined at the ultrastructural level (Fig. 4). Ultrastructural analysis also revealed the presence of extensive cytoplasmic vacuoles that are seldom observed in dying neurons of control embryos. Although the degeneration of neurons in the caspase-deficient embryos included swelling and dilation of mitochondria and rough endoplasmic reticulum (RER) (Fig. 4), similar changes have also been observed during the normal apoptotic PCD of avian and mouse neurons (Pilar and Landmesser, 1976; Chu-Wang and Oppenheim, 1978; Clarke, 1990; Li et al.,1998). For this reason, we cannot exclude the possibility that these changes may reflect a sampling bias. A more detailed analysis of the ultrastructure of caspase-3- and caspase-9-deficient and control neuronal degeneration is in progress.
Fig. 2.
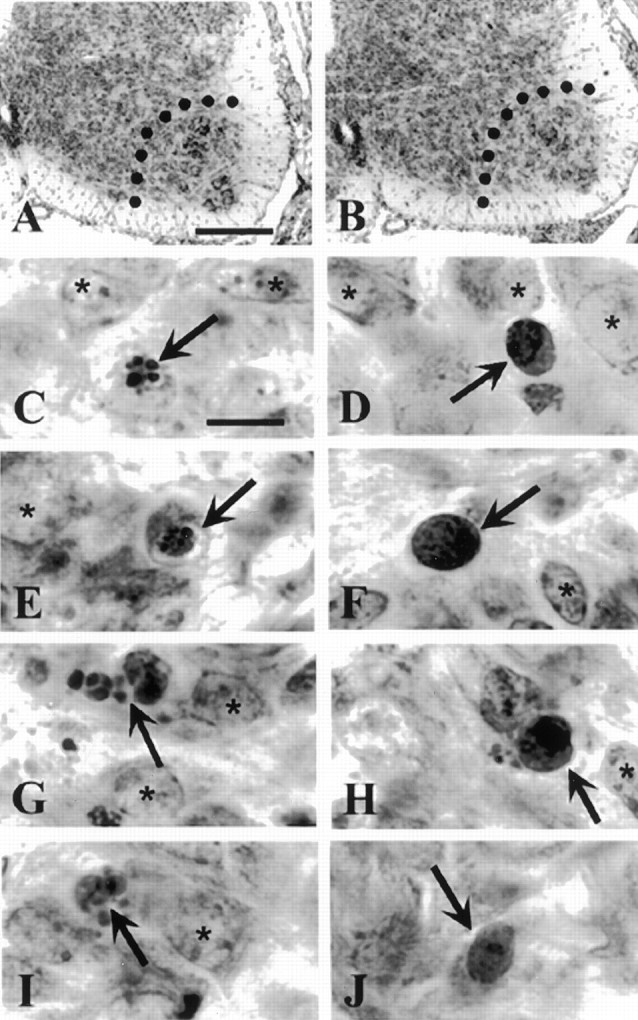
Nissl-stained transverse sections of E16.5 lumbar spinal cord of homozygous littermate control (+/+) (A) and caspase-9-deficient −/− (B) embryos. Dotted lines inA and B indicate the medial border of the ventral horn. Scale bar, 100 μm. C, E, G, I, Degenerating neurons (arrows) from E14.5 caspase-3 (C, E) and caspase-9 (G, I) control (+/+) embryos. Note fragmented apoptotic bodies inG and I. Scale bar, 10 μm. D, F, H, J, Degenerating neurons (arrows) from caspase-3-deficient (D, F) and caspase-9-deficient (H, J) (−/−) embryos. Note the lack of prominent pyknosis and apoptotic bodies and the increased cytoplasmic density (compare G andH) in caspase-deficient dying neurons.Asterisks indicate normal non-dying cells.
Fig. 4.
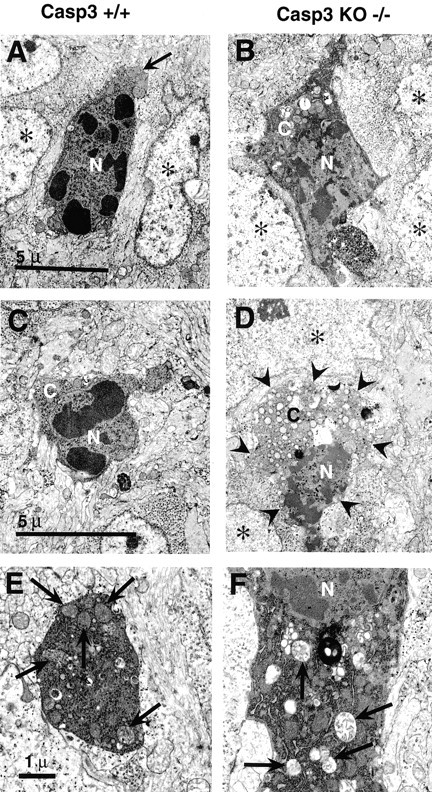
Photomicrographs of degenerating spinal cord neurons from E14.5 caspase-3 (Casp3) +/+ (A, C, E) and caspase-3 (Casp3) KO −/− (B, D, F) littermate embryos showing the distinct morphology of dying neurons in the caspase KO. These cells exhibit reduced chromatin condensation and nuclear pyknosis, marked cytoplasmic vacuolization, and dilation of mitochondria and RER compared with neurons from the caspase-3 +/+ embryos. C, Cytoplasm/soma;N, nuclei. Asterisks indicate normal non-dying neurons. The arrow in Aindicates the cytoplasm, the arrows in Eand F indicate mitochondria, and thearrowheads in D indicate the cell boundary of this degenerating motoneuron. Note the numerous vacuoles and abnormal organelles in the cytoplasm of the cells inD and F. Only part of the cytoplasm of a dying cell is shown in E. The scale bars inA, C, and E are the same for B, D, and F, respectively.
Neurons exhibiting similar aberrant morphologies in avian embryos that have been treated with caspase inhibitors in vivo, when examined with the electron microscope, are also characterized by condensation and increased electron density of cytoplasm, dilated mitochondria, cytoplasmic vacuoles, and crystallized ribosomes, but with only a modest aggregation of nuclear chromatin (Shiraiwa et al., 2001). This morphology is most consistent with the type 3B cytoplasmic type of PCD according to the classification of Clarke (1990), whereas the more typical apoptotic morphology described above is classified as type 1 by Clarke (1990). Because of the similarity in morphology between these avian neurons, which we have proven to be degenerating cells that are phagocytized (Shiraiwa et al., 2001), and the neurons with aberrant morphology in caspase-3- and caspase-9-deficient embryos (McCarthy et al., 1997; Hakem et al., 1998; Woo et al., 1998; Tanabe et al., 1999), these cells have been included in our quantitative assessment of PCD in control and caspase-deficient embryos (Fig.5). Few degenerating neurons would have been counted in caspase-deficient embryos if these profiles had been excluded. In addition, because we know from the data presented here on the number of surviving neurons that comparable amounts of cell death occur in control and caspase-deficient embryos, we consider it highly likely that, despite their distinct morphology, these cells represent neurons undergoing PCD. By including them in our quantitative analysis (Fig. 5), we find that there are similar numbers of degenerating MNs in caspase-3- and caspase-9-deficient embryos when compared with the more typical, frankly pyknotic (apoptotic) neurons in control embryos (Figs. 2, 4). In contrast to MNs, the number of degenerating spinal interneurons in the caspase-3- and caspase-9-deficient embryos on E16.5 was increased compared with control values [caspase-deficient, 10.3 ± 2.5 (mean ± SD) per section, n = 4 vs wild-type control, 6.1 ± 2.1, n = 4; p < 0.05, ttest]. From these data, we conclude that the PCD of MNs occurs to the same extent in caspase-3- and caspase-9-deficient embryos as it does in control embryos, albeit with a distinct morphology, whereas the increased number of dying interneurons in mutant embryos on E16.5 may reflect a delayed kinetics of PCD (see Discussion).
Fig. 5.

The number (mean ± SD) of degenerating MNs in the lumbar spinal cord of control +/+ (CON), caspase-3 −/− (Casp3−/−), and caspase-9 −/− (Casp9−/−) embryos on E14.5 and E16.5 expressed as the number per 1000 surviving MNs. The sample size (number of embryos) is indicated in thebars. In all groups, there was a significant (p < 0.01) reduction between E14.5 and E16.5.
TUNEL
In contrast to our failure to observe a decrease in the number of degenerating neurons (type 1 and type 3B PCD) between control and caspase-3- or caspase-9-deficient embryos, the number of cells labeled by TUNEL was reduced in the spinal cord of caspase-3-deficient embryos on E14.5 (WT control, mean of 50, n = 2 vs KO, mean of 20, n = 2), consistent with previous reports from caspase-9 KO embryos (Kuida et al., 1998). Even those neurons that exhibit TUNEL in KO embryos have less apparent chromatin breakdown and condensation (Fig. 6). Because TUNEL is primarily a means for detecting DNA fragmentation, these data are consistent with the suggestion that neuronal PCD in caspase-3- and caspase-9-deficient embryos occurs without or with reduced DNA fragmentation, as is also indicated by the distinct morphology of the apparently dying neurons in Nissl- and bisbenzimide-stained material from these embryos (Figs. 2, 6). Therefore, TUNEL may not always be a reliable marker for the PCD of developing neurons, because as shown here, in some situations neurons can degenerate in vivowithout TUNEL-positive DNA degradation.
Fig. 6.
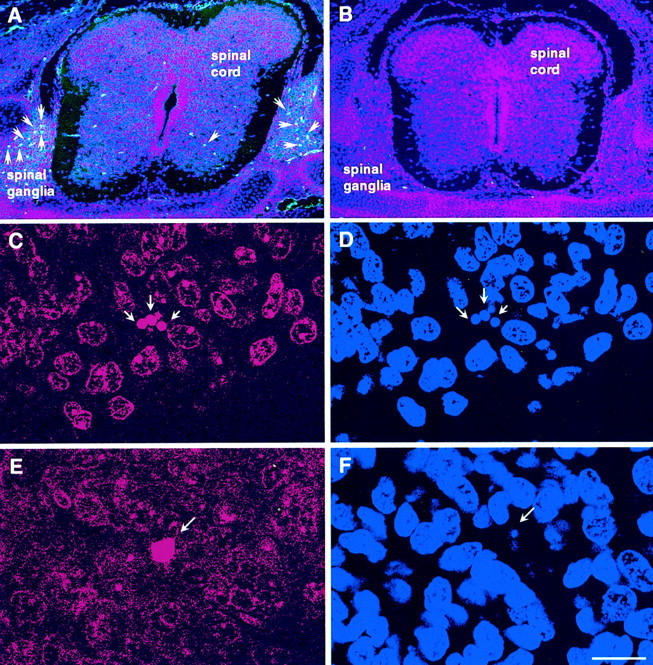
Caspase-3 immunocytochemistry (A, B), TUNEL (C, E), and bisbenzimide staining (D, F) in E14.5 wild-type (A, C, D) and caspase-3 KO (B, E, F) lumbar spinal cord. A, Immunocytochemistry shows the staining of a cleaved (activated) caspase-3 subunit in the spinal motor neuron column and spinal ganglia in E14.5 wild-type mouse spinal cord (arrowheads), indicating the activation of caspase-3. B, In contrast, no staining of activated caspase-3 subunit is found in E14.5 caspase-3-deficient spinal cord. However, when the TUNEL method is used to detect cell death (C, E), it reveals positive staining in both wild-type (C) and caspase-3-deficient (E) spinal cord, although the staining patterns are distinct. The TUNEL-positive profiles in C andE represent the nucleus of an individual neuron. In wild-type spinal cord, there are more TUNEL-positive nuclei showing punctuate nuclear staining (C), which is correlated with condensed chromatin as revealed by bisbenzimide staining (D). In contrast, there are fewer TUNEL-positive cells in caspase-3-deficient spinal cord (see Results). Moreover, these cells typically show a distinct kind of TUNEL staining (E) and no clear evidence of chromatin condensation as indicated by the size of TUNEL-positive nuclei and reduced bisbenzimide-stained chromatin (F). Although not obvious in this photomicrograph, the nucleus indicated by the arrow inF is weakly labeled with bisbenzimide, indicating little, if any, chromatin condensation. Scale bars: A, B, 400 μm; C–F, 20 μm.
Assays for caspase activation
The normal development of the neuronal populations examined here in caspase-KO mice raises the question of whether caspase-3 or caspase-9 is involved in the normal PCD of these cells in control animals. To test this possibility, we examined caspase activity in the spinal cord of control and caspase KO embryos. Using an antibody specific for activated caspase-3, numerous positive cells were observed in control spinal cord and peripheral sensory ganglia (Fig. 6). In contrast, labeling was never observed in neurons from caspase-3 KO embryos. These results indicate that caspase-3 is involved in but not indispensable for normal PCD in these neuronal populations.
DISCUSSION
Caspases are considered to be one of the most highly conserved and ubiquitous families of pro-apoptotic molecules involved in PCD. In the nematode, CED-3 and CED-4 are required for virtually all developmental cell deaths (Ellis et al., 1991; Horvitz, 1994), and in vertebrates, interactions of their counterparts (caspase-3, caspase-9, and Apaf-1) are also thought to be essential for the PCD of many cell types, especially neurons. Null mutant mice lacking each of these genes exhibit a remarkably similar CNS malformation (exencephaly) that has been attributed in part to a failure of apoptotic PCD of neurons (Kuida et al., 1996, 1998; Cecconi et al., 1998; Hakem et al., 1998; Yoshida et al., 1998; Roth et al., 1999). This explanation of the exencephaly is primarily based on direct evidence for decreased TUNEL-positive and pyknotic cells and increased numbers of mitotically active cells in the forebrain (Yoshida et al., 1998). However if TUNEL or a typical pyknotic morphology is not always a reliable marker for PCD in these mutants, then it is possible that it is the specific kind of DNA degradation detected by these methods, but not cell death per se, that is reduced or absent in the mutants. Accordingly, it seems likely that other aspects of CNS development may be abnormal (e.g., proliferation) and may contribute to the exencephaly in the forebrain. Although the cellular mechanisms are not well understood, a failure of neural fold elevation appears to be involved in virtually all cases of exencephaly in mice (Harris and Juriloff, 1999).
There is increasing evidence that TUNEL is not a reliable index of PCDin vitro for either neuronal or non-neuronal cells that lack caspase-3 (knock-outs) or after reductions in caspase-3 or other caspases by treatment with peptide inhibitors (Xiang et al., 1996;Jänicke et al., 1998a,b; Stefanis et al., 1998, 1999; Weil et al., 1998; Woo et al., 1998; Zheng et al., 1998; Bortner and Cidlowski, 1999; Cregan et al., 1999; Ferrer, 1999; Xue et al., 1999;D'Mello et al., 2000; Keramaris et al., 2000; Sperandio et al., 2000). The dying cells in these cases are reported to degenerate in the absence of the typical nuclear changes that are considered to be hallmarks of apoptosis, including pyknosis and the DNA degradation recognized by the TUNEL technique. Although the death of cells in this situation is reported to be delayed (Cregan et al., 1999; Keramaris et al., 2000; Williams et al., 2000), they exhibit degradative changes in the cytoplasm when they do degenerate, and these changes appear to be distinct from those occurring during classic apoptosis, including extensive vacuolation (Xiang et al., 1996; Woo et al., 1998;Sperandio et al., 2000).
Although our results suggest that caspase-3 is normally involved in the PCD of developing postmitotic neurons in vivo, data from caspase-3- and caspase-9-deficient animals, together with other evidence (Shiraiwa et al., 2001), clearly indicates that in the absence of these caspases the extent of cell death in many neuronal populations is normal. However, the mode of degeneration includes reduced TUNEL and nuclear and cytoplasmic changes that are distinct from classic apoptosis. Because the neuronal populations examined here by us all undergo PCD as postmitotic cells, it is interesting that we have not observed the kind of malformations (exencephaly, spina bifida) in the spinal cord or hindbrain that occur in more rostral, mitotically active regions of the brain of caspase-3- and caspase-9-deficient mice. This is consistent with the suggestion that it may be primarily the delay in PCD and the subsequent increased proliferation of these rostral forebrain precursor cells that is responsible for the forebrain abnormality (Kuan et al., 2000). Although additional detailed studies of the kinetics of neuronal degeneration in caspase-3- and caspase-9-deficient embryos are needed to determine whether there is in fact a delay, as occurs in caspase-3-deficient cultured cells (D'Mello et al., 2000) and in chick embryo neurons in vivo (Shiraiwa et al., 2001), the evidence presented here for spinal interneurons is consistent with this possibility. If there is in fact a delay in the kinetics of cell death in the mutant E16.5 embryos, it is more likely to be detected in spinal interneurons, which are at the beginning of their normal period of cell death on E16.5 (Lawson et al., 1997;Grieshammer et al., 1998), compared with MNs, which are near the end of their cell-death period at this age (Oppenheim et al., 1986) and therefore may have had sufficient time to undergo even a delayed cell death. A delay in the degeneration of caspase-deficient neurons suggests that caspase-mediated neuronal death is a more efficient means of PCD than caspase-independent PCD.
We have also observed that the extent of PCD of spinal and cranial motoneurons and DRG sensory cells is normal in Apaf-1-deficient embryos (Oppenheim et al., 2001), consistent with the report that the hindbrain and spinal cord in these mutants appear normal (Cecconi et al., 1998). This raises the question of what molecular pathways are being used in place of Apaf-1, caspase-9, and caspase-3 for mediating PCD in these neuronal populations. One possibility is that these neurons have the capacity to switch to another, caspase-independent, degradative pathway of PCD. Interestingly, cultured PC12 cells and peripheral sensory neurons in which caspase activity is inhibited do in fact switch to a death-inducing pathway that uses lysosomal proteases (cathepsins) instead of caspases for PCD, and they exhibit a morphology similar to that described here for caspase-3- and caspase-9-deficient embryos (Isahara et al., 1999; Agerman et al., 2000). It is also possible that neuronal death in these situations involves the recently described caspase-independent pathway that uses the apoptosis-inducing factor which activates an apoptotic-like mode of degeneration (Joza et al., 2001). Alternatively, other caspases may be able to compensate for the actions of caspase-3 and caspase-9 in mutant mice (Cryus and Yuan, 1998; Hakem et al., 1998; Stefanis et al., 1998; Woo et al., 1998; Zheng et al., 1998, 2000). It has been reported recently that cultured mammalian MNs may use Fas receptors and caspase-8 instead of caspase-9 during PCD (Raoul et al., 1999). However, because caspase-3 is thought to be the major downstream caspase in the Fas pathway, this mode of cell death would not be available for compensation in the caspase-3 KO animals. Cultured NGF-deprived sympathetic neurons treated with the pan-caspase inhibitor bok–asp–fmk (BAF) also continue to undergo cell death, albeit by an apparent autophagic (Clarke type 2), nonapoptotic pathway (Xue et al., 1999). The dying neurons in the chick embryo after caspase inhibition by BAF (Shiraiwa et al., 2001) and those observed here in caspase-deleted mice do not appear to be undergoing an autophagic cell death because the cytoplasmic vacuoles in these cases appear to be empty of lysosomal material. Rather, we suggest that cell death in this situation more closely fits the type 3B “cytoplasmic” mode of embryonic neuronal death reported by Clarke (1990) during normal development. The cytoplasmic vacuoles observed in type 3B PCD appear to reflect the extreme dilation and swelling of cytoplasmic organelles, including mitochondria (Pilar and Landmesser, 1976; Chu-Wang and Oppenheim, 1978). In some respects, dying cells in caspase-deficient animals resemble necrosis (Hirsch et al., 1997; Lemaire et al., 1998). However, consistent with several other studies, we consider this form of degeneration to be a nonapoptotic form of normal PCD (Pilar and Landmesser, 1976; Chu-Wang and Oppenheim, 1978; Clarke, 1990; Sperandio et al., 2000). In a recent study of thymocyte apoptosis, it was shown that during phagocytosis, macrophage-derived lysosomal proteases can induce cell death and DNA fragmentation in thymocytes by a caspase-independent, cell-nonautonomous pathway (McIlroy et al., 2000). Studies are in progress to more fully characterize the mechanisms involved and the morphological changes that occur in degenerating avian and mouse neurons after the loss of caspases.
We find it rather remarkable that in the absence of two of the major pro-apoptotic caspases thought to be required for neuronal cell death, not only do neurons in the diverse populations examined here continue to undergo PCD, but they do so in a highly regulated manner such that the same proportion of cells undergo PCD as do cells in the presence of caspase-3 or caspase-9. Although we assume that all of these populations undergo normal PCD in caspase-9-deficient animals, because we have not been able to examine embryos older than E16.5, we cannot entirely exclude the possibility that later stages of PCD may not be normal. Because this seems unlikely, however, our results indicate that the control of cell number in these neuronal populations by cell death is regulated at a point upstream of the specific machinery used for actually killing the cell. Although we have not examined the release of cytochrome c during the degeneration of caspase-deficient neurons, several studies have shown that caspase deficiency does not affect cytochrome c release in dying sympathetic, cortical, and motor neurons in vitro (Desmukh et al., 2000; Keramaris et al., 2000; Li et al., 2001).
In summary, many populations of developing postmitotic mammalian neurons are able to exhibit normal amounts of PCD in the absence of either caspase-3 or caspase-9. However, the morphology of these degenerating neurons differs from the more typical apoptotic type of cell death and the kinetics of their degeneration may be delayed. These data, together with several other lines of evidence reviewed here, demonstrate that the use of nuclear changes alone for assessing PCD can often be misleading. For example, the apparent discrepancy between the report by Miller et al. (1997) and that by Eldadah et al. (2000)regarding the role of caspase-3 in the death of cultured cerebellar granule neurons may be explained by a primary reliance on nuclear changes alone on the one hand (Eldadah et al., 2000) versus the use of nuclear changes as well as other markers on the other hand (Miller et al., 1997; D'Mello et al., 2000) for assessing the death of neurons. Similarly, whereas the use of TUNEL indicates a significant reduction in the PCD of DRG cells in E12.5 caspase-9-deficient embryos (Zaidi et al., 2001), we find no differences in the number of surviving DRG cells in these mutants on E16.5. Together, these and other recent reports (see the introductory remarks) as well as our own in vivoobservations here and elsewhere (Shiraiwa et al., 2001) support the growing idea that distinct caspases as well as other caspase-independent pathways may mediate neuronal death in specific brain regions and cell types, at specific ages, in different species, and after particular kinds of perturbations (Agerman et al., 2000; Kuan et al., 2000; Zaidi et al., 2001). Finally, these data suggest a note of caution in attempts to use caspase inhibition as a therapeutic strategy for preventing cell death in neurodegenerative diseases (Kermer et al., 1999; Schulz et al., 1999), because compensatory mechanisms for cell death may be activated when caspase-dependent pathways are blocked.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NS20402 (R.W.O.), by the Howard Hughes Medical Institute (R.A.F.), and by NIH Grant NS14841 (P.R.). We thank Carol FloresDevalgaz, J. Bao, and J. Musco for technical assistance and T. F. Haydar for confocal microscopy.
Correspondence should be addressed to Dr. Ronald W. Oppenheim, Department of Neurobiology and Anatomy, Wake Forest University School of Medicine, Medical Center Boulevard, Winston-Salem, NC 27157-1010. E-mail: roppenhm@wfubmc.edu.
REFERENCES
- 1.Agerman K, Baudet C, Fundin B, Willson C, Ernfors P. Attenuation of a caspase-3-dependent cell death in NT-4 and p75-deficient embryonic sensory neurons. Mol Cell Neurosci. 2000;16:258–268. doi: 10.1006/mcne.2000.0875. [DOI] [PubMed] [Google Scholar]
- 2.Bortner CD, Cidlowski JA. Caspase-independent/dependent regulation of K+, cell shrinkage, and mitochondrial membrane potential during lymphocyte apoptosis. J Biol Chem. 1999;274:21953–21962. doi: 10.1074/jbc.274.31.21953. [DOI] [PubMed] [Google Scholar]
- 3.Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 1998;94:727–737. doi: 10.1016/s0092-8674(00)81732-8. [DOI] [PubMed] [Google Scholar]
- 4.Chautan M, Chazal G, Cecconi F, Gruss P, Golstein P. Interdigital cell death can occur through a necrotic and caspase-independent pathway. Curr Biol. 1999;9:967–970. doi: 10.1016/s0960-9822(99)80425-4. [DOI] [PubMed] [Google Scholar]
- 5.Chu-Wang IW, Oppenheim RW. Cell death of motoneurons in the chick embryo spinal cord. J Comp Neurol. 1978;177:59–86. doi: 10.1002/cne.901770106. [DOI] [PubMed] [Google Scholar]
- 6.Clarke PGH. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- 7.Clarke PGH, Oppenheim RW. Neuron death in vertebrate development: in vivo methods. Methods Cell Biol. 1995;46:277–321. [PubMed] [Google Scholar]
- 8.Cregan SP, MacLaurin JG, Craig CG, Robertson GS, Nicholson DW, Park DS, Slack RS. Bax-dependent caspase-3 activation is a key determinant in p53-induced apoptosis in neurons. J Neurosci. 1999;19:7860–7869. doi: 10.1523/JNEUROSCI.19-18-07860.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cryus V, Yuan J. Proteases to die for. Genes Dev. 1998;12:1551–1570. doi: 10.1101/gad.12.11.1551. [DOI] [PubMed] [Google Scholar]
- 10.Desmukh M, Kuida K, Johnson EM. Caspase inhibition extends the commitment to neuronal death beyond cytochrome-c release to the point of mitochondrial depolarization. J Cell Biol. 2000;150:131–143. doi: 10.1083/jcb.150.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D'Mello SR, Kuan C-Y, Flavell R, Rakic P. Caspase-3 is required for apoptosis-associated DNA fragmentation but not for cell death in neurons deprived of potassium. J Neurosci Res. 2000;59:24–31. [PubMed] [Google Scholar]
- 12.Eldadah BA, Ren RF, Faden AI. Ribozyme-mediated inhibition of caspase-3 protects cerebellar granule cells from apoptosis induced by serum-potassium deprivation. J Neurosci. 2000;20:179–186. doi: 10.1523/JNEUROSCI.20-01-00179.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellis RE, Yuan J, Horvitz HR. Mechanisms and functions of cell death. Annu Rev Cell Biol. 1991;7:663–698. doi: 10.1146/annurev.cb.07.110191.003311. [DOI] [PubMed] [Google Scholar]
- 14.Ferrer I. Role of caspases in ionizing radiation-induced apoptosis in the developing cerebellum. J Neurobiol. 1999;41:549–558. doi: 10.1002/(sici)1097-4695(199912)41:4<549::aid-neu10>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 15.Grieshammer U, Lewandoski M, Prevette D, Oppenheim RW, Martin GR. Muscle-specific cell ablation conditional upon cre-mediated DNA recombination in transgenic mice leads to massive spinal and cranial motoneuron loss. Dev Biol. 1998;197:234–247. doi: 10.1006/dbio.1997.8859. [DOI] [PubMed] [Google Scholar]
- 16.Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas M, Elia A, de la Pompa JL, Kagi D, Khoo W, Potter J, Yoshida R, Kaufman SA, Lowe SW, Penninger JM, Mak TW. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94:339–352. doi: 10.1016/s0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- 17.Harris MJ, Juriloff DM. Mini-review: toward understanding mechanisms of genetic neural tube defects in mice. Teratology. 1999;60:292–295. doi: 10.1002/(SICI)1096-9926(199911)60:5<292::AID-TERA10>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 18.Hirsch T, Marchetti P, Susin SA, Dallaporta B, Zamzami N, Marzo I, Geuskens M, Kroemer G. The apoptosis-necrosis paradox: apoptogenic proteases activated after mitochondrial permeability transition determine the mode of cell death. Oncogene. 1997;15:1573–1581. doi: 10.1038/sj.onc.1201324. [DOI] [PubMed] [Google Scholar]
- 19.Horvitz HR. The genetics of programmed cell death in the nematode Caenorhabditis elegans. Cold Spring Harb Symp Quant Biol. 1994;59:377–385. doi: 10.1101/sqb.1994.059.01.042. [DOI] [PubMed] [Google Scholar]
- 20.Isahara K, Ohsawa Y, Kanamori S, Shibata M, Waguri S, Sato N, Gotow T, Watanabe T, Momoi T, Urase K, Kominami E, Uchiyama Y. Regulation of a novel pathway for cell death by lysosomal aspartic and cysteine proteinases. Neuroscience. 1999;91:233–249. doi: 10.1016/s0306-4522(98)00566-1. [DOI] [PubMed] [Google Scholar]
- 21.Jänicke RU, Sprengart ML, Wati MR, Porter AG. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem. 1998a;273:9357–9360. doi: 10.1074/jbc.273.16.9357. [DOI] [PubMed] [Google Scholar]
- 22.Jänicke RU, Ng P, Sprengart ML, Porter AG. Caspase-3 is required for α-fodrin cleavage but dispensable for cleavage of other death substrates in apoptosis. J Biol Chem. 1998b;273:15540–15545. doi: 10.1074/jbc.273.25.15540. [DOI] [PubMed] [Google Scholar]
- 23.Joza N, Susin SA, Dangas E, Stanford WL, Cho SK, Li CY, Sasaki T, Elia AJ, Cheng H-Y, Ravagnan L, Ferri KF, Zamzami N, Wakeham A, Hakem R, Yoshida H, Kong Y-Y, Mak TW, Zùniga-Pflücker JC, Kroemer G, Penninger JM. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature. 2001;410:549–554. doi: 10.1038/35069004. [DOI] [PubMed] [Google Scholar]
- 24.Keramaris E, Stefanis L, MacLaurin J, Harada N, Kazuaki T, Ishikawa T, Taketo MM, Robertson GS, Nicholson DW, Slack RS, Park DS. Involvement of caspase 3 in apoptotic death of cortical neurons evoked by DNA damage. Mol Cell Neurosci. 2000;15:368–379. doi: 10.1006/mcne.2000.0838. [DOI] [PubMed] [Google Scholar]
- 25.Kermer P, Klöcker N, Bähr M. Long-term effects of inhibition of ced-3-like caspases on the survival of axotomized retinal ganglion cells. Exp Neurol. 1999;158:202–205. doi: 10.1006/exnr.1999.7094. [DOI] [PubMed] [Google Scholar]
- 26.Kuan C-Y, Roth KA, Flavell RA, Rakic P. Mechanisms of programmed cell death in the developing brain. Trends Neurosci. 2000;23:291–297. doi: 10.1016/s0166-2236(00)01581-2. [DOI] [PubMed] [Google Scholar]
- 27.Kuida K, Zheng TS, Na S, Kuan C-Y, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 28.Kuida K, Haydar TF, Kuan C-Y, Gu Y, Taya C, Karasuyama H, Su MS-S, Rakic P, Flavell RA. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- 29.Lawson SJ, Davies HJ, Bennett JP, Lowrie MB. Evidence that spinal interneurons undergo programmed cell death postnatally. Eur J Neurosci. 1997;9:794–799. doi: 10.1111/j.1460-9568.1997.tb01428.x. [DOI] [PubMed] [Google Scholar]
- 30.Lemaire C, Andreau K, Souvannavong V, Adam A. Inhibition of caspase activity induces a switch from apoptosis to necrosis. FEBS Lett. 1998;425:266–270. doi: 10.1016/s0014-5793(98)00252-x. [DOI] [PubMed] [Google Scholar]
- 31.Li H, Yuan J. Deciphering the pathways of life and death. Curr Opin Cell Biol. 1999;11:261–266. doi: 10.1016/s0955-0674(99)80035-0. [DOI] [PubMed] [Google Scholar]
- 32.Li L, Oppenheim RW, Lei M, Houenou LF. Neurotrophic agents prevent motoneuron death following sciatic nerve section in the neonatal mouse. J Neurobiol. 1994;25:759–766. doi: 10.1002/neu.480250702. [DOI] [PubMed] [Google Scholar]
- 33.Li L, Houenou LJ, Wu W, Lei M, Prevette DM, Oppenheim RW. Characterization of spinal motoneuron degeneration following different types of peripheral nerve injury in neonatal and adult mice. J Comp Neurol. 1998;396:158–168. [PubMed] [Google Scholar]
- 34.Li L, Oppenheim RW, Milligan CE. Characterization of the execution pathway of developing motoneurons deprived of trophic support. J Neurobiol. 2001;46:249–264. doi: 10.1002/1097-4695(200103)46:4<249::aid-neu1006>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 35.McCarthy NJ, Whyte MKB, Gilbert CS, Evan GI. Inhibition of Ced-3/ICE-related proteases does not prevent cell death induced by oncogenes, DNA damage, or the Bcl-2 homologue Bak. J Cell Biol. 1997;136:215–227. doi: 10.1083/jcb.136.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McIlroy D, Tanaka M, Sakahira H, Fukuyama H, Suzuki M, Yamamura K, Ohsawa Y, Uchiyama Y, Nagata S. An auxiliary mode of apoptotic DNA fragmentation provided by phagocytes. Genes Dev. 2000;14:549–558. [PMC free article] [PubMed] [Google Scholar]
- 37.Miller TM, Moulder KL, Knudson CM, Creedon DJ, Desmukh M, Korsmeyer SJ, Johnson EM. Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J Cell Biol. 1997;139:205–217. doi: 10.1083/jcb.139.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oppenheim RW. Programmed cell death. In: Zigmond MJ, Bloom FE, Landis SC, Roberts JL, Squire LR, editors. Fundamental neuroscience. Academic; New York: 1999. pp. 581–610. [Google Scholar]
- 39.Oppenheim RW, Houenou LJ, Pinon-Raymond M, Powell JA, Rieger F, Standish LJ. The development of motoneurons in the embryonic spinal cord of the mouse mutant, muscular dysgenesis (mdg/mdg): survival, morphology, and biochemical differentiation. Dev Biol. 1986;114:426–436. doi: 10.1016/0012-1606(86)90207-1. [DOI] [PubMed] [Google Scholar]
- 40.Oppenheim RW, Prevette D, Ethell D (2001) Normal programmed cell death of developing neurons in the absence of Apaf-1. Soc Neurosci Abstr, in press.
- 41.Pilar G, Landmesser L. Ultrastructural differences during embryonic cell death in normal and peripherally deprived ciliary ganglia. J Cell Biol. 1976;68:339–356. doi: 10.1083/jcb.68.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raoul C, Henderson CE, Petteman B. Programmed cell death of embryonic motoneurons triggered through the Fas death receptor. J Cell Biol. 1999;147:1049–1061. doi: 10.1083/jcb.147.5.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roth KA, Yuan CY, Haydar TF, D'Sa-Eipper C, Shindler KS, Zheng TS, Kuida K, Flavell RA, Rakic P. Epistatic and independent functions of caspase-3 and Bcl-XL in developmental programmed cell death. Proc Natl Acad Sci USA. 1999;97:466–471. doi: 10.1073/pnas.97.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schulz JB, Weller M, Moskowitz MA. Caspases as treatment targets in stroke and neurodegenerative diseases. Ann Neurol. 1999;45:421–429. doi: 10.1002/1531-8249(199904)45:4<421::aid-ana2>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 45.Shiraiwa N, Shimada T, Nishiyama K, Hong J, Wang S, Momoi T, Uchiyama Y, Oppenheim RW, Yaginuma H (2001) A novel role for caspase-3 activity in early motoneuron death in the chick embryo cervical spinal cord. Mol Cell Neurosci, in press. [DOI] [PubMed]
- 46.Sperandio S, deBelle I, Bredeson DE. An alternative, nonapoptotic form of programmed cell death. Proc Natl Acad Sci USA. 2000;97:14376–14381. doi: 10.1073/pnas.97.26.14376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stefanis L, Troy CM, Qi H, Shelanski ML, Greene LA. Caspase-2 (Nedd-2) processing and death of trophic factor-deprived PC12 cells and sympathetic neurons occur independently of caspase-3 (CPP32)-like activity. J Neurosci. 1998;18:9204–9215. doi: 10.1523/JNEUROSCI.18-22-09204.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stefanis L, Park SD, Friedman WJ, Greene LA. Caspase-dependent and -independent death of camptothecin-treated embryonic cortical neurons. J Neurosci. 1999;19:6235–6247. doi: 10.1523/JNEUROSCI.19-15-06235.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tanabe K, Nakanish H, Meada H, Nishioku T, Hashimoto K, Liou S-Y, Akamine A, Yamamoto K. A predominant apoptotic death pathway of neuronal PC12 cells induced by activated microglia is displaced by a non-apoptotic death pathway following blockage of caspase-3-dependent cascade. J Biol Chem. 1999;274:15725–15731. doi: 10.1074/jbc.274.22.15725. [DOI] [PubMed] [Google Scholar]
- 50.Weil M, Jacobson MD, Raff MC. Are caspases involved in the death of cells with a transcriptionally inactive nucleus? Sperm and chicken erythrocytes. J Cell Sci. 1998;111:2707–2715. doi: 10.1242/jcs.111.18.2707. [DOI] [PubMed] [Google Scholar]
- 51.Williams JA, Barrios A, Gatchalian C, Rubin L, Wilson SW, Holder N. Programmed cell death in zebrafish Rohon-Beard neurons is influenced by TrkC1/NT-3 signaling. Dev Biol. 2000;226:220–230. doi: 10.1006/dbio.2000.9860. [DOI] [PubMed] [Google Scholar]
- 52.Woo M, Hakem R, Soengas MS, Duncan GS, Shahinian A, Kägi D, Hakem A, McCurrach M, Khoo W, Kaufman SA, Senaldi G, Howard T, Lowe SW, Mak TW. Essential contribution of caspase 3/CPP32 to apoptosis and its associated nuclear changes. Genes Dev. 1998;12:806–819. doi: 10.1101/gad.12.6.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wright LL, Cunningham TJ, Smolen AJ. Developmental neuron death in the rat superior cervical sympathetic ganglion: cell counts and ultrastructure. J Neurocytol. 1983;12:727–738. doi: 10.1007/BF01258147. [DOI] [PubMed] [Google Scholar]
- 54.Xiang J, Choa DT, Korsmeyer SJ. BAX-induced cell death may not require interleukin 1β-converting enzyme-like proteases. Proc Natl Acad Sci USA. 1996;93:14559–14563. doi: 10.1073/pnas.93.25.14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xue L, Fletcher GC, Tolkovsky AM. Autophagy is activated by apoptotic signaling in sympathetic neurons: an alternative mechanism of death execution. Mol Cell Neurobiol. 1999;14:180–198. doi: 10.1006/mcne.1999.0780. [DOI] [PubMed] [Google Scholar]
- 56.Yaginuma H, Tomita M, Takashita N, McKay SE, Cardwell C, Yin QW, Oppenheim RW. A novel type of programmed neuronal death in the cervical spinal cord of the chick embryo. J Neurosci. 1996;16:3685–3703. doi: 10.1523/JNEUROSCI.16-11-03685.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoshida H, Kong Y-Y, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- 58.Zaidi AU, D'Sa-Eipper C, Brenner J, Kuida K, Zheng TS, Flavell RA, Rakic P, Roth KA. Bcl-XL-caspase-9 interactions in the developing nervous system: evidence for multiple death pathways. J Neurosci. 2001;21:169–175. doi: 10.1523/JNEUROSCI.21-01-00169.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zheng TS, Schlosser SF, Dao T, Hingorani R, Crispe IN, Boyer JL, Flavell RA. Caspase-3 controls both cytoplasmic and nuclear events associated with Fas-mediated apoptosis in vivo. Proc Natl Acad Sci USA. 1998;95:13618–13623. doi: 10.1073/pnas.95.23.13618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zheng TS, Hunot S, Kuida K, Momoi T, Srinivasan A, Nicholson DW, Lazebnik Y, Flavell RA. Deficiency in caspase-9 or caspase-3 induces compensatory caspase activation. Nat Med. 2000;6:1241–1247. doi: 10.1038/81343. [DOI] [PubMed] [Google Scholar]