

Summary
The gut microbiota can be altered by dietary interventions to prevent and treat various diseases. However, the mechanisms by which food products modulate commensals remain largely unknown. We demonstrate that plant-derived exosomes-like nanoparticles (ELNs) are taken up by the gut microbiota and contain RNAs that alter microbiome composition and host physiology. Ginger ELNs (GELNs) are preferentially taken up by Lactobacillaceae in a GELN lipid-dependent manner and contain microRNAs that target various genes in Lactobacillus rhamnosus (LGG). Among these, GELN mdo-miR7267-3p-mediated targeting of the LGG monooxygenase ycnE yields increased indole-3-carboxaldehyde (I3A). GELN RNAs or I3A, a ligand for aryl hydrocarbon receptor (AHR), are sufficient to induce production of IL-22, which is linked to barrier function improvement. These functions of GELN RNAs can ameliorate mouse colitis via IL-22-dependent mechanisms. These findings reveal how plant products and their effects on the microbiome may be used to target specific host processes to alleviate disease.
Keywords: Ginger exosome-like nanoparticle, small RNA, lipid targeting, gut microbiota composition, miRNA/mRNA interaction, tryptophan metabolites, IL-22, LexA, LGG adherence
Graphical Abstract
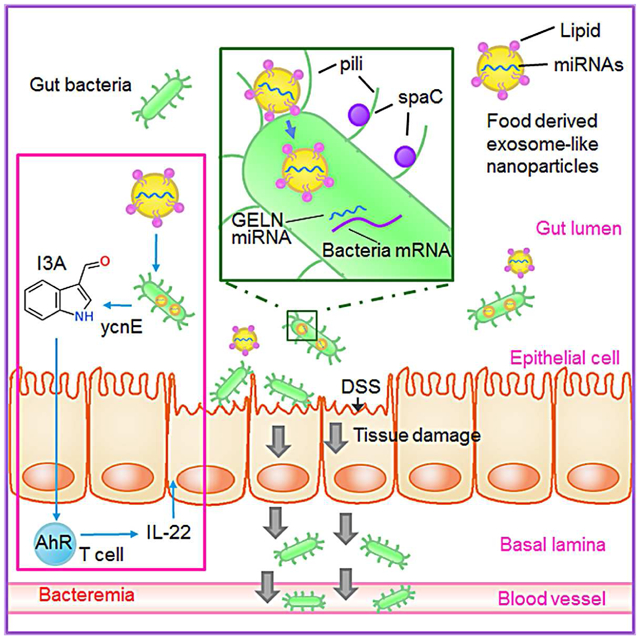
Teng et al. show that exosomes-like nanoparticles (ELNs) from edible plants such as ginger are preferentially taken up by gut bacteria in an ELN lipid dependent manner. ELN RNAs regulate gut microbiota composition and localization as well as host physiology, notably enhancing gut barrier function to alleviate colitis.
Introduction
Diet has been demonstrated to have a considerable effect on the composition of the gut microbiota throughout the human lifespan (Sonnenburg et al., 2016). Different human populations can have vastly different intestinal microbiomes, and changes in diet lead to changes in microbiota composition (Maslowski and Mackay, 2011). Owing to the essential role of the gut microbiota in maintaining host physiology, its alteration as a result of an unhealthy diet can trigger a wide range of physiological disorders, including metabolic disorders (Chassaing and Gewirtz, 2014). Vegetables are mostly made of cells that contain high levels of nucleic acids, including RNA (Garcia-Segura et al., 2013). Although it is known that food, including edible plants, are the main carbon and energy source for gut microbes, whether the expression of gut bacterial genes is affected by edible plant RNA is not known. A number of diet-derived metabolites promote activation of the aryl hydrocarbon receptor (AHR)-mediated pathway (Bessede et al., 2014; Li et al., 2011). Activation of the AHR pathway results in induction of IL-22 (Monteleone et al., 2011). IL-22 prevents systemic dissemination of intestinal microbiota (Sonnenberg and Artis, 2012). However, it is not known whether edible plant-derived exosome-like nanoparticles (ELNs) RNA has a role in induction of IL-22 via activation of AHR.
Our published data indicate that ELN-host cell interactions can prevent alcohol-induced liver damage (Zhuang et al., 2015) and dextran sulfate sodium (DSS)-induced colitis in mouse models (Deng et al., 2017). Those initial findings encouraged us to further determine (1) whether ELNs can be taken up by gut bacteria and (2) upon taking up the ELNs, whether the diet-derived ELN RNA affects the biology of gut bacteria.
Here, we show that ELNs, such as ginger ELNs (GELNs), are preferentially taken up by gut bacteria, resulting in changes in the composition and localization of bacteria, as well as in host physiology. Phosphatidic acid (PA)-enriched GELNs send a signal to LGG, resulting in preferential uptake of GELNs. Once GELNs are taken up by LGG, GELN RNAs interact with a panel of LGG genes, altering the composition of the gut microbiota. Meanwhile, metabolites released from GELN-RNA-treated LGG in turn regulate the growth of other gut bacteria. Collectively and as proof of concept, these findings indicate a mechanism that underlies selective uptake of GELNs by LGG and opens up avenues for studying molecular pathway(s) that underlie how diet through ELNs regulate cross-talk between gut microbiota and host cells.
Results
GELN RNA shapes gut microbiota
A number of edible nanoparticles have been characterized based on electron microscopy examination (Mu et al., 2014). In this study, in addition to electron microscopy (EM) imaging (Figure S1A), GELNs were characterized based on size distribution (Figure S1B) and the RNAs (Figure S1C) and proteins (Figure S1D) present. Gel electrophoresis demonstrated the presence of substantial amounts of small-sized RNAs (less than 300 nucleotides). The yield and number of GELNs per gram of ginger tissue and yield of GELN RNA per mg of GELNs were also quantitatively analyzed (Figure S1E). The purity of GELN preparations was determined by comparing the ratio of GELN counts to protein concentration (Figure S1F). Next-generation sequencing analysis of GELN RNA (Table S1) further suggested that GELNs contained miRNAs. At a sequencing depth of 20 million reads, 93,679 of the miRNA reads were mapped to 109 mature miRNAs from the NCBI plant miRNA database (Table S1). A number of bacterial mRNAs that could be potentially bound by a seed sequence (7-8 NTs) of GELN miRNAs are listed in Figure S1G.
It is obvious that ELN RNA stability in the gut is required for potential interaction of ELN RNA with gut bacterial mRNA. Analysis of the tissue distribution of ELNs indicated that grapefruit ELNs preferentially migrated to the liver, but GELNs were more likely to stay in the intestine (Figure S2A). The evidence of GELNs in the gut and feces over a 6-hour period was further confirmed by quantitative PCR (qPCR) analysis of GELN miRNA aly-miR319a-3p and grapefruit miRNA vvi-miR166c (Figure S2B). The PCR results (Figure S2C) indicate that in fewer than 30 PCR cycles, only aly-miR319a was detected in the feces of mice fed GELNs, whereas vvi-miR166c was only detected in the feces of mice fed with grapefruit ELNs, suggesting that qPCR analysis of fecal samples provides a specific and sensitive approach to investigating the stability of ELNs in the gut.
The above findings led us to investigate whether GELNs cross-talk with gut microbiota and regulate their composition. We collected fecal samples from C57BL/6 mice that were administered GELNs for a week and then analyzed the microbial composition via 16S rRNA gene (v1-v3 regions) sequencing (the sequencing data were deposited in NCBI Sequence Read Archive (SRA), accession number SRP121341). Further downstream analysis was performed using QIIME 1.9.1 pipeline tools (Caporaso et al., 2010). Microbial alignment and classification was performed using the Green Genes reference database (gg_13_8_otus) as described in the Star Methods section. We found an increase in Lactobacillaceae and Bacteroidales S24-7 and a decrease in Clostridiaceae in GELN-treated mice in comparison with mice treated with PBS (Figure 1A, Table S2). The Lactobacillaceae percentage increased from 0.25 ± 0.15% to 24.80 ± 5.41% (p<0.001) in mice that received GELNs by gavage. The sequencing data were subsequently verified using a qPCR assay (Figure 1B). We next estimated the effect of GELNs at a high dose of 10 mg/25 g of body weight and at a low dose of 0.5 mg/25 g of body weight (a physiologically relevant dose for human intake (Schwertner et al., 2006)) on the composition of gut bacteria. The results generated from both higher and lower doses of GELNs supported the conclusion that GELN treatment increases Lactobacillaceae and Bacteroidaceae and decreases Clostridiaceae compared with PBS (Figure S2D). Neither the higher nor lower doses of GELNs induced any abnormalities in treated mice compared with PBS control mice, based on serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) (Figure S2E). We next investigated whether the impact of GELNs on gut microbiota composition is associated with sex. Gene sequencing analysis of 16S rRNA indicated that although the percentage of the abundance of the bacteria analyzed were variable between male and female mice in response to GELNs, the overall trend affecting the increase or decrease in bacteria is the same regardless of sex. Notably, GELNs induce Lactobacillaceae and Bacteroidaceae and decrease Clostridiaceae in both males and females to varying degrees (Figure S3A, Table S2).
Figure 1. GELNs shape gut microbiota and uptake by gut bacteria.

(A) GELNs or PBS were fed to C57BL/6 mice every other day for a total of 3 times. Bacterial DNA from feces evaluated using 16S rRNA gene sequencing (n=5). The bar graph shows the percentage of each bacteria sequences in all sequence reads at the level of family.
(B) Selected bacteria identified by qPCR in feces of mice. Sequencing results (left); qPCR results (right); GELNs vs PBS, *P<0.05.
(C) Heat map depicting the mouse gut microbiota using a qPCR array; red and blue represent high and low levels of bacteria, respectively.
(D) Healthy subjects treated with GELNs (n=28) or 0.9% NaCl (n=30). Bacteria in feces were evaluated via 16S rRNA gene sequencing after being randomly pooled into three groups. The bar graph shows the percentage of each bacteria sequence in all sequence reads (bottom left). The level of selected bacteria verified with qPCR (bottom right).
(E) A representative confocal microscopy image of a fecal sample from mice fed PKH26-labeled GELNs (left, scale bar: 10 μm) and quantitative FACS analysis (right).
(F) Schematic representation of the treatment schedule for PKH2-labeled GELN uptake by gut bacteria in mice (n=5 mice per group).
(G) The bar graph shows the results of 16S rRNA sequencing at the level of family for bacteria with a preference for taking up GELNs. The data are representative of three independent experiments (error bars, SD).
To address whether ELN RNAs influence gut microbiota composition, ELN RNAs from ginger, grapefruit and carrot were extracted from purified ELNs and encapsulated in GELN nanovectors (GNVs) made with GELN-derived lipid. The results indicated that the mice gavaged with ginger, grapefruit or carrot ELN RNAs exhibited a change in the composition of gut microbiota (Figure 1C), which would suggest that edible plant ELN RNAs have an effect on the gut microbiota composition in general. Ginger ELN RNAs induced several species of Lactobacillus identical to GELNs when compared with GNVs as a control. However, carrot ELN RNAs seem to have no effect on the Lactobacillus level. Cluster analysis using R software (Rosselli et al., 2016) indicated that PBS and GNVs are grouped in the same cluster (Figure 1C), GNVs/grapefruit ELN RNAs and GNVs/carrot ELN RNAs are in the same cluster, and GNVs/GELN-RNAs have the least similarity with the other two clusters.
To further determine whether our findings described above can be translated into clinical application, 16S rRNA gene sequencing data generated from stool samples of healthy subjects after oral GELN administration for a week were quantitatively analyzed (Figure 1D top panel). To exclude the bias of sex in the study results, bacterial DNA from both males and females in each treatment group was pooled into three groups prior to 16S rRNA sequencing. The percentage of each bacterial species sequence in all sequence reads indicated an increase in Lactobacillaceae, Bacteroidaceae and Bacteroidales S24-7 and a decrease in Clostridiaceae and Ruminococcaceae in GELN-treated subjects in comparison with particle vehicle (0.9% NaCl)-treated subjects (Figure 1D bottom left panel, Table S2). The sequencing data from healthy subjects were subsequently verified with a qPCR assay (Figure 1D bottom, right panel).
Considering the direct contact of food ELNs and numerous bacteria in the gut, we hypothesized that food ELNs might be taken up by bacteria and the contents of food ELN RNAs could directly regulate gene expression in bacteria. To test our hypothesis, PKH26-labeled GELNs were administered to C57BL/6 mice via oral gavage. Confocal imaging analysis indicated that the GELNs were taken up by gut bacteria (Figure 1E, left panel), and this result was further confirmed by quantitative fluorescence-activated cell sorting (FACS) analysis of PKH26+ GELNs (Figure 1E, right panel). To determine whether the changes in gut bacteria composition were associated with preferential uptake of GELNs by specific gut microbiota, PKH26+ GELNs were administered to mice via oral gavage. The PKH26+ bacteria from fecal samples of mice were sorted by FACS (Figure 1F) followed by 16S rRNA gene sequencing. The results showed that 31.54 (± 7.92%) of the GELNs/PKH26-positive gut bacteria were Lactobacillaceae (Figure 1G, Table S3). Next, we determined whether the concept that edible plant exosomes can preferentially target bacteria in the intestine can be generalized. ELNs from turmeric, which belongs to the same family as ginger, garlic and grapefruit, were used as proof of concept. We generated a 16S ribosomal rRNA gene library and applied 16S sequencing analysis of fecal samples from mice administered PKH26+ ELNs from garlic, turmeric and grapefruit (Figure S3B, Table S3). The analysis of GELN+ bacteria suggested that all three types of ELNs were preferentially taken up by Bacteroidales S24-7. Interestingly, turmeric, from the same Zingiberaceae family as ginger, was also preferentially taken up by Lactobacillaceae. In contrast, garlic- and grapefruit-derived ELNs were preferentially taken up by Ruminococcaceae. These data established a foundation for further study of the molecular basis underlying preferential uptake by gut microbiota.
Our data show that Lactobacillaceae numbers increased in GELN-administered mice (Figure 1A) and that GELNs were preferentially taken up by Lactobacillaceae (Figure 1G). The results generated from in vitro cultures of LGG with GELNs indicated that GELNs directly promoted LGG growth (Figure S4A-S4D), whereas grapefruit-derived ELNs reduced LGG growth (Figure S4D). To determine whether GELNs also have an effect on the growth of other Lactobacillus species and other families of bacteria, the growth of Lactobacillus reuteri (L. reuteri), Lactobacillus murinus (L. murinus), B. fragilis, E. coli and Ruminococcaceae sp. (TSD-27) was evaluated after incubation with GELNs for 8 h. We found that GELNs also induced L. reuteri and L. murinus growth (Figure S4E). GELNs had no effect on B. fragilis or E. coli growth, but we observed that GELNs inhibited Ruminococcaceae growth (Figure S4E). It is known that the metabolites released from one species of gut bacteria can have an effect on the growth of other species. Our data show that the metabolic products from GELN-treated LGG inhibited Listeria, E. coli, and B. fragilis growth in vitro (Figures S4F, S4G) and in vivo (Figures S4F, S4H) but had no effect on LGG growth. Collectively, these data suggest that GELNs can regulate LGG metabolites and prompted us to select LGG for further analysis of the molecular mechanisms underlying GELN-mediated biological effects on LGG.
GELNs are selectively taken up by gut bacteria and regulate the expression of LGG mRNA and protein.
To explore the mechanism by which ELNs are preferentially taken up by specific bacteria, we next assessed comparative lipid profiles generated from mass spectrometry (MS) analysis (Table S4). We found that GELN and turmeric ELN-derived lipids were enriched with phosphatidic acids (PAs, 35.2% and 34.4%, respectively), primarily 1,2-dilinoleoyl-sn-glycero-3-phosphate, C18:1/C18:3 (36:4) and 1-palmitoyl-2-linoleoyl-sn-glycero-3-phosphate, C16:0/C18:2 (34:2), whereas PAs in grapefruit and garlic represented only 3.5% and 5.5%, respectively, of the total lipid content (Figure 2A). In contrast, the majority of the lipid in grapefruit and garlic was phosphatidylcholine (PC, 36.2% and 52.6%, respectively). We hypothesized that GELN PA lipids may serve as a signal for preferential uptake by LGG. To generate PA-depleted GELN lipids, GELN lipids were isolated with chloroform and separated via thin-layer chromatography (TLC) (Figure 2B). The band containing PA was identified based on standard PA migration in TLC and then removed. The results generated from GNVs made from PA-depleted GELN lipids (Figure 2C) indicated that depletion of GELN PA lipids leads to a significant reduction in GNV-positive LGG, whereas addition of PA 34:2 or PA 36:4 back into PA-depleted GNVs rescued the uptake of GNVs, and grapefruit ELN lipid-derived nanovector (GFNV) uptake by LGG was minimal (Figure 2C). These data suggest that PA is required for GELN uptake by LGG. ELN lipid-dependent uptake was also demonstrated in grapefruit ELNs. FACS analysis indicated that PC-enriched grapefruit GFNVs were preferentially taken up by Ruminococcaceae sp. (TSD-27). PC depletion in grapefruit GFNVs resulted in reduced uptake by Ruminococcaceae, and the uptake was rescued by addition of PC 34:2 back into the PC-depleted grapefruit GNVs (Figure 2D). To further determine whether lipids also play a role in tissue targeting in vivo, mice were gavaged with DiR-labeled GELNs, PA-depleted GELNs, GELNs plus PC 34:2, grapefruit ELNs and PC-depleted grapefruit ELNs. Analysis of imaging signals in mouse intestines and livers was performed at 1 h and 6 h after the gavage. The results suggest that PA lipids play a role in maintaining the duration and amount of ELN accumulation in the gut. PC lipid enhances migration of ELNs from the intestine to the liver (Figure 2E).
Figure 2. PA-enriched GELNs are preferentially taken up by LGG and regulate the expression of LGG mRNA and protein.

(A) Lipid extracted from ELNs from ginger, turmeric, garlic and grapefruit. Lipid composition was determined using a triple quadrupole mass spectrometer. The bar graph shows the percentage of each lipid in all the lipids.
(B) GELN- and grapefruit ELN-derived lipids were separated by thin-layer chromatography (TLC). PA and PC loaded was as a standard marker.
(C) GNVs generated with whole lipids, PA-depleted lipids from GELNs and grapefruit ELNs, and supplementary PA with depleted lipids. PKH26-labeled GNVs exposed to LGG. PKH26-positive bacteria were quantitatively analyzed vis FACS.
(D) FACS analysis of LGG incubated with PKH26-labeled GNVs from whole grapefruit ELN lipids with or without PC depletion and supplementary PC with depleted lipids.
(E) A representative image of the duodenum, colon and liver from mice (n=5) receiving a gavage of DiR dye-labeled GNVs with or without PA or PC lipid (left); quantification of fluorescence intensity (right). Ginger GNVs vs ginger GNVs/PC+, *P<0.05; grapefruit GNVs vs grapefruit GNVs/PC−, # P<0.05
(F) PKH26-labeled GELNs were incubated with 1×107 colony-forming units (CFU) of LGG, and uptake of PKH26-labeled GELNs was visualized using confocal microscopy.
(G) Frequency of PKH67-labeled LGG and PKH26-labeled GELNs assessed using flow cytometry. Numbers in quadrants indicate the percentage of LGG in each.
(H) A heat map showing the effect of GELNs on LGG mRNA expression determined by next-generation sequencing.
(I) Venn diagram of all mRNAs detected in LGG. The numbers in brackets indicate the in vitro results.
(J) A heat map based on LC-MS data showing the effect of GELNs on LGG proteins.
(K) Venn diagram of all the proteins detected in LGG.
(L) qPCR of LexA expression in LGG treated with gma-miR396e. *P<0.05 (two-tailed t-test).
(M) Analysis of LGG proliferation after treatment with gma-miR396e. *P<0.05 (two-tailed t-test). The data are representative of three independent experiments (error bars, SD).
To determine whether GELNs have an effect on gene expression and protein production in LGG, we first sought to determine the efficiency of PKH26-labeled GELN uptake by LGG in an in vitro culture assay. LGG was incubated with PKH26-labeled GELNs for 1 h at 22°C, and uptake of GELNs by LGG was visualized with confocal microscopy. We then evaluated GELN uptake in vivo. Briefly, two hours after mice were gavaged with PKH67 fluorescence-labeled LGG (1×109), the mice were administered PKH26-labeled GELNs (500 mg/kg of body weight in 300 μl PBS). Twelve hours after the last gavage, analysis of the PKH67+PKH26+ double-positive bacteria suggested that LGG took up the GELNs. In vitro confocal (Figure 2F) and in vivo FACS analysis (Figure 2G) data suggested that LGG took up the GELNs. We then obtained an LGG gene expression profile using next-generation mRNA sequencing (Figure 2H, 2I) ((SRA) accession number SRP121341) and protein profiles using liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Figure 2J, 2K) of sorted (via FACS) PKH67+PKH26+ LGG. The RNA sequencing (Figure 2I) and proteomic (Figure 2K) data analyses indicated that 398 mRNAs and 149 proteins were predominantly present in GELN-treated LGG. In addition, 249 LGG mRNAs and 133 proteins were reduced in GELN-treated LGG. The top 50 LGG mRNAs and proteins with altered expression are listed in Table S5 [mRNAs] and Table S5 [proteins]. Among the LGG genes affected by GELN RNAs, the transcription repressor LexA was reduced due to GELN RNA treatment at both the transcript and protein level (Figures 2H, 2J; Figure S4I). An alignment of nucleotide sequences using BLAST indicated that LGG LexA mRNA is one of the potential target genes of GELN gma-miR396e (Figure S1G, Figure S4J). LGG treated with gma-miR396e had a lower level of LexA expression (Figure 2L) and grew faster than LGG treated with scrambled miRNA (Figure 2M).
GELN RNAs enhance LGG-mediated inhibition of mouse colitis by inducing the IL-22 expression via activation of the AHR pathway
RNA sequence analysis further revealed that GELN RNAs harboring the complementary seed-matching sequence of LGG mRNA have the potential for binding gut bacterial mRNA (Table S1, Figure S1G). The evidence indicating a similarity in regulating the composition of gut microbiota of mice fed with GELNs and GELN RNA (Figure S5A, Table S6) prompted us to further examine whether GELN RNAs could modulate bacterial function and in turn host biology. First, evidence showing PKH26-labeled GNVs encapsulating GELN-RNAs were present in LGG was visualized using confocal microscopy (Figure 3A). LGG growth in MRS broth was induced by GELN RNAs but not by scrambled RNAs (Figure 3B). The results indicated that mice fed GNV/GELN-RNAs had superior protection against DSS-induced mouse colitis comparted with mice fed GNV/scrambled RNA (Figure 3C-G), suggesting that the protective effect on DSS-induced mouse colitis is GELN RNA specific. The results generated from DSS-induced colitis in germ-free mice further demonstrated that LGG is required for better protection of mice against DSS-induced colitis because germ-free mice administered GNVs/GELN-RNAs without LGG did not experience a reduction in colitis severity (Figure 3H-J).
Figure 3. GELN RNAs enhance LGG-mediated protection against mouse colitis.

(A) GELN RNAs (1 μg) were packed in 100 nM ginger-derived lipid to form nanovectors (GNVs/GELN-RNAs) and were incubated with 1×107 CFU of LGG. Uptake of PKH26-labeled GNVs/GELN-RNAs was visualized using confocal microscopy; 200x magnification; scale bar: 10 μm.
(B) Proliferation of LGG treated with GNVs/GELN-RNAs over time. *P<0.05.
(C) Schematic representation of the treatment schedule for DSS-induced colitis.
(D) Body weight. GELN-RNAs vs scrambled RNAs, *P<0.05 and **P<0.01.
(E) Survival of mice after administration of 2.5% DSS in drinking water.
(F) Representative colons from mice treated as labeled in the figure (left); quantification of colon length (right). *P<0.05.
(G) H&E-stained sections of colon (400x magnification) from mice treated as indicated in the figure.
(H-J) Representative colons (H) from germ-free mice (n=5) treated as labeled in the figure (left); quantification of colon length (right). (I) Body weight. (J) H&E-stained sections of colon (400x magnification). *P<0.05; **P<0.01.
(K) Wild-type (WT) and AHR knockout (KO) mice (n=5) supplied with 2.5% DSS after gavage with GELN RNAs or scrambled RNAs. ELISA analysis of IL-22, IL-1β and TNFα levels in colon mucus. The data are representative of three independent experiments (n=5; error bars, SD). *P<0.05; ** P<0.01 (two-tailed t-test); NS, not significant; (error bars, SD).
Related to Figure S4.
Cytokine array analysis (Figures S6A-C) indicated that the levels of the majority of proinflammatory cytokines/chemokines were decreased in the colon tissue of germ-free mice treated with LGG+GNVs/GELN-RNAs (Figures S6B, S6C) in comparison with germ-free mice treated with LGG alone. In addition to confirmation of a reduction in the TNFα level in pathogen-free (SPF) mice, ELISA analysis indicated that GELN-RNA treatment inhibited the induction of the proinflammatory cytokines IL-1β and TNFα and promoted the production of IL-22 in colon mucus from DSS-treated SPF mice (Figure 3K). Failure of IL-22 induction by GELN-RNAs in germ-free mice suggested that IL-22 induction by GELN-RNAs was bacteria-dependent. It is known that the aryl hydrocarbon receptor (AHR) pathway contributes to induction of IL-22 (Monteleone et al., 2011). We next tested whether the GELN-mediated inhibition of IL-1β and TNFα and the increase in IL-22 occurs through the AHR pathway. Knockout (KO) of AHR led to neither inhibition of IL-1β and TNFα expression nor an increase in the expression of IL-22 in AHR KO mice with DSS-induced colitis (Figure 3K).
Next, we investigated the molecular basis of LGG+GNV/GELN-RNA-mediated induction of IL-22. LGG metabolizes tryptophan to indole derivates, including I3A, which acts as a ligand for AHR, inducing local production of IL-22. High-performance liquid chromatography (HPLC) analysis indicated that the level of I3A in the feces of GELN-RNA-treated C57BL/6 mice dramatically increased (Figure 4A) compared with that in feces from mice treated with GELN-scrambled RNA, whereas the level of indole-3 acetamide (I3AM), another metabolite of tryptophan, decreased (Figure 4B). Interestingly, indole-3-acetaldehyde (IAAld), which is an intermediate metabolite product for synthesis of I3A was also induced in the feces of GELN-RNA-treated mice (Figure 4A). We then determined the role of I3AM in generation of I3A. HPLC analysis indicated that addition of I3AM to LGG cultures significantly inhibited GELN-RNA-mediated induction of the I3A precursor, IAAId (Figure 4C-E). To further determine the inhibitory effect of I3AM on the production of LGG I3A, the quantity of IAAld (Figure 4F, left panel) and I3A (Figure 4F, right panel) in MRS media of LGG treated with different concentrations of I3AM was analyzed. We found that I3AM inhibited the production of IAAld and I3A in a dose-dependent manner, and the inhibitory effect of I3AM on the production of I3A was cancelled by additional IAAld (Figure 4F), suggesting that the I3AM-mediated inhibitory effect on I3A production occurs upstream of IAAId. Additional IAAId also cancelled the I3AM-mediated inhibitory effort on IL-22 production (Figure 4G, left panel). This inhibitory effect did not occur in AHR KO mice treated with LGG supernatant exposed to I3AM (Figure 4G, right panel), indicating that the inhibitory effect of I3AM on LGG I3A-mediated induction of IL-22 occurs via the AHR pathway.
Figure 4. GELN RNAs mediate induction of IL-22 via inhibition of I3AM production.

(A-B) The C57BL/6 mouse (n=5) treatment schedule was the same as that described in Figure 3C. Representative HPLC analysis of (A) indole-3-carboxaldehyde (I3A) and indole-3-acetaldehyde (IAAld) via detection of UV absorbance at 300 nm and (B) tryptophan and indole-3-acetamide (I3AM) using a fluorescence detector. Arrows point to the peak of the standard. *P<0.05; **P<0.01.
(C-D) LGG grown in MRS medium with I3AM and GNV/scrambled RNAs or GNV/GELN-RNAs for 6 h. HPLC analysis of IAAld and I3A in the MRS medium. The concentrations of the metabolites listed in the figure were quantified (D). *P<0.05.
(E) HPLC analysis of I3A in LGG cultures in the presence of I3AM at the time indicated in the figure. GNV/GELN-RNAs vs GNV; GNV/I3AM vs GNV; *P<0.05.
(F) HPLC analysis of IAAld and I3A in MRS medium from LGG treated with I3AM at the different concentrations indicated in the figure without or with IAAld. *P<0.05; **P<0.01.
(G) ELISA analysis of IL-22 in colon mucus of mice (WT, left; AHR KO, right) gavaged with MRS medium from LGG treated with I3AM. The data are representative of three independent experiments (error bars, SD). * P<0.05; ** P<0.01.
Related to Figure 5.
Protein LC-MS/MS and mRNA sequencing analysis indicated that the expression of monooxygenase ycnE (embl-cds:CAR87039) in LGG (Figure 5A) was inhibited by GELN-RNA (Figures 2J-K). Monooxygenase is a key enzyme that catalyzes tryptophan to I3AM (Kosuge et al., 1966; Stutz, 1958). We found that GELN-derived mdo-miR7267-3p has a potential binding site for mRNA encoding LGG monooxygenase ycnE (Figure 5B). qPCR data generated from LGG treated with mdo-miR7267-3p indicated that ycnE gene expression was indeed inhibited (Figure 5C), and HPLC analysis indicated that mdo-miR7267-3p treatment leads to inhibition of I3AM and induction of I3A (Figure 5D). Tryptophan is metabolized into I3A and I3AM in LGG (Figure 5E). Production of I3AM and I3A is regulated by LGG ycnE. After chemical equilibrium is reached, the net amount of accumulated tryptophan is dependent on ycnE enzyme activity. Our data indicated that tryptophan was accumulated due to GELN-RNA-mediated blocking of I3AM production and was not utilized for I3A production at 100%.
Figure 5. GELN RNAs protect mice from colitis by initiating regulation of monooxygenase ycnE expression in LGG.

(A) LC-MS spectra of monooxygenase ycnE from LGG.
(B) Schematic diagram of the putative binding sites of mdo-miR7267-3p in the monooxygenase ycnE.
(C) qPCR analysis of ycnE expression in LGG treated with mdo-miR7267-3p or scrambled miRNA.
(D) HPLC analysis of I3AM and I3A in LGG cultures treated with mdo-miR7267-3p.
(E) Hypothetical model of GELN RNA regulation of LGG I3A induction. I3AM inhibits the production of IAAId, which is a precursor of I3A synthesis. GELN-RNA pretreatment leads to a reduction in I3AM production.
(F) Mice were treated with antibiotics in drinking water for one week followed by an oral gavage treatment, as indicated in the figure (n=5). HPLC analysis of I3A in feces.
(G) Representative Western blot of AHR, phosphorylated AHR (pAHR) at Ser36, CYP1A1 and GAPDH (loading control) in colon lymphocytes.
(H) Representative FACS analysis of IL-22, CD3 and RORγt expression in colon lymphocytes. The numbers in quadrants indicate the percentage of cells in each.
(I) Colon lymphocytes isolated from WT and AHR KO mice (n=5) and incubated with I3A or supernatant from LGG treated with the agents listed in the figure. ELISA analysis of IL-22 in cell supernatants.
(J) Representative colons from WT or IL-22 KO mice treated as listed in the figure (left); quantification of colon length (right).
(K) H&E-stained sections of colon (400x magnification) from WT and IL-22 KO mice treated as indicated in the figure. The data (C, D, F, I, J) are representative of three independent experiments (error bars, SD). * P<0.05 and ** P<0.01 (two-tailed t-test); NS, not significant.
To validate that the induction of I3A by GELN-RNAs is gut bacteria-dependent, broad-spectrum antibiotic treatment was used to deplete gut bacteria prior to GELN-RNA administration. The I3A level in feces of antibiotic-treated mice was decreased, and GELN-RNAs no longer induced I3A until additional LGG was administered (Figure 5F). We next investigated whether the AHR pathway in gut lymphocytes of mice gavaged with GELN-RNAs is activated by evaluating phosphorylation of AHR and induction of IL-22. Mice were gavaged with GELN-RNA one week prior to being treated with 2.5% DSS in drinking water for one additional week. Lymphoid cells were then isolated from the colon of treated mice. Western blotting analysis indicated that the levels of cytochrome P450 1A1 (CYP1A1) and phosphorylated AHR increased as a result of GELN-RNA treatment without affecting the total amount of AHR (Figure 5G). FACS analysis indicated that CD3+ RORγt+ cells from GELN-RNA-treated mice exhibited increased IL-22 expression (Figure 5H). However, the induction of IL-22 in CD3+ RORγt+ cells was abolished in AHR KO mice (Figure 5H). To further address whether the metabolites of LGG treated with GELN-RNAs have an impact on the induction of IL-22 via the AHR pathway, colon lymphocytes from naïve B6 mice were incubated for 3 h with I3A, MRS supernatant of LGG treated with GELN-RNAs or GNVs only. ELISA results indicated that I3A induced expression of IL-22, and more IL-22 was induced when the colon lymphocytes were treated with LGG supernatant (Figure 5I). In contrast, there was no evidence of an impact of I3A and LGG supernatant on the induction of IL-22 in colon lymphocytes from AHR KO mice (Figure 5I). Collectively, our data suggest that metabolites from the supernatants of LGG treated with GELN-RNAs induce IL-22 via the AHR pathway.
Dysfunction of the gut epithelial barrier is a hallmark of inflammatory intestinal diseases. The intestinal epithelial barrier is maintained by tight junctions that connect adjacent epithelial cells and seal the paracellular space. IL-22 is critical for maintenance of the intestinal barrier function. We next tested whether GELN-RNA-mediated induction of IL-22 plays a causative role in protecting mice against colitis. Indeed, unlike WT mice, IL-22 KO led to a loss of GELN-RNA-mediated protection against DSS-induced colon injury (Figures 5J, 5K). This result agreed with the fact that GELN-RNA reduced gut permeability, shown by a fluorescein-5-isothiocyanate (FITC)-conjugated dextran assay (Figure S5B), and increased the expression of Gjb, Cldn and Jam2 genes, which play a role in regulating gut epithelial tight junction stability (Figure S5C). Collectively, these data suggest that the preventive effect of GELN-RNAs on mouse colitis is partially IL-22 dependent.
GELN ath-miR167a prevents LGG accumulation in gut mucosa by down-regulating the LGG pili gene, SpaC
LGG that took up GELN-RNAs exhibited reduced migration into the bloodstream and liver of DSS-treated SPF (Figures 6A, 6C) and germ-free (Figure 6D) mice compared with LGG alone. The mucosa-associated LGG was decreased, particularly in the colon (Figure S5D). These findings were corroborated with confocal microscopy (Figures 6B, 6E), showing that most of the PKH26-labeled LGG stayed in the intestinal lumen of mice that had been fed LGG prepulsed with GNVs/GELN-RNAs. Moreover, the data generated from in vitro assays via colony-forming unit (cfu) quantification of LGG entry into C57BL/6 murine colon MC38 cells (Figures 6F, 6H) and human colon epithelial Caco2 cells (Figures 6G, 6I) and confocal examination of colocalization of LGG with GNVs/GELN-RNAs in MC38 cells (Figure 6J) further supported the notion that GELN-RNAs prevent LGG entry into both mouse and human gut epithelial cells.
Figure 6. GELN RNA treatment reduces bloodstream infections in DSS-induced colitis.

(A) C57BL/6 mice supplemented with 2.5% DSS following gavage with 109 CFU LGG treated with GNV/GELN-RNA or GNV/scrambled RNA. Representative numbers of bacteria colonies in the blood and liver cultured on MRS agar plates (uppers) and quantified (bottoms) as bacteria CFU; each symbol represents an individual mouse (n=5). *P<0.05 and **P<0.01 (two-tailed t-test).
(B) Representative imaging of location of PKH26-labeled LGG (red) treated with PKH67-labeled GNVs (green) in the colon using confocal microscopy; 200x magnification; scale bar: 10 μm.
(C) Blood cultured on MRS agar plates at different time points after gavage, as indicated in the figure, quantified (right) as CFU. *P<0.05.
(D) Germ-free mice treated using the same procedure described in (A). *P<0.05.
(E) Germ-free mice treated using the same procedure described in (B) and visualized with confocal microscopy; 200x magnification; scale bar: 25 μm.
(F) MC38 cells and (G) Caco-2 cells were inoculated with LGG treated with GNVs/GELN-RNAs, and the frequency of PKH26-labeled LGG and PKH67-labeled GNVs/GELN-RNAs was assessed using flow cytometry. The numbers in quadrants indicate the percentage of LGG in each.
(H, I) The number of intracellular bacteria was determined by plating on MRS plates. Quantification of CFU in H and I (right). *P<0.05.
(J) Visualization via confocal microscopy of MC38 cells presented with LGG and LGG/GELN-RNAs. Arrows in red indicate LGG/PKH26; arrows in green indicate GNVs/GELN-RNAs/PKH67. Scale bars, 10 μm. 3D images showing colocalization of LGG/PKH26 and GNVs/GELN-RNAs/PKH67 in MC38 cells. The data are representative of three independent experiments (error bars, SD).
Related to Figure S5.
The LGG pilus-specific protein SpaC was down-regulated at both the transcriptional and protein levels when LGG was treated with GELNs (Figures 2H, 2J). Experimentally, based on array data (Figures 2H, 2J) and in vitro transmigration of colon epithelial cell data (Figures 6F-I), we suggest that GELN-RNAs prevent the invasion of LGG into gut epithelial cells. To further investigate the mechanism underlying how GELN-RNAs prevent LGG migration, nucleotide sequences were aligned using BLAST, and the results indicated that the GELN miRNA ath-miR167a might directly bind to the LGG pilus protein SpaC mRNA and regulate SpaC expression (Figure S1G). MC38 cells exposed to LGG treated with seven specifically selected GELN-derived miRNAs indicated that ath-miR167a-5p, ath-miR842 and ath-miR827 can prevent the entry of LGG into gut epithelial cells (Figures 7A, 7B). Considering the critical role of SpaC in LGG migration and that ath-miR167a-5p has eight complementary bases with SpaC mRNA, we hypothesized that GELN ath-miR167a regulates the expression of SpaC in LGG (Figure 7C). Investigation of the effect of GELN miRNA on LGG migration in vivo indicated that ath-miR167a significantly reduced LGG translocation into the peripheral blood (Figure 7D), and LGG remained on mucosal surfaces (Figure 7E). LGG treated with ath-miR167a had significantly down-regulated SpaC mRNA (Figure 7F) and protein (Figure 7G) expression compared with LGG treated with a scrambled miRNA. TEM images showed that much less SpaC protein could be detected on pili of LGG treated with ath-miR167a compared with those exposed to PBS or scrambled miRNA (Figure 7H). A mutation that disrupted the binding site for ath-miR167a entirely restored GFP expression (Figure 7I). To further determine whether SpaC is specifically targeted by miR167a-5p, we first analyzed the effects of SpaC on entry into gut epithelial cells and mucosa-associated LGG and miR167a-5p-treated LGG growth. Immuno-TEM demonstrated that SpaC deleted (SpaCdel) LGG had no detectable SpaC (Figure S7A). SpaCdel LGG was less efficient in entering MC38 cells than wild-type LGG (Figure S7B). Although the gene encoding for SpaC was not detected in SpaCdel LGG (Figure S7C, left panel) and there was no difference in the in vitro growth between wild-type LGG and SpaCdel LGG (Figure S7C, middle panel), the in vivo results showed that the fewer SpaCdel LGG were present on the mucosa than wild-type LGG (Figure S7C, right panel). miR167a-5p treatment had no effect on the growth of SpaCdel LGG compared with scrambled miRNA (Figure S7D). Then, to further demonstrate the role of endogenous miR167a-5p in the context of GELN RNA, we depleted miR167a-5p from the pooled GELN RNAs with biotinylated anti-sense miR167a-5p. Then, GELN RNA or scrambled miRNA was encapsulated in GNVs. qPCR data indicated that miR167a-5p was successfully depleted from the pooled GELN RNA (Figure S7E). GELN RNAs with depletion of miR167a-5p had no effect on the expression of the LGG SpaC gene in comparison with GNV-scrambled miRNA-treated samples (Figure S7F). The depletion of GELN miR167a-5p had no effect on the level of mucosa-associated LGG compared with data generated from GNV-scrambled miRNA-treated LGG (Figure S7G). Collectively, these data suggest that after exposure to GELN-RNA, the LGG SpaC gene is targeted and down-regulated by GELN miR167a-5p.
Figure 7. GELN miR-167a targets and down-regulates SpaC in LGG.

(A) MC38 cells were inoculated with 107 LGG prior to being presented with selected ginger ELN miRNAs. The number of intracellular LGG was determined by FACS (top) and the CFU number on MRS plates (bottom).
(B) Quantification of CFU in A. *P<0.05.
(C) Schematic diagram of the putative binding sites of ath-miR167a-5p in the WT SpaC gene. The ath-miR167a-5p seed matches in the SpaC gene are mutated at the positions indicated.
(D) LGG were treated with ath-miR167a and administered via oral gavage to mice supplemented with 2.5% DSS. Representative numbers of bacterial colonies in the diluted aliquots of blood cultured on MRS agar.
(E) qPCR quantification of LGG on mucus. *P<0.05.
(F) ath-miR167a mimic knock-down of SpaC in LGG evaluated by qPCR. *P<0.05.
(G) ath-miR167a mimic reduced SpaC protein in LGG, determined by Western blotting.
(H) Immunogold labeling and electron microscopy of LGG using SpaC antiserum with protein A-10-nm gold particles. The location of gold particles (black arrow) on the pilus structure is indicated.
(I) A portion of the sequence of the SpaC gene containing the potential miR-167a binding site was constructed into a GFP reporter vector to obtain pSpaC. A binding site mutant construct (pSpaCm-GFP) served as the control. *P<0.05 (two-tailed t-test). The data are representative of three independent experiments (error bars, SD).
Discussion
Extensive research has shown that diet modulates the composition and function of the gut microbiota in humans and other mammals. Despite the body of knowledge that exists on fecal RNA, the impact of gut RNA on the microbiota is particularly poorly understood. Here, we identified small RNAs and miRNAs from ELNs and found that they modulate the composition of gut microbiota and their metabolites and inhibit mouse colitis. At a molecular level, our findings support the idea that (1) lipid-enriched ELNs send a signal that causes their uptake by gut bacteria; (2) ELN small RNAs mediate the cross-talk between gut microbiota and the host immune system, shaping the homeostatic balance between immunity and gut microbiota; and (3) ELN RNA regulates the composition, metabolites, growth, and localization of gut microbiota. Specifically, we found that gut probiotic LGG I3A induced by GELN-RNA promotes the expression of IL-22 through activation of the AHR signaling pathway, eliciting antimicrobial immunity and tissue repair at barrier surfaces.
A given miRNA may have hundreds of different bacterial mRNA targets (Hausser and Zavolan, 2014), several hundred miRNAs might be encapsulated in a given edible plant ELN, and ELNs from one type of edible plant could have a different miRNA profile than those from other plants. Therefore, it is conceivable that unlike endogenous miRNA released from host intestinal epithelial cells (Liu et al., 2016), which release a limited number of miRNAs, a large number and variety of food-derived ELN miRNAs can be taken up by gut microbiota. In addition, in terms of gut bacterial targeting, the ELNs from one type of edible plant could also have a different lipid profile, resulting in a targeting signal that is distinct from other ELNs. Therefore, a large variety of ELN-derived miRNAs and lipids may meet the requirement for potential regulation of the more than two million genes present in the gut microbiota through dietary-derived ELNs in a lipid targeting and a sequence-specific manner.
All organisms possess a diverse set of genetic programs that are used to alter cellular physiology in response to environmental cues. LexA acts as a transcriptional repressor of the SOS response genes coding primarily for error-prone DNA polymerases, DNA repair enzymes and cell division inhibitors (Miller et al., 2004). Our finding that GELN gma-miR396e promotes LGG growth at least partly through inhibition of LexA expression opens up a paradigm for further study of whether ELNs from different types of food have a role in regulating LexA-mediated SOS activity in the intestinal microenvironment. In addition, since SOS activity has a direct effect on the bacterial cell cycle and survival in general and ELNs from different types of food have preferential bacterial hosts, it is conceivable that the SOS activity in different species of gut bacteria is dependent on what types of food are eaten. Therefore, our current study provides a foundation for further examination of whether ELN-dependent SOS activity has an effect on the composition of gut microbiota through regulating the bacterial cell cycle and bacteria survival.
Ingestion of probiotics, beneficial molecules or microbes, is designed to deliver a health benefit to the host by increasing the numbers of beneficial microbes or their products within the gut. From a therapeutic application aspect, using ginger ELN-derived liposomes, we were successful in orally delivering miRNA to target gut bacteria for treatment of mouse colitis. This strategy could provide an alternate approach for gene therapy in gut dysbiosis-related disease and provide a rationale for ELN-based oral delivery of therapeutic miRNA for treatment of disease due to dysbiosis. It is conceivable that gut bacterial activity regulated by miRNA that interacts with bacterial mRNA in a gene-specific manner will have many advantages over other approaches such as chemotherapy drugs, which induce gut dysbiosis, and antibiotic treatment, which drives rapid development of resistant strains.
A variety of host-derived factors, such as antimicrobial peptides, play a crucial role in selecting and maintaining the stable diversity of the gut microbiota. Edible plant-derived factors that selectively regulate the stability of mucosa-associated microbiota have not been studied in detail. In this study, we found that the ratios of LGG among other species of mucosa-associated microbiota can remain stable, with a shift from mucosal-associated LGG to the majority of the LGG accumulating in the lumen. In zebrafish, an increased level of lumen-associated LGG has a more beneficial effect on gut barrier function than mucosal LGG (He et al., 2017); the role of lumen-associated LGG in mammals is less well understood. Our results explain how GELN small RNAs contribute to gut health via the LGG pilus-specific protein SpaC. Since all of the diets we have tested in the past contain ELN small RNAs, we propose the concept that spatial niche partitioning could be governed by diet ELN small RNAs, and this may help to explain both the long-term persistence of relatively stable numbers and the resilience of the microbiota, as well as the resistance to colonization by pathogens. Furthermore, the immune system has an active role in allowing only beneficial species to access these locations during homeostasis, as demonstrated in this study by IL-22. Our study opens up avenues for investigating whether other factors, such as GELN lipids and ELNs from other diets, may also participate in selection of which particular bacterial species are close to the epithelium and the creation of stable reservoirs for microorganisms to persist in the face of rapidly changing conditions in the gut lumen. Thus, through localized, immune-facilitated and adherence-dependent ELN selection, the host can maintain the stability of a diverse community of microbial symbionts.
In conclusion, given the importance of gut microbiota in human physiology, our findings reveal an important molecular mechanism underlying how diet ELN miRNAs can cross-talk with gut microbiota to maintain gut health. Because the composition of diet-derived ELN miRNAs and lipids is different among diets and each ELN miRNA has targets specific bacterial mRNA, this feature could be utilized for specific manipulation of the microbiome for human health and treatment of dysbiosis-related disease. However, the concept that food-derived ELNs are selectively taken up by the gut microbiome and host cells is relatively new in the field of gut physiology and health, and we are just beginning to define the individual steps in this process.
STAR ★ METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Huang-Ge Zhang (h0zhan17@louisville.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Eight- to twelve-week-old male specific-pathogen-free (SPF) C57BL/6 mice and IL-22 knockout mice (C57BL/6-IL-22tm1.1(icre)Stck/J) were purchased from the Jackson Laboratory (Bar Harbor, ME). AHR knockout mice were purchased from Taconic Biosciences. All mice were housed under specific-pathogen-free conditions. Germ-free mice were purchased from the National Gnotobiotic Rodent Resource Center (University of North Carolina, NC, Chapel Hill, NC) and maintained in flexible film isolators (Taconic Farm) at the Clean Mouse Facility of the University of Louisville. Animal care was performed following the Institute for Laboratory Animal Research (ILAR) guidelines, and all animal experiments were conducted in accordance with protocols approved by the University of Louisville Institutional Animal Care and Use Committee (Louisville, KY).
Human Subjects
The study involved 58 healthy volunteers between the ages of 25 and 46 years (30 males, 28 females) who were randomly assigned to a GELN group (14 males, 14 females) or a control group (16 males, 14 females) using simple randomization(Kim and Shin, 2014) (Kim and Shin, 2014). The sample size for human subjects was determined by a one-way ANOVA-based power analysis (http://www.biostathandbook.com/power.html) (Given a power of 0.8, effect size of 0.4 and significance level of 0.05, the sample size needed in each group was 25.52458 (rounded = 26) and resource equation method(Festing and Altman, 2002). The participants in the two groups were matched for age and sex. All clinical fecal samples from healthy volunteers were collected in the Department of Surgery, Huai'an First People's Hospital, Huai'an, Jiangsu, China with written informed consent from patients. Approval for the study was granted by the Institutional Research Ethics Committee at the Health Department of Huai’an. All subjects provided signed informed consent for participation in the study. Volunteers were recruited from the population in 2017 in Huai’an, Jiangsu, China. No subjects had a history of chronic gastrointestinal disease, antibiotic use within three months prior to testing, alcohol abuse, or smoking. To prevent bias in study results, participants were kept blinded to the allocation. Both researchers and participants were kept blinded to the treatment groups. Before taking GELNs, volunteers provided fecal samples at day 0. Participants drank GELNs in the amount of 200 mg in 10 ml of sterile 0.9% sodium chloride (GELNs group, n=28) or 10 ml of sterile 0.9% sodium chloride only (control group, n=30) every other day for 6 days. The feces of all enrolled subjects were collected at day 7.
Cells
C57BL/6 murine colon adenocarcinoma MC-38 cells (sex is unknown, Kerafast) or human epithelial colorectal adenocarcinoma Caco-2 cells (male, American Type Culture Collection, ATCC) were grown in tissue culture plates with Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37Ό in a 5% CO2 atmosphere.
Bacteria
Lactobacillus rhamnosus (LGG, ATCC# 53103, Manassas, VA) was The SpaC-deleted LGG was cultured in Lysogeny broth (LB) (ISC BioExpress, Kaysville, UT) at 37°C. Ruminococcaceae sp. (ATCC, TSD-27) was grown in GS2 + cellobiose medium (Amy Biddle, et al. Diversity 2013). Listeria monocytogenes (Listeria, ATCC# 15313) and Bacteroides fragilis (Fragilis, ) were cultured in brain heart infusion (BHI) broth (Hardy Diagnostics, Santa Maria, CA) as described. LGG was grown in MRS broth media at 37°C in anaerobic conditions for 14– 16 h to an OD600 of 0.8–1.0. Bacterial viability and concentration were checked by MRS agar platting. Cultures were centrifuged and the bacterial pellet was diluted in MRS for in vitro experiments and in PBS for gavaging at 109 CFU/mouse per day.
METHOD DETAILS
Preparation of Plant ELNs
To prepare plant exosome-like nanoparticles (ELNs), peeled Hawaiian ginger roots (Simply ginger, PLU#:4612), carrot, garlic, turmeric roots and grapefruit were used for isolation and purification of ELNs using a previously described method (Mu et al., 2014). Briefly, the plants listed above were peeled and then homogenized in a high-speed blender for 1 min. The juice was collected after net filtration. The supernatant was collected after centrifugation at 1,000x g for 10 min, 2,000x g for 20 min, 4,000x g for 30 min, and 10,000x g for 1 h. The pellets containing nanoparticles derived from each plant were spun down at 100,000x g for 1.5 h at 4°C. The isolated exosomes were further purified in a sucrose gradient (8, 30, 45, and 60% sucrose in 20 mM Tri-Cl, pH 7.2), followed by centrifugation at 100,000x g for 1.5 h at 4°C. Purified GELNs were fixed in 2% paraformaldehyde and imaged under a Zeiss EM 900 electron microscope using a previously described method (Mu et al., 2014).
The purity of GELNs was evaluated by calculating the ratio of particle to protein (Webber and Clayton, 2013). The size distribution of GELNs was analyzed using a Zetasizer Nano ZS (Malvern Instrument, UK) at a flow rate of 0.03 ml per min. The protein concentration of GELNs was determined using a BioRad Protein Quantitation Assay kit with bovine serum albumin as the standard.
RNA Extraction
Total RNA containing miRNA was isolated from ELNs and murine tissue using a miRNeasy mini kit (Qiagen) according to the manufacturer's instructions. In brief, 50 mg of plant-derived ELNs or tissue was disrupted in QIAzol Lysis Reagent. Tissue was homogenized using a tissue grinder before disruption. The homogenate was mixed with 140 μl of chloroform and centrifuged. The upper aqueous phase was mixed with 1.5 volumes of ethanol and loaded into an RNeasy spin column. The flow-through was discarded after centrifugation, and the column was washed with RWT and RPE sequentially. Total RNA was eluted with RNase-free water. Bacterial mRNA was isolated using RiboPure Bacteria and MICROBExpress kits (Thermo Fisher Scientific) according to the manufacturers’ instructions. The quality and quantity of the isolated RNA were analyzed using a NanoDrop spectrophotometer and Agilent Bioanalyzer.
Preparation of Plant Nanovectors
To prepare GELN nanovectors (GNVs) and grapefruit ELN nanovectors (GFNVs), the GELN or grapefruit ELN-derived lipids were extracted with chloroform and dried under vacuum. To generate GNVs and GFNVs, 200 nmol of lipid was suspended in 200-400 μl of 155 mM NaCl with or without 10 μg of ELN-derived RNA. After UV irradiation at 500 mJ/cm2 in a Spectrolinker crosslinker (Spectronic Corp.) and a bath sonication (FS60 bath sonicator, Fisher Scientific) for 30 min, the pelleted particles were collected by centrifugation at 100,000x g for 1 h at 4°C,. The RNA encapsulation efficiency of GNVs (68 ± 5%) was determined using a previously described method (Teng et al., 2016).
Plant ELN Distribution in Vivo
Plant ELNs labeled with DiR dye were administered to C57BL/6 mice (n=5) by oral gavage at 500 mg/kg. The labeled plant ELNs in the gut of mice were visualized using an Odyssey CLx Imaging System (Licor Biosciences).
Migration Assays
MC-38 or Caco-2 cells (ATCC) were seeded at 1×105 cells per well in a 12-well tissue culture plate with Dulbecco's modified Eagle's medium (DMEM) without antibiotics. The cells were inoculated with 1 χ 107 bacteria per well for 90 minutes at 37°C in a 5% C O2 atmosphere to allow bacterial adhesion and entry. The number of intracellular bacteria was quantified using a previously described method (Zhang and Wang, 1998).
DSS Colitis Model
Colitis was induced by addition of dextran sulfate sodium (DSS, MP Biomedicals, Santa Ana, CA) to autoclaved drinking water at 2.5%. Colitis development was monitored daily by assessing body weight and presence of blood in the stool.
Bacterial Translocation
Bacterial translocation from the murine gut to peripheral blood and liver was determined at the indicated times presented in the relevant figures after oral bacterial administration. Fifty microliters of anticoagulant blood were cultured on MRS agar for 48 h at 37°C in an anaerobic chamber. Liver tissue sam ples were homogenized in 0.5% Triton X-100/PBS, and 100 μl of the homogenates were cultured on MRS agar plates. After 48 h of incubation, CFUs were counted, and the results are expressed as number of bacteria detected/mL of blood or gram of liver.
Labeling of Bacteria and Nanoparticles
Bacteria, ELNs or ELN nanovectors were labeled with PKH26 or PKH67 Fluorescent Cell Linker Kits (Sigma) in accordance with the manufacturer's instructions. After a wash with PBS, bacteria pellets, ELNs or ELN nanovectors were suspended in 250-500 μl of diluent C with 2-4 μl of PKH26/67 and subsequently incubated for 30 min at room temperature. After centrifugation for 5 minutes at 13,000x g, labeled LGG, ELNs or ELN nanovectors were resuspended for further experiments.
Bacteria GELN Uptake Assay
Briefly, 1×107 LGG cells were incubated for 30 min at room temperature with 1 mg of PKH26-labeled GELNs or 1 μg of GELN RNA encapsulated in GNVs. After two washes with PBS, LGG uptake of GELNs was visualized using a confocal microscope. To exclude the possibility of detecting GELNs remaining (adhering) on the outside of bacteria, the bacteria were washed three times with medium and treated with 100 μl of 0.5% Triton X-100 for eight minutes, followed by the immediate addition of bacteria broth to wash bacteria 2x before the bacteria were imaged using confocal microscopy. (Note: 0.5% Triton X-100 did not affect bacterial viability for at least 30 minutes after addition).
Immunogold Labeling of LGG Pili and TEM
To visualize LGG pili via transmission electron microscopy (TEM), LGG cultures were grown overnight (OD600<1.0) and washed once with PBS. Formvar-carbon-coated copper grids (Electron Microscopy Sciences, PA) first were floated for 1 h on droplets of the diluted LGG at 107/mL in PBS, washed several times with 0.02 M glycine in PBS, and then treated with a blocking solution of 1% bovine serum albumin (BSA) in PBS. The grids were then floated for 1 h on droplets of anti-SpaC serum (1:100) in blocking solution. After a wash with 0.1% BSA in PBS, the grids were incubated for 20 min with protein A-conjugated 10-nm-diameter gold particles (Cytodiagnostics, Canada) diluted 1:55 in blocking solution. After one wash in PBS, the grids were fixed with 1% glutaraldehyde, washed with distilled water, and then stained with 1% ammonium molybdate on the surface of the EM grid. After excess ammonium molybdate was removed from the grid, images were visualized using a Thermo-Fisher TEM Tecnai Spirit at 80 kV, and images were collected with an AMT XR60 digital camera.
Quantitative Real-Time PCR for RNA Expression
The quantity of mature miRNAs was determined with quantitative real-time PCR (qPCR) using a miScript II RT kit (Qiagen) and miScript SYBR Green PCR Kit (Qiagen) with Qiagen 3’ universal primers. The 5’ specific miRNA primers used are listed in Table S7. For analysis of gene mRNA expression, 1 μg of total RNA was reverse transcribed using SuperScript III reverse transcriptase (Invitrogen), and quantitation was performed using SsoAdvancedTM Universal SYBR Green Supermix (BioRad) and the listed primers (Table S7) with SsoAdvancedTM Universal SYBR Green Supermix (BioRad). qPCR was performed using a BioRad CFX96 qPCR System with each reaction run in triplicate. Analysis and fold-changes were determined using the comparative threshold cycle (Ct) method. Changes in miRNA or mRNA expression was calculated as fold-change.
Quantification of Gut Bacteria Using QPCR
For gut bacteria identification, qPCR was performed from gut microbiota-derived DNA extracted with a QIAamp DNA Stool Mini Kit (Qiagen). All kits were used according to the manufacturer's instructions. Quantitation was performed using SsoAdvancedTM Universal SYBR Green Supermix (BioRad), and the bacterial specific primers are listed in Table S7. qPCR was performed using the BioRad CFX96 qPCR System with each reaction run in triplicate. Analysis and fold-change were determined using the comparative threshold cycle (Ct) method.
Plasmid Construction and Mutagenesis
The SpaC fragment (339-800, LGG_RS02140) spanning the sequences of the potential target (710-717) of GELN ath-miR167a was obtained by PCR with cDNA from LGG RNA. PCR was performed using a BioRad thermal cycler T100. The 462-bp PCR product amplified by the primer pair SpaC-pGFPuv-F: GCGCATGCCTGCAACTAATTTTGTCGCAAACG and SpaC-pGFPuv-R: CCTCTAGAACAGTTTTCAGCAGGCATCC was ligated into the SphI and XbaI restriction enzyme sites of a pGFPuv vector (Clontech) to obtain a green fluorescent protein (GFP) expression reporter. SpaC-pGFPuv fused with the SpaC gene fragment, which can be expressed in prokaryotic cells. To generate mutants of SpaC, the oligonucleotide primers SpaCMut-F, CTGTAGGTGCTGTAACTGCCTGAATACCGTAATAC, and SpaCMut-R, GTATTACGGT ATTCAGGCAG TTACAGCACC TACAG, were designed to specifically disrupt the putative ath-miR167a binding site. A Geneart® Site-Directed Mutagenesis System (#A13282, Invitrogen) was used in conjunction with specific primers to introduce a SpaC mutation in the pGFPuv construct according to the manufacturer's instructions. After mutant strand synthesis (using T4 DNA polymerase) and ligation, the resultant plasmids were introduced into E. coli, and transformants were selected using ampicillin resistance. Further restriction endonuclease SphI and XbaI analysis was performed to screen clones, and all of the constructs were confirmed by DNA sequencing.
Microbiota 16S rRNA Gene Sequencing
GELNs were administered by gavage to C57BL/6 mice (500 mg/kg of body weight) three times in seven days (n=5). To identify bacterial strains that preferentially take up GELNs, PKH26-labeled GELNs were administered by gavage, and PKH26-positive microbiota from fecal samples were sorted using a BD FACSAria™ III cell sorter (BD Biosciences, San Jose, CA). Bacterial DNA from fecal samples was isolated with QIAamp DNA Stool Mini Kits (Qiagen), and bacterial strains were investigated using 16S rRNA gene sequencing. DNA (15 ng) was used as a template to amplify the 16S rRNA gene using a High Fidelity PCR system kit (Roche). The v1-v3 regions of 16S ribosomal RNA gene were amplified using 27f (AGAGTTTGATCCTGGCTCAG) and 534r (ATTACCGCGGCTGCTGG) primers (1 μM). The primers were anchored with adaptor (adopter A: 5’ CCATCTCATCCCTGCGTGTCTCCGACTCAG 3’ and adopter B: 5’ CCTATCCCCTGTGTGCCTTGGCAGTCTCAG 3’) and Multiplex Identifiers (MIDs; 10 bp long). The multiplexed amplicons were purified using a QIAquick Gel Extraction Kit (Qiagen). The amplicon sequence was conducted using the 454 Jr. Sequencing platform. The 16S rRNA gene sequences were analyzed using QIIME platform scripts (www.qiime.org). The microbial classification was performed with the GreenGenes reference data base (gg_otus-13_8) using QIIME tools (Caporaso et al., 2010). By applying hierarchical clustering algorithms (HCAs), we determined the species clustering based on the operational taxonomic unit (OTU) using amplicon sequencing of 16S RNA. The reference sequences allowed sorting of the results into OTUs by clustering 97% sequence similarity (uclust) and classification according to various taxonomic ranks (phylum, order, class, family, genus, and species). The percentage of each bacterial species was virtualized with Interactive Tree Of Life (iTOL) and R software (Letunic and Bork, 2007).
Mouse Cytokine Array
To investigate effects of GELN RNAs on the regulation of cytokine expression in colon epithelia, germ-free mice with DSS-induced colitis were administered 109 LGG pretreated with GNVs or GNVs with GELN RNAs. The colon tissue extracts were prepared in modified radioimmunoprecipitation assay (RIPA) buffer (Sigma) with the addition of protease and phosphatase inhibitors (Roche). Cytokine proteins were analyzed with a Proteome Profiler Mouse XL Cytokine Array Kit (R&D Systems, ARY028). Quantification of the spot intensity in the arrays was conducted with background subtraction using ImageJ.
Proteomic LGG Sample Preparation
Briefly, 1×107 LGG were incubated with 1 mg GELNs for 2 h and then harvested by centrifugation at 13,000x g for 5 min. Bacteria were suspended in lysis buffer (2% SDS, 100 mM DTT, 20 mM Tris-HCl pH 8.8) for 20 min at 95°C. LGG lysate was collected from supernatants after centrifugation, and concentrations were estimated using an RC DC Protein Assay Kit (BioRad, Hercules, CA). Protein aliquots (50 μg) were diluted into 4% SDS / 0.1 M Tris-HCl pH 8.5 and 1 M DTT and processed according to the filter-aided sample preparation (FASP) method as described previously (Teng et al., 2017). The digested, ultrafiltered samples were trap-cleaned with C18 PROTOTM, 300 A Ultra MicroSpin columns; lyophilized by vacuum centrifugation; and redissolved in 16 μl of 2% v/v acetonitrile. Concentrations were estimated based on absorption at 205 nm using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, San Jose, CA, USA).
LC-MS Analysis of LGG Protein
Liquid chromatography–mass spectrometry (LC-MS) was carried out using a method described previously (Teng et al., 2017). Proteome Discoverer v1.4.1.114 (Thermo) was used to analyze the data collected by MS. The database used in Mascot v2.5.1 and SequestHT searches was the 2/17/2017 version of the LGG proteome from UniprotKB (Proteome ID UP000000955). Scaffold was used to calculate the false discovery rate using the Peptide and Protein Prophet algorithms. Proteins were grouped to satisfy the parsimony principle. The proteins were clustered based on differential expression, and heat maps representing differentially regulated proteins by GELNs were generated using R software.
Coomassie Blue Staining
The LGG proteins were separated on a 10% SDS-PAGE gel. The gel was fixed and stained for imaging using 0.1% Coomassie Brilliant Blue R-250 (BioRad).
Western Blotting
LGG cells were treated as indicated in the individual figure legends and harvested from 100-350 ml cultures by centrifugation at 5,000x g for 10 minutes. The cells were resuspended on ice for 45 min in 1 ml of TE buffer (100 mM Tris-Cl, 10 mM EDTA, pH 8.0) supplemented with protease inhibitor cocktail (Roche) and lysozyme (1 mg/mL). The cell lysates were sonicated on ice using three 10-second bursts at medium intensity and then frozen in liquid nitrogen. The lysates were quickly thawed at 37°C, and two more rapid sonication-freeze-thaw cycles were performed. Proteins were separated on 10% SDS-PAGE gels and transferred to PVDF membranes (BioRad Laboratories, Inc., Hercules, CA). Mouse anti-SpaC antibody was purchased from PodiCeps (PODI-0063, Netherland) and anti-DnaK antibody was purchased from Abcam (ab69617). After the Alexa Fluor-647 (Invitrogen) conjugated secondary antibody incubation, the bands were visualized and analyzed using an Odyssey Imager (LiCor Inc, Lincoln, NE). For immunoblotting of tissue, mice were treated as indicated in the figure legends, and lysates were prepared in modified RIPA buffer (Sigma) with the addition of protease and phosphatase inhibitors (Roche). Proteins were separated on 10% SDS-PAGE gels and transferred to PVDF membranes (BioRad Laboratories, Inc., Hercules, CA). Dual-color precision protein MW markers (BioRad) were separated in parallel. Antibodies were purchased as follows: AHR (PA5-25447) and phosphorylated AHR (PA5-38404) antibodies from Thermo Fisher, CYP1A1 (ab79819) and GAPDH (ab9485) antibodies from Abcam. The secondary antibodies conjugated to Alex Fluor-488 or Alex Fluor-594 were purchased from Invitrogen (Eugene, OR). The bands were visualized using an Odyssey Imager (LiCor Inc, Lincoln, NE).
GELN RNA Libraries and Sequencing
Small RNA libraries were generated with 100 ng of total RNA and TruSeq Small RNA Library Preparation Kits (Illumina) according to the manufacturer’s instructions. Following PCR amplification (16 cycles), libraries between 140 and 160 bp in size were gel purified and resuspended in 11 μl of ultrapure water. Equal amounts of libraries were pooled and sequenced on the Illumina HiSeq 2500, followed by demultiplexing and fastq generation with CASAVA v1.8.4. Raw fastqs were adapter and quality score trimmed with cutadapt v1.10. with a minimum length of 15 nt. MicroRNAs were identified using sRNABench Pipeline software (version 05/14). A core set of plant miRNAs from miRBase v21 were used as the reference, and this set included all 14 plant species with at least 200 mature microRNA sequences annotated in miRBase. Within the sRNABench pipeline, mapping was performed with bowtie software (v0.12.9), and microRNA folding was predicted with RNAfold from the Vienna package (v2.1.6).
LGG mRNA Sequencing
LGG cells were treated with fluorescent dye PKH26-labeled GELNs or PBS as indicated in the individual figure legends and harvested from 100-350 ml cultures by centrifugation at 4000x g for 10 minutes. PKH26-positive LGG were sorted with FACS. mRNA was isolated from bacteria using RiboPure Bacteria and MICROBExpress (Thermo Fisher Scientific). For each RNA sample, double-stranded cDNA was synthesized from 10 ng mRNA using a SMARTer Universal Low Input RNA Kit (Takara, catalog# 634940) for sequencing, which included a 16-cycle PCR. Following quantitation with Qubit dsDNA HS Reagent (Thermo Scientific, cat. # Q32854), 10 ng of dscDNA/sample was fragmented with an E220 Focused-ultrasonicator (Covaris). The fragmented cDNA was then prepared into libraries using a KAPA Hyper Prep Kit (KAPA Biosystems, cat. # KK8504). Libraries were then combined into equimolar pools, which were then measured for size and concentration. The pools were clustered onto a paired-end flowcell with a 20% v/v PhiX spike-in and sequenced on an Illumina HighSeq 2500 sequencer. The first and second reads were each 83 bases.
Species Alignment and Analysis
The mRNA sequencing data were demultiplexed and converted to fastqs with CASAVA v1.8.4, and 7 nt were trimmed from R1 and R2 raw fastqs with cutadapt v1.10 as recommended by the SMARTer kit. Transcript abundance was estimated with salmon v7.2 with the following options: -libType A -num, Bootstraps 100, -seqBias -gcBias - dumpEq -geneMap. For LGG abundance, the transcriptome fasta and annotation from EnsemblBacteria (Genome Assembly ASM2650v1) were used. For Zingiber officinale (ginger) abundance, the ESTs from NCBI were used as transcriptome input for salmon. Ginger transcript sequence similarity was determined using NCBI blast v.2.2.26, keeping the top hit against the nucleotide (nt) database with a maximum e-value of 0.001.
Predicting GELN miRNA Targeting to LGG mRNA
After downloading eleven gut bacterial genomes from the NCBI RefSeq database (ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/bacteria/), bacterial mRNAs potentially targeted by ginger miRNAs were identified by enrichment analysis of the reverse complement of the miRNA seed sequence in the 300 bp region near the coding sequence (CDS) start site (200 bp before and 100 bp after the site). For the enrichment analysis, two seed subsequences were used: a 7-mer (nt 2-8) and an 8-mer (nt 1-8). The enrichment analysis adopted a framework that utilizes the 1st order Markov model (MM). In this framework, the observed k-mer count in the 300-bp region of each bacterial mRNA was compared against the background count derived from the 1st order Markov model. A P-value was then calculated for each miRNA-mRNA pair to estimate the likelihood of having a functional pair. Once all p-values were calculated, the false discovery rate (FDR) was obtained using the Benjamini–Hochberg method (Benjamini and Hochberg, 1995) for multiple P-value correction.
Microbial DNA QPCR Arrays
To determine whether ELN RNA has an effect on the composition of major gut microbial species, mice were gavaged with GELN RNAs and grapefruit and carrot ELN RNAs encapsulated in the GELN lipid-derived liposomes (GNVs) (500 mg/kg mouse weight in 100 μl PBS; n=5). The GNVs were given to mice once every other day for seven days. Three hours after the last dose, the mice were sacrificed, fecal DNA was extracted, and a qPCR array was performed using Qiagen Custom Microbial DNA qPCR Arrays (cat# 330161) on an Applied Biosystems ViiA™ 7 Real-Time PCR System. Normalization to Pan Bacteria (BPCL00362A) was performed using a threshold cycle (Ct) to correct for potential DNA input or RT efficiency biases. DNA qPCR array data generated from the fecal samples were analyzed using SPSS 16.0 software and are based on fold-changes compared with PBS as a control. Heat maps generated from qPCR data using software R reflect the abundance of the microbial species analyzed.
Thin-Layer Chromatography (TLC) Analysis
Lipids from ELNs were extracted and quantitatively analyzed using a method previously described (Wang et al., 2013). TLC was performed (Zhuang et al., 2015). Briefly, HPTLC-plates (silica gel 60 with a concentrating zone, 20 cm × 10 cm; Merck) were used for the separation. After aliquots of concentrated lipid samples were extracted from plant ELNs, they were separated on a plate that had been developed with chloroform/methanol/acetic acid (190:9:1, by vol). After drying in air, the plates were sprayed with a 10% copper sulfate and 8% phosphoric acid solution and then charred by heating at 180°C for 5 min. The bands of lipid on the plate were imaged using an Odyssey Scanner (Licor Bioscience, Lincoln NE).
Lipidomic Analysis with Mass Spectrometry
Lipid samples extracted from ELNs were submitted to the Lipidomics Research Center, Kansas State University (Manhattan, KS) for analysis using a method previously described. In brief, the lipid composition was determined using triple quadrupole MS (Applied Biosystems Q-TRAP, Applied Biosystems, Foster City, CA). The protocol has been previously described (Wang et al., 2013). The data are reported as the concentration (nmol/mg ELNs) and percentage of each lipid within the total signal for the molecular species determined after normalization of the signals to internal standards of the same lipid class.
Histological Analysis
For hematoxylin and eosin (H&E) staining, tissues were fixed with buffered 10% formalin solution (SF93–20; Fisher Scientific, Fair Lawn, NJ) overnight at 4°C. Dehydration was achieved by sequential immersion in a graded ethanol series of 70%, 80%, 95%, and 100% ethanol for 40 min each. Tissues were embedded in paraffin and subsequently cut into ultrathin slices (5 μm) using a microtome. Tissue sections were deparaffinized in xylene (Fisher), rehydrated in decreasing concentrations of ethanol in PBS, and stained with H&E, and the slides were scanned with an Aperio ScanScope. For frozen sections, tissues were fixed with periodate-lysine-paraformaldehyde (PLP) and dehydrated with 30% sucrose in PBS overnight at 4°C, and nuclei were stained with 4',6-diamidino- 2-phenylindole dihydrochloride (DAPI). The slides were scanned using an Aperio ScanScope or visualized via confocal laser scanning microscopy (Nikon, Melville, NY) as previously described (Teng et al., 2017).
HPLC Analysis of Tryptophan Metabolites
The fecal samples and LGG MRS broth were diluted with an equal volume of methanol. After centrifugation at 10,000x g for 30 min, 50 μl of supernatant was injected for high-performance liquid chromatography (HPLC) analysis. The HPLC analysis was performed on an Agilent 1260 Infinity system equipped with an Agilent ZORBAX SB-C18 column (4.6 × 150 mm, 3.5 μm), with following parameters: mobile phase A: 5 mM NH4Ac in water modified with 0.1% formic acid (v/v); mobile phase B: 5 mM NH4Ac in 90% acetonitrile modified with 0.1% formic acid (v/v); gradient: 5% B in first 5 min, 5–20% B for 10 min, hold 20% B for 5 min, 20%-50% B for 5 min, hold 50% B for 5 min, 50%-100% B for 5 min, hold 100% B for 10 min, 100–5% B for 5 min; flow rate: 1.0 ml/min; temperature: 30°C. UV detection at 300 nm was used to monitor indole-3-carboxaldehyde (I3A) and indole-3-acetaldehyde (IAAld); FLD (ex=280 nm, em=350 nm) was used for detection of tryptophan and indole-3-acetamide (I3AM). The standard for I3A (cat#: 129445-5g), I3AM (cat#: 286281-1G), IAAld (cat#: I1000-25MG) and tryptophan (Cat: T0254-25g) were purchased from Sigma.
Enzyme-Linked Immunosorbent Assay (ELISA)
The cytokine IL-22, IL-1β and TNFα levels in cell culture supernatants or mouse colon mucus were quantified using ELISA kits (eBioscience) according to the manufacturer’s instructions. Briefly, a microtiter plate was coated with anti-mouse IL-22, IL-1β and TNFα antibody at 1:200 overnight at 4°C. Excess binding sites were blocked with 200 μl of 1x ELISA/ELISPoT Diluent (eBioscience) for 1 h at 22°C. After washing three times with PBS containing 0.05% Tween 20, the plate was incubated with detection antibody in blocking buffer for 1 h at 22°C. After three washes, avidin conjugated with horseradish peroxidase and substrate were each added sequentially for 1 h and 30 min at 22°C. An analysis of absorbance at 405 nm using a microtiter plate reader (BioTek Synergy HT) followed.
Isolation of Lymphoid Cells in the Colon
Intestinal lymphoid cells were isolated from the intestine by incubation in PBS supplemented with 1 mM EDTA, 15 mM HEPES and 10% FCS for 30 min at 37°C. Supernatants were discarded, and tissues were then incubated in RPMI supplemented with 15 mM HEPES and 300 units/mL collagenase type VIII (Sigma) for 30 min with gentle shaking. Lysates were gently pressed through nylon cell strainers (70 μm in diameter, Fisher Scientific), and mononuclear cells were isolated on a 40%/80% colloidal silica particle (Percoll) gradient. Lymphocytes were recovered from the interface and washed twice.
Antibiotic Treatment
Six- to eight-week-old male mice were provided sterile drinking water supplemented with vancomycin (0.5 mg/mL), streptomycin (1 mg/mL), neomycin (1 mg/mL), chloramphenicol (0.5 mg/mL) or metronidazole (1 mg/mL) for 3 weeks before the beginning of GELN or GNV treatment.
Flow Cytometry
Isolated lymphocytes from colon tissue were seeded into 6-well plates and stimulated for 6 h with LPS (10 μg/mL) in the presence of brefeldin A (5 μg/mL; Invitrogen). Washed cells were stained for 40 min at 4°C with the appropriate fluorochrome-conjugated antibodies in PBS with 2% FBS. Characterization and phenotyping of the various lymphocyte subsets from the liver or spleen were performed using flow cytometry. Data were acquired using a BD FACSCalibur flow cytometer (BD Biosciences, San Jose, CA) and analyzed using FlowJo software (Tree Star Inc., Ashland, OR). To visualize bacteria stained with PKH26/67 and transformed with SpaC-pGFPuv plasmid, the PKH26/67-positive and GFP-positive bacteria were detected by flow cytometry using a BD FACSCalibur flow cytometer (BD Biosciences, San Jose, CA), and data were analyzed using FlowJo software (Tree Star Inc., Ashland, OR).
In Vivo Intestinal Permeability Assay
For in vivo intestinal permeability studies, fluorescein-5-isothiocyanate (FITC)-conjugated dextran (MW 4000; Sigma-Aldrich, St. Louis, MO) was administered by oral gavage at a concentration of 60 mg/100 g of body weight. Serum was collected retro-orbitally five hours later, and fluorescence intensity was determined with a fluorescence spectrophotometer (BioTek) at emission and excitation wavelengths of 485 nm and 528 nm, respectively. FITC concentration was measured from standard curves generated by serial dilution of FITC-dextran.
Quantification and Statistical Analysis
Unless otherwise indicated, all statistical analyses in this study were performed with SPSS 16.0 software. The data are presented as values with standard deviation (as the mean ± SD). The significance of differences in mean values between two groups was analyzed using Student’s t-test. Differences between individual groups were analyzed via one- or two-way ANOVA. Differences between percentages of bacterial composition were analyzed with a chi-square test. Differences were considered significant when the P-value was less than 0.05 or 0.01. A P value greater than 0.05 was considered not significant (NS). Both animals and human subjects were randomly assigned to a control group and different experimental condition groups matched for age and sex using simple randomization. Double-blinded studies were used for animal and human subjects studies. Unless otherwise indicated, the mice used in the in vivo study were male C57BL/6 strain mice. Using one-way ANOVA comparing up to four groups, for a power of 0.7, a large effect size (0.75) and a significance level of 0.05, the minimum sample size needed in each group was 4.992 (rounded = 5)(Festing and Altman, 2002). The reported “n” in animal and human studies represents the number of animals and human subjects. Data are representative of three independent experiments.
Data and Software Availability
Data Resources
The sequencing data and mass spectrometry data were deposited in NCBI Sequence Read Archive (SRA) with accession number SRP121341.
Supplementary Material
Table S1 (related to Figure 1 and Figure 2) List of GELN miRNAs sequencing and prediction target genes expressed in LGG
Table S4 (related to Figure 2) Concentration of plant ELNs lipids analyzed with mass spec (nmol/mg ELNs)
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse AHR | Thermo Fisher | Cat# PA5-25447 |
| phosphorylated AHR | Thermo Fisher | Cat# PA5-38404 |
| Anti-mouse CYP1A1 | Abcam | Cat# ab79819 |
| Anti-mouse GAPDH | Abcam | Cat# ab9485 |
| Mouse anti-DnaK | Abcam | Cat# ab69617 |
| Anti-mouse lgG Alexa Fluor 647 | Invitrogen | Cat# A21236 |
| Anti-mouse lgG Alexa Fluor 488 | Invitrogen | Cat# A32723 |
| Anti-mouse lgG Alexa Fluor 594 | Invitrogen | Cat# A11005 |
| Mouse anti-SpaC | PodiCeps | Cat# PODI-0063 |
| Anti-SpaC serum | (von Ossowski et al., 2013) | N/A |
| Bacterial and Virus Strains | ||
| Lactobacillus rhamnosus (LGG) | ATCC | Cat# 53103 |
| SpaC-deleted LGG | (von Ossowski et al., 2013) | N/A |
| Escherichia coli | Inserm | Cat# U1048 |
| Ruminococcaceae sp. | ATCC | Cat# TSD-27 |
| Listeria monocytogenes | ATCC | Cat# 15313 |
| Bacteroides fragilis | NCTC | Cat# 9343 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MRS broth | Hardy Diagnostics | Cat# C5931 |
| BHI broth | Hardy Diagnostics | Cat# C5141 |
| Lysogeny broth | Gibco | Cat# 10855-001 |
| PKH26 Fluorescent Cell Linker Kit | Sigma | Cat# PKH26GL |
| PKH67 Fluorescent Cell Linker Kit | Sigma | Cat# PKH67GL |
| Dextran Sulfate Sodium Salt (DSS) | MP Biomedicals | Cat# 0216011090 |
| FITC-conjugated dextran (MW 4000) | Sigma | Cat# 46944 |
| Indole-3-carboxaldehyde | Sigma | Cat# 129445 |
| Indole-3-acetaldehyde | Sigma | Cat# I1000 |
| Tryptophan | Sigma | Cat# T0254 |
| Indole-3-acetamide | Sigma | Cat# 286281 |
| PA 18:1 | Avanti Polar Lipids | Cat# 840875 |
| PA 18:2 | Avanti Polar Lipids | Cat# 840885 |
| PA 16:0-18:1 | Avanti Polar Lipids | Cat# 840857 |
| PA 16:0-18:2 | Avanti Polar Lipids | Cat# 840858 |
| PC 16:0-18:2 | Avanti Polar Lipids | Cat# 850458 |
| Critical Commercial Assays | ||
| miRNeasy mini kit | Qiagen | Cat# 217004 |
| QIAamp DNA Stool Mini Kits | Qiagen | Cat# 51504 |
| Microbial DNA qPCR Arrays | Qiagen | Cat# 330161 |
| MICROBExpress™ Bacterial mRNA Enrichment Kit | Invitrogen | Cat# AM1905 |
| RiboPure™ RNA Purification Kit, bacteria | Invitrogen | Cat# AM1925 |
| Protein A conjugated gold particles | Cytodiagnostics | Cat# AC-10-05 |
| Proteome Profiler Mouse XL Cytokine Array Kit | R&D Systems | Cat# ARY028 |
| TruSeq Small RNA Library Preparation Kits | Illumina | Cat# RS-200-0024 |
| ELISA kits | eBioscience | Cat# 88732477 |
| Deposited Data | ||
| Next generation mRNA sequencing | SRA database | SRP121341 |
| Liquid chromatography-tandem mass spectrometry | SRA database | SRP121341 |
| Experimental Models: Cell Lines | ||
| MC-38 | Kerafast | Cat# ENH204 |
| Caco-2 | ATCC | Cat# HTB-37 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | Jackson Laboratory Stock | Cat# 000664 |
| Mouse: C57BL/6-IL-22tm1.1(icre)Stck | Jackson Laboratory Stock | Cat# 027524 |
| Mouse: AHR KO | Taconic Biosceinces | Cat# 9166 |
| Mouse: Germ free | National Gnotobiotic Rodent Resource Center | N/A |
| Oligonucleotides | ||
| See Table S7 | N/A | N/A |
| Recombinant DNA | ||
| pGFPuv vector | Clontech | Cat# 632312 |
| Software and Algorithms | ||
| QIIME2 | J Gregory Caporaso, 2010 | https://qiime2.org |
| GreenGenes | T. Z. DeSantis, 2006 | http://greengenes.lbl.gov |
| SPSS 16.0 | Marija Norusis 2008 | https://spss-64bits.en.softonic.com/ |
| R | Douglas Bates, 2017 | https://www.r-project.org/ |
| Bowtie | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Other | ||
| miRNA sequence data | MiRBase | http://www.mirbase.org/ |
| Bacterial genomes | RefSeq database | ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/bacteria/ |
Highlights.
Plant exosomes-like nanoparticles (ELNs) are taken up by gut bacteria
The lipid composition of ELNs determines uptake by specific bacteria
ELN RNAs affect bacterial genes, notably impacting Lactobacillus production of I3A
ELN-mediated I3A alterations affect IL-22 production, resulting in ameliorated colitis
Acknowledgments
We thank Dr. D. Wilkey for mass spectrometry analysis; Dr. Willem de Vos for invaluable discussions regarding LGG biology and for providing the anti-SpaC serum and LGG-SpaC mutant; Ms. Mary Roth and Dr. Ruth Welt for lipid analysis; and Dr. J. Ainsworth for editorial assistance. Funding: This work was supported by a grant from the National Institutes of Health (NIH) (UH3TR000875, R01AT008617) and the Robley Rex VA Medical Center Merit Review Grant (H-GZ). Huang-Ge Zhang is supported by a Research Career Scientist (RCS) Award. Yun Teng is partially supported by a pilot project funded through the 1P30GM106396 Molecular Targets Phase III COBRE grant and Zhong-Bin Deng is partially supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM113226-6174. J.P. is supported by the National Institute of General Medical Sciences of the National Institutes of Health (NIH) grant P20GM103436.
Footnotes
Competing interests: The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- Benjamini Y, and Hochberg Y (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the royal statistical society. Series B (Methodological). 289–300. [Google Scholar]
- Bessede A, Gargaro M, Pallotta MT, Matino D, Servillo G, Brunacci C, Bicciato S, Mazza EM, Macchiarulo A, Vacca C, et al. (2014). Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature. 511(7508), 184–190. DOI: 10.1038/nature13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nature methods. 7(5), 335–336. DOI: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassaing B, and Gewirtz AT (2014). Gut microbiota, low-grade inflammation, and metabolic syndrome. Toxicologic pathology. 42(1), 49–53. DOI: 10.1177/0192623313508481. [DOI] [PubMed] [Google Scholar]
- Deng Z, Rong Y, Teng Y, Mu J, Zhuang X, Tseng M, Samykutty A, Zhang L, Yan J, Miller D, et al. (2017). Broccoli-Derived Nanoparticle Inhibits Mouse Colitis by Activating Dendritic Cell AMP-Activated Protein Kinase. Molecular therapy : the journal of the American Society of Gene Therapy. 25(7), 1641–1654. DOI: 10.1016/j.ymthe.2017.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festing MF, and Altman DG (2002). Guidelines for the design and statistical analysis of experiments using laboratory animals. ILAR journal / National Research Council, Institute of Laboratory Animal Resources. 43(4), 244–258. [DOI] [PubMed] [Google Scholar]
- Garcia-Segura L, Perez-Andrade M, and Miranda-Rios J (2013). The emerging role of MicroRNAs in the regulation of gene expression by nutrients. Journal of nutrigenetics and nutrigenomics. 6(1), 16–31. DOI: 10.1159/000345826. [DOI] [PubMed] [Google Scholar]
- Hausser J, and Zavolan M (2014). Identification and consequences of miRNA-target interactions--beyond repression of gene expression. Nature reviews. Genetics. 15(9), 599–612. DOI: 10.1038/nrg3765. [DOI] [PubMed] [Google Scholar]
- He S, Ran C, Qin C, Li S, Zhang H, de Vos WM, Ringo E, and Zhou Z (2017). Anti-Infective Effect of Adhesive Probiotic Lactobacillus in Fish is Correlated With Their Spatial Distribution in the Intestinal Tissue. Scientific reports. 7(1), 13195 DOI: 10.1038/s41598-017-13466-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, and Shin W (2014). How to do random allocation (randomization). Clin Orthop Surg. 6(1), 103–109. DOI: 10.4055/cios.2014.6.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosuge T, Heskett MG, and Wilson EE (1966). Microbial synthesis and degradation of indole-3-acetic acid. I. The conversion of L-tryptophan to indole-3-acetamide by an enzyme system from Pseudomonas savastanoi. The Journal of biological chemistry. 241(16), 3738–3744. [PubMed] [Google Scholar]
- Letunic I, and Bork P (2007). Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. 23(1), 127–128. DOI: 10.1093/bioinformatics/btl529. [DOI] [PubMed] [Google Scholar]
- Li Y, Innocentin S, Withers DR, Roberts NA, Gallagher AR, Grigorieva EF, Wilhelm C, and Veldhoen M (2011). Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell. 147(3), 629–640. DOI: 10.1016/j.cell.2011.09.025. [DOI] [PubMed] [Google Scholar]
- Liu S, da Cunha AP, Rezende RM, Cialic R, Wei Z, Bry L, Comstock LE, Gandhi R, and Weiner HL (2016). The Host Shapes the Gut Microbiota via Fecal MicroRNA. Cell host & microbe. 19(1), 32–43. DOI: 10.1016/j.chom.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslowski KM, and Mackay CR (2011). Diet, gut microbiota and immune responses. Nature immunology. 12(1), 5–9. DOI: 10.1038/ni0111-5. [DOI] [PubMed] [Google Scholar]
- Miller C, Thomsen LE, Gaggero C, Mosseri R, Ingmer H, and Cohen SN (2004). SOS response induction by beta-lactams and bacterial defense against antibiotic lethality. Science. 305(5690), 1629–1631. DOI: 10.1126/science.1101630. [DOI] [PubMed] [Google Scholar]
- Monteleone I, Rizzo A, Sarra M, Sica G, Sileri P, Biancone L, MacDonald TT, Pallone F, and Monteleone G (2011). Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology. 141(1), 237–248, 248 e231. DOI: 10.1053/j.gastro.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Mu J, Zhuang X, Wang Q, Jiang H, Deng ZB, Wang B, Zhang L, Kakar S, Jun Y, Miller D, et al. (2014). Interspecies communication between plant and mouse gut host cells through edible plant derived exosome-like nanoparticles. Molecular nutrition & food research. 58(7), 1561–1573. DOI: 10.1002/mnfr.201300729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosselli R, Romoli O, Vitulo N, Vezzi A, Campanaro S, de Pascale F, Schiavon R, Tiarca M, Poletto F, Concheri G, et al. (2016). Direct 16S rRNA-seq from bacterial communities: a PCR-independent approach to simultaneously assess microbial diversity and functional activity potential of each taxon. Scientific reports. 6, 32165 DOI: 10.1038/srep32165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwertner HA, Rios DC, and Pascoe JE (2006). Variation in concentration and labeling of ginger root dietary supplements. Obstetrics and gynecology. 107(6), 1337–1343. DOI: 10.1097/01.AOG.0000217697.33787.8c. [DOI] [PubMed] [Google Scholar]
- Sonnenberg GF, and Artis D (2012). Innate lymphoid cell interactions with microbiota: implications for intestinal health and disease. Immunity. 37(4), 601–610. DOI: 10.1016/j.immuni.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg ED, Smits SA, Tikhonov M, Higginbottom SK, Wingreen NS, and Sonnenburg JL (2016). Diet-induced extinctions in the gut microbiota compound over generations. Nature. 529(7585), 212–215. DOI: 10.1038/nature16504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutz RE (1958). Enzymatic Formation of Indole-3-Carboxaldehyde from Indole-3-Acetic Acid. Plant physiology. 33(3), 207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y, Mu J, Hu X, Samykutty A, Zhuang X, Deng Z, Zhang L, Cao P, Yan J, Miller D, et al. (2016). Grapefruit-derived nanovectors deliver miR-18a for treatment of liver metastasis of colon cancer by induction of M1 macrophages. Oncotarget. 7(18), 25683–25697. DOI: 10.18632/oncotarget.8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y, Ren Y, Hu X, Mu J, Samykutty A, Zhuang X, Deng Z, Kumar A, Zhang L, Merchant ML, et al. (2017). MVP-mediated exosomal sorting of miR-193a promotes colon cancer progression. Nature communications. 8, 14448 DOI: 10.1038/ncomms14448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zhuang X, Mu J, Deng ZB, Jiang H, Zhang L, Xiang X, Wang B, Yan J, Miller D, et al. (2013). Delivery of therapeutic agents by nanoparticles made of grapefruit-derived lipids. Nature communications. 4, 1867 DOI: 10.1038/ncomms2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber J, and Clayton A (2013). How pure are your vesicles? Journal of extracellular vesicles. 2 DOI: 10.3402/jev.v2i0.19861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LF, and Wang C (1998). Induction of cytokine messenger RNA transcripts in mouse macrophages by Listeria monocytogenes isolated from channel catfish. American journal of veterinary research. 59(6), 717–721. [PubMed] [Google Scholar]
- Zhuang X, Deng ZB, Mu J, Zhang L, Yan J, Miller D, Feng W, McClain CJ, and Zhang HG (2015). Ginger-derived nanoparticles protect against alcohol-induced liver damage. Journal of extracellular vesicles. 4, 28713 DOI: 10.3402/jev.v4.28713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 (related to Figure 1 and Figure 2) List of GELN miRNAs sequencing and prediction target genes expressed in LGG
Table S4 (related to Figure 2) Concentration of plant ELNs lipids analyzed with mass spec (nmol/mg ELNs)
Data Availability Statement
Data Resources
The sequencing data and mass spectrometry data were deposited in NCBI Sequence Read Archive (SRA) with accession number SRP121341.