

Abstract
A fundamental question in synaptic physiology is whether the unitary strength of a synapse can be regulated by presynaptic characteristics and, if so, what those characteristics might be. Here, we characterize a newly proposed mechanism for altering the strength of glutamatergic synapses based on the recently identified vesicular glutamate transporter VGLUT1. We provide direct evidence that filling in isolated synaptic vesicles is subject to a dynamic equilibrium that is determined by both the concentration of available glutamate and the number of vesicular transporters participating in loading. We observe that changing the number of vesicular transporters expressed at hippocampal excitatory synapses results in enhanced evoked and miniature responses and verify biophysically that these changes correspond to an increase in the amount of glutamate released per vesicle into the synaptic cleft. In addition, we find that this modulation of synaptic strength by vesicular transporter expression is endogenously regulated, both across development to coincide with a maturational increase in vesicle cycling and quantal amplitude and by excitatory and inhibitory receptor activation in mature neurons to provide an activity-dependent scaling of quantal size via a presynaptic mechanism. Together, these findings underscore that vesicular transporter expression is used endogenously to directly regulate the extent of glutamate release, providing a concise presynaptic mechanism for controlling the quantal efficacy of excitatory transmission during synaptic refinement and plasticity.
Keywords: VGLUT1, glutamate release, quantal size, homeostatic, excitatory, vesicular transporter
Introduction
A continuing goal of synaptic physiology is to uncover the features of synaptic structure that most dramatically influence synaptic strength. In excitatory glutamatergic synapses, remarkable progress has been made in detailing the postsynaptic factors that regulate synaptic efficacy (Malenka and Nicoll, 1999; Sheng and Kim, 2002). However, comparatively less has been done to identify corresponding presynaptic mechanisms that influence postsynaptic activation by governing the amount of transmitter released (Atwood and Karunanithi, 2002; Liu, 2003). Nevertheless, recent studies of central synapses have demonstrated that normal transmitter release can be insufficient to fully activate postsynaptic receptors (Bekkers et al., 1990; Liu and Tsien, 1995; Silver et al., 1996; Forti et al., 1997; Liu et al., 1999; Mainen et al., 1999; McAllister and Stevens, 2000), and variability in excitatory quantal size is increasingly considered to be significantly attributable to presynaptic fluctuations in transmitter release (Liu et al., 1999; Hanse and Gustafsson, 2001; Franks et al., 2003). Therefore, it is worthwhile to consider what presynaptic molecular mechanisms might influence the amount of glutamate released into the synaptic cleft and whether such mechanisms might be endogenously controlled to provide additional modes of synaptic regulation.
Recent work has demonstrated that artificial perturbations can alter excitatory transmission by modulating the amount of glutamate released from synaptic vesicles. In particular, either disrupting electrochemical proton gradients that drive the loading of transmitter into vesicles before release (Zhou et al., 2000) or making additional glutamate available for loading (Ishikawa et al., 2002; Yamashita et al., 2003) are both effective at changing the degree of postsynaptic activation during transmission.
We hypothesize that postsynaptic activation is similarly controlled by endogenous presynaptic mechanisms via the expression of the vesicular glutamate transport protein (VGLUT1), which is now known to underlie the transport of glutamate into excitatory vesicles (Bellocchio et al., 2000; Takamori et al., 2000). Functional work has recently demonstrated that perturbations to VGLUT1 expression can influence the strength of excitatory transmission, with knocking out VGLUT1 in particular leading to a profound reduction in excitatory signaling (Fremeau et al., 2004; Wojcik et al., 2004). VGLUT1 thus seems well poised as a control point for regulating synaptic transmission, although its precise mode of action in synaptic transmission remains uncharacterized, and its propensity for meaningful regulation is unreported. Indeed, although properties of cholinergic (Song et al., 1997) and aminergic (Pothos et al., 2000) vesicular transporters have now been examined as well, whether the expression of any vesicular transporter can be controlled endogenously to facilitate activity-dependent plasticity has not been explored.
The present work thus aims to more fully characterize the manner by which VGLUT1 expression is able to influence synaptic function and to elucidate whether neurons actually make use of this machinery endogenously, to presynaptically modulate the function of excitatory signaling. The characterizations of VGLUT1 described below, along with the specific expression of VGLUT1 in brain areas associated with adult plasticity (Fremeau et al., 2001; Liu, 2003), suggest an intriguing role for the regulation of vesicular transport in facilitating the functional dynamic range of glutamatergic synapses.
Materials and Methods
Synaptic vesicle purification and uptake. Synaptic vesicles were purified from 30 g of adult rat cerebral cortex (including hippocampus), using the procedure of Hell et al. (1988), and stored at -80°C until use. Vesicles were thawed on ice and diluted in sucrose buffer (320 mm sucrose, 10 mm HEPES, pH 7.4) to a protein concentration of 250 μg/ml (Pierce Bradford assay using BSA as a standard; Pierce, Rockford, IL). Aliquots (100 μl) were mixed with 50 μl of KCl solution (8 mm) in uptake buffer (110 mm potassium tartrate, 20 mm HEPES, 2 mm MgS04, pH 7.4) and preincubated for 5 min at 37°C in the presence or absence of the specific inhibitors Rose Bengal (RB) or trypan blue (TB). Uptake was initiated by the addition of 50 μl of uptake buffer containing 20 mm Na2-ATP, 0.9 μm 3H-glutamate (51 Ci/mmol; NEN, Boston, MA), and various concentrations of unlabeled glutamate and allowed to proceed at 37°C for 0.5-15 min. To terminate the uptake, tubes were placed in an ice/water slurry, and the vesicle suspensions were filtered under vacuum through GF/F filters prewetted with uptake buffer and washed with 8 ml of ice-cold uptake buffer. The radioactivity bound to the filter was solubilized in 1 ml of 1% SDS before adding 5 ml of EcoScint scintillation fluid.
VGLUT1 overexpression and functional analysis. Sequences encoding VGLUT1 (GenBank accession number U07609) (Ni et al., 1994) were cloned (Varoqui et al., 2002) and amplified using a 5′ primer containing an EcoRI site (ggaattccaccatggagttccggcaggaggagtttcgg) and a 3′ primer containing an SalI site to remove the stop codon (actggtcgaccagtagtcccggacagggggtggggg). After digestion with EcoRI and SalI, the PCR product was subcloned in frame into pEGFP-N1 to produce a transporter tagged on its C terminus with enhanced green fluorescent protein (EGFP). The cDNA fusion insert was subcloned into pCDNA3.1 using EcoRI and NotI for transient heterologous expression using the vaccinia T7 hybrid system in PC12 cells as described previously (Varoqui et al., 2002). A light population of membranes containing synaptic-like microvesicles was isolated from VGLUT-expressing PC12 cells and from mock-transfected cells as described previously (Varoqui et al., 2002). Western blot analysis was then also performed as described previously (Varoqui et al., 2002). VGLUT1 transport activity was assessed by measuring 3H-glutamate uptake under optimal conditions (Mg2+-ATP, 5 mm; chloride ion, 4 mm) for 5 min at 32°C in the presence and absence of the H+-uncoupler carbonyl cyanide m-chlorophenylhydrazone (CCCP; 50 μm) as in previous studies (Schafer et al., 2002; Varoqui et al., 2002).
Cultured hippocampal neurons and transfection. Hippocampi were dissected from postnatal day 1 Sprague Dawley rat pups and cultured as described previously (Liu et al., 1999). A pCDNA3.1-VGLUT1 construct (Varoqui et al., 2002) was used to transfect cells at 6-11 d in vitro (DIV) using the calcium phosphate method, with neurons transfected at least 2 d before observation. VGLUT-enhanced neurons were compared with control neurons from the same batches of culture, with evoked recordings age-matched from 9-11 DIV, miniature recordings measured at 10 DIV, glutamate release experiments performed at 14-16 DIV, and activity-dependence experiments at 16 DIV after 2 d of drug treatment. All experiments involving animals were approved by the Massachusetts Institute of Technology Committee on Animal Care.
Imaging and immunohistochemistry. All imaging was performed using an Olympus Fluoview confocal microscope (Olympus, Melville, NY) with a 40× planapochromatic water immersion lens (1.15 numerical aperture). Cell cultures were fixed with 4% paraformaldehyde and 4% sucrose in 1× PBS, and permeabilized with 0.5% Triton X-100 for 30 min at 22-24°C. Primary antibodies against VGLUT1 (Chemicon, Temecula, CA), synaptotagmin (American Qualex, San Clemente, CA), or synapsin I (Chemicon) were applied, followed by rinses in PBS and visualization with Alexa 488-, 546-, or 633-conjugated secondary antibodies (1/400; Molecular Probes, Eugene, OR). AM1-43 (Biotium, Richmond, CA) is a form of the styryl dye FM1-43 commonly used to visualize vesicle fusion activity (Renger et al., 2001), but with an additional aldehyde-reactive amino group at the hydrophilic end, rendering it less sensitive to fixation. Coverslips containing cultured neurons were bathed for 120 s at 22-24°C in high-K+ solution (plus 8 μm AM1-43) and placed into Tyrode's solution [see above; plus 1 μm tetrodotoxin (TTX)] for 15 min. They were then transferred for quenching to the sulfonated b-cyclodextrin derivative ADVASEP-7 (125 μm; Biotium) for 4 min at 22-24°C. Cells were fixed immediately after staining in FSB (see above) and 0.02% glutaraldehyde for 60 min, rinsed in 1× PBS, and blocked with 0.4% saponin and 5% serum in 1× PBS. First and secondary antibodies were then applied as above. All images were collected at 1280 × 1024 pixel resolution and 4× software zoom, using a z series projection of 8-11 images taken at 0.8 μm depth intervals. An average image contained ∼250 synapses.
Whole-cell recording. Whole-cell patch-clamp recordings were performed as described previously (Renger et al., 2001). Patch pipettes (3-6 MΩ) contained the following (in mm): 120 potassium gluconate, 3 KCl, 10 HEPES, 8 NaCl, 0.5 CaCl2, 5 EGTA, 2 Mg2+-ATP, and 0.3 GTP, pH was adjusted to 7.3 with NaOH. Perforated-patch pipettes were front-filled with a solution containing the following (in mm): 130 potassium gluconate, 4 KCl, 10 HEPES, 8 NaCl, 0.4 EGTA, pH was adjusted to 7.2 with KOH and then back-filled with the same solution containing 150 ng/ml amphotericin B (Sigma, St. Louis, MO). Extracellular solution was based on Tyrode's solution containing the following (in mm): 145 NaCl, 3 KCl, 15 glucose, 10 HEPES, 1.3-2.6 MgCl2, and 1.3-2.6 CaCl2 and 50 μm picrotoxin (Sigma), pH adjusted to 7.4 with NaOH. Signals were recorded under voltage clamp (-60 mV) using a MultiClamp 700A amplifier (Molecular Devices, Foster City, CA), digitized at 10 kHz, filtered at 1 kHz, and recorded using in-house software. 1,2,3,4-Tetrahydro-6-nitro-2,3-dioxo-benzo [f] quinoxaline-7-sulfonamide (NBQX; 5 μm; Sigma) was used to verify AMPA currents. TTX (1 μm; Biotium) was added for miniature EPSC (mEPSC) recordings.
Dual-cell evoked recordings. For dual cell-evoked EPSCs, a second whole-cell patch or perforated patch was achieved, and action potentials were stimulated by injecting 100-300 pA of current over 10 ms (current clamp) (see Fig. 5, or 30-80 mV over 0.5 ms (voltage clamp) (see Fig. 8). For glutamate antagonist experiments, 600 μm γ-d-glutamylglycine (γ-DGG) (Tocris Cookson, Ellisville, MO) was bath applied. For tuning of synaptic failure rates, varying concentrations of calcium (0.6-1.3 mm) and magnesium (2.6-3.4 mm) were bath applied for each connection until the apparent probability of release was low (0.4-0.1). Data was then collected after a stable baseline at a consistent failure rate was established.
Figure 5.
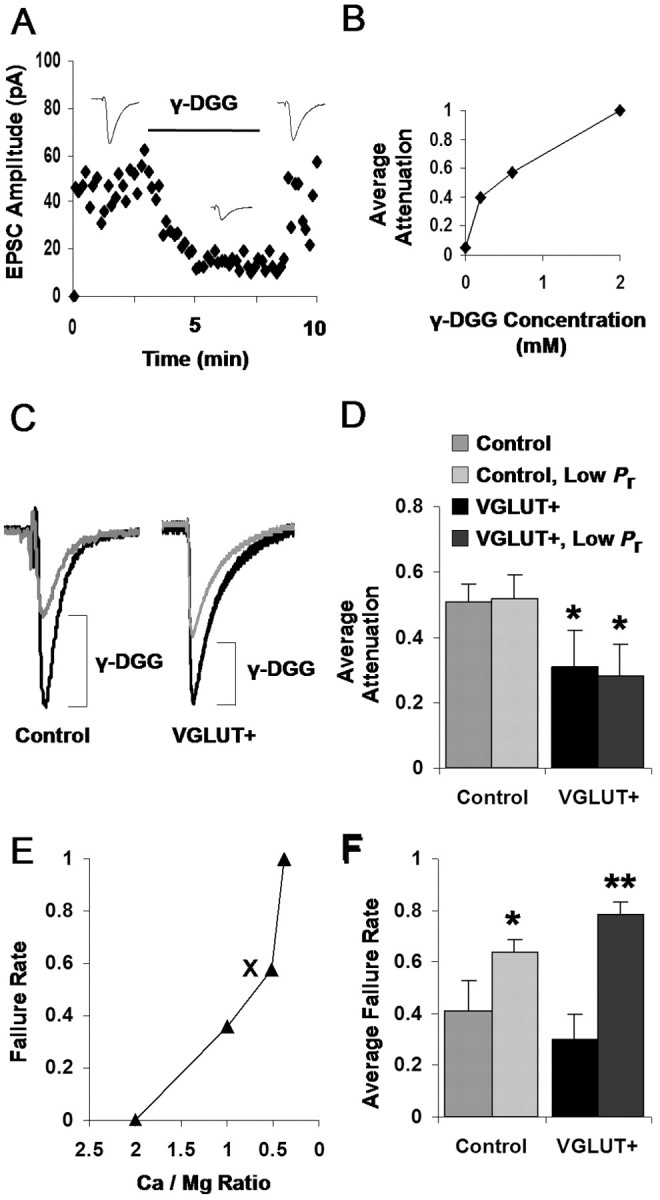
Enhanced per vesicle glutamate release via VGLUT1 overexpression. A, Sample application of the competitive glutamate antagonist γ-DGG (600 μm) while evoking EPSCs at 0.3 Hz between a pair of neurons. Dots represent an average of three EPSCs. B, Dose-response curve measuring the degree of EPSC attenuation by different concentrations of the competitive glutamate antagonist γ-DGG. C, Typical attenuation of control versus VGLUT-mediated EPSCs by 600 μm γ-DGG. Median traces from single connections, normalized by predrug amplitudes for direct comparison, are shown. Bottom traces depict the γ-DGG normalized to the height of their predrug counterpart for comparison of kinetics. D, Summary data of γ-DGG attenuation across all observed connections: VGLUT-mediated transmission is significantly less attenuated than control-mediated transmission (*p < 0.05; unpaired t test; control vs VGLUT+ bars). Summary data for γ-DGG attenuation acquired under intentionally reduced probability of release conditions are also shown (Control, Low Pr vs VGLUT+, Low Pr bars; see Results). Here, VGLUT-mediated transmission is again significantly less attenuated than control-mediated transmission (*p < 0.05; unpaired t test). Average failure rates used during each condition are depicted in F. E, Examples of tuning the failure rate of synaptic transmission by varying divalent cation concentrations for low Pr experimental conditions. The ratio of calcium/magnesium concentrations is varied while failure rate is measured until failure rate is sufficiently high to ensure a significant number of single vesicle events. The marker (X) denotes the condition in which γ-DGG attenuation data were acquired. F, Average failure rates used during normal Pr and low Pr experiments summarized in D. Failure rates were intentionally increased during the low Pr experiments for both control (*p < 0.05; unpaired t test) and VGLUT+ (**p < 0.01; unpaired t test) conditions.
Figure 8.

Activity-dependent plasticity of VGLUT1 expression and glutamate release. A, Double immunostaining for VGLUT1 and the vesicle protein synapsin I after 48 h of incubation with either BIC to block GABAA receptors and increase neuronal activity or AP-5 to block NMDA receptors and decrease neuronal calcium flux. B, Mean VGLUT1 expression (VGLUT1 IOD normalized by synapsin I IOD at the same synapse) was reduced following chronic GABAA receptor blockade (*p = 0.02; unpaired t test; values scaled to control) and enhanced by chronic NMDA receptor blockade (***p < 0.001; unpaired t test; values scaled to control). C, Comparison of EPSC amplitude attenuation by 600 μm γ-DGG in control, BIC, and AP-5-treated cultures. Each data point represents an average of three adjacent sampled EPSCs (0.2 Hz). Representative pre-DGG and post-DGG baselines, normalized to their respective predrug levels for comparison, are shown. D, Typical attenuation of control, BIC, and AP-5-treated EPSCs by 600 μm γ-DGG in the paired connection exhibiting median attenuation for the group. Traces are EPSCs averaged from multiple samples of a single connection, normalized by predrug amplitudes for direct comparison. E, Summary data of γ-DGG attenuation across multiple connections: BIC-treated transmission is significantly more at tenuated than nontreated control transmission (*p < 0.05; unpaired t test; values scaled to control). AP-5-treated transmission is significantly less attenuated than nontreated control transmission (p < 0.01; unpaired t test; values scaled to control). F, γ-DGG attenuation measured in mEPSCs as an additional control. BIC-treated transmission is significantly more attenuated by γ-DGG application than nontreated control mEPSCs (*p < 0.05; unpaired t test), indicating that observed changes in glutamate release correspond to differences in per vesicle glutamate release. G, Sample mEPSCs from bicuculline-treated, control, or AP-5-treated cultures. Calibration: 15 pA, 150 ms.H, Mean mEPSC amplitude is significantly reduced in BIC-treated cultures compared with nontreated control cultures and is significantly elevated in AP-5-treated cultures (*p < 0.05; unpaired t test). CTRL, Control; Syn I, synapsin I.
Estimation of the potential for multivesicular release. The potential for multivesicular release (MVR) in our measurements was determined while assuming, first, ∼10 release sites per bouton (Schikorski and Stevens, 1997) and, second, independent release between vesicles. Binomial statistics were used in which the number of vesicles undergoing fusing (N) relates to the probability that a given vesicle will fuse (p) and the number of vesicles able to fuse per bouton (Nmax). We determined the probability of event failure as the likelihood that zero vesicles could undergo fusion: PN=0 = (1 - P)N max, using observed failure rates to estimate p. The probability of exactly one vesicle fusing, in turn, would be: PN=1 = Nmax (1 - P)N max - 1 p. As such, the probability of multivesicular release would be the likelihood per action potential that there is neither a failure nor a single release but rather that several vesicles release: PN≥2 = 1 - PN=1 - PN=0, or 1 - Nmax (1 - p)N max-1p(1 - P)N max.
Chronic drug treatments of cultures. In treatments to chronically shift activity levels, cultures were treated continuously beginning at 14 DIV and observed at 16-17 DIV. NBQX (5 μm; Sigma), dl-2-Amino-5-phosphonopentanoic acid (AP-5; 50 μm; Tocris Cookson), or bicuculline methobromide (20 μm; Sigma) were used to block AMPA, NMDA, and GABAA receptors.
Data analysis. Analysis of electrophysiological data was performed in MiniAnalysis (Synaptosoft, Leonia, NJ) for mEPSCs, and custom scripts were written in C++ and MatLab (MathWorks, Natick, MA) for EPSCs evoked presynaptically and iontophoretically. EPSC failure rates were estimated using a threshold of ∼4 pA. Such a threshold was also invoked for determining mean amplitude before γ-DGG attenuation, and an equal proportion of events was then compared after drug application to determine mean attenuation (consistent probability of release was assumed). Traces for which access resistance varied substantially or were >25 MΩ were rejected from analysis. Image analysis was performed via custom scripts written in ImagePro Plus (Media Cybernetics, Carlsbad, CA) for puncta localization and quantification. Pixels within each punctum were assigned intensity values, and the combined sum of the intensity values of a punctum, or integrated optical density (IOD), was taken as the total intensity of the punctum. VGLUT1 total intensity was measured against that of synapsin I or synaptotagmin in the same punctum. All comparisons involving multiple conditions were quantified first using ANOVA, followed by unpaired t tests between adjacent groups as reported, and all error bars report SEM unless otherwise noted.
Results
Loading in excitatory synaptic vesicles depends on glutamate concentration and number of available transporters
To characterize the nature by which VGLUT1 expression might impact synaptic transmission, it was first important to demonstrate that varying the parameters of glutamate transport could actually modulate the extent of vesicle filling. A possible scenario is that although different transport conditions might influence the speed of filling, vesicles eventually fill to a “set point” that is dictated by some parameter independent of transport, such as vesicle size. In contrast, if the amount of glutamate loaded into vesicles was to be specified by a dynamic equilibrium process that could be stoichiometrically influenced by the components of the loading reaction, it would allow for the modulation of vesicle filling and the consequent release of glutamate by endogenous characteristics such as transporter expression.
Previous work has assayed the loading of glutamate into PC12 cells (Bellocchio et al., 2000), the BON line of human endocrine cells (Takamori et al., 2000), and even purified synaptic vesicles at specific external glutamate concentrations (Maycox et al., 1988; Wolosker et al., 1996). However, given a recent set of insightful studies that has begun to elucidate the previously unknown range of the cytosolic glutamate concentration (Ishikawa et al., 2002; Yamashita et al., 2003), we were interested in systematically measuring the glutamate uptake equilibrium around variants of this physiological range of conditions as well as assessing the influence of transporter number in defining this equilibrium.
We therefore quantified the time course and final equilibrium of glutamate transport under different conditions using VGLUT1 synaptic vesicles isolated from rat cerebral cortex and hippocampus (Fig. 1). Observing the uptake of the vesicles of 50 μm 3H-glutamate (Fig. 1A) revealed that vesicular transport in our system reached an equilibrium balance between transmitter influx and efflux along a similar time course to that reported previously (Maycox et al., 1988; Wolosker et al., 1996). To examine whether the number of transporters participating in loading could influence its final equilibrium, we also compared our equilibrium uptake to that occurring in the presence of Rose Bengal, an antagonist of VGLUT1 loading that is believed to interact allosterically with the transporter (Ogita et al., 2001). Because the drug is thought to noncompetitively disrupt glutamate transport at the site of loading, it offered a pharmacological means for reducing the effective number of transporters participating in uptake to examine whether the size of the available transporter pool can help determine the final amount of vesicle filling. Loading in the presence of Rose Bengal indeed exhibited a reduced equilibrium of filling (Fig. 1A), a property that was also observed under treatment with trypan blue, the most potent known competitive antagonist of vesicular glutamate transport (Roseth et al., 1998). Both drugs were applied at approximate IC50 concentrations, whereby 2 μm at either 5 or 10 min preincubation achieved complete inhibition of loading (data not shown).
Figure 1.

VGLUT1 loading in synaptic vesicles depends on glutamate concentration and the number of available transporters. A, Uptake time course of 50 μm 3H-glutamate by purified synaptic vesicles (•) reaches equilibrium in ∼10 min. CON, Control. Uptake in the presence of either the competitive VGLUT1 inhibitor trypan blue (□) or the noncompetitive VGLUT1 inhibitor Rose Bengal (○) both fill along a similar time course but to smaller equilibrium amounts. B, Uptake time course for 1 mm 3H-glutamate by synaptic vesicles (•) occurs along a similar time course as in A but to a dramatically increased final equilibrium amount. Equilibrium is again reduced in the presence of either trypan blue (□) or Rose Bengal (○). C, Saturation isotherm of 3H-glutamate (0.06-6 mm) accumulation at 10 min in the presence and absence of Rose Bengal (○) or trypan blue (□). In the presence of >1 mm glutamate, the competitive trypan blue inhibition is lost while the noncompetitive Rose Bengal inhibition is maintained. D, Time courses of 3H-glutamate (Glu) uptake at four subsaturating concentrations of glutamate show that steady-state vesicular glutamate levels depend on the concentration of glutamate present in the medium.
By repeating these treatments under a more physiological concentration of extravesicular glutamate (1 mm) (Fig. 1B), we were next able to observe whether transporter activity can also influence vesicle filling at a glutamate concentration closer to what has endogenously been estimated in functional studies (Ishikawa et al., 2002). At this higher concentration of glutamate, the vesicles now filled to a higher steady-state concentration, indicating that the final extent of VGLUT1 filling depends directly on the concentration of glutamate around the vesicle and does not have an independent set point. Equilibrium filling was again reduced by the presence of either RB or TB (Fig. 1B), demonstrating that, also at this higher concentration, the equilibrium can be shifted by reducing the number of active transporters contributing to the loading process.
Varying the external glutamate concentration along a continuum while measuring 10 min equilibrium uptake levels (Fig. 1C) reveals that maximal uptake increases linearly up to 1 mm exogenous glutamate and begins to saturate at ∼4-6 mm. Even at these concentrations, the extent of transporter availability is critical in determining the equilibrium value, because RB attenuates the final concentration reached for all values of external glutamate in a similar manner to TB, although competitive antagonism of TB is eventually overwhelmed as glutamate concentration is increased. Together, the results lead us to conclude that VGLUT1 vesicles are subject to a flexible degree of filling that can be influenced by both the concentration of glutamate outside of the vesicle as well as the effective number of available transporters. Aligning the time courses of loading observed under various glutamate concentrations (Fig. 1D) demonstrates that both the magnitude and, to a lesser degree, the rate of uptake are influenced by alterations in transport conditions.
We thus observe three properties of glutamate uptake into synaptic vesicles at physiological terminal glutamate concentrations. First, additional increases to external glutamate concentrations can enhance the amount of transmitter loaded, as predicted by the aforementioned functional work (Ishikawa et al., 2002; Yamashita et al., 2003). Second, the number of transporters available for loading can influence the final loading equilibrium, even at glutamate concentrations above the physiological range. Third, vesicle filling is a slow process, requiring minutes to reach completion, which could indicate an importance of changes to the vesicle loading rate, in addition to the loading amount, in determining physiological synaptic transmission.
Transporter overexpression in PC12 cells and hippocampal synapses
If VGLUT1 vesicles do not merely accumulate transmitter until full but rather have their speed and amount of filling determined by an equilibrium process that is subject to the available concentration of transmitter and transporter, we were interested in examining whether this equilibrium could also be shifted in a more physiological context. Therefore, we next attempted to vary the amount of vesicular transporters expressed in the synaptic terminals of neurons to examine the resulting consequences for glutamate release in endogenous synaptic transmission.
To characterize the functional relationship between VGLUT1 expression and excitatory synaptic transmission, we attempted to overexpress the transporter at hippocampal synaptic terminals. We created two expression constructs for delivery into neurons: one for wild-type VGLUT1 (WT) and another tagging VGLUT1 with the reporter GFP at its C terminus. To verify the effectiveness of our constructs in enhancing VGLUT1 expression, PC12 cells were transiently transfected as described previously for VGLUT2 (Varoqui et al., 2002) and the recently identified VGLUT3 (Schafer et al., 2002). A Western blot analysis (Fig. 2A) using a guinea pig antibody specific for VGLUT1 (Schafer et al., 2002) reveals that although mock-transfected cells (M) did not express detectable levels of VGLUT1, single immunoreactive bands of appropriate size were observed for PC12 cells transfected with either the wild-type VGLUT1 or the VGLUT-GFP fusion (+GFP) constructs. The fusion protein had an ∼26 kb higher molecular weight than wild-type VGLUT1 because of the additional mass of the fused EGFP. To check the functionality of these newly synthesized proteins, we observed whether transfection of our constructs into PC12 cells gave rise to glutamate transport by assaying for radioactive glutamate uptake in the presence of magnesium-ATP and chloride ions to activate transporter function (Fig. 2B). Here, both WT and GFP-tagged (+GFP) constructs demonstrated successful transport of 3H-glutamate into a light population of membranes, a function that was found to be inhibited by the proton uncoupler CCCP. CCCP-sensitive uptake in this and similar preparations of vesicles is considered a valid indicator of the H+-dependent uptake of glutamate (Takamori et al., 2000, 2002). Although notable levels of uptake were not observed in mock-transfected cells (Mock), uptake capacity for both constructs was found to be comparable with those reported previously for VGLUT1 (Bellocchio et al., 2000; Takamori et al., 2000) as well as for VGLUT2 and VGLUT3 (Schafer et al., 2002; Varoqui et al., 2002). We therefore concluded that our constructs were effective for increasing VGLUT1 protein levels and enhancing glutamate transport.
Figure 2.
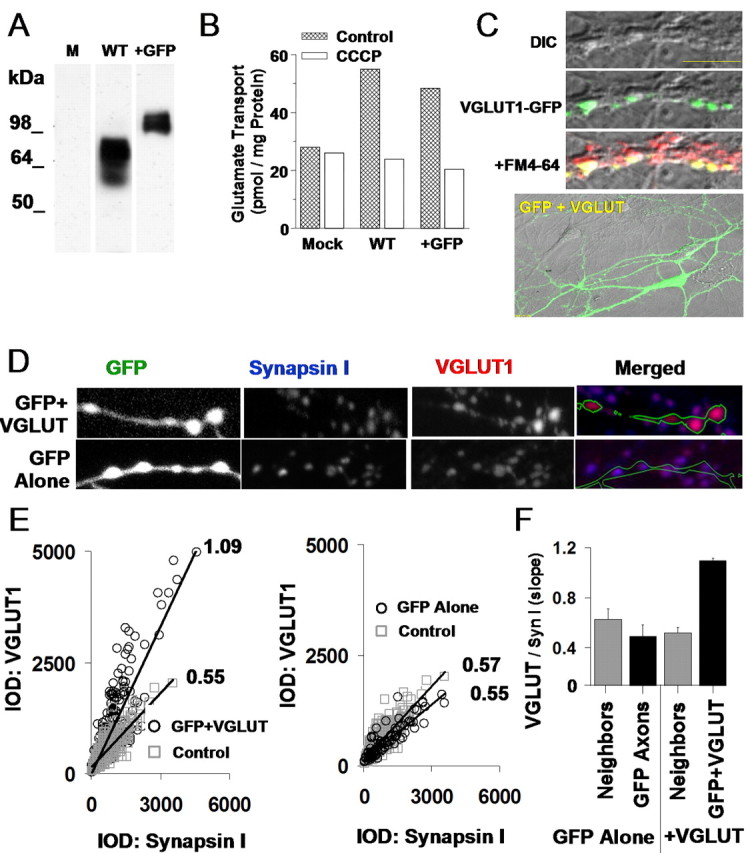
Overexpression of VGLUT1 in PC12 cells and hippocampal synapses. A, Expression of both wild-type VGLUT1 (WT) and GFP-tagged VGLUT1 (+GFP) constructs in PC12 cells reveals single immunoreactive bands of appropriate size for wild-type and EGFP-tagged proteins as visualized by Western blotanalysis. Mock-transfected cells (M) did not contain immunoreactivity. B, Both constructs succeeded in facilitating CCCP-sensitive glutamate loading into PC12 cells, whereas mock transfection (Mock) did not. C, Top, Overexpressed VGLUT1-GFP fusion protein localized to synaptic terminals (top) as shown by colocalization of the fusion protein (green) with FM4-64 (red). Scale bar, 10 μm. Bottom, Wild-type VGLUT1 cotransfected with GFP clearly labels the morphology of transfected neurons. DIC, Differential interference contrast. D, Double-immunostaining for synapsin I and VGLUT1 along GFP-labeled hippocampal axons. VGLUT1 intensity was higher in axons expressing GFP plus VGLUT (top images) than in axons expressing GFP alone (bottom images). E, Ratio of VGLUT1 IOD to synapsin I IOD at individual synapses. Ratios were higher along axons transfected with GFP plus VGLUT (left; 278 transfected synapses, 576 control synapses) than axons transfected with GFP alone or adjacent nontransfected synapses from the same area (right; 92 transfected synapses, 422 control synapses). F, Summary data across multiple cover slips (total, n = 8) for data in E. GFP plus VGLUT1 cotransfection results in augmented VGLUT1 protein levels per synapse.
We next examined whether VGLUT1 protein levels could be enhanced at the synaptic terminals of hippocampal neurons. This requires not only an increase in the production of VGLUT1 protein in transfected neurons but also that the VGLUT1 generated from our constructs contain the correct targeting sequence to be transferred to synaptic terminals effectively. We examined VGLUT1 targeting by transfecting neurons with the VGLUT1-GFP fusion construct and visualizing protein localization. Here, confocal imaging of the fluorescent-tagged protein (Fig. 2C, top) showed that new protein was successfully targeted to the synaptic terminals, where it accumulated in discrete puncta colocalized with the presynaptic bouton label FM4-64 and was not observable in dendrites. In contrast, when GFP was cotransfected separately from VGLUT1 to fully label neurons overexpressing the protein for later experiments (Fig. 2C, bottom), the GFP diffused evenly throughout the cell, with no specific targeting to synaptic terminals. We therefore conclude that overexpressed VGLUT1 can be targeted to hippocampal synapses.
Although the fusion construct proved useful for establishing the targeting of overexpressed protein to synaptic terminals, we chose to use wild-type VGLUT1 for the subsequent functional characterizations to avoid any modification of VGLUT1 structure that could perturb insertion at the vesicular membrane or disrupt potential endogenous regulation at the terminal. It was then necessary to verify that our VGLUT1/GFP cotransfection also succeeded in increasing total VGLUT1 protein levels at the synaptic terminal. Figure 2D shows immunolabeled VGLUT1 occurring within synaptic terminals marked with synapsin I along axons cotransfected with GFP and VGLUT1 (top images) or transfected with GFP alone (bottom images). We found that VGLUT1 puncta appeared more intense in synapses occurring on the GFP-labeled transfected axon than in neighboring puncta occurring off the GFP axon (top images). As an additional control demonstrating that GFP transfection alone did not enhance VGLUT1 levels per synaptic terminal, the above comparison was repeated with GFP axons lacking VGLUT1 cotransfection, again versus neighboring GFP-negative terminals (Fig. 2D, bottom images). In this case, there did not appear to be an appreciable difference in the size or intensities of VGLUT1 puncta occurring on or off the GFP-labeled axon.
The absolute amount of VGLUT1 per synapse is determined by both the number of synaptic vesicles and the average amount of VGLUT1 protein per vesicle. It was important to discern whether any increase in VGLUT1 levels that we observed was indeed attributable to a specific change in the concentration of VGLUT1 across the synapse and not just a general increase in the size or density of the vesicle pool. Because synapsin I intensity provides a measure of the concentration of a second protein associated with vesicles, we used the ratio of VGLUT1 to synapsin I to determine the available amount of transporter protein independently of fluctuations in the number of vesicles or the synaptic area. Figure 2E demonstrates a quantification of this measure, which utilizes the IOD of VGLUT1 and synapsin I to compare the expression levels of the two proteins. Synapses from axons transfected with both GFP and VGLUT1 show a nearly twofold increase in the VGLUT1/synapsin I IOD ratio compared with neighboring synaptic terminals (Fig. 2E, GFP+VGLUT, left plot). GFP alone axons, in contrast, did not show noticeably larger VGLUT1/synapsin I IOD ratios than in neighboring terminals (Fig. 2E, GFP Alone, right plot). Repeating this measure across multiple coverslips demonstrates a consistent increase in the targeting of VGLUT1 protein to synapses as a result of GFP plus VGLUT1 cotransfection (Fig. 2F), with no increase in VGLUT1 protein at neighboring synapses in the same neuronal network. We therefore conclude that our constructs were capable of effectively targeting elevated levels of VGLUT1 protein to the synaptic terminals of our hippocampal cultures.
VGLUT1 overexpression enhances AMPA receptor-mediated responses and decreases evoked failure rate
Having demonstrated a successful elevation of VGLUT1 protein levels in synaptic terminals, we examined the physiological consequences for functional synaptic transmission. Traditional electrophysiological methods record events postsynaptically, such that postsynaptic genetic manipulations have the possibility of influencing all recorded events. Presynaptic modifications, however, are more difficult to assay, because they require strict differentiation over which terminals are contributing to measured events. To solve this problem, we used dual whole-cell patch clamp, evoking an action potential in a presynaptic cell under current clamp while recording the resulting EPSCs mediated by AMPA receptors in a neighboring postsynaptic cell under voltage clamp (Fig. 3A, traces). Using confocal microscopy (Fig. 3A, image), we could distinguish presynaptic cells that were transfected with VGLUT1 by their coexpressed GFP label, as opposed to control neurons that did not express GFP. A common paradigm was to elicit transmission from a VGLUT1 neuron to a control neuron and then, as an independent control, evoke synaptic transmission under reverse configuration to compare reciprocal transmission. Figure 3B shows evoked synaptic transmission in a reciprocally connected pair of neurons (top), where stimulating a VGLUT-enhanced neuron evoked an EPSC that was an average of 92% larger than that evoked from the control neuron in the reverse direction (left traces). Normalizing these traces to the same peak amplitude (Fig. 3B, right traces) revealed a similar average time course. Visualizing the distribution of events evoked from this reciprocal pair (Fig. 3C) reveals a rightward shift in the VGLUT+ distribution relative to control (p < 0.0001; Kolmogorov-Smirnov test), and, on average, control events were consistently exceeded by their VGLUT+ counterparts in EPSC amplitude (12.5 ± 2.9 vs 20.5 ± 3.4, respectively; p < 0.05, unpaired t test; n = 7 pairs control, 7 pairs VGLUT+) (Fig. 3D).
Figure 3.
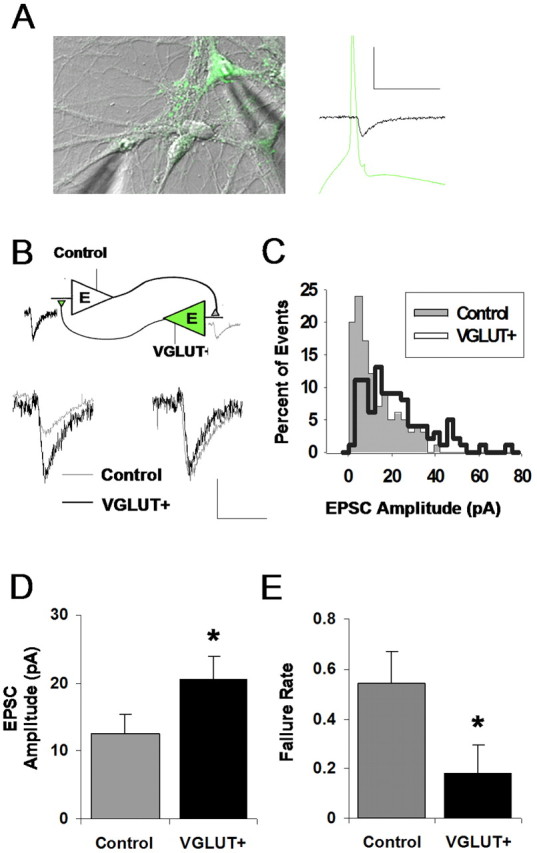
Enhanced excitatory synaptic transmission via VGLUT1 overexpression. A, Experimental paradigm: dual whole-cell patch clamp is achieved between a VGLUT1-overexpressing VGLUT+ neuron cotransfected with GFP (green) and a nontransfected control neuron (clear). An action potential is evoked in one neuron of the pair under current clamp at 0.3 Hz, and the resulting EPSC is recorded in the other neuron under voltage clamp. Calibration: 50 pA, 50 ms. B, EPSC responses evoked in a reciprocally connected pair of VGLUT+ and control neurons. Average EPSCs (left traces) contrast the responses evoked from each type of presynaptic neuron. Calibration: 10 pA, 20 ms. Normalizing these traces to the same peak amplitude (right traces) reveals a similar time course. C, Distribution of EPSC amplitudes observed in the reciprocal pair shown in B. Events from a VGLUT+ presynaptic neuron tend to comprise a right-shifted distribution that differs in shape from its control counterpart. D, Summary data for the effect of presynaptic VGLUT1 overexpression on EPSC amplitude: mean amplitude measured across multiple pairs is significantly higher for VGLUT+ (*p < 0.05; unpaired t test; n = 7 control; n = 7 VGLUT+). E, Summary data for the effect of overexpression on failure rate of evoked transmission; probability of receiving a postsynaptic response to presynaptic stimulation is increased by VGLUT1 overexpression (*p = 0.03; unpaired t test).
Of potential interest, VGLUT+ transmission also exhibited an unexpected decrease in the failure rate of synaptic transmission (0.54 ± 0.11 vs 0.18 ± 0.13, respectively; p = 0.03 unpaired t test) (Fig. 3E), suggesting that transporter overexpression leads to an increase in either of the following: (1) the number of release events that are large enough to elicit detectable AMPA receptor-mediated responses, or (2) the probability that a vesicle will be released after an action potential.
VGLUT1 overexpression enhances quantal synaptic transmission
Increases in evoked synaptic transmission after VGLUT1 overexpression could be a result of changes in the number of synaptic terminals, the probability of release per terminal, or the individual quantal size of response following each release. Therefore, we next examined whether quantal transmission is impacted by VGLUT1 overexpression by monitoring miniature synaptic transmission under bath application of tetrodotoxin to block action potentials and ensure spontaneous, single-vesicle release in control versus VGLUT1-overexpressing neurons. The difficulty in interpreting this experiment, of course, is that not all presynaptic terminals in a recorded neuron can be successfully transfected. Our strategy then was to rely first on a high-transfection rate (∼25%) and, from this population, select areas of the coverslip unusually dense in transfected cells and their axons (Fig. 4A, bottom image). Under this condition, for a given recorded neuron, a perceptible proportion of presynaptic terminals should contain higher levels of VGLUT1. Comparing responses observed here (VGLUT+) with responses collected in a completely nontransfected coverslip (control), we attempted to assay for detectable changes in quantal transmission.
Figure 4.

Effects of VGLUT1 overexpression on quantal excitatory transmission. A, mEPSCs were recorded postsynaptically via a whole-cell patch clamp in the presence of TTX in either a network completely lacking VGLUT1 overexpression (Control) or from a subregion of a transfected network visually determined to be dense in overexpressing neurons (VGLUT+). B, Distributions of mEPSC amplitudes observed in the neurons shown in A. VGLUT+ events tend to be right-shifted relative to the control event distribution. C, Sample mEPSCs from cells recorded in control and VGLUT+, which in this case are an average of 48% larger in the VGLUT+ recording. Calibration: 25 pA, 150 ms. D, Average mEPSC amplitude for neurons recorded in each group shows an increase in VGLUT+ mean event amplitude (*p = 0.04; unpaired t test). mEPSC data are aligned with the EPSC amplitude data from Figure 3C. E, No significant difference between control and VGLUT+ neurons is observed for mEPSC frequency (shown here) or rise and decay time kinetics (data not shown). Error bars indicate SEM.
mEPSCs recorded from neurons in areas rich in VGLUT+ terminals were larger than those recorded in cells devoid of enhanced terminals. Comparing distributions from sample VGLUT+ and control neurons indicated a rightward shift of amplitudes in the VGLUT+ cultures (Fig. 4B) (p < 0.001; Kolmogorov-Smirnov test), whereas aligning sample and average traces (Fig. 4C) further emphasized the difference. On average, the size of mEPSCs from the neurons with VGLUT1 transfection was 31% larger than that of neurons in the control group (13.6 ± 0.6 vs 17.8 ± 2.2, respectively; p = 0.04 unpaired t test; n = 5 cells control, 8 cells VGLUT+) (Fig. 4D). In Figure 4D, we also compared the effects of VGLUT transfection in miniature versus evoked synaptic transmission. Control mEPSCs and EPSCs were similar in amplitude, suggesting that evoked EPSCs most likely originated from single synapses. Interestingly, although EPSCs evoked from transfected terminals were 64% larger than control, VGLUT+ mEPSCs only increased by 31%. This difference is expected from the fact that the VGLUT+ mean mEPSC amplitude was calculated from a population of events that included both VGLUT-enhanced and control terminals, and also given likely differences in detectability between evoked and spontaneous transmission (Zhou et al., 2000). Examining mEPSC frequency between the two pools of cells revealed no significant change (1.89 ± 0.54 vs 1.55 ± 0.56, respectively; p = 0.33; unpaired t test) (Fig. 4E). Similarly to what we observed in evoked transmission, no change was observed in the time course of control and VGLUT-enhanced spontaneous transmission (data not shown), suggesting that VGLUT1 overexpression did not induce detectable changes in postsynaptic receptor properties.
VGLUT-enhanced transmission results from increased glutamate release per vesicle
Changes in evoked and quantal transmission after VGLUT1 overexpression could be a result of either presynaptic changes in cleft glutamate concentration or postsynaptic changes in receptor number or properties. Therefore, we next investigated whether the observed enhancement to evoked and miniature excitatory transmission following VGLUT1 overexpression was a result of an increased amount of glutamate released from synaptic vesicles. To assess the extent of glutamate release during synaptic transmission, we made use of a previously established pharmacological approach (Liu et al., 1999; Wadiche and Jahr, 2001). The low-affinity antagonist γ-DGG competes with glutamate for postsynaptic AMPA receptors, such that it partially attenuates glutamatergic release events at hippocampal synapses. Because of the competitive nature of its interaction with glutamate, the degree of its attenuation is inversely related to the amount of glutamate being released, such that it can be used as a quasidirect indicator of the extent of glutamate release (Liu et al., 1999). By recording EPSCs and determining the extent of γ-DGG attenuation, we could obtain a relative metric of the amount of glutamate involved in a population of synaptic release events.
Therefore, we repeated our dual whole-cell patch-clamp procedure (Fig. 3), this time with perforated patches to obtain long-duration recordings. We evoked EPSCs from either control or VGLUT-enhanced presynaptic neurons and then applied γ-DGG to measure the impact of the antagonist on the transmission. By repeatedly evoking transmission at a regular interval, it was possible to establish a baseline amplitude that could then be reliably attenuated by the perfusion of 600 μm γ-DGG (Fig. 5A), a concentration that we found optimal for partial attenuation of typical transmission (Fig. 5B). A comparison of average traces from before and after antagonist application (Fig. 5C) at both control synapses (left traces) and VGLUT-enhanced synapses (right traces) depicts sample attenuation of control and VGLUT-enhanced transmission. As shown, the efficacy of γ-DGG in EPSC attenuation was weaker at synapses containing higher levels of VGLUT1. On the whole, γ-DGG reduced the size of control EPSCs by 51%, in contrast to 31% in VGLUT-transfected pairs (Fig. 5D) (control vs VGLUT+; 51 ± 5 vs 31 ± 11%, respectively; p < 0.05, unpaired t test; n = 10 pairs control, 6 pairs VGLUT+). This line of experiments provides direct evidence that the VGLUT-enhanced synaptic transmission that we observed is indeed a result of enhanced presynaptic glutamate release from synaptic terminals.
The amount of glutamate released by synaptic terminals can be varied by changes to the amount of glutamate released per vesicle, which is likely because VGLUT1 can impact vesicle loading (Fig. 1). However, we cannot rule out the possibility that VGLUT1 overexpression is instead altering the probability of release of the terminals, which in turn could impact the likelihood of multiple vesicles fusing simultaneously at the same terminal (multivesicular release) (Tong and Jahr, 1994; Oertner et al., 2002). To directly distinguish between these two possibilities, we repeated the above γ-DGG attenuation experiments in a low probability of release condition, in which divalent cation concentrations were systematically varied until the observed transmission evoked not only approximated the quantal size (Fig. 4D) but also exhibited a high-failure rate. If a low proportion of stimuli results in the release of vesicles, then the proportion of stimuli that fosters multivesicular release will be exceedingly low. If vesicles release independently of one another as is widely assumed and synaptic boutons could be comprised of ∼10 vesicles (Schikorski and Stevens, 1997), then the probability of a given EPSC involving two or more vesicles can be determined using binomial statistics (see Materials and Methods), and MVR will be minimized when probability of release is low and failure rates are high. Evoked synaptic transmission in control and VGLUT+ pairs was thus tuned for a high level of baseline failures in each individual pair using different ratios of calcium and magnesium concentrations, as depicted in Figure 5E. As shown in Figure 5F, the average failure rate of transmission used in these experiments was 0.64 for control pairs and 0.79 for VGLUT+ pairs. Therefore, during transmission in these experiments, an upper bound for the incidence of MVR (assuming synapses with 10 sites) was ∼7 and 3%, respectively, and the majority of synaptic transmission should have been mediated by single vesicle release events. If the synapses in our study had even fewer release sites, then the likelihood of MVR would have been still further reduced.
Once such a low probability of release (Pr) condition had been achieved in each connection, we measured γ-DGG attenuation. In this new condition, favoring the release of single vesicles, γ-DGG attenuation in control versus VGLUT+ pairs were again significantly disparate (Fig. 5D) (control, low Pr vs VGLUT+, low Pr, 52 ± 7 vs 29 ± 14%, respectively; p < 0.05, unpaired t test; n = 5 pairs control, 5 pairs VGLUT+). Notably, even when the failure rates were made to differ dramatically between control and control, low Pr (0.41 ± 0.12 vs 0.64 ± 0.05; p < 0.05; unpaired t test) (Fig. 5F, left bars) and between VGLUT+ and VGLUT+, low Pr (0.30 ± 0.10 vs 0.79 ± 0.05; p < 0.01; unpaired t test) (Fig. 5F, right bars), similar degrees of γ-DGG attenuation were observed regardless of these manipulations to failure rate. Thus, VGLUT1 overexpression impacts the amount of glutamate released from single vesicles.
VGLUT1 expression is developmentally regulated
Although the above results demonstrate the potential of VGLUT1 regulation to alter synaptic transmission and establish the molecule as a limiting step for controlling glutamate release, we were interested in ascertaining whether this control point was used endogenously as a physiological mechanism for regulating synaptic transmission. In particular, our previous work (Renger et al., 2001) indicates that developing synapses exhibit several presynaptic functional differences from mature ones, including a reduced number of docking vesicles and less active vesicle cycling, that are rectified during maturation. Given that VGLUT1 can influence transmitter release (Fig. 5), we were interested in determining whether an additional maturational change might involve a developmental increase in VGLUT1 expression.
To monitor the developmental regulation of VGLUT1, quantitative immunohistochemistry was used to determine the amount of transporter expressed at each stage of development (Fig. 6). Immunostaining in mature hippocampal cultures (>14 DIV) depicts the transporter as discrete puncta that are tightly colocalized with synaptotagmin (Fig. 6A). Repeating this procedure across various stages of development reveals that expression of the transporter appears to markedly increase at later stages compared with earlier ones. This maturation results in an increased proportion of synapses expressing detectable levels of VGLUT1 (Fig. 6B) (n = 11 images for each group; 6 vs 9 DIV, p < 0.001, unpaired t test; 9 vs 20 DIV, p < 0.01; no significant change between 20 and 27 DIV). Of the synapses expressing detectable levels of the transporter, the ratio of VGLUT1 and synaptotagmin IODs per synapse also exhibits a graded increase across development (Fig. 6C) (6 vs 9 DIV, p < 0.01, unpaired t test; 9 vs 20 DIV, p < 0.01; no significant change between 20 and 27 DIV), indicating that the amount of VGLUT1 protein expressed per synaptic terminal is developmentally upregulated.
Figure 6.

VGLUT1 expression is developmentally regulated and coincides with a developmental change in quantal amplitude. A, Immunolabeling of VGLUT1 and the synaptic marker synaptotagmin Ia across development. B, Average proportion, at various developmental stages, of synaptotagmin-labeled puncta that exhibit detectable levels of VGLUT1 protein. Average proportion of VGLUT synapses increases significantly from 6 to 9 DIV (***p < 0.001; unpaired t test) and from 9 to 20 DIV (**p < 0.01). C, Average IOD for synapses already expressing detectable levels of VGLUT1. The ratio of VGLUT1 IOD to synapsin I IOD per synapse increases significantly across development from 6 to 9 DIV (**p < 0.01; unpaired t test) and from 9 to 20 DIV. D, Miniature excitatory currents (mEPSCs) recorded at different stages of development. Calibration: 15 pA, 150 ms. Bottom traces represent an average of a population of events acquired from a single cell at each developmental stage. Calibration: 5 pA, 25 ms. E, Mean mEPSC amplitude (n = 8, 8, and 6 cells at 8, 10, and 15 DIV, respectively) increases significantly during development (*p < 0.05; unpaired t test). F, Mean mEPSC frequency also increases significantly between 10 and 15 DIV (*p < 0.05).
Given that VGLUT1 is upregulated within synapses across development, we were interested in identifying functional changes that could potentially be related to its gradual expression. One of our observations is that the peak amplitude of quantal excitatory synaptic currents seemed to undergo a strengthening as synapses matured during development (Fig. 6D), a trend that was significant across all populations of recorded neurons. mEPSC mean amplitude increased from 7.9 ± 1.3 pA at 8 DIV to 11.1 ± 0.9 pA at 10 DIV to 14.3 ± 0.9 pA by 15 DIV (Fig. 6E) (n = 8, 8, and 6 cells, respectively; p < 0.05; unpaired t test), thus mirroring a developmental increase that has been reported in the brainstem for AMPA receptor-mediated mEPSCs (Ishikawa et al., 2002). Although this effect could also coincide with changes in the number and conductance properties of postsynaptic receptors, the significant increase of VGLUT1 protein expressed in synaptic terminals during this period (Fig. 6C) could also contribute, at least in part, to this observed maturational change. Although a significant developmental increase was observed for mEPSC frequency between 10 and 15 DIV (8 DIV, 0.20 ± 0.004 Hz; 10 DIV, 0.31 ± 0.10 Hz; 15 DIV, 1.20 ± 0.45 Hz; p < 0.05; unpaired t test) (Fig. 6F), given our findings on the lack of influence of VGLUT1 expression on mEPSC frequency (Fig. 4E), we cannot necessarily conclude that VGLUT1 participates in this phenomenon.
VGLUT1 expression is coordinated with the onset of functional vesicle cycling
Previous work in our group (Renger et al., 2001) has shown that silent synapses can arise in part from incomplete activation of postsynaptic AMPA receptors by a diminished presynaptic release of glutamate. The work demonstrated that conversion from silent to functional transmission could correspond to the developmentally regulated onset of “functional” vesicle cycling and the uptake of the FM1-43 styryl dye (Betz and Bewick, 1992). Thus, there is a developmental delay in FM uptake at early stages of development and now a delay in VGLUT1 expression as well (Fig. 6). We therefore asked whether the delays were coordinated, and whether the developmental time course of VGLUT1 expression might be synchronized with the maturation of vesicle cycling. It seemed sensible that as vesicle cycling matures, the functional capacity of transmitter loading needs to be upregulated accordingly to prevent the release of synaptic vesicles with insignificant transmitter content.
To examine whether such features of presynaptic maturation might be coordinated, it was necessary to concurrently compare the functional capacity of presynaptic terminals with their structural protein content. We made use of AM1-43, a fixable version of FM1-43, to label the functional presynaptic terminals, and followed up with an immunostaining procedure to check the level of desired presynaptic proteins (Renger et al., 2001). It has been shown that the amount FM dye taken up during stimulation can provide an index of the functional capacity of presynaptic terminals. The ability to visualize a fixable version together with immunolabeled protein allowed us to simultaneously measure both VGLUT1 expression levels across development and the developmental onset of functional vesicle cycling. As shown by imaging VGLUT1, AM1-43, and the synaptic vesicle protein synapsin I (Syn I) at the same synaptic terminals (Fig. 7A), AM1-43 uptake begins appearing at a similar stage of development as VGLUT1 expression. Figure 7B-D quantifies these developmental changes in a population of synapses, again using IOD as a measure of protein expression. Despite a relatively constant level of synapsin I staining across development (Fig. 7B), VGLUT1 expression (Fig. 7C) appeared to increase in tandem with the onset of AM1-43 labeled functional vesicle cycling (Fig. 7D). In general, the ratio of VGLUT1/Syn I increases from 8 to 15 DIV (p < 0.001; unpaired t test) and from 10 to 15 DIV (p < 0.001) in parallel with the ratio of AM1-43/Syn I (p < 0.01; P < 0.05) measured at the same synapses (Fig. 7E). We thus conclude that the maturation of transporter expression and delivery is jointly coordinated with functional vesicle cycling, a coupling that could be designed to help guarantee the efficient loading of transmitter as vesicle cycling becomes more active.
Figure 7.

VGLUT1 expression is coordinated with the onset of functional vesicle cycling. A, Triple immunostaining for VGLUT1, the functional bouton marker AM1-43, and the vesicle protein synapsin I. B, IOD per synapse for synapsin I, an additional vesicle protein, remains relatively stable through development. C, D, Synaptic IOD for VGLUT1 and AM1-43 appears to undergo a developmental change. E, Mean synaptic integrated optical density per image. Comparing VGLUT1 and AM1-43 intensity levels (with each normalized to synapsin I levels in the same synapse) demonstrates a significant and coordinated increase in VGLUT1 and AM1-43 staining across development. (***p < 0.001; **p < 0.01; *p < 0.05; unpaired t tests). Syn I, Synapsin I.
Activity-dependent plasticity of glutamate release and VGLUT1 expression in mature circuits
Exciting work by others (Turrigiano et al., 1998; Burrone et al., 2002; Thiagarajan et al., 2002) has demonstrated recently that excitatory synapses undergo activity-dependent plasticity in quantal size in response to global changes in network activity. Mechanisms have been proposed to begin to explain this change in quantal size, including changes in postsynaptic receptor number (O'Brien et al., 1998; Watt et al., 2000; Wierenga et al., 2005). Given that molecules responsible for specifying the quantal size postsynaptically, such as AMPA receptors, appear to be critically regulated during activity-dependent synaptic scaling, we decided to examine whether VGLUT1 expression might provide a complementary presynaptic mechanism by which quantal size can be coordinately regulated via endogenous, homeostatic control.
We therefore measured VGLUT1 expression levels per synaptic terminal by again taking the IOD ratio of VGLUT1 to synapsin I at each immunolabeled synapse as before but now comparing between untreated control cultures versus cultures that were preincubated for 48 h in 20 μm bicuculline (BIC) for the purpose of chronically increasing activity levels by blocking GABAA receptor-mediated inhibitory currents (Turrigiano et al., 1998) (Fig. 8A). We observed that chronic increases in activity levels (BIC) led to a significant reduction in synaptic VGLUT1 expression relative to control values (0.75 ± 0.06 control vs 0.95 ± 0.03 BIC; p = 0.02; unpaired t test; n = 8 images, 4482 synapses control; n = 7 images, 4464 synapses BIC) (Fig. 8B). We then attempted the exact opposite condition, chronically blocking AMPA and NMDA-type excitatory receptors by applying either 5 μm NBQX or 50 μm AP-5 to effectively decrease activity levels through each one of these pathways (Thiagarajan et al., 2002; Liu, 2004) and measuring the resulting expression levels of VGLUT1. Here, although we observed a robust increase in VGLUT1 in response to chronic NMDA receptor blockade (Fig. 8A, B), a less prominent increase and a surprising variability were disclosed in response to AMPA receptor blockade (supplementary data, available at www.jneurosci.org as supplemental material). This dichotomy was confirmed by replicating the experiments for NBQX and AP-5 many times. NMDA receptor blockade, however, resulted in increased VGLUT1 expression across every instance tested and, in the experiment presented in Figure 8, A and B, increased VGLUT1/Syn I levels by 35 ± 4% relative to controls (p < 0.001; unpaired t test; n = 10 images, 6120 synapses control; n = 8 images, 6532 synapses AP-5).
Thus, VGLUT1 expression appears to be capable of bidirectional endogenous regulation in response to changes in activity patterns, which could provide a mechanism for presynaptic terminals to coordinate functionally with activity-dependent changes to their postsynaptic partners. To verify that such changes to expression levels indeed had functional implications, we next measured whether glutamate release was also changing as a result of the activity-dependent VGLUT1 regulation. To examine activity-driven changes in glutamate release, we repeated our competitive antagonism protocol applied previously (Fig. 5), again in mature hippocampal cultures, except that in this case, one set of cultures was preincubated for 48 h in either 20 μm BIC or 50 μm AP-5. In these experiments, the cleft glutamate concentration at nontreated synapses appeared to be similar to that observed previously (Fig. 5D), with an ∼50% reduction in evoked postsynaptic current in response to 600 μm γ-DGG (Fig. 8C, gray symbols, D, middle traces). However, bicuculline-treated synapses were significantly more sensitive to the antagonist (Fig. 8C, white symbols, D, left traces) and led to 47% more attenuation than control (76 ± 4 vs 51 ± 3%; p = 0.03; unpaired t test; n = 4 pairs BIC, 4 pairs control) (Fig. 8E), indicating that the bicuculline-treated terminals released less glutamate, thus rendering their transmission more susceptible to the effects of the antagonist. To examine whether a comparable change in glutamate release was also observed in the opposite direction following a treatment that upregulates VGLUT1, we repeated this comparison with chronically treated AP-5 cultures. Here, we observed that the antagonist was significantly less effective on AP-5-treated transmission (Fig. 8C, black symbols, D, right traces), whereby the γ-DGG block was reduced by 65% in AP-5 synapses compared with control transmission (20 ± 8% AP-5 vs 61 ± 5% control; p < 0.01; unpaired t test; n = 6 pairs AP-5, 6 pairs control) (Fig. 8E).
Because the activity-dependent changes that altered vesicular glutamate release (Fig. 8C-E) also impacted VGLUT1 expression (Fig. 8A, B), and because our previous data demonstrate a causal relationship between VGLUT1 expression and glutamate release, we interpret our observed changes in glutamate release under BIC and AP-5 treatments to occur primarily as a direct consequence of the altered VGLUT1 expression. However, if synaptic terminals can at times undergo multivesicular release from single release sites, then the cleft glutamate concentration could also be altered in part by changes to the number of vesicles released from single terminals. This is possible given evidence that the probability of release, and thus the prevalence of MVR-mediated changes to cleft glutamate concentration, can be modified by perturbations to network activity levels (Murthy et al., 2001). As an additional control to further verify that these changes correspond to changes to the loading of single vesicles, we also measured the cleft glutamate concentration during spontaneous, miniature EPSCs in the bicuculline-treated transmission (Fig. 8F). Here, we observed that median γ-DGG attenuation was significantly increased relative to controls (Fig. 8F) (25 ± 4% control vs 39 ± 2% BIC; p = 0.02; unpaired t test), although the attenuations observed in the mEPSCs were less than those in the EPSCs, presumably because of the detection problems associated with spontaneous transmission when it is pharmacologically reduced (Zhou et al., 2000), which likely serves to underestimate the attenuation in the control condition and, to a greater degree, the smaller amplitude bicuculline-treated condition. Even with this detection problem, significant changes in the amount of glutamate released from single vesicles were observed between the two treatments.
To verify that activity-induced changes to VGLUT1 expression were also accompanied by changes to quantal synaptic efficacy, we quantified the amplitude of spontaneous mEPSCs. Here, we observed that in bicuculline-treated transmission, mEPSC amplitude was significantly reduced compared with control transmission (Fig. 8G, top trace, H) (n = 6 control; n = 4 BIC; p = 0.04; unpaired t test), whereas in AP-5-treated transmission, mEPSC amplitude was increased by 52% from control transmission (Fig. 8G, bottom trace, H) (n = 7 control; n = 6 AP-5; p = 0.03; unpaired t test). Thus, in both cases, activity-induced changes to VGLUT1 expression are accompanied by changes to quantal synaptic efficacy.
As such, chronic changes to either levels of activity or calcium flux alter vesicular transporter expression in a manner that is accompanied by relevant changes to physiological transmission. Endogenous control over VGLUT1 provides one mechanism by which an observed presynaptic scaling of glutamate release could be coordinated with a concomitant scaling of postsynaptic sensitivity (Watt et al., 2000).
Discussion
For neurons to be able to make use of VGLUT1 expression to modulate transmission makes specific predictions on a set of issues that remain controversial at glutamatergic synapses. By following the impact of transporter number through vesicle loading (Fig. 1), glutamate release (Fig. 5), and postsynaptic receptor activation (Figs. 3, 4), we provide new evidence in support of a flexibility in excitatory vesicle filling (Atwood and Karunanithi, 2002) and a nonsaturation of hippocampal synapses (Auger and Marty, 2000; Liu, 2003). In addition, although it remains undemonstrated whether presynaptic mechanisms endogenously contribute to regulating the quantal efficacy of excitatory transmission, we show that VGLUT1 expression can confer some properties of this regulation to excitatory synapses.
Vesicle loading depends on the number of available VGLUT1 transporters
It is imaginable that vesicles set in a low concentration of glutamate might simply continue filling until they have reached a set point level of saturation (Sulzer and Pothos, 2000), such that adding transporters could influence the speed but not the final concentration of glutamate uptake. The work presented here indicates that VGLUT1 vesicles do not fill to an invariant set point, and that reducing the pool of participating transporters (Fig. 1) reduces the equilibrium value of uptake, although it still cannot be resolved whether this is accomplished by changing the intravesicular concentration of transmitter or, instead, the structural capacity of the vesicle (Bruns et al., 2000; Colliver et al., 2000; Sulzer and Edwards, 2000; Karunanithi et al., 2002).
VGLUT1 overexpression enhances excitatory synaptic transmission via an increase in glutamate released per vesicle
We then found that it was possible to traffic additional functional VGLUT1 protein to synaptic terminals (Fig. 2), where we consequently observed larger evoked and spontaneous excitatory currents (Figs. 3, 4) as a result of an increased cleft glutamate concentration (Fig. 5). We provide direct verification that vesicular transport is able to exploit receptor nonsaturation at the level of single vesicles via a shift in the cleft glutamate concentration. An additional finding of some surprise was that VGLUT1 overexpression resulted in a decrease in evoked failure rate (Fig. 3E). This could suggest that in addition to enhancing the amount of glutamate deposited per release event, VGLUT1 expression might also be able to influence the likelihood that such release events will occur. Interestingly, another recent study reports that loss of VGLUT1 expression results in a substantially depleted pool of synaptic vesicles at excitatory terminals (Fremeau et al., 2004). Such a relationship between VGLUT1 expression and likelihood of release could depend on either the presence of the transporter itself or on the extent of filling that it facilitates.
It is also important to consider whether VGLUT1 expression could contribute to synaptic efficacy by influencing not only the amount of filling but also the rate (Sulzer and Pothos, 2000). This is especially plausible given two recent studies into the nature of vesicle cycling (Aravanis et al., 2003; Gandhi and Stevens, 2003), which confirmed a “kiss-and-run” type of release that involves the brief fusion and rapid reuse of synaptic vesicles. Such efficiency in cycling would place high demands on vesicle filling to prevent the “firing of blanks” via the exocytosis of “empty” vesicles. Although the precise speed of vesicle filling in the endogenous context can only be estimated, loading in vesicles isolated from the brain (Fig. 1) (Maycox et al., 1988; Wolosker et al., 1996) as well as loading in BON and PC12 cell lines (Bellocchio et al., 2000; Takamori et al., 2000) have been shown to involve a relatively slow process (several minutes), compared with the rapid process of vesicle reuse (∼20 s) (Aravanis et al., 2003). A rapid turnover of vesicles could thus result in a depleted synaptic response, and an increasing degree of transporter expression may be important for ensuring a match between the speed of transmitter loading and the functional capacity of the release machinery. Indeed, VGLUT1 is upregulated in close register with the increase of vesicle cycling that occurs during synaptic maturation (Fig. 7) and downregulated during activity-dependent scaling (Fig. 8) in parallel with a reduction in probability of vesicle release (Murthy et al., 2001).
Vesicular transporter expression is endogenously regulated
We found that transporter levels are modified endogenously across development (Fig. 6) and by levels of activity (Fig. 8). If vesicular transporters do contribute meaningfully to synaptic transmission, it would not be surprising to find them amid a molecular ensemble of similar versatility to that in the postsynaptic density (PSD) (Sheng and Kim, 2002). Molecular studies at inhibitory synapses, for example, have recently uncovered a direct association between the vesicular GABA transporter, VGAT, and the synthetic enzyme GAD65 (Jin et al., 2003), suggesting that transmitter synthesis may be functionally coupled to vesicle loading. The fact that VGLUT1 expression is coordinated with the onset of functional vesicle cycling (Fig. 7) could, in turn, suggest a conjunctive signal that couples fusion with adequate filling to ensure that increasingly active vesicles are able to release meaningful quantities of transmitter, perhaps in coordination with a growing pool of receptors postsynaptically.
Similarly, a negative-feedback relationship between certain forms of synaptic activity and VGLUT1 expression, which adjusts glutamate release in response to either circuit excitability or calcium influx (Fig. 8), could provide a presynaptic means for maintaining a stable degree of activity in the face of changing input levels (Turrigiano and Nelson, 2000) in a manner congruent with other recently uncovered postsynaptic mechanisms (O'Brien et al., 1998; Watt et al., 2000; Thiagarajan et al., 2002). Chronic blockade or enhancement of synaptic activity has been shown previously to lead to compensatory shifts in the size of the readily releasable pool and the number of presynaptic active zones (Murthy et al., 2001). This observation has supported a “size principle” (Harris and Stevens, 1989; Pierce and Lewin, 1994; Schikorski and Stevens, 1997) in which functional changes in morphology can correspond to changes in the size of the presynaptic terminal or PSD. It is possible that an activity-driven scaling of presynaptic terminal size (Murthy et al., 2001) is similarly accompanied by alterations in transporter expression per vesicle (Fig. 8) to provide a presynaptic shift in quantal efficacy that coincides with the known activity-driven changes in probability of release. In contrast, although blocking inhibitory receptors was effective at scaling VGLUT1, we were surprised to find that blocking NMDA receptors triggered a more profound regulation than that observed while blocking AMPA receptors. This suggests that VGLUT1 regulation, at least in hippocampal synapses, may be more sensitive to calcium flux than postsynaptic membrane depolarization, although other regimes of regulation may be more prominent in cortical synapses (Watt et al., 2000; Pratt et al., 2003) depending on the layer of cortex and which vesicular transporter is expressed.
Most likely, additional work will elucidate a range of distinct modes by which vesicular transport contributes to the dynamics of synaptic efficacy as well as distinct isoforms by which those contributions are made. Independent regulation of VGLUT1 and its recently identified isoforms VGLUT2 (Fremeau et al., 2001; Takamori et al., 2001; Varoqui et al., 2002) and VGLUT3 (Fremeau et al., 2002; Schafer et al., 2002) could offer, in particular, exciting clinical possibilities. The first mutations to the VGLUT1 ortholog eat-4 in Xenopus have revealed the importance of vesicular transport in both short-term (Rankin and Wicks, 1995) and long-term (Rose et al., 2002) memory. Future studies into the regulation of transmitter loading and release should thus discern new principles by which adaptations on both sides of the synapse are coordinated to ensure a cohesive robustness of memory.
Footnotes
This work was supported by The Institute of Physical and Chemical Research (RIKEN)-Massachusetts Institute of Technology Neuroscience Center Grant MH58880 to G.L. and by National Institutes of Health Grants NS37342 (G.L.), 1P29RR16816 (H.V.), and NS36936 (J.D.E.). N.R.W. was supported by a predoctoral fellowship from the National Science Foundation. We thank Dr. Bing Li for culture assistance, Dr. Yasunori Hayashi for insightful discussions, and Dr. Morgan Sheng for helpful suggestions.
Correspondence should be addressed to Dr. Guosong Liu, Picower Center for Learning and Memory, E18-218, Massachusetts Institute of Technology, Cambridge, MA 02139-4307. E-mail: liu@mit.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/256221-14$15.00/0
References
- Aravanis AM, Pyle JL, Tsien RW (2003) Single synaptic vesicles fusing transiently and successively without loss of identity. Nature 423: 643-647. [DOI] [PubMed] [Google Scholar]
- Atwood HL, Karunanithi S (2002) Diversification of synaptic strength: presynaptic elements. Nat Rev Neurosci 3: 497-516. [DOI] [PubMed] [Google Scholar]
- Auger C, Marty A (2000) Quantal currents at single-site central synapses. J Physiol (Lond) 526: 3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM, Richerson GB, Stevens CF (1990) Origin of variability in quantal size in cultured hippocampal neurons and hippocampal slices. Proc Natl Acad Sci USA 87: 5359-5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellocchio EE, Reimer RJ, Fremeau Jr RT, Edwards RH (2000) Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science 289: 957-960. [DOI] [PubMed] [Google Scholar]
- Betz WJ, Bewick GS (1992) Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science 255: 200-203. [DOI] [PubMed] [Google Scholar]
- Bruns D, Riedel D, Klingauf J, Jahn R (2000) Quantal release of serotonin. Neuron 28: 205-220. [DOI] [PubMed] [Google Scholar]
- Burrone J, O'Byrne M, Murthy VN (2002) Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature 420: 414-418. [DOI] [PubMed] [Google Scholar]
- Colliver TL, Pyott SJ, Achalabun M, Ewing AG (2000) VMAT-mediated changes in quantal size and vesicular volume. J Neurosci 20: 5276-5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forti L, Bossi M, Bergamaschi A, Villa A, Malgaroli A (1997) Loose-patch recordings of single quanta at individual hippocampal synapses. Nature 388: 874-878. [DOI] [PubMed] [Google Scholar]
- Franks KM, Stevens CF, Sejnowski TJ (2003) Independent sources of quantal variability at single glutamatergic synapses. J Neurosci 23: 3186-3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremeau RT, Troyer MD, Pahner I, Nygaard GO, Tran CH, Reimer RJ, Bellocchio EE, Fortin D, Storm-Mathisen J, Edwards RH (2001) The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron 31: 247-260. [DOI] [PubMed] [Google Scholar]
- Fremeau Jr RT, Burman J, Qureshi T, Tran CH, Proctor J, Johnson J, Zhang H, Sulzer D, Copenhagen DR, Storm-Mathisen J, Reimer RJ, Chaudhry FA, Edwards RH (2002) The identification of vesicular glutamate transporter 3 suggests novel modes of signaling by glutamate. Proc Natl Acad Sci USA 99: 14488-14493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremeau Jr RT, Kam K, Qureshi T, Johnson J, Copenhagen DR, Storm-Mathisen J, Chaudhry FA, Nicoll RA, Edwards RH (2004) Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science 304: 1815-1819. [DOI] [PubMed] [Google Scholar]
- Gandhi SP, Stevens CF (2003) Three modes of synaptic vesicular recycling revealed by single-vesicle imaging. Nature 423: 607-613. [DOI] [PubMed] [Google Scholar]
- Hanse E, Gustafsson B (2001) Quantal variability at glutamatergic synapses in area CA1 of the rat neonatal hippocampus. J Physiol (Lond) 531: 467-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Stevens JK (1989) Dendritic spines of CA 1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J Neurosci 9: 2982-2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW, Maycox PR, Stadler H, Jahn R (1988) Uptake of GABA by rat brain synaptic vesicles isolated by a new procedure. EMBO J 7: 3023-3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Sahara Y, Takahashi T (2002) A single packet of transmitter does not saturate postsynaptic glutamate receptors. Neuron 34: 613-621. [DOI] [PubMed] [Google Scholar]
- Jin H, Wu H, Osterhaus G, Wei J, Davis K, Sha D, Floor E, Hsu CC, Kopke RD, Wu JY (2003) Demonstration of functional coupling between gamma-aminobutyric acid (GABA) synthesis and vesicular GABA transport into synaptic vesicles. Proc Natl Acad Sci USA 100: 4293-4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunanithi S, Marin L, Wong K, Atwood HL (2002) Quantal size and variation determined by vesicle size in normal and mutant Drosophila glutamatergic synapses. J Neurosci 22: 10267-10276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G (2003) Presynaptic control of quantal size: kinetic mechanisms and implications for synaptic transmission and plasticity. Curr Opin Neurobiol 13: 324-331. [DOI] [PubMed] [Google Scholar]
- Liu G (2004) Local structural balance and functional interaction of excitatory and inhibitory synapses in hippocampal dendrites. Nat Neurosci 7: 373-379. [DOI] [PubMed] [Google Scholar]
- Liu G, Tsien RW (1995) Properties of synaptic transmission at single hippocampal synaptic boutons. Nature 375: 404-408. [DOI] [PubMed] [Google Scholar]
- Liu G, Choi S, Tsien RW (1999) Variability of neurotransmitter concentration and nonsaturation of postsynaptic AMPA receptors at synapses in hippocampal cultures and slices. Neuron 22: 395-409. [DOI] [PubMed] [Google Scholar]
- Mainen ZF, Malinow R, Svoboda K (1999) Synaptic calcium transients in single spines indicate that NMDA receptors are not saturated. Nature 399: 151-155. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA (1999) Long-term potentiation—a decade of progress? Science 285: 1870-1874. [DOI] [PubMed] [Google Scholar]
- Maycox PR, Deckwerth T, Hell JW, Jahn R (1988) Glutamate uptake by brain synaptic vesicles. Energy dependence of transport and functional reconstitution in proteoliposomes. J Biol Chem 263: 15423-15428. [PubMed] [Google Scholar]
- McAllister AK, Stevens CF (2000) Nonsaturation of AMPA and NMDA receptors at hippocampal synapses. Proc Natl Acad Sci USA 97: 6173-6178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy VN, Schikorski T, Stevens CF, Zhu Y (2001) Inactivity produces increases in neurotransmitter release and synapse size. Neuron 32: 673-682. [DOI] [PubMed] [Google Scholar]
- Ni B, Rosteck Jr PR, Nadi NS, Paul SM (1994) Cloning and expression of a cDNA encoding a brain-specific Na+-dependent inorganic phosphate cotransporter. Proc Natl Acad Sci USA 91: 5607-5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL (1998) Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron 21: 1067-1078. [DOI] [PubMed] [Google Scholar]
- Oertner TG, Sabatini BL, Nimchinsky EA, Svoboda K (2002) Facilitation at single synapses probed with optical quantal analysis. Nat Neurosci 5: 657-664. [DOI] [PubMed] [Google Scholar]
- Ogita K, Hirata K, Bole DG, Yoshida S, Tamura Y, Leckenby AM, Ueda T (2001) Inhibition of vesicular glutamate storage and exocytotic release by Rose Bengal. J Neurochem 77: 34-42. [DOI] [PubMed] [Google Scholar]
- Pierce JP, Lewin GR (1994) An ultrastructural size principle. Neuroscience 58: 441-446. [DOI] [PubMed] [Google Scholar]
- Pothos EN, Larsen KE, Krantz DE, Liu Y, Haycock JW, Setlik W, Gershon MD, Edwards RH, Sulzer D (2000) Synaptic vesicle transporter expression regulates vesicle phenotype and quantal size. J Neurosci 20: 7297-7306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt KG, Watt AJ, Griffith LC, Nelson SB, Turrigiano GG (2003) Activity-dependent remodeling of presynaptic inputs by postsynaptic expression of activated CaMKII. Neuron 39: 269-281. [DOI] [PubMed] [Google Scholar]
- Rankin CH, Wicks SR (1995) Mutations of the Caenorhabditis elegans brain-specific inorganic phosphate transporter eat-4 affect habituation of the tap-withdrawal response without affecting the response itself. J Neurosci 15: 2434-2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renger JJ, Egles C, Liu G (2001) A developmental switch in neurotransmitter flux enhances synaptic efficacy by affecting AMPA receptor activation. Neuron 29: 469-484. [DOI] [PubMed] [Google Scholar]
- Rose JK, Kaun KR, Rankin CH (2002) A new group-training procedure for habituation demonstrates that presynaptic glutamate release contributes to long-term memory in Caenorhabditis elegans Learn Mem 9: 130-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseth S, Fykse EM, Fonnum F (1998) Uptake of L-glutamate into synaptic vesicles: competitive inhibition by dyes with biphenyl and amino- and sulphonic acid-substituted naphthyl groups. Biochem Pharmacol 56: 1243-1249. [DOI] [PubMed] [Google Scholar]
- Schafer MK, Varoqui H, Defamie N, Weihe E, Erickson JD (2002) Molecular cloning and functional identification of mouse vesicular glutamate transporter 3 and its expression in subsets of novel excitatory neurons. J Biol Chem 277: 50734-50748. [DOI] [PubMed] [Google Scholar]
- Schikorski T, Stevens CF (1997) Quantitative ultrastructural analysis of hippocampal excitatory synapses. J Neurosci 17: 5858-5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Kim MJ (2002) Postsynaptic signaling and plasticity mechanisms. Science 298: 776-780. [DOI] [PubMed] [Google Scholar]
- Silver RA, Cull-Candy SG, Takahashi T (1996) Non-NMDA glutamate receptor occupancy and open probability at a rat cerebellar synapse with single and multiple release sites. J Physiol (Lond) 494: 231-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Ming G, Fon E, Bellocchio E, Edwards RH, Poo M (1997) Expression of a putative vesicular acetylcholine transporter facilitates quantal transmitter packaging. Neuron 18: 815-826. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Edwards R (2000) Vesicles: equal in neurotransmitter concentration but not in volume. Neuron 28: 5-7. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Pothos EN (2000) Regulation of quantal size by presynaptic mechanisms. Rev Neurosci 11: 159-212. [DOI] [PubMed] [Google Scholar]
- Takamori S, Rhee JS, Rosenmund C, Jahn R (2000) Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature 407: 189-194. [DOI] [PubMed] [Google Scholar]
- Takamori S, Rhee JS, Rosenmund C, Jahn R (2001) Identification of differentiation-associated brain-specific phosphate transporter as a second vesicular glutamate transporter (VGLUT2). J Neurosci 21: RC182(1-6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, Malherbe P, Broger C, Jahn R (2002) Molecular cloning and functional characterization of human vesicular glutamate transporter 3. EMBO Rep 3: 798-803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajan TC, Piedras-Renteria ES, Tsien RW (2002) α- and βCaMKII. Inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron 36: 1103-1114. [DOI] [PubMed] [Google Scholar]
- Tong G, Jahr CE (1994) Multivesicular release from excitatory synapses of cultured hippocampal neurons. Neuron 12: 51-59. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB (2000) Hebb and homeostasis in neuronal plasticity. Curr Opin Neurobiol 10: 358-364. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB (1998) Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391: 892-896. [DOI] [PubMed] [Google Scholar]
- Varoqui H, Schafer MK, Zhu H, Weihe E, Erickson JD (2002) Identification of the differentiation-associated Na+/PI transporter as a novel vesicular glutamate transporter expressed in a distinct set of glutamatergic synapses. J Neurosci 22: 142-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadiche JI, Jahr CE (2001) Multivesicular release at climbing fiber-Purkinje cell synapses. Neuron 32: 301-313. [DOI] [PubMed] [Google Scholar]
- Watt AJ, van Rossum MC, MacLeod KM, Nelson SB, Turrigiano GG (2000) Activity coregulates quantal AMPA and NMDA currents at neocortical synapses. Neuron 26: 659-670. [DOI] [PubMed] [Google Scholar]
- Wierenga CJ, Ibata K, Turrigiano GG (2005) Postsynaptic expression of homeostatic plasticity at neocortical synapses. J Neurosci 25: 2895-2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik SM, Rhee JS, Herzog E, Sigler A, Jahn R, Takamori S, Brose N, Rosenmund C (2004) An essential role for vesicular glutamate transporter 1 (VGLUT1) in postnatal development and control of quantal size. Proc Natl Acad Sci USA 101: 7158-7163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolosker H, de Souza DO, de Meis L (1996) Regulation of glutamate transport into synaptic vesicles by chloride and proton gradient. J Biol Chem 271: 11726-11731. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Ishikawa T, Takahashi T (2003) Developmental increase in vesicular glutamate content does not cause saturation of AMPA receptors at the calyx of held synapse. J Neurosci 23: 3633-3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Petersen CC, Nicoll RA (2000) Effects of reduced vesicular filling on synaptic transmission in rat hippocampal neurones. J Physiol (Lond) 525: 195-206. [DOI] [PMC free article] [PubMed] [Google Scholar]