

Abstract
Many studies have reported the roles played by regulated proteolysis in synaptic plasticity and memory, but the role of autophagy in neurons remains unclear. In mammalian cells, autophagy functions in the clearance of long-lived proteins and organelles and in adaptation to starvation. In neurons, although autophagy-related proteins (ATGs) are highly expressed, autophagic activity markers, autophagosome (AP) number, and light chain protein 3-II (LC3-II) are low compared with other cell types. In contrast, conditional knock-out of ATG5 or ATG7 in mouse brain causes neurodegeneration and behavioral deficits. Therefore, this study aimed to test whether autophagy is especially regulated in neurons to adapt to brain functions. In cultured rat hippocampal neurons, we found that KCl depolarization transiently increased LC3-II and AP number, which was partially inhibited with APV, an NMDA receptor (NMDAR) inhibitor. Brief low-dose NMDA, a model of chemical long-term depression (chem-LTD), increased LC3-II with a time course coincident with Akt and mammalian target of rapamycin (mTOR) dephosphorylation and degradation of GluR1, an AMPA receptor (AMPAR) subunit. Downstream of NMDAR, the protein phosphatase 1 inhibitor okadaic acid, PTEN inhibitor bpV(HOpic), autophagy inhibitor wortmannin, and short hairpin RNA-mediated knockdown of ATG7 blocked chem-LTD-induced autophagy and partially recovered GluR1 levels. After chem-LTD, GFP-LC3 puncta increased in spines and in dendrites when AP–lysosome fusion was blocked. These results indicate that neuronal stimulation induces NMDAR-dependent autophagy through PI3K–Akt–mTOR pathway inhibition, which may function in AMPAR degradation, thus suggesting autophagy as a contributor to NMDAR-dependent synaptic plasticity and brain functions.
Introduction
Protein degradation plays crucial roles in neuronal physiology and pathology (Bingol and Sheng, 2011). Early evidence in Aplysia linked synaptic plasticity and memory to neural activity-regulated proteolysis via the ubiquitin–proteasome system (Hegde et al., 1993, 1997), which degrades short-lived proteins and is now known as an important modulator of synaptic plasticity, learning and memory, and neurodegeneration (Yao et al., 2007; Yi and Ehlers, 2007; Lee et al., 2008; Tai and Schuman, 2008). Neurons also use the lysosome system, which involves endocytosis to degrade proteins, to affect synaptic plasticity through receptor trafficking, particularly of AMPA-type glutamate receptors (AMPARs) (Ehlers, 2000; Collingridge et al., 2004; Lee et al., 2004; Hirling, 2009).
Macro-autophagy (hereafter autophagy) is another major protein degradation pathway in which isolated membrane sequesters part of the cytoplasm to form a double-membrane vesicle, called autophagosome (AP), that fuses with lysosomes for the degradation of its contents. Basally, autophagy functions in clearance of long-lived cytoplasmic proteins or damaged organelles to maintain normal cell homeostasis. Inducible autophagy in response to some physiological stress conditions also functions in survival during starvation, tumor suppression, and protection against neurodegenerative diseases and oxidative stress (Kuma et al., 2004; Mathew et al., 2009; Yue et al., 2009; Lee and Koh, 2010).
Autophagy-related proteins (ATGs) mediate AP formation. Several ATG proteins function in light chain protein 3 (LC3, or ATG8) conjugation to phosphatidylethanolamine, thereby converting the inactive form, LC3-I, to the lipidated active form, LC3-II. This activation process occurs on isolated membranes at the initial stages of vesicle formation and ends with LC3-II bound to the outer and inner layers of the complete AP double-membrane vesicle, representing a marker for AP or other autophagic compartments derived from AP (Mizushima et al., 2010). AP initiation requires PI3K class III activity and is inhibited by mammalian target of rapamycin (mTOR) kinase activity. Therefore, AP formation can be modulated by mTOR-dependent pathways (such as in starvation) or, in some cases, by mTOR-independent pathways (Ravikumar et al., 2009).
Autophagy function in mature neurons remains controversial. In rodent brains, although ATG proteins are highly expressed, autophagic activity markers (AP number and LC3-II) are low compared with other organs. Although it was difficult to detect autophagy induction after starvation in GFP-LC3 transgenic mouse brains compared with other organs (Mizushima et al., 2004), recent more sensitive methods could detect such induction (Alirezaei et al., 2010; Kaushik et al., 2011). In contrast, conditional knock-out of ATG5 or ATG7, which are essential for AP formation, in mouse brains causes neurodegeneration and behavioral deficits (Hara et al., 2006; Komatsu et al., 2006), and blockade of AP–lysosomal fusion induces AP accumulation in cultured cortical neurons, indicating efficient constitutive autophagy (Boland et al., 2008).
Few reports have indicated that autophagy may contribute to synapse remodeling. In Caenorhabditis elegans, endocytosed GABAA receptors, but not acetylcholine receptors, are targeted to autophagosomes (Rowland et al., 2006). In Drosophila, autophagy promotes synapse growth (Shen and Ganetzky, 2009). Whether autophagy has such contributions in mammals remains unclear.
This study investigated whether autophagy is a component of neural activity-regulated proteolysis with a physiological role in synaptic plasticity using primary cultured hippocampal neurons, used previously to model synaptic remodeling, through various neuronal stimulation protocols (Okubo-Suzuki et al., 2008; Okada et al., 2009).
Materials and Methods
Chemical reagents
Potassium chloride (KCl), NMDA, wortmannin (WRT), d-(−)-2-amino-5-phosphonovaleric acid (APV), and bafilomycin A (Baf) were from Sigma-Aldrich. Okadaic acid (Oka) and dipotassium bisperoxo(5-hydroxypyridine-2-carboxyl) oxovanadate V [bpV(HOpic)] were from Calbiochem. Propidium iodide (PI) solution was from BD Biosciences, and 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI), wheat germ agglutinin conjugated to Alexa Fluor 488 (WGA-Alex488), and LysoTracker Red were from Invitrogen.
Primary cultured hippocampal neurons
Culture was performed as described previously with slight modifications (Okubo-Suzuki et al., 2008). Hippocampi were dissected from Wistar/ST rats of either sex on embryonic day 18 and dissociated using the Sumilon Nerve Cell Dissociation kit (Sumitomo Bakelite). Neurons were suspended in MEM (Invitrogen) supplemented with 5% horse serum, 5% fetal calf serum, 2 mm l-glutamine, and 1 mm pyruvic acid. The dissociated cells were plated onto poly-l-lysine-coated six-well plates at a density of 4 × 104 cells/cm2 for lysate preparation or onto glass coverslips in six-well plates at a density of 7 × 104 cells/cm2 for microscopic examination. On day in vitro 1 (DIV1), the medium was replaced with MEM containing B27 supplement (Invitrogen) and 0.5 mm l-glutamine (MEM/B27) and maintained until DIV20–26.
Neuronal stimulation treatment
For KCl depolarization, neurons were treated with MEM/B27 containing 60 mm KCl for 10 min, followed by washing and medium exchange with fresh MEM/B27 (without KCl). Neurons were returned to a CO2 incubator at 37°C for 1, 2, 6, or 18 h (as indicated). In inhibition experiments, 500 nm WRT or 100 μm APV was added 15 min before KCl depolarization with continued exposure until analysis. For chemical long-term depression (chem-LTD), neurons were treated with MEM/B27 containing 20, 50, or 300 μm NMDA for 5 min, followed by washing and medium exchange with fresh MEM/B27. Neurons were returned to a CO2 incubator at 37°C for 15 or 30 min or 2, 6, or 18 h (as indicated). In inhibition experiments, before chem-LTD, inhibitors were added for 15 min for 500 nm WRT, 30 min for 100 nm Baf, or 1 h for 20 nm Oka or 50 nm bpV(HOpic), with continued exposure until analysis.
Lentiviral Atg7 knockdown
To generate small hairpin RNA (shRNA) against Atg7 (shATG7), the sequence of siRNA (Accell SMARTpool, A-095596-15, LOC312647 rat; Thermo Fisher Scientific) was used, and for scrambled shRNA (shScrmb), the following sequence (sense-loop-antisense) was used: 5′-GATCCGGATCGCTGGCGCTATCTAGACCACTCTTCCTGTCAGAAGTGGTCTAGATAGCGCCAGCGATCCTTTTTG-3′ (sense) and 5′-AATTCAAAAAGGATCGCTGGCGCTATCTAGACCACTTCTGACAGGAAGAGTGGTCTAGATAGCGCCAGCGATCCG-3′ (antisense). Oligonucleotides were annealed and cloned into pSIH-H1 lentivector according to the manufacturer's instructions (System Biosciences). Production of VSV-G pseudotyped lentivirus was performed according to the manufacturer's instructions (Invitrogen), with some modifications. 293FT producer cells (Invitrogen) were seeded to a 100 mm dish with culture medium (DMEM with 1 mm sodium pyruvate/4 mm l-glutamine containing 10% FBS, 2 mm l-glutamine, 0.1 mm MEM nonessential amino acids, 500 μg/ml Geneticin, and penicillin/streptomycin) at 24 h before transfection. Four hours before transfection, the culture medium was replaced with new culture medium without Geneticin. Five micrograms of the lentiviral vectors (shATG7 or shScrmb) were cotransfected with 12 μg of the mixture of the packaging vectors (pLP1, pLP2, and pLP/VSVG; Invitrogen) into the 293FT cells, using 36 μl of Lipofectamine2000 (Invitrogen). The medium was replaced at 24 h after transfection with UltraCULTURE medium (Lonza) containing 4 mm l-glutamine, 2 mm GlutaMAX, 0.1 m nonessential amino acids, 1 mm sodium pyruvate, and penicillin/streptomycin. After 72 h from transfection, supernatants were collected by low-speed centrifugation (2000 rpm, 15 min), filtered through 0.45 mm filters (Millipore), and concentrated with PEGit Virus Precipitation Solution (System Biosciences). Solutions were titrated in HeLa cells by GFP expression from transgene of lentiviral vector encoding shRNAs. Cultured neurons were infected on DIV17, and treatment was done on DIV22 as described previously.
Immunoblot analysis
After the indicated treatments, neuronal cells were harvested in RIPA buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 2 mm EDTA, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mm NaF) containing 1× protease inhibitor mixture, complete ULTRA tablets, and, in the case of phosphorylated protein detection, phosphatase inhibitor mixture (PhosSTOP tablets; Roche Applied Science). Samples were then vortexed at 4°C for 15 min and centrifuged at 14,000 rpm for 15 min at 4°C, and supernatants were stored at −30°C until use. After measurement of protein concentration using a protein assay bicinchoninate kit (Nacalai Tesque), lysates were subjected to SDS-PAGE and immunoblotting using the iBlot Dry Blotting System (Invitrogen). The appropriate primary antibodies, rabbit polyclonal anti-LC3 (1:1000; Abcam), rabbit polyclonal anti-Akt (1:1000; Cell Signaling Technology), mouse monoclonal anti-phospho-Akt (Ser473; 1:1000; Cell Signaling Technology), rabbit polyclonal anti-mTOR (1:1000; Cell Signaling Technology), rabbit polyclonal anti-phospho-mTOR (Ser2448; 1:1000; Cell Signaling Technology), rabbit polyclonal anti-GluR1 (1:1000; Millipore), rabbit polyclonal anti-ATG7 (1:1000; Cell Signaling Technology), and rabbit polyclonal anti-α-tubulin (1:1000; Cell Signaling Technology), and the corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies (1:5000) were diluted in TBST–5% skim milk, applied sequentially to the blots and analyzed with the ECL Plus chemiluminescence detection system (GE Healthcare). An ImageQuant LAS 4000 mini imager (GE Healthcare) was used for blot imaging and quantification. The anti-Akt and anti-mTOR antibodies were applied to the same blots after stripping of anti-phospho-Akt and anti-phospho-mTOR antibodies, respectively. In Figure 5, total GluR1 and ATG7 are normalized to α-tubulin and/or total Akt as loading controls. The data are represented relative to the data for the control treatment. In Figures 1 and 3D, we also calculated the LC3-II/GAPDH ratio, which gave us similar results (data not shown).
Figure 5.

Autophagy induced by chem-LTD contributes to AMPAR degradation. A, Representative immunoblots and quantitation of the time course of GluR1 and total Akt (loading control) after chem-LTD (n = 3; *p < 0.05; ***p < 0.001; one-way ANOVA with Tukey's post hoc test compared with the control). B, Representative immunoblots and quantitation of GluR1 and tubulin (loading control) 2 h after 20, 50, and 300 μm NMDA for 5 min (n = 3; *p < 0.05; **p < 0.01; ***p < 0.001; one-way ANOVA with Tukey's post hoc test compared with control unless where indicated). C, Representative immunoblots and quantitation of GluR1 and total Akt (loading control) 2 h after chem-LTD alone or with 20 nm Oka or 50 nm bpV(HOpic) (bpV) (n = 3; *p < 0.05; ***p < 0.001; one-way ANOVA with Tukey's post hoc test compared with control unless where indicated). D, Representative immunoblots and quantitation of GluR1 and tubulin (loading control) 2 h after chem-LTD alone or with 500 nm WRT (n = 3; ***p < 0.001, one-way ANOVA with Tukey's post hoc test compared with control unless where indicated). E, Representative immunoblots and quantitation of ATG7, LC3, GluR1, and total Akt (loading control) 2 h after chem-LTD of neurons infected by lentivirus encoding shScrmb or shRNA against ATG7 (shATG7) (n = 3; *p < 0.05; **p < 0.01; ***p < 0.001; one-way ANOVA with Tukey's post hoc test compared with control unless where indicated). We also calculated NMDA/Ctrl per trial for each shRNA (shScrmb, 0.39 ± 0.04; shATG7, 0.56 ± 0.07; p < 0.05, paired t test; n = 3). Error bars indicate mean ± SE. n.s., No significant difference; Ctrl, control.
Figure 1.
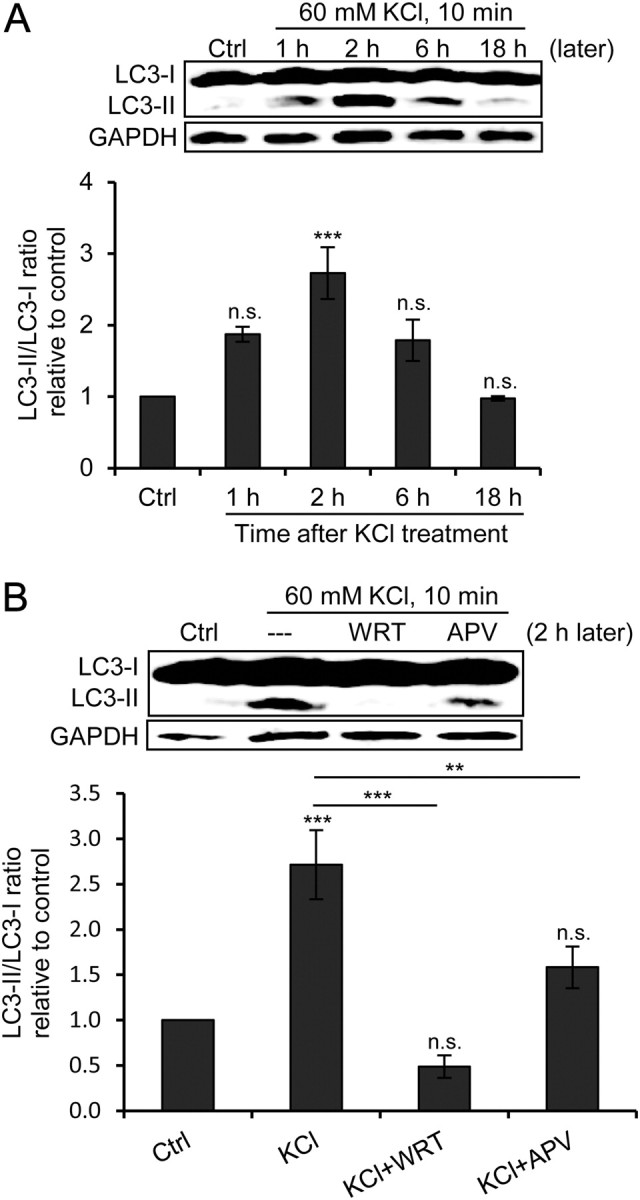
KCl depolarization transiently increases LC3-II level in an NMDAR-dependent manner. A, Representative LC3 and GAPDH (loading control) immunoblots of cultured hippocampal neurons treated with 60 mm KCl for 10 min and from which lysates were prepared 1, 2, 6, or 18 h after treatment (later). The graph shows the results of densitometric quantitation of immunoblots represented as the LC3-II/LC3-I ratio relative to the control (n = 3; ***p < 0.001, one-way ANOVA with Tukey's post hoc test compared with control). B, Representative LC3 and GAPDH (loading control) immunoblots of cultured hippocampal neurons treated with 60 mm KCl for 10 min alone or with 500 nm WRT or 100 μm APV; lysates were prepared 2 h after treatment (2 h later). The graph shows the results of densitometric quantitation of immunoblots represented as the LC3-II/LC3-I ratio relative to the control (n = 3; **p < 0.01; ***p < 0.001; one-way ANOVA with Tukey's post hoc test compared with control unless indicated). Error bars indicate mean ± SE. n.s., No significant difference; Ctrl, control.
Figure 3.

Chemical LTD increases LC3-II level. A, Representative LC3 and GAPDH (loading control) immunoblots of cultured hippocampal neurons treated with 50 μm NMDA for 5 min (chem-LTD) and from which lysates were prepared 15 or 30 min or 2, 6, or 18 h after treatment (later). The graph shows the results of densitometric quantitation of immunoblots represented as the LC3-II/LC3-I ratio relative to the control (n = 3; **p < 0.01, one-way ANOVA with Tukey's post hoc test compared with the control). B, Representative LC3 and tubulin (loading control) immunoblots of cultured hippocampal neurons treated with 20, 50, or 300 μm NMDA for 5 min and from which lysates were prepared 2 h after treatment (2 h later). The graph shows the results of densitometric quantitation of immunoblots represented as the LC3-II/LC3-I ratio relative to the control (n = 3; *p < 0.05; ***p < 0.001; one-way ANOVA with Tukey's post hoc test compared with the control unless where indicated). C, Percentage of neurons showing PI uptake, indicating cellular injury, 18 h after treatment with 20, 50, or 300 μm NMDA for 5 min or directly after continuous (cont.) treatment with 50 μm NMDA for 18 h (n = 24 fields from 3 independent experiments; *p < 0.05; ***p < 0.001; one-way ANOVA with Tukey's post hoc test compared with the control unless where indicated). D, Representative LC3 and GAPDH (loading control) immunoblots of cultured hippocampal neurons treated with 50 μm NMDA for 5 min alone, combined with 100 nm Baf, or with Baf alone, from which lysates were prepared 2 h after NMDA treatment. The graph shows the results of densitometric quantitation of immunoblots represented as the LC3-II/LC3-I ratio relative to the control (n = 3; *p < 0.05; ***p < 0.001; one-way ANOVA with Tukey's post hoc test compared with control unless where indicated). E, Representative LC3 and tubulin (loading control) immunoblots of cultured hippocampal neurons treated with 50 μm NMDA for 5 min alone or with 500 nm WRT, from which lysates were prepared 2 h after NMDA treatment. The graph shows the results of densitometric quantitation of immunoblots represented as the LC3-II/LC3-I ratio relative to the control (n = 3; ***p < 0.001, one-way ANOVA with Tukey's post hoc test compared to control). Error bars indicate mean ± SE. n.s., No significant difference; Ctrl, control.
Plasmids
Construction of pEGFP-LC3B.
cDNA encoding rat LC3B (Wu et al., 2006) (GenBank accession number AY392036) was obtained by RT-PCR from rat total cDNA with LC3BF1 primer (5′-TATAGATCTGCCGCCATGCCGTCCGAGAAGACCTTC-3′) and LC3BR1 primer (5′-GGAATTCTTACACAGCCAGTGCTGTCCCGA-3′). To obtain the pEGFP-LC3B plasmid, the LC3 cDNA product was directly inserted into BglII and EcoRI sites of the pEGFP-C1 vector (Clontech), and the sequence was confirmed with an ABI Prism 3700 DNA Sequencer (Applied Biosystems).
Construction of pmCherry-β-actin.
Rat β-actin cDNA prepared from rat PC-12 cell mRNAs was kindly provided by Prof. M. Takahashi (Kitasato University, Kanazawa, Japan). The β-actin open-reading-frame cDNA was subcloned into EcoRI and BamHI sites of pEGFP-C1 vector (Clontech) to produce pEGFP-β-actin. mCherry cDNA was kindly provided by Prof. R. Tsien (University of California at San Diego, La Jolla, CA) and amplified using 5′mCherry FW Eco47IV primer (5′-AAAGCGCTACCGGTCGCCACCATGGTGAGCAAGGGCGAGGAGGAT-3′) and 3′mCherry RV BglII primer (5′-AAAGATCTGAGTCCGGCCGGACTTGTACAGCTCGTCCATGCCGCC-3′). EGFP of pEGFP–β-actin was replaced by mCherry cDNA through Eco47IV and BglII sites to produce pmCherry-β-actin.
Analysis of autophagosome and lysosome number
For autophagosome detection, neurons were cotransfected with 1 μg of pEGFP-LC3B or pEGFP-N1 and 1 μg of pDsRed2-N1 plasmids (Clontech) for the KCl depolarization experiment, or cotransfected with 1 μg of pEGFP-LC3B and 1 μg of pmCherry-β-actin for the chem-LTD experiment. Transfection was performed using Lipofectamine 2000 (Invitrogen) on DIV9, and neurons were treated on DIV20–26. For lysosome detection, nontransfected neurons (DIV20–26) were exposed to 200 nm LysoTracker Red for 1 h before chem-LTD treatment. Neurons were then washed and incubated with fresh MEM/B27 containing WGA-Alexa488 (to monitor cell morphology) for 15 min. In both cases, neurons were fixed with 4% paraformaldehyde in PBS, pH 7.4, for 10 min at 4°C, washed three times with cold Dulbecco's PBS (Invitrogen), and mounted with ProLong Gold Antifade Reagent (Invitrogen) before examination with an LSM700 confocal laser-scanning microscope (Carl Zeiss). Stacked images of consecutive focal planes at 0.4 μm intervals (z-series sections) were reconstructed into a 2D projection image using the LSM Image Browser software (Carl Zeiss).
Analysis of cell death
Treated cultured neurons (DIV20–26) were washed with warm DBPS and incubated with cell death assay mixture [PI, WGA-Alex488, and DAPI (40:4:1) in DBPS] for 10 min at 37°C. Images of the neurons were taken within 15 min by a BioRevo microscope (KEYENCE Corporation). PI-positive nuclei were manually counted and represented as a percentage of the total number of DAPI-positive nuclei. WGA-Alex488 was used to monitor cell morphology.
Statistical analysis
All statistical analyses were performed using InStat version 3.1 (GraphPad Software). Comparisons between data from two groups were analyzed by the two-tailed Student's t test. Multiple-group comparisons were assessed using a one-way ANOVA, followed by Tukey's or Fisher's PLSD post hoc tests. The null hypothesis was rejected at p < 0.05. Quantitative data are presented as mean ± SEM.
Results
KCl depolarization of hippocampal neurons causes a transient NMDAR-dependent increase in LC3-II/LC3-I ratio
Neuronal stimulation with KCl causes depolarization, activation of voltage-gated receptors, and induction of immediate-early genes simulating physiological stimulation (Bading et al., 1993). Primary cultured hippocampal neurons (DIV20–26) treated with 60 mm KCl for 10 min (KCl depolarization) showed a significant increase in the LC3-II/LC3-I ratio 2 h after treatment, which returned to the control level within 18 h (Fig. 1A). When APV, a NMDAR inhibitor, was added 15 min before and throughout KCl depolarization treatment, it partially blocked the increase in the LC3-II/LC3-I ratio observed 2 h after KCl treatment, reducing it to approximately half. Moreover, the addition of WRT, a PI3K inhibitor and common autophagy inhibitor, completely blocked the increase in the LC3-II/LC3-I ratio in a similar manner (Fig. 1B). The partial blockade by APV indicates the involvement of NMDAR in addition to other voltage-gated receptors.
KCl depolarization increases GFP-LC3 puncta in hippocampal pyramidal neurons
Primary cultured hippocampal neurons are mainly neurons with a small fraction of glial cells. To confirm that neuronal stimulation-induced autophagy occurred in neurons, we focused on pyramidal cells, the main neuron type involved in hippocampal memory function and easily morphologically identifiable in the mature state by their multiple spiny dendrites (Sekiguchi et al., 2009). Primary cultured hippocampal neurons cotransfected with pGFP fused to LC3 (GFP-LC3), as an AP marker, and pDsRed, as a morphology marker, were observed 2 h after KCl depolarization. KCl significantly increased the average number of GFP-LC3 puncta in pyramidal cell soma, which was partially blocked by APV (Fig. 2A,B). Moreover, in some KCl-stimulated pyramidal neurons (∼3 of 30 neurons), we noticed GFP-LC3 puncta in dendritic shafts and spines (Fig. 2C), indicating that AP may increase in dendrites after KCl depolarization. However, the scarcity of such AP hindered quantitation of the results. These results indicate that neuronal stimulation via KCl depolarization induces autophagy in pyramidal neurons.
Figure 2.
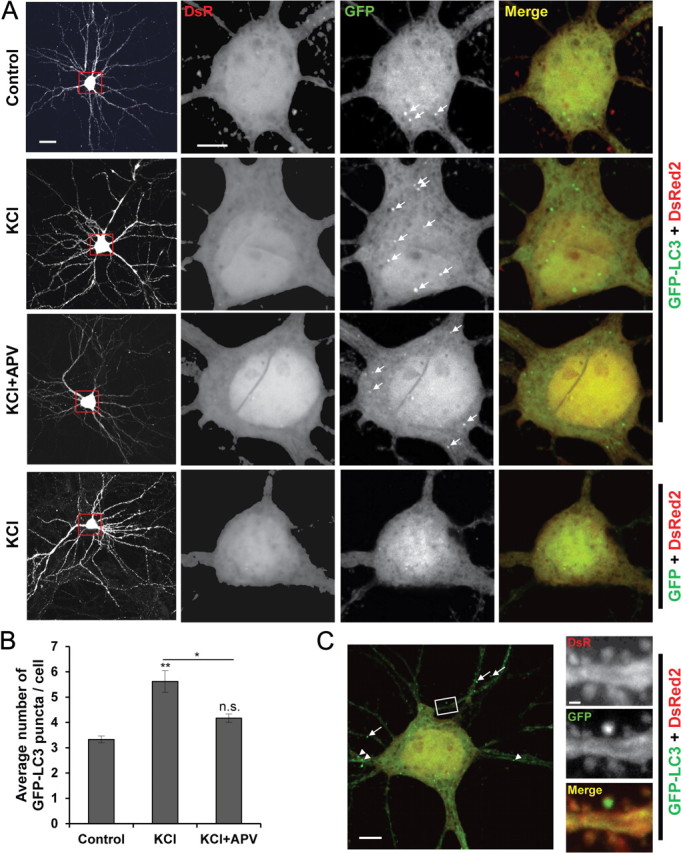
KCl depolarization increases GFP-LC3 puncta in pyramidal neurons. Cultured hippocampal neurons were cotransfected at DIV9 with pDsRed2 and pGFP-LC3 or pGFP-C1, and treatments were performed at DIV20–26. A, Representative images of fully mature pyramidal neurons (left; scare bar, 50 μm) and magnified cell soma (scale bar, 5 μm) showing GFP-LC3 puncta as a marker of AP (arrows) in control or 2 h after treatment with 60 mm KCl for 10 min alone or with 100 μm APV. B, Quantitation of the average number of GFP-LC3 puncta/pyramidal cell soma (n = 3 independent experiments, each representing an average of 10 neurons per treatment; *p < 0.05; **p < 0.01; one-way ANOVA with Fisher's PLSD post hoc compared with control except where indicated; n.s., no significant difference; error bars indicate mean ± SE). C, A representative cell treated with KCl showing GFP-LC3 puncta in dendrites (arrowheads) and spines (arrows and magnified inset). Scare bars: 5 μm; inset, 0.5 μm.
Brief low-dose NMDA (chem-LTD) induces autophagy
How does activation of NMDAR induce autophagy? NMDAR activation in neurons can elicit two opposing forms of synaptic modifications: long-term potentiation (LTP) and LTD (Bliss and Collingridge, 1993; Malenka and Nicoll, 1999; Collingridge et al., 2010). LTP is associated with an increase in synaptic strength as well as spine formation and enlargement (Fukazawa et al., 2003; Abraham and Williams, 2008; Bramham, 2008). In contrast, LTD is associated with weakening in synaptic strength and spine shrinkage and pruning, suggesting a role for protein degradation (Segal, 2005; Bingol and Sheng, 2011). We tested whether NMDAR-dependent LTD induces autophagy by stimulating neurons with brief low-dose NMDA, an agonist of NMDAR, as a model of chem-LTD in hippocampal neurons (Hsin et al., 2010; Li et al., 2010). As observed with KCl depolarization, bath application of 50 μm NMDA for 5 min (hereafter, chem-LTD refers to 50 μm of NMDA for a 5 min treatment, unless where otherwise described) significantly increased the LC3-II/LC3-I ratio 2 h after treatment, which returned to normal levels within 18 h (Fig. 3A).
High doses and/or long incubation of NMDA are excitotoxic to neurons, and autophagy may be induced under these conditions in response to excitotoxicity (Borsello et al., 2003; Shacka et al., 2007; Chakrabarti et al., 2009; Sadasivan et al., 2010). However, the chem-LTD protocol was previously reported not to be excitotoxic (Li et al., 2010). To confirm that brief low-dose, NMDA-induced autophagy was not a consequence of excitotoxicity, increasing doses of NMDA were applied for 5 min, and their effects on the LC3-II/LC3-I ratio and PI uptake (as a measure of cell membrane injury) 2 h (data not shown) and 18 h after exposure were compared. Low doses of NMDA (20 and 50 μm) induced dose-dependent significant increases in the LC3-II/LC3-I ratio with a nonsignificant change in PI uptake. However, 300 μm NMDA did not further increase the LC3-II/LC3-I ratio while leading to a significant increase in PI uptake (Fig. 3B,C). These data indicate that the brief low-dose, NMDA-induced autophagy is a consequence of NMDAR activation, rather than secondary to cellular injury.
Monitoring autophagic flux is recommended, because the increase in the LC3-II/LC3-I ratio may be attributable to either the induction of AP formation or the blockade of AP–lysosomal fusion (Mizushima and Yoshimori, 2007). Therefore, 100 nm Baf (saturating dose; data not shown), which blocks AP–lysosomal fusion, was applied starting 30 min before and throughout the chem-LTD protocol. The combined treatment resulted in an additive increase of the LC3-II/LC3-I ratio over that of Baf alone, thus indicating increased AP production (Fig. 3D). Finally, WRT completely blocked the chem-LTD-induced autophagy (Fig. 3E). Collectively, these results indicate that neuronal stimulation via chem-LTD induces autophagy.
Dephosphorylation of Akt and mTOR by protein phosphatases mediates chem-LTD- induced autophagy
Next we aimed to determine which signaling pathway downstream of NMDAR leads to autophagy induction. The inhibition of mTOR activity represents the mTOR-dependent autophagy induction pathway (Ravikumar et al., 2009). NMDAR-dependent LTD-inducing stimuli activate protein phosphatase 1 (PP1), a key enzyme in LTD, which dephosphorylates Akt and in turn may inactivate mTOR (Collingridge et al., 2010). In addition, PTEN, a known regulator of Akt phosphorylation, is recruited at synapses by NMDAR-dependent LTD-inducing stimuli, and its activity is necessary for LTD (Jurado et al., 2010). Therefore, we investigated the possibility that chem-LTD induces autophagy by inhibiting the PI3K–Akt–mTOR pathway.
In the time course of Akt and mTOR phosphorylation after chem-LTD, phosphorylation of both proteins significantly decreased 15 and 30 min after NMDA treatment. The decrease in mTOR was maintained for 2 h, whereas both phosphorylated proteins returned to normal levels within 18 h (Fig. 4A,B). This time course coincided with that of the increase in the LC3-II/LC3-I ratio after chem-LTD. Moreover, we tested the effect of Oka, a specific inhibitor of PP1, and bpV(HOpic), a specific inhibitor of PTEN, on chem-LTD-induced autophagy. Both reagents inhibited NMDAR-dependent LTD when induced electrophysiologically in hippocampal slices (Mulkey et al., 1993; Jurado et al., 2010). The addition of either inhibitor 1 h before and throughout chem-LTD recovered the phosphorylation of Akt (at 15 min) and mTOR and its substrate p70S6K (at 2 h) after NMDA treatment (Fig. 4C). Interestingly, each inhibitor alone, especially Oka, could completely recover the phosphorylation levels. Oka or bpV(HOpic) completely blocked the increase in the LC3-II/LC3-I ratio 2 h after NMDA treatment (Fig. 4D). These data indicate that chem-LTD induces mTOR-dependent autophagy by activating protein phosphatases.
Figure 4.
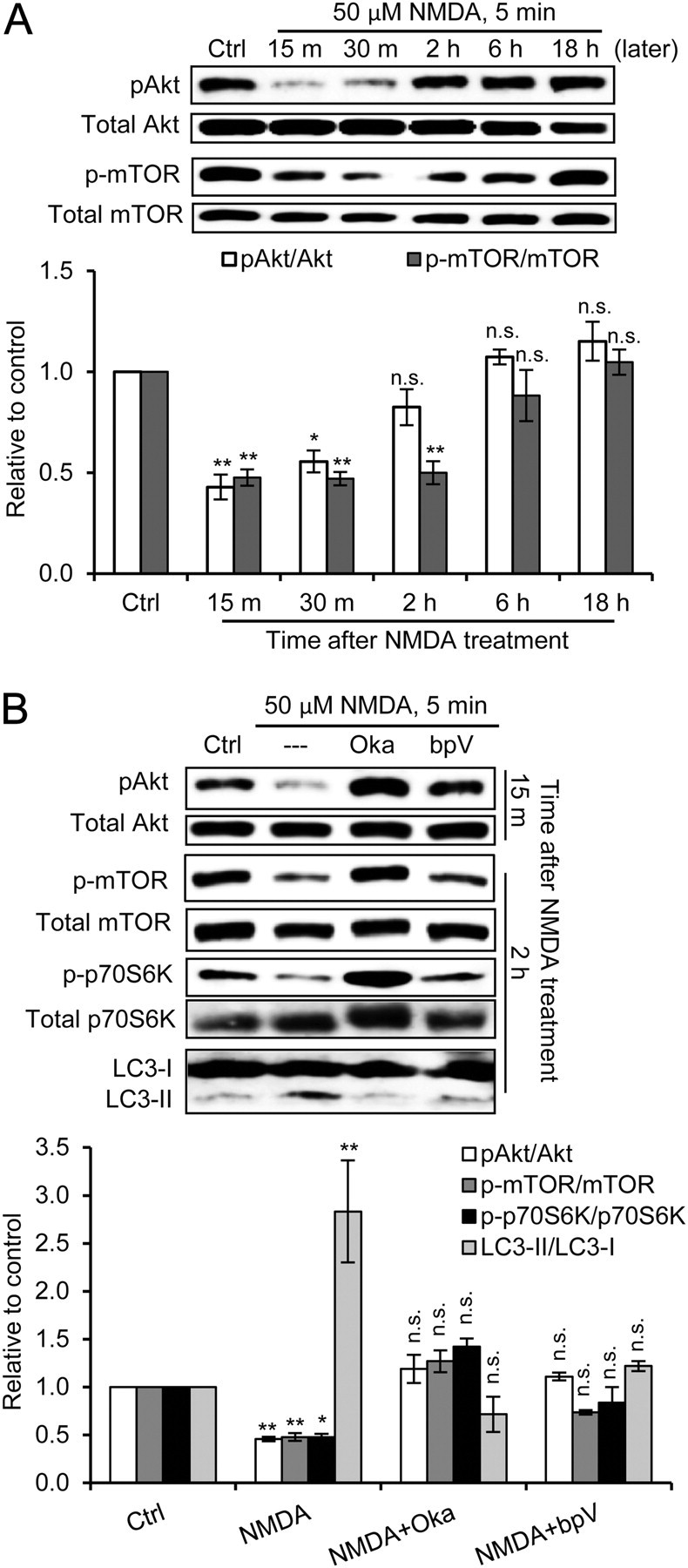
Akt and mTOR dephosphorylation by protein phosphatases mediates chem-LTD-induced autophagy. A, Representative immunoblots and quantitation for the time course of phospho-Ser473-Akt (pAkt), total Akt, phospho-Ser2448-mTOR (p-mTOR), and total mTOR after chem-LTD in cultured hippocampal neurons (n = 3; *p < 0.05; **p < 0.01; one-way ANOVA with Tukey's post hoc test compared with the control). B, Representative immunoblots and quantitation of pAkt, and total Akt 15 min after chem-LTD, and of p-mTOR, total mTOR, phospho-Thr389-p70S6K (p-p70S6K), total p70S6K, and LC3 2 h after chem-LTD alone or with 20 nm Oka or 50 nm bpV(HOpic) (bpV) (n = 3 per time point; *p < 0.05; **p < 0.01, one-way ANOVA with Tukey's post hoc test compared with control). Error bars indicate mean ± SE. n.s., No significant difference; Ctrl, control.
Autophagy induction by chem-LTD contributes to AMPAR degradation
To gain some insight into possible functions of neuronal stimulation-induced autophagy, we investigated its relationship to AMPAR degradation. After LTD stimulation, AMPARs are internalized by endocytosis and are then either redirected back to the synapse via recycling endosomes or taken up by lysosomes for final degradation (Collingridge et al., 2004; Hirling, 2009). AMPARs are heterotetrameric complexes composed of various combinations of four subunits (GluR1–4). Two major subtypes of AMPAR are present in adult hippocampus: GluR1/2 and GluR2/3 heteromeric receptors. The GluR1 subunit acts dominantly over other subunits to drive AMPAR to the surface in response to NMDAR stimulation, resulting in synaptic potentiation (Shi et al., 2001). GluR1/2 addition to synapses is believed to occur during plasticity, whereas GluR2/3 continuously cycles between synapse and intracellular compartments. GluR2 is thought to control the recycling or lysosomal degradation of AMPAR after internalization (Lee et al., 2004). Therefore, monitoring GluR1/2 by detecting GluR1 degradation is more informative concerning how AMPAR sorting in response to NMDAR stimulation relates to synaptic plasticity (Ehlers, 2000; Lee et al., 2004).
The observation of the time course of GluR1 degradation after chem-LTD revealed that GluR1 levels began to decrease significantly 30 min after NMDA treatment, reaching a maximum decrease by 2 h (Fig. 5A), which was concomitant with the maximum increase in the LC3-II/LC3-I ratio (Fig. 3A). Comparing different doses of NMDA 2 h after chem-LTD, we found that 20 and 50 μm NMDA induced a significant dose-dependent decrease in GluR1 levels, whereas 300 μm NMDA did not lead to any further decrease (Fig. 5B). These results coincided with the increase in the LC3-II/LC3-I ratio, but not with PI uptake using the same NMDA doses (Fig. 3B,C), indicating that autophagy induction correlates with GluR1 degradation but not with cellular injury. Moreover, WRT, Oka, and bpV(HOpic), which inhibited chem-LTD-induced autophagy (Figs. 3E, 4D), partially recovered GluR1 levels 2 h after chem-LTD. These findings strongly suggest that chem-LTD-induced autophagy at least contributes to AMPAR degradation.
Although the addition of Oka or bpV(HOpic) resulted in complete recovery of Akt, mTOR, and p70S6K phosphorylation (Fig. 4C), chem-LTD-induced AMPAR degradation was only partially recovered. This observation possibly implies the involvement of alternative pathways other than protein phosphatase signaling in AMPAR degradation. WRT inhibits autophagy by inhibiting PI3K III, but it also nonspecifically inhibits PI3K class I. PI3K I inhibition decreases Akt phosphorylation and signals for AMPAR internalization and degradation. These two opposing effects may account for the partial recovery of GluR1 levels by WRT after chem-LTD.
To specifically inhibit autophagy, lentiviral shRNA was used to knockdown ATG7, which is necessary for LC3-I conversion to LC3-II. Neurons infected with shRNA against ATG7 (shATG7) showed ATG7 levels about half compared with neurons infected with shScrmb. Two hours after chem-LTD, the increase in the LC3-II/LC3-I ratio was blocked by shATG7, and GluR1 levels were partially recovered (Fig. 5E). The ratio of normalized GluR1 level in chem-LTD treated (NMDA) to control neurons were higher in shATG7 compared with shScrmb-infected neurons (shScrmb, 0.39 ± 0.04; shATG7, 0.56 ± 0.07; p < 0.05, paired t test; n = 3). The aforementioned results demonstrate that one function of neuronal stimulation-induced autophagy is contribution to AMPAR degradation after chem-LTD.
GFP-LC3 puncta increase in the spines and dendritic shafts after chem-LTD
We tested whether AP can be detected in dendritic shaft and spines to facilitate AMPAR lysosomal degradation. Neurons were cotransfected with pGFP-LC3 and pmCherry-β-actin as spine marker. F-actin is a major cytoskeletal structure in dendritic spines, and β-actin conjugated to fluorescent proteins allows visualization of spines (Fischer et al., 1998). GFP-LC3 puncta were rarely detected in dendritic shaft or spines in control neurons. After NMDA treatment, we detected significant increase of GFP-LC3 puncta colocalized with mCherry-β-actin signal but only slight a nonsignificant increase of GFP-LC3 puncta in dendritic shafts after NMDA treatment (Fig. 6A,B). The rapid dynamics of AP, i.e., their rapid formation and fusion to lysosomes, may account for this rareness (Boland et al., 2008). Treatment of neurons with Baf for only 2.5 h allowed the detection of a considerable number of GFP-LC3 puncta in the dendritic shafts of pyramidal neurons even in the absence of neuronal stimulation. This observation indicates that APs are present in dendritic shafts even under control conditions, but their rapid fusion to lysosomes hinders their detection. NMDA treatment in the presence of Baf led to a significant increase in GFP-LC3 puncta in the dendritic shaft compared with treatment with Baf alone (Fig. 6A,B). In addition, an increase in lysosome number in the dendritic shaft was detected after NMDA treatment (Fig. 6C), implying that the increase in lysosomes after NMDA treatment maintains the rapid dynamics of AP degradation. Interestingly, the number of GFP-LC3 puncta colocalized with the mCherry-β-actin signal did not increase after Baf alone or when NMDA treatment was combined with Baf (Fig. 6A,B).
Figure 6.

Detection of GFP-LC3 puncta in dendrites and spines after chem-LTD. Cultured hippocampal neurons were cotransfected at DIV9 with pmCherry-β-actin and pGFP-LC3, and treatments were performed at DIV20–26. A, Representative images of dendrites from fully mature pyramidal neurons after control, 2 h after 5 min of 20 μm NMDA, 100 nm bafilomycin A for 2.5 h (Baf), or combined NMDA and Baf (NMDA+Baf) treatments. Scale bars, 2 μm. B, Quantitation of results in A represented as the average number of GFP-LC3 puncta in the dendritic shaft or spines (colocalized with mCherry-β-actin)/10 μm dendritic length [n = 16–31 from 3 independent experiments each representing the average of 3–5 proximal dendrites (20–50 μm) per neuron; *p < 0.05; **p < 0.01; ***p < 0.001; one-way ANOVA with Tukey's post hoc test compared with control except where indicated]. C, Representative image of a neuronal dendritic shaft and lysosomes 2 h after 5 min of 20 μm NMDA (NMDA) or under control conditions (control). Lysosomes were detected by LysoTracker Red (LysoTR), and dendrite morphology was visualized using WGA-Alexa488 (WGA). Scale bar, 5 μm. Results were quantitated as the average number of LysoTR puncta in dendritic shaft/10 μm dendritic length [control, 1.17 ± 0.09; NMDA, 1.49 ± 0.06; p < 0.01, unpaired t test; n = 20–22 from 3 independent experiments, each representing the average of 3–8 dendrites (50–180 μm) per neuron]. Error bars indicate mean ± SE. n.s., No significant difference; Ctrl, control.
Discussion
This study demonstrated that KCl depolarization, as a general neuronal stimulation protocol, and the specific chem-LTD protocol induced autophagy with the involvement of NMDAR. However, our data do not exclude the induction of autophagy in response to other neuronal activity-related receptors or other forms of plasticity, such as LTP. In fact, we present some evidence that other voltage-gated receptors, e.g., voltage-gated calcium channel receptors, may induce autophagy, as APV only partially inhibited KCl-induced autophagy (Fig. 1B). Additional studies are needed to clarify precisely how neural activity regulates autophagy.
mTOR is known to contribute to synaptic plasticity and memory through the regulation of local translation (Hoeffer and Klann, 2010). In addition, here we demonstrate that mTOR also regulates chem-LTD-induced autophagy (Fig. 7A). Therefore, mTOR regulates protein turnover in neurons by functioning at the intersection between protein synthesis and degradation. Pharmacological reagents, such as rapamycin or WRT, modulate mTOR activity and are known to have roles in synaptic plasticity and memory (Dash et al., 2002; Cammalleri et al., 2003; Hoeffer et al., 2008). Their effects have been explained only on the basis of local translation, although these reagents are common autophagy modulators. Therefore, we suggest that mTOR may act as the switch between protein synthesis and degradation depending on the neural function involved, a speculation that requires further investigation in the context of synaptic plasticity and memory.
Figure 7.
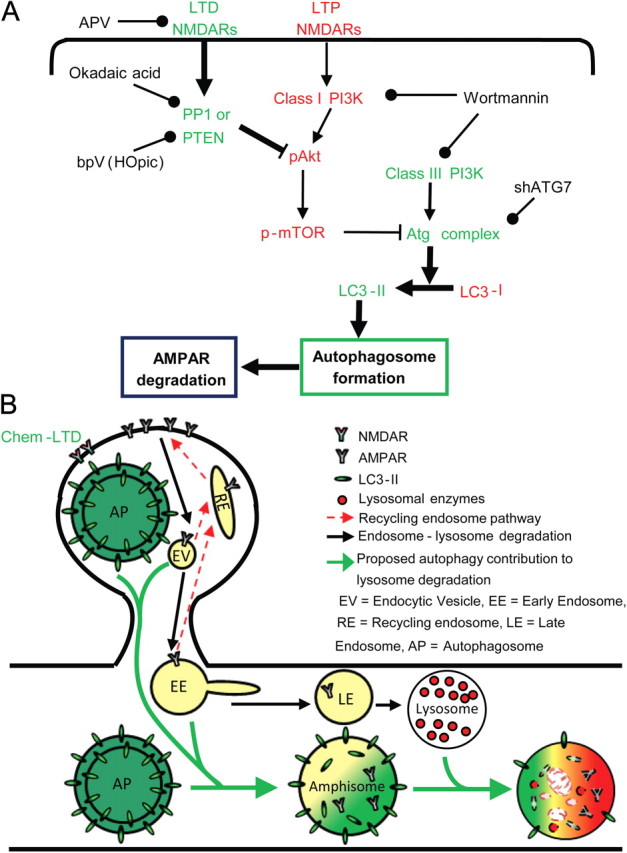
Proposed model for neuronal stimulation-induced autophagy. A, Regulation of chem-LTD autophagy induction; during LTD, NMDA receptors activate protein phosphatases, including protein phosphatase 1 and PTEN, which in turn dephosphorylate Akt and mTOR. This process deactivates mTOR and releases the inhibition of the Atg complex, thus increasing autophagosome formation, which may have a role in AMPAR degradation. Proteins printed in red share in inhibiting autophagy, and those in green share in autophagy induction. B, Possible scenario for autophagy contribution to AMPAR degradation after chem-LTD. Black arrows represent the known pathway for AMPAR degradation after endocytosis, where endocytic vesicles (EV) proceed to early endosomes (EE) and then late endosomes (LE) before final fusion to lysosomes and degradation by acid hydrolases. Red arrows represent the possible recycling of AMPAR from EV or EE back to the synapse through recycling endosomes (RE). Green arrows represent autophagosomes (AP) fusing with EV or EE to form amphisomes, before their final fusion to lysosomes, resulting in the formation of autolysosomes and degradation of the inner contents. Increased AP in dendrites and spines was observed after chem-LTD. AP may fuse with endosomes to increase their targeting for lysosome degradation (bold green arrows) and to decrease their possible recycling (dashed red arrows).
Despite having a common fate, endosomes and autophagosomes possess different origins and pathways, indicating that their role in synaptic plasticity should not be functional redundancy. However, dissociating their endosomal and autophagosomal roles, as in AMPAR degradation, may be challenging because of the lack of pharmacological inhibitors specific for each pathway. Most inhibitors used to study the endosomal pathway are also known to be autophagy inhibitors, such as chloroquine, ammonium chloride, and leupeptin (Klionsky et al., 2008). Previous studies revealed that these reagents rescued AMPAR from lysosomal degradation; however, they interpreted these data only on the basis of the endosomal role, probably because autophagy involvement in such processes had not yet been proposed (Ehlers, 2000; Lee et al., 2004). However, in the context of the work presented in this study, these previous studies may now be interpreted as an indirect proof of the contribution of autophagy to AMPAR degradation. The present study introduced genetic manipulation to specifically inhibit autophagy through knockdown of ATG7, which is involved in the autophagosome formation process but excluded from endocytosis. ATG7 knockdown resulted in partial recovery of GluR1 levels after chem-LTD, which was attributed to decreased autophagic activity as measured by LC3-II levels (Fig. 5E). These data indicate that autophagy takes a part in AMPAR degradation after chem-LTD. However, the fact that LC3-II levels returned to basal levels after ATG7 knockdown, and yet AMPAR degradation was only partially inhibited, suggests an autophagy-independent contribution and the possibility that autophagy contributes by enhancing the endosome–lysosome AMPAR degradation, as will be discussed below.
Furthermore, we demonstrated an increase in GFP-LC3 puncta in dendritic shafts and spines after chem-LTD that may account for the involvement of autophagy in the degradation of AMPAR. One possible scenario for this involvement may be a change in the kinetics of endosome cycling. AP fuses with endosomes to form amphisomes (Eskelinen, 2005; Mizushima, 2007), which dictate the final fate of endosome-to-lysosomal degradation (Fig. 7B). Increasing the number of AP decreases the possibility for recycling endosome formation and directs more AMPAR-containing endosomes to lysosomal degradation. Another possibility may involve p62 protein, also known as sequestosome 1 (SQSTM1), which is commonly found in inclusion bodies containing polyubiquitinated protein aggregates and is a common target of the AP (Klionsky et al., 2008; Mizushima et al., 2010). Interestingly, p62 is also important for LTP and spatial memory (Ramesh Babu et al., 2008). It interacts with AMPAR and is required for its trafficking (Jiang et al., 2009). It is thus probable that increased levels of AP trap AMPAR via p62 interaction with GluR1, a role that may accompany the previously suggested change in endosome recycling rate or work independently to mediate AMPAR degradation. This scenario could provide a deeper insight into the underlying mechanism if proven true in future studies.
In conclusion, we demonstrated that autophagy is regulated by neuronal activity and is a new form of regulated proteolysis. After neuronal stimulation by chem-LTD, autophagosome formation increases in pyramidal neuron dendrites and spines, contributes to directing internalized AMPAR toward lysosome degradation, and thus may be involved in the maintenance of LTD. Therefore, our study adds to the understanding of the physiological role of autophagy in neurons and proposes that autophagy is involved in synaptic plasticity. Whether autophagy plays such a physiological role in synaptic plasticity needs proof using electrophysiological experiments in slices or in vivo. Finally, because autophagy is now a candidate for the treatment of neurodegenerative diseases, autophagic inducers may be applied clinically for this purpose (Cheung and Ip, 2009; García-Arencibia et al., 2010). Understanding the precise mechanisms underlying autophagy in the brain and its effects on memory are of great clinical importance.
Footnotes
This work was supported by the CREST program of the Japan Science and Technology Agency; Grants-in-Aid for Scientific Research from the Ministry of Education, Science, Sports, Culture, and Technology of Japan; the Mitsubishi Foundation; and the Uehara Memorial Foundation to K.I. We thank Prof. M. Takahashi (Kitasato University, Kanazawa, Japan) for providing the rat β-actin cDNA and Prof. R. Tsien (University of California at San Diego, La Jolla, CA) for providing mCherry cDNA. We thank Prof. N. Mizushima (Tokyo Medical and Dental University, Tokyo, Japan) for valuable advice and discussions and Dr. T. Kitamura (University of Toyama, Toyama, Japan) for helpful support.
The authors declare no competing financial interests.
References
- Abraham WC, Williams JM. LTP maintenance and its protein synthesis-dependence. Neurobiol Learn Mem. 2008;89:260–268. doi: 10.1016/j.nlm.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Alirezaei M, Kemball CC, Flynn CT, Wood MR, Whitton JL, Kiosses WB. Short-term fasting induces profound neuronal autophagy. Autophagy. 2010;6:702–710. doi: 10.4161/auto.6.6.12376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- Bingol B, Sheng M. Deconstruction for reconstruction: the role of proteolysis in neural plasticity and disease. Neuron. 2011;69:22–32. doi: 10.1016/j.neuron.2010.11.006. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci. 2008;28:6926–6937. doi: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsello T, Croquelois K, Hornung JP, Clarke PG. N-methyl-d-aspartate-triggered neuronal death in organotypic hippocampal cultures is endocytic, autophagic and mediated by the c-Jun N-terminal kinase pathway. Eur J Neurosci. 2003;18:473–485. doi: 10.1046/j.1460-9568.2003.02757.x. [DOI] [PubMed] [Google Scholar]
- Bramham CR. Local protein synthesis, actin dynamics, and LTP consolidation. Curr Opin Neurobiol. 2008;18:524–531. doi: 10.1016/j.conb.2008.09.013. [DOI] [PubMed] [Google Scholar]
- Cammalleri M, Lütjens R, Berton F, King AR, Simpson C, Francesconi W, Sanna PP. Time-restricted role for dendritic activation of the mTOR-p70S6K pathway in the induction of late-phase long-term potentiation in the CA1. Proc Natl Acad Sci U S A. 2003;100:14368–14373. doi: 10.1073/pnas.2336098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti L, Eng J, Ivanov N, Garden GA, La Spada AR. Autophagy activation and enhanced mitophagy characterize the Purkinje cells of pcd mice prior to neuronal death. Mol Brain. 2009;2:24. doi: 10.1186/1756-6606-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung ZH, Ip NY. The emerging role of autophagy in Parkinson's disease. Mol Brain. 2009;2:29. doi: 10.1186/1756-6606-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Isaac JT, Wang YT. Receptor trafficking and synaptic plasticity. Nat Rev Neurosci. 2004;5:952–962. doi: 10.1038/nrn1556. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Peineau S, Howland JG, Wang YT. Long-term depression in the CNS. Nat Rev Neurosci. 2010;11:459–473. doi: 10.1038/nrn2867. [DOI] [PubMed] [Google Scholar]
- Dash PK, Mach SA, Blum S, Moore AN. Intrahippocampal wortmannin infusion enhances long-term spatial and contextual memories. Learn Mem. 2002;9:167–177. doi: 10.1101/lm.50002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Eskelinen EL. Maturation of autophagic vacuoles in mammalian cells. Autophagy. 2005;1:1–10. doi: 10.4161/auto.1.1.1270. [DOI] [PubMed] [Google Scholar]
- Fischer M, Kaech S, Knutti D, Matus A. Rapid actin-based plasticity in dendritic spines. Neuron. 1998;20:847–854. doi: 10.1016/s0896-6273(00)80467-5. [DOI] [PubMed] [Google Scholar]
- Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron. 2003;38:447–460. doi: 10.1016/s0896-6273(03)00206-x. [DOI] [PubMed] [Google Scholar]
- García-Arencibia M, Hochfeld WE, Toh PP, Rubinsztein DC. Autophagy, a guardian against neurodegeneration. Semin Cell Dev Biol. 2010;21:691–698. doi: 10.1016/j.semcdb.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Hegde AN, Goldberg AL, Schwartz JH. Regulatory subunits of cAMP-dependent protein kinases are degraded after conjugation to ubiquitin: a molecular mechanism underlying long-term synaptic plasticity. Proc Natl Acad Sci U S A. 1993;90:7436–7440. doi: 10.1073/pnas.90.16.7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde AN, Inokuchi K, Pei W, Casadio A, Ghirardi M, Chain DG, Martin KC, Kandel ER, Schwartz JH. Ubiquitin C-terminal hydrolase is an immediate-early gene essential for long-term facilitation in Aplysia. Cell. 1997;89:115–126. doi: 10.1016/s0092-8674(00)80188-9. [DOI] [PubMed] [Google Scholar]
- Hirling H. Endosomal trafficking of AMPA-type glutamate receptors. Neuroscience. 2009;158:36–44. doi: 10.1016/j.neuroscience.2008.02.057. [DOI] [PubMed] [Google Scholar]
- Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffer CA, Tang W, Wong H, Santillan A, Patterson RJ, Martinez LA, Tejada-Simon MV, Paylor R, Hamilton SL, Klann E. Removal of FKBP12 enhances mTOR-Raptor interactions, LTP, memory, and perseverative/repetitive behavior. Neuron. 2008;60:832–845. doi: 10.1016/j.neuron.2008.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsin H, Kim MJ, Wang CF, Sheng M. Proline-rich tyrosine kinase 2 regulates hippocampal long-term depression. J Neurosci. 2010;30:11983–11993. doi: 10.1523/JNEUROSCI.1029-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Parameshwaran K, Seibenhener ML, Kang MG, Suppiramaniam V, Huganir RL, Diaz-Meco MT, Wooten MW. AMPA receptor trafficking and synaptic plasticity require SQSTM1/p62. Hippocampus. 2009;19:392–406. doi: 10.1002/hipo.20528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado S, Benoist M, Lario A, Knafo S, Petrok CN, Esteban JA. PTEN is recruited to the postsynaptic terminal for NMDA receptor-dependent long-term depression. EMBO J. 2010;29:2827–2840. doi: 10.1038/emboj.2010.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S, Rodriguez-Navarro JA, Arias E, Kiffin R, Sahu S, Schwartz GJ, Cuervo AM, Singh R. Autophagy in hypothalamic AgRP neurons regulates food intake and energy balance. Cell Metab. 2011;14:173–183. doi: 10.1016/j.cmet.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, Bi X, Biard-Piechaczyk M, Blum JS, Bredesen DE, Brodsky JL, Brumell JH, Brunk UT, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- Lee SH, Simonetta A, Sheng M. Subunit rules governing the sorting of internalized AMPA receptors in hippocampal neurons. Neuron. 2004;43:221–236. doi: 10.1016/j.neuron.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Lee SH, Choi JH, Lee N, Lee HR, Kim JI, Yu NK, Choi SL, Kim H, Kaang BK. Synaptic protein degradation underlies destabilization of retrieved fear memory. Science. 2008;319:1253–1256. doi: 10.1126/science.1150541. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Koh JY. Roles of zinc and metallothionein-3 in oxidative stress-induced lysosomal dysfunction, cell death, and autophagy in neurons and astrocytes. Mol Brain. 2010;3:30. doi: 10.1186/1756-6606-3-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Jo J, Jia JM, Lo SC, Whitcomb DJ, Jiao S, Cho K, Sheng M. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell. 2010;141:859–871. doi: 10.1016/j.cell.2010.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Long-term potentiation–a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, Dipaola RS, Karantza-Wadsworth V, White E. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–1075. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey RM, Herron CE, Malenka RC. An essential role for protein phosphatases in hippocampal long-term depression. Science. 1993;261:1051–1055. doi: 10.1126/science.8394601. [DOI] [PubMed] [Google Scholar]
- Okada D, Ozawa F, Inokuchi K. Input-specific spine entry of soma-derived Vesl-1S protein conforms to synaptic tagging. Science. 2009;324:904–909. doi: 10.1126/science.1171498. [DOI] [PubMed] [Google Scholar]
- Okubo-Suzuki R, Okada D, Sekiguchi M, Inokuchi K. Synaptopodin maintains the neural activity-dependent enlargement of dendritic spines in hippocampal neurons. Mol Cell Neurosci. 2008;38:266–276. doi: 10.1016/j.mcn.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Ramesh Babu J, Lamar Seibenhener M, Peng J, Strom AL, Kemppainen R, Cox N, Zhu H, Wooten MC, Diaz-Meco MT, Moscat J, Wooten MW. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J Neurochem. 2008;106:107–120. doi: 10.1111/j.1471-4159.2008.05340.x. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Futter M, Jahreiss L, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Narayanan U, Renna M, Jimenez-Sanchez M, Sarkar S, Underwood B, Winslow A, Rubinsztein DC. Mammalian macroautophagy at a glance. J Cell Sci. 2009;122:1707–1711. doi: 10.1242/jcs.031773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland AM, Richmond JE, Olsen JG, Hall DH, Bamber BA. Presynaptic terminals independently regulate synaptic clustering and autophagy of GABAA receptors in Caenorhabditis elegans. J Neurosci. 2006;26:1711–1720. doi: 10.1523/JNEUROSCI.2279-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadasivan S, Zhang Z, Larner SF, Liu MC, Zheng W, Kobeissy FH, Hayes RL, Wang KK. Acute NMDA toxicity in cultured rat cerebellar granule neurons is accompanied by autophagy induction and late onset autophagic cell death phenotype. BMC Neurosci. 2010;11:21. doi: 10.1186/1471-2202-11-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal M. Dendritic spines and long-term plasticity. Nat Rev Neurosci. 2005;6:277–284. doi: 10.1038/nrn1649. [DOI] [PubMed] [Google Scholar]
- Sekiguchi M, Hayashi F, Tsuchida K, Inokuchi K. Neuron type-selective effects of activin on development of the hippocampus. Neurosci Lett. 2009;452:232–237. doi: 10.1016/j.neulet.2009.01.074. [DOI] [PubMed] [Google Scholar]
- Shacka JJ, Lu J, Xie ZL, Uchiyama Y, Roth KA, Zhang J. Kainic acid induces early and transient autophagic stress in mouse hippocampus. Neurosci Lett. 2007;414:57–60. doi: 10.1016/j.neulet.2006.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Ganetzky B. Autophagy promotes synapse development in Drosophila. J Cell Biol. 2009;187:71–79. doi: 10.1083/jcb.200907109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S, Hayashi Y, Esteban JA, Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 2001;105:331–343. doi: 10.1016/s0092-8674(01)00321-x. [DOI] [PubMed] [Google Scholar]
- Tai HC, Schuman EM. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat Rev Neurosci. 2008;9:826–838. doi: 10.1038/nrn2499. [DOI] [PubMed] [Google Scholar]
- Wu J, Dang Y, Su W, Liu C, Ma H, Shan Y, Pei Y, Wan B, Guo J, Yu L. Molecular cloning and characterization of rat LC3A and LC3B–two novel markers of autophagosome. Biochem Biophys Res Commun. 2006;339:437–442. doi: 10.1016/j.bbrc.2005.10.211. [DOI] [PubMed] [Google Scholar]
- Yao I, Takagi H, Ageta H, Kahyo T, Sato S, Hatanaka K, Fukuda Y, Chiba T, Morone N, Yuasa S, Inokuchi K, Ohtsuka T, Macgregor GR, Tanaka K, Setou M. SCRAPPER-dependent ubiquitination of active zone protein RIM1 regulates synaptic vesicle release. Cell. 2007;130:943–957. doi: 10.1016/j.cell.2007.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi JJ, Ehlers MD. Emerging roles for ubiquitin and protein degradation in neuronal function. Pharmacol Rev. 2007;59:14–39. doi: 10.1124/pr.59.1.4. [DOI] [PubMed] [Google Scholar]
- Yue Z, Friedman L, Komatsu M, Tanaka K. The cellular pathways of neuronal autophagy and their implication in neurodegenerative diseases. Biochim Biophys Acta. 2009;1793:1496–1507. doi: 10.1016/j.bbamcr.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]