

Abstract
Mutations in human parkin have been identified in familial Parkinson's disease and in some sporadic cases. Here, we report that expression of mutant but not wild-type human parkin in Drosophila causes age-dependent, selective degeneration of dopaminergic (DA) neurons accompanied by a progressive motor impairment. Overexpression or knockdown of the Drosophila vesicular monoamine transporter, which regulates cytosolic DA homeostasis, partially rescues or exacerbates, respectively, the degenerative phenotypes caused by mutant human parkin. These results support a model in which the vulnerability of DA neurons to parkin-induced neurotoxicity results from the interaction of mutant parkin with cytoplasmic dopamine.
Keywords: dopaminergic, Drosophila, monoamine, aging (ageing), neuroprotection, neuronal death
Introduction
The clinical features of Parkinson's disease (PD), including bradykinesia, rigidity, resting tremor, and postural impairment, are primarily resulting from the loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNc) (Fahn, 2003). Although the vast majority of PD cases are sporadic, insights from studies of rare inherited forms have provided opportunities to investigate the pathogenesis of idiopathic disease. Mutations in parkin are thought to be the second most common genetic cause of sporadic PD, after LRRK2/dardarin (Kitada et al., 1998; Foroud et al., 2003; Klein et al., 2003; Lincoln et al., 2003; Hedrich et al., 2004; Gilks et al., 2005; Hernandez et al., 2005). Parkin mutations originally were identified in families with autosomal recessive juvenile parkinsonism (AR-JP) (Kitada et al., 1998).
Although neuropathology is not limited to the nigrostriatal system, ablation of DA neurons in the SNc remains the hallmark of sporadic PD; yet, the reason that DA neurons are selectively vulnerable is unresolved. Dopamine is a highly reactive amine, and its oxidation generates neurotoxic quinones and reactive oxygen species (Stokes et al., 1999; Sulzer and Zecca, 2000). DA quinone spontaneously reacts with reduced sulfhydryl groups of cysteine, and recent results indicate that covalent modification of parkin by DA quinone reduces its E3 ligase activity and also decreases solubility (LaVoie et al., 2005). Cell culture models also suggest that dopamine and other potential oxidants modify parkin solubility and may reduce its neuroprotective effects (Sriram et al., 2005; Wang et al., 2005a,b). These data support the long-standing suspicion that dopamine itself may be the basis for the regional vulnerability seen in some forms of PD (Cookson, 2005). Mouse and Drosophila models used to test this hypothesis show variable degrees of pathology in DA neurons (Bonifati et al., 2003; Goldberg et al., 2003, 2005; Von Coelln et al., 2004; Chen et al., 2005; Menzies et al., 2005; Meulener et al., 2005; Perez and Palmiter, 2005).
Here, we use Drosophila to explore whether parkin mutations identified in familial PD can exert cell-specific toxic effects in vivo, suspending conventional notions regarding the genetics of parkin-linked AR-JP. Using histological and behavioral analysis, we demonstrate that expression of mutant but not wild-type human parkin in Drosophila induces progressive, age-dependent degeneration of DA neurons as well as motor dysfunction. Manipulation of dopamine storage using the Drosophila vesicular monoamine transporter (DVMAT) has profound effects on mutant parkin-induced toxicity. These in vivo data support a neurotoxic mechanism in which interaction of mutant parkin with dopamine underlies selective vulnerability of DA neurons in parkin-associated familial PD.
Materials and Methods
Molecular biology.
Plasmid cDNAs encoding wild-type human parkin, including an N-terminal Myc tag and two different FLAG-tagged mutant forms (Q311Stop and T240R) (Shimura et al., 2000), were subcloned as PmeI fragments into the StuI site using Exelixis (San Francisco, CA) modification of the Drosophila upstream activation sequence (UAS) expression vector (pEx-UAS) (Ollmann et al., 2000). The identity of all constructs was confirmed by sequencing. UAS-DVMAT has been described previously (Chang et al., 2006). To generate the DVMAT interference (DVMATi) construct, a modified hairpin-loop RNA interference (RNAi) procedure (Kalidas and Smith, 2002) was used. Two PCR products were ligated in head-to-tail (genomic DNA) and tail-to-head (cDNA) configurations. The genomic DVMAT fragment corresponded to exon 2–intron 2–exon 3–intron 3–exon 4–intron 4 (forward primer, 5′-tgcccatcatacccgagttcctg-3′; reverse primer, 5′-ctaagaaaaggttatgggttcaa-3′), whereas the cDNA used exons 2–4 (forward primer, 5′-tctagatgcccatcatacccgagttc-3′, underline indicates an XbaI site; reverse primer, 5′-ttatcgttgagaggaacatatcac-3′). These were engineered using PCR and subcloned into the pCRII-topoisomerase I (TOPO) vector (Invitrogen, San Diego, CA). The pCRII-TOPO–VMAT cDNA–exon 2–4 construct was then cut with XbaI and NotI and inserted into the pCRII-TOPO–VMAT genomic construct. The orientation and junctions of the inserts were confirmed by sequencing. A partial digest using EcoRI and XbaI yielded the complete VMATi construct, which was then subcloned into pEx-UAS.
Drosophila genetics.
Germline transformation followed standard methods (Rubin and Spradling, 1982; Spradling and Rubin, 1982), and multiple transgenic lines for each construct were obtained. Both immunohistochemical and immunoblotting studies confirmed increased expression using UAS-DVMAT (Chang et al., 2006) as well as decreased expression of DVMAT using DVMATi (supplemental Fig. 7, available at www.jneurosci.org as supplemental material). DOPA decarboxylase (ddc)-GAL4 was generously provided by Jay Hirsh (University of Virginia, Charlottesville, VA). UAS–green fluorescent protein (GFP)-based Ca2+ sensor (G-CaMP) was a gift from Richard Axel (Columbia University, New York, NY). UAS-mCD8-GFP and UAS–tetanus neurotoxin light chain (TNT) stocks were obtained from Larry Zipursky (University of California, Los Angeles, CA). Leo Pallanck (University of Washington, Seattle, WA) furnished the dpark mutants. appl-GAL4 was a kind gift from Kalpana White (Brandeis University, Waltham, MA). The UAS-Q108 stock was from Leslie Michels Thompson and Larry Marsh (University of California, Irvine, CA). Serge Birman (Universite de la Mediterranee, Marseille, France) generously provided the tyrosine hydroxylase (TH)-GAL4 line. The recombined choline acetyltransferase (chat)-GAL4, UAS-GFP chromosome, as well as 24B-GAL4, were from the Bloomington Drosophila Stock Center (Bloomington, IN). All experiments were performed at room temperature (∼24°C).
Immunohistochemistry.
Immunohistochemistry and TRITC (tetramethylrhodamine isothiocyanate)-phalloidin (Sigma, St. Louis, MO) staining were performed using whole-mount preparations as described previously (Greer et al., 2005; Sang et al., 2005; Chang et al., 2006). Each brain was scanned using optical sections covering 40 μm that included DA neurons of both dorsomedial (DM) and dorsolateral (DL) clusters. Each brain was examined with the examiner blinded to the genotype. The collected Z-series from each brain were then projected into a three-dimensional animation to precisely quantitate DA neuron numbers. We defined neuronal loss as a complete loss of somatic TH staining. To avoid confocal settings in different batches of samples that might artificially affect data interpretation, age-matched brains from different genotypes were examined simultaneously. Experiments in Figures 4 D and 7, D and E, as well as supplemental Figures 5B and 7, A and B (available at www.jneurosci.org as supplemental material), used alignment of one genotype in a row adjacent to samples of another genotype. Primary antibodies used were mouse anti-TH (1:100; Immunostar, Hudson, WA), rabbit anti-human parkin 2132 (1:50; Cell Signaling Technology, Danvers, MA), or mouse anti-parkin PRK8 (1:100; Abcam, Cambridge, MA), rat anti-5-hydroxytryptamine (5-HT; 1:50; Millipore, Bedford, MA), chicken anti-GFP (1:100; Millipore), rabbit anti-DVMAT (Chang et al., 2006), and mouse anti-FLAG M5 (1:50; Sigma). Fluorochrome-conjugated antibodies included FITC, Cy3, and Cy5 (1:100; Jackson ImmunoResearch, West Grove, PA) corresponding to the appropriate species of primary antibodies used. NIH Image was used to quantitate pixel intensity for fluorescence images. Toxicity of mutant compared with wild-type forms of parkin was confirmed using blinded analysis, and at least two independently transgenic insertions for each transgene were studied to avoid potential confounds resulting from positional effects (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material).
Figure 4.
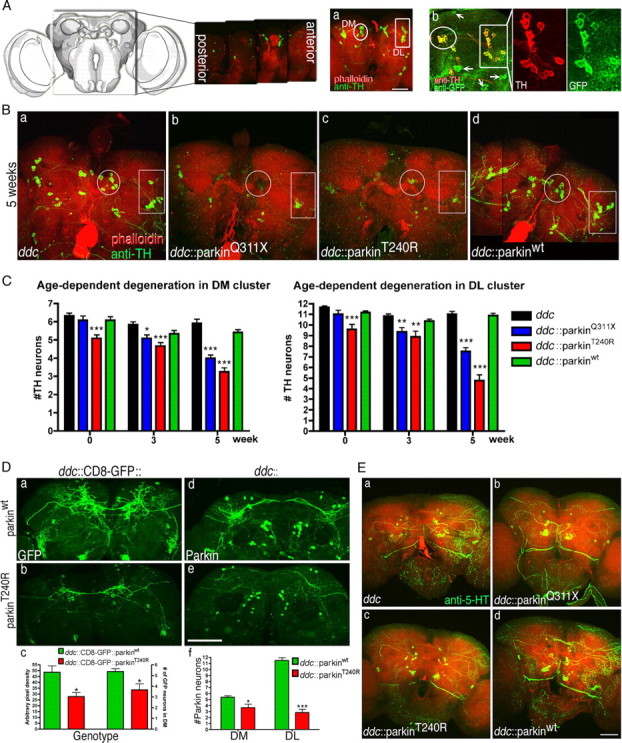
Expression of mutant human parkin in Drosophila DA neurons causes age-dependent neurodegeneration. A, Schematic representation of DA neurons in the central brain of Drosophila. a, The projected confocal image shows anti-TH and phalloidin staining spanning optical sections 40 μm in depth from posterior to anterior. Two major DA clusters, DM (circles) and DL (rectangles), are indicated. b, A projected image shows anti-TH (red) and anti-GFP (green) colocalized in all DA neurons in both DM and DL (enlarged images to the right) clusters. Arrows indicate 5-HT neurons expressing GFP but not TH. B, Reductions in TH immunoreactivity at 5 weeks after eclosion are apparent in ddc::parkinQ311X (b) and ddc::parkinT240R (c) but not ddc control (a) or ddc::parkinwt (d) brains. Circles, DM; rectangles, DL. C, Quantification of DA neurons at 0, 3, and 5 weeks after eclosion in DM and DL clusters. Progressive loss of TH-immunoreactive neurons is induced in both clusters by ddc::parkinQ311X (blue bars) and ddc::parkinT240R (red bars) compared with the ddc control (black bars) and ddc::parkinwt (green bars). Values represent the mean ± SEM; n = 12. *p < 0.05, **p < 0.01, ***p < 0.001 relative to the ddc control. D, At 4 weeks, mCD8-GFP signal (green) is reduced in parkinT240R (b) compared with parkinwt (a) brain, which is similar to changes observed in corresponding images showing reduced TH immunoreactivity. Measurement of GFP pixel density (corresponding to the left y-axis) and GFP cell counts in the DM cluster (corresponding to the right y-axis) from 4-week-old brains of both genotypes showed a significant decrease (unpaired t test; n = 5; *p < 0.05) of GFP signal in parkinT240R brains compared with parkinwt (c). The GFP signal at eclosion was indistinguishable between parkinT240R and parkinwt (data not shown). d, e, Confocal images of ddc::parkinT240R (e) and ddc::parkinwt (d) brains stained with an anti-parkin antibody (green) reveals reduced parkin signals in ddc::parkinT240R compared with ddc::parkinwt brains. f, Analysis of brains at 4 weeks indicates that cell counts stained with anti-parkin for parkinT240R are decreased compared with parkinwt (unpaired t test; n = 6; *p < 0.05 for DM; ***p < 0.001 for DL). Parkin staining at eclosion was indistinguishable between parkinT240R and parkinwt (data not shown). a, b, d, and e are confocal images of adjacent brains imaged simultaneously in the same slide but reoriented for presentation. E, Anti-5-HT staining at 5 weeks after eclosion shows reduced neuritic immunoreactivity in 5-HT neurons expressing mutant parkin (b, c) compared with those expressing wild-type parkin (d) or controls (a). Scale bar, 40 μm.
Figure 7.

DVMAT modulates mutant parkin-induced degeneration and motor phenotypes. A, Rotarod performance reveals early onset of postural instability in ddc::parkinT240R::DVMATi flies at 2 weeks (gray lines) compared with ddc::parkinT240R (red lines) and ddc::parkinT240R::DVMAT (green lines). At least six flies were scored for each genotype. B, The righting reflex demonstrates earlier onset of postural instability in ddc::parkinT240R::DVMATi flies (gray bars at 1 week) compared with ddc::parkinT240R (red bar at 1 week). At 2 weeks, the progressively impaired righting ability of ddc::parkinT240R (red bar) was ameliorated in ddc::parkinT240R::DVMAT flies (green bar), whereas ddc::parkinT240R::DVMATi flies continued to show a more pronounced motor deficit compared with ddc::parkinT240R (red bar). The values represent the mean ± SEM; n = 20. *p < 0.05, **p < 0.01 relative to control ddc::parkinT240R by one-way ANOVA with Bonferroni's multiple comparison correction. All genotypes at individual stages were compared with ddc::parkinT240R. C, Modulation of DVMAT affects the incomplete pupal lethality phenotype produced using two copies of ddc-GAL4 driver to express mutant parkin. Overexpression of parkinT240R (solid red bar) or parkinQ311X (solid blue bar) causes incomplete pupal lethality, whereas parkinwt (solid green bar) produces an ∼100% eclosion rate. Knockdown of DVMAT significantly worsens the parkinT240R lethality phenotype (red checked bar; *p < 0.05), whereas overexpressing DVMAT significantly suppresses both parkinT240R-induced (red striped bar; **p < 0.01) and parkinQ311X-induced (blue striped bar; *p < 0.05) pupal lethality. Modulation of DVMAT expression has no effect on wild-type parkin (green bars; one-way ANOVA with Bonferroni multiple comparison test; n ≥ 4). D, Confocal images of ddc::parkinT240R (a, b) or ddc::parkinwt (d, e) coexpressed with either DVMAT (a, d) or DVMATi (b, e) brains at 2 weeks. A marked reduction of TH immunoreactivity is observed in ddc::parkinT240R::DVMATi (b) compared with ddc::parkinT240R::DVMAT (a). At this stage, ddc::parkinT240R::DVMAT and ddc::parkinT240R brains are indistinguishable (data not shown). Quantitation of TH-positive neurons shows significant decreases in both DM and DL clusters in mutant parkin brains coexpressing DVMATi (c) (mean ± SEM; unpaired t test; n = 4; *p < 0.05; **p < 0.01). Modulation of DVMAT expression had no evident effect on wild-type parkin brains (d–f). Confocal images were reoriented from adjacent brains imaged simultaneously for paired comparison (a, b, d, e). E, Confocal images of ddc::parkinT240R (a) coexpressed with DVMAT (b) brains at 4 weeks. Significant increases in TH immunoreactivity are observed in ddc::parkinT240R::DVMAT (b) compared with ddc::parkinT240R (a). Quantitation of TH-positive neurons showed significant increases in both DM and DL clusters in mutant parkin brains coexpressing DVMAT (c) (mean ± SEM; unpaired t test; n = 8; **p < 0.01; ***p < 0.001). Confocal images were reoriented from adjacent brains imaged simultaneously for paired comparison.
Behavior.
Flies used for behavioral analysis were precisely aged and selected randomly. Control groups were included in each test. We used the startle-induced negative geotactic response and a countercurrent apparatus (Benzer, 1967) to test the climbing ability of the flies. For each time point, at least five cohorts of 16–18 flies from each genotype were scored. After transfer of flies to 14 ml Falcon plastic tubes, each tube was inserted into the countercurrent apparatus. Flies were gently knocked down to the bottom of tubes and allowed 10 s to climb up to the top receiver tubes. Flies failing to climb to the receiver tubes were trapped in the bottom tubes; flies were counted, and each test was repeated three times to deduce the percentage of flies that were able to reach the top. For the righting reflex assay, flies were transferred individually to small chambers [5 mm (diameter) × 2 mm (height)] and digitally recorded for 2 min after a 2 h recovery from CO2 anesthesia. Video clips documenting righting reflex episodes were then analyzed in a frame-by-frame manner. The function of the righting reflex was defined as the time between falling from the chamber roof and resumption of upright posture in 1/100 s units.
The Drosophila rotarod consists of six 25-mm-diameter glass cylinders that can be rotated between 3 and 15 rpm under the control of a motor, which requires flies to maintain an upside-down position while rotating. There are five infrared beams placed at 60, 120, 180, 240, and 300° around the tube that shine axially along the circumference of each cylinder. Beam breaks were automatically recorded using DAMSystem software (Trikinetics, Waltham, MA). The readout for flies that can adhere to the walls of the cylinder for a complete turn consists of five beam breaks. Flies with postural impairment generally fall before reaching the 180° beam and thus fail to break the beam at 180° and beyond. They may break 60 and 120° beams more frequently because they will encounter these beams more often during a complete rotation cycle after falls. For each test, individual flies were placed into chambers and recorded for 30 min, and the average number of beam breaks at each position was divided by the average frequency of beam breaks at all angles to obtain an arbitrary index. Data plotting and statistics were performed using Prism software (GraphPad Software, San Diego, CA).
Immunoblotting.
Protein preparation from Drosophila heads and SDS-PAGE was performed as described previously (Sang et al., 2005). Blots were probed using rabbit anti-parkin (1:2000; Cell Signaling Technology), mouse anti-parkin (PRK8, 1:2000; Abcam) (Pawlyk et al., 2003), mouse anti-FLAG M2 (1:200), and mouse anti-β-tubulin (1:1000; Accurate Chemical and Scientific, Westbury, NY). Immunoblots for DVMAT used anti-DVMAT (Chang et al., 2006) (1:2000) or anti-Drosophila vesicular glutamate transporter (1:10,000; provided by Aaron DiAntonio, Washington University, St. Louis, MO). Films from immunoblots were scanned with a Powerlook 1000 transmissive flatbed scanner (UMAX, Dallas, TX). Densities of bands were measured with NIH ImageJ (http://rsb.info.nih.gov/ij). Graphical and statistical analyses were performed using SigmaPlot 9.0 and SigmaStat 3.1 (Systat, San Jose CA.).
Neurochemical analysis.
Fly heads (three per sample) were manually collected and homogenized in 0.1 m perchloric acid containing 0.1% EDTA using a glass-on-glass microtissue grinder (Kimble/Kontes, Vineland, NJ). Insoluble debris were sedimented by centrifugation, and the supernatant was filtered through a Millipore MC cartridge. The filtrate was diluted 10-fold before analysis, and 5 μl of the diluted sample was analyzed using HPLC with electrochemical detection (Antec Leyden, Leiden, The Netherlands) using a mobile phase consisting of sodium acetate (75 mm), sodium dodecane sulfonate (0.75 mm), EDTA (10 mm), triethylamine (0.01%), acetonitrile (12%), methanol (12%), and tetrahydrofuran (1%), pH 5.5, pumped at a rate of 200 ml/min (model LC-10AD; Shimadzu, Columbia, MD) through a 100 × 2 mm column (3 μm, Hypersil C18; Keystone Scientific, Bellefonte, PA). The system was calibrated at regular intervals and provided a limit of detection of 0.5 fmol for a 5 μl injection of sample. The data were collected and analyzed using ChromPerfect software (Justice Innovations, Mountain View, CA).
Results
Generation of human parkin transgenic Drosophila
To test whether the expression of PD-linked parkin alleles might exert toxicity in DA neurons, parkin cDNAs encoding wild type and two mutant forms derived from familial PD, Gln311Stop (Q311X) and Thr240Arg (T240R) (Fig. 1) (Shimura et al., 2000), were subcloned into a Drosophila UAS expression vector, and transgenic lines were derived. To facilitate their detection, we generated transgenes expressing wild-type parkin tagged with either the myc or FLAG epitope. FLAG- and myc-tagged lines expressing wild-type parkin were behaviorally and histologically indistinguishable from each other and from controls (see Fig. 4 and supplemental Fig. 1, available at www.jneurosci.org as supplemental material), indicating that neither tag generates a neurotoxic phenotype and thus allowing the tags to be used interchangeably in transgenes expressing human parkin.
Figure 1.

Domain structure of human parkin and Myc-tagged wild-type and FLAG-tagged mutant constructs used to generate transgenic flies.
To drive expression in both DA and 5-HT neurons, we expressed parkin under the control of a ddc-GAL4 driver (Li et al., 2000) to generate ddc::parkin progeny. Importantly, 5-HT neurons in ddc::parkin flies provide an internal control for the neurochemical specificity of potential neurotoxic effects. Immunoblotting demonstrated expression for multiple lines of both wild-type (ddc::parkinwt) and mutant parkin (ddc::parkinQ311X and ddc::parkinT240R) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The expression level of the lines used was higher for parkinT240R than parkinwt for the majority of the experiments performed. Additional experiments using different lines with similar expression also obtained toxicity with T240R but no effect with wild-type parkin. ddc::parkinwt-41 (supplemental Fig. 2, lane 7, available at www.jneurosci.org as supplemental material) was expressed at levels comparable to ddc::parkinT240R-147 (supplemental Fig. 2, lane 10, available at www.jneurosci.org as supplemental material); however, this wild-type line was nontoxic, whereas the comparably expressed T20R line induced loss of DA neurons (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material).
Expression of mutant but not wild-type human parkin in aminergic neurons causes age-dependent motor deficits
Motor behavior has been used as an assay for DA cell function in other Drosophila models of neurodegeneration (Feany and Bender, 2000; Auluck and Bonini, 2002; Whitworth et al., 2005). Therefore, to assess potentially neurotoxic effects of parkin, we first subjected the parkin-expressing lines to a battery of three behavioral tests: (1) negative geotaxis to test climbing ability, (2) a righting reflex assay to test coordination, and (3) a novel Drosophila rotarod test to further quantitate motor function. As a positive control, we used ddc-GAL4 to express TNT to block aminergic neurotransmission; as expected, this caused severe motor phenotypes in multiple behavioral tests (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Using negative geotaxis, flies expressing wild-type and mutant parkin showed comparable climbing ability immediately after eclosion. However, between 3 and 6 weeks after eclosion, flies expressing parkinT240R or parkinQ311X showed dramatic declines in climbing ability relative to control ddc flies or flies expressing parkinwt (Fig. 2 A).
Figure 2.

Mutant parkin produces age-dependent deficits in motor performance. Three independent assays were used to analyze motor behavior. A, Using the ddc-GAL4 driver, parkinQ311X and parkinT240R produce age-dependent impairments in climbing performance compared with control and parkinwt. For each genotype at each time point, at least five cohorts consisting of 16–18 flies each were tested. B, At 5 weeks, the righting reflex test demonstrates postural instability of parkinQ311X and parkinT240R flies compared with parkinwt or control. Twenty flies of each genotype were analyzed. C, Rotarod performance demonstrates normal postural stability for all genotypes at 2 weeks. D, By 4 weeks, postural instability is apparent in parkinQ311X and parkinT240R compared with parkinwt and control flies; the <1 score for the arbitrary index at higher angles indicates flies that fall more frequently. For each genotype at each time point, ≥12 flies were tested. Values shown represent mean ± SEM. **p < 0.01, ***p < 0.001 relative to control ddc [two-way ANOVA with Bonferroni's multiple comparison test (A, C, D) or unpaired t test (B)].
We next tested motor behavior using the righting reflex, which measures the time between a spontaneous fall from the testing chamber lid and successful attainment of a normal posture (Leal and Neckameyer, 2002). Consistent with the climbing assay, aged ddc::parkinT240R and ddc::parkinQ311X flies showed greater difficulty in postural control and coordination compared with control and ddc::parkinwt flies (Fig. 2 B). As an automated assay for postural impairment, we designed a Drosophila rotarod (supplemental Fig. 4, available at www.jneurosci.org as supplemental material) (see Materials and Methods). Briefly, the Drosophila rotarod comprises six small cylinders rotated at 10 rpm in which the flies stand on the internal surface. Flies that successfully adhere to the chamber wall for a complete turn break five successive infrared beams, generating a readout consisting of a flat line at the arbitrary index of 1.0. Flies that fall can either break the beams prematurely or fail to break them; both generate a readout that deviates from 1.0 at the angle (shown on the x-axis; see Materials and Methods for additional details). At 2 weeks, the frequency of beam breaks at different angles was identical, indicating that young flies expressing both wild-type and mutant parkin performed equivalently in the rotarod assay (Fig. 2 C). However, by 4 weeks the behavior of ddc::parkinT240R and ddc::parkinQ311X was markedly impaired relative to ddc::parkinwt (Fig. 2 D). These data indicate that mutant but not wild-type parkin causes a defect in motor behavior when expressed in aminergic cells. Moreover, this deficit was not manifest until a minimum of 2–3 weeks after eclosion, suggesting a degenerative rather than a developmentally induced defect.
To determine more specifically whether these behavioral deficits were caused by changes in the function of aminergic cells, we first assayed cell function at early time points, before the manifestation of behavioral deficits. In PD, it is likely that neuronal dysfunction precedes cell loss, but it has been difficult to assess this possibility experimentally. The fluorescent calcium probe G-CaMP has been used to monitor neuronal activity in the Drosophila olfactory system in vivo (Wang et al., 2003). We therefore used this technique to investigate the function of aminergic neurons in flies expressing mutant parkin. We observed G-CaMP fluorescence in 1-week-old brains expressing G-CaMP with either parkinT240R or parkinwt. Compared with the ddc::G-CaMP control (Fig. 3 A), ddc::parkinT240R::G-CaMP brains showed decreased fluorescence, indicating reduced neuronal activity (Fig. 3 B,D). In contrast, the signal in ddc::parkinwt::G-CaMP brains was indistinguishable from controls (Fig. 3 A,D). These data confirm behavioral tests showing that wild-type parkin does not adversely affect the function of aminergic neurons in the fly. More importantly, they indicate that mutant parkin causes dysfunction of these cells, consistent with the behavioral deficits we observe.
Figure 3.
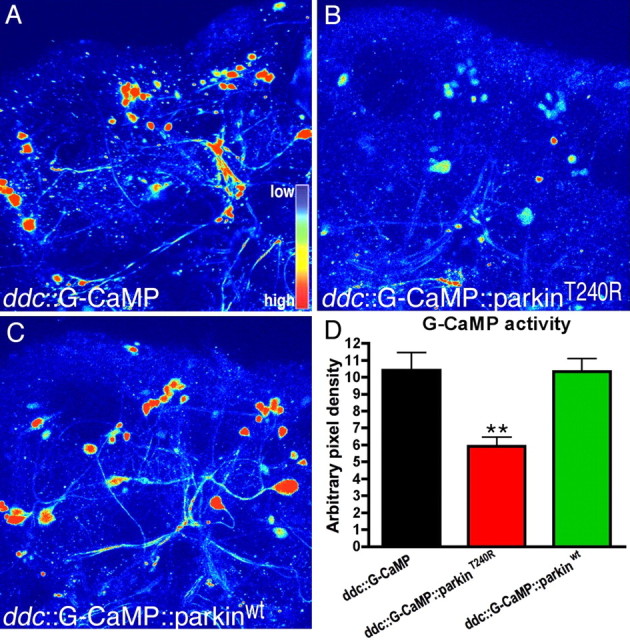
Analysis of activity-dependent G-CaMP signal in living brains at 1 week after eclosion. GFP signal intensity was converted into thermally coded color images. A, In the control ddc::G-CaMP brain, intense GFP signal was detected. B, parkinT240R expressed in ddc::G-CaMP flies resulted in markedly reduced GFP signal. C, Expression with parkinwt resembled the control. D, Analysis of GFP pixel density of at least five brains for each genotypes showed decreased G-CaMP activity in the parkinT240R brain (one-way ANOVA with Bonferroni's multiple comparison test; n ≥ 5; **p < 0.01). The images are represented using a pseudocolor scale as indicated in A, where blue is low intensity and yellow–red is high intensity. Scale bar, 40 μm.
The hallmark of PD and the gold standard for most animal models of PD is the degeneration of DA neurons. To assay for DA cell death in flies expressing wild-type and mutant parkin, we used whole-mount staining of adult brains in conjunction with confocal analysis and three-dimensional reconstructions. We focused on DA neurons in the DM and DL clusters, which are more easily counted than other more numerous or widely dispersed clusters, and have been used extensively in other Drosophila models of PD (Feany and Bender, 2000; Auluck et al., 2002; Yang et al., 2003; Chen and Feany, 2005; Meulener et al., 2005; Whitworth et al., 2005). Using an anti-GFP antibody to recognize mCD8-GFP (Lee and Luo, 2001), we found that ddc-GAL4 drives expression in all DA neurons in both DM and DL clusters (Fig. 4 A), indicating that all of these cells have the potential to be affected by mutant parkin.
We monitored the viability of DA neurons in brains expressing mutant and wild-type parkin driven by ddc-GAL4 in three ways: (1) TH staining, (2) coexpression of mCD8-GFP, and (3) staining with an antibody to human parkin. The collected Z-series from brains were projected into a single plane image (Fig. 4 Aa), and additional three-dimensional animations were conducted to precisely quantify DA neuron numbers. Consistent with our behavioral results, TH immunoreactivity was similar in flies expressing wild-type and mutant parkin at eclosion, albeit with a modest but significant decrease in DA cell counts in flies expressing parkinT240R. In contrast, beginning 3 weeks after eclosion and increasing with age, we observed robust and consistent declines in TH staining in brains expressing mutant parkin compared with parkinwt transgenics or ddc-GAL4 controls (Fig. 4 Ba–Bd,C) (supplemental Fig. 5A, available at www.jneurosci.org as supplemental material). Similarly, using the TH-GAL4 driver, which is expressed solely in DA neurons (Friggi-Grelin et al., 2003), we observed a decline in TH-staining flies expressing parkinT240R but not parkinwt (supplemental Fig. 5B, available at www.jneurosci.org as supplemental material). Consistent with the relative severity of their behavioral effects, parkinT240R (using either ddc- or TH-GAL4) caused more pronounced DA neuronal loss than parkinQ311X. We also observed a loss of DA neurons in clusters other than DM and DL (data not shown). However, the relatively tight apposition of the cells in these clusters limited our ability to precisely quantitate cell loss.
The loss of TH immunoreactivity is commonly interpreted as an indication of cell loss. However, TH is subject to extensive post-translational regulation (Flatmark et al., 2002). Therefore, to determine whether the decrease in TH staining that we observed was the result of cell loss or TH downregulation, we used an independent label for DA neurons. We expressed mCD8-GFP in ddc::parkinwt and ddc::parkinT240R flies, and brains were examined side by side on the same slide. Quantification of fluorescent pixels revealed a significant decrease in GFP signal by 4 weeks in parkinT240R-expressing DA neurons compared with those expressing parkinwt (Fig. 4 Da–Dc), indicating that the decrease in TH staining was indeed the result of cell loss. We also quantitated the number of mCD8-GFP-expressing neurons in the DM cluster and observed significant cell loss for ddc::parkinT240R compared with ddc::parkinwt; results obtained using cell counts correlated well with those that used pixel intensity (Fig. 4 Dc). Finally, we found that ddc::parkinT240R but not ddc::parkinwt flies showed an age-dependent reduction in anti-parkin staining in DA neurons 4 weeks after eclosion (Fig. 4 Dd–Df). Together, these data suggest that expressing mutant human parkin in Drosophila causes DA cells to lose function and eventually degenerate.
DA neurons are selectively vulnerable to mutant human parkin
Having established that mutant parkin causes degeneration of DA neurons, we next determined whether this model might also show selective vulnerability as seen in PD. Because ddc-GAL4 drives expression of the parkin transgenes in 5-HT as well as DA neurons, we first examined whether mutant parkin might also affect 5-HT neurons. Brains staged 5 weeks after eclosion and expressing either wild-type or mutant parkin were stained with an anti-5-HT antibody. Small declines in 5-HT immunoreactivity were observed in brains expressing mutant parkin, with some reductions in the staining intensity of neuritic processes (Fig. 4 E). However, in contrast to the profound degeneration of DA neuronal somata observed at this stage (Fig. 4 B), the somata of 5-HT neurons in brains expressing mutant parkin resembled controls. Thus, 5-HT neurons are somewhat susceptible to mutant parkin, but to a lesser extent than DA neurons.
We also studied expression of parkin using the pan-neuronal driver appl-GAL4 (Torroja et al., 1999). Flies expressing either wild-type or mutant parkin under control of appl-GAL4 behaved normally at eclosion. However, by 5 weeks, the rotarod assay showed significant motor impairment of appl::parkinT240R flies, with more modest changes in appl::parkinQ311X flies compared with appl::parkinwt and controls (Fig. 5 A). Analysis of brains at this stage revealed reductions in TH immunoreactivity for appl::parkinT240R in both DM and DL clusters compared with appl::parkinwt and control brains (Fig. 5 Ba–Bd). No obvious pathology such as vacuolization, which is commonly observed in fly models of neurodegenerative disease (Jackson, 2000; Wittmann et al., 2001; Jackson et al., 2002), was observed in brains when parkinT240R was expressed using this pan-neural driver. Changes in TH staining in appl::parkinQ311X could not be detected, consistent with the lower level of toxicity seen with this mutant using the ddc-GAL4 driver. Given that appl-GAL4 also drives expression in photoreceptor neurons, retinal morphology was examined at 5 weeks, but degenerative changes were not detected (Fig. 5 Be–Bh), suggesting that these histaminergic photoreceptor neurons (Stuart, 1999) are resistant to mutant parkinT240R compared with DA neurons in the central brain.
Figure 5.

Pan-neuronal expression of parkinT240R induces motor dysfunction and degeneration of DA neurons, whereas photoreceptor neurons are resistant. A, Rotarod assays at 5 weeks demonstrate postural instability of appl:: parkinT240R flies, which fall more frequently at higher angle positions compared with the appl-GAL4 control and appl::parkinwt. appl:: parkinQ311X flies show a modest trend toward postural instability. For each genotype at each time point, >12 flies were scored. The values shown represent mean ± SD. ***p < 0.001 relative to control appl (two-way ANOVA with Bonferroni's multiple-comparison test). Ba–Bd, Confocal images of brains aged 5 weeks stained with anti-TH (green) and phalloidin (red). Circles, DM; rectangles, DL. Reductions in TH immunoreactivity are apparent in the DL cluster in appl::parkinT240R brain (c) compared with appl (a), appl::parkinQ311X (b), and appl::parkinwt (d). Arrowheads indicate reduced TH-immunoreactive processes (c) in brains expressing mutant parkin compared with wild type (d). Be–Bh, No differences in morphology of rhabdomeres within individual ommatidia morphology occur in ddc control (e) or flies expressing parkinQ311X (f), parkinT240R (g), or parkinwt (h). Scale bar, 40 μm.
Also, to test whether DA neurons were selectively susceptible to degeneration, we used chat-GAL4 to drive transgene expression in cholinergic cells (Yasuyama and Salvaterra, 1999). As a positive control for neurodegeneration, flies expressing an expanded polyglutamine peptide, Q108 (Marsh et al., 2000) were examined in parallel. chat::Q108 flies died at a late pupal stage, and brains showed dramatically decreased GFP-labeled cells (Fig. 6 Ae–Af). In contrast, chat::parkinQ311X and chat::parkinT240R flies showed normal behavior at eclosion, similar to flies expressing parkinwt. Furthermore, as late as 6 weeks after eclosion, brains expressing either mutant or wild-type parkin showed comparable GFP-labeled cholinergic neurons compared with the chat::GFP control (Fig. 6 Aa–Ad). Similarly, using GMR-GAL4 (Freeman, 1996), we observed degeneration of neuronal, histaminergic photoreceptor cells expressing Q108 (Marsh et al., 2000) but no effects in histaminergic photoreceptors (Stuart, 1999) expressing either mutant or wild-type parkin (data not shown). The lack of mutant parkin-induced degeneration using chat-GAL4 or GMR-GAL4 drivers was not attributable to lower expression, because quantitative immunoblots demonstrate higher expression of parkinT240R in histaminergic and cholinergic neurons compared with the ddc driver (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). These data indicate that the toxicity of mutant parkin is either limited to DA neurons or significantly more potent in these cells.
Figure 6.
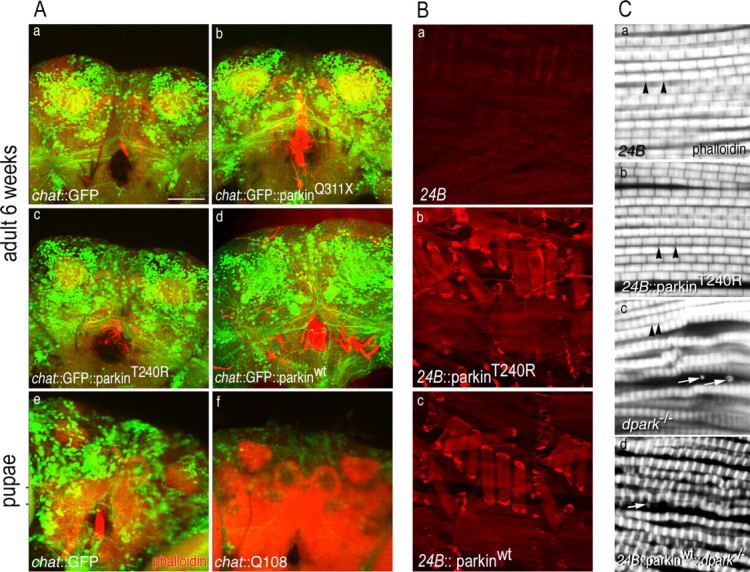
Cholinergic neurons and indirect flight muscle are resistant to mutant parkin. A, Images show signals for TRITC (tetramethylrhodamine isothiocyanate)-phalloidin (red) and GFP (green). a–d, Confocal images of chat::GFP brains coexpressing mutant or wild-type parkin transgenes at 6 weeks. Expression of either parkinQ311X (b) or parkinT240R (c) has no effect on GFP signal that is distinguishable from the expression of parkinwt (d) or control (a). e, f, In contrast to the robust fluorescence of mutant parkin brains (a–d), expression of a toxic polyglutamine construct, Q108 (f), markedly reduces GFP signal in midpupal brains compared with controls (e). (Images show pupal brains for Q108 as expression of this transgene is late pupal lethal.) B, Staining of larval muscle verified expression of human parkin under control of 24B-GAL4. a–c, The driver-alone control (a) shows only background staining with PRK8 monoclonal antibody, whereas a robust signal in larval muscle is observed for both mutant (b) and wild-type human parkin transgenes (c). C, Confocal image of sarcomeres from indirect flight muscle stained with phalloidin. a, b, 24B-GAL4 control (a) and 24B::parkinT240R (b) show normal organization of sarcomeres. Black arrowheads show normal Z-bands. c, The control dpark null mutant shows abnormal deposition of actin-containing debris in indirect fly muscle (white arrows) with irregularly organized sarcomeres. d, 24B-GAL4::parkinwt fails to rescue the muscle phenotype of the dpark null mutant. The white arrow shows abnormal debris. Scale bar, 40 μm.
Despite the cell-type specificity we observe, one potential mechanism for the degenerative phenotypes caused by mutant parkin is transdominant inhibition of the endogenous fly homolog. To test this hypothesis, we used the driver 24B-GAL4 (Brand and Perrimon, 1993) to express both mutant parkinT240R (24B::parkinT240R) and parkinQ311X (24B::parkinQ311X) in muscle cells. We verified robust expression of both wild-type and mutant transgenes in third instar larval muscle (Fig. 6 Ba–Bc). Unlike dpark−/− adult escapers, which were unable to fly, 24B::parkinT240R and 24B::parkinQ311X flies showed normal flight behavior (data not shown). Furthermore, as opposed to the abnormal morphology of indirect flight muscle from dpark mutants (Fig. 6 Cc), 24B::parkinT240R muscle showed normal myofibril organization (Fig. 6 Cb), indicating that misexpression of mutant human parkin in muscle does not interfere with function of endogenous fly parkin. Interestingly, in contrast to a report showing that expression of wild-type Drosophila parkin using 24B-GAL4 could rescue the dpark null phenotype (Greene et al., 2003; Pesah et al., 2004), we found that wild-type human parkin did not rescue either the muscle phenotype (Fig. 6 Cd) or flightless behavior of dpark null flies (data not shown). These data suggest potentially important functional differences between fly and human parkin.
Modulation of VMAT expression affects mutant parkin-induced neurodegeneration and motor behaviors
Given the selective vulnerability of DA neurons to mutant parkin, we hypothesized that mutant human parkin-induced toxicity may depend on dopamine or its metabolites and that alterations in DA homeostasis could modulate parkin-induced phenotypes. VMAT is known to play a crucial role in regulating the amount of dopamine that is stored in synaptic vesicles or remains in the cytosol to participate in oxidative and potentially neurotoxic processes (Fumagalli et al., 1999; Pothos et al., 2000; Sulzer et al., 2000; Weingarten and Zhou, 2001; Hansen et al., 2002; Choi et al., 2005). Similar to mammalian orthologs, DVMAT is responsible for transporting dopamine and other monoamines into synaptic vesicles (Greer et al., 2005; Chang et al., 2006). Furthermore, overexpression of DVMAT causes an increase in DVMAT levels and behavioral phenotypes consistent with an increase in dopamine release (Chang et al., 2006). To complement the UAS-DVMAT lines, we made a DVMAT RNAi construct and confirmed the efficacy of ddc::DVMAT-RNAi in specifically reducing endogenous DVMAT expression (supplemental Fig. 7, available at www.jneurosci.org as supplemental material). ddc::DVMAT also produced a modest but significant decrease in total dopamine concentrations as assessed using HPLC, whereas ddc::DVMATi knockdown markedly increased total dopamine in extracts of fly heads (supplemental Fig. 8, available at www.jneurosci.org as supplemental material). These and other previously published data on the phenotype of DVMAT-overexpressing flies confirm that DVMAT causes changes in dopamine storage and release (Chang et al., 2006).
We first tested whether changes in DVMAT expression also modified mutant parkin behavioral phenotypes. We found that flies coexpressing DVMAT RNAi with mutant parkin (ddc::parkinT240R::DVMATi) showed an earlier onset of postural impairment as assessed by rotarod performance (Fig. 7 A). The righting reflex proved to be the most sensitive behavioral assay to detect modulation of mutant parkin effects by DVMAT (Fig. 7 B). At 1 week, ddc::parkinT240R::DVMATi flies (Fig. 7 B, gray bar) took longer to right themselves compared with those expressing mutant parkin alone (red bar). Flies expressing DVMATi alone (pink bar) did not differ significantly from those expressing mutant parkin alone. At 2 weeks, overexpression of DVMAT provided protection against effects of mutant parkin without significantly affecting behavior on its own; the impaired righting ability of ddc::parkinT240R flies (red bar) was suppressed in ddc::parkinT240R::DVMAT flies (green bar). In contrast, overexpression of DVMAT alone (blue bar) did not affect performance in this assay and was similar to the rescued parkin phenotype (green bar).
Dramatic changes in aminergic function can be lethal in Drosophila (Neckameyer and Quinn, 1989; McClung and Hirsh, 1998; Stathakis et al., 1999). Consistent with this observation, we found that expressing high levels of parkinT240R or parkinQ311X using two rather than one copy of ddc-GAL4 causes incomplete pupal lethality. This phenotype is easily quantitated and was therefore used to further evaluate the genetic interaction between DVMAT and parkin. Importantly, overexpression or knockdown of DVMAT alone using two copies of ddc-GAL4 had no detectable effects on lethality (data not shown). Changes in DVMAT expression had no effect on flies expressing wild-type parkin (Fig. 7 C, green bars). In contrast, overexpression of DVMAT significantly suppressed both parkinT240R- and parkinQ311X-induced pupal lethality; conversely, DVMAT knockdown enhanced parkinT240R-induced lethality (Fig. 7 C, red and blue bars). These data support a genetic interaction between DVMAT and mutant parkin and suggest that DA homeostasis might be an important factor mediating the neurotoxic effects of mutant parkin.
Others, using rotenone (Pendleton et al., 2002; Coulom and Birman, 2004) or α-synuclein to model PD in the fly, have shown that enhancing DA synthesis, and presumably release, can rescue the phenotype caused by DA cell loss without necessarily decreasing the degeneration of DA cells (Coulom and Birman, 2004). In these cases, as in the symptomatic treatment of PD, increased release by the remaining cells presumably compensates for the death of other DA neurons. Therefore, to assess more specifically the effects of DVMAT on DA cell degeneration, we performed a quantitative immunohistochemical analysis of flies coexpressing parkin and VMAT transgenes. Importantly, neither UAS-DVMAT nor DVMAT RNAi alone altered the survival of DA neurons in aged flies (data not shown). Furthermore, manipulation of DVMAT expression using either UAS-DVMAT or DVMAT RNAi had no effect on the number of DA neurons in aged flies expressing wild-type human parkin (ddc::parkinwt in Fig. 7 Dd–Df). In contrast, coexpressing DVMATi and parkinT240R using ddc-GAL4 potentiated the neurodegenerative phenotype of parkinT240R; the number of TH-positive neurons was significantly reduced in ddc::parkinT240R::DVMATi at 2 weeks compared with those without DVMATi or overexpressing DVMAT (Fig. 7 Da–Dc). In addition, we observed that increased DVMAT expression in mutant parkin brains suppressed mutant parkin-induced DA neurodegeneration; the loss of TH-positive neurons evident in ddc::parkinT240R brain at 4 weeks (Fig. 7 Ea,Ec) was rescued by coexpression of parkinT240R with UAS-DVMAT (Fig. 7 Eb,Ec). Thus, modulation of VMAT activity may significantly affect mutant parkin-induced degeneration of DA neurons, and such genetic interaction might, at least in part, underlie regional vulnerability in PD.
Discussion
Here, we report that expression of mutant but not wild-type human parkin in Drosophila results in progressive, age-dependent dysfunction and degeneration of DA neurons. Degeneration is less marked in 5-HT neurons and does not occur in histaminergic or cholinergic neurons expressing mutant parkin. The selective vulnerability of DA neurons to mutant parkin is likely to be associated with dopamine itself and/or its metabolites, because genetic manipulation of VMAT affects mutant parkin-induced neurodegeneration and motor dysfunction.
Relationship between parkin mutations and cell death
The discovery of monogenic forms of PD has opened new avenues toward understanding disease pathogenesis. The linkage of parkin to PD was originally described in families with inherited AR-JP (Kitada et al., 1998). Although the vast majority of parkin-related PD cases are inherited recessively, an increasing number of reports identify a proportion of PD patients in which only a single copy is mutated (Leroy et al., 1998; Maruyama et al., 2000; Farrer et al., 2001; Lucking et al., 2001; West et al., 2002; Foroud et al., 2003; Tan et al., 2003; Mata et al., 2004). Given the complexity of analyzing the 1.3 Mb parkin genomic locus, it is possible that a proportion of these apparent dominant cases represent a failure to ascertain a second site mutation. Nonetheless, several reports have identified abnormalities in Fluoro-DOPA positron emission tomography (Hilker et al., 2001; Khan et al., 2002, 2005; Scherfler et al., 2004) and 99mTc-TRODAT-1 single-photon emission computed tomography scans (Shyu et al., 2005) in heterozygous parkin mutant “carriers.” Intriguingly, functional magnetic resonance imaging has also identified motor reorganization in otherwise asymptomatic parkin mutation carriers (Buhmann et al., 2005). One plausible explanation for such findings is that heterozygosity for parkin mutations increases the susceptibility of DA neurons to other environmental or genetic factors; however, this remains to be proven. Nonetheless, it is clear that heterozygous parkin mutations are an important determinant of age of onset (Sun et al., 2006). The observation of affected PD patients carrying a combination of both normal and mutant parkin alleles suggests that haploinsufficiency might serve as a risk factor for disease. An alternative explanation for single-copy parkin mutations that give rise to symptomatic PD is that certain mutations confer dominant-negative or toxic gain of function properties.
Cell culture and in vitro studies indicate that parkin functions as an E3 ligase (Giasson and Lee, 2001, 2003; Hattori and Mizuno, 2004; Moore et al., 2005), although additional roles in microtubule-based transport (Ren et al., 2003) and regulation of dopamine transporter activity have been suggested (Jiang et al., 2004). It is generally thought that the loss of E3 ligase activity is involved in the pathogenesis of parkin-linked PD. Recently, Brice and colleagues (Lesage et al., 2007) identified a family with compound heterozygous and homozygous deletions of the parkin promoter, resulting in a complete absence of parkin transcripts; these patients are clinically indistinguishable from those bearing other severe mutations in the parkin gene. Clearly, DA cell death in humans can result from a complete absence of parkin activity. However, additional mechanisms may contribute to at least some neurotoxic processes associated with mutant forms of parkin. It is interesting to note that, apart from the neurodegeneration in locus ceruleus identified in parkin exon 7 knock-out mice reported by Dawson and coworkers (Von Coelln et al., 2004), a number of mouse genetic strategies have failed to recapitulate the neuronal cell death that is characteristic of PD (Goldberg et al., 2003; Perez and Palmiter, 2005). Second, many PD-linked parkin mutants, including parkinT240R and parkinQ311X, retain substantial E3 ligase activity, and some show a surprising increase in ligase activity (Zhang et al., 2000; Chung et al., 2001; Wang et al., 2005a). Third, although some proposed substrates such as Pael-R accumulate in postmortem AR-JP brains (Imai et al., 2001; Shimura et al., 2001), parkin knock-out mice do not show elevated steady-state concentrations of other substrates such as CDCrel-1 and synphilin-1 (Von Coelln et al., 2004). Fourth, many parkin mutations show altered solubility and form aggresome-like structures in transfected cells (Ardley et al., 2003; Cookson et al., 2003; Gu et al., 2003; Muqit et al., 2004; Henn et al., 2005; Wang et al., 2005a), thereby conferring potentially toxic properties.
Our data suggest that although a dominant mechanism could contribute to the pathological phenotypes caused by mutant parkin in Drosophila, it is unlikely that the phenotypes we observe are the result of a dominant-negative effect. Others have found that the Drosophila parkin homolog (dparkin) loss-of-function (null) flies show degeneration of indirect flight muscle (Greene et al., 2003). If human parkin mutants were capable of exerting a dominant negative effect on the endogenous fly ortholog, their misexpression in muscle would likely phenocopy the effects of the dparkin null mutation. However, we failed to detect any histological abnormalities in indirect flight muscle from 24B::parkinT240R flies. Although it is possible that the use of a different driver line or multiple copies of the parkin transgene could produce effects in muscle, this experiment argues against a transdominant effect of mutant human parkin. Moreover, overexpression of wild-type fly parkin cannot rescue the pupal lethality phenotype of ddcX2::parkinT240R (data not shown), and wild-type human parkin cannot rescue the muscle phenotype of dparkin mutant animals. Although additional effort is required to completely exclude the possibility that misexpression of human parkin affects the function of endogenous parkin in the fly, our observations suggest that mutations of endogenous fly parkin and the expression of mutant human forms of parkin that we report here may confer cytotoxicity by different mechanisms.
Role of dopamine in mutant parkin-associated neurodegeneration
Most motor symptoms of PD are the result of progressive degeneration of DA neurons originating in the substantia nigra. The molecular pathways that lead to the death of this population of DA neurons are not known, and understanding the basis of selective mesencephalic DA neuron vulnerability may aid the rational design of therapeutics. Because all identified PD-linked genes are expressed ubiquitously in the CNS, it is unclear why such mutations give rise to selective pathology in the nigrostriatal system. It has been suggested that the pathogenesis of monogenic and perhaps idiopathic PD might involve proteins or neurochemicals that are particularly abundant in DA neurons. In vertebrates, nigral DA neurons are characterized by a distinct set of proteins that play a role in DA synthesis and metabolism, such as TH, Ddc, monoamine oxidase (MAO), and the plasma membrane dopamine transporter, as well as other proteins such as VMAT that may be differentially expressed (Vernier et al., 2004). And of course, unlike other cells, DA neurons also store and release dopamine. The neurotoxic effects of dopamine may be mediated through general oxidative effects and perhaps more specific interactions with proteins implicated in familial PD. Yankner and colleagues (Xu et al., 2002) observed that α-synuclein-induced neurotoxicity is observed in primary cultures of DA neurons but not in non-DA cortical neurons. Moreover, mutant forms of α-synuclein mutation form neurotoxic adducts with DA quinone (Conway et al., 2001). In addition, the oxidative effects of dopamine may increase protein nitrosylation, and parkin activity is altered by this modification both in vitro and in vivo (Chung et al., 2004; Yao et al., 2004).
Fruit flies use similar sets of genes for dopamine synthesis and transport, but they metabolize dopamine differently from mammals. Insects do not express MAO (Roelofs and Van Haastert, 2001) and are thought to use conjugation as the primary route for amine degradation. This difference may be related to the unique use of dopamine for the hardening and pigmentation of cuticles in insects and other arthropods (Wright, 1987). Indeed, the need to maintain adequate levels of cytosolic dopamine for these pathways may account for the somewhat surprising effects of DVMAT in total head concentrations of dopamine. We find that overexpression of DVMAT decreases total dopamine and that inhibition of DVMAT using RNAi increases total tissue dopamine. Overexpression of mammalian VMAT2 in cultured cells decreases cytosolic dopamine (Sulzer et al., 2000), and decreased VMAT2 activity in mammals reduces total tissue dopamine (Fon et al., 1997; Takahashi et al., 1997; Wang et al., 1997; Mooslehner et al., 2001). We suggest that the cytoplasmic pool of dopamine may account for a larger proportion of total tissue dopamine in Drosophila, perhaps because of the requirement of dopamine for cuticle formation in other tissues. It is possible that this may render DA neurons in the fly particularly sensitive to neurotoxic mechanisms involving the conjugation of dopamine to cytoplasmic targets.
Based on previous findings in mammals, we speculate that dopamine and/or oxidized derivatives might contribute to mutant parkin-induced degeneration. If so, mutant parkin phenotypes should be relatively specific for DA neurons and sensitive to modulation of cytoplasmic dopamine levels. Indeed, we observe that DA but not cholinergic or histaminergic neurons degenerate in response to expression of mutant parkin. Interestingly, we observe a more limited degenerative phenotype in 5-HT neurons, consistent with the loss of other aminergic cell types in PD (Braak et al., 2003). We find that DVMAT knockdown enhances mutant parkin phenotypes, suggesting that an increase in cytoplasmic dopamine increases the vulnerability of neurons to mutant parkin. Conversely, increasing DVMAT partially rescues pupal lethality and DA neuron degeneration, presumably by reducing cytoplasmic dopamine. These data therefore suggest that mutations in parkin either increase susceptibility of neurons to dopamine or its metabolites or that dopamine is permissive for the toxic effects of parkin. Our model, which provides robust behavioral and neuropathological phenotypes, suggests that some mutations may give rise to disease by dominant mechanisms. Moreover, our model may prove a useful addition to other genetically based vertebrate and invertebrate models aimed at understanding PD and identifying potential therapeutic targets.
Footnotes
This work was supported by National Institutes of Health Grants NS046489 and AG016570 (G.R.J.), NS03836 (N.J.M.), and MH01709, DK60857, and ES012078 (D.E.K.), and by the American Parkinson's Disease Association and Parkinson's Disease Foundation (G.R.J.). We thank Richard Axel, Serge Birman, Jay Hirsh, Larry Marsh, Leo Pallanck, Leslie Michels Thompson, Kalpana White, and Larry Zipursky for fly strains. We also thank Larry Zipursky and Matthew LaVoie for critical comments on this manuscript and Mark Spencer for the rotarod.
References
- Ardley HC, Scott GB, Rose SA, Tan NG, Markham AF, Robinson PA. Inhibition of proteasomal activity causes inclusion formation in neuronal and non-neuronal cells overexpressing Parkin. Mol Biol Cell. 2003;14:4541–4556. doi: 10.1091/mbc.E03-02-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auluck PK, Bonini NM. Pharmacological prevention of Parkinson disease in Drosophila . Nat Med. 2002;8:1185–1186. doi: 10.1038/nm1102-1185. [DOI] [PubMed] [Google Scholar]
- Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science. 2002;295:865–868. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- Benzer S. Behavioral mutants of Drosophila isolated by countercurrent distribution. Proc Natl Acad Sci USA. 1967;58:1112–1119. doi: 10.1073/pnas.58.3.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Buhmann C, Binkofski F, Klein C, Buchel C, van Eimeren T, Erdmann C, Hedrich K, Kasten M, Hagenah J, Deuschl G, Pramstaller PP, Siebner HR. Motor reorganization in asymptomatic carriers of a single mutant Parkin allele: a human model for presymptomatic parkinsonism. Brain. 2005;128:2281–2290. doi: 10.1093/brain/awh572. [DOI] [PubMed] [Google Scholar]
- Chang HY, Grygoruk A, Brooks ES, Ackerson LC, Maidment NT, Bainton RJ, Krantz DE. Overexpression of the Drosophila vesicular monoamine transporter increases motor activity and courtship but decreases the behavioral response to cocaine. Mol Psychiatry. 2006;11:99–113. doi: 10.1038/sj.mp.4001742. [DOI] [PubMed] [Google Scholar]
- Chen L, Feany MB. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci. 2005;8:657–663. doi: 10.1038/nn1443. [DOI] [PubMed] [Google Scholar]
- Chen L, Cagniard B, Mathews T, Jones S, Koh HC, Ding Y, Carvey PM, Ling Z, Kang UJ, Zhuang X. Age-dependent motor deficits and dopaminergic dysfunction in DJ-1 null mice. J Biol Chem. 2005;280:21418–21426. doi: 10.1074/jbc.M413955200. [DOI] [PubMed] [Google Scholar]
- Choi HJ, Lee SY, Cho Y, Hwang O. Inhibition of vesicular monoamine transporter enhances vulnerability of dopaminergic cells: relevance to Parkinson's disease. Neurochem Int. 2005;46:329–335. doi: 10.1016/j.neuint.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Chung KK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, Ross CA, Dawson VL, Dawson TM. Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nat Med. 2001;7:1144–1150. doi: 10.1038/nm1001-1144. [DOI] [PubMed] [Google Scholar]
- Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, Dawson TM. S-nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science. 2004;304:1328–1331. doi: 10.1126/science.1093891. [DOI] [PubMed] [Google Scholar]
- Conway KA, Rochet JC, Bieganski RM, Lansbury PT., Jr Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science. 2001;294:1346–1349. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- Cookson MR. The biochemistry of Parkinson's disease. Annu Rev Biochem. 2005;74:29–52. doi: 10.1146/annurev.biochem.74.082803.133400. [DOI] [PubMed] [Google Scholar]
- Cookson MR, Lockhart PJ, McLendon C, O'Farrell C, Schlossmacher M, Farrer MJ. RING finger 1 mutations in Parkin produce altered localization of the protein. Hum Mol Genet. 2003;12:2957–2965. doi: 10.1093/hmg/ddg328. [DOI] [PubMed] [Google Scholar]
- Coulom H, Birman S. Chronic exposure to rotenone models sporadic Parkinson's disease in Drosophila melanogaster . J Neurosci. 2004;24:10993–10998. doi: 10.1523/JNEUROSCI.2993-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahn S. Description of Parkinson's disease as a clinical syndrome. Ann NY Acad Sci. 2003;991:1–14. doi: 10.1111/j.1749-6632.2003.tb07458.x. [DOI] [PubMed] [Google Scholar]
- Farrer M, Chan P, Chen R, Tan L, Lincoln S, Hernandez D, Forno L, Gwinn-Hardy K, Petrucelli L, Hussey J, Singleton A, Tanner C, Hardy J, Langston JW. Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol. 2001;50:293–300. doi: 10.1002/ana.1132. [DOI] [PubMed] [Google Scholar]
- Feany MB, Bender WW. A Drosophila model of Parkinson's disease. Nature. 2000;404:394–398. doi: 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- Flatmark T, Almas B, Ziegler MG. Catecholamine metabolism: an update on key biosynthetic enzymes and vesicular monoamine transporters. Ann NY Acad Sci. 2002;971:69–75. doi: 10.1111/j.1749-6632.2002.tb04436.x. [DOI] [PubMed] [Google Scholar]
- Fon EA, Pothos EN, Sun BC, Killeen N, Sulzer D, Edwards RH. Vesicular transport regulates monoamine storage and release but is not essential for amphetamine action. Neuron. 1997;19:1271–1283. doi: 10.1016/s0896-6273(00)80418-3. [DOI] [PubMed] [Google Scholar]
- Foroud T, Uniacke SK, Liu L, Pankratz N, Rudolph A, Halter C, Shults C, Marder K, Conneally PM, Nichols WC. Heterozygosity for a mutation in the parkin gene leads to later onset Parkinson disease. Neurology. 2003;60:796–801. doi: 10.1212/01.wnl.0000049470.00180.07. [DOI] [PubMed] [Google Scholar]
- Freeman M. Reiterative use of the EGF receptor triggers differentiation of all cell types in the Drosophila eye. Cell. 1996;87:651–660. doi: 10.1016/s0092-8674(00)81385-9. [DOI] [PubMed] [Google Scholar]
- Friggi-Grelin F, Coulom H, Meller M, Gomez D, Hirsh J, Birman S. Targeted gene expression in Drosophila dopaminergic cells using regulatory sequences from tyrosine hydroxylase. J Neurobiol. 2003;54:618–627. doi: 10.1002/neu.10185. [DOI] [PubMed] [Google Scholar]
- Fumagalli F, Gainetdinov RR, Wang YM, Valenzano KJ, Miller GW, Caron MG. Increased methamphetamine neurotoxicity in heterozygous vesicular monoamine transporter 2 knock-out mice. J Neurosci. 1999;19:2424–2431. doi: 10.1523/JNEUROSCI.19-07-02424.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Lee VM. Parkin and the molecular pathways of Parkinson's disease. Neuron. 2001;31:885–888. doi: 10.1016/s0896-6273(01)00439-1. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Lee VM. Are ubiquitination pathways central to Parkinson's disease? Cell. 2003;114:1–8. doi: 10.1016/s0092-8674(03)00509-9. [DOI] [PubMed] [Google Scholar]
- Gilks WP, Abou-Sleiman PM, Gandhi S, Jain S, Singleton A, Lees AJ, Shaw K, Bhatia KP, Bonifati V, Quinn NP, Lynch J, Healy DG, Holton JL, Revesz T, Wood NW. A common LRRK2 mutation in idiopathic Parkinson's disease. Lancet. 2005;365:415–416. doi: 10.1016/S0140-6736(05)17830-1. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem. 2003;278:43628–43635. doi: 10.1074/jbc.M308947200. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Pisani A, Haburcak M, Vortherms TA, Kitada T, Costa C, Tong Y, Martella G, Tscherter A, Martins A, Bernardi G, Roth BL, Pothos EN, Calabresi P, Shen J. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron. 2005;45:489–496. doi: 10.1016/j.neuron.2005.01.041. [DOI] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer CL, Grygoruk A, Patton DE, Ley B, Romero-Calderon R, Chang HY, Houshyar R, Bainton RJ, Diantonio A, Krantz DE. A splice variant of the Drosophila vesicular monoamine transporter contains a conserved trafficking domain and functions in the storage of dopamine, serotonin, and octopamine. J Neurobiol. 2005;64:239–258. doi: 10.1002/neu.20146. [DOI] [PubMed] [Google Scholar]
- Gu WJ, Corti O, Araujo F, Hampe C, Jacquier S, Lucking CB, Abbas N, Duyckaerts C, Rooney T, Pradier L, Ruberg M, Brice A. The C289G and C418R missense mutations cause rapid sequestration of human Parkin into insoluble aggregates. Neurobiol Dis. 2003;14:357–364. doi: 10.1016/j.nbd.2003.08.011. [DOI] [PubMed] [Google Scholar]
- Hansen JP, Riddle EL, Sandoval V, Brown JM, Gibb JW, Hanson GR, Fleckenstein AE. Methylenedioxymethamphetamine decreases plasmalemmal and vesicular dopamine transport: mechanisms and implications for neurotoxicity. J Pharmacol Exp Ther. 2002;300:1093–1100. doi: 10.1124/jpet.300.3.1093. [DOI] [PubMed] [Google Scholar]
- Hattori N, Mizuno Y. Pathogenetic mechanisms of parkin in Parkinson's disease. Lancet. 2004;364:722–724. doi: 10.1016/S0140-6736(04)16901-8. [DOI] [PubMed] [Google Scholar]
- Hedrich K, Djarmati A, Schafer N, Hering R, Wellenbrock C, Weiss PH, Hilker R, Vieregge P, Ozelius LJ, Heutink P, Bonifati V, Schwinger E, Lang AE, Noth J, Bressman SB, Pramstaller PP, Riess O, Klein C. DJ-1 (PARK7) mutations are less frequent than Parkin (PARK2) mutations in early-onset Parkinson disease. Neurology. 2004;62:389–394. doi: 10.1212/01.wnl.0000113022.51739.88. [DOI] [PubMed] [Google Scholar]
- Henn IH, Gostner JM, Lackner P, Tatzelt J, Winklhofer KF. Pathogenic mutations inactivate parkin by distinct mechanisms. J Neurochem. 2005;92:114–122. doi: 10.1111/j.1471-4159.2004.02854.x. [DOI] [PubMed] [Google Scholar]
- Hernandez D, Paisan Ruiz C, Crawley A, Malkani R, Werner J, Gwinn-Hardy K, Dickson D, Wavrant Devrieze F, Hardy J, Singleton A. The dardarin G 2019 S mutation is a common cause of Parkinson's disease but not other neurodegenerative diseases. Neurosci Lett. 2005;389:137–139. doi: 10.1016/j.neulet.2005.07.044. [DOI] [PubMed] [Google Scholar]
- Hilker R, Klein C, Ghaemi M, Kis B, Strotmann T, Ozelius LJ, Lenz O, Vieregge P, Herholz K, Heiss WD, Pramstaller PP. Positron emission tomographic analysis of the nigrostriatal dopaminergic system in familial parkinsonism associated with mutations in the parkin gene. Ann Neurol. 2001;49:367–376. [PubMed] [Google Scholar]
- Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of parkin. Cell. 2001;105:891–902. doi: 10.1016/s0092-8674(01)00407-x. [DOI] [PubMed] [Google Scholar]
- Jackson GR. Modeling neurodegenerative diseases in the fruit fly. In: Chesselet MF, editor. Molecular mechanisms of neurodegenerative diseases. Totowa, NJ: Humana; 2000. pp. 373–406. [Google Scholar]
- Jackson GR, Wiedau-Pazos M, Sang TK, Wagle N, Brown CA, Massachi S, Geschwind DH. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila . Neuron. 2002;34:509–519. doi: 10.1016/s0896-6273(02)00706-7. [DOI] [PubMed] [Google Scholar]
- Jiang H, Jiang Q, Feng J. Parkin increases dopamine uptake by enhancing the cell surface expression of dopamine transporter. J Biol Chem. 2004;279:54380–54386. doi: 10.1074/jbc.M409282200. [DOI] [PubMed] [Google Scholar]
- Kalidas S, Smith DP. Novel genomic cDNA hybrids produce effective RNA interference in adult Drosophila . Neuron. 2002;33:177–184. doi: 10.1016/s0896-6273(02)00560-3. [DOI] [PubMed] [Google Scholar]
- Khan NL, Brooks DJ, Pavese N, Sweeney MG, Wood NW, Lees AJ, Piccini P. Progression of nigrostriatal dysfunction in a parkin kindred: an [18F]dopa PET and clinical study. Brain. 2002;125:2248–2256. doi: 10.1093/brain/awf237. [DOI] [PubMed] [Google Scholar]
- Khan NL, Scherfler C, Graham E, Bhatia KP, Quinn N, Lees AJ, Brooks DJ, Wood NW, Piccini P. Dopaminergic dysfunction in unrelated, asymptomatic carriers of a single parkin mutation. Neurology. 2005;64:134–136. doi: 10.1212/01.WNL.0000148725.48740.6D. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Klein C, Hedrich K, Wellenbrock C, Kann M, Harris J, Marder K, Lang AE, Schwinger E, Ozelius LJ, Vieregge P, Pramstaller PP, Kramer PL. Frequency of parkin mutations in late-onset Parkinson's disease. Ann Neurol. 2003;54:415–416. doi: 10.1002/ana.10737. author reply 416–417. [DOI] [PubMed] [Google Scholar]
- LaVoie MJ, Ostaszewski BL, Weihofen A, Schlossmacher MG, Selkoe DJ. Dopamine covalently modifies and functionally inactivates parkin. Nat Med. 2005;11:1214–1221. doi: 10.1038/nm1314. [DOI] [PubMed] [Google Scholar]
- Leal SM, Neckameyer WS. Pharmacological evidence for GABAergic regulation of specific behaviors in Drosophila melanogaster . J Neurobiol. 2002;50:245–261. doi: 10.1002/neu.10030. [DOI] [PubMed] [Google Scholar]
- Lee T, Luo L. Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci. 2001;24:251–254. doi: 10.1016/s0166-2236(00)01791-4. [DOI] [PubMed] [Google Scholar]
- Leroy E, Anastasopoulos D, Konitsiotis S, Lavedan C, Polymeropoulos MH. Deletions in the Parkin gene and genetic heterogeneity in a Greek family with early onset Parkinson's disease. Hum Genet. 1998;103:424–427. doi: 10.1007/s004390050845. [DOI] [PubMed] [Google Scholar]
- Lesage S, Magali P, Lohmann E, Lacomblez L, Teive H, Janin S, Cousin PY, Durr A, Brice A. Deletion of the parkin and PACRG gene promoter in early-onset parkinsonism. Hum Mutat. 2007;28:27–32. doi: 10.1002/humu.20436. [DOI] [PubMed] [Google Scholar]
- Li H, Chaney S, Roberts IJ, Forte M, Hirsh J. Ectopic G-protein expression in dopamine and serotonin neurons blocks cocaine sensitization in Drosophila melanogaster . Curr Biol. 2000;10:211–214. doi: 10.1016/s0960-9822(00)00340-7. [DOI] [PubMed] [Google Scholar]
- Lincoln SJ, Maraganore DM, Lesnick TG, Bounds R, de Andrade M, Bower JH, Hardy JA, Farrer MJ. Parkin variants in North American Parkinson's disease: cases and controls. Mov Disord. 2003;18:1306–1311. doi: 10.1002/mds.10601. [DOI] [PubMed] [Google Scholar]
- Lucking CB, Bonifati V, Periquet M, Vanacore N, Brice A, Meco G. Pseudo-dominant inheritance and exon 2 triplication in a family with parkin gene mutations. Neurology. 2001;57:924–927. doi: 10.1212/wnl.57.5.924. [DOI] [PubMed] [Google Scholar]
- Marsh JL, Walker H, Theisen H, Zhu YZ, Fielder T, Purcell J, Thompson LM. Expanded polyglutamine peptides alone are intrinsically cytotoxic and cause neurodegeneration in Drosophila . Hum Mol Genet. 2000;9:13–25. doi: 10.1093/hmg/9.1.13. [DOI] [PubMed] [Google Scholar]
- Maruyama M, Ikeuchi T, Saito M, Ishikawa A, Yuasa T, Tanaka H, Hayashi S, Wakabayashi K, Takahashi H, Tsuji S. Novel mutations, pseudo-dominant inheritance, and possible familial affects in patients with autosomal recessive juvenile parkinsonism. Ann Neurol. 2000;48:245–250. [PubMed] [Google Scholar]
- Mata IF, Lockhart PJ, Farrer MJ. Parkin genetics: one model for Parkinson's disease. Hum Mol Genet 13 Spec No. 2004;1:R127–R133. doi: 10.1093/hmg/ddh089. [DOI] [PubMed] [Google Scholar]
- McClung C, Hirsh J. Stereotypic behavioral responses to free-base cocaine and the development of behavioral sensitization in Drosophila . Curr Biol. 1998;8:109–112. doi: 10.1016/s0960-9822(98)70041-7. [DOI] [PubMed] [Google Scholar]
- Menzies FM, Yenisetti SC, Min KT. Roles of Drosophila DJ-1 in survival of dopaminergic neurons and oxidative stress. Curr Biol. 2005;15:1578–1582. doi: 10.1016/j.cub.2005.07.036. [DOI] [PubMed] [Google Scholar]
- Meulener M, Whitworth AJ, Armstrong-Gold CE, Rizzu P, Heutink P, Wes PD, Pallanck LJ, Bonini NM. Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with Parkinson's disease. Curr Biol. 2005;15:1572–1577. doi: 10.1016/j.cub.2005.07.064. [DOI] [PubMed] [Google Scholar]
- Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson's disease. Annu Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- Mooslehner KA, Chan PM, Xu W, Liu L, Smadja C, Humby T, Allen ND, Wilkinson LS, Emson PC. Mice with very low expression of the vesicular monoamine transporter 2 gene survive into adulthood: potential mouse model for parkinsonism. Mol Cell Biol. 2001;21:5321–5331. doi: 10.1128/MCB.21.16.5321-5331.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muqit MM, Davidson SM, Payne Smith MD, MacCormac LP, Kahns S, Jensen PH, Wood NW, Latchman DS. Parkin is recruited into aggresomes in a stress-specific manner: over-expression of parkin reduces aggresome formation but can be dissociated from parkin's effect on neuronal survival. Hum Mol Genet. 2004;13:117–135. doi: 10.1093/hmg/ddh012. [DOI] [PubMed] [Google Scholar]
- Neckameyer WS, Quinn WG. Isolation and characterization of the gene for Drosophila tyrosine hydroxylase. Neuron. 1989;2:1167–1175. doi: 10.1016/0896-6273(89)90183-9. [DOI] [PubMed] [Google Scholar]
- Ollmann M, Young LM, Di Como CJ, Karim F, Belvin M, Robertson S, Whittaker K, Demsky M, Fisher WW, Buchman A, Duyk G, Friedman L, Prives C, Kopczynski C. Drosophila p53 is a structural and functional homolog of the tumor suppressor p53. Cell. 2000;101:91–101. doi: 10.1016/S0092-8674(00)80626-1. [DOI] [PubMed] [Google Scholar]
- Pawlyk AC, Giasson BI, Sampathu DM, Perez FA, Lim KL, Dawson VL, Dawson TM, Palmiter RD, Trojanowski JQ, Lee VM. Novel monoclonal antibodies demonstrate biochemical variation of brain parkin with age. J Biol Chem. 2003;278:48120–48128. doi: 10.1074/jbc.M306889200. [DOI] [PubMed] [Google Scholar]
- Pendleton RG, Parvez F, Sayed M, Hillman R. Effects of pharmacological agents upon a transgenic model of Parkinson's disease in Drosophila melanogaster . J Pharmacol Exp Ther. 2002;300:91–96. doi: 10.1124/jpet.300.1.91. [DOI] [PubMed] [Google Scholar]
- Perez FA, Palmiter RD. Parkin-deficient mice are not a robust model of parkinsonism. Proc Natl Acad Sci USA. 2005;102:2174–2179. doi: 10.1073/pnas.0409598102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesah Y, Pham T, Burgess H, Middlebrooks B, Verstreken P, Zhou Y, Harding M, Bellen H, Mardon G. Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development. 2004;131:2183–2194. doi: 10.1242/dev.01095. [DOI] [PubMed] [Google Scholar]
- Pothos EN, Larsen KE, Krantz DE, Liu Y, Haycock JW, Setlik W, Gershon MD, Edwards RH, Sulzer D. Synaptic vesicle transporter expression regulates vesicle phenotype and quantal size. J Neurosci. 2000;20:7297–7306. doi: 10.1523/JNEUROSCI.20-19-07297.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, Zhao J, Feng J. Parkin binds to alpha/beta tubulin and increases their ubiquitination and degradation. J Neurosci. 2003;23:3316–3324. doi: 10.1523/JNEUROSCI.23-08-03316.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelofs J, Van Haastert PJ. Genes lost during evolution. Nature. 2001;411:1013–1014. doi: 10.1038/35082627. [DOI] [PubMed] [Google Scholar]
- Rubin GM, Spradling AC. Genetic transformation of Drosophila with transposable element vectors. Science. 1982;218:348–353. doi: 10.1126/science.6289436. [DOI] [PubMed] [Google Scholar]
- Sang TK, Li C, Liu W, Rodriguez A, Abrams JM, Zipursky SL, Jackson GR. Inactivation of Drosophila Apaf-1 related killer suppresses formation of polyglutamine aggregates and blocks polyglutamine pathogenesis. Hum Mol Genet. 2005;14:357–372. doi: 10.1093/hmg/ddi032. [DOI] [PubMed] [Google Scholar]
- Scherfler C, Khan NL, Pavese N, Eunson L, Graham E, Lees AJ, Quinn NP, Wood NW, Brooks DJ, Piccini PP. Striatal and cortical pre- and postsynaptic dopaminergic dysfunction in sporadic parkin-linked parkinsonism. Brain. 2004;127:1332–1342. doi: 10.1093/brain/awh150. [DOI] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson's disease. Science. 2001;293:263–269. doi: 10.1126/science.1060627. [DOI] [PubMed] [Google Scholar]
- Shyu WC, Lin SZ, Chiang MF, Pang CY, Chen SY, Hsin YL, Thajeb P, Lee YJ, Li H. Early-onset Parkinson's disease in a Chinese population: 99mTc-TRODAT-1 SPECT, Parkin gene analysis and clinical study. Parkinsonism Relat Disord. 2005;11:173–180. doi: 10.1016/j.parkreldis.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Spradling AC, Rubin GM. Transposition of cloned P elements into Drosophila germ line chromosomes. Science. 1982;218:341–347. doi: 10.1126/science.6289435. [DOI] [PubMed] [Google Scholar]
- Sriram SR, Li X, Ko HS, Chung KK, Wong E, Lim KL, Dawson VL, Dawson TM. Familial-associated mutations differentially disrupt the solubility, localization, binding and ubiquitination properties of parkin. Hum Mol Genet. 2005;14:2571–2586. doi: 10.1093/hmg/ddi292. [DOI] [PubMed] [Google Scholar]
- Stathakis DG, Burton DY, McIvor WE, Krishnakumar S, Wright TR, O'Donnell JM. The catecholamines up (Catsup) protein of Drosophila melanogaster functions as a negative regulator of tyrosine hydroxylase activity. Genetics. 1999;153:361–382. doi: 10.1093/genetics/153.1.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes AH, Hastings TG, Vrana KE. Cytotoxic and genotoxic potential of dopamine. J Neurosci Res. 1999;55:659–665. doi: 10.1002/(SICI)1097-4547(19990315)55:6<659::AID-JNR1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Stuart AE. From fruit flies to barnacles, histamine is the neurotransmitter of arthropod photoreceptors. Neuron. 1999;22:431–433. doi: 10.1016/s0896-6273(00)80699-6. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Zecca L. Intraneuronal dopamine-quinone synthesis: a review. Neurotox Res. 2000;1:181–195. doi: 10.1007/BF03033289. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Bogulavsky J, Larsen KE, Behr G, Karatekin E, Kleinman MH, Turro N, Krantz D, Edwards RH, Greene LA, Zecca L. Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc Natl Acad Sci USA. 2000;97:11869–11874. doi: 10.1073/pnas.97.22.11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Latourelle JC, Wooten GF, Lew MF, Klein C, Shill HA, Golbe LI, Mark MH, Racette BA, Perlmutter JS, Parsian A, Guttman M, Nicholson G, Xu G, Wilk JB, Saint-Hilaire MH, DeStefano AL, Prakash R, Williamson S, Suchowersky O, et al. Influence of heterozygosity for parkin mutation on onset age in familial Parkinson disease: the GenePD study. Arch Neurol. 2006;63:826–832. doi: 10.1001/archneur.63.6.826. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Miner LL, Sora I, Ujike H, Revay RS, Kostic V, Jackson-Lewis V, Przedborski S, Uhl GR. VMAT2 knockout mice: heterozygotes display reduced amphetamine-conditioned reward, enhanced amphetamine locomotion, and enhanced MPTP toxicity. Proc Natl Acad Sci USA. 1997;94:9938–9943. doi: 10.1073/pnas.94.18.9938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan LC, Tanner CM, Chen R, Chan P, Farrer M, Hardy J, Langston JW. Marked variation in clinical presentation and age of onset in a family with a heterozygous parkin mutation. Mov Disord. 2003;18:758–763. doi: 10.1002/mds.10432. [DOI] [PubMed] [Google Scholar]
- Torroja L, Chu H, Kotovsky I, White K. Neuronal overexpression of APPL, the Drosophila homologue of the amyloid precursor protein (APP), disrupts axonal transport. Curr Biol. 1999;9:489–492. doi: 10.1016/s0960-9822(99)80215-2. [DOI] [PubMed] [Google Scholar]
- Vernier P, Moret F, Callier S, Snapyan M, Wersinger C, Sidhu A. The degeneration of dopamine neurons in Parkinson's disease: insights from embryology and evolution of the mesostriatocortical system. Ann NY Acad Sci. 2004;1035:231–249. doi: 10.1196/annals.1332.015. [DOI] [PubMed] [Google Scholar]
- Von Coelln R, Thomas B, Savitt JM, Lim KL, Sasaki M, Hess EJ, Dawson VL, Dawson TM. Loss of locus coeruleus neurons and reduced startle in parkin null mice. Proc Natl Acad Sci USA. 2004;101:10744–10749. doi: 10.1073/pnas.0401297101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Tan JM, Ho MW, Zaiden N, Wong SH, Chew CL, Eng PW, Lim TM, Dawson TM, Lim KL. Alterations in the solubility and intracellular localization of parkin by several familial Parkinson's disease-linked point mutations. J Neurochem. 2005a;93:422–431. doi: 10.1111/j.1471-4159.2005.03023.x. [DOI] [PubMed] [Google Scholar]
- Wang C, Ko HS, Thomas B, Tsang F, Chew KC, Tay SP, Ho MW, Lim TM, Soong TW, Pletnikova O, Troncoso J, Dawson VL, Dawson TM, Lim KL. Stress-induced alterations in parkin solubility promote parkin aggregation and compromise parkin's protective function. Hum Mol Genet. 2005b;14:3885–3897. doi: 10.1093/hmg/ddi413. [DOI] [PubMed] [Google Scholar]
- Wang JW, Wong AM, Flores J, Vosshall LB, Axel R. Two-photon calcium imaging reveals an odor-evoked map of activity in the fly brain. Cell. 2003;112:271–282. doi: 10.1016/s0092-8674(03)00004-7. [DOI] [PubMed] [Google Scholar]
- Wang YM, Gainetdinov RR, Fumagalli F, Xu F, Jones SR, Bock CB, Miller GW, Wightman RM, Caron MG. Knockout of the vesicular monoamine transporter 2 gene results in neonatal death and supersensitivity to cocaine and amphetamine. Neuron. 1997;19:1285–1296. doi: 10.1016/s0896-6273(00)80419-5. [DOI] [PubMed] [Google Scholar]
- Weingarten P, Zhou QY. Protection of intracellular dopamine cytotoxicity by dopamine disposition and metabolism factors. J Neurochem. 2001;77:776–785. doi: 10.1046/j.1471-4159.2001.00263.x. [DOI] [PubMed] [Google Scholar]
- West A, Periquet M, Lincoln S, Lucking CB, Nicholl D, Bonifati V, Rawal N, Gasser T, Lohmann E, Deleuze JF, Maraganore D, Levey A, Wood N, Durr A, Hardy J, Brice A, Farrer M. Complex relationship between Parkin mutations and Parkinson disease. Am J Med Genet. 2002;114:584–591. doi: 10.1002/ajmg.10525. [DOI] [PubMed] [Google Scholar]
- Whitworth AJ, Theodore DA, Greene JC, Benes H, Wes PD, Pallanck LJ. Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson's disease. Proc Natl Acad Sci USA. 2005;102:8024–8029. doi: 10.1073/pnas.0501078102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293:711–714. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- Wright TR. The genetics of biogenic amine metabolism, sclerotization, and melanization in Drosophila melanogaster . Adv Genet. 1987;24:127–222. [PubMed] [Google Scholar]
- Xu J, Kao SY, Lee FJ, Song W, Jin LW, Yankner BA. Dopamine-dependent neurotoxicity of alpha-synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat Med. 2002;8:600–606. doi: 10.1038/nm0602-600. [DOI] [PubMed] [Google Scholar]
- Yang Y, Nishimura I, Imai Y, Takahashi R, Lu B. Parkin suppresses dopaminergic neuron-selective neurotoxicity induced by Pael-R in Drosophila . Neuron. 2003;37:911–924. doi: 10.1016/s0896-6273(03)00143-0. [DOI] [PubMed] [Google Scholar]
- Yao D, Gu Z, Nakamura T, Shi ZQ, Ma Y, Gaston B, Palmer LA, Rockenstein EM, Zhang Z, Masliah E, Uehara T, Lipton SA. Nitrosative stress linked to sporadic Parkinson's disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc Natl Acad Sci USA. 2004;101:10810–10814. doi: 10.1073/pnas.0404161101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuyama K, Salvaterra PM. Localization of choline acetyltransferase-expressing neurons in Drosophila nervous system. Microsc Res Tech. 1999;45:65–79. doi: 10.1002/(SICI)1097-0029(19990415)45:2<65::AID-JEMT2>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM. Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci USA. 2000;97:13354–13359. doi: 10.1073/pnas.240347797. [DOI] [PMC free article] [PubMed] [Google Scholar]