

Abstract
The aristaless-related homeobox (ARX) gene has been implicated in a wide spectrum of disorders ranging from phenotypes with severe neuronal migration defects, such as lissencephaly, to mild forms of X-linked mental retardation without apparent brain abnormalities. To better understand its role in corticogenesis, we used in utero electroporation to knock down or overexpress ARX. We show here that targeted inhibition of ARX causes cortical progenitor cells to exit the cell cycle prematurely and impairs their migration toward the cortical plate. In contrast, ARX overexpression increases the length of the cell cycle. In addition, we report that RNA interference-mediated inactivation of ARX prevents cells from acquiring multipolar morphology in the subventricular and intermediate zones, resulting in decreased neuronal motility. In contrast, ARX overexpression appears to promote the development of tangentially oriented processes of cells in the subventricular and intermediate zones and affects radial migration of pyramidal neurons. We also demonstrate that the level of ARX expression is important for tangential migration of GABA-containing interneurons, because both inactivation and overexpression of the gene impair their migration from the ganglionic eminence. However, our data suggest that ARX is not directly involved in GABAergic cell fate specification. Overall, these results identify multiple and distinct cell-autonomous roles for ARX in corticogenesis.
Keywords: ARX, lissencephaly, neuronal migration, RNA interference, interneuron, cell cycle
Introduction
Mental retardation and epilepsy have a variety of acquired and genetic causes. Recently, genetic investigations of X-linked mental retardation (XLMR) disorders have identified aristaless-related homeobox (ARX) as the causative gene in X-linked infantile spasms, Partington syndrome, and in certain families with nonsyndromic XLMR (Bienvenu et al., 2002; Scheffer et al., 2002; Strømme et al., 2002a,b). More severe mutations in ARX, resulting in premature termination of the protein, have been reported in X-linked lissencephaly with abnormal genitalia (XLAG) (Dobyns et al., 1999; Kitamura et al., 2002). Lissencephaly (smooth brain), characterized by massive disorganization of neurons throughout the cortex, is thought to result from a defect in neuronal migration (Dobyns et al., 1996). In addition to ARX, four other genes have so far been shown to be responsible for type I lissencephaly: LIS1, doublecortin (DCX), reelin (RELN), and α-1 tubulin (TUBA3) (Reiner et al., 1993; des Portes et al., 1998; Gleeson et al., 1998; Hong et al., 2000; Keays et al., 2007).
Interest in the cellular and molecular mechanisms responsible for the migration of the neuronal cell types of the cortex has never been greater. Pyramidal neurons, the projection cells of the cortex, are generated directly from radial glial cells in the germinal ventricular zone (VZ) [for review, see Rakic (1990), Nadarajah and Parnavelas (2002), and Kriegstein and Noctor, (2004)] or indirectly from intermediate progenitors in the subventricular zone (SVZ) (Noctor et al., 2001, 2004). After cell division at the ventricular surface, newborn neurons ascend to the SVZ–intermediate zone (IZ), where they assume a multipolar morphology as their cell bodies move slowly (Tabata and Nakajima, 2003). After ∼24 h, they convert to a bipolar shape and resume glia-directed migration to take up their positions in the cortex in an orderly manner. The majority of the other neuronal cell type of the cortex, the GABA-containing interneurons, are generated in the ganglionic eminences of the ventral forebrain and reach the cortex by tangential migration [for review, see Marin and Rubenstein (2003), Metin et al. (2006), and Nakajima (2007)].
In mouse, Arx is mainly expressed in telencephalic structures, and more specifically in the mantle zones of the developing lateral ganglionic eminence (LGE) and medial ganglionic eminence (MGE). In the developing cortex, Arx expression is observed in progenitor cells of the VZ as well as in migrating interneurons (Colombo et al., 2004; Poirier et al., 2004; Friocourt et al., 2006). Mutant mice for Arx display a phenotype very similar to the human XLAG (Kitamura et al., 2002). However, the plethora of defects has made it difficult to distinguish between direct and secondary roles of ARX in the developing brain. For this reason, we used a combination of in utero electroporation (Saito and Nakatsuji, 2001; Tabata and Nakajima, 2001) and RNA interference (RNAi)-mediated loss-of-function or overexpression experimental approaches, which revealed that ARX directly affects the proliferation of cortical progenitor cells as well as their radial migration. In addition, inactivation and overexpression of ARX by focal electroporation of the MGE in whole telencephalic hemisphere cultures showed that it plays a cell-autonomous role in tangential migration of cortical interneurons. However, the expression of this gene is not sufficient to induce the GABAergic phenotype in these cells. Overall, our findings identify multiple distinct roles for ARX in corticogenesis.
Materials and Methods
Plasmids and RNAi constructs.
For overexpression experiments, the mouse arx cDNA was subcloned into the CAG-IRES-EGFP expression vector (a kind gift from Dr. M. Hoshino, Kyoto University, Kyoto, Japan), which has a modified chicken β-actin promoter with a cytomegalovirus-immediate-early enhancer (CAG) promoter (Niwa et al., 1991). For RNAi experiments, we designed three different oligonucleotides, targeting specific regions of mouse arx cDNA [S1 specifically recognizes nucleotides 130–153; S2, nucleotides 594–616; and S3 targets nucleotides 1933–1950 in the 3′-untranslated region (UTR) (GenBank accession number NM_007492.3)]. Three oligonucleotides targeting the corresponding regions of rat arx cDNA (sequence submitted to GenBank) were used in dissociated cell culture experiments.
Annealed oligonucleotides were cloned in the psiStrike U6 promoter-driven vector according to the manufacturer's instructions (Promega). As controls, we used short hairpin RNAs (shRNAs) targeting the same regions, but containing three point mutations, and thus not affecting the stability of ARX mRNA. The efficiency of the three different shRNAs in targeting ARX mRNA was determined by using the psiCheck2 vector (Promega). This vector contains both a synthetic version of Renilla luciferase reporter gene that monitors RNAi activity, and a synthetic firefly luciferase gene that permits normalization of the changes in Renilla luciferase expression to the expression of firefly luciferase. COS-7 cells were seeded into 96-well plates at a density of 3000 cells per well. After overnight incubation, cells were cotransfected with psiCheck2 containing mouse arx cDNA and the different shRNAs at a ratio 1:4, using Lipofectamine 2000 (Invitrogen). Forty-eight hours after transfection, Renilla and firefly luciferase activities were measured using the Dual-Glo Luciferase Assay System (Promega).
Dissociated cell cultures and transfections.
All animal procedures were performed in accordance with institutional guidelines. Brains of embryos, removed from pregnant Sprague Dawley albino rats at embryonic day 16 (E16) (E1, day vaginal plug was found), were used for the preparation of dissociated cortical cell cultures and immunohistochemistry. The brains were removed, and their cortices or GEs were microdissected and placed in a solution containing 0.05% trypsin and 100 μg/ml DNase1 in Neurobasal medium. After incubation in 5% CO2 at 37°C for 15 min, they were washed in Neurobasal medium with 10% fetal bovine serum and dissociated with a fire-polished Pasteur pipette. The resulting suspension was centrifuged at 3000 rpm for 3 min. The cell pellet was resuspended in Neurobasal medium containing 2 mm l-glutamine, penicillin/streptomycin, and B27 (diluted 1:50). A total of 2 × 105 cells were plated onto each 13-mm-diameter glass coverslip coated with laminin and poly-l-lysine (100 μg/ml). Dissociated cells were transfected 2 d after plating using Lipofectamine 2000 according to the guidelines of the manufacturer (Invitrogen). The cultures were processed for immunohistochemistry 48 h after transfection. All materials were purchased from Invitrogen unless otherwise stated.
In utero electroporation.
In utero electroporations were performed as described previously (Tabata and Nakajima, 2001) using pregnant ICR mice (Japan SCL). Briefly, 1–2 μl of plasmid DNA (5 μg/μl) were injected into the lateral ventricles of E13.5 brains (E0 was defined as the day of confirmation of vaginal plug) and then introduced into VZ cells by delivering four electric pulses at 30 V for 50 ms at 950 ms intervals through the uterine wall. Embryos were then allowed to develop for 3–5 d, and the positions and morphology of transfected cells were analyzed by fluorescence microscopy. Plasmids used for in utero electroporation were prepared using the EndoFree Plasmid kit (Qiagen). Because the green fluorescent protein (GFP) gene in the psiStrike vector was under the control of the cytomegalovirus (CMV) promoter, its expression in electroporated brains was too faint to be detected. To visualize transfected cells, the shRNAs were cotransfected with a DsRed expression vector (DsRed-Express; Clontech) containing a modified CAG promoter (Niwa et al., 1991). To ensure that all red cells expressed the shRNA, the plasmids were cotransfected at a ratio 2:1.
Antibodies and immunohistochemistry.
The primary antibodies used were anti-ARX (rabbit polyclonal, 1:200) (Poirier et al., 2004), anti-calbindin (rabbit polyclonal, 1:1000; Swant), anti-GABA (rabbit polyclonal, 1:1000; Sigma), anti-caspase 3 (rabbit polyclonal, 1:200; Millipore Bioscience Research Reagents), anti-phospho-histone H3 (P-H3) (rabbit polyclonal, 1:200; Millipore), anti-Ki67 (rabbit polyclonal, 1:1000; Novocastra), anti-BrdU (rat monoclonal, 1:200; Abcam), anti-5-iodo-2′-deoxyuridine (IdU)/5-bromo-2′-deoxyuridine (BrdU) (mouse monoclonal, 1:50; BD Biosciences), anti-β3-tubulin (clone Tuj1, mouse monoclonal, 1:1000; Sigma), and anti-GFP (mouse monoclonal, 1:1000; Roche Molecular Biochemicals).
To visualize ARX-, Ki67-, P-H3-, calbindin- or GABA-positive cells, immunolabeling was performed on free-floating sections. Electroporated mouse brains were removed from the skulls and fixed with 4% paraformaldehyde overnight at 4°C. For GABA labeling, electroporated E18.5 mice were first perfused through the heart with a phosphate-buffered (0.1 m) fixative containing 4% paraformaldehyde and 0.1% glutaraldehyde. Brains were then embedded in 4% agarose and sectioned at 60–80 μm with a Vibratome (VT1000S; Leica Microsystems). Sections were blocked with 10% normal goat serum in PBS and 0.2% Triton X-100 for 1 h, and incubated in primary antibody (see above) overnight at 4°C. After three washes in PBS, sections were incubated with either FITC- or Alexa Fluor 633-conjugated secondary antibody (Invitrogen) in a 1:400 dilution at room temperature for 2 h, washed, and mounted with Citifluor anti-fading solution (Agar). Images were acquired using a Leica TCS SP2 confocal system. All cells were counted for statistical analysis using ImageJ (NIH).
To perform ARX/GABA and ARX/calbindin double labeling using two rabbit polyclonal antibodies, we used the Renaissance TSA Fluorescence Systems (NEN Life Science Products). Sections were first incubated with H2O2 (final concentration of 0.3%) in PBS at room temperature for 15 min and, after three washes in PBS for 5 min, were blocked for 1 h as described above. Incubation with rabbit anti-ARX antibody (1:10,000) was performed overnight at 4°C, followed by biotinylated goat anti-rabbit IgG (1:200; Vector Laboratories) for 90 min and streptavidin-HRP (1:100; Renaissance TSA Fluorescence Systems) at room temperature for 30 min. This was followed by 5 min of amplification with FITC-tyramide diluted 1:100 in amplification diluent (Renaissance TSA Fluorescence Systems). After several washes, the sections were processed for the other rabbit antibody (GABA or calbindin) using a standard protocol.
Cell proliferation experiments.
E13.5 embryos were electroporated with either CAG-IRES-EGFP or CAG-IRES-EGFP-ARX, and their mothers were injected with IdU (Sigma; 50 μg/g of body weight) 3 d later. Three hours after IdU injection, 50 μg of BrdU (Sigma) per gram of body weight was also administered into the mother, which was killed 2 h later. Brains were then fixed with 4% paraformaldehyde and 16- to 20-μm-thick sections were cut with a Cryostat. For BrdU and IdU immunolabeling, sections were first incubated with 2N HCl at 37°C for 30 min to unmask the antigen, followed by three washes in PBS. The remaining of the procedure was performed as described above.
Focal electroporation of the MGE in whole telencephalic hemisphere cultures.
Telencephalic hemispheres from E14.5 mouse embryos were dissected in PBS, and electroporations were performed as previously described (Yozu et al., 2005). Briefly, 0.1 μl of the plasmid solution (5 μg/μl) was injected into the MGE using a glass micropipette. The telencephalic hemispheres were placed between a platinum plate electrode and a tungsten needle electrode, and electric pulses (100 V; 5 ms) were given twice at intervals of 5 ms using an electroporator (CUY21E; Nepa Gene). Brains were then cultured for 40 h in DMEM/F12 (Sigma) containing N2-supplement (Invitrogen) in rotating glass bottles with continuous gas flow (95% O2 and 5% CO2 at 37°C). Brains were fixed in 4% paraformaldehyde overnight, embedded in 4% agarose, and sectioned at 100 μm with a Vibratome. To visualize transfected cells, the shRNAs were cotransfected with a DsRed expression vector at a ratio 2:1.
Results
ARX inactivation using RNA interference
A previous study that used Arx knock-out mice documented a number of brain defects (Kitamura et al., 2002). However, because it has been difficult to differentiate between primary and secondary effects of ARX inactivation, we decided to apply the RNAi technique to investigate the role(s) of this gene in corticogenesis. A major advantage of this method is that it allows us to explore cell-autonomous defects in a wild-type background. Thus, we designed three RNAi sequences (called S1–S3) that target three different regions within ARX mRNA (Fig. 1A). To test the effectiveness of our shRNA constructs on ARX expression, we transfected cells dissociated from the GE of E16 rat embryos. As a control, we used a mutated shRNA directed against the 3′UTR of DCX (3UTR3mhp) (Bai et al., 2003; Friocourt et al., 2007). Cells were fixed 48 h after transfection and immunolabeled with anti-ARX antibody. We observed that all cells transfected with the control shRNA expressed ARX in the nucleus as previously described (Poirier et al., 2004), but the vast majority of cells expressing the S1, S2, or S3 shRNAs showed reduced ARX immunolabeling (Fig. 1B). Similar results were obtained when COS-7 cells were transfected with both a mouse Arx construct, expressed under a CMV promoter, and different shRNAs. Cells were lysed 48 h after transfection and immunoblotted using anti-ARX antibody (Fig. 1C). Most of the ARX protein was absent compared with control cells that were transfected with triple point mutant shRNAs (S1m–S3m) (Fig. 1A), or the shRNA targeting the 3′UTR part of DCX, respectively (Fig. 1C,D).
Figure 1.

Suppression of ARX protein expression by three shRNAs targeting different regions of ARX mRNA. A, Sequences of specific and triple-point-mutated shRNAs targeting mouse ARX mRNA. B, Representative confocal images of ARX protein expression in dissociated cell cultures of E16 rat ganglionic eminences transfected either with S2 or a control shRNA (DCX 3UTR3mhp). Each shRNA plasmid also encodes EGFP, enabling identification of transfected cells. Forty-eight hours after transfection, cells expressing the control shRNA were still immunopositive for ARX, whereas most cells transfected with ARX RNAi showed reduced ARX expression (arrows). C, COS-7 cells were transfected with an ARX-expressing plasmid and different shRNA constructs. Two days after transfection, cell lysates were subjected to Western blot analysis using ARX antibody. ARX levels in cells transfected with specific shRNAs (S1–S3) were significantly lower than those in cells transfected with triple-point-mutated shRNAs (S1m–S3m) or a control shRNA (DCX 3UTR3mhp). α-Tubulin detection shows that similar amounts of proteins were loaded on the gel. D, Representative confocal images of ARX protein expression in COS-7 cells transfected with CMV-ARX and either S2 RNAi or a control RNAi (S2m or DCX 3UTR3mhp). E, Effect of the three shRNAs on ARX mRNA stability. COS-7 cells were transfected with psiCheck2-ARX and different shRNAs. Forty-eight hours after transfection, Renilla and firefly luciferase activities were measured. The histogram shows Renilla luciferase data normalized to firefly luciferase measurements. Data represent the mean of 10 wells. Error bars indicate SEM. ***p < 0.001, Student's t test. Scale bars: B, 20 μm; D, 50 μm.
To quantify the efficiency of our shRNAs on the stability of ARX mRNA, we used the psiCheck-2 vector (Promega). This vector contains both a synthetic version of Renilla luciferase reporter gene to monitor RNAi activity and a synthetic firefly luciferase gene that permits normalization of changes in Renilla luciferase expression to the expression of firefly luciferase. We cloned arx mouse cDNA into this vector, downstream to the synthetic Renilla luciferase gene and its translational stop codon. When this construct is transfected into a mammalian cell line, a fusion of the Renilla gene and ARX is transcribed. Functional Renilla luciferase is then translated from the intact transcript. If a specific shRNA binds to ARX mRNA and effectively initiates the RNAi process, the fused Renilla-ARX mRNA sequence is degraded, resulting in reduced Renilla luciferase activity. COS-7 cells were transfected with psiCheck2-ARX and the different shRNAs at a ratio 1:4. Forty-eight hours after transfection, Renilla and firefly luciferase activities were measured. Figure 1E shows the Renilla luciferase measurements normalized to the firefly luciferase ones. ARX expression was knocked down from ∼53% for S3 to 74% for S2. Together, these results show that all three shRNAs efficiently target ARX mRNA and thus reduce its expression. However, because S3 shRNA was the least efficient of the three, we did not use it in further experiments.
ARX affects progenitor cell proliferation
To examine the effects of ARX silencing on cell proliferation, S2 shRNA or a control shRNA (S2m or DCX 3UTR3mhp) were introduced together with a DsRed vector into neural progenitor cells of mouse neocortex by in utero electroporation at E13.5. We found that ARX inactivation resulted in a marked decrease, sometimes even a complete absence, of DsRed-positive cells in the VZ 3 d after transfection (Fig. 2A,B). To establish that this effect was not caused by increased cell death, we labeled sections with anti-caspase 3 antibody. These experiments showed no change in cell death after ARX inactivation compared with controls (data not shown) and suggested that silencing ARX promotes the exit of progenitors from the cell cycle. To test this hypothesis, we used P-H3 antibody, a marker of mitotic cells, to examine a potential reduction of the mitotic-cell fraction in ARX-silenced progenitors (Fig. 2C,D). Three days after electroporation, ∼8.4 ± 1.5% of DsRed-labeled cells in the VZ/SVZ of controls were P-H3 positive (106 cells/1253 DsRed-positive cells). However, when ARX expression was silenced, only 1.2 ± 0.8% DsRed-labeled cells in the VZ/SVZ were P-H3 positive (9 cells/756 DsRed-positive cells). A similar result was obtained with Ki67, another marker of proliferating cells (data not shown). To analyze the identities of DsRed-labeled cells, cortical cells were dispersed 24 h after electroporation and immunolabeled using the neuronal marker Tuj1. When control shRNA construct was introduced, ∼36.9 ± 5.7% of DsRed-labeled cells expressed Tuj1 (101 cells/274 DsRed-positive cells). In contrast, when S2 shRNA was introduced, DsRed and Tuj1 double-positive cells increased to ∼64.7 ± 4.6% (270 cells/417 DsRed-positive cells). These results suggest that ARX inactivation leads progenitors to exit the cell cycle prematurely and adopt the neuronal fate.
Figure 2.
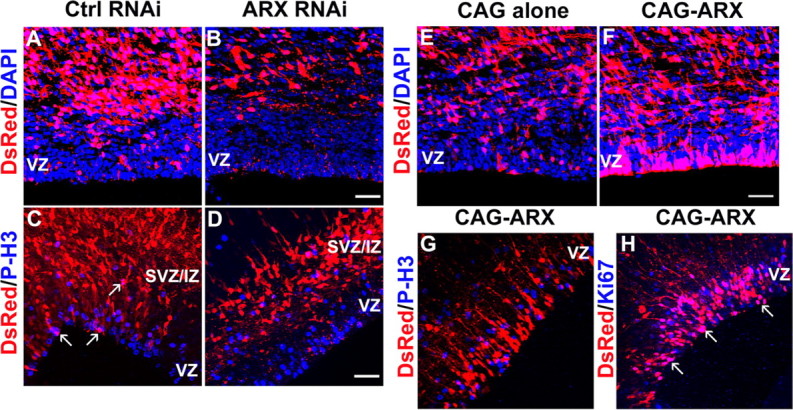
Effect of ARX inactivation or overexpression on cell proliferation. A, B, Examination of E16.5 coronal sections of mouse brains electroporated at E13.5 with S2 shRNA or a control (S2m or DCX 3UTR3mhp) shRNA together with a DsRed-expressing vector. Sections were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI). C, D, Brain sections were stained with an antibody that recognizes the mitotic marker P-H3 (blue). DsRed-positive cells labeled with P-H3 remain active in the cell cycle (arrows). E, F, Examination of E16.5 coronal sections of mouse brains electroporated at E13.5 with an ARX-overexpressing construct or an empty vector together with a DsRed plasmid. G, H, Brain sections were stained with antibodies against Ki67 or the mitotic marker P-H3 (blue). Scale bars, 30 μm.
Conversely, to test the effect of ARX overexpression on cell proliferation, we introduced an ARX-overexpressing construct together with the DsRed vector in cortical progenitor cells using in utero electroporation. Contrary to ARX inactivation, there were substantially more DsRed-positive cells lining the ventricle 3 d after electroporation with CAG-IRES-EGFP-ARX compared with control (Fig. 2E,F). To establish whether these cells were still proliferating, we labeled sections with P-H3 and Ki67 antibodies (Fig. 2G,H). Whereas Ki67 is a marker for proliferating cells from S-phase through M-phase of the cell cycle, P-H3 only labels cells that are in late G2- or M-phase. When the control plasmid was introduced, ∼8.6 ± 2% of DsRed-labeled cells in the VZ/SVZ were P-H3 positive (68 cells/793 DsRed-positive cells), and 10.4 ± 5.1% were Ki67 positive (14 cells/135 DsRed-positive cells). On the contrary, when ARX was overexpressed, only 2 ± 0.8% of the DsRed-positive cells in the VZ/SVZ were P-H3 positive (21 cells/1036 DsRed-positive cells), whereas 65.8 ± 6.6% of the DsRed-positive cells were Ki67 positive (131 cells/199 DsRed-positive cells). These results show that most ARX-overexpressing cells remained in the cell cycle, although they were not dividing. They also suggest the possibility that ARX overexpression increases the length of the cell cycle, and particularly the S- and/or G2 phase.
To further verify this hypothesis, embryos were electroporated with either the ARX-expressing plasmid or the GFP vector alone at E13.5, injected with IdU at E16.5, and then pulse labeled with BrdU 3 h later, before being killed 2 h later (Fig. 3A). Both IdU and BrdU are thymidine analogues, which are incorporated into DNA during the S-phase of the cell cycle. Sections were then labeled with either anti-IdU antibody, which recognizes both BrdU- and IdU-labeled cells, or with an anti-BrdU antibody that recognizes specifically BrdU-positive cells (Fig. 3A). GFP+/IdU+/BrdU+ cells represent electroporated cells that were in S-phase in the interval of 5 h between the IdU injection and the animals being killed, and DsRed+/BrdU+ cells that were in S-phase in the interval of 2 h before the animals were killed. Thus, this experiment allowed us to determine the length of the cell cycle (Tc) and the length of the S-phase (Ts) as explained herein. If we assume that (1) cells comprising the proliferative population are asynchronously distributed in the cell cycle, (2) all cells have similar cycle times, and (3) they are uniformly distributed in the cell cycle, we can then conclude that the number of cells in each phase of the cell cycle is proportional to the ratio of the length of that phase (Fig. 3B), which means that, if we fix the brains just after an injection (t = T0), the proportion of cells in S-phase at that time and, thus, labeled (LI0), is equal to Ts/Tc (Nowakowski et al., 1989) (Fig. 3C). Because only the fraction Ts/Tc of proliferating cells are labeled by one injection of IdU or BrdU, the labeling index reaches a maximum when all cells in S-phase have been labeled by a series of injections. The time it takes to reach this maximum is Tc − Ts, which is the time it takes for a cell that was just at the end of S-phase at the time of the first injection to pass through G2, M, and G1, and then reenter S-phase (Fig. 3B). In this manner, we obtained the graph shown in Figure 3C if we consider that all cells in the VZ are proliferating cells [which means that the growth fraction (GF) equals 1] (Nowakowski et al., 1989). From the slope of the line in this graph, we can also deduce Tc (Fig. 3C).
Figure 3.
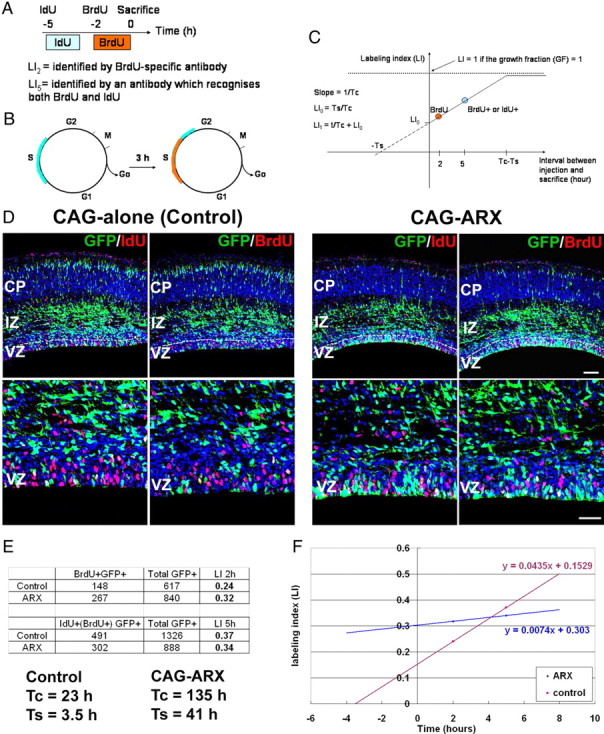
Estimation of the length of the cell cycle of cortical progenitors after ARX overexpression. A, E13.5 mouse embryos were electroporated with either the CAG-IRES-EGFP vector alone or CAG-IRES-EGFP-ARX. IdU was administrated 5 h before the mice were killed at E16.5 and BrdU 3 h later. The availability of BrdU and IdU for incorporation in dividing cells is estimated at ∼2 h. B, The time it takes to label all cells is Tc − Ts, which is the time it takes for a cell that was just at the end of the S-phase at the time of the first injection to pass through G2, M, and G1 and then reenter S-phase. C, Estimation of the proportion of GFP+ cells that were proliferating 2 or 5 h before the mice were killed allows us to estimate Tc and Ts. D–F, Brain sections were stained with antibodies recognizing GFP and either both BrdU- and IdU-positive or specifically BrdU-positive cells (red). Counting the number of GFP cells in the VZ that are BrdU+ or BrdU+/IdU+ showed that ARX overexpression increases the length of the cell cycle compared with control. Sections were counterstained with DAPI (4′,6′-diamidino-2-phenylindole; blue). Scale bars: top, 100 μm; bottom, 50 μm.
We thus counted the proportion of GFP-positive cells in the VZ that were BrdU+ or BrdU+/IdU+ (Fig. 3D,E) and recorded the values obtained on a graph (Fig. 3F). We found that, in the brains electroporated with the control vector, Tc was ∼23 h and Ts = 3.5 h in the VZ (Fig. 3E). These results are very similar to what had been described by Miyama et al. (1997), who estimated 18.4 h for Tc and 4 h for Ts in the dorsal cortical VZ at the same stage (E16). However, when ARX was overexpressed, we obtained much longer times (Tc = 135 h and Ts = 41 h), confirming that ARX overexpression increases the length of the cell cycle.
Both inactivation and overexpression of ARX affect radial migration
In the developing cortex, ARX is expressed in tangentially arranged corridors in the MZ and IZ, the paths populated by migrating GABAergic interneurons arising in the GE (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In addition, it is localized in proliferating cells in the cortical VZ, but not in radially migrating neurons (Colombo et al., 2004; Poirier et al., 2004; Friocourt et al., 2006). Because some defects in the organization of pyramidal neurons have been reported in Arx mutant mice (Kitamura et al., 2002), we examined the effects of ARX silencing in neuronal progenitors on radial migration. We performed experiments similar to those described above, and the proportion of DsRed-positive cells was established in the VZ/SVZ, IZ and cortical plate (CP), 3 or 5 d after electroporation (Fig. 4). At day 3, ARX RNAi led to an accumulation of cells in the IZ (79.1 ± 1.4%; n = 8 embryos) and a decrease in the number of cells in the VZ/SVZ (6.2 ± 0.8%) and CP (14.7 ± 1.2%) compared with controls (VZ/SVZ: 14.7 ± 1%; IZ: 50.2 ± 1.5%; CP: 35.1 ± 1.4; n = 4 embryos) (Fig. 4A–C). Interestingly, the results obtained with the negative control S2m shRNA were somewhat different from those obtained with the CAG-IRES-EGFP empty vector (Fig. 4, compare A, G), for which approximately one-half of DsRed-positive control cells (57 ± 1.4%; n = 4 embryos) had reached the CP at day 3 (Fig. 4I). To ensure that this accumulation of cells in the IZ was not an off-target effect of our RNAi construct, we used DCX 3UTR3mhp as another negative control. Both controls showed very similar cell distribution (data not shown), suggesting that the psiStrike vector itself may have some nonspecific effect on radial cell migration. However, because the results obtained with S2 shRNA were statistically different from both controls, we consider that the effect of ARX RNAi is most likely genuine. Five days after transfection, 64.9% of ARX-inactivated cells (n = 4 embryos) had finally reached the CP, suggesting that ARX suppression delayed rather than fully inhibited radial migration (data not shown).
Figure 4.
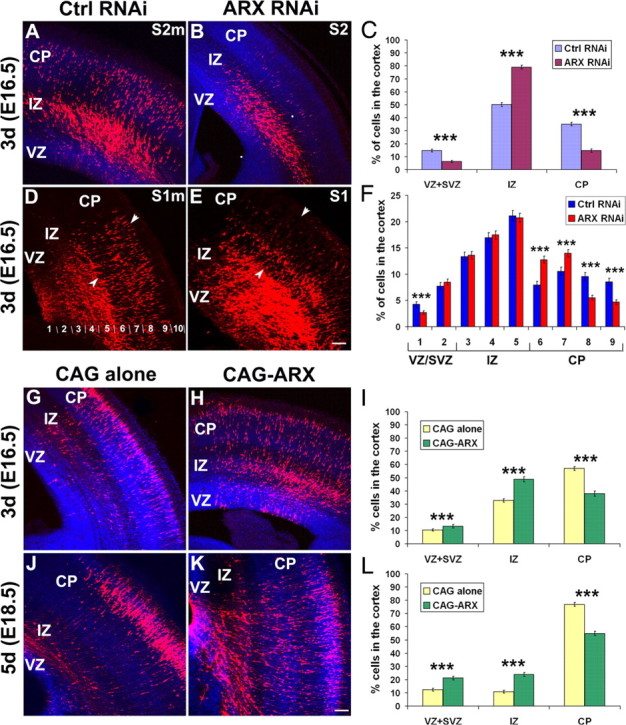
Disruption of radial migration in the neocortex after ARX silencing or overexpression. A–F, Examination of coronal sections of mouse brains electroporated at E13.5 with specific or control shRNAs. A–C, Three days after electroporation, cells expressing S2 shRNA were found primarily in the IZ (79.1 ± 1.4% compared with 50.2 ± 1.5% for S2m shRNA), with only a subset having reached the CP (14.7 ± 1.2% compared with 35.1 ± 1.4% for S2m shRNA). D–F, Although the effect of S1 shRNA on cell migration was less severe, there was an increased percentage of cells transfected with S1 in the deeper part of the CP (bins 6 and 7) and a subsequent decrease in the upper layers (bins 8 and 9) compared with the control. The limits of the CP are indicated by arrowheads. G–L, Examination of coronal sections of mouse brains electroporated at E13.5 with an empty vector or an ARX-overexpressing construct. G–I, By day 3, embryos electroporated with an ARX-overexpressing plasmid showed greater cell dispersion and fewer cells in the CP compared with controls (CAG-ARX: 38 ± 2%, vs 57 ± 1.4% for the control). J–L, By day 5, a large majority of control cells had reached the CP (76.9 ± 1.4%). In contrast, only one-half of the cells expressing ARX were located in the CP (54.9 ± 1.7%). Sections were counterstained with DAPI (4′,6′-diamidino-2-phenylindole; blue). Error bars indicate SEM. ***p < 0.001, χ2 test. Scale bars, 100 μm.
To rule out the possibility that the observed defect in radial migration was a nonspecific effect of S2 shRNA, we used the S1 RNAi construct targeting another region of ARX (Fig. 4D–F). Although there was no major difference between the overall distribution of ARX-inactivated cells (VZ/SVZ: 11.2 ± 0.6%; IZ: 51.8 ± 1%; CP: 38.1 ± 1; n = 4 embryos) compared with control S1m (VZ/SVZ: 12.1 ± 0.8%; IZ: 51.4 ± 1.2%; CP: 36.5 ± 1.2; n = 4 embryos), we noted an accumulation of cells in the deeper aspect of the CP 3 d after electroporation (Fig. 4D,E). Thus, we examined cell distribution in the two conditions by dividing the cortex into 10 equally spaced divisions (bin 1 corresponding to the VZ of the cortex and bin 10 to the MZ) (Fig. 4D), and estimated the percentage of labeled cells in each bin. These results confirmed the accumulation of cells in the deeper part of the CP when ARX expression was silenced (Fig. 4F, bins 6 and 7). This finding strongly suggests that ARX inactivation in cortical progenitors has a direct effect on radial migration during cortical development.
Because ARX inactivation in progenitor cells seems to impair radial migration, we also studied the effect of ectopic ARX expression in radially migrating cells. Three days after transfection, only 38 ± 1.9% of ARX-expressing cells had reached the CP (n = 5 embryos), and 48.8 ± 2% were still in the IZ, compared with 32.6 ± 1.3% (n = 4 embryos) for the vector alone (Fig. 4G–I). By day 5, the majority of control cells had migrated to the CP (76.9 ± 1.4%; n = 3 embryos) compared with only 54.9 ± 1.7% of ARX-expressing cells (n = 4 embryos) (Fig. 4J–L). We also noted that the distribution of ARX-expressing cells was impaired not only in dorsal cortex, but also in lateral cortex (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Together, these results suggest that ARX expression is necessary for proper radial migration of pyramidal neurons during cortical development.
Morphology of radially migrating cells after inactivation or overexpression of ARX in cortical progenitors
To better understand the observed defect in radial migration, we examined the morphology of migrating cells. When ARX expression was knocked down using S2 shRNA, cells accumulated in the IZ (Figs. 4A–C, 5A,B). Although there was also some accumulation in the control, the morphology of the cells was different between S2 and the control shRNA (S2m or DCX 3UTR3mhp). The majority of control cells in the SVZ and the middle and lower parts of the IZ exhibited the previously described multipolar morphology (Fig. 5C, arrows and insets), whereas cells in the upper part of the IZ and in the CP had a bipolar form (Tabata and Nakajima, 2003). In contrast, ARX-silenced cells in the SVZ and IZ appeared more round, with very few or no visible processes (Fig. 5B,D). Only a few cells in the upper IZ had a normal bipolar morphology as they migrated toward the CP (Fig. 5D). These results suggest that loss of ARX expression in cortical progenitors leads to an alteration in the morphology of radially migrating neurons, resulting in their accumulation in the SVZ/IZ and impairment of their migration into the CP.
Figure 5.
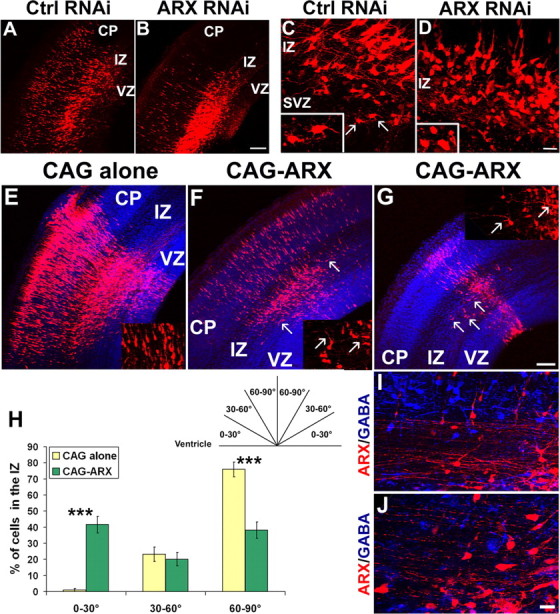
Morphology of radially migrating neurons after ARX inactivation or overexpression in cortical progenitors. A–D, Morphology of cells in the SVZ/IZ of E16.5 mouse brains electroporated 3 d earlier with a specific (S2) or a control (S2m or DCX 3UTR3mhp) shRNA together with a DsRed-expressing plasmid. Many control cells migrated to the IZ and CP, where they became bipolar, whereas most ARX-inactivated cells in the IZ appeared round, showing very few or no processes (see insets). E–G, Examination of coronal sections of E16.5 mouse brains electroporated at E13.5 with an empty vector or an ARX-overexpressing construct. Tangentially orientated cells migrating away from the site of injection were detectable in the IZ of sections electroporated with ARX (see arrows). Some of these cells had long and complex processes, orientated tangentially (see insets). H, These cells represented ∼41.6 ± 5.1% of the total number of cells with visible processes in the IZ, versus 0.9 ± 1% for the control. I, J, Labeling of sections with anti-GABA antibody (blue) showed that none of these cells was GABA positive. Error bars indicate SEM. ***p < 0.001, χ2 test. Scale bars: A, B, E–G, 100 μm; C, D, I, J, 50 μm.
Because ectopic expression of ARX in migrating neurons also impairs radial migration, we examined the morphology of ARX-overexpressing cells. We found that the brains of E16.5 mice electroporated with CAG-IRES-EGFP-ARX contained several cells that appeared to migrate tangentially in the SVZ and lower IZ, and away from the site of injection; such cells were not present in control brains (Fig. 5E–G, arrows). In addition, most of these cells exhibited complex branching, and some had very long processes (Fig. 5F,G, insets). To quantify the number of these tangentially orientated neurons, we counted cells with visible processes in the SVZ and lower IZ of injected animals and separated them into three categories according to orientation of their main process (Fig. 5H). We found that, 3 d after electroporation, most of the control cells were radially orientated (75.9 ± 4.6%; n = 3 embryos), and only a few were orientated tangentially (0.9 ± 1%), whereas ARX-overexpressing animals displayed an increased percentage of tangentially orientated cells (41.6 ± 5.1%; n = 3 embryos) and, consequently, a decrease in the proportion of radially migrating neurons (38.2 ± 5.1%) (Fig. 5H). To rule out the possibility that these tangentially orientated cells were GABAergic interneurons electroporated while in the VZ, we stained a number of sections with GABA antibody. However, none of the DsRed-positive cells expressed GABA (Fig. 5I,J), which confirms that interneurons migrating into the cortex from the subpallium are not electroporated with this technique, as previously demonstrated (Tabata and Nakajima, 2003). Similar results were obtained using anti-calbindin antibody (data not shown). Together, these results suggest that ARX may have a role in cell morphology and particularly in the formation of processes, which may explain the radial migration defects.
The level of ARX expression is important for tangential migration
Several studies have reported the expression of ARX in interneurons migrating from the MGE to the cortex during embryonic development (Kitamura et al., 2002; Poirier et al., 2004; Colombo et al., 2004; Friocourt et al., 2006). To assess whether this gene also plays a cell-autonomous role in tangential migration, we isolated E14.5 mouse telencephalic hemispheres and performed focal electroporation into the MGE (Yozu et al., 2005). As previously, S2 and control RNAi constructs were electroporated together with the DsRed vector to visualize the transfected cells (Fig. 6). After electroporation, the telencephalic hemispheres were cultured for 40 h and cut into 100-μm-thick coronal sections, and the positions of the electroporated cells were noted. In control brains, DsRed-positive cells appeared dispersed predominantly in the GE, with a few also present in the cortex (Fig. 6A). However, in ARX-inactivated brains, most of the cells were still located in the MGE and had not migrated away from the site of injection (indicated by an asterisk). Although some cells had initiated their migration, a very small number had actually entered the cortex (Fig. 6B). To quantify this defect in migration, we estimated the proportion of DsRed-positive cells that had reached the cortex. We observed that in control brains, ∼11.3 ± 1.2% cells (n = 3 embryos) had crossed the corticostriatal boundary and had entered the cortex, whereas sections taken from brains electroporated with S2 shRNA had only 3.1 ± 0.7% cells in the cortex (n = 3 embryos). These results suggest that ARX inactivation impairs tangential migration in a cell-autonomous way. Interestingly, we did not observe any difference in cell morphology between control and ARX-inactivated cells (Fig. 6E).
Figure 6.
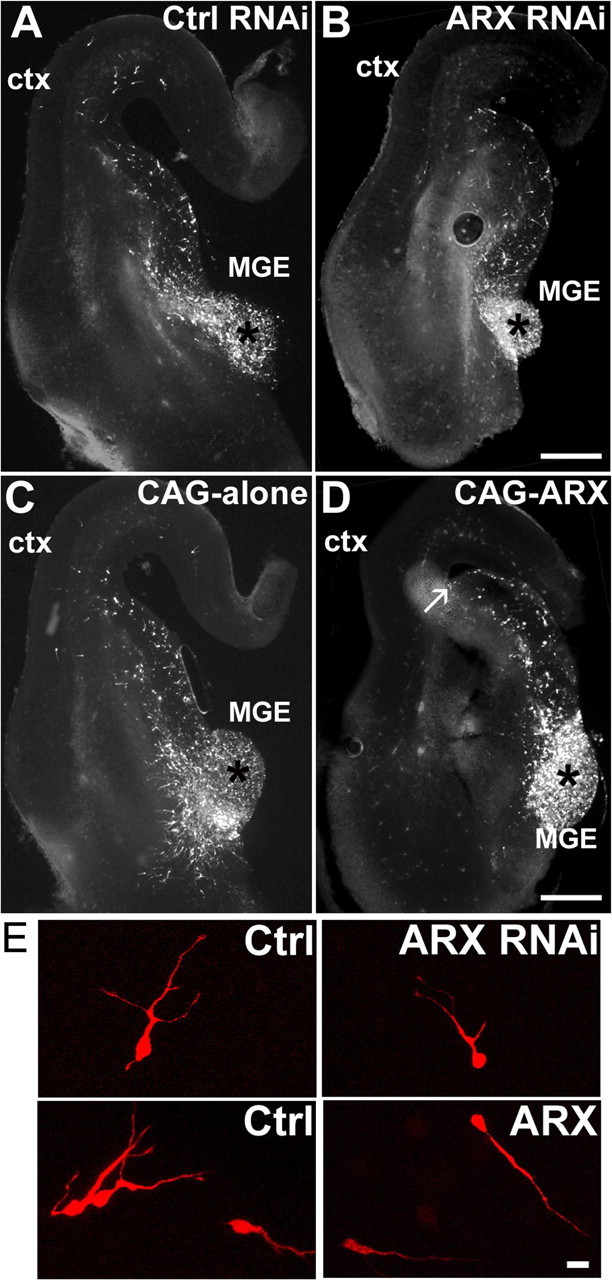
ARX inactivation or overexpression impairs interneuron migration to the neocortex. A, B, Representative images of 100-μm-thick sections of E14.5 brains electroporated with the RNAi constructs together with a DsRed plasmid and cultured for 40 h. Some DsRed-positive cells migrated from the site of injection in the MGE (indicated by an asterisk) to the cortex (ctx), but there were fewer cells in ARX-inactivated cortex compared with control. C, D, Representative images of 100-μm-thick sections of E14.5 brains electroporated with CAG-IRES-EGFP or CAG-ARX together with a DsRed plasmid. ARX overexpression resulted in labeled cells dispersing within the GE, but not entering the cortex. Neurons seemed to accumulate at the corticostriatal boundary (see arrow). E, Morphology of the migrating cells. Although there was no difference in morphology between control cells and ARX-silenced cells, we observed that several ARX-overexpressing neurons appeared bipolar with a uniquely long process. Scale bars: A–D, 200 μm; E, 20 μm.
We then performed similar experiment to assess the effect of ARX overexpression on tangential migration. Surprisingly, the overexpression of ARX seemed to have an even stronger effect on tangential migration. Although there was dispersion from the site of injection, most of the migrating cells only reached the corticostriatal boundary without crossing it (Fig. 6C,D). The proportion of cells that had entered the cortex was estimated and was found to be even lower than with ARX RNAi [1.8 ± 0.7 for CAG-ARX (n = 6 embryos), compared with 10.7 ± 2.4 (n = 4 embryos) for the vector alone]. To establish that this effect was not a consequence of a proliferation defect, we stained a few sections with Ki67 antibody but did not observe any difference with the control, suggesting that, in contrast to the cortex, ARX does not play any role in the regulation of cell proliferation in the basal forebrain (data not shown). We then examined the morphology of migrating cells and found that most had a bipolar appearance with a very long process, different from the typical morphology of migrating interneurons seen in controls (Fig. 6E). Taken altogether, these results indicate that ARX is necessary for tangential migration of interneurons, possibly by regulating cell morphology and process formation during migration.
ARX does not control the expression of GABA in cortical interneurons
The high degree of colocalization that exists between ARX and GABA in embryonic and adult brains and the observation that ARX expression is regulated by Dlx genes, which are strong candidates for regulating the differentiation of most, if not all, telencephalic GABAergic neurons (Stühmer et al., 2002b), have led to the suggestion that this gene might be involved in the specification of the GABAergic phenotype (Cobos et al., 2005). To test this hypothesis, we examined the degree of colocalization between ARX and GABA. We first performed immunohistochemistry on E16 rat cortical sections (Fig. 7A1–A3). As previously shown, ARX antibody labels cells in the VZ of the developing cortex and in streams of migrating interneurons that express GABA. Although the vast majority of these cells expressed both markers, we noted the presence of ARX-positive cells that were GABA negative (Fig. 7A1–A3, arrows). To further verify the degree of colocalization of these two molecules, we performed double immunolabeling experiments on dissociated cell cultures prepared from E16 rat cortices and kept in culture for 3, 6, or 9 d (Fig. 7B1–B3). We noticed at the three stages that, although most of the ARX-expressing cells were GABA-positive, not all GABAergic cells expressed ARX (Fig. 7B1–B3, arrowheads). Similar experiments were performed in ventral forebrain neurons. As previously described, our ARX antibody does not label the VZ of the MGE and LGE, suggesting that ARX is only expressed in postmitotic neurons in the ventral forebrain (Friocourt et al., 2006) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). However, to avoid any potential contamination with progenitors that might express ARX but not GABA, we prepared dissociated cultures from E16 striatum without the VZ/SVZ and left them in culture for 3, 6, or 9 d (Fig. 7C1–C3). Similarly, we observed that only a proportion of ARX-positive neurons contained GABA and vice versa (61–65% of ARX-positive cells expressed GABA and 58–72% of GABAergic cells expressed ARX in dissociated cultures from rat E16 striatum) (Fig. 7D). These results suggest that ARX is not fully associated with the expression of GABA in both cortical and subpallial neurons.
Figure 7.
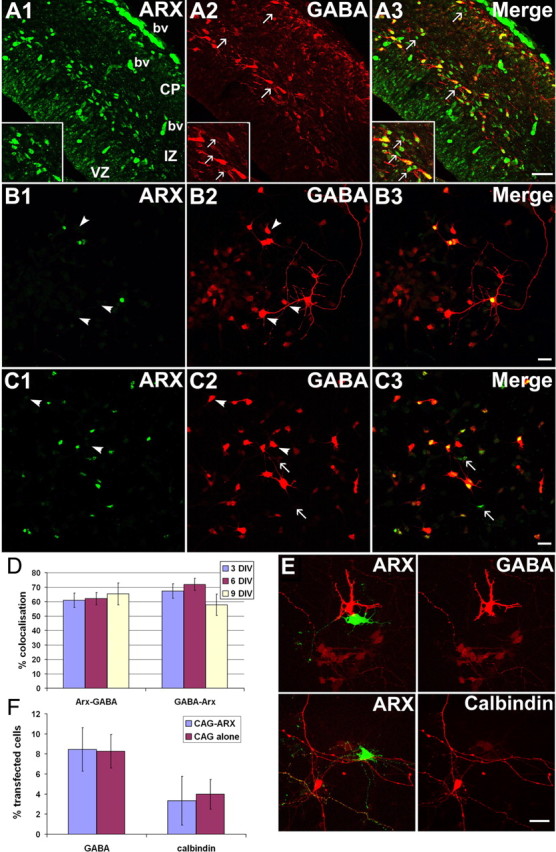
ARX does not control GABAergic cell specification. A1–A3, Colabeling ARX/GABA on E16 rat cortical sections. Some ARX-positive cells appear GABA− (see arrows and insets). bv, Blood vessels. B1–B3, Colabeling ARX/GABA in dissociated E16 rat cortical cultures. Not all GABAergic cells express ARX (arrowheads). C1–C3, Colabeling ARX/GABA in dissociated E16 cultures from striatum. A few GABAergic cells negative for ARX (arrowheads) and some ARX-positive cells not expressing GABA (arrows) were observed. D, Quantification of the colocalization between ARX and GABA in dissociated E16 cultures from striatum kept for 3, 6, or 9 d in vitro. E, ARX overexpression does not induce GABA or calbindin expression in rat E16 dissociated cultures from cortex or GE. F, Percentage of GFP+ cells expressing GABA or calbindin after transfection with CAG-ARX or the vector alone. Scale bars: A1–A3, 100 μm; B1–C3, E, 40 μm.
To further test whether ARX regulates GABAergic cell differentiation, rat E16 cortical and GE cell cultures were transfected with either CAG-IRES-EGFP-ARX or the GFP-vector alone, and immunolabeled with interneuron markers such as GABA or calbindin. Two days after transfection, we found that ARX overexpression did not affect the expression of these markers in cortical or GE cultures (Fig. 7E,F). Similar results were obtained after fixing the culture 4 or 6 d after transfection (data not shown). Conversely, to assess the effect of ARX silencing, GE cultures were transfected with RNAi constructs and labeled with the same markers. As previously, we did not observe any difference in GABA or calbindin expression (data not shown). Altogether, these results suggest that ARX is neither necessary nor sufficient for the expression of the neurotransmitter GABA in the forebrain.
Discussion
Several defects in corticogenesis have been attributed to absence of ARX based on analysis of mutant mice (Kitamura et al., 2002) (for review, see Friocourt et al., 2006). However, the plethora of defects has made it difficult to differentiate between primary and secondary functions of this gene in the developing brain. For this reason, we used a combination of in utero electroporation (Saito and Nakatsuji, 2001; Tabata and Nakajima, 2001) and RNAi-mediated silencing or overexpression approaches to dissect the role(s) of ARX in cortical development. Our results have shown that ARX plays distinct cell-autonomous roles in (1) the regulation of the cell cycle of cortical progenitor cells; (2) the morphology and radial migration of pyramidal neurons; and (3) the tangential migration of interneurons from the ventral telencephalon; however, ARX does not control the development of their GABAergic phenotype.
Role of ARX in progenitor cell proliferation
Several studies have reported that progenitor cells in the cerebral cortex express ARX, which is later downregulated as cells exit the VZ (Kitamura et al., 2002; Colombo et al., 2004; Poirier et al., 2004; Friocourt et al., 2006). Here, we demonstrated that targeted inhibition of ARX in these cells caused their premature exit from the cell cycle and differentiation into neurons. From our results, it is difficult to determine whether the effect of ARX inactivation was primarily to drive progenitors prematurely out of the cell cycle or to induce their neuronal differentiation. It is, indeed, possible that once they become postmitotic, these cells adopt a neuronal fate as a default response to environmental cues. However, the finding that ARX overexpression increased the length of the cell cycle favors a role for this gene in maintaining progenitor proliferation.
Our observations are also consistent with the microcephaly (small brain) phenotype observed in Arx mutant mice and patients with XLAG (Kitamura et al., 2002; Kato et al., 2004). This suggests that the level of ARX expression is important for the control of production of cortical neurons and the regulation of cerebral cortical size, similar to what has been described for β-catenin (Chenn and Walsh, 2003; Woodhead et al., 2006). Mutations in several genes are responsible for microcephaly in humans, including ASPM (abnormal spindle in microcephaly), microcephalin, CDK5RAP2, and CenPJ (Woods, 2004). All these genes are associated with key elements of mitosis, such as the centrosome and mitotic spindles. Although the genes regulated by ARX have not yet been identified, it would be interesting to test whether it acts on one of these elements.
Role of ARX in radial migration
Earlier studies have indicated that Arx is strongly expressed in migrating GABAergic interneurons but is absent from radially migrating cells (Colombo et al., 2004; Poirier et al., 2004; Friocourt et al., 2006). This raised the question as to what causes the misplacement of pyramidal neurons observed in the cortex of Arx knock-out mice (Kitamura et al., 2002). To establish whether ARX has a direct role in radial migration, we studied the effects of its inactivation or overexpression by using in utero electroporation. In both cases, we observed defects in neuronal migration, with cells accumulating in the IZ. We also noticed that ARX-inactivated cell bodies were oval or round in shape, with very few or no processes, very different from the multipolar morphology normally exhibited when cells exit the VZ and enter the lower IZ (Tabata and Nakajima, 2003). Since the first description of the multipolar stage during radial migration, several studies have suggested that the transition into and out of this stage is particularly vulnerable and that it is disrupted in several disorders of neocortical development, including lissencephaly (LoTurco and Bai, 2006). Cell morphology defects, very similar to those induced by RNAi-mediated inactivation of ARX, have been reported after Filamin A (Nagano et al., 2004), Rac1, or Cdk5/p27 (Kawauchi et al., 2003, 2006) inactivation. Interestingly, all these proteins interact with the actin cytoskeleton, which suggests that ARX could play a previously unsuspected role in cell morphology through actin regulation.
It is difficult to determine from our findings whether neurons fail to undergo appropriate radial migration as a consequence of the observed proliferation defect or whether neurons require a factor whose expression is controlled by ARX for their radial migration. It is important to note that, because ARX is not normally expressed in radially migrating neurons, it is not the absence of ARX itself that accounts for the migration defect. The observed changes in morphology after gain or loss of function tend to support two distinct roles for ARX. Neurogenesis and neuronal migration have in some cases appeared as two independent processes, as is the case in mice lacking the cell cycle inhibitors p19Ink4d and p27kip1, in which a population of cortical neurons fail to exit the cell cycle, but migrate to the cortex (Zindy et al., 1999). It should also be pointed out that we did not observe any change in the overall orientation of radial glial processes or their morphology (data not shown), which suggests that the neuronal migration defects are distinct from the proliferation ones.
Role of ARX in tangential migration
A number of studies have documented the absence of interneurons in the cortex of XLAG patients (Bonneau et al., 2002; Forman et al., 2005) and a defect in tangential migration of cortical interneurons in Arx mutant mice (Kitamura et al., 2002; Colombo et al., 2007), suggesting the importance of ARX in this process. To analyze the tangential migration of GABAergic interneurons from the ventral telencephalon to the developing neocortex, we used a technique that combines focal electroporation of the MGE and whole-hemisphere cultures (Yozu et al., 2005). In these experiments, we observed that both inactivation and overexpression of ARX resulted in impairment of cortical interneuron migration from the MGE. We were unable to assess whether this defect was attributable to a delay in migration rather than cessation of cell movement. The fact that there was reduced cell dispersion from the site of injection, especially for ARX-silenced cells, suggests that the motility of neurons was affected. When we examined cell morphology, we noted that, unlike control-electroporated cells, many neurons that overexpressed ARX had one unusually long process. However, we did not detect any difference in cell morphology between ARX-inactivated cells and control cells. Interestingly, a recent study found that interneurons derived from Arx mutant mice have longer processes and increased branching while migrating in a Matrigel matrix (Colombo et al., 2007). This discrepancy may be attributable to differences in experimental methods applied (RNAi vs genetic deletion). Indeed, because our shRNAs are not 100% effective, some amount of ARX protein is still present in cells targeted with RNAi.
Role of ARX in cell differentiation
The expression of ARX in regions rich in GABAergic cells as well as the diminution of cortical interneurons in patients with XLAG have prompted speculation that this gene may be involved in the specification of the GABAergic phenotype of cortical interneurons. However, we present here several arguments against this hypothesis: (1) the colocalization of ARX and GABA in cortex or GE is not complete; (2) ARX overexpression in cortical progenitors in vivo does not induce GABA or calbindin expression, even 5 d after electroporation; (3) ARX overexpression does not induce GABA expression in dissociated cultures from E16 rat cortex or GE. In addition, the observation that the introduction of an ARX-overexpressing construct into the VZ of the dorsal telencephalon induces tangential migration within the IZ, although these cells do not express GABA, suggests that ARX may be involved in migration rather than cell specification or differentiation. We thus present a model that suggests a way the ARX and Dlx genes regulate the specification and migration of cortical GABAergic interneurons (Fig. 8). Although ARX is downstream of the Dlx genes, it may only regulate migration, whereas the Dlx genes may also control the expression of GABA through other genes (Anderson et al., 1997, 1999; Stühmer et al., 2002a).
Figure 8.

Model of the relationship between ARX and the Dlx genes that control the specification and migration of GABAergic neurons. A, The Dlx genes have been found to be both necessary and sufficient for the control of the cortical GABAergic phenotype. In contrast, our results suggest a role of ARX in migration of these neurons rather than in their specification. This model thus explains why Dlx1/2 knock-out mice lack most of GABAergic cells in the cortex and the olfactory bulb (Anderson et al., 1997). In the case of Arx mutant mice, these neurons may fail to differentiate correctly because, as a result of impaired migration, they do not receive the correct cues from their environment (Kitamura et al., 2002). B, Concerning the basal telencephalon, other genes such as Mash1 can probably replace Dlx1 and Dlx2 in controlling GABAergic specification, which explains why in the Dlx1/2 mutant animals, many of the cells that accumulate within the striatal SVZ still express GABA and GAD67 despite their failure to migrate into the striatum (Anderson et al., 1999). In addition, ARX may also be necessary for cell migration in the striatum because the SVZ of the MGE and LGE has been reported to be enlarged in the absence of ARX, with a marked decrease in the number of GABAergic cells in the striatum and the ventral pallidum (Colombo et al., 2007).
However, we cannot exclude a potential role for ARX in the specification of distinct subsets of GABAergic neurons in the subpallium. Indeed, two different studies have reported a decrease in cholinergic neurons in the basal forebrain of Arx mutants, suggesting that ARX, uniquely or in combination with other transcription factors, may play a role in the specification of at least one subpopulation of GABAergic cells (Kitamura et al., 2002; Colombo et al., 2007). In addition, the observation that ARX is still expressed in GABAergic cells in the adult brain suggests that it may also have a role in more mature neurons.
Pathophysiology of phenotypes resulting from ARX mutations
Work to date has shown that ∼10 different syndromes, with or without brain malformations, are associated with mutations of ARX. Several studies have shown that there is a connection between altered neuronal positioning and susceptibility to epilepsy (Kriegstein, 2005). Moreover, subtle alterations in interneuron position and/or function have been reported to contribute to disorders such as dyslexia, schizophrenia, or mental retardation (Kamiya et al., 2005; Galaburda et al., 2006). Our data are consistent with these findings, because both ARX inactivation and overexpression affect cell proliferation and neuronal migration, suggesting that a precise balance in the expression of this gene is necessary for the correct development of the cortex.
In the future, a better understanding of ARX-related disorders will require the identification of the pathways involving this gene. Because it is implicated in several processes that are crucial for cortical development, it is likely that some of the genes regulated by ARX may be potential candidates for neurological diseases such as epilepsy, mental retardation, schizophrenia, dyslexia, autism, and cortical malformations (such as lissencephaly and microcephaly or macrocephaly). Interestingly, a recent study has shown that mutant mice for N-cofilin, an F-actin-depolymerizing factor, display impaired radial and tangential migration, as well as an increased cell cycle exit of neuronal progenitors in the VZ (Bellenchi et al., 2007). These results, similar to what we have shown for ARX, demonstrate that mutations affecting regulators of the actin cytoskeleton contribute to the pathology of cortex development, and open new prospects concerning the pathways regulated by ARX.
Footnotes
This work was supported by a Wellcome Trust Programme Grant 074549 (J.G.P.) and by the Japan Society for the Promotion of Science, the Ministry of Education, Culture, Sports, Science, and Technology, Japan, the Ichiro Kanehara Foundation, the Tokyo Biochemical Research Foundation, the Sumitomo Foundation, the Uehara Memorial Foundation, the Novartis Foundation, and the Brain Science Foundation (K.N.). G.F. was supported in part by a Prix Jeune Chercheur from the Fondation Bettencourt-Schueller. We are grateful to Karine Poirier and David Hunt for advice with the cloning of the rat ARX gene, Kathy Tamai and David Whitmore for their help with the luminometer, Mary Rahman for technical help, and Clare Faux and William Andrews for critical reading of this manuscript.
References
- Anderson S, Mione M, Yun K, Rubenstein JL. Differential origins of neocortical projection and local circuit neurons: role of Dlx genes in neocortical interneuronogenesis. Cereb Cortex. 1999;9:646–654. doi: 10.1093/cercor/9.6.646. [DOI] [PubMed] [Google Scholar]
- Anderson SA, Eisenstat DD, Shi L, Rubenstein JLR. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science. 1997;278:474–476. doi: 10.1126/science.278.5337.474. [DOI] [PubMed] [Google Scholar]
- Bai J, Ramos RL, Ackman JB, Thomas AM, Lee RV, LoTurco JJ. RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat Neurosci. 2003;6:1277–1283. doi: 10.1038/nn1153. [DOI] [PubMed] [Google Scholar]
- Bellenchi GC, Gurniak CB, Perlas E, Middei S, Ammassari-Teule M, Witke W. N-cofilin is associated with neuronal migration disorders and cell cycle control in the cerebral cortex. Genes Dev. 2007;21:2347–2357. doi: 10.1101/gad.434307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienvenu T, Poirier K, Friocourt G, Bahi N, Beaumont D, Fauchereau F, Ben Jeema L, Zemni R, Vinet MC, Francis F, Couvert P, Gomot M, Moraine C, van Bokhoven H, Kalscheuer V, Frints S, Gecz J, Ohzaki K, Chaabouni H, Fryns JP, et al. ARX, a novel Prd-class-homeobox gene highly expressed in the telencephalon, is mutated in X-linked mental retardation. Hum Mol Genet. 2002;11:981–991. doi: 10.1093/hmg/11.8.981. [DOI] [PubMed] [Google Scholar]
- Bonneau D, Toutain A, Laquerrière A, Marret S, Saugier-Veber P, Barthez MA, Radi S, Biran-Mucignat V, Rodriguez D, Gélot A. X-linked lissencephaly with absent corpus callosum and ambiguous genitalia (XLAG): clinical, magnetic resonance imaging, and neuropathological findings. Ann Neurol. 2002;51:340–349. doi: 10.1002/ana.10119. [DOI] [PubMed] [Google Scholar]
- Chenn A, Walsh CA. Increased neuronal production, enlarged forebrains and cytoarchitectural distortions in beta-catenin overexpressing transgenic mice. Cereb Cortex. 2003;13:599–606. doi: 10.1093/cercor/13.6.599. [DOI] [PubMed] [Google Scholar]
- Cobos I, Broccoli V, Rubenstein JLR. The vertebrate ortholog of Aristaless is regulated by Dlx genes in the developing forebrain. J Comp Neurol. 2005;483:292–303. doi: 10.1002/cne.20405. [DOI] [PubMed] [Google Scholar]
- Colombo E, Galli R, Cossu G, Gécz J, Broccoli V. Mouse orthologue of ARX, a gene mutated in several X-linked forms of mental retardation and epilepsy, is a marker of adult neural stem cells and forebrain GABAergic neurons. Dev Dyn. 2004;231:631–639. doi: 10.1002/dvdy.20164. [DOI] [PubMed] [Google Scholar]
- Colombo E, Collombat P, Colasante G, Bianchi M, Long J, Mansouri A, Rubenstein JL, Broccoli V. Inactivation of Arx, the murine ortholog of the X-linked lissencephaly with ambiguous genitalia gene, leads to severe disorganization of the ventral telencephalon with impaired neuronal migration and differentiation. J Neurosci. 2007;27:4786–4798. doi: 10.1523/JNEUROSCI.0417-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- des Portes V, Pinard JM, Billuart P, Vinet MC, Koulakoff A, Carrie A, Gelot A, Dupuis E, Motte J, Berwald-Netter Y, Catala M, Kahn A, Beldjord C, Chelly J. A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell. 1998;92:51–61. doi: 10.1016/s0092-8674(00)80898-3. [DOI] [PubMed] [Google Scholar]
- Dobyns WB, Andermann E, Andermann F, Czapansky-Beilman D, Dubeau F, Dulac O, Guerrini R, Hirsch B, Ledbetter DH, Lee NS, Motte J, Pinard JM, Radtke RA, Ross ME, Tampieri D, Walsh CA, Truwit CL. X-linked malformations of neuronal migration. Neurology. 1996;47:331–339. doi: 10.1212/wnl.47.2.331. [DOI] [PubMed] [Google Scholar]
- Dobyns WB, Berry-Kravis E, Havernick NJ, Holden KR, Viskochil D. X-linked lissencephaly with absent corpus callosum and ambiguous genitalia. Am J Med Genet. 1999;86:331–337. [PubMed] [Google Scholar]
- Forman MS, Squier W, Dobyns WB, Golden JA. Genotypically defined lissencephalies show distinct pathologies. J Neuropathol Exp Neurol. 2005;64:847–857. doi: 10.1097/01.jnen.0000182978.56612.41. [DOI] [PubMed] [Google Scholar]
- Friocourt G, Poirier K, Rakic S, Parnavelas JG, Chelly J. The role of ARX in cortical development. Eur J Neurosci. 2006;23:869–876. doi: 10.1111/j.1460-9568.2006.04629.x. [DOI] [PubMed] [Google Scholar]
- Friocourt G, Liu JS, Antypa M, Rakic S, Walsh CA, Parnavelas JG. Both doublecortin and doublecortin-like kinase play a role in cortical interneuron migration. J Neurosci. 2007;27:3875–3883. doi: 10.1523/JNEUROSCI.4530-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galaburda AM, LoTurco J, Ramus F, Fitch RH, Rosen GD. From genes to behavior in developmental dyslexia. Nat Neurosci. 2006;9:1213–1217. doi: 10.1038/nn1772. [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, Cooper EC, Dobyns WB, Minnerath SR, Ross ME, Walsh CA. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92:63–72. doi: 10.1016/s0092-8674(00)80899-5. [DOI] [PubMed] [Google Scholar]
- Hong SE, Shugart YY, Huang DT, Shahwan SA, Grant PE, Hourihane JO, Martin ND, Walsh CA. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet. 2000;26:93–96. doi: 10.1038/79246. [DOI] [PubMed] [Google Scholar]
- Kamiya A, Kubo K, Tomoda T, Takaki M, Youn R, Ozeki Y, Sawamura N, Park U, Kudo C, Okawa M, Ross CA, Hatten ME, Nakajima K, Sawa A. A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nat Cell Biol. 2005;7:1167–1178. doi: 10.1038/ncb1328. [DOI] [PubMed] [Google Scholar]
- Kato M, Das S, Petras K, Kitamura K, Morohashi K, Abuelo DN, Barr M, Bonneau D, Brady AF, Carpenter NJ, Cipero KL, Frisone F, Fukuda T, Guerrini R, Iida E, Itoh M, Feldman Lewanda A, Nanba Y, Oka A, Proud VK, et al. Mutations of ARX are associated with striking pleiotropy and consistent genotype-phenotype correlation. Hum Mutat. 2004;23:147–159. doi: 10.1002/humu.10310. [DOI] [PubMed] [Google Scholar]
- Kawauchi T, Chihama K, Nabeshima Y, Hoshino M. The in vivo roles of STEF/Tiam1, Rac1 and JNK in cortical neuronal migration. EMBO J. 2003;22:4190–4201. doi: 10.1093/emboj/cdg413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi T, Chihama K, Nabeshima Y, Hoshino M. Cdk5 phosphorylates and stabilizes p27kip1 contributing to actin organization and cortical neuronal migration. Nat Cell Biol. 2006;8:17–26. doi: 10.1038/ncb1338. [DOI] [PubMed] [Google Scholar]
- Keays DA, Tian G, Poirier K, Huang GJ, Siebold C, Cleak J, Oliver PL, Fray M, Harvey RJ, Molnar Z, Pinon MC, Dear N, Valdar W, Brown SD, Davies KE, Rawlins JN, Cowan NJ, Nolan P, Chelly J, Flint J. Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell. 2007;128:45–57. doi: 10.1016/j.cell.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura K, Yanazawa M, Sugiyama N, Miura H, Iizuka-Kogo A, Kusaka M, Omichi K, Suzuki R, Kato-Fukui Y, Kamiirisa K, Matsuo M, Kamijo SI, Kasahara M, Yoshioka H, Ogata T, Fukuda T, Kondo I, Kato M, Dobyns WB, Yokoyama M, et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet. 2002;32:359–369. doi: 10.1038/ng1009. [DOI] [PubMed] [Google Scholar]
- Kriegstein AR. Constructing circuits: neurogenesis and migration in the developing neocortex. Epilepsia. 2005;46(Suppl 7):15–21. doi: 10.1111/j.1528-1167.2005.00304.x. [DOI] [PubMed] [Google Scholar]
- Kriegstein AR, Noctor SC. Patterns of neuronal migration in the embryonic cortex. Trends Neurosci. 2004;27:392–399. doi: 10.1016/j.tins.2004.05.001. [DOI] [PubMed] [Google Scholar]
- LoTurco JJ, Bai J. The multipolar stage and disruptions in neuronal migration. Trends Neurosci. 2006;29:407–413. doi: 10.1016/j.tins.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Marin O, Rubenstein JL. Cell migration in the forebrain. Annu Rev Neurosci. 2003;26:441–483. doi: 10.1146/annurev.neuro.26.041002.131058. [DOI] [PubMed] [Google Scholar]
- Metin C, Baudoin JP, Rakic S, Parnavelas JG. Cell and molecular mechanisms involved in the migration of cortical interneurons. Eur J Neurosci. 2006;23:894–900. doi: 10.1111/j.1460-9568.2006.04630.x. [DOI] [PubMed] [Google Scholar]
- Miyama S, Takahashi T, Nowakowski RS, Caviness VS., Jr A gradient in the duration of the G1 phase in the murine neocortical proliferative epithelium. Cereb Cortex. 1997;7:678–689. doi: 10.1093/cercor/7.7.678. [DOI] [PubMed] [Google Scholar]
- Nadarajah B, Parnavelas JG. Modes of neuronal migration in the developing cerebral cortex. Nat Rev Neurosci. 2002;3:423–432. doi: 10.1038/nrn845. [DOI] [PubMed] [Google Scholar]
- Nagano T, Morikubo S, Sato M. Filamin A and FILIP (Filamin A-interacting protein) regulate cell polarity and motility in neocortical subventricular and intermediate zones during radial migration. J Neurosci. 2004;24:9648–9657. doi: 10.1523/JNEUROSCI.2363-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K. Control of tangential/non-radial migration of neurons in the developing cerebral cortex. Neurochem Int. 2007;51:121–131. doi: 10.1016/j.neuint.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR. Neurons derived from radial glial cells establish radial units in neocortex. Nature. 2001;409:714–720. doi: 10.1038/35055553. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martinez-Cerdeno V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7:136–144. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- Nowakowski RS, Lewin SB, Miller MW. Bromodeoxyuridine immunohistochemical determination of the lengths of the cell cycle and the DNA-synthetic phase for an anatomically defined population. J Neurocytol. 1989;18:311–318. doi: 10.1007/BF01190834. [DOI] [PubMed] [Google Scholar]
- Poirier K, Van Esch H, Friocourt G, Saillour Y, Bahi N, Backer S, Souil E, Castelnau-Ptakhine L, Beldjord C, Francis F, Bienvenu T, Chelly J. Neuroanatomical distribution of ARX in brain and its localisation in GABAergic neurons. Mol Brain Res. 2004;122:35–46. doi: 10.1016/j.molbrainres.2003.11.021. [DOI] [PubMed] [Google Scholar]
- Rakic P. Principles of neural cell migration. Experientia. 1990;46:882–891. doi: 10.1007/BF01939380. [DOI] [PubMed] [Google Scholar]
- Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB, Caskey CT, Ledbetter DH. Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature. 1993;364:717–721. doi: 10.1038/364717a0. [DOI] [PubMed] [Google Scholar]
- Saito T, Nakatsuji N. Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev Biol. 2001;240:237–246. doi: 10.1006/dbio.2001.0439. [DOI] [PubMed] [Google Scholar]
- Scheffer IE, Wallace RH, Phillips FL, Hewson P, Reardon K, Parasivam G, Stromme P, Berkovic SF, Gecz J, Mulley JC. X-linked myoclonic epilepsy with spasticity and intellectual disability: mutation in the homeobox gene ARX. Neurology. 2002;59:348–356. doi: 10.1212/wnl.59.3.348. [DOI] [PubMed] [Google Scholar]
- Strømme P, Mangelsdorf ME, Shaw MA, Lower KM, Lewis SM, Bruyere H, Lütcherath V, Gedeon AK, Wallace RH, Scheffer IE, Turner G, Partington M, Frints SG, Fryns JP, Sutherland GR, Mulley JC, Gécz J. Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat Genet. 2002a;30:441–445. doi: 10.1038/ng862. [DOI] [PubMed] [Google Scholar]
- Strømme P, Mangelsdorf ME, Scheffer IE, Gécz J. Infantile spasms, dystonia, and other X-linked phenotypes caused by mutations in Aristaless related homeobox gene, ARX. Brain Dev. 2002b;24:266–268. doi: 10.1016/s0387-7604(02)00079-7. [DOI] [PubMed] [Google Scholar]
- Stühmer T, Anderson SA, Ekker M, Rubenstein JL. Ectopic expression of the Dlx genes induces glutamic acid decarboxylase and Dlx expression. Development. 2002a;129:245–252. doi: 10.1242/dev.129.1.245. [DOI] [PubMed] [Google Scholar]
- Stühmer T, Puelles L, Ekker M, Rubenstein JL. Expression from a Dlx gene enhancer marks adult mouse cortical GABAergic neurons. Cereb Cortex. 2002b;12:75–85. doi: 10.1093/cercor/12.1.75. [DOI] [PubMed] [Google Scholar]
- Tabata H, Nakajima K. Efficient in utero gene transfer system to the developing mouse brain using electroporation: visualization of neuronal migration in the developing cortex. Neuroscience. 2001;103:865–872. doi: 10.1016/s0306-4522(01)00016-1. [DOI] [PubMed] [Google Scholar]
- Tabata H, Nakajima K. Multipolar migration: the third mode of radial neuronal migration in the developing cerebral cortex. J Neurosci. 2003;23:9996–10001. doi: 10.1523/JNEUROSCI.23-31-09996.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhead GJ, Mutch CA, Olson EC, Chenn A. Cell-autonomous β-catenin signaling regulates cortical precursor proliferation. J Neurosci. 2006;26:12620–12630. doi: 10.1523/JNEUROSCI.3180-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods CG. Human microcephaly. Curr Opin Neurobiol. 2004;14:112–117. doi: 10.1016/j.conb.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Yozu M, Tabata H, Nakajima K. The caudal migratory stream: a novel migratory stream of interneurons derived from the caudal ganglionic eminence in the developing mouse forebrain. J Neurosci. 2005;25:7268–7277. doi: 10.1523/JNEUROSCI.2072-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zindy F, Cunningham JJ, Sherr CJ, Jogal S, Smeyne RJ, Roussel MF. Postnatal neuronal proliferation in mice lacking Ink4d and Kip1 inhibitors of cyclin-dependent kinases. Proc Natl Acad Sci USA. 1999;96:13462–13467. doi: 10.1073/pnas.96.23.13462. [DOI] [PMC free article] [PubMed] [Google Scholar]