

Abstract
Circulating monocytoid cells have the ability to infiltrate nervous tissue, differentiate into microglia, and clear amyloid-β (Aβ) from the brain of mouse models of Alzheimer's disease. Interaction between the chemokine CCL2 and its CC chemokine receptor 2 (CCR2) plays a critical role in the recruitment of inflammatory monocytes into the injured/diseased brain. Here, we show that CCR2 deficiency aggravates mnesic deficits and amyloid pathology in transgenic mice expressing the chimeric mouse/human β-amyloid precursor protein and presenilin 1 (APPSwe/PS1). Indeed, memory impairment was accelerated and enhanced in APPSwe/PS1/CCR2−/− mice. Apparition of cognitive decline occurred earlier (i.e., at 3 months of age before plaque formation) and correlated with intracellular accumulation of soluble oligomeric forms of Aβ. Memory deficits worsened with age and were aggravated in APPSwe/PS1/CCR2−/− mice compared with their respective control groups. Soluble Aβ assemblies increased significantly in APPSwe/PS1 mice in a context of CCR2 deficiency, whereas the plaque load remained relatively similar in the brain of aging APPSwe/PS1 and APPSwe/PS1/CCR2−/− mice. However, CCR2 deficiency stimulated the expression of TGF-β1, TGF-β receptors, and CX3CR1 transcripts in plaque-associated microglia, a pattern that is characteristic of an antiinflammatory subset of myeloid cells. A decreased expression of CCR2 could play a potential role in the etiology of Alzheimer's disease, a neurodegenerative pathology that could be treated by a genetic upregulation of the transgene in monocytoid cells.
Introduction
Alzheimer's disease (AD), the most common cause of dementia in the elderly, is characterized by a progressive decline in memory. The pathological hallmarks of AD are senile plaques, intracellular neurofibrillary tangles, accumulation of activated microglia and astrocytes around β-amyloid (Aβ) plaques, and degenerative neurons (Selkoe, 2002). The Aβ peptide is produced by amyloid precursor protein (APP) cleavage, and two main Aβ isoforms exist (i.e., Aβ1-40 and Aβ1-42). Aβ can form different oligomeric assemblies, such as monomers, dimers, or Aβ*56, to finally generate amyloid plaques (Lesné et al., 2006; Haass and Selkoe, 2007). These senile plaques contain densely packed Aβ fibrils and a large number of molecules and cellular elements. In particular, microglia, the mononuclear phagocytes of the brain (Perry and Gordon, 1988), are attracted around amyloid plaques and originate in part from the periphery (Malm et al., 2005; Stalder et al., 2005; Simard et al., 2006).
Monocytes constitutively express several chemokine receptors, which induce leukocyte migration and attraction by binding to their specific chemotactic cytokines, or chemokines (Luster, 1998; Charo and Ransohoff, 2006; Auffray et al., 2009). CC chemokine receptor 2 (CCR2) is mainly expressed on the surface of monocytes (Mack et al., 2001). In mice, two principal subsets of monocytes are defined by CCR2 expression, CX3CR1loCCR2+GR1+ inflammatory and CX3CR1hiCCR2−GR1− resident monocytes (Geissmann et al., 2003). CCR2 is involved in the brain infiltration of monocytes in a mouse model for multiple sclerosis (Izikson et al., 2000), at sites of axonal injury in the hippocampus (Babcock et al., 2003) and in systemic organs during inflammation (D'Mello et al., 2009). The main CCR2 chemokine ligand is monocyte chemotactic protein-1 (MCP-1) [CCL2 (CC chemokine ligand 2)] and is expressed in many cells, including endothelial cells (Cushing et al., 1990; Rollins et al., 1990), astrocytes (Glabinski et al., 1996; Smits et al., 2002), and microglia (Meda et al., 1995; El Khoury et al., 2003; Deshmane et al., 2009).
MCP-1 is upregulated in the brain of AD patients in mature senile plaques, microglia (Ishizuka et al., 1997), and microvessels (Grammas and Ovase, 2001). MCP-1 expression is also increased in the brain of AD transgenic mice (Simard et al., 2006; El Khoury et al., 2007) and Aβ stimulates MCP-1 production in cultures of microglia (Meda et al., 1995; El Khoury et al., 2003) and astrocytes (Smits et al., 2002). Together, these data strongly suggest a predominant role of CCR2 in AD, since CCR2 is the key signaling receptor for MCP-1 (Deshmane et al., 2009). This was recently evidenced by a study using APP Tg2576 mice, a mouse model of AD developing the hallmarks of the disease tardily (9–10 months of age) (Hsiao et al., 1996). Unfortunately, their breeding with CCR2−/− mice produced APP-CCR2−/− mice dying prematurely (i.e., before Aβ plaque formation or microglia recruitment around Aβ deposits) (El Khoury et al., 2007), severely limiting the in vivo study of disease development. To overcome this limitation, we used another APP transgenic mouse model, the APPSwe/PS1 mice, a well established model of AD (Borchelt et al., 1997; Simard et al., 2006; Richard et al., 2008; Boissonneault et al., 2009).
The main objective of this study was to follow progression of the disease in a context of CCR2 gene deficiency. All our results converge into a very important beneficial role of CCR2-expressing cells in AD.
Materials and Methods
Transgenic mice.
We used adult male C57BL/6 mice [wild type (WT)], CCR2−/− (B6.129S4-Ccr2tm1Ifc/J), and APP transgenic mice (APPSwe/PS1) harboring the chimeric mouse/human β-amyloid precursor protein (APP695swe) and the human presenilin I (A246E variant) under the control of independent mouse prion protein promoter elements [B6C3-Tg(APP695)3Dbo Tg(PSEN1)5Dbo/J]. APPSwe/PS1 were bred with the CCR2−/− mouse strain for three or more generations to generate APPSwe/PS1/CCR2−/− triple transgenic animals. All mouse strains were purchased from The Jackson Laboratory and maintained in a C57BL/6J background. All newborn pups were genotyped as described in The Jackson Laboratory protocol. A total of 201 mice were used to study behavioral and biochemical modifications from 3 to 12 months of age. Mice were housed at three to five per cage and acclimated to standard laboratory conditions (12 h light/dark cycle; lights on at 7:00 A.M. and off at 7:00 P.M.) with ad libitum access to mouse chow and water. Animal breeding and experiments were conducted according to Canadian Council on Animal Care guidelines, as managed by the Laval University Animal Care Committee.
Water T-maze.
Mice were tested during the “lights-on” phase of the day. The behavioral experimenter was blinded to the genetic and treatment status of the animals. To assess hippocampal-dependent spatial learning and memory, mice were trained in the water T-maze task. In this paradigm, we evaluate the mouse's ability to remember the spatial location of a submerged platform. The T-maze apparatus (length of stem, 64 cm; length of arms, 30 cm; width, 12 cm; height of walls, 16 cm) was made of clear fiberglass and filled with water (23 ± 1°C) at a height of 12 cm. A platform (11 × 11 cm) was placed at the end of the target arm and was submerged 1 cm below the surface. The acquisition phase allows to evaluate animals for left–right spatial learning. During the first two trials, platforms were placed on each arms of the maze to test the spontaneous turning preference of the mouse. After these two trials, the least chosen arm was reinforced by the escape platform. The mice were placed in the stem of the T-maze and chose to swim either left or right until they found the submerged platform and escaped to it, to a maximum of 60 s. After reaching the platform, the mice remained on it for 20 s and then were immediately put back in the maze. If the animals did not find the platform within this time limit, they were gently guided onto it. Repeated trials were presented on the same day up to a maximum of 48 trials. A rest period of at least 10–15 min was allowed between each block of 10 trials. A mouse was considered to have learned the task when it made no errors in a block of five consecutive trials. The reversal learning phase was then conducted 48 h later. During this phase, the same protocol was repeated, except that the mice were trained to find the escape platform on the side opposite to that on which they had learned during the acquisition phase. The number of trials needed to reach the criterion (five of five correct choices made on consecutive trials) was noted as well as the latency for finding the escape platform.
Passive avoidance test.
Based on the animal's natural tendency to prefer the dark environment, mice were also evaluated on their retention of nonspatial memory for one-trial passive avoidance task. The passive avoidance apparatus (Ugo Basile) was divided into two sections, one illuminated (start compartment) and the other dark (escape compartment). The floor of each compartment contained a grid, with only the dark compartment being electrified by a generator. On the training day, mice were placed into the lighted (start) compartment for a 60 s acclimation period. The guillotine door was then opened, and the latency to enter the dark side was recorded. Immediately after entering the dark compartment, the door was closed and an electric shock (0.5 mA for 2 s) was delivered. The mouse was kept in the dark compartment for 10 s before being returned to its home cage. On the next day, the mice were again placed in the lighted compartment, and the step-through latency time to enter the dark side was measured for up to 300 s.
Tissue analyses.
Transgenic mice were anesthetized under isoflurane and blood was drawn via cardiac puncture before decapitation. Brains were rapidly removed from the skulls and placed in cold PBS solution. Then hemibrains were separated and olfactory bulbs and cerebellum were removed. One hemibrain was rapidly frozen in liquid nitrogen and stored at −80°C for protein analysis. The other one was postfixed for 2–4 d in 4% paraformaldehyde, pH 9.5, at 4°C, and then placed in a paraformaldehyde solution containing 10% sucrose overnight at 4°C. The frozen brains were mounted on a microtome (Reichert-Jung) and cut into 25 μm coronal sections. The slices were collected in cold cryoprotectant solution (0.05 m sodium phosphate buffer, pH 7.3, 30% ethylene glycol, and 20% glycerol) and stored at −20°C until immunocytochemistry or in situ hybridization histochemistry.
In situ hybridization and immunohistochemistry.
Every 12th section of brain slices, starting from the end of the olfactory bulb to the end of the cerebral cortex, was mounted on Colorfrost/Plus microscope slides (Thermo Fisher Scientific) for APPSwe/PS1 and APPSwe/PS1/CCR2−/− mice (3–12 months of age; n = 5–12 per group). Histochemical localization of CX3CR1, TGF-β1, and TGF-β receptors R1 and R2 by in situ hybridization was performed using 35S-labeled cRNA probes. Plasmids were linearized, and sense and antisense cRNA probes were synthesized with the appropriate RNA polymerase, as described in Table 1. Riboprobe synthesis and preparation, as well as in situ hybridization were performed according to a protocol described previously (Laflamme et al., 1999; Nadeau and Rivest, 2000; Laflamme and Rivest, 2001; Naert et al., 2009).
Table 1.
Plasmids and enzymes used for the synthesis of the cRNA probes
| Plasmid | Vector | Insert (bp) | Antisense probe | Sense probe | Source |
|---|---|---|---|---|---|
| Mouse CX3CR1 | PCR II Topo | 1132 | BamHI/T7 | XhoI/Sp6 | Cloned by PCR |
| Mouse MCP-1 | pGEM-1 | 578 | BamHI/T7 | SacI/Sp6 | Dr. S. C. Williams (Texas Tech University, Lubbock, TX) |
| Mouse TGF-β1 R1 | PCR II Topo | 1093 | NotI/Sp6 | SpeI/T7 | Cloned by PCR |
| Mouse TGF-β1 R2 | PCR II Topo | 933 | XhoI/Sp6 | BamHI/T7 | Cloned by PCR |
| Mouse TGF-β1 | pBSKS+ | 1173 | EcoRI/T3 | XhoI/T7 | Dr. S. Lacroix (Laval University, Quebec City, Quebec, Canada) |
Dual labeling combining immunocytochemistry and in situ hybridization was performed as described previously (Nadeau and Rivest, 2000; Laflamme and Rivest, 2001) to detect CX3CR1, TGF-β1, TGF-β R1, and TGF-β R2 transcripts in microglia (iba-1+-staining). We used polyclonal rabbit anti-ionized calcium binding adaptor molecule 1 (iba-1) (1:3000; Wako Chemicals).
The expression level of each transcript was estimated by qualitative analysis using a Nikon Eclipse 80i microscope over the whole-brain sections for 5–10 animals/group. Quantitative analysis of hybridization signal for CX3CR1 mRNA was performed on images captured using a Nikon Eclipse 80i microscope equipped with a digital camera (QImaging) and the ImageJ software. Three coronal sections were chosen for prefrontal cortex at +2.58, +2.34, and +2.10 mm from the bregma according to a stereotaxic atlas (Paxinos and Franklin, 2001) and four sections for hippocampus/cortex at −1.70, −1.94, −2.46, and −2.92 mm. For each structure, labeling area was measured for three to four animals and means were used for statistical analysis (mean ± SEM).
All images were captured using a Nikon Eclipse 80i microscope equipped with a digital camera (QImaging), processed to enhance contrast and sharpness using Adobe Photoshop 7 (Adobe Systems), and then assembled using Adobe Illustrator (Adobe Systems). The images depicted by the different panels are representative of the signal detected on the slides for each group of mice.
Stereological analysis.
An observer who was blind to the treatment status of the material did all quantitative histological analyses. To count Aβ plaques and plaque-associated microglia, sections of APPSwe/PS1 and APPSwe/PS1/CCR2−/− mice (3–12 months of age, n = 7–15 per group) were immunostained for Aβ (polyclonal mouse anti-Aβ 6E10; 1:3000; Covance) and iba-1 proteins with DAPI (4′,6′-diamidino-2-phenylindole), as previously reported (Simard et al., 2006; Richard et al., 2008). Two sections were chosen for prefrontal cortex at +2.34 and +2.10 mm from the bregma according to a stereotaxic atlas (Paxinos and Franklin, 2001) and four sections for hippocampus/cerebral cortex at −1.70, −1.94, −2.46, and −2.92 mm. Unbiased stereological analysis was performed as described previously (Simard et al., 2006; Richard et al., 2008; Boissonneault et al., 2009). Briefly, the contours of the prefrontal cortex, the hippocampus, and the cortex areas were traced as virtual overlay on the streamed images and areas were calculated. For 3- and 6-month-old APPSwe/PS1 mice, the area occupied by all Aβ-labeled plaques was determined as well as the number of plaque-associated microglia in each structure. For 9- and 12-month-old APPSwe/PS1 mice, the Stereo Investigator software (MicroBrightField) sequentially chose counting frames (670 × 500 mm) at every 2000 μm along the x-axis and every 1000 μm along the y-axis while automatically moving the motorized stage into the previously delimited zones in the prefrontal cortex, the hippocampus, and the cortex. The areas of analyzed structures and areas occupied by plaques were calculated, as well as the number of plaque-associated microglia in these counting frames. All parameters analyzed were reported to the whole section.
Protein extraction and detection of total Aβ levels by Western blot.
Proteins from hemi-forebrains were extracted using a modification of the procedure described by Lesné et al. (2006). All manipulations were done on ice to minimize protein degradation. One hemi-forebrain was placed in a 1 ml syringe with a 20 G needle. Five hundred microliters of buffer A [50 mm Tris-HCl, pH 7.6, 0.01% NP-40, 150 mm NaCl, 2 mm EDTA, 0.1% SDS, 1 mm phenylmethylsulfonyl fluoride (PMSF), protease inhibitor mixture] were added and 10 up-and-down passages were made to homogenize the tissue, followed by a 5 min centrifugation at 3000 rpm at 4°C. The supernatant (extracellular protein-enriched fraction) was then collected and frozen at −80°C. The insoluble pellet was suspended in 500 μl of TNT buffer (buffer B; 50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 0.1% Triton X-100, 1 mm PMSF, protease inhibitor mixture), followed by a 90 min centrifugation at 13,000 rpm at 4°C. The supernatant (cytoplasmic protein-enriched fraction) was then collected and frozen at −80°C. The pellet was suspended in 500 μl of buffer C (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.5% Triton X-100, 1 mm EGTA, 3% SDS, 1% deoxycholate, 1 mm PMSF, protease inhibitor mixture) and incubated at 4°C, 50 rpm for 1 h. The samples were centrifuged for 90 min at 13,000 rpm and 4°C, and the supernatant (membrane protein-enriched fraction) was collected and frozen at −80°C. The protein concentration in each fraction was determined using the Quantipro bicinchoninic acid assay kit (Sigma-Aldrich) according to the manufacturer's protocol.
For total Aβ detection, 10–30 μg of extracellular, cytoplasmic, and membrane protein fractions were separated on a precast 10–20% SDS polyacrylamide Tris-Tricine gel (Bio-Rad). Separated proteins were then transferred onto polyvinylidene fluoride membranes (PerkinElmer Life and Analytical Sciences) and detected by Western blotting. Blots were probed with a mouse anti-Aβ protein monoclonal antibody clone 6E10 (1:1000; Covance) in a buffer containing 1 m Tris-HCl, pH 8.0, 5 m NaCl, 5% skim milk, and 0.05% Tween 20. Blots were visualized using an anti-mouse secondary antibody tagged with horseradish peroxidase (1:1000; Jackson ImmunoResearch) and enhanced chemiluminescence (PerkinElmer Life and Analytical Sciences). Membranes were stripped in 25 mm glycine-HCl, pH 2.0, containing 1% SDS to detect β-actin using first a mouse β-actin antibody (MAB1501; 1:5000; Millipore Bioscience Research Reagents) and then a goat anti-mouse peroxidase-conjugated secondary antibody (1:1000; Jackson ImmunoResearch).
Quantification was done by determining integrated density of the bands using a gel imaging system (scanner Agfa Arcus II; NIH ImageJ software, version 1.32j), and background values were subtracted. Optical values were normalized relatively to the actin loading control. Results are expressed as mean ± SEM.
Statistical analysis.
Results are expressed as mean ± SEM. Statistical analysis was performed by one- or two-way ANOVA, followed by either Bonferroni's or Thamane's tests as appropriate, as post hoc comparisons (SPSS software). Data were analyzed using standard two-tailed unpaired t test for comparisons between two groups. Correlations were estimated by linear regression analysis and the Spearman's correlation coefficient (GraphPad Software) with p and r values and 95% confidence intervals included in the graph. A value of p < 0.05 was considered statistically significant.
Results
CCR2 deficiency aggravates cognitive impairment in APPSwe/PS1 mice
To determine whether CCR2 deficiency affects disease progression, we bred APPSwe/PS1 mice with CCR2-deficient animals. APPSwe/PS1/CCR2−/− mice appeared healthy and had a survival curve comparable with APPSwe/PS1 mice, reaching >12 months of age (Fig. 1). Mice of each genotype from 3 to 12 months of age were submitted to hippocampus-based spatial learning in a water T-maze (Fig. 1a–d) and displayed similar swimming capacity and motivation to escape from water. During this acquisition phase, the means of trials (Fig. 1a) and the latency (Fig. 1c) to reach the criterion were similar between mice independently of genotype and age. During the reversal phase, 3- to 12-month-old WT and CCR2−/− mice were able to learn the novel platform location. APPSwe/PS1 mice exhibited memory impairment starting at 6 months of age and this deficit worsened with age (Fig. 1b,d). The spatial memory was affected earlier and to a greater extent in APPSwe/PS1/CCR2−/− mice, which exhibited a significant increase in the number of trials made (14.2 ± 1.8 vs 4.6 ± 1.1 for APPSwe/PS1; p < 0.001) starting at 3 months of age, and the time spent to reach the platform (15.7 ± 1.4 vs 11.3 ± 0.7 for APPSwe/PS1; p < 0.05). The larger decline of spatial memory seen in APPSwe/PS1/CCR2−/− mice progressed gradually with age, as evidenced by the number of trials at 6 and 9 months of age (22.9 ± 2.4 vs 14.5 ± 1.6 for APPSwe/PS1; p < 0.05 and 26.9 ± 3.1 vs 16.8 ± 1.5 for APPSwe/PS1; p < 0.05, respectively) and by the increased latency at 6 months (18.5 ± 1.8 vs 13.0 ± 0.5 for APPSwe/PS1; p < 0.05). At 12 months of age, no difference in spatial memory was shown with the water T-maze task, suggesting that memory impairment had reached a plateau at that age in both groups of APPSwe/PS1 mice.
Figure 1.

CCR2 deficiency accelerates the onset of spatial and contextual memory deficits and aggravates cognitive impairment in APPSwe/PS1 mice. Spatial memory was assessed using the water T-maze test (a–d) in wild-type, CCR2−/−, APPSwe/PS1, and APPSwe/PS1/CCR2−/− mice aged from 3 to 12 months. The numbers of trials (a, b) and the latency (c, d) to accomplish the task were determined during the acquisition (a, c) and reversal learning phases (b, d). APPSwe/PS1/CCR2−/− mice exhibited an earlier and greater spatial memory decline than APPSwe/PS1 mice. The passive avoidance test was used to establish contextual memory deficit (e, f). The latency was determined at 3–12 months of age during the acquisition phase (e) and 24 h after the conditioning test (f) for each group. The latency to enter the dark compartment was already decreased at 6 months of age in APPSwe/PS1/CCR2−/− mice compared with the control groups of mice. Results are expressed as mean ± SEM; n = 8–16 per group; *p < 0.05, **p < 0.01, and ***p < 0.001. *Versus WT at same age; °versus CCR2−/− at same age; #versus APPSwe/PS1 at same age. (Two-way ANOVA was performed and, for each parameter analysis, revealed a significant interaction between the factors of age and genotype. Then, one-way ANOVA at each level of age was performed using Bonferroni's or Tamhane's post hoc test.)
To assess contextual memory, mice were submitted to the passive avoidance test. All animals were trained to avoid a dark compartment and presented similar latency to enter the dark side from 3 to 12 months of age during the acquisition phase (Fig. 1e). Twenty-four hours after the conditioning test, both APPSwe/PS1 and APPSwe/PS1/CCR2−/− mice exhibited a decreased latency, although it appeared earlier in APPSwe/PS1/CCR2−/− mice (Fig. 1f). At 6 months, a significantly decreased latency was observed only in APPSwe/PS1/CCR2−/− mice (103 ± 15 vs 239 ± 26 for WT; p < 0.001). This contextual memory impairment reached a significant level at 9 months in APPSwe/PS1 mice (107 ± 20 vs 283 ± 9 for WT; p < 0.001). This cognitive decline was maintained with age and was similar at 9 and 12 months between the two groups of APPSwe/PS1 mice.
These results demonstrate a greater cognitive impairment in APPSwe/PS1/CCR2−/− mice, which appeared earlier than in APPSwe/PS1 mice, suggesting that CCR2 deficiency has deleterious effects on memory and learning processes.
CCR2 deficiency increases levels of soluble Aβ oligomers without changing Aβ deposition
The major hallmark of AD is Aβ accumulation in brain, which correlates strongly with mnesic impairment (Selkoe, 2002). We first quantified the plaque load by stereological analyses on coronal sections stained with the 6E10 antibody specific for the 1–17 fragment of the human Aβ protein. Similar Aβ plaque burden was found in APPSwe/PS1 and APPSwe/PS1/CCR2−/− mice from 3 to 9 months of age in hippocampus (Fig. 2), prefrontal cortex (Fig. 3, left column), and cerebral cortex (Fig. 3, right column). Plaque number, plaque size, and area occupied by Aβ plaques increased with age in both groups. At 12 months of age, the area occupied by plaques in prefrontal cortex (Fig. 3i) and hippocampus (Fig. 2t) was larger in APPSwe/PS1/CCR2−/− mice (2.67 ± 0.22%, p < 0.05, and 1.24 ± 0.17%, p < 0.001, respectively) than APPSwe/PS1 littermates (1.87 ± 0.21 and 0.57 ± 0.12%, respectively).
Figure 2.
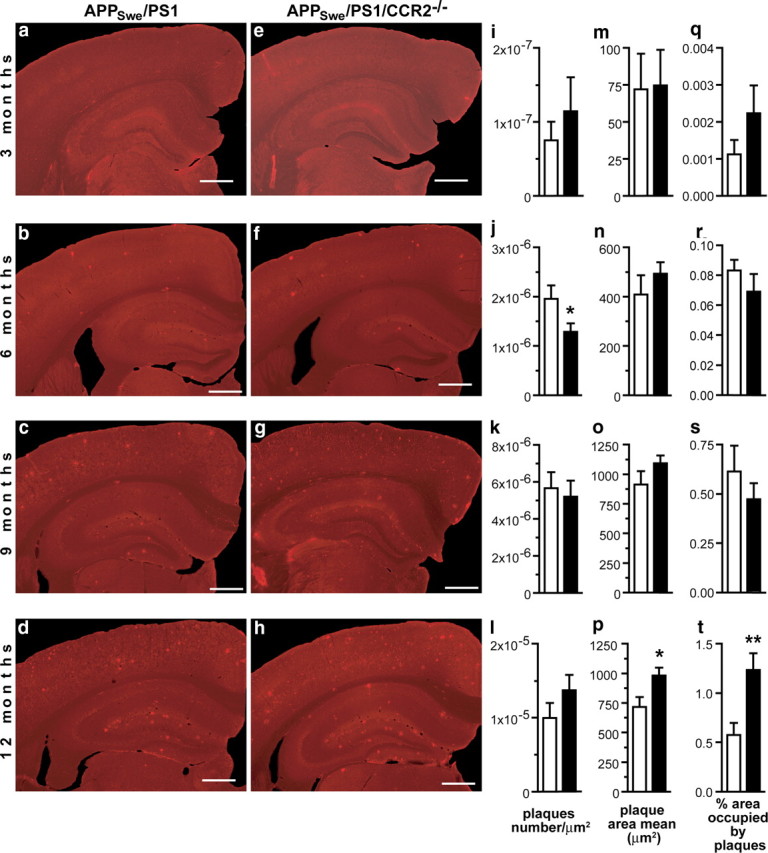
Amyloid plaque burden in the hippocampus of APPSwe/PS1 and APPSwe/PS1/CCR2−/− mice. Brain sections of 3- to 12-month-old APPSwe/PS1 (a–d) and APPSwe/PS1/CCR2−/− mice (e–h) were immunostained for Aβ. Unbiased stereological analysis of plaques was performed in hippocampus to determine plaque density (i–l), plaque size (m–p), and the area percentage occupied by plaques (q–t) of 3- to 12-month-old APPSwe/PS1 (open bar) and APPSwe/PS1/CCR2−/− mice (solid bar). APPSwe/PS1/CCR2−/− and APPSwe/PS1 mice had similar amyloid plaque formation at 3, 6, and 9 months of age. However, a significant higher Aβ load was found in the hippocampus of 12-month-old APPSwe/PS1/CCR2−/− mice. Results are expressed as mean ± SEM; n = 7–15 per group; Student's t test; *p < 0.05 and **p < 0.01 versus APPSwe/PS1 at same age. Scale bar, 500 μm.
Figure 3.
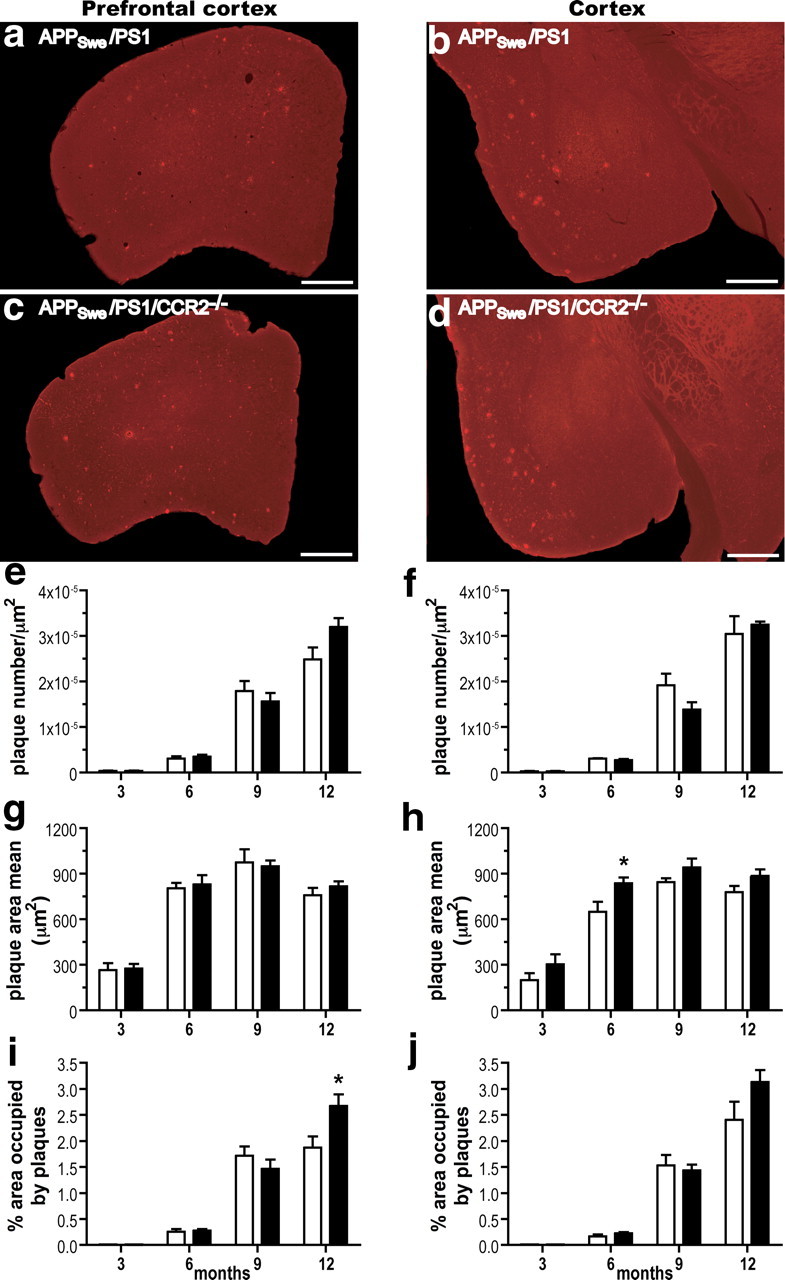
Amyloid plaque formation in the prefrontal cortex and cerebral cortex of APPSwe/PS1 and APPSwe/PS1/CCR2−/− mice. Anti-Aβ immunoreactivity is depicted in the prefrontal cortex (left column) and cerebral cortex (right column) of APPSwe/PS1 (a, b) and APPSwe/PS1/CCR2−/− mice (c, d) at 9 months of age. A detailed analysis of plaque quantification was performed to determine plaque density (e, f), plaque size (g, h), and the area percentage occupied by plaques (i, j) of 3- to 12-month-old APPSwe/PS1 (open bar) and APPSwe/PS1/CCR2−/− mice (solid bar). Amyloid plaque formation was similar in both groups at 3, 6, and 9 months of age, although Aβ plaque load significantly increased in the prefrontal cortex of 12-month-old APPSwe/PS1/CCR2−/− mice. Results are expressed as mean ± SEM; n = 7–15; Student's t test; *p < 0.05 vs APPSwe/PS1 at the same age. Scale bar, 500 μm.
These results do not support a significant correlation between plaque formation and the occurrence of cognitive impairment in a context of CCR2 deficiency. It is important to mention that the severity of dementia in AD patients correlates more strongly with cortical levels of soluble Aβ species than with insoluble Aβ plaque burden (Lue et al., 1999; McLean et al., 1999). Moreover, memory deficits also correlate with soluble Aβ oligomers instead of Aβ plaque in mouse models of AD (Lesné et al., 2006; Cheng et al., 2007). Therefore, we next determined levels of soluble Aβ in the extracellular- and intracellular-enriched, and membrane-associated protein fractions by Western blot, using the method published by Lesné et al. (2006).
At 3 months of age, similar levels of Aβ species were observed in extracellular-enriched and membrane-associated protein fractions between APPSwe/PS1 and APPSwe/PS1/CCR2−/− mice (Fig. 4a,b; e,f, respectively). By contrast, APPSwe/PS1/CCR2−/− mice exhibited increased levels of Aβ species in the intracellular-enriched fraction comparatively to APPSwe/PS1 mice (Fig. 4c,d). To establish whether these higher levels of intracellular Aβ species contribute to the appearance of mnesic deficit, we determined the correlation between spatial memory decline (water T-maze test) and soluble intracellular Aβ (Fig. 4g–m). We noted a significant positive correlation between the levels of most Aβ species and impairment of spatial memory, with a better correlation for high-molecular-mass assemblies (Fig. 4g–j).
Figure 4.
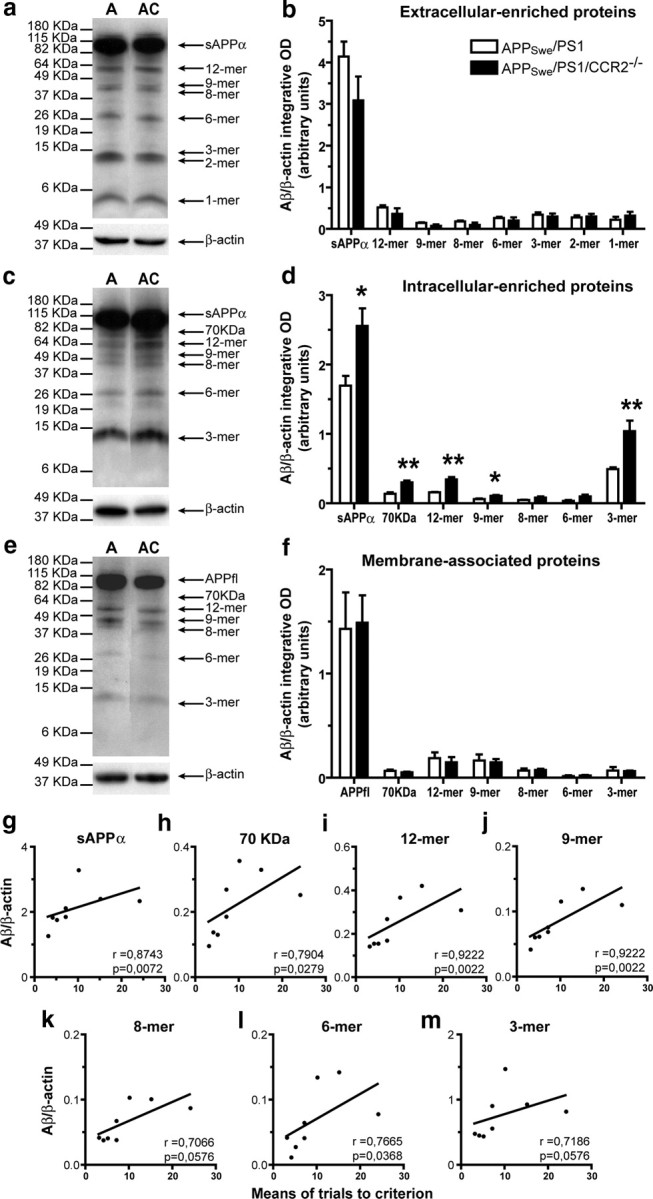
Soluble intracellular Aβ species are increased in 3-month-old APPSwe/PS1/CCR2−/− mice and are strongly correlated with spatial memory decline. Hemibrain extracts from soluble extracellular (a), intracellular (c), and membrane-associated proteins (e) of 3-month-old APPSwe/PS1 (A) and APPSwe/PS1/CCR2−/− (AC) mice were assessed by Western blot on a 10–20% Tris-Tricine denaturing polyacrylamide gel to separate Aβ species using anti-Aβ antibody 6E10. The intensity of each band was quantified by densitometric analysis and normalized per β-actin values. Ratios of Aβ species (Aβ/β-actin) are represented for extracellular (b), intracellular (d), and membrane-associated proteins (f). Please note the increase of soluble intracellular Aβ species in the brain of APPSwe/PS1/CCR2−/− mice. Positive significant correlations exist between spatial memory decline (water T-maze test) and soluble intracellular Aβ species (g–m) and, more importantly, for high-molecular mass products (g–j). Results are expressed as mean ± SEM; n = 4; Student's t test; *p < 0.05 and **p < 0.01, versus APPSwe/PS1. Correlation test was performed using the Spearman's correlation coefficient.
Although Aβ levels in extracellular-enriched and membrane-associated fractions remained comparable in 6-month-old APPSwe/PS1 and APPSwe/PS1/CCR2−/− mice (Fig. 5a,b; e,f, respectively), higher intracellular trimer Aβ levels were found in the brain of APPSwe/PS1/CCR2−/− (Fig. 5c,d). At 9 months of age, levels of soluble Aβ species were significantly increased in extracellular-, intracellular-enriched, and membrane-associated protein fractions of APPSwe/PS1/CCR2−/− (Fig. 5). These changes in extracellular and intracellular soluble Aβ vanished at 12 months of age, but APPSwe/PS1/CCR2−/− brains still had significantly higher 9-, 6-, and 5-mer levels in membrane-associated fractions (Fig. 5f).
Figure 5.

Age-dependent increase of soluble Aβ species in extracellular, intracellular, and membrane-associated fractions extracted from the brain of APPSwe/PS1/CCR2−/− mice. Hemibrain extracts from soluble extracellular (a), intracellular (c), and membrane-associated proteins (e) of 6- to 12-month-old APPSwe/PS1 (A) and APPSwe/PS1/CCR2−/− (AC) mice were assessed by Western blot. The intensity of each band was quantified by densitometric analysis and normalized per β-actin values. Aβ species ratios (Aβ/β-actin) are represented for extracellular (b), intracellular (d), and membrane-associated proteins (f). Soluble Aβ species levels were generally higher in the brains of APPSwe/PS1/CCR2−/− mice than those of APPSwe/PS1 mice. Results are expressed as mean ± SEM; n = 4–5; Student's t test; *p < 0.05, **p < 0.01, and ***p < 0.001. *Versus APPSwe/PS1 at the same age.
These data provide evidence that levels of soluble Aβ oligomers in extracellular-, intracellular-enriched, and membrane-associated fractions increased in the brains of APPSwe/PS1/CCR2−/− mice and intracellular Aβ correlated strongly with early cognitive deficits in a context of CCR2 deficiency.
Enhanced microglial expression of TGF-β1 and TGF-β receptors in APPSwe/PS1/CCR2−/− mice
In addition to Aβ production and accumulation, microglia are attracted to amyloid deposits, are activated by Aβ, and express inflammatory molecules. In particular, TGF-β1, a pleiotropic cytokine with a central role in immune suppression (Li et al., 2006), was shown to contribute to Aβ deposition in a transgenic mouse model of AD (Wyss-Coray et al., 1997). We found a robust signal for TGF-β1 and its receptors from 6 to 12 months of age in APPSwe/PS1 and APPSwe/PS1/CCR2−/− mice (Fig. 6). The signal was mainly associated to senile plaques in prefrontal cortex, hippocampus, and cerebral cortex and all mRNAs are expressed in iba1-positive microglial cells (Fig. 6c,f,i). Qualitative analysis demonstrated an increase in the number of positive cells and the signal intensity for TGF-β1 and TGF-β-R2 mRNA in APPSwe/PS1/CCR2−/− mice, whereas TGF-β-R1 mRNA levels were lower in 9- and 12-month-old APPSwe/PS1/CCR2−/− mice (Fig. 6j).
Figure 6.
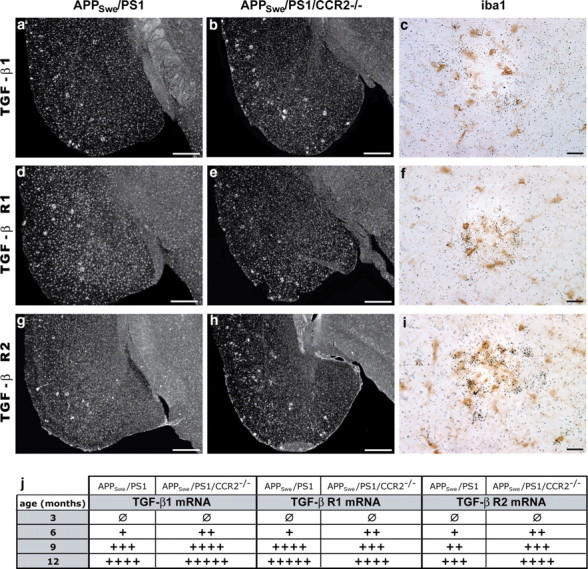
TGF-β1 and TGF-β receptor 1 and 2 mRNA levels are strongly increased in microglia surrounding plaques of CCR2-deficient APPSwe/PS1 mice. Representative dark-field photomicrographs of in situ hybridization showing the cortical expression of TGF-β1 (a, b), TFG-β-R1 (d, e), and TGF-β-R2 mRNA (g, h) in the brain of 9-month-old APPSwe/PS1 (left column) and APPSwe/PS1/CCR2−/− mice (middle column). Brain sections of 12-month-old APPSwe/PS1/CCR2−/− mice were stained with an anti-iba1 antibody and peroxidase-conjugated secondary antibody, and thereafter hybridized with a mouse TGF-β1 (c), TGF-β-R1 (f), or TGF-β-R2 (i) cRNA probe. Each transcript (agglomeration of silver grains) is clearly expressed in microglia associated to plaques (brown cells). Qualitative quantification was performed for each transcript in the whole brain of 3- to 12-month-old APPSwe/PS1 and APPSwe/PS1/CCR2−/− mice (n = 5–10 per group). CCR2 deficiency clearly increased microglial expression of TGF-β1 and TGF-β-R2 around Aβ plaques of APPSwe/PS1 mice. Scale bars: Left and middle panels, 500 μm; right panel, 20 μm.
Enhanced recruitment of CX3CR1 microglia surrounding Aβ plaques in APPSwe/PS1/CCR2−/− mice
Stereological analyses were performed on coronal brain sections immunostained for iba1 and Aβ to count plaque-associated microglia. At 3 months of age, APPSwe/PS1 and APPSwe/PS1/CCR2−/− mice had a similar number of microglia around the few Aβ plaques (Fig. 7f–h). Microglia recruitment in the plaque vicinity reached a plateau at 6 months in APPSwe/PS1 and APPSwe/PS1/CCR2−/− mice and more plaque-associated microglia were found in the cortex of APPSwe/PS1/CCR2−/− mice (0.0045 ± 0.0002 vs 0.0019 ± 0.0002 microglia/μm2 for APPSwe/PS1; p < 0.05) (Fig. 7h). This enhanced recruitment of microglia became more pronounced in 9-month-old APPSwe/PS1/CCR2−/− mice and was significantly different from APPSwe/PS1 mice for the prefrontal cortex (0.0027 ± 0.0003 vs 0.0038 ± 0.0002 microglia/μm2; p < 0.001), hippocampus (0.0034 ± 0.0003 vs 0.0024 ± 0.0002 microglia/μm2; p < 0.01), and cerebral cortex (0.0026 ± 0.0001 vs 0.0017 ± 0.0002 microglia/μm2; p < 0.001) (Fig. 7f–h). At 12 months of age, both groups of mice exhibited a similar recruitment in all regions studied. This increased recruitment was concomitantly observed with an enhanced expression of MCP-1. In fact, in situ hybridization was performed for MCP-1 mRNA (Fig. 7a–d) and showed a strong increase of MCP-1 signal in APPSwe/PS1/CCR2−/− mice from 3 to 12 months of age (Fig. 7e). In APPSwe/PS1 mice, MCP-1 expression was induced later at 6 months and also increased with age (Fig. 7e). In addition, this increased recruitment correlated with a strong expression of CX3CR1 mRNA in microglia (Fig. 8). Although the transcript was constitutively expressed in microglia in both groups of mice, APPSwe/PS1/CCR2−/− mice exhibited higher CX3CR1 mRNA levels than APPSwe/PS1 mice in the prefrontal cortex (data not shown) and cerebral cortex at 9 months of age (1.15 ± 0.10 vs 0.65 ± 0.17% in APPSwe/PS1; p < 0.05) (Fig. 8f).
Figure 7.
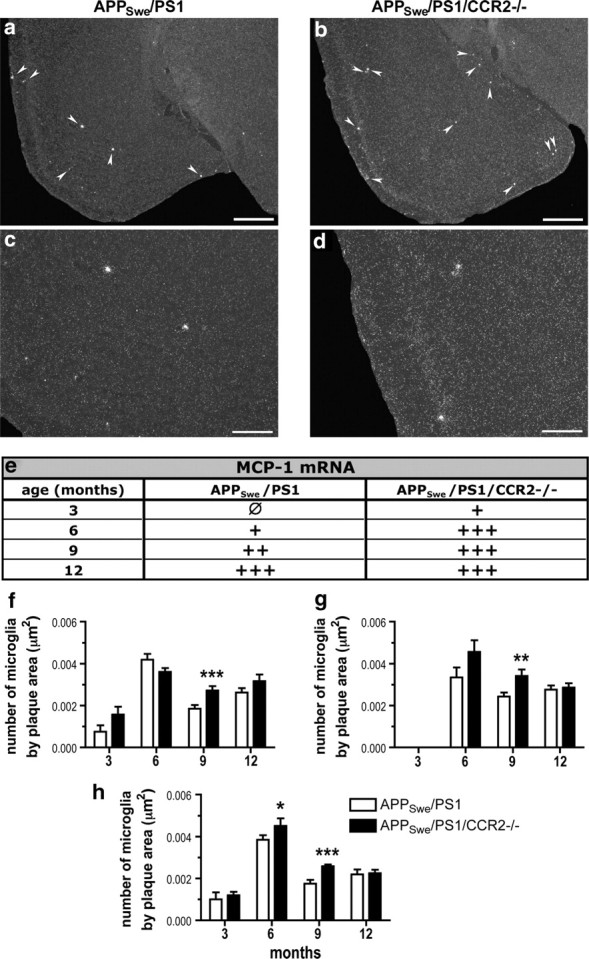
CCR2 deficiency increases MCP-1 expression and microglial recruitment around Aβ plaque formation in APPSwe/PS1 mice. Representative dark-field photomicrographs of in situ hybridization showing MCP-1 expression in brains of 9-month-old APPSwe/PS1 (left column) and APPSwe/PS1/CCR2−/− mice (right column). Qualitative analysis of brains hybridized with a MCP-1 probe from 3- to 12-month-old mice (n = 5–10 per group) shows an enhanced expression in APPSwe/PS1/CCR2−/− mice (e). Increased expression of MCP-1 is concomitant with an increase recruitment of microglia in the plaque vicinity in prefrontal cortex (f), hippocampus (g), and cortex (h). Results are expressed as mean ± SEM; n = 7–15; Student's t test; *p < 0.05, **p < 0.01, and ***p < 0.001. *Versus APPSwe/PS1 at same age. Arrowheads, MCP-1 positive signal. Scale bars: a, b, 500 μm; c, d, 200 μm.
Figure 8.
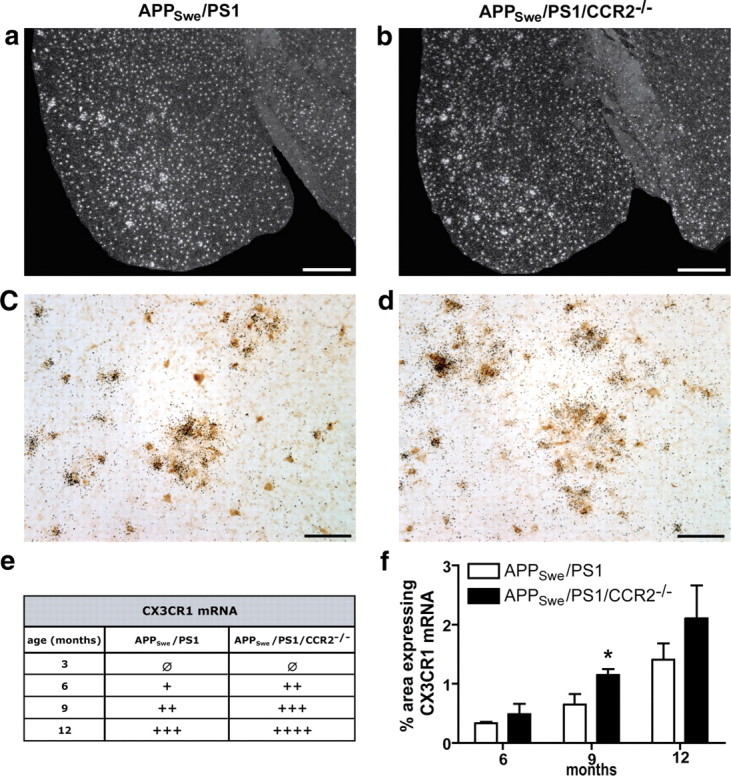
Increased expression of CX3CR1 mRNA in microglia surrounding Aβ plaques of APPSwe/PS1/CCR2−/− mice. Representative dark-field photomicrographs of cortical brain sections hybridized with radioactive cRNA probe for CX3CR1 mRNA of 9-month-old APPSwe/PS1 (a) and APPSwe/PS1/CCR2−/− mice (b). CX3CR1 mRNA signal always colocalized with iba1-immunoreactive cells (brown cells), as shown by double-labeled cells in 12-month-old APPSwe/PS1 (c) and APPSwe/PS1/CCR2−/− mice (d). Note the particular ring formation of microglia strongly expressing CX3CR1 mRNA. Qualitative analysis of CX3CR1 indicated an increased expression signal from 3 to 12 months of age in APPSwe/PS1/CCR2−/− mice (e) (n = 3–5). Such a tendency was validated by measuring the area containing CX3CR1-positive message in cortex on dark-field photomicrographs (f). Results are expressed as mean ± SEM; n = 3–4; Student's t test; *p < 0.05 versus APPSwe/PS1 at same age. Scale bars: a, b, 500 μm; c, d, 50 μm.
We have shown that, surprisingly, microglia recruitment in and around senile plaque is increased, strongly suggesting that a compensatory process takes place in the brain to counteract CCR2 deficiency.
Discussion
Our data offer new insight on the importance and role of microglia in the etiology of AD. We demonstrate that CCR2 deficiency in a mouse model of AD accelerated the onset of disease and aggravated mnesic deficits and Aβ pathology.
AD brain contains soluble and insoluble assemblies of Aβ, both of which have been hypothesized to underlie dementia. However, the correlation between parenchymal amyloid deposition and cognitive decline remains controversial. The amyloid hypothesis has been revised recently to include the notions that smaller soluble Aβ oligomers may play an early and significant role in AD and correlate with the impairment in cognitive function, learning, and memory (Lue et al., 1999; McLean et al., 1999; Lesné et al., 2006, 2008; Cheng et al., 2007; Selkoe, 2008). In this regard, we found no significant variation in Aβ deposits despite an early aggravation of memory impairment in APPSwe/PS1/CCR2−/−. Cognitive impairment occurs at 3 months of age when there are only few scattered plaques. By contrast, brain levels of soluble intracellular Aβ species are drastically increased at this age, suggesting a rapid accumulation of Aβ species within neurons. The significant correlation between cognitive deficit and intracellular Aβ levels supports the hypothesis that intracellular Aβ impairs neuronal functions (Wirths et al., 2004; LaFerla et al., 2007). These data confirm previous studies showing intraneuronal Aβ accumulation in different mouse models of AD (Billings et al., 2005; Cruz et al., 2006; Oakley et al., 2006; Knobloch et al., 2007) and in postmortem brain tissues from AD patients (Gouras et al., 2000, 2005). Intracellular Aβ species may therefore be responsible for the early AD-related cognitive deficits before Aβ plaque formation.
APPSwe/PS1/CCR2−/− mice exhibited increased intracellular levels of all Aβ species, suggesting the increase of Aβ production in their brain. In addition, inhibition of Aβ degradation could also participate in Aβ accumulation in the brain, since the expression of neprilysin/Mme, a major Aβ-degrading enzyme, is decreased in a context of CCR2 deficiency (El Khoury et al., 2007). Moreover, synaptic activity can reduce intraneuronal Aβ by Mme action and protect against Aβ-related synaptic alterations (Tampellini et al., 2009). In this regard, the expression of synaptic genes (e.g., EGR1, PSD95, and NR2A) is downregulated in the brain of 3-month-old APPSwe/PS1/CCR2−/− mice (data not shown). Since Aβ can be phagocytosed by microglia (Mandrekar et al., 2009) and astrocytes (Matsunaga et al., 2003; Nielsen et al., 2009), CCR2 deficiency can also affect Aβ clearance by preventing the recruitment of CCR2+ monocytes and contributing therefore to soluble Aβ accumulation. Such a mechanism is supported by the soluble extracellular Aβ levels (including 12-, 6-, 2-, and 1-mers), which were significantly higher in the brain of APPSwe/PS1/CCR2−/− mice.
Many soluble Aβ assemblies have been shown to induce cognitive impairment, such as extracellular Aβ*56 (Lesné et al., 2006) or soluble Aβ dimer and trimer (Cleary et al., 2005). Moreover, Mme overexpression fails to prevent learning and memory deficits despite restricting Aβ deposition in cortex and hippocampus. Interestingly, soluble Aβ trimers and Aβ*56 levels were not reduced in the brain of these APP mice (Meilandt et al., 2009). In the Tg2576 model, memory impairment begins when Aβ dimers start to accumulate (i.e., ∼6 months before plaque formation) (Kawarabayashi et al., 2004). In AD patients, soluble intracellular and membrane-associated Aβ in temporal neocortex is more closely related to AD symptoms than other Aβ species (Steinerman et al., 2008). Aβ binding to cell membranes is therefore not negligible and could disrupt their fluidity and, therefore, synaptic activity and neurotransmission. Another important aspect of the amyloid cascade is that extracellular-, intracellular-, and membrane-associated pools of Aβ are tightly interconnected. For example, extracellular Aβ can increase its own production and then induce Aβ accumulation within neurons (Peters et al., 2009; Tampellini et al., 2009). Moreover, animals with amyloid plaques have normal memory function throughout an episode of reduced extracellular Aβ oligomers (including Aβ*56), which occurs during a period of accelerated amyloid plaque formation (Lesné et al., 2008). These data strongly suggest that the Aβ entities responsible for memory decline and disruption of synaptic plasticity in AD pathology are variable, but always associated to soluble fractions. Our study further reinforces this concept, because soluble Aβ accumulation occurs before plaque formation in cognitively impaired young CCR2-deficient mice.
We expected to find a lower number of plaque-associated microglia in CCR2-deficent mice because of the inability of CX3CR1loCCR2+GR1+ monocytes to infiltrate the brain despite enhanced MCP-1 gene expression in AD mouse models (Simard et al., 2006; El Khoury et al., 2007). Surprisingly, we found more microglia recruited around Aβ plaques in APPSwe/PS1/CCR2−/− mice than APPSwe/PS1. Microglia are known to react quickly to Aβ production and subsequently migrate toward plaques (Bolmont et al., 2008). Despite similar Aβ deposits, microglia recruitment was enhanced in APPSwe/PS1/CCR2−/− mice from 6 months of age, suggesting a compensatory mechanism induced by the lack of CCR2-expressing cells. As CCR2−/− cells cannot migrate in response to MCP-1 (El Khoury et al., 2007), the chemokine responsible for this strengthened attraction has yet to be identified. These plaque-associated microglia expressed strongly TGF-β1 and TGF-β1 receptor-R1 and -R2 transcripts, which were increased in the brain of APPSwe/PS1/CCR2−/− mice. Enhanced TGF-β1 levels have already been described in AD patients (van der Wal et al., 1993; Wyss-Coray et al., 1997; Rota et al., 2006) and in our mouse model of AD (Richard et al., 2008). Although the role of TGF-β1 in AD pathology remains controversial, this cytokine binds Aβ with high affinity and enhances its oligomerization in vitro (Mousseau et al., 2003). Moreover, TGF-β1 overexpression accelerates and promotes amyloidogenesis in AD mice (Wyss-Coray et al., 1997, 2000, 2001). Interruption of TGF-β1 signaling in bone marrow-derived microglia/macrophages mitigates cognitive impairment and attenuates amyloidosis in AD mice (Town et al., 2008). Indeed, macrophage lacking TGF-β1 signaling phagocyte Aβ monomers and oligomers. By contrast, TGF-β1 in brain dampens microglial activation (Brionne et al., 2003). This effect of TGF-β1 could suppress the ability of macrophage/microglia to eliminate or restrict soluble Aβ oligomers and consequently aggravate memory deficits. These results further support previous studies suggesting a detrimental role of TGF-β system on cognitive impairment and Aβ accumulation.
Microglia surrounding Aβ plaques also strongly expressed CX3CR1 transcripts. CX3CR1 expression is enhanced in APPSwe/PS1/CCR2−/− mice, correlating with the degree of microglial recruitment. CX3CL1 is primarily produced by neurons and signals via CX3CR1 within microglia and macrophages (Harrison et al., 1998; Cardona et al., 2006). CX3CR1 deficiency is neurotoxic in mouse models of Parkinson's disease, amyotrophic lateral sclerosis, and systemic lipopolysaccharide administration, suggesting that CX3CL1–CX3CR1 signaling in microglia is beneficial to neurons (Cardona et al., 2006). By contrast, memory impairment and Aβ pathology are worsened in APPSwe/PS1/CCR2−/− mice, a phenomenon associated with increased recruitment of CX3CR1-positive microglia and upregulation of CX3CR1 transcript levels. These data point toward a detrimental role of CX3CR1 in APPSwe/PS1 mice. Indeed, a recent study demonstrated that CX3CR1 deficiency prevents neuronal loss in a mouse model of AD (Fuhrmann et al., 2010). This discrepancy might be explained by a phenotypic change of microglia in a context of CCR2 deficiency. Indeed, the robust expression of TGF-β1, TGF-β1 receptors, and CX3CR1 in microglia associated to plaques supports the idea that CCR2 deficiency gives rise to a phenotype of alternatively activated microglia (Mosser and Edwards, 2008), which are apparently unable to counteract AD pathology.
CCR2 is required for monocytes to emigrate from bone marrow (Serbina and Pamer, 2006; Tsou et al., 2007; Engel et al., 2008; Jia et al., 2008). Adoptive transfer studies with Ly-6Chigh monocytes have shown that they rapidly return to bone marrow in the absence of active recruitment to inflammatory sites (Varol et al., 2007, 2009). Therefore, CCR2 deficiency drastically decreases the frequency of monocytes in blood, which could contribute to the detrimental effects of CCR2 deficiency in APPSwe/PS1. A “peripheral sink hypothesis” exists, based on the findings of active Aβ transport from the CNS into the periphery across the blood–brain barrier via the low-density lipoprotein receptor (Deane et al., 2004), and from the periphery into CNS via the receptor for advanced glycation end products (Shibata et al., 2000; Deane et al., 2003). The peripheral sink hypothesis proposes that even low levels of free Aβ in the blood may alter the dynamics of transport and thereby contribute to a net efflux of Aβ from the brain (DeMattos et al., 2001; Holtzman et al., 2002; Lemere et al., 2003). It would then be possible that CCR2-competent mononuclear cells participate in the clearance of blood Aβ and are part of such a “sink hypothesis” by carrying soluble Aβ species out of the brain.
In conclusion, we demonstrated that CCR2 deficiency in APPSwe/PS1 mice provokes a rapid cognitive decline closely correlated with brain accumulation of soluble Aβ oligomers and a robust mRNA expression of TGF-β1, TGF-β-R2, and CX3CR1 in microglia. Our study supports a neuroprotective role of CCR2-competent cells in the pathophysiology of AD. We propose that such innate immune mechanisms may be deficient or suboptimal in AD patients and, therefore, that improving the availability of CCR2+ monocytes has considerable therapeutic potential.
Footnotes
This work was supported by the Canadian Institutes of Health Research and Neuroscience Canada (Brain Repair Program). G.N. is supported by a postdoctoral fellowship from the Alzheimer Society of Canada. S.R. holds a Canadian Research Chair in Neuroimmunology. We acknowledge the contribution of Dr. Mohammed Filali, Martine Lessard, Paul Préfontaine, Dr. Denis Soulet, and Marie-Michèle Plante for technical help.
References
- Auffray C, Fogg DK, Narni-Mancinelli E, Senechal B, Trouillet C, Saederup N, Leemput J, Bigot K, Campisi L, Abitbol M, Molina T, Charo I, Hume DA, Cumano A, Lauvau G, Geissmann F. CX3CR1+ CD115+ CD135+ common macrophage/DC precursors and the role of CX3CR1 in their response to inflammation. J Exp Med. 2009;206:595–606. doi: 10.1084/jem.20081385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock AA, Kuziel WA, Rivest S, Owens T. Chemokine expression by glial cells directs leukocytes to sites of axonal injury in the CNS. J Neurosci. 2003;23:7922–7930. doi: 10.1523/JNEUROSCI.23-21-07922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Boissonneault V, Filali M, Lessard M, Relton J, Wong G, Rivest S. Powerful beneficial effects of macrophage colony-stimulating factor on beta-amyloid deposition and cognitive impairment in Alzheimer's disease. Brain. 2009;132:1078–1092. doi: 10.1093/brain/awn331. [DOI] [PubMed] [Google Scholar]
- Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M, Calhoun ME. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. 2008;28:4283–4292. doi: 10.1523/JNEUROSCI.4814-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- Brionne TC, Tesseur I, Masliah E, Wyss-Coray T. Loss of TGF-beta 1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron. 2003;40:1133–1145. doi: 10.1016/s0896-6273(03)00766-9. [DOI] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- Cheng IH, Scearce-Levie K, Legleiter J, Palop JJ, Gerstein H, Bien-Ly N, Puoliväli J, Lesné S, Ashe KH, Muchowski PJ, Mucke L. Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem. 2007;282:23818–23828. doi: 10.1074/jbc.M701078200. [DOI] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Cruz JC, Kim D, Moy LY, Dobbin MM, Sun X, Bronson RT, Tsai LH. p25/cyclin-dependent kinase 5 induces production and intraneuronal accumulation of amyloid-β in vivo. J Neurosci. 2006;26:10536–10541. doi: 10.1523/JNEUROSCI.3133-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushing SD, Berliner JA, Valente AJ, Territo MC, Navab M, Parhami F, Gerrity R, Schwartz CJ, Fogelman AM. Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. Proc Natl Acad Sci U S A. 1990;87:5134–5138. doi: 10.1073/pnas.87.13.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2001;98:8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. 2009;29:313–326. doi: 10.1089/jir.2008.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Mello C, Le T, Swain MG. Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J Neurosci. 2009;29:2089–2102. doi: 10.1523/JNEUROSCI.3567-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khoury JB, Moore KJ, Means TK, Leung J, Terada K, Toft M, Freeman MW, Luster AD. CD36 mediates the innate host response to beta-amyloid. J Exp Med. 2003;197:1657–1666. doi: 10.1084/jem.20021546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- Engel DR, Maurer J, Tittel AP, Weisheit C, Cavlar T, Schumak B, Limmer A, van Rooijen N, Trautwein C, Tacke F, Kurts C. CCR2 mediates homeostatic and inflammatory release of Gr1high monocytes from the bone marrow, but is dispensable for bladder infiltration in bacterial urinary tract infection. J Immunol. 2008;181:5579–5586. doi: 10.4049/jimmunol.181.8.5579. [DOI] [PubMed] [Google Scholar]
- Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, Haass C, LaFerla FM, Kretzschmar H, Herms J. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer's disease. Nat Neurosci. 2010;13:411–413. doi: 10.1038/nn.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- Glabinski AR, Balasingam V, Tani M, Kunkel SL, Strieter RM, Yong VW, Ransohoff RM. Chemokine monocyte chemoattractant protein-1 is expressed by astrocytes after mechanical injury to the brain. J Immunol. 1996;156:4363–4368. [PubMed] [Google Scholar]
- Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouras GK, Almeida CG, Takahashi RH. Intraneuronal Abeta accumulation and origin of plaques in Alzheimer's disease. Neurobiol Aging. 2005;26:1235–1244. doi: 10.1016/j.neurobiolaging.2005.05.022. [DOI] [PubMed] [Google Scholar]
- Grammas P, Ovase R. Inflammatory factors are elevated in brain microvessels in Alzheimer's disease. Neurobiol Aging. 2001;22:837–842. doi: 10.1016/s0197-4580(01)00276-7. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, Streit WJ, Salafranca MN, Adhikari S, Thompson DA, Botti P, Bacon KB, Feng L. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci U S A. 1998;95:10896–10901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Paul SM, DeMattos RB. Abeta immunization and anti-Abeta antibodies: potential therapies for the prevention and treatment of Alzheimer's disease. Adv Drug Deliv Rev. 2002;54:1603–1613. doi: 10.1016/s0169-409x(02)00158-8. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Ishizuka K, Kimura T, Igata-yi R, Katsuragi S, Takamatsu J, Miyakawa T. Identification of monocyte chemoattractant protein-1 in senile plaques and reactive microglia of Alzheimer's disease. Psychiatry Clin Neurosci. 1997;51:135–138. doi: 10.1111/j.1440-1819.1997.tb02375.x. [DOI] [PubMed] [Google Scholar]
- Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med. 2000;192:1075–1080. doi: 10.1084/jem.192.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia T, Serbina NV, Brandl K, Zhong MX, Leiner IM, Charo IF, Pamer EG. Additive roles for MCP-1 and MCP-3 in CCR2-mediated recruitment of inflammatory monocytes during Listeria monocytogenes infection. J Immunol. 2008;180:6846–6853. doi: 10.4049/jimmunol.180.10.6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawarabayashi T, Shoji M, Younkin LH, Wen-Lang L, Dickson DW, Murakami T, Matsubara E, Abe K, Ashe KH, Younkin SG. Dimeric amyloid-β protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. 2004;24:3801–3809. doi: 10.1523/JNEUROSCI.5543-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobloch M, Konietzko U, Krebs DC, Nitsch RM. Intracellular Abeta and cognitive deficits precede beta-amyloid deposition in transgenic arcAbeta mice. Neurobiol Aging. 2007;28:1297–1306. doi: 10.1016/j.neurobiolaging.2006.06.019. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer's disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- Laflamme N, Rivest S. Toll-like receptor 4: the missing link of the cerebral innate immune response triggered by circulating gram-negative bacterial cell wall components. FASEB J. 2001;15:155–163. doi: 10.1096/fj.00-0339com. [DOI] [PubMed] [Google Scholar]
- Laflamme N, Lacroix S, Rivest S. An essential role of interleukin-1β in mediating NF-κB activity and COX-2 transcription in cells of the blood–brain barrier in response to a systemic and localized inflammation but not during endotoxemia. J Neurosci. 1999;19:10923–10930. doi: 10.1523/JNEUROSCI.19-24-10923.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemere CA, Spooner ET, LaFrancois J, Malester B, Mori C, Leverone JF, Matsuoka Y, Taylor JW, DeMattos RB, Holtzman DM, Clements JD, Selkoe DJ, Duff KE. Evidence for peripheral clearance of cerebral Abeta protein following chronic, active Abeta immunization in PSAPP mice. Neurobiol Dis. 2003;14:10–18. doi: 10.1016/s0969-9961(03)00044-5. [DOI] [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Lesné S, Kotilinek L, Ashe KH. Plaque-bearing mice with reduced levels of oligomeric amyloid-beta assemblies have intact memory function. Neuroscience. 2008;151:745–749. doi: 10.1016/j.neuroscience.2007.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luster AD. Chemokines—chemotactic cytokines that mediate inflammation. N Engl J Med. 1998;338:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- Mack M, Cihak J, Simonis C, Luckow B, Proudfoot AE, Plachý J, Brühl H, Frink M, Anders HJ, Vielhauer V, Pfirstinger J, Stangassinger M, Schlöndorff D. Expression and characterization of the chemokine receptors CCR2 and CCR5 in mice. J Immunol. 2001;166:4697–4704. doi: 10.4049/jimmunol.166.7.4697. [DOI] [PubMed] [Google Scholar]
- Malm TM, Koistinaho M, Pärepalo M, Vatanen T, Ooka A, Karlsson S, Koistinaho J. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to beta-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis. 2005;18:134–142. doi: 10.1016/j.nbd.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Mandrekar S, Jiang Q, Lee CY, Koenigsknecht-Talboo J, Holtzman DM, Landreth GE. Microglia mediate the clearance of soluble Aβ through fluid phase macropinocytosis. J Neurosci. 2009;29:4252–4262. doi: 10.1523/JNEUROSCI.5572-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga W, Shirokawa T, Isobe K. Specific uptake of Abeta1–40 in rat brain occurs in astrocyte, but not in microglia. Neurosci Lett. 2003;342:129–131. doi: 10.1016/s0304-3940(03)00240-4. [DOI] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- Meilandt WJ, Cisse M, Ho K, Wu T, Esposito LA, Scearce-Levie K, Cheng IH, Yu GQ, Mucke L. Neprilysin overexpression inhibits plaque formation but fails to reduce pathogenic Aβ oligomers and associated cognitive deficits in human amyloid precursor protein transgenic mice. J Neurosci. 2009;29:1977–1986. doi: 10.1523/JNEUROSCI.2984-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousseau DD, Chapelsky S, De Crescenzo G, Kirkitadze MD, Magoon J, Inoue S, Teplow DB, O'Connor-McCourt MD. A direct interaction between transforming growth factor (TGF)-betas and amyloid-beta protein affects fibrillogenesis in a TGF-beta receptor-independent manner. J Biol Chem. 2003;278:38715–38722. doi: 10.1074/jbc.M304080200. [DOI] [PubMed] [Google Scholar]
- Nadeau S, Rivest S. Role of microglial-derived tumor necrosis factor in mediating CD14 transcription and nuclear factor-κB activity in the brain during endotoxemia. J Neurosci. 2000;20:3456–3468. doi: 10.1523/JNEUROSCI.20-09-03456.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naert G, Laflamme N, Rivest S. Toll-like receptor 2-independent and MyD88-dependent gene expression in the mouse brain. J Innate Immun. 2009;1:480–493. doi: 10.1159/000225990. [DOI] [PubMed] [Google Scholar]
- Nielsen HM, Veerhuis R, Holmqvist B, Janciauskiene S. Binding and uptake of A beta1-42 by primary human astrocytes in vitro. Glia. 2009;57:978–988. doi: 10.1002/glia.20822. [DOI] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Ed 2. San Diego: Academic; 2001. [Google Scholar]
- Perry VH, Gordon S. Macrophages and microglia in the nervous system. Trends Neurosci. 1988;11:273–277. doi: 10.1016/0166-2236(88)90110-5. [DOI] [PubMed] [Google Scholar]
- Peters I, Igbavboa U, Schütt T, Haidari S, Hartig U, Rosello X, Böttner S, Copanaki E, Deller T, Kögel D, Wood WG, Müller WE, Eckert GP. The interaction of beta-amyloid protein with cellular membranes stimulates its own production. Biochim Biophys Acta. 2009;1788:964–972. doi: 10.1016/j.bbamem.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard KL, Filali M, Préfontaine P, Rivest S. Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid-β 1-42 and delay the cognitive decline in a mouse model of Alzheimer's disease. J Neurosci. 2008;28:5784–5793. doi: 10.1523/JNEUROSCI.1146-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins BJ, Yoshimura T, Leonard EJ, Pober JS. Cytokine-activated human endothelial cells synthesize and secrete a monocyte chemoattractant, MCP-1/JE. Am J Pathol. 1990;136:1229–1233. [PMC free article] [PubMed] [Google Scholar]
- Rota E, Bellone G, Rocca P, Bergamasco B, Emanuelli G, Ferrero P. Increased intrathecal TGF-β1, but not IL-12, IFN-γ and IL-10 levels in Alzheimer's disease patients. Neurol Sci. 2006;27:33–39. doi: 10.1007/s10072-006-0562-6. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Biochemistry and molecular biology of amyloid beta-protein and the mechanism of Alzheimer's disease. Handb Clin Neurol. 2008;89:245–260. doi: 10.1016/S0072-9752(07)01223-7. [DOI] [PubMed] [Google Scholar]
- Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. 2006;7:311–317. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV. Clearance of Alzheimer's amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Smits HA, Rijsmus A, van Loon JH, Wat JW, Verhoef J, Boven LA, Nottet HS. Amyloid-beta-induced chemokine production in primary human macrophages and astrocytes. J Neuroimmunol. 2002;127:160–168. doi: 10.1016/s0165-5728(02)00112-1. [DOI] [PubMed] [Google Scholar]
- Stalder AK, Ermini F, Bondolfi L, Krenger W, Burbach GJ, Deller T, Coomaraswamy J, Staufenbiel M, Landmann R, Jucker M. Invasion of hematopoietic cells into the brain of amyloid precursor protein transgenic mice. J Neurosci. 2005;25:11125–11132. doi: 10.1523/JNEUROSCI.2545-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinerman JR, Irizarry M, Scarmeas N, Raju S, Brandt J, Albert M, Blacker D, Hyman B, Stern Y. Distinct pools of beta-amyloid in Alzheimer disease-affected brain: a clinicopathologic study. Arch Neurol. 2008;65:906–912. doi: 10.1001/archneur.65.7.906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tampellini D, Rahman N, Gallo EF, Huang Z, Dumont M, Capetillo-Zarate E, Ma T, Zheng R, Lu B, Nanus DM, Lin MT, Gouras GK. Synaptic activity reduces intraneuronal Aβ, promotes APP transport to synapses, and protects against Aβ-related synaptic alterations. J Neurosci. 2009;29:9704–9713. doi: 10.1523/JNEUROSCI.2292-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman RS, Flavell RA. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14:681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, Charo IF. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. 2007;117:902–909. doi: 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wal EA, Gómez-Pinilla F, Cotman CW. Transforming growth factor-beta 1 is in plaques in Alzheimer and Down pathologies. Neuroreport. 1993;4:69–72. doi: 10.1097/00001756-199301000-00018. [DOI] [PubMed] [Google Scholar]
- Varol C, Landsman L, Fogg DK, Greenshtein L, Gildor B, Margalit R, Kalchenko V, Geissmann F, Jung S. Monocytes give rise to mucosal, but not splenic, conventional dendritic cells. J Exp Med. 2007;204:171–180. doi: 10.1084/jem.20061011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varol C, Yona S, Jung S. Origins and tissue-context-dependent fates of blood monocytes. Immunol Cell Biol. 2009;87:30–38. doi: 10.1038/icb.2008.90. [DOI] [PubMed] [Google Scholar]
- Wirths O, Multhaup G, Bayer TA. A modified beta-amyloid hypothesis: intraneuronal accumulation of the beta-amyloid peptide—the first step of a fatal cascade. J Neurochem. 2004;91:513–520. doi: 10.1111/j.1471-4159.2004.02737.x. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Masliah E, Mallory M, McConlogue L, Johnson-Wood K, Lin C, Mucke L. Amyloidogenic role of cytokine TGF-beta1 in transgenic mice and in Alzheimer's disease. Nature. 1997;389:603–606. doi: 10.1038/39321. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Sanan DA, Mucke L, Masliah E. Chronic overproduction of transforming growth factor-beta1 by astrocytes promotes Alzheimer's disease-like microvascular degeneration in transgenic mice. Am J Pathol. 2000;156:139–150. doi: 10.1016/s0002-9440(10)64713-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L, Masliah E, Mucke L. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7:612–618. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]