

Abstract
Numerous loss-of-function mutations in the progranulin (GRN) gene cause frontotemporal lobar degeneration with ubiquitin and TAR–DNA binding protein 43-positive inclusions by reduced production and secretion of GRN. Consistent with the observation that GRN has neurotrophic properties, pharmacological stimulation of GRN production is a promising approach to rescue GRN haploinsufficiency and prevent disease progression. We therefore searched for compounds capable of selectively increasing GRN levels. Here, we demonstrate that four independent and highly selective inhibitors of vacuolar ATPase (bafilomycin A1, concanamycin A, archazolid B, and apicularen A) significantly elevate intracellular and secreted GRN. Furthermore, clinically used alkalizing drugs, including chloroquine, bepridil, and amiodarone, similarly stimulate GRN production. Elevation of GRN levels occurs via a translational mechanism independent of lysosomal degradation, autophagy, or endocytosis. Importantly, alkalizing reagents rescue GRN deficiency in organotypic cortical slice cultures from a mouse model for GRN deficiency and in primary cells derived from human patients with GRN loss-of-function mutations. Thus, alkalizing reagents, specifically those already used in humans for other applications, and vacuolar ATPase inhibitors may be therapeutically used to prevent GRN-dependent neurodegeneration.
Introduction
Frontotemporal lobar degeneration (FTLD) is the second most abundant form of dementia in people under the age of 60 years after Alzheimer's disease (Graff-Radford and Woodruff, 2007). Although ∼40% of FTLD patients are pathologically characterized by tau positive inclusions, the remaining patients present with tau and α-synuclein-negative, ubiquitin-positive nuclear or cytoplasmic inclusions [frontotemporal lobar degeneration with ubiquitin-positive inclusions (FTLD-U)] (Mackenzie and Rademakers, 2007; Cruts and Van Broeckhoven, 2008). Deposited proteins observed in FTLD-U brains include the TAR–DNA binding protein 43 [TDP-43 (FTLD–TDP) (Neumann et al., 2006)] and the fused in sarcoma protein [FUS (FTLD–FUS)] (Neumann et al., 2009). Genetic linkage studies and/or mutation screenings identified loss-of-function mutations in the progranulin gene (GRN) in patients with familial FTLD–TDP (Baker et al., 2006; Cruts et al., 2006; Cruts and Van Broeckhoven, 2008; Gijselinck et al., 2008). Of the mutations reported to date (http://www.molgen.vib-ua.be/FTDMutations/), most are loss-of-function mutations leading to GRN haploinsufficiency (Gijselinck et al., 2008), which results in a severe reduction of GRN levels in tissues and biological fluids of patients (Ghidoni et al., 2008; Finch et al., 2009; Sleegers et al., 2009). Additionally, missense mutations (Schymick et al., 2007; van der Zee et al., 2007; Brouwers et al., 2008) lead to cytoplasmic missorting and degradation of GRN (Mukherjee et al., 2008; Shankaran et al., 2008) or to reduced secretion probably attributable to misfolding (Shankaran et al., 2008). Reduced GRN levels in biological fluids, such as CSF and plasma or serum, are not only sensitive biomarkers but also predict GRN mutations and a significantly enhanced risk for FTLD–TDP (Ghidoni et al., 2008; Finch et al., 2009; Sleegers et al., 2009). Because GRN is known to have neurotrophic properties (Van Damme et al., 2008), these findings strongly indicate that GRN haploinsufficiency is causally linked to neurodegeneration. We therefore searched for compounds that are capable of stimulating GRN production and/or secretion and may be used to restore physiological levels of GRN in FTLD–TDP patients with GRN haploinsufficiency.
Materials and Methods
Cell culture.
Human cervical carcinoma (HeLa) cells, human embryonic kidney (HEK 293T) cells, and mouse embryonic fibroblasts (MEFs) from autophagy-related gene-5 (ATG-5) knock-out and wild type (wt) mice (Mizushima et al., 2001) were cultured in DMEM with Glutamax I (Invitrogen). Lymphoblasts, immortalized by Ebstein Barr virus transformation of lymphocytes collected from whole blood on lithium heparin according to standard procedures (Brouwers et al., 2007; Gijselinck et al., 2008), were cultured in RPMI 1640 medium (Invitrogen) with glutamine (Invitrogen). Mouse neuroblastoma cells (N2a) were cultured in modified Eagle's medium (MEM) with glutamine. Human neuroblastoma (SH-SY5Y) cells were cultured in DMEM/F-12 with glutamine (Cambrex) supplemented with non-essential amino acids (Invitrogen). All media were supplemented with 10% (v/v) fetal calf serum (Invitrogen) and penicillin/streptomycin (PAA Laboratories).
Organotypic slice culture of mouse neocortex.
Organotypic slice cultures of mouse neocortex were prepared according to the protocol of Del Turco and Deller (2007) with minor modifications. Male or female mouse pups (GRN+/−,GRN+/+, GRN−/−) at postnatal days 3–5 were decapitated, brains were gently removed, and neocortex was dissected and cut on a chopper (McIlwain Tissue Chopper; Mickle Laboratory Engineering) into sections of 400 μm. Four sections were transferred to one membrane insert (Millicell, 30 mm, 0.4 μm pore size; Millipore Corporation) and cultured in a six-well plate. Cultures were kept in a humidified incubator (95% air, 5% CO2, 35°C) and allowed to adjust to culture conditions for 3 d. Thereafter, media were exchanged for media supplemented with compounds and incubated for 48 h. Supernatants were collected, immediately frozen, and stored at −80°C. Slice culture medium contained the following: 50% MEM, 25% heat inactivated normal horse serum, 25% Basal Medium Eagle, 25 mm HEPES, 2 mm Glutamax I, 0.65% glucose, 0.1 mg/ml streptomycin, 100 U/ml penicillin, and 0.15% sodium bicarbonate, pH 7.3.
Inhibitors and reagents.
The following inhibitors were used: bafilomycin A1 (BafA1), pepstatin A, antipain, ALLN (N-acetyl-Leu-Leu-Nle-CHO) (all Merck, Calbiochem), MG132 (carbobenzoxy-l-leucyl-l-leucyl-l-leucinal), epoxomicin (Biomol), concanamycin A, bepridil, amiodarone (all Sigma), archazolid B, and apicularen A were dissolved in DMSO. Leupeptin (Merck, Calbiochem), chloroquine (CQ) NH4Cl, EDTA, and phenanthroline (Sigma) were dissolved in H2O. Trans-Epoxysucciny-l-leucyl-amido(4-guanidino) butane (E64) (Biomol) was dissolved in 50% ethanol, and actinomycin D and cycloheximide (Sigma) were dissolved in methanol. Concentrations and duration of treatment are indicated in the figure legends.
Antibodies.
The following antibodies were used for immunoblotting: rabbit polyclonal antibody to human GRN (1:700; Invitrogen), sheep polyclonal antibody to mouse GRN (1:1000; R & D Systems), mouse monoclonal antibody to β-actin (1:2000; Sigma), rabbit polyclonal antibody to ATG-5 (1:2000; Cell Signaling Technology), and a mouse monoclonal antibody to LC3 (1:1200; Nanotools). Secondary antibodies were HRP-conjugated goat anti-mouse, goat anti-rabbit IgG (1:10,000; Promega), or anti-sheep IgG (1:5000; Santa Cruz Biotechnology). For immunocytochemistry, mouse monoclonal antibody to GRN (1:500; R & D Systems), rabbit polyclonal antibody to calnexin (1:500), Alexa Fluor-488- and Alexa Fluor-647-conjugated monoclonal antibodies to cis-Golgi matrix protein (GM-130) (1:10; BD Pharmingen) and LAMP-1 (1:50; Santa Cruz Biotechnology), respectively, were used. The rabbit antibody to TMEM59 (1:300) was a generous gift from Dr. Lichtenthaler (German Center for Neurodegenerative Diseases, Munich, Germany) and have been described previously (Ullrich et al., 2010).
Immunocytochemistry.
HeLa cells were grown on poly-lysine-coated coverslips, fixed for 20 min with 4% paraformaldehyde and 4% sucrose in PBS, permeabilized for 10 min with 0.2% Triton X-100 and 50 mm NH4Cl in PBS, and subsequently blocked for 1 h in PBS with 5% BSA. Cells were then double stained with the indicated antibodies for 2 h. After washing repeatedly with PBS, cells were incubated with Alexa Fluor-488 and Alexa Fluor-555 (Invitrogen) coupled secondary anti-mouse, anti-rat, or anti-rabbit antibodies for 1 h. Subsequently, cells were washed with PBS, and the coverslips were mounted on glass slides using Mowiol (Hoechst) supplemented with 0.5% 1,4-diazabicyclo(2.2.2)octane (Sigma). Images were obtained on a Carl Zeiss confocal laser scanning microscope (LSM 510 META) using an oil-immersion 100×/1.4 objective and the LSM software version 4.2 (Carl Zeiss). For LC3 staining, the GFP–LC3 cDNA construct (Schmid et al., 2007) was transfected into HeLa cells grown on coverslips, using Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturer. At 24 h after transfection, cells were subjected to BafA1 treatment (30 nm) for 16 h. Immunocytochemistry was performed as described above. LysoSensor DND-189 and LysoTracker DND-99 (Invitrogen) dyes were used for labeling acidic cell organelles. Therefore, cells were incubated with the indicated dye for 30 min according to the instructions of the manufacturer. Cells were imaged directly after incubation with the indicated dye, using an oil-immersion 40×/1.3 objective or a 10× objective.
Metabolic labeling and TCA precipitation on filter.
To analyze total protein secretion, HeLa cells were incubated for 16 h with 5 MBq/ml 35S-methionine/cysteine (Hartmann Analytic) in methionine-, cysteine-, and serum-free medium, in the presence of DMSO, BafA1, or CQ at the indicated concentrations. Conditioned media, 10 μl, were pipetted on Whatman filter paper, and proteins were precipitated by boiling the filter in 5% TCA for 10 min, followed by extensive washing in acetone. Quantification was performed in a scintillation counter (Beckman Coulter).
Preparation of conditioned media, cell lysates, and immunoblotting.
Conditioned media were collected, immediately cooled down, and centrifuged at 15,000 × g for 15 min at 4°C. Supernatants were either directly or after TCA precipitation subjected to standard 10% SDS-PAGE. For cell lysates, cells were washed twice with PBS, scraped off, and pelleted at 1000 × g, 5 min. Cell pellets were lysed for 15 min in ice-cold STEN lysis buffer (150 mm NaCl, 50 mm Tris-HCl, pH 7.6, 2 mm EDTA, and 1% NP-40), freshly supplemented with protease inhibitor cocktail (Sigma), and clarified by centrifugation at 4°C for 30 min at 15,000 × g. Equal amounts of protein were separated by SDS-PAGE and transferred onto polyvinylidene difluoride membranes. For detection, the indicated antibodies were used. Bound antibodies were visualized by horseradish peroxidase-conjugated secondary antibody using enhanced chemiluminescence technique (GE Healthcare).
Quantifying mRNA with real time reverse transcription-PCR.
For quantitative reverse transcription (qRT)-PCR total RNA was prepared using the RNeasy kit (Qiagen). RNA preparations were treated with DNase (DNase I, RNase-free; Qiagen) and 1 μg of total RNA was used for reverse transcription with oligo-dT primer and Moloney murine leukemia virus reverse transcriptase (Ambion) according to the protocols of the manufacturer. qRT-PCR was performed on a 7500 Fast Real-Time PCR System (Applied Biosystems) with TaqMan technology, using primer sets from Applied Biosystems: for mouse GRN, Mm00433848_m1 (exon boundary 4–5); for mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 4352339E; for human GRN, Hs00173570_m1 (exon boundary 1–2); and for human GAPDH, 4326317E. Each sample was analyzed in triplicate, and levels of GRN cDNA were normalized to GAPDH cDNA according to the ΔΔCt method using the equation 2−(CtGRN − CtGAPDH)treatment − (CtGRN− CtGAPDH)control.
Northern blotting.
For Northern blot analysis, quality of total RNA was controlled using the Agilent 2100 Bioanalyser (data not shown). Total RNA, 3 μg, were separated on a formaldehyde-containing agarose gel. Transfer onto a HyBond N membrane (GE Healthcare) and hybridization were performed as described previously (Lammich et al., 2004). Templates of GRN and GAPDH for generating the radioactive probes were amplified by PCR using following the primer pairs: for GRN, 5′-GAGCTCGGATCCGTCGACCCACGCGTCCGCAAGGTAC-3′ and 5-AAACTGCAGTGGAAGCCCCGTGGGCAGCAG-3′; for GAPDH, 5′-GGAAGCTTGTCATCAATGG-3′ and 5′-CAGGGATGATGTTCTGGAG-3′. [32P]dCTP (Hartmann Bioanalytics)-labeled probes were generated using the Random Primers DNA Labeling System (Invitrogen). Labeled RNA was detected by exposure of the blot to Super RX film (Fuji) and quantified by PhosphorImager (Molecular Dynamics).
ELISA for human and mouse GRN.
Secreted GRN in conditioned media was quantified by a sandwich immunoassay using the Meso Scale Discovery Sector Imager 2400. Streptavidin-coated 96-well multi-array plates were blocked in blocking buffer (0.5% bovine serum albumin and 0.05% Tween 20 in PBS, pH 7.4) overnight. For detection of human GRN, plates were incubated for 1 h at room temperature with a biotinylated goat anti-human GRN capture antibody (R & D Systems) diluted 1:100 in blocking buffer. Plates were washed four times with washing buffer (0.05% Tween 20 in PBS) before addition of the samples or the standards (GenWay Biotech) and the first detection antibody (mouse monoclonal anti-human GRN antibody; 1:2000 diluted in blocking buffer; R & D Systems). Plates were incubated at room temperature for 2 h, followed by three washing steps. For detection, a SULFO-TAG-labeled secondary anti-mouse antibody (1:1000; Meso Scale Discovery) was added, and plates were incubated for 1 h in the dark. After three washes, Meso Scale Discovery Read buffer was added, and the light emission at 620 nm after electrochemical stimulation was measured using the Meso Scale Discovery Sector Imager 2400 reader. For detection of mouse GRN, mouse-specific anti-GRN antibodies and the appropriate secondary detection antibody were used: biotinylated sheep anti-mouse GRN antibody (1:200; R & D Systems), rat anti-mouse GRN antibody (1:1000; R & D Systems), and a SULFO-TAG-labeled secondary anti-rat antibody diluted 1:500 [SULFO-TAG was coupled to anti-rat IgG (Sigma) using Meso Scale Discovery SULFO-TAG-NHS-ester according to the protocol of the manufacturer, respectively].
Animal husbandry.
All experiments were performed in compliance with the guidelines of the German Council on Animal Care.
Results
Bafilomycin A1 increases intracellular and secreted GRN
Strong evidence supports the finding that GRN haploinsufficiency is causally associated with neurodegeneration observed in all patients carrying a loss-of-function mutation in GRN. Increasing GRN levels by influencing its turnover or production is consequently a promising therapeutic approach. We specifically screened for compounds capable of inhibiting proteolytic degradation of GRN, because GRN may be metabolized during its passage through the secretory pathway or during receptor-mediated uptake. To analyze whether GRN is subject for degradation, cells were treated with a variety of protease inhibitors, and subsequently cell lysates as well as conditioned media were analyzed for an increase in GRN levels. In two different cell lines, HeLa cells and neuronal SH-SY5Y cells, inhibitors of lysosomal proteases such as pepsatin A, E64, and leupeptin, had no effect on the amount of intracellular (Fig. 1A) or secreted (Fig. 1B) GRN, except the alkalizing reagent BafA1, which significantly increased intracellular (Fig. 1A) as well as secreted (Fig. 1B) GRN. In addition, inhibition of the ubiquitin proteasome system with MG132 or epoxomicin failed to increase GRN levels in cell lysates or conditioned media (Fig. 1A,B). Moreover, inhibition of the neutrophil elastase, an enzyme involved in processing of progranulin into granulin peptides (Bateman and Bennett, 2009), had no effect on GRN levels (data not shown). Thus, BafA1 was the only compound found to significantly elevate intracellular and secreted GRN levels.
Figure 1.
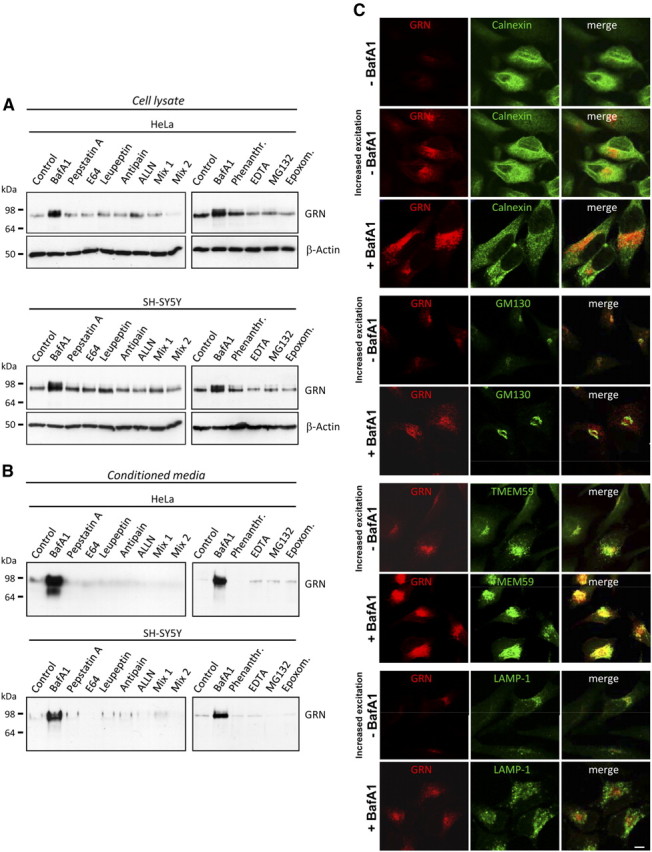
BafA1 but none of the tested protease inhibitors increase intracellular and secreted GRN levels. A, B, HeLa and SH-SY5Y cells were treated for 16 h with DMSO, BafA1 (50 nm), phenanthroline (10 mm), EDTA (15 mm), MG132 (10 μm), epoxomicin (1 μm), pepstatin A (1 μm), E64 (10 μm), antipain (5 μm), ALLN (5 μm), mix 1 (E64, leupeptin, and antipain), and mix 2 (mix 1 plus pepstatin A and ALLN). Cell lysates (A) and conditioned media (B) were analyzed for GRN by immunoblotting. Equal loading of cell lysates was confirmed by probing for β-actin (A). C, HeLa cells treated with 25 nm BafA1 or DMSO were stained with a monoclonal anti-GRN antibody (red) and costained with antibodies against calnexin (green, ER), GM-130 (green, cis-Golgi) TMEM59 (green, Golgi) and LAMP-1 (green, lysosomes). The BafA1-mediated increase of GRN does not affect intracellular GRN localization. Increased excitations are shown to visualize the low levels of GRN under control conditions. Scale bar, 10 μm.
Bafilomycin A1 increases GRN independent of lysosomal and autophagosomal degradation
BafA1 selectively inhibits the vacuolar ATPase (v-ATPase), which among other cellular consequences leads to impaired lysosomal degradation (Bowman et al., 2004). Surprisingly, all inhibitors of lysosomal proteases had no effect on GRN levels (Fig. 1A,B), suggesting that the BafA1-induced GRN increase is independent of lysosomal degradation. To confirm that GRN does not accumulate in lysosomes during BafA1 treatment, we investigated the subcellular localization of GRN in the presence and absence of BafA1. Without BafA1 treatment, low levels of endogenous GRN were only detectable after increased laser excitation (Fig. 1C). GRN was localized in a vesicular compartment that costained with Golgi marker antibodies to GM-130 and TMEM59 (Ullrich et al., 2010), respectively, and to some extend with the endoplasmic reticulum (ER) marker antibody to calnexin (Fig. 1C). During BafA1 treatment, we observed a significant increase in intracellular GRN within the ER and the Golgi network, so that images could be acquired with reduced laser excitation. However, BafA1 treatment caused no change in intracellular GRN distribution and did not result in a colocalization of GRN with the lysosomal marker LAMP-1 (Fig. 1C, bottom). These findings suggest that BafA1 does not cause an accumulation of GRN in lysosomes but instead causes an increase in intracellular GRN levels early within the secretory pathway.
To further prove that BafA1 increases GRN levels independently of impaired lysosomal degradation, we blocked protein transport beyond the Golgi complex by incubating cells at 20°C (Griffiths et al., 1989). In control cells kept at 37°C, BafA1 treatment led to the expected intracellular and extracellular increase of GRN (Fig. 2A). In cells incubated at 20°C, GRN secretion was abolished (Fig. 2A, top) as expected. However, GRN still accumulated intracellularly during BafA1 treatment (Fig. 2A, middle), confirming that the intracellular GRN accumulation is not attributable to impaired endocytosis or lysosomal degradation of GRN.
Figure 2.
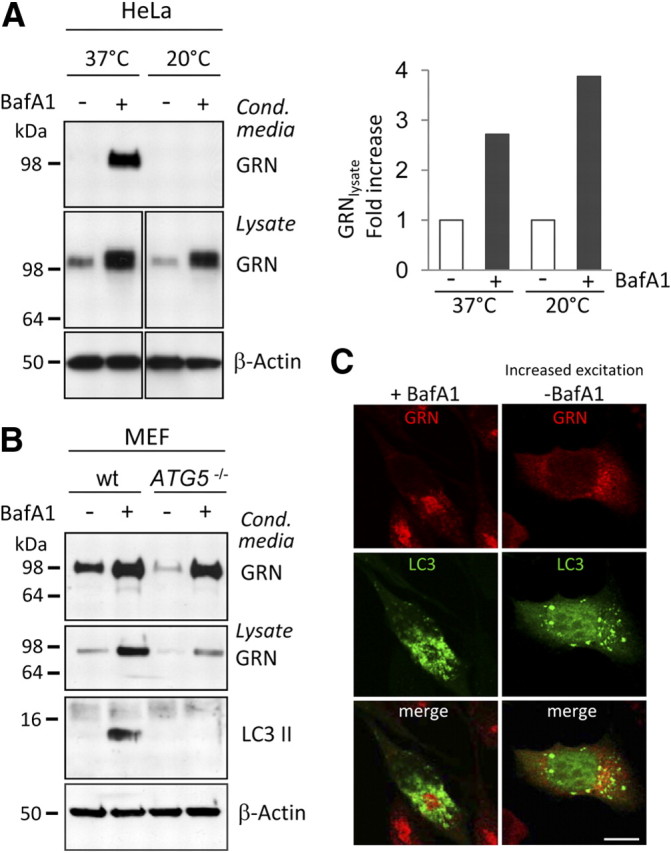
The BafA1-mediated GRN increase is independent of lysosomal degradation and autophagy. A, BafA1treatment was performed at 20°C to block forward transport through the secretory pathway and under control conditions (37°C). Media and cell lysates were subjected to GRN immunoblotting, followed by quantification. GRN levels were normalized to untreated control cells. Note that at 20°C no secretion is observed; therefore, no GRN is detected in conditioned media. B, MEFs from ATG-5 knock-out and wt mice (Mizushima et al., 2001) were treated with and without 25 nm BafA1 for 20 h. The complete loss of autophagosome formation was verified by LC3 immunoblotting. Note that inhibition of autophagy does not affect GRN secretion neither under control conditions nor during BafA1 treatment. The absolute GRN levels of the ATG-5 knock-out and the wt MEF cells are not directly comparable because primary MEF cells are of different origin. C, HeLa cells transfected with a GFP–LC3 (green) fusion construct were immunostained for GRN in the absence and presence of BafA1. No colocalization of GRN with autophagosomes was observed. Scale bar, 10 μm.
Besides inhibition of lysosomal degradation, blocking of vesicle acidification also leads to impaired autophagy. To investigate whether GRN undergoes autophagic degradation, we investigated GRN metabolism in the presence and absence of BafA1 in mouse embryonic fibroblasts derived from ATG-5 knock-out mice (Mizushima et al., 2001) and in control fibroblasts. A deficiency in ATG-5, a gene essential for autophagy, caused no a priori increase in GRN levels (Fig. 2B). However, absence of autophagosome formation still allowed the BafA1-induced increase in intracellular and extracellular levels of GRN (Fig. 2B), suggesting that increased GRN levels are not caused by an impaired autophagic degradation of GRN. Immunohistochemistry demonstrates that GRN does not colocalize with transiently transfected GFP–LC3, a marker protein of autophagosomes, with or without BafA1 treatment (Fig. 2C), confirming that autophagy is not involved in the BafA1-mediated increase of GRN.
Bafilomycin A1 increases GRN levels independent of transcription
Recently, it has been demonstrated that, under aberrant lysosomal storage conditions, GRN mRNA among many others is transcriptionally upregulated (Sardiello et al., 2009). Furthermore, it has been shown that, under extracellular acidic conditions, GRN mRNA is increased up to twofold in primary rat skin fibroblast cells (Guerra et al., 2007). We therefore investigated whether transcriptional mechanisms are responsible for the increase in GRN during treatment with BafA1. In HeLa cells, GRN mRNA levels were not significantly changed, whereas in N2a cells, a mouse neuroblastoma cell line, a twofold increase in GRN mRNA was detected during treatment with BafA1 (Fig. 3A). However, in contrast to the twofold increased mRNAs levels in N2a cells, BafA1 caused an approximately 25-fold increase of GRN in conditioned media (Fig. 3A,B). Moreover, GRN protein still increased several-fold during BafA1 treatment when transcription was blocked by actinomycin D (Fig. 3A,B), demonstrating that posttranscriptional mechanisms are sufficient to cause the significant increase in secreted GRN. Finally, the generation of alternatively spliced mRNAs could be excluded, because we detected only one mRNAs species of identical length with and without BafA1 treatment (Fig. 3C). Thus, besides a moderate and probably cell line-dependent transcriptional upregulation, posttranscriptional mechanisms most likely cause the significant increase in GRN expression and secretion during BafA1 treatment.
Figure 3.

BafA1 causes a posttranscriptional increase of GRN expression and secretion. A, Quantification of GRN mRNA in BafA1 (25 nm; 16 h) treated and untreated HeLa and N2a cells by qRT-PCR. GRN mRNA levels were normalized to GAPDH mRNA and are presented as the ratio to the untreated control. Parallel experiments were performed in the presence of the transcription inhibitor actinomycin D (ActD; 1 μm). B, Conditioned media of the samples used for RNA extraction in A were analyzed for protein levels of GRN by immunoblotting and subsequently quantification. C, Northern blot of BafA1 (25 nm; 16 h) treated and nontreated HeLa cells probed for GRN and reprobed for GAPDH. Quantification of triplicates was performed with a PhosphorImager. For A–C, normalized values are shown as means ± SD (n = 3) of independent experiments (***p < 0.001; ****p < 0.0001; *****p < 0.00001 by Student's two-tailed, unpaired t test).
Alkalizing reagents increase GRN
Consistent with the neutralizing effect of BafA1 on acidic cellular compartments, monitoring of the intracellular pH in BafA1-treated and untreated cells proved that BafA1 rapidly and reversibly affects the cellular pH. Whereas in control cells lysosomes and other acidic vesicles were readily stained with LysoSensor, no labeling was observed immediately after 16 h treatment with BafA1 (time point 0 h in Fig. 4A). However, during withdrawal of BafA1, the intracellular pH rapidly normalized, as proven by the robust vesicular staining with LysoSensor after 4, 8, and 24 h (Fig. 4A). Thus, BafA1 prevents vesicular acidification in our cellular system, and this effect is fully reversible. Consistent with the alkalizing activity of BafA1, other alkalizing reagents, such as NH4Cl and CQ, also elevated endogenous GRN levels in cell lysates and in conditioned media of HeLa cells (Fig. 4B) and all analyzed neuronal and non-neuronal cells (data not shown). BafA1 reaches its maximal effect on GRN at 25–50 nm without affecting general protein secretion (Fig. 4C). CQ increased GRN levels dose dependently up to 50 μm (Fig. 4D). Even at high doses of CQ, no general effect on total protein secretion was observed (Fig. 4D). Together, impaired vesicle acidification, obtained during BafA1, CQ, or NH4Cl treatment, is sufficient to increase intracellular and consequently secreted GRN without affecting general secretion.
Figure 4.
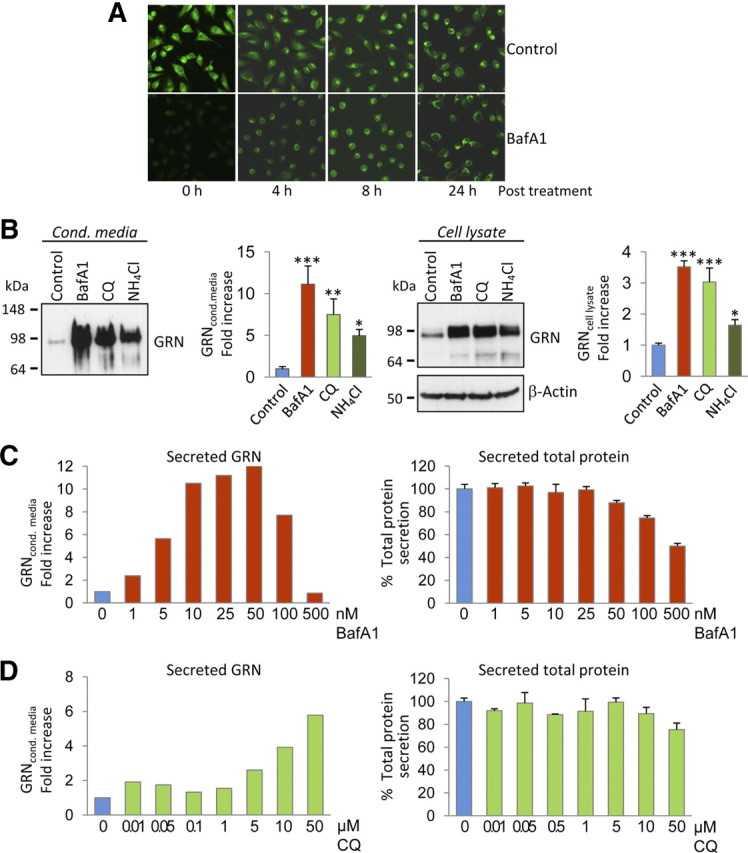
Alkalizing reagents increase intracellular and secreted GRN levels. A, HeLa cells pretreated with BafA1 (16 h, 30 nm) and nontreated control cells were incubated with 100 nm LysoSensor, at indicated time points after BafA1 treatment. Note that the inhibition of lysosomal acidification by BafA1 is reversible. B, HeLa cells were treated with BafA1 (50 nm), CQ (50 μm), and NH4Cl (25 mm). Conditioned media (left) and cell lysates (right) were analyzed for GRN expression by immunoblotting. GRN increase was quantified in conditioned media and cell lysates. Data are expressed as fold increase of untreated control cells and shown as means ± SD (n = 3) (*p < 0.05; **p < 0.01; ***p < 0.001 by one-way ANOVA post hoc Dunnett's test). For dose–response curves, HeLa cells were treated for 16 h with BafA1 (C) or CQ (D) at the indicated concentrations. Secreted GRN was analyzed by immunoblot and quantified (C, D, left). Potential effects of BafA1 or CQ treatment on total protein production and secretion were quantified by metabolic labeling. Secreted 35S-methionine-labeled proteins were measured after TCA precipitation by scintillation counting. Data were normalized to untreated cells (means ± SD, n = 3) (C, D, right).
v-ATPase is the cellular target of BafA1
v-ATPase was confirmed as the cellular target of BafA1 by treatment of cells with three independent and highly selective v-ATPase inhibitors, namely concanamycin A, archazolid B, and apicularen A (Huss et al., 2005). All three v-ATPase inhibitors increased intracellular and extracellular levels of GRN to a similar extent as BafA1 in HeLa cells (Fig. 5) and all analyzed neuronal and non-neuronal cells (data not shown).
Figure 5.

v-ATPase inhibitors increase intracellular and secreted GRN levels. HeLa cells were treated with the highly specific v-ATPase inhibitors concanamycin A (ConA; 50 nm), archazolid B (ArcB; 50 nm), and apicularen A (ApiA; 100 nm). Conditioned media and cell lysates were analyzed for GRN expression by immunoblotting. GRN increase was quantified in conditioned media and cell lysates. Data are expressed as fold increase of untreated control cells and shown as means ± SD (n = 3) (**p < 0.01; ***p < 0.001 by one-way ANOVA post hoc Dunnett's test).
Interestingly, inhibition of v-ATPase not only led to a robust intracellular and extracellular increase of GRN but also had sustained effects. Even 72 h after the initial overnight treatment with BafA1, significantly increased levels of GRN were still observed in cell lysates and conditioned media compared with control-treated cells (Fig. 6A). Importantly, 72 h after BafA1 treatment was terminated, LysoTracker staining revealed robust labeling of acidic vesicles, demonstrating that BafA1 was washed out and that lysosomal pH was normalized again (Fig. 6B). Thus, the long-lasting posttreatment increase of GRN secretion is independent of the actual lysosomal pH, consistent with the data presented above.
Figure 6.
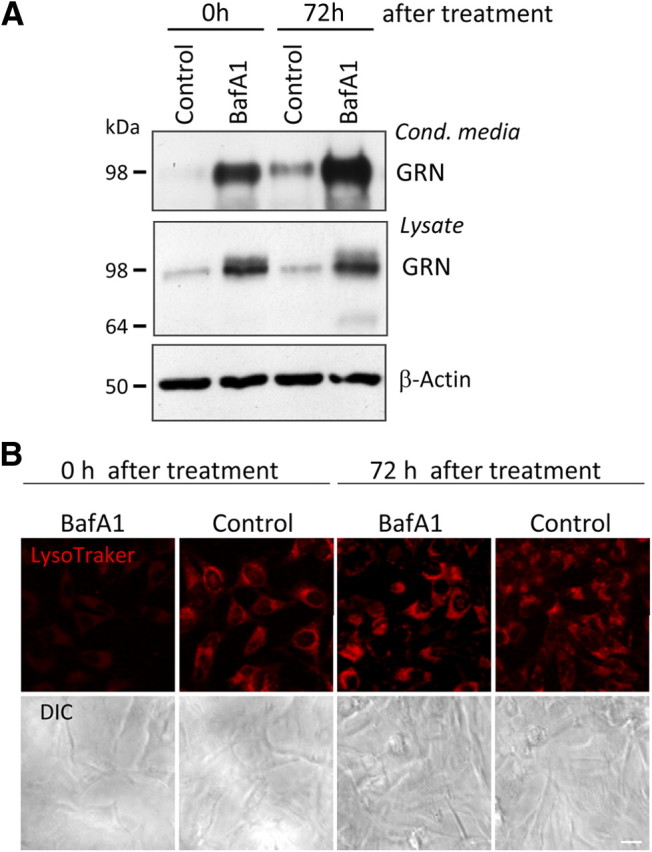
Sustained effects of BafA1 on GRN secretion. A, HeLa cells were treated with BafA1 (30 nm) for 16 h. In a first set of cultures, GRN in conditioned media and lysates was analyzed directly after BafA1 treatment (time point 0 h). After terminating BafA1 treatment, a second set of cultures were kept without BafA1 for additional 72 h. Conditioned media were collected during the last 16 h of this time period. Control cells were treated with DMSO for the same time interval (control). B, Vesicle acidification was monitored by LysoTracker staining (30 min). Corresponding differential interference contrast (DIC) images are shown below. Impairment of vesicular acidification is observed immediately after BafA1 treatment but not 72 h after treatment. Scale bar, 20 μm.
Targeting of v-ATPase or increasing intracellular pH rescues GRN deficiency in cortical slice cultures from GRN knock-out mice and in primary cells from human patients
Next we investigated whether reduced GRN levels attributable to disease-causing GRN haploinsufficiency could be elevated to physiological levels by alkalizing reagents. The only currently available animal model for GRN deficiency is the heterozygous GRN knock-out mouse (Kayasuga et al., 2007; Yin et al., 2010). We used organotypic cortical slice cultures to monitor GRN levels in the presence and absence of BafA1. As expected, we observed an ∼60–70% reduction of GRN in conditioned media from organotypic cortical slice cultures derived from GRN+/− mice (Fig. 7A) similar to human patients with GRN mutations (Ghidoni et al., 2008; Finch et al., 2009; Sleegers et al., 2009). During BafA1 treatment of organotypic cortical slice cultures derived from GRN+/− mice, an increase in GRN in the culture media was achieved (Fig. 7B), whereas a smaller but still significant stimulation was observed with CQ (Fig. 7B). Again, the effects were long lasting, and an even more pronounced increase was detected after BafA1 treatment was terminated (Fig. 7C). To test whether GRN can be increased to physiological levels in a disease setting, we treated lymphoblasts derived from four healthy controls and six patients with confirmed familial FTLD–TDP carrying a GRN loss-of-function mutation (Brouwers et al., 2007; Gijselinck et al., 2008) with BafA1 and CQ. CQ is of special interest for therapy because it is frequently used for malaria prophylaxis and treatment, in autoimmune disorders, and in sensitizing cancer therapy (Solomon and Lee, 2009). As expected, absolute levels of GRN were reduced in conditioned media obtained from cells with GRN loss-of-function mutations compared with cells derived from healthy controls (Fig. 8). Levels of GRN were significantly increased during stimulation with BafA1 to levels at least similar to untreated cells of healthy control subjects (Fig. 8A). CQ treatment led to a less pronounced but still significant increase of GRN secretion (Fig. 8B). Finally, we also investigated the potential of bepridil and amiodarone to increase GRN levels. Both drugs are weak bases and are clinically used for treatment of angina pectoris or arrhythmias (Hollingshead et al., 1992; Siddoway, 2003). Moreover, both drugs were shown recently to modulate amyloid β-peptide production (Mitterreiter et al., 2010). When the above described lymphoblast cells from healthy controls and FTLD–TDP patients carrying a GRN loss-of-function mutation were treated with bepridil or amiodarone, both drugs significantly increased GRN levels in conditioned media (Fig. 8C,D). Thus, our data demonstrate that GRN deficiency can be rescued with clinically used drugs in lymphoblasts derived from patients with familial FTLD–TDP and in brain slices of GRN+/− mice, two independent models that recapitulate disease associated GRN deficiency.
Figure 7.

Rescue of reduced GRN levels in organotypic cortical slice cultures of heterozygous GRN knock-out mice. Cortical slice cultures (n = 6 for each mouse), GRN+/+ (n = 1) or GRN+/− (n = 8), were prepared. After 72 h, media were collected (A, days 0–3) and exchanged with drug- or DMSO-containing media. Two sets of cortical slice cultures of each mouse were treated with BafA1 (25 nm), CQ (10 μm), or DMSO for 48 h (B, days 3–5). After 48 h, media were replaced and secreted proteins were collected for 96 h in the absence of drugs (C, days 5–9). GRN levels were measured in triplicates using a sandwich ELISA specific for mouse GRN (means ± SD, n = 3).
Figure 8.

BafA1 and CQ treatment rescues reduced GRN levels in primary human lymphoblasts. Lymphoblasts derived from loss-of-function mutation carriers (DR lines) and control lymphoblasts from non-mutation carriers (CR lines) were cultured at equal cell density. Triplicates of each DR-cell line were treated with BafA1 (25 nm) (A), CQ (10 μm) (B), or the alkalizing drugs amiodarone (5 μm) (C) and bepridil (5 μm) (D) for 16 h. For controls, triplicates of each CR cell line were treated with DMSO for 16 h. GRN levels in conditioned media were determined by ELISA. For CR cell lines, GRN levels of untreated cells are shown. Data represent means ± SD. Note that all cell lines derived from GRN mutation carriers displayed a significant increase of GRN during treatment with both drugs.
Discussion
GRN haploincufficiency is the cause of FTLD–TDP in patients with GRN-associated loss-of function mutations. Increasing production of GRN from the remaining wt allele is therefore a logical therapeutic approach. Based on the currently available data, the full-length GRN protein rather than the proteolytically processed granulin peptides is expected to exert the described beneficial neurotrophic and anti-inflammatory function (Kessenbrock et al., 2008; Van Damme et al., 2008; Ryan et al., 2009). However, even if GRN-associated FTLD–TDP would be caused by lack of GRN peptides, rather than by the GRN holoprotein, elevating their precursor would be beneficial, because this should also restore the amount of its cleavage products.
Here, we demonstrate that alkalizing compounds, including three clinically used drugs, as well as inhibitors of the v-ATPase are capable to restore GRN levels in an animal model with reduced GRN and in cells from patients with loss-of function GRN mutations. The pH of intracellular compartments and the extracellular space is carefully controlled by the proton pump v-ATPase, a multiprotein complex, which is ubiquitously expressed in all cell types and is localized in cellular organelles such as Golgi, lysosomes, and endosomes and at the plasma membrane (summarized by Forgac, 2007). We demonstrate that specific inhibitors of the v-ATPase, such as the well known plecomacrolides BafA1 and concanamycin A as well as the novel inhibitors archazolid B and apicularen A (summarized by Huss and Wieczorek, 2009), increase intracellular and thus consequently extracellular GRN levels at nanomolar concentrations. Off-target effects beyond v-ATPase inhibition are unlikely because inhibitors of different chemical classes consistently increase GRN expression. Moreover, apicularen A and plecomacrolides are known to bind to distinct sites of the v-ATPase (Huss et al., 2005). We can also rule out that inhibitors of v-ATPase affect putative receptor-mediated uptake of GRN, because GRN accumulates during treatment with BafA1 not only in conditioned media but also in cell lysates. Moreover, when cellular transport through the secretory pathway beyond the Golgi was blocked by low temperature and consequently traffic to lysosomes and secrection is abolished, GRN still accumulates within cells during BafA1 treatment. In addition, GRN does not accumulate in lysosomes or autophagosomes during treatment with BafA1. Finally, the effect on increased intracellular and extracellular GRN still persists even when BafA1 is washed out and the lysosomal acidification is restored to physiological conditions. Together, these findings demonstrate that impaired lysosomal degradation or reduced receptor-mediated uptake of GRN is not causative for the GRN increase during BafA1 treatment. Finally, we also exclude that transcriptional upregulation of GRN is required for the strong increase in protein levels of GRN during inhibition of v-ATPase. Our findings therefore suggest a translational upregulation of GRN initiated by the intracellular pH changes induced by v-ATPase inhibitors or alkalizing drugs. We demonstrate that drug-induced GRN increase can compensate for reduced levels of GRN in lymphoblasts of GRN-associated FTLD–TDP patients and in organotypic slice cultures derived from a mouse model for GRN deficiency. We therefore suggest that specific v-ATPase inhibitors, such as those currently discussed in cancer therapy (Fais et al., 2007), or alkalizing drugs, such as the clinically used CQ, bepridil, and amiodarone, may be further developed or improved to increase GRN levels in FTLD–TDP patients with GRN mutations to physiologically normal levels. Such drugs may be tolerated without major adverse side effects, as shown for CQ, a frequently used malaria drug (Solomon and Lee, 2009). Drug concentrations used in this study are therapeutically achievable in plasma of patients during treatment (Sanchez et al., 2007). For treatment of FTLD–TDP, it is of obvious interest that the potential drugs are able to cross the blood–brain barrier. CQ and amiodarone have been reported to cross the blood–brain barrier (Riva et al., 1982; Koreeda et al., 2007) to some extent. Moreover, CQ is used in ongoing studies on brain tumor therapy (Solomon and Lee, 2009). To ensure correct dosing of the drugs, established ELISAs are available for convenient monitoring of GRN levels in plasma and CSF (Ghidoni et al., 2008; Finch et al., 2009; Sleegers et al., 2009).
Footnotes
This work was supported by the Center for Integrated Protein Science Munich, the Sonderforschungsbereich Molecular Mechanisms of Neurodegeneration (SFB 596), the Competence Network for Neurodegenerative Diseases of the Bundesministerium für Bildung und Forschung, and a European Molecular Biology Organization postdoctoral fellowship (D.D.). C.H. is supported by a “Forschungsprofessur” of the Ludwig Maximilians University. The Antwerp site was in part funded by Fund for Scientific Research—Flanders (FWO-V), the Foundation for Alzheimer Research, the Medical Foundation Queen Elisabeth, the Interuniversity Attraction Poles Program P6/43 of the Belgian Science Policy Office, and a Methusalem Excellence Grant of the Flemish Government, Belgium. N.B., I.G., and K.S. are receiving a postdoctoral fellowship of the FWO-V. We acknowledge the contribution of the clinical neurologists and the research nurses to the biosampling of the patients (DR numbers) and the control individuals (CR numbers). We are also grateful for the support of the personnel of the Antwerp Biobank at the Institute Born Bunge and the Genetic Service Facility at VIB. We thank M. Nishihara for providing the GRN knock-out mice, N. Mizushima for the ATG-5 knock-out MEF cells, S. Lichtenthaler and Anna Münch for providing the antibody against TMEM59, and R. Page and D. Edbauer for critical reading and valuable discussion.
References
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- Bateman A, Bennett HP. The granulin gene family: from cancer to dementia. Bioessays. 2009;31:1245–1254. doi: 10.1002/bies.200900086. [DOI] [PubMed] [Google Scholar]
- Bowman EJ, Graham LA, Stevens TH, Bowman BJ. The bafilomycin/concanamycin binding site in subunit c of the V-ATPases from Neurospora crassa and Saccharomyces cerevisiae. J Biol Chem. 2004;279:33131–33138. doi: 10.1074/jbc.M404638200. [DOI] [PubMed] [Google Scholar]
- Brouwers N, Nuytemans K, van der Zee J, Gijselinck I, Engelborghs S, Theuns J, Kumar-Singh S, Pickut BA, Pals P, Dermaut B, Bogaerts V, De Pooter T, Serneels S, Van den Broeck M, Cuijt I, Mattheijssens M, Peeters K, Sciot R, Martin JJ, Cras P, Santens P, Vandenberghe R, De Deyn PP, Cruts M, Van Broeckhoven C, Sleegers K. Alzheimer and Parkinson diagnoses in progranulin null mutation carriers in an extended founder family. Arch Neurol. 2007;64:1436–1446. doi: 10.1001/archneur.64.10.1436. [DOI] [PubMed] [Google Scholar]
- Brouwers N, Sleegers K, Engelborghs S, Maurer-Stroh S, Gijselinck I, van der Zee J, Pickut BA, Van den Broeck M, Mattheijssens M, Peeters K, Schymkowitz J, Rousseau F, Martin JJ, Cruts M, De Deyn PP, Van Broeckhoven C. Genetic variability in progranulin contributes to risk for clinically diagnosed Alzheimer disease. Neurology. 2008;71:656–664. doi: 10.1212/01.wnl.0000319688.89790.7a. [DOI] [PubMed] [Google Scholar]
- Cruts M, Van Broeckhoven C. Loss of progranulin function in frontotemporal lobar degeneration. Trends Genet. 2008;24:186–194. doi: 10.1016/j.tig.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- Del Turco D, Deller T. Organotypic entorhino-hippocampal slice cultures: a tool to study the molecular and cellular regulation of axonal regeneration and collateral sprouting in vitro. Methods Mol Biol. 2007;399:55–66. doi: 10.1007/978-1-59745-504-6_5. [DOI] [PubMed] [Google Scholar]
- Fais S, De Milito A, You H, Qin W. Targeting vacuolar H+-ATPases as a new strategy against cancer. Cancer Res. 2007;67:10627–10630. doi: 10.1158/0008-5472.CAN-07-1805. [DOI] [PubMed] [Google Scholar]
- Finch N, Baker M, Crook R, Swanson K, Kuntz K, Surtees R, Bisceglio G, Rovelet-Lecrux A, Boeve B, Petersen RC, Dickson DW, Younkin SG, Deramecourt V, Crook J, Graff-Radford NR, Rademakers R. Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain. 2009;132:583–591. doi: 10.1093/brain/awn352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol. 2007;8:917–929. doi: 10.1038/nrm2272. [DOI] [PubMed] [Google Scholar]
- Ghidoni R, Benussi L, Glionna M, Franzoni M, Binetti G. Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology. 2008;71:1235–1239. doi: 10.1212/01.wnl.0000325058.10218.fc. [DOI] [PubMed] [Google Scholar]
- Gijselinck I, Van Broeckhoven C, Cruts M. Granulin mutations associated with frontotemporal lobar degeneration and related disorders: an update. Hum Mutat. 2008;29:1373–1386. doi: 10.1002/humu.20785. [DOI] [PubMed] [Google Scholar]
- Graff-Radford NR, Woodruff BK. Frontotemporal dementia. Semin Neurol. 2007;27:48–57. doi: 10.1055/s-2006-956755. [DOI] [PubMed] [Google Scholar]
- Griffiths G, Fuller SD, Back R, Hollinshead M, Pfeiffer S, Simons K. The dynamic nature of the Golgi complex. J Cell Biol. 1989;108:277–297. doi: 10.1083/jcb.108.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra RR, Kriazhev L, Hernandez-Blazquez FJ, Bateman A. Progranulin is a stress-response factor in fibroblasts subjected to hypoxia and acidosis. Growth Factors. 2007;25:280–285. doi: 10.1080/08977190701781222. [DOI] [PubMed] [Google Scholar]
- Hollingshead LM, Faulds D, Fitton A. Bepridil. A review of its pharmacological properties and therapeutic use in stable angina pectoris. Drugs. 1992;44:835–857. doi: 10.2165/00003495-199244050-00009. [DOI] [PubMed] [Google Scholar]
- Huss M, Wieczorek H. Inhibitors of V-ATPases: old and new players. J Exp Biol. 2009;212:341–346. doi: 10.1242/jeb.024067. [DOI] [PubMed] [Google Scholar]
- Huss M, Sasse F, Kunze B, Jansen R, Steinmetz H, Ingenhorst G, Zeeck A, Wieczorek H. Archazolid and apicularen: novel specific V-ATPase inhibitors. BMC Biochem. 2005;6:13. doi: 10.1186/1471-2091-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayasuga Y, Chiba S, Suzuki M, Kikusui T, Matsuwaki T, Yamanouchi K, Kotaki H, Horai R, Iwakura Y, Nishihara M. Alteration of behavioural phenotype in mice by targeted disruption of the progranulin gene. Behav Brain Res. 2007;185:110–118. doi: 10.1016/j.bbr.2007.07.020. [DOI] [PubMed] [Google Scholar]
- Kessenbrock K, Fröhlich L, Sixt M, Lämmermann T, Pfister H, Bateman A, Belaaouaj A, Ring J, Ollert M, Fässler R, Jenne DE. Proteinase 3 and neutrophil elastase enhance inflammation in mice by inactivating antiinflammatory progranulin. J Clin Invest. 2008;118:2438–2447. doi: 10.1172/JCI34694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koreeda A, Yonemitsu K, Kohmatsu H, Mimasaka S, Ohtsu Y, Oshima T, Fujiwara K, Tsunenari S. Immunohistochemical demonstration of the distribution of chloroquine (CQ) and its metabolites in CQ-poisoned mice. Arch Toxicol. 2007;81:471–478. doi: 10.1007/s00204-007-0185-6. [DOI] [PubMed] [Google Scholar]
- Lammich S, Schöbel S, Zimmer AK, Lichtenthaler SF, Haass C. Expression of the Alzheimer protease BACE1 is suppressed via its 5′-untranslated region. EMBO Rep. 2004;5:620–625. doi: 10.1038/sj.embor.7400166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Rademakers R. The molecular genetics and neuropathology of frontotemporal lobar degeneration: recent developments. Neurogenetics. 2007;8:237–248. doi: 10.1007/s10048-007-0102-4. [DOI] [PubMed] [Google Scholar]
- Mitterreiter S, Page RM, Kamp F, Hopson J, Winkler E, Ha HR, Hamid R, Herms J, Mayer TU, Nelson DJ, Steiner H, Stahl T, Zeitschel U, Rossner S, Haass C, Lichtenthaler SF. Bepridil and amiodarone simultaneously target the Alzheimer's disease beta- and gamma-secretase via distinct mechanisms. J Neurosci. 2010;30:8974–8983. doi: 10.1523/JNEUROSCI.1199-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657–668. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee O, Wang J, Gitcho M, Chakraverty S, Taylor-Reinwald L, Shears S, Kauwe JS, Norton J, Levitch D, Bigio EH, Hatanpaa KJ, White CL, Morris JC, Cairns NJ, Goate A. Molecular characterization of novel progranulin (GRN) mutations in frontotemporal dementia. Hum Mutat. 2008;29:512–521. doi: 10.1002/humu.20681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009;132:2922–2931. doi: 10.1093/brain/awp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riva E, Gerna M, Neyroz P, Urso R, Bartosek I, Guaitani A. Pharmacokinetics of amiodarone in rats. J Cardiovasc Pharmacol. 1982;4:270–275. doi: 10.1097/00005344-198203000-00016. [DOI] [PubMed] [Google Scholar]
- Ryan CL, Baranowski DC, Chitramuthu BP, Malik S, Li Z, Cao M, Minotti S, Durham HD, Kay DG, Shaw CA, Bennett HP, Bateman A. Progranulin is expressed within motor neurons and promotes neuronal cell survival. BMC Neurosci. 2009;10:130. doi: 10.1186/1471-2202-10-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez AM, Thomas D, Gillespie EJ, Damoiseaux R, Rogers J, Saxe JP, Huang J, Manchester M, Bradley KA. Amiodarone and bepridil inhibit anthrax toxin entry into host cells. Antimicrob Agents Chemother. 2007;51:2403–2411. doi: 10.1128/AAC.01184-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- Schmid D, Pypaert M, Münz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. 2007;26:79–92. doi: 10.1016/j.immuni.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schymick JC, Yang Y, Andersen PM, Vonsattel JP, Greenway M, Momeni P, Elder J, Chiò A, Restagno G, Robberecht W, Dahlberg C, Mukherjee O, Goate A, Graff-Radford N, Caselli RJ, Hutton M, Gass J, Cannon A, Rademakers R, Singleton AB, Hardiman O, Rothstein J, Hardy J, Traynor BJ. Progranulin mutations and amyotrophic lateral sclerosis or amyotrophic lateral sclerosis-frontotemporal dementia phenotypes. J Neurol Neurosurg Psychiatry. 2007;78:754–756. doi: 10.1136/jnnp.2006.109553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankaran SS, Capell A, Hruscha AT, Fellerer K, Neumann M, Schmid B, Haass C. Missense mutations in the progranulin gene linked to frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions reduce progranulin production and secretion. J Biol Chem. 2008;283:1744–1753. doi: 10.1074/jbc.M705115200. [DOI] [PubMed] [Google Scholar]
- Siddoway LA. Amiodarone: guidelines for use and monitoring. Am Fam Physician. 2003;68:2189–2196. [PubMed] [Google Scholar]
- Sleegers K, Brouwers N, Van Damme P, Engelborghs S, Gijselinck I, van der Zee J, Peeters K, Mattheijssens M, Cruts M, Vandenberghe R, De Deyn PP, Robberecht W, Van Broeckhoven C. Serum biomarker for progranulin-associated frontotemporal lobar degeneration. Ann Neurol. 2009;65:603–609. doi: 10.1002/ana.21621. [DOI] [PubMed] [Google Scholar]
- Solomon VR, Lee H. Chloroquine and its analogs: a new promise of an old drug for effective and safe cancer therapies. Eur J Pharmacol. 2009;625:220–233. doi: 10.1016/j.ejphar.2009.06.063. [DOI] [PubMed] [Google Scholar]
- Ullrich S, Münch A, Neumann S, Kremmer E, Tatzelt J, Lichtenthaler SF. The novel membrane protein TMEM59 modulates complex glycosylation, cell surface expression, and secretion of the amyloid precursor protein. J Biol Chem. 2010;285:20664–20674. doi: 10.1074/jbc.M109.055608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Van Hoecke A, Lambrechts D, Vanacker P, Bogaert E, van Swieten J, Carmeliet P, Van Den Bosch L, Robberecht W. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J Cell Biol. 2008;181:37–41. doi: 10.1083/jcb.200712039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Zee J, Le Ber I, Maurer-Stroh S, Engelborghs S, Gijselinck I, Camuzat A, Brouwers N, Vandenberghe R, Sleegers K, Hannequin D, Dermaut B, Schymkowitz J, Campion D, Santens P, Martin JJ, Lacomblez L, De Pooter T, Peeters K, Mattheijssens M, Vercelletto M, Van den Broeck M, Cruts M, De Deyn PP, Rousseau F, Brice A, Van Broeckhoven C. Mutations other than null mutations producing a pathogenic loss of progranulin in frontotemporal dementia. Hum Mutat. 2007;28:416. doi: 10.1002/humu.9484. [DOI] [PubMed] [Google Scholar]
- Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T, Ma X, Ma Y, Iadecola C, Beal MF, Nathan C, Ding A. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J Exp Med. 2010;207:117–128. doi: 10.1084/jem.20091568. [DOI] [PMC free article] [PubMed] [Google Scholar]